
Accelerated cellular on- and off-target screening of bioactive compounds using microarrays†
Jiaqi
Fu
a,
Zhenkun
Na
a,
Bo
Peng
a,
Mahesh
Uttamchandani
*ab and
Shao Q.
Yao
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: chmyaosq@nus.edu.sg; Fax: +65 6770169; Tel: +65 65162925
bDefence Medical and Environmental Research Institute DSO National Laboratories, 27 Medical Drive, Singapore 117510, Singapore. E-mail: mahesh@nus.edu.sg
First published on 16th November 2015
Abstract
In situ proteome labeling was carried out with 9 drug-like probes in live mammalian cells, with the corresponding cellular targets captured on microarrays and simultaneously screened using a diverse set of antibodies, revealing potential on- and off-targets.
Introduction
Drug discovery is a long-drawn and expensive process, partly due to the unexpected behaviour and off-target binding spectrum of drug candidates.1 Often, the side-effects become evident only at the late stages beyond lead development, particularly during pre-clinical and clinical trials, when failures are costly. The ability to perform broad-based, proteome-wide screening at the outset of drug discovery introduces a new paradigm, where the target binding spectrum and key biological pathways affected are revealed and, if necessary, tuned upfront. Such a drug development approach will enhance candidate success, and also provide an informative toolbox for pre-empting multi-drug reactions and even facilitating combination drug therapy.2 In the last several years, by borrowing from concepts in the field of activity-based protein profiling (ABPP),3in situ drug profiling has become possible using drug-like chemicals minimally modified from their parental compounds. Such drug-like probes enable the large-scale interrogation of protein–small molecule interactions and can rapidly identify potential cellular targets, both on- and off-targets, in live cells.4 One of the main challenges, however, is with the target validation step (Fig. 1A; workflow on the right); upon protein labelling, pull-down (PD) experiments are usually required, followed by western blot (WB) analysis, to confirm the genuine in situ target–probe interactions. The process is cumbersome, expensive and low-throughput.5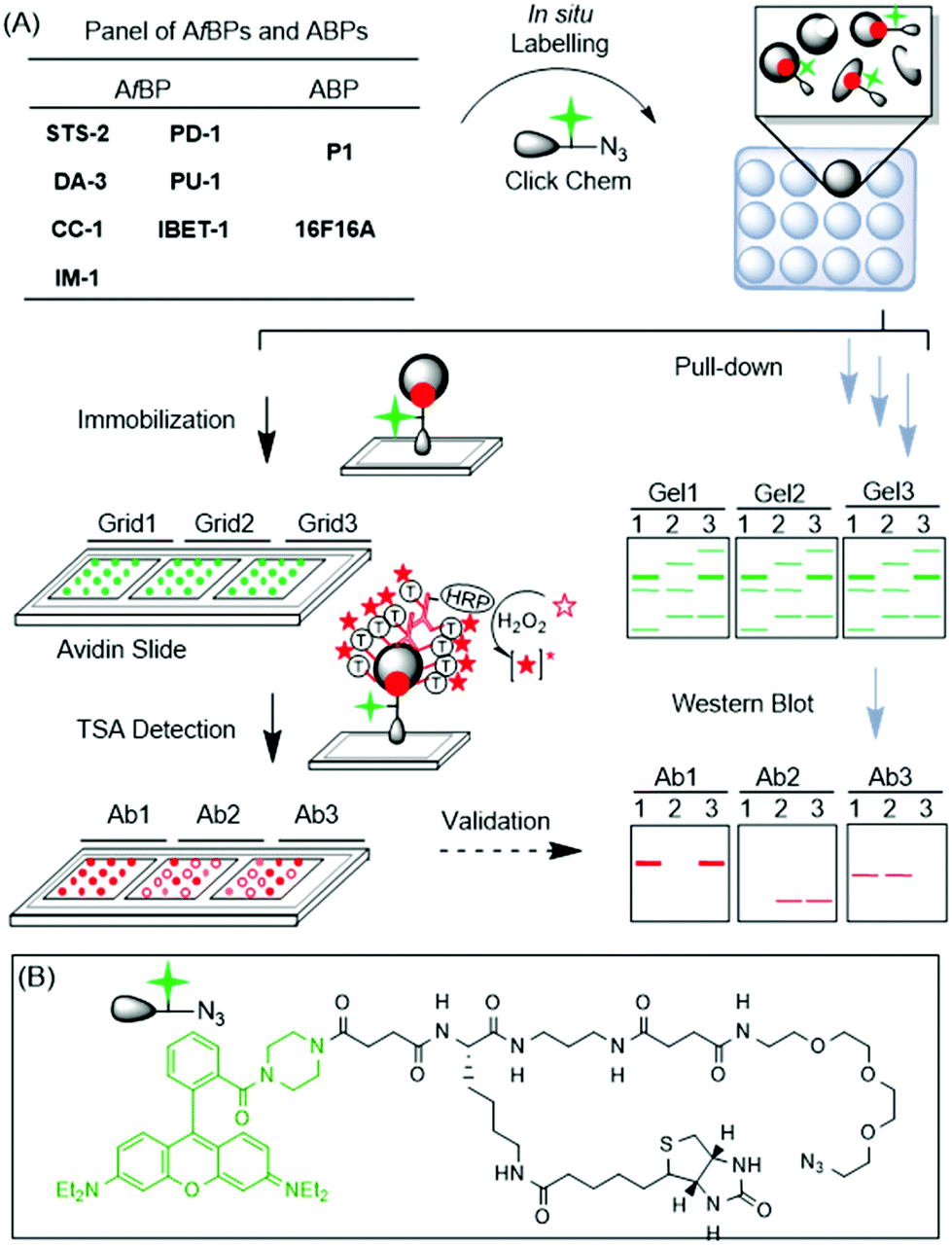 | ||
