
Oxacalix[2]arene[2]triazine based ion-pair transporters†
Xu-Dong
Wang
,
Sen
Li
,
Yu-Fei
Ao
,
Qi-Qiang
Wang
,
Zhi-Tang
Huang
and
De-Xian
Wang
*
Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, CAS, Beijing, 100190, China. E-mail: dxwang@iccas.ac.cn
First published on 24th November 2015
Abstract
Heteracalixaromatics are a new generation of macrocyclic hosts showing a unique structure and versatile recognition properties towards various guests. Amazingly, the application of heteracalixaromatics as membrane transporters or ion channels has never been explored. We reported herein the elaborated design of a series of oxacalix[2]arene[2]triazine-based derivatives 1–10 and their ion transport properties. Among these compounds, 3, 8–10 can mediate the transport of chloride across the lipid bilayer of EYPC with activity (EC50) ranging from 0.43 to 8.23 μM. These compounds serve as ion carriers during the transport process, and the transport activity is both anion- and cation-dependent, suggesting a Cl−/M+ ion-pair transport model. Structure–activity studies indicate that hydrogen bonding, electron deficiency of the triazine rings, lipophilicity and macrocyclic frameworks are essential for ion transport.
Introduction
Ion transport across membranes catalyzed by proteins that serve as ion channels or carriers is one of the most important processes in living cells.1 The desire to understand the function and mechanism of proteins has inspired the development of synthetic ion channel models.2 Owing to their unique structure and facile functionalization, calixarenes and the related resorcinarenes have a rich history as transmembrane ion channels or transporters.3 Early work demonstrated the use of resorcinarenes and calixarenes as cation transporters or channels.3a,b Later on, several groups including Davis, Izzo and Tecilla reported the transport of anions through the phospholipid bilayer membrane with 1,3-alternate or paco calix[4]arene derivatives.2a,4 Very recently, Matile reported ditopic ion transport systems based on anion–π interactions and halogen bonds by taking cone calix[4]arene as a molecular platform.5 Besides the largely explored classical calixarenes, other calixaromatics such as calixpyrroles, in which pyrrole instead of benzene units are incorporated into the macrocyclic framework, have been intensively studied as anion transporters by the Gale and Sessler groups.6 In addition to the meta-bridged macrocycles, Hou and coworkers7 designed novel and versatile transmembrane channel models on the basis of pillararenes, a new type of para-bridged macrocycle emerged in 2008.8Heteracalixaromatics, or heteroatom bridged calix(het)arenes are a new type of macrocyclic molecule.9 In the traditional calixarene the cavity itself shows almost no recognition ability. The introduction of heteroatoms as bridging linkages and heteroaromatic moieties into heteracalixaromatics endowed the fine-tuned cavities and versatile recognition abilities. For example, these hosts can recognize various guest species, from metal ions and clusters,10 neutral molecules11 to anions.12 As the representative examples of heteracalixaromatics, oxacalix[2]arene[2]triazines, 1,3-alternate shape-persistent macrocyclic molecules bearing electron-deficient triazine rings, have been shown as unique anion acceptors based on anion–π interactions.12 Despite their versatile recognition properties, the application of heteracalixaromatics as transmembrane models has never been explored. Herein we report the use of a series of oxacalix[2]arene[2]triazine derivatives (Fig. 1) as transmembrane transporters. Based on the fluorescence assays, we present that compounds 3, 8–10 can mediate the ion pair transport across the lipid bilayers of EYPC. Hydrogen bonding, electron deficiency of the triazine rings, lipophilicity and macrocyclic frameworks are considered to be essential factors in order to mediate the effective ion transport.
