
Synthesis of 3-substituted isoindolin-1-ones via a tandem desilylation, cross-coupling, hydroamidation sequence under aqueous phase-transfer conditions†
Socrates B.
Munoz
a,
Alexandra N.
Aloia
a,
Alexander K.
Moore
a,
Attila
Papp
a,
Thomas
Mathew
a,
Santos
Fustero
b,
George A.
Olah
a and
G. K.
Surya Prakash
*a
aLoker Hydrocarbon Research Institute and Department of Chemistry, University of Southern California, University Park, Los Angeles, CA-90089-1661, USA. E-mail: gprakash@usc.edu
bDepartamento de Quimica Organica, Universidad de Valencia and Laboratorio de Moleculas Organicas, Centro de Investigacion Principe Felipe, E-46012 Valencia, Spain
First published on 24th November 2015
Abstract
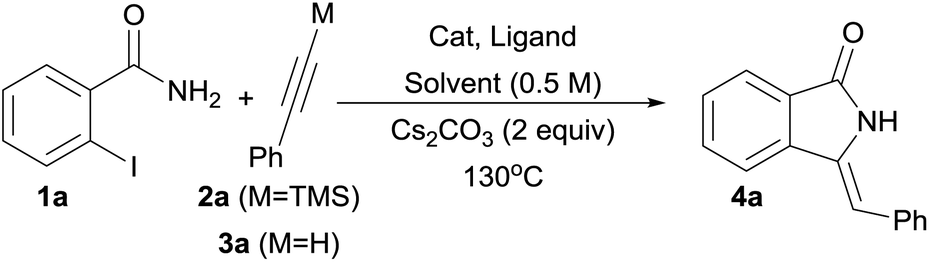
A simple and expedient method for the synthesis of 3-methylene-isoindolin-1-ones 4 under aqueous phase-transfer conditions has been developed. Starting from 2-iodobenzamides 1 and (silyl)alkynes, the products are obtained in high yields and short reaction times (30 min) with the use of inexpensive CuCl/PPh3 catalyst system in the presence of n-Bu4NBr (TBAB) as a phase-transfer agent. Terminal alkynes are conveniently “unmasked” upon in situ desilylation under the reaction conditions. Alkynes possessing heterocyclic moieties were also found as amenable substrates. Furthermore, a one-pot process starting from 2-iodobenzamides 1, aryl halides (bromides or iodides) and trimethylsilylacetylene (TMSA) as a convenient acetylene surrogate was also shown to be feasible under Pd/Cu catalysis.
Introduction
Isoindolin-1-ones are preeminent amongst the heterocyclic scaffolds found in both naturally occurring products1 as well as designed medicinal agents.2 They possess many biological activities varying from antihypertensive,3 antipsychotic,4 anesthetic5 to anxiolytic, antiviral,6 and antileukemic7 effects (Fig. 1).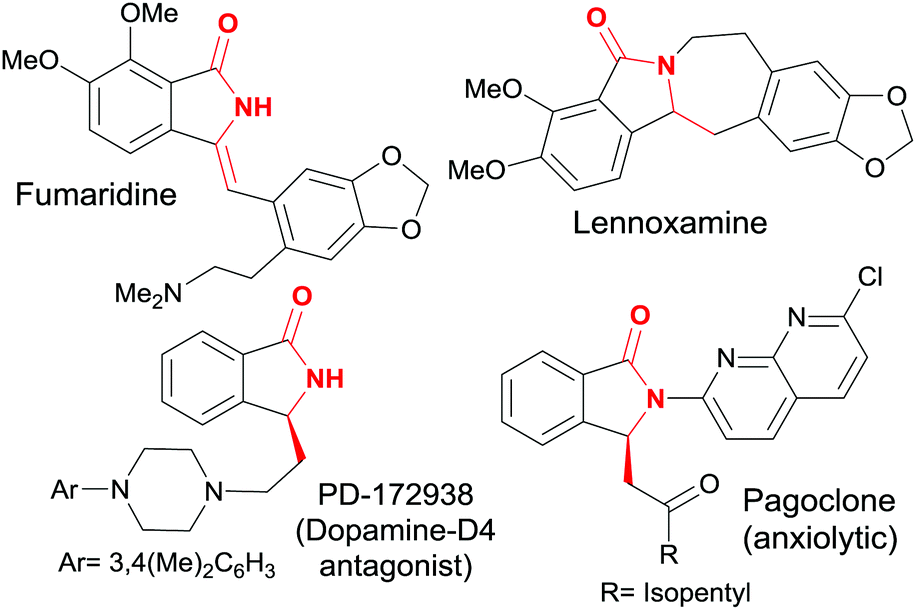 | ||
| Fig. 1 Structure of some biologically relevant isoindolin-1-ones. | ||
Accordingly, several methods for their preparation have been developed, some of which include the Wittig reaction of phtalimide derivatives with phosphorous ylides,8 organometallic reagents9 or benzylic radical donors10 and subsequent dehydration of the obtained 3-hydroxy-isoindolinone. A condensation of aldehydes with a preassembled phosphorylated derivative (prepared in three steps) via a Wittig–Horner reaction was also reported by Couture and coworkers.11 A more recent approach involves the Pd catalyzed elimination–cyclization–Suzuki coupling sequence.12 However, the preparation of the required o-gem-dibromovinylbenzamides is not straightforward (Scheme 1, path A).