| Fig. 1 (A) Overall strategy of the microarray-guided, target profiling/validation of drug-like compounds. (Left) Following click reactions and immobilizations onto microarrays, the samples were probed with multiple antibodies (Abs) and detected by tyramide signal amplification (TSA). (Right) Traditional PD/WB workflow for target validation. (B) Structure of the tri-functional linker containing rhodamine, azide and biotin units. | ||
Microarrays are ideal for selectivity screening, because of their low cost, vast scalability and throughput.6,7 However, there exist no approaches for microarrays to contribute to functional drug selectivity screening in situ. Herein we report a variant of reverse phase protein microarrays,8 to unite in situ drug profiling and microarray approaches for the first time, in order to rapidly identify and/or validate on- and off-targets of bioactive compounds (Fig. 1A; left).
The proposed microarray target screening approach is depicted in Fig. 1A. 9 bioactive compounds were first minimally tagged with an alkyne handle, with or without an alkyl diazirine photo-crosslinker to generate the corresponding affinity-based probes (AfBPs) or activity-based probes (ABPs), respectively (Scheme 1).5,9 The AfBPs/ABPs were incubated with recombinant proteins, spiked cell lysates or living cells, to initiate protein labelling. Thereafter, the labelled reaction was clicked with an azide-containing trifunctional linker (Fig. 1B), using copper-catalyzed alkyne–azide cycloaddition (CuAAC).10 As the trifunctional linker also contained rhodamine and biotin units (for imaging and protein capture, respectively), the resulting samples could be directly spotted onto avidin-coated slides, where the labelled proteins were captured across multiple sub-grids. The arrays were then probed with antibodies (Abs), representing putative targets. To ensure high-sensitivity detection, tyramide signal amplification (TSA) was employed.11 Potential advantages of this strategy include (1) scalability and throughput in probing drug–target interactions in vivo, (2) simultaneous and quantitative comparisons across various dimensions (protein targets, compounds and cell types), and (3) ease of use with minimal quantities of reagents and antibodies required. Similar experiments, if conducted by classical PD experiments, followed by in-gel fluorescence scanning and WB analysis (Fig. 1A; right), remain cumbersome, and require large quantities of expensive antibodies.