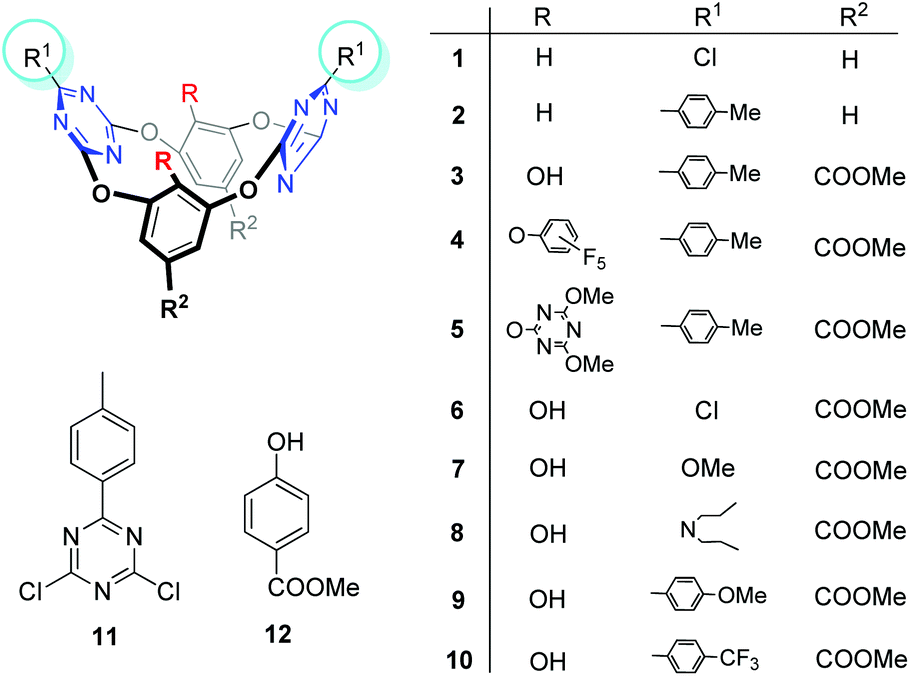 | ||
| Fig. 1 Structures of tetraoxacalix[2]arene[2]triazine derivatives 1–10, and the control compounds 11 and 12. | ||
Results and discussion
Ion transport activity measurements
Oxacalix[2]arene[2]triazine derivatives 1–10 (Fig. 1) were synthesized according to our reported procedures or as described in the Experimental part (ESI†).11b,13 To evaluate the anion transport activities of these compounds, we chose the fluorescence assays with the halide selective dye lucigenin as the probe.2a Briefly, the egg yolk phosphatidylcholine (EYPC) lipid film was rehydrated with an aqueous solution containing 10 mM HEPES, 1 mM lucigenin and 100 mM sodium nitrate. After freeze–thaw cycles, extrusions through a membrane with 100 nm pores and size exclusion chromatography to remove the external lucigenin, EYPC large unilamellar vesicles (LUVs) containing lucigenin were prepared (for experimental details, see the ESI†). A NaCl solution (100 mM) was added to the extravesicular buffer to create a chloride gradient. Upon addition of compounds 1–10, the fluorescence emission of intravesicular lucigenin was measured over time.Effect of substituents on the ion transport activity
The ion transport activity, as illustrated in Fig. 2, closely depends on the structure of these compounds. For example, compounds 1 and 2, which have been previously reported as good anion-π receptors,12a,b,14 show little ion transport activity. The lack of activity for 1 probably owes to its poor lipophilicity and thus it can't be inserted into the lipid membrane, as indicated by precipitation that occurred when 1 was injected into the buffer solution. Compound 2, with enhanced lipophilicity by introduction of the p-tolyl groups and no precipitation observed, still can't mediate the ion transport. In contrast, compound 3 with hydroxyl groups attached on the lower rim of benzene rings shows significant ion transport activity. Hill analysis of the dose–response curve gives an effective concentration EC50 = 4.31 μM and the Hill coefficient n = 2.9 (Table 1 and ESI†). According to our previous results,13c with OH groups on the lower rim, 3 can form cooperative hydrogen bonding and anion–π interactions with chloride and lead to an enhanced binding affinity. This outcome could contribute to the activity observed. | ||
| Fig. 2 Evaluation of the ion transport activity of transporters 1–10 by lucigenin⊂EYPC fluorescence assays. (A) All compounds and blank DMF, with the concentration for each compound is 5 μM except for 9 is 7.5 μM, (B) 3, (C) 8, (D) 9 and (E) 10 in varied concentrations. | ||
EC50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (μM) a (μM) |
n | 1H NMRb (ppm) | E (kcal mol−1) | K a(1:1) (L mol−1) | Retention timec (min) | |
|---|---|---|---|---|---|---|
|
a Effective concentration, the molar ratios of transporters to lipids are: 1:40 for 3, 1:60 for 8, 1:20 for 9, 1:400 for 10.