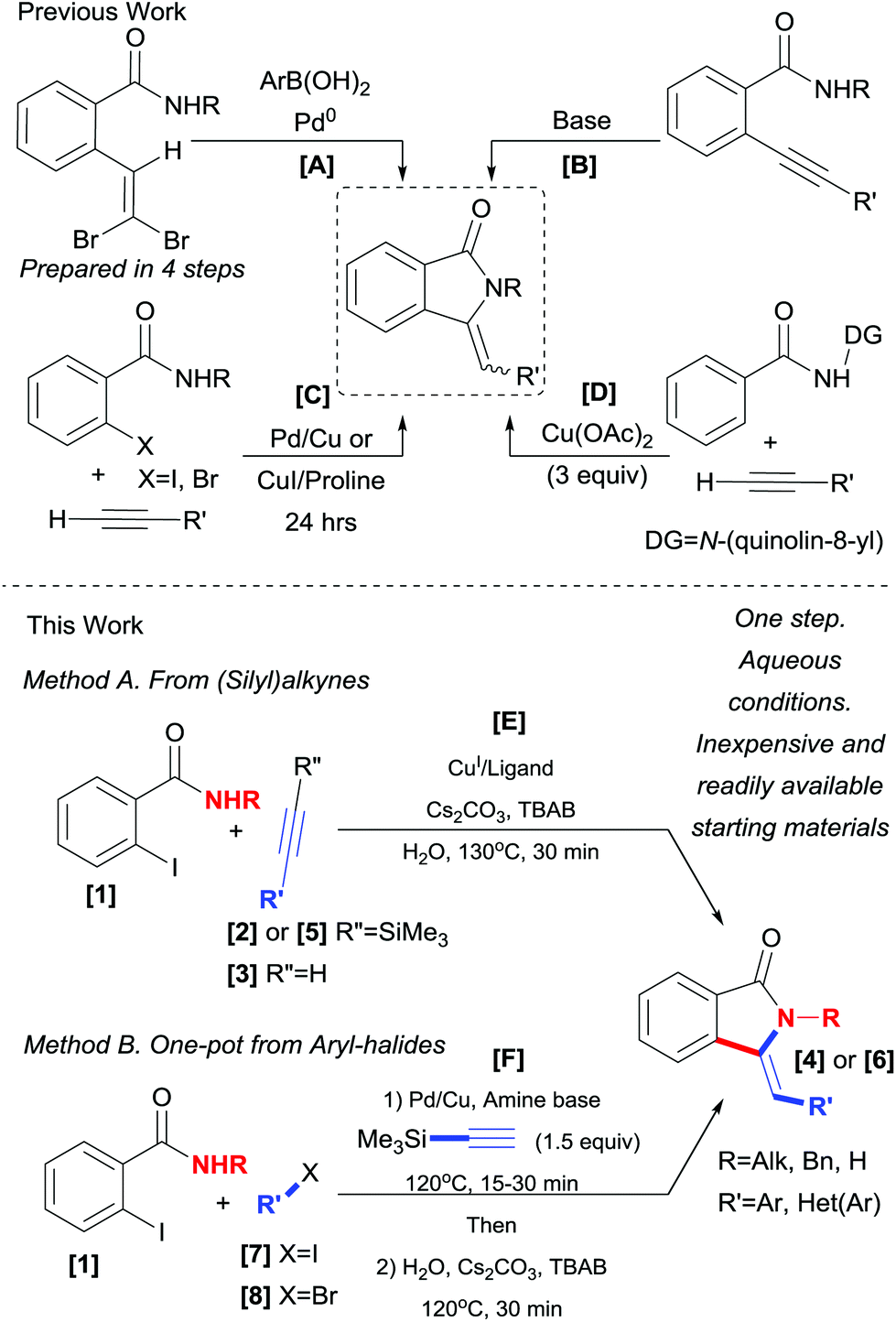 | ||
| Scheme 1 Selected methods for the synthesis of 3-methylene-Isoindolinone and present work. | ||
An alternative strategy for the assembly of the target molecules was first introduced by Kondo et al. in which an o-alkynylbenzamide was subjected to a Pd catalyzed oxidative cyclization–alkoxycarbonylation sequence to obtain 3-[(alkoxycarbonyl)methylene]-isoindolin-1-ones in modest yields (55%).13 Very recently, Fustero and coworkers,14 disclosed a Pd catalyzed aminocarbonylation approach for the asymmetric synthesis of fluorinated isoindolinones and in 2008 Alper and coworkers15 delineated an elegant four component approach in which an o-alkynylbenzamide was the likely key intermediate. In fact, due to their ease of access, o-alkynylbenzamide moieties have been exploited by several groups to access unsaturated isoindolinones upon treatment with strong bases.16 However, this approach suffers from lack of regio- and stereoselectivity (6-endo-dig cyclization product, isoquinolin-1(2H)-ones, could also be obtained in some cases). In addition, the need to pre-synthesize and isolate the necessary o-alkynylbenzamides has rendered these protocols less practical (Scheme 1, path B).
To circumvent these limitations, several tandem cross-coupling cyclization sequences have appeared in the literature. Starting from o-halobenzamides (X = Br or I) and terminal alkynes, several Pd/Cu,17 Pd,18 and Cu19 catalyzed/meditated protocols have been reported (Scheme 1, path C). Among these methods, reports using inexpensive copper-based catalysts are quite attractive. Nevertheless, these methods still require the use of organic media and/or long reaction times (24–36 h).
Recently, an important protocol was reported by You and coworkers19c in which N-(quinolin-8-yl)benzamides were cross-coupled with terminal alkynes under oxidative conditions mediated by a Cu(II) salt. While this method has the advantages of direct C–H bond functionalization, it is nonetheless, exclusively applied to benzamides bearing N-(quinolin-8-yl) directing group, which is not easily removable. In addition to long reaction times (24 h), super-stoichiometric amounts of Cu(OAc)2 (3 equiv.) were required to achieve good yields (Scheme 1, path D).