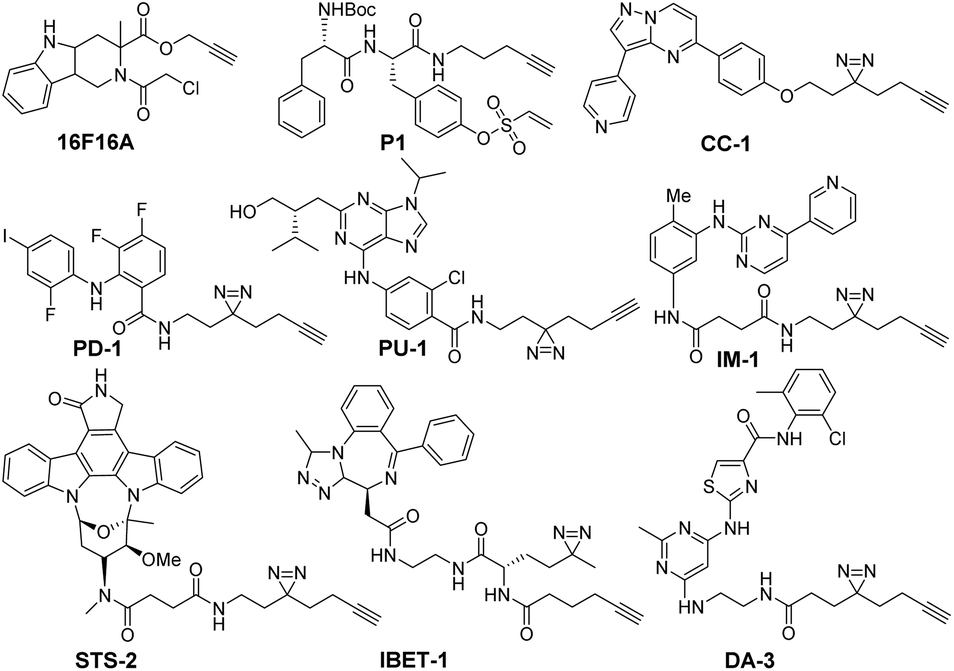 | ||
| Scheme 1 Structures of 9 minimally modified drug-like compounds for target profiling. All probes were previously reported.5c,9 | ||
The 9 probes used in this study were derivatives of bioactive compounds targeting protein kinases (STS-2, DA-3, PU-1, PD-1, IM-1, CC-1),5c protein disulfide isomerases (P1, 16F16A) and bromodomains (IBET-1) (Scheme 1).9 These classes of proteins are vital targets for a host of human diseases, ranging from cancer to neurodegenerative disorders. The compounds were modified minimally from the original inhibitor using an aliphatic alkyne linker, which in the case of reversible inhibitors also included a diazirine moiety to facilitate the covalent and UV dependent target capture. The linker was inserted at the solvent accessible site, which preserved the target binding properties of the compounds (Table S1, ESI†).5c,9
Results and discussion
To prove the concept, we first selected a protein tyrosine kinase, c-Src and its corresponding antibody, together with 3 AfBPs (STS-2, DA-3 and IBET-1). c-Src was expected to bind both STS-2 and DA-3 while IBET-1 was expected to exhibit low or negligible affinity towards this biological target. c-Src, at a concentration of 1 mg ml−1, was incubated with the probes. Following UV initiation and the click reaction,5 the excess unreacted probes were removed by acetone precipitation. The labelled samples were serially diluted and spotted onto avidin-coated glass slides in a concentration dependent manner (1, 10, 100, 1000, and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ng ml−1).12 Following a 2 h incubation, the slides were extensively washed with TBST (0.5% Tween20), probed with the anti-c-Src antibody, and visualized with the TSA-Cy5 detection system.13 The set of experiments were conveniently replicated across multiple subgrids on the same slide, with good reproducibility (Fig. 2A). The array results were also plotted graphically (right); c-Src was shown to be captured most effectively by STS-2, followed by DA-3, with readable signals even at the lowest 1 ng ml−1 concentration. At the highest concentration of c-Src used (i.e. 10000 ng ml−1), only slight signals were detectable with the IBET-1 probe. Nevertheless, there was a clear concentration-dependent trend (Fig. 2A). These binding profiles with each of the 3 probes were consistent with the in-gel fluorescence scanning results (Fig. 2A; bottom, Fig. S2†), which were obtained by detecting rhodamine from the trifunctional linker. While this initial experiment was able to produce good sensitivity, at the concentration ranges of proteins we might expect in vivo, we anticipated that the performance could vary in response to the sensitivity and specificity of antibodies used, and were hence selective in the choice of the antibodies we used (see Table S2, ESI†).