b Measured in DMSO-d6.
c Determined by HPLC, ODS column, mobile phase is acetonitrile containing 0.1% formic acid.
|
||||||
| 3 | 4.31 ± 0.60 | 2.9 ± 1.1 | 10.90 | 20.1 | 2.4 × 104 | 19.7 |
| 8 | 2.65 ± 0.62 | 1.9 ± 0.7 | 10.36 | 15.8 | 3.6 × 103 | 20.6 |
| 9 | 8.23 ± 0.68 | 2.8 ± 0.7 | 10.86 | 18.3 | 1.2 × 104 | 16.9 |
| 10 | 0.43 ± 0.19 | 1.5 ± 0.7 | 11.00 | 27.8 | 3.6 × 104 | 20.4 |
Encouraged by the transport activity observed for 3, the influence of the macrocyclic host structure on transport activity was further systematically evaluated. For example, when OH groups in 3 are replaced with perfluorophenoxy (4) or 3,5-dimethoxyl-1-triazinoxyl (5) the activity is negligible, suggesting that the OH groups are probably essential to mediate the ion transport. However, this speculation is argued by the little activity observed for 6 and 7, which still contain OH groups but the p-tolyl groups are replaced with –Cl and –OMe respectively. Considering the p-tolyl groups are more lipophilic than –Cl and –OCH3, it is envisioned that ion transport occurs when both hydrogen bonding and lipophilicity take effects. To verify this hypothesis, compound 8 with N,N-dipropyl groups attached on the triazine rings was investigated. 8 is more lipophilic than 3 as suggested by a longer retention time (t = 20.57 vs. 19.69 min) on an ODS-HPLC column (Table 1). Accordingly the obtained effective concentration show that 8 is more active than 3 (EC50 = 2.56 vs. 4.31 μM).
The influence of the electronic nature of the substituents on ion transport activity was found to be significant. For example, when electron donating groups such as p-OMe-phenyl were used to replace the p-tolyl groups in 3, the resulting compound 9 shows decreased activity (EC50 = 8.23 μM). However, with electron withdrawing groups such as p-CF3-phenyl, compound 10 gives increased activity with an EC50 value as low as 0.43 μM, about one order of magnitude lower than 3. According to our previous report,12a the substituent properties regulate the electron density of the triazine rings. With electron withdrawing substituents the electron density of triazine decreases and accordingly anion–π interactions strengthen. As listed in Table 1 and Fig. S12,† the surface potential of the triazine rings increases from the electron donating to electron withdrawing substituents (from 9, 3 to 10). This clearly demonstrates the considerable substituent effect on the π-deficiency of triazine rings. The association constants between 9, 3, 10 and chloride (as tetrabutylammonium salt, in acetonitrile) are 1.2 × 104, 2.4 × 104 and 3.6 × 104 M−1 respectively (Fig. S13–16†), following the order of the surface potentials of the triazine rings. As the hydrogen bonding ability of the hydroxyl groups on the benzene rings is less affected by the substituents on the triazine rings (could be judged from the very similar OH chemical shifts, δ = 10.86, 10.90, 11.00 ppm for 9, 3, 10, respectively, Table 1), the increased association constant hence mainly benefits from the electron deficiency of triazine rings. These outcomes indicate that the anion–π interaction probably makes one of the main contributions to the efficient ion transport activity for 10. In addition to the electron withdrawing nature, the hydrophobicity of CF3 is also considerable (retention time = 20.37 min), hence the contribution of lipophilicity to the high activity could not be excluded.