000 ng ml−1).12 Following a 2 h incubation, the slides were extensively washed with TBST (0.5% Tween20), probed with the anti-c-Src antibody, and visualized with the TSA-Cy5 detection system.13 The set of experiments were conveniently replicated across multiple subgrids on the same slide, with good reproducibility (Fig. 2A). The array results were also plotted graphically (right); c-Src was shown to be captured most effectively by STS-2, followed by DA-3, with readable signals even at the lowest 1 ng ml−1 concentration. At the highest concentration of c-Src used (i.e. 10000 ng ml−1), only slight signals were detectable with the IBET-1 probe. Nevertheless, there was a clear concentration-dependent trend (Fig. 2A). These binding profiles with each of the 3 probes were consistent with the in-gel fluorescence scanning results (Fig. 2A; bottom, Fig. S2†), which were obtained by detecting rhodamine from the trifunctional linker. While this initial experiment was able to produce good sensitivity, at the concentration ranges of proteins we might expect in vivo, we anticipated that the performance could vary in response to the sensitivity and specificity of antibodies used, and were hence selective in the choice of the antibodies we used (see Table S2, ESI†).
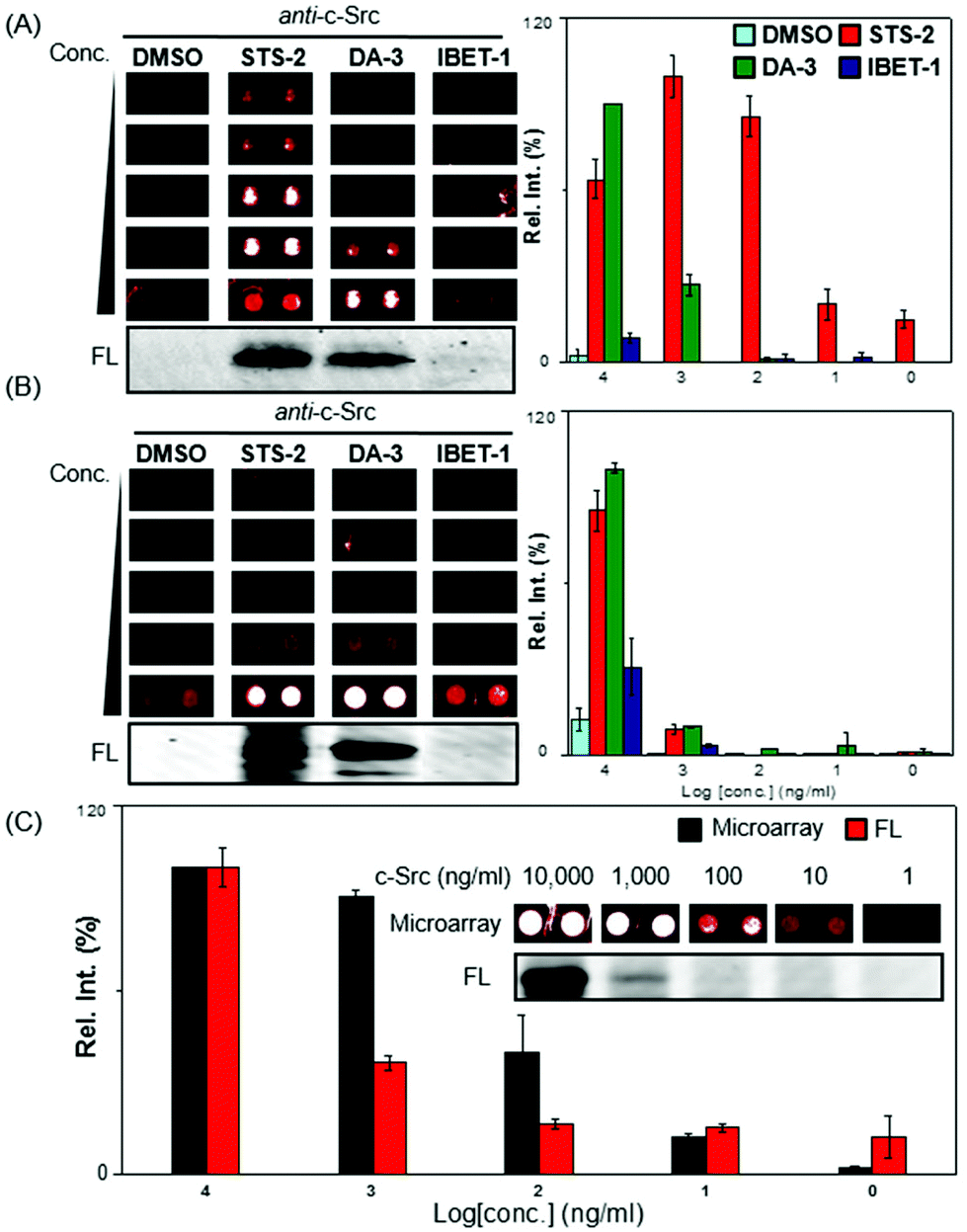 | ||
| Fig. 2 Probe binding profiles with c-Src on microarrays. (A) Recombinant c-Src was labelled with three probes (STS-2, DA-3, IBET-1) and a DMSO control, captured on the microarray and visualized with anti-c-Src Abs and a TSA Cy5 kit. Bottom to top (ng ml−1): 10000, 1000, 100, 10, and 1. The array was scanned under PMT 140. In-gel fluorescence scanning of the samples was analysed in a gel format (bottom left). (B) Identical experiments were carried out with HepG2 lysates (1 mg ml−1) spiked with the same concentration gradient of recombinant c-Src (10000, 1000, 100, 10, and 1 ng ml−1). The array was scanned under PMT 140. (C) Concentration-dependent gel and microarray results of recombinant c-Src labelled with DA-3. The corresponding fluorescent gel (inset) was imaged in the Cy3 channel. The error bars represent the standard deviations across two independent experimental replicates. The array was scanned under PMT 180. | ||
We next performed spike-in experiments, to assess the potential to perform this capture and profiling within the context of a complex cellular proteome. HepG2 lysates were spiked with recombinant c-Src at varying concentrations (1, 10, 100, 1000, and 10000 ng ml−1). Each of these mixtures was treated with 3 different probes respectively (Fig. 2B). Encouragingly, the results were generally consistent with those obtained with pure protein and in-gel fluorescence scanning. The background signals were evident with DMSO at 10000 ng ml−1 c-Src spiked concentrations, due to non-specific capture of proteins on the array even after extensive washing. However, the background was negligible at lower protein target concentrations (Fig. 2B). Both DA-3 and STS-2 were able to capture c-Src from complex cell lysates, with signals diminished with decreasing quantities of spiked protein. Under a higher PMT to improve the signal to noise ratio, it was observed that as low as 10 ng ml−1 of c-Src in the lysate could successfully be detected with DA-3 by using the array strategy (Fig. 2C).
We next compared the sensitivity of the microarray system with that of the fluorescent gel. The sensitivity attained on the microarray was around 10 ng mL−1, with spots distinguishable at this concentration (Fig. 2C, inset). This appeared to be one order of magnitude higher than the detectable bands imaged on fluorescent gels (Fig. 2C and S3, ESI†), and is likely attributable to the TSA strategy adopted, which offered signal enhancement. In addition, the spectrum of the c-Src profiles in lysate screened with the other 6 probes remained were largely within expectations. Another kinase probe (IM-1) was able to capture c-Src and showed detectable signals on the arrays (Fig. S4A and B, ESI†). In addition, P1 a probe that was designed for PDIs, was capable of binding c-Src within cell lysates, while PU-1 and 16F16A presented weaker binding with c-Src. PD-1 and CC-1 generally did not bind c-Src, even at high protein concentrations.
Having demonstrated that we could successfully adopt this strategy in vitro with c-Src, we went ahead also to test this approach in live cells. To increase the captured targets as much as possible, we used our compounds at a high concentration (10 μM), with HepG2 cells under standard growth conditions. The cells were then isolated and lysed. The resulting labelled proteome was immobilized onto the slides at an optimal concentration (2 mg ml−1) and detected against a repertoire of six antibodies. The complete array result is summarized as a coloured heatmap (Fig. S5, ESI†). Based on these results, some initial conclusions could be drawn.
Firstly, each probe displayed unique binding affinity to the corresponding targets. Secondly, most probes exhibited a potent binding ability with their potential targets. For instance, PU-1 and DA-3 successfully captured their respective targets CDK1 and c-Src (Fig. 3). STS-2 appeared to capture a range of kinases (PKA, CDK1, c-Src and MEK1), as should be expected.1416F16A successfully bound to its target PDI, as did P1. CC-1 consistently exhibited weak binding against the range of protein targets.