Based on the above structure-dependent ion transport activity, we believe that hydrogen bonding, lipophilicity and anion–π interactions are all essential driving forces for mediating effective ion transport. Although it's hard to distinguish which contribution is dominant, these contributions should take effect in a subtly balanced and cooperative way. Moreover, when control compounds 11 and 12 were used negligible ion transport activity was observed (Fig. S1†). This highlights the function of a cavity of this type of macrocyclic transporter.
Mechanism of the transport process
In order to investigate the transport mechanism, we first set up an assay using vesicles prepared with dipalmitoyl phosphatidylcholine (DPPC). As DPPC has a gel to liquid crystalline transition phase at 41 °C, transport assays at 45 °C and 25 °C were performed respectively. The dramatically reduced transport activity caused by the decrease of membrane fluidity of DPPC at 25 °C suggests that the ion transport process is governed by a carrier mechanism (Fig. S9a†).Then we performed a carboxyfluorescein (CF) release experiment to understand the transport process. CF is a self-quenched dye at a high concentration and hence it is weakly fluorescent when entrapped in the vesicles. Enhancement of CF fluorescence could be detected if the transporters cause the membrane defects of LUVs and lead to the release of CF to the diluted extravesicular buffer solution. As illustrated in Fig. S7,† addition of transporters 3, 8–10 to the suspension of LUVs containing CF shows no fluorescence enhancement at all, indicating that the transporters do not induce the membrane defects. To investigate whether the transport is through a Cl−/NO3− anti-port process, we prepared the vesicles containing lucigenin and sodium chloride. Sodium nitrate was added to the extravesicular buffer solution. In the control experiment, sodium sulfate was applied as the external salt solution. As shown in Fig. S8,† in both cases the intensity of quenched lucigenin is recovered with the release of chloride to the extravesicular buffer. Hence, the chloride efflux mediated by the transporters is independent of NO3− or SO42− contained in the external solution. As sulfate is an extremely hydrophilic anion and it is normally impossible to be transported from the aqueous solution to the lipid bilayer membrane, the results thus indicate less possibility of a Cl−/NO3− anti-port exchange.
The influence of the external cations (alkali metal) and anions (Cl− and Br−) on the transport activity was further studied. As illustrated in Fig. S6,† the transport activity (EC50) of 3, 8–10 is both anion- and cation-dependent. For example, the chloride transport is more active than bromide except for 9. For different cations, the activities of 3, 9 and 10 follow an overall sequence of Li+ < Na+ < K+ < Rb+ ≈ Cs+. Noticeably, 8 gives the almost reversed activity sequence. Although the exact reason is not very clear, one can speculate that it might be difficult for the larger cation to enter the cavity of 8 owing to the steric hindrance by the substituents. These outcomes indicate that the ion transport is most probably through an X−/M+ ion-pair process (Fig. 3). We also investigated the ion transport activity at pH 7.6 by taking compound 3 as a representative example. In principle, a higher pH value would facilitate the deprotonation of OH groups, and hence the enhancement of binding with cations. If the ion transport is dominated by cations, a decreased effective concentration should be obtained at a higher pH value (pH = 7.6). However, as shown in Fig. S10,† the calculated EC50 values are almost the same at pH 7.6 and pH 7.0, respectively (EC50 = 4.59 vs. 4.31 μM), suggesting less possibility of cation-dominated ion-pair transport. In combination with the aforementioned investigations, we postulated the ion-pair transport entity as: the transporters form a complex with the anion through cooperative anion–π interactions and hydrogen bonding, the cation as the counter ion forming a compact ion-pair with the complex (Fig. 3).