 | ||
| Fig. 3 (A) Microarray image of 6 probes profiling against 4 selective antibodies. (B) Pull-down result of the corresponding probes for target validation. (C) Preliminary off-target validation for P1 (left) and PU-1 (right). Concentration dependent labelling of P1 and PU-1 with recombinant PKA. | ||
To further evaluate the accuracy of the heatmap obtained from microarrays, in situ labelling and PD/WB target identification with 4 of the antibodies and six of the probes were carried out under the same conditions (Fig. 3A and B). The results were largely consistent, as visualized in the similar heatmap patterns. For instance, both STS-2 and DA-3 exhibited interactions with c-Src. This was consistent with the spike-in experiments. Apart from c-Src, STS-2, also interacted with PKA and CDK1, as is characteristic of staurosporine, being a broad-spectrum kinase inhibitor. P1 and 16F16A, as the strongest PDI probes based on the microarray results, exhibited a strong interaction with PDI in the PD experiment while the rest of the probes consistently showed weak binding to PDI, confirming the microarray result. In addition, P1 also exhibited interactions with various kinases including PKA, c-Src and CDK1. P1 displayed a much wider spectrum of interactions than that initially expected, potentially due to its electrophilic phenyl vinyl sulfonate. Similarly, the results of PD-1 and PU-1 from WB matched those from the microarray. In particular, PD-1 bound CDK1. PU-1 as a probe for CDK1, exhibited binding interactions with PKA and c-Src as well. With these preliminary results, we picked up both ABP (P1) and AfBP (PU-1) for further validation. As for P1, in vitro concentration dependent labelling of recombinant PKA was carried out. As shown in Fig. 3C, P1 was able to label PKA, with signals detectable even at low concentrations of 0.1 μM, suggesting strong interactions between P1 and PKA. Similarly, PU-1 bound with recombinant PKA at a low concentration of 0.2 μM (Fig. 3).
Conclusions
In conclusion, we have developed the first microarray-based platform to our knowledge, capable of rapidly identifying on- and off-targets of drug-like compounds from live cells. Compared to gel-based scans and WB, microarrays consume fewer samples but can sensitively detect endogenous proteins. In this work, we have successfully profiled a panel of drug-like ABP and AfBP bioactive compounds against various protein targets, and have likely identified the off-targets of ABP (P1) and AfBP (PU-1). It should be highlighted that the quality of the antibodies used are critical to ensure robust interpretations of only the target proteins in the array. Overall, this microarray-based strategy would enhance and complement high-content screening approaches, for early stage lead-optimization, and off-target screening, in drug discovery applications. A single experiment thus performed on a microarray slide could replace dozens of WBs/PDs, and consequently, accelerate target profiling.Experimental details
General information
All probes were synthesized as reported.5,9 Other chemicals were purchased from vendors and used without further purification, unless indicated otherwise. HBTU, HOBT, and EDC were purchased from GL Biochem (China). Tyramide signal amplification kits were purchased from Invitrogen (USA). Gels were scanned on a Typhoon fluorescence gel scanner (GE Healthcare, USA). Microarray slides were scanned using a Tecan Launch LS Reloaded Microarray Scanner (Tecan Trading AG, Switzerland) installed with suitable lasers: Cy3: λEx/Em = 532/575 nm; and Cy5: λEx/Em = 633/692 nm.Expression and labelling of proteins
Proteins were purified from bacterial cell lysates, as described.15 The pre-cultures of LB with a single colony containing c-Src (32 kDa) were grown at 37 °C to reach OD600 0.6–0.8. The expression was then induced by using 0.1 mM IPTG and the cultures were further grown at 18 °C for 18 h. The harvested pellets were resuspended in lysis buffer and incubated for 20 min. After sonication, the insoluble cell debris was removed by centrifugation. The resulting supernatant was incubated with Ni-NTA beads for 2 h at 4 °C. After washing with wash buffer, the target proteins were eluted in elution buffer. The desired proteins were dialysed with a Microcon® centrifuge filter device and stored at −20 °C in Hepes containing 20% glycerol.Pure protein labelling experiment
1 mg ml−1 pure protein (c-Src) was incubated with 10 μM probes for 2 h at room temperature following UV irradiation for 20 min for AfBPs. The resulting proteins were tagged with TER-biotin-N3via click chemistry. After 2 h of the click reaction, the labelled proteins were washed with acetone (1×) and methanol (2×) to remove the excess dye. After washing, 2% SDS in TBS was added to dissolve proteins; the final concentration was around 1 mg ml−1. In-gel fluorescence scanning was used to visualize the labelled protein bands. Both in-gel fluorescence scanning (FL) and Coomassie brilliantblue staining (CBB) were always carried out on the gels, following separation of the labelled samples by SDS-PAGE.Spike in experiment and in situ proteome labelling
The procedure is similar to our previous reported methods but with further modification as described.9 For spike-in labelling, the probes were added to a final concentration of 10 μM into 1 mg ml−1 of a fresh cell lysate with spiked proteins and incubated for 2 h at room temperature. For activity-based probes, 20 min UV irradiation was required. The resulting lysate was tagged with TER-biotin-N3via click chemistry. The reactions were further incubated for 2 h at room temperature with gentle mixing before the reaction was terminated by addition of acetone. The precipitated proteins were subsequently washed with methanol twice to remove the excess dye followed by dissolving in TBS with 2% SDS. Around 20 μg proteins were separated by SDS-PAGE gel and then visualized by in-gel fluorescence scanning. For in situ labelling, the cells were grown between 80% and 90% confluence. The growth medium was removed and the cells were washed with PBS twice. Then the cells were treated with DMEM containing 10 μM probes. After 5 h incubation at 37 °C/5% CO2, the medium was aspirated and the cells with activity-based probes were irradiated by UV for 20 min on ice. The cells were trypsinized and spun down. The resulting cell pellets were resuspended in lysis buffer and homogenized by sonication, and brought to a final concentration of 4 mg ml−1 in Hepes buffer. All subsequent procedures were performed in the same way as described for the in vitro labelling experiments.Array-based target identification and validation
Avidin slides were prepared as previously reported.11 The slides were blocked with 1% BSA for 1 h at room temperature before the sample was spotted to reduce non-specific binding as much as possible. The labelled proteins were dissolved in Spotting Buffer (20% DMSO in TBS) in a 1:1 ratio and spotted onto the avidin slides. Spike-in and in situ labelling samples were spotted onto slides with a final concentration of 2 mg ml−1. After incubation for 2 h, the avidin slides were washed with 0.5% TBST 6 times, 5 min per time. The slides with three sub-grids were first incubated with different antibodies separately for 1.5 h at room temperature. After washing with TBST, the avidin slides were incubated with a secondary antibody for 1 h at room temperature followed by washing with TBST. The targets were identified by using Cy5 tyramide signal amplification kits according to the manufacturer's protocol with slight modifications. Generally, tyramide was diluted into 1:400 with amplification buffer containing 0.015% H2O2. A 200 μl reagent volume was applied onto each slide under a cover slip with a 15 min exposure time at room temperature. After development, the resulting slides were washed with TBST 5 times and scanned by using a Tecan Launch LS Reloaded Microarray Scanner in the Cy5 channel. The slides were generally scanned under a PMT setting of 140.
Data extraction and analysis
Microarray data was extracted using the Array-Pro®software. The values from the duplicated points were background subtracted and averaged. For each antibody, the signals were first normalized by its mean and subtracted by DMSO treated controls. The coloured array heatmaps were produced using Cluster 3.0 and Java Treeview 3.0 (http://sourceforge.net/projects/jtreeview/files).Pull-down experiment
To identify the potential cellular targets and off-targets of the probes and their parental compounds, pull-down (PD) experiments were carried out, followed by western blotting (WB). The general pull-down procedure was based on previously reported procedures.5In situ labelling was carried out as described above. After labelling, the reaction was reacted by click chemistry with TER-biotin-N3 before acetone precipitated, washed with methanol and resolubilized in 2% SDS in TBS with brief sonication. This resuspended sample was then incubated with avidin-agarose beads (100 μl per mg protein) for 3 h at room temperature. After centrifugation, the supernatants were removed. The beads were washed with 5% SDS once and 1% SDS PBS four times. After washing, the beads were boiled in 1× SDS loading buffer (200 mM Tris pH 6.8, 400 mM DTT, 8% SDS) for 15 min. Control PD using DMSO was carried out concurrently. WB experiments were carried out as previously described using the corresponding antibodies. The results are shown in Fig. 3B.Recombinant PKA labelling
1 μg ml−1 pure protein (PKA) was incubated with probes (P1 and PU-1) of varying concentrations (0.1, 0.2, 0.5, 1, 2, and 5 μM) for 2 h at room temperature before UV irradiation for 20 min for AfBPs. The resulting proteins were tagged with TER-biotin-N3via click chemistry for 2 h. SDS-PAGE gel profilings were carried out for the sample without further purification before silver staining. The results are shown in Fig. 3C.Acknowledgements
Funding support was provided by the Singapore National Medical Research Council (CBRG/0038/2013) and the Ministry of Education (MOE2013-T2-1-048).Notes and references
- S. Ziegler, V. Pries, C. Hedberg and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 2744 CrossRef PubMed.