 | ||
| Fig. 3 Proposed schematic description of the ion-pair transport. | ||
To verify our postulation and get deep insights into the ion transport mechanism at the molecular level, we cultivated the single crystals of the complexes of the compounds and halide anions.‡ Fortunately, single crystals of the complex between 7 and bromide were obtained through slow evaporation of the solvent. Although 7 shows no transport activity, its structural similarity with the active transporters could provide detailed information on the host–guest interactions. As revealed by the crystal structure (Fig. 4A and B), 7 interacts with bromide through cooperative anion–π interactions and hydrogen bonding. Bromide locates over one of the triazine rings with the distances to the plane and centroid of the triazine ring being 3.266 Å (dBr1–plane) and 3.315 Å (dBr1–centroid) respectively, forming typical non-covalent anion–π interactions. Meanwhile the two hydroxyl groups interact with bromide through multiple hydrogen bonds. The tetraethylammonium cation locates outside the cavity and shows a short contact with bromide and oxygen atoms of one of the hydroxyl groups. Two such complexes, with the aid of intermolecular C–H⋯Br hydrogen bonds between OCH3 on the triazine rings and bromides, self-assemble into a dimeric structure (2 + 2) (Fig. 4B). Based on the crystal structure, we set up DFT modeling at the B3LYP/6-31G* level to speculate the complex structure of 3 and NaCl. DFT models give an energy favorable ion-pair complex (Fig. 4D). The optimized model shows that 3 binds Cl− through the aforementioned cooperative anion–π and hydrogen bonding interactions, meanwhile sodium as the counter cation seats at the outer rim of the cavity and interacts with one of oxygen atoms (dNa–O = 2.325 Å) through dipole-ion interactions (Fig. 4C). It is worth addressing that the ion-pair complex structure observed in both the crystal structure and DFT calculation all support our hypothesized ion-pair transport entity. Moreover, from the dimeric crystal structure and the Hill coefficient (n) obtained around 2 from Hill analysis of the dose–response curve, it is likely that the transporters mediate the transport of ions through a 2 + 2 dimeric complex.
 | ||
| Fig. 4 Crystal structure of (A) the complex of 7 and tetraethylammonium bromide (Et4N+(7·Br−)), (B) dimeric structure, DFT optimized structure of (C) side view and (D) top view of the complex of 3 and NaCl. | ||
Conclusions
We have designed a series of oxacalix[2]arene[2]triazine derivatives and studied their capabilities as ion transporters through the double layer membrane of EYPC. The fluorescence assays demonstrated that compounds 3, 8–10 can mediate the ion pair transport and act as transmembrane carriers. Hydrogen bonding, electron deficiency of the triazine rings, lipophilicity and macrocyclic frameworks were revealed to be essential for mediating ion transport. This work hence provides new dimensions of heteracalixaromatics as a molecular model of ion transporters or ion channels. Design of heteracalixaromatics-based ion channels is underway in our laboratory.Acknowledgements
We thank Prof. Stefan Matile and Dr Naomi Saikai from the University of Geneva for their helpful discussions. This work was financially supported by MOST (2013CB834504) and NNSFC (21272239, 91427301, 21521002).Notes and references
- B. Hille, Ionic Channels of Excitable Membranes, Sinauer, Sunderland, MA, 2nd edn, 1992 Search PubMed.
- For recent reviews on ion channels, see: (a) J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC; (b) H. Valkenier and A. P. Davis, Acc. Chem. Res., 2013, 46, 2898–2909 CrossRef CAS PubMed; (c) J. K. W. Chui and T. M. Fyles, Chem. Soc. Rev., 2012, 41, 148–175 RSC; (d) C. J. E. Haynes and P. A. Gale, Chem. Commun., 2011, 47, 8203–8209 RSC; (e) B. Gong and Z. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef CAS PubMed; (f) Y. Zhao, H. Cho, L. Widanapathirana and S. Zhang, Acc. Chem. Res., 2013, 46, 2763–2772 CrossRef CAS PubMed; (g) N. Sakai and S. Matile, Langmuir, 2013, 29, 9031–9040 CrossRef CAS PubMed.