- (a) G. M. Simon, M. J. Niphakis and B. F. Cravatt, Nat. Chem. Biol., 2013, 9, 200 CrossRef PubMed; (b) A. C. Dar, T. K. Das, K. M. Shokat and R. L. Cagan, Nature, 2012, 486, 80 CrossRef CAS PubMed; (c) B. R. Lanning, L. R. Whitby, M. M. Dix, J. Douhan, A. M. Gilbert, E. C. Hett, T. O. Johnson, C. Joslyn, J. C. Kath, S. Niessen, L. R. Roberts, M. E. Schnute, C. Wang, J. J. Hulce, B. Wei, L. O. Whiteley, M. M. Hayward and B. F. Cravatt, Nat. Chem. Biol., 2014, 10, 760 CrossRef CAS PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383 CrossRef CAS PubMed.
- (a) J. Eder, R. Sedrani and C. Wiesmann, Nat. Rev. Drug Discovery, 2014, 13, 577 CrossRef CAS PubMed; (b) Y. Su, J. Ge, B. Zhu, Y. Zheng, Q. Zhu and S. Q. Yao, Curr. Opin. Chem. Biol., 2013, 17, 768 CrossRef CAS PubMed; (c) C. Harrison, Nat. Rev. Drug Discovery, 2014, 13, 102 CrossRef PubMed.
- (a) P.-Y. Yang, K. Liu, M. H. Ngai, M. J. Lear, M. Wenk and S. Q. Yao, J. Am. Chem. Soc., 2010, 132, 656 CrossRef CAS PubMed; (b) H. Shi, C. Zhang, G. Y. J. Chen and S. Q. Yao, J. Am. Chem. Soc., 2012, 134, 3001 CrossRef CAS PubMed; (c) Z. Li, P. Hao, L. Li, C. Y. J. Tan, X. Cheng, G. Y. J. Chen, S. K. Sze, H. Shen and S. Q. Yao, Angew. Chem., Int. Ed., 2013, 52, 8551 CrossRef CAS PubMed.
- H. Sun, G. Y. J. Chen and S. Q. Yao, Chem. Biol., 2013, 20, 685 CrossRef CAS PubMed.
- S. A. Sieber, T. S. Mondala, S. R. Head and B. F. Cravatt, J. Am. Chem. Soc., 2004, 126, 15640 CrossRef CAS PubMed.
- M. Sevecka and G. MacBeath, Nat. Methods, 2006, 3, 825 CrossRef CAS PubMed.
- (a) J. Ge, C. Zhang, L. Li, X. Wu, P. Hao, S. K. Sze and S. Q. Yao, ACS Chem. Biol., 2013, 8, 2577 CrossRef CAS PubMed; (b) Z. Li, D. W. L. Li, S. Pan, Z. Na, C. Y. J. Chen and S. Q. Yao, J. Am. Chem. Soc., 2014, 136, 9990 CrossRef CAS PubMed.
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed; (b) K. A. Kalesh, H. Shi, J. Ge and S. Q. Yao, Org. Biomol. Chem., 2010, 8, 1749 RSC.
- H. Wu, J. Ge, P.-Y. Yang, J. Wang, M. Uttamchandani and S. Q. Yao, J. Am. Chem. Soc., 2011, 133, 1946 CrossRef CAS PubMed.
- M. Uttamchandani, W. L. Lee, J. Wang and S. Q. Yao, J. Am. Chem. Soc., 2007, 129, 13110 CrossRef CAS PubMed.
- http://www.perkinelmer.com/ .
- M. A. Fabian, W. H. Biggs, D. K. Treiber, C. E. Atteridge, M. D. Azimioara, M. G. Benedetti, T. A. Carter, P. Ciceri, P. T. Edeen, M. Floyd, J. M. Ford, M. Galvin, J. L. Gerlach, R. M. Crotzfeld, S. Herrgard, D. E. Insko, M. A. Insko, A. G. Lai, J. M. Lelias, S. A. Mehta, Z. V. Milanov, A. M. Velasco, L. M. Wodicka, H. K. Patel, P. P. Zarrinkar and D. J. Lockhart, Nat. Biotechnol., 2005, 3, 329 CrossRef PubMed.
- K. Liu, K. A. Kalesh, L. B. Ong and S. Q. Yao, ChemBioChem, 2008, 9, 1883 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, gel and microarray images, list of antibodies and probe designs. See DOI: 10.1039/c5ob02233a |
| This journal is © The Royal Society of Chemistry 2016 |