- (a) Y. Tanaka, Y. Kobuke and M. Sokabe, Angew. Chem., Int. Ed. Engl., 1995, 34, 693–694 CrossRef CAS; (b) J. de Mendoza, F. Cuevas, P. Prados, E. S. Meadows and G. W. Gokel, Angew. Chem., Int. Ed., 1998, 37, 1534–1537 CrossRef CAS; (c) P. Schmitt, P. D. Beer, M. G. B. Drew and P. D. Sheen, Angew. Chem., Int. Ed. Engl., 1997, 36, 1840–1842 CrossRef CAS; (d) K. S. J. Iqbal and P. J. Cragg, Dalton Trans., 2007, 26–32 RSC.
- (a) V. Sidorov, F. W. Kotch, G. Abdrakhmanova, R. Mizani, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc., 2002, 124, 2267–2278 CrossRef CAS PubMed; (b) P. V. Santacroce, O. A. Okunola, P. V. Zavalij and J. T. Davis, Chem. Commun., 2006, 3246–3248 RSC; (c) J. L. Seganish, P. V. Santacroce, K. J. Salimian, J. C. Fettinger, P. Zavalij and J. T. Davis, Angew. Chem., Int. Ed., 2006, 45, 3334–3338 CrossRef CAS PubMed; (d) O. A. Okunola, J. L. Seganish, K. J. Salimian, P. Y. Zavalij and J. T. Davis, Tetrahedron, 2007, 63, 10743–10750 CrossRef CAS; (e) I. Izzo, S. Licen, N. Maulucci, G. Autore, S. Marzocco, P. Tecilla and F. De Riccardis, Chem. Commun., 2008, 2986–2988 RSC; (f) S. Licen, V. Bagnacani, L. Baldini, A. Casnati, F. Sansone, M. Giannetto, P. Pengo and P. Tecilla, Supramol. Chem., 2013, 25, 9–11 CrossRef.
- A. V. Jentzsch, D. Emery, J. Mareda, P. Metrangolo, G. Resnati and S. Matile, Angew. Chem., Int. Ed., 2011, 50, 11675–11678 CrossRef PubMed.
- (a) C. C. Tong, R. Quesada, J. L. Sessler and P. A. Gale, Chem. Commun., 2008, 6321–6323 RSC; (b) M. G. Fisher, P. A. Gale, J. R. Hiscock, M. B. Hursthouse, M. E. Light, F. P. Schmidtchen and C. C. Tong, Chem. Commun., 2009, 3017–3019 RSC; (c) P. A. Gale, C. C. Tong, C. J. E. Haynes, O. Adeosun, D. E. Gross, E. Karnas, E. M. Sedenberg, R. Quesada and J. L. Sessler, J. Am. Chem. Soc., 2010, 132, 3240–3241 CrossRef CAS PubMed; (d) M. Yano, C. C. Tong, M. E. Light, F. P. Schmidtchen and P. A. Gale, Org. Biomol. Chem., 2010, 8, 4356–4363 RSC; (e) S. J. Moore, M. G. Fisher, M. Yano, C. C. Tong and P. A. Gale, Dalton Trans., 2011, 40, 12017–12020 RSC.
- (a) W. Si, L. Chen, X.-B. Hu, G. Tang, Z. Chen, J.-L. Hou and Z.-T. Li, Angew. Chem., Int. Ed., 2011, 50, 12564–12568 CrossRef CAS PubMed; (b) X.-B. Hu, Z. Chen, G. Tang, J.-L. Hou and Z.-T. Li, J. Am. Chem. Soc., 2012, 134, 8384–8387 CrossRef CAS PubMed; (c) L. Chen, W. Si, L. Zhang, G. Tang, Z.-T. Li and J.-L. Hou, J. Am. Chem. Soc., 2013, 135, 2152–2155 CrossRef CAS PubMed; (d) W. Si, Z.-T. Li and J.-L. Hou, Angew. Chem., Int. Ed., 2014, 53, 4578–4581 CrossRef CAS PubMed.
- T. Ogoshi, S. Kanai, T. A. Fujinami and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- For reviews on heteracalixaromatics see: (a) M.-X. Wang, Chem. Commun., 2008, 4541–4551 RSC; (b) W. Maes and W. Dehaen, Chem. Soc. Rev., 2008, 37, 2393–2402 RSC; (c) H. Tsue, K. Ishibashi and R. Tamura, Top. Heterocycl. Chem., 2008, 17, 73–96 CAS; (d) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed; (e) J. Thomas, W. V. Rossom, K. V. Hecke, L. V. Meervelt, M. Smet, W. Maes and W. Dehaen, Chem. Commun., 2012, 48, 43–45 RSC; (f) B. König and M. H. Fonseca, Eur. J. Inorg. Chem., 2000, 2303–2310 CrossRef.
- (a) H.-Y. Gong, Q.-Y. Zheng, X.-H. Zhang, D.-X. Wang and M.-X. Wang, Org. Lett., 2006, 8, 4895–4898 CrossRef CAS PubMed; (b) H.-Y. Gong, D.-X. Wang, Q.-Y. Zheng and M.-X. Wang, Tetrahedron, 2009, 65, 87–92 CrossRef CAS; (c) C.-Y. Gao, L. Zhao and M.-X. Wang, J. Am. Chem. Soc., 2011, 133, 8448–8451 CrossRef CAS PubMed; (d) C.-Y. Gao, L. Zhao and M.-X. Wang, J. Am. Chem. Soc., 2012, 134, 824–827 CrossRef CAS PubMed.
- (a) H.-Y. Gong, D.-X. Wang, J.-F. Xiang, Q.-Y. Zheng and M.-X. Wang, Chem. – Eur. J., 2007, 13, 7791–7802 CrossRef CAS PubMed; (b) Q.-Q. Wang, D.-X. Wang, H.-B. Yang, Z.-T. Huang and M.-X. Wang, Chem. – Eur. J., 2010, 16, 7265–7275 CrossRef CAS PubMed.
- (a) D.-X. Wang, Q.-Y. Zheng, Q.-Q. Wang and M.-X. Wang, Angew. Chem., Int. Ed., 2008, 47, 7485–7488 CrossRef CAS PubMed; (b) D.-X. Wang and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 892–897 CrossRef CAS PubMed; (c) D.-X. Wang, Q.-Q. Wang, Y. Han, Y. Wang, Z.-T. Huang and M.-X. Wang, Chem. – Eur. J., 2010, 16, 13053–13057 CrossRef CAS PubMed.
- (a) M.-X. Wang and H.-B. Yang, J. Am. Chem. Soc., 2004, 126, 15412–15422 CrossRef CAS PubMed; (b) S. Li, S.-X. Fa, Q.-Q. Wang, D.-X. Wang and M.-X. Wang, J. Org. Chem., 2012, 77, 1860–1867 CrossRef CAS PubMed; (c) S. Li, D.-X. Wang and M.-X. Wang, Tetrahedron Lett., 2012, 53, 6226–6229 CrossRef CAS; (d) X.-D. Wang, D.-X. Wang, Z.-T. Huang and M.-X. Wang, Supramol. Chem., 2014, 26, 601–606 CrossRef CAS.
- W. Liu, Q.-Q. Wang, Z.-T. Huang and D.-X. Wang, Tetrahedron Lett., 2014, 55, 3172–3175 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1432620. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02291f |
| ‡ Crystallographic data for 2([7·Br−]Et4+) (CHCl3) (C65H77Br2Cl3N14O24): Mr = 1704.58, monoclinic, space group P2(1)/n, a = 21.688(6), b = 17.152(4), c = 22.729(5) Å, α = 90.00°, β = 115.043°, γ = 90.00°, V = 7660(3) Å3, T = 173(2) K, full-matrix least-squares refinement on F2 converged to RF = 0.1495 [I > 2σ(I)], 0.1745 (all data) and Rw(F2) = 0.3675 [I > 2σ(I)], 0.3798 (all data), goodness of fit 1.882. CCDC 1432620. |
| This journal is © The Royal Society of Chemistry 2016 |