Since the pioneering work by Hiyama20 and Denmark,21 owing in part to their stability, non-toxicity and natural abundance of silicon, much attention has been paid to the organic chemistry of organosilicon compounds (silanes and silanols), particularly to their utilization in metal catalyzed cross-coupling reactions and accordingly, several publications have emerged.22,23
While several reports on the synthesis of 3-methylene-isoindolin-1-ones 4via tandem cross-coupling and cyclization sequences utilizing terminal alkynes 3 have been reported,17–19 the use of silylated derivatives 2 (TMS), to the best of our knowledge, has not been reported thus far. Herein, we describe a triple tandem desilylation, cross-coupling and hydroamidation approach for the synthesis of 3-methylene-isoindolin-1-ones 4 under aqueous phase-transfer conditions from (silyl)alkynes (Scheme 1, path E). A one-pot protocol starting from Ar–X (X = I, Br) is also disclosed (Scheme 1, path F).
Alkynylsilanes are conveniently and inexpensively accessed by the Sonogashira24 coupling of Aryl–X (X = I, Br, Cl, OTf)25 with easy-to-handle trialkylsilylacetylene derivatives R3Si–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH TMSA (R = Me) or TIPSA (R = iPr) which serve as convenient liquid acetylene surrogates. Terminal alkynes are then “unmasked” upon treatment with inorganic base or fluoride in a protic solvent and can be further subjected to cross-coupling conditions (Scheme 2).
CH TMSA (R = Me) or TIPSA (R = iPr) which serve as convenient liquid acetylene surrogates. Terminal alkynes are then “unmasked” upon treatment with inorganic base or fluoride in a protic solvent and can be further subjected to cross-coupling conditions (Scheme 2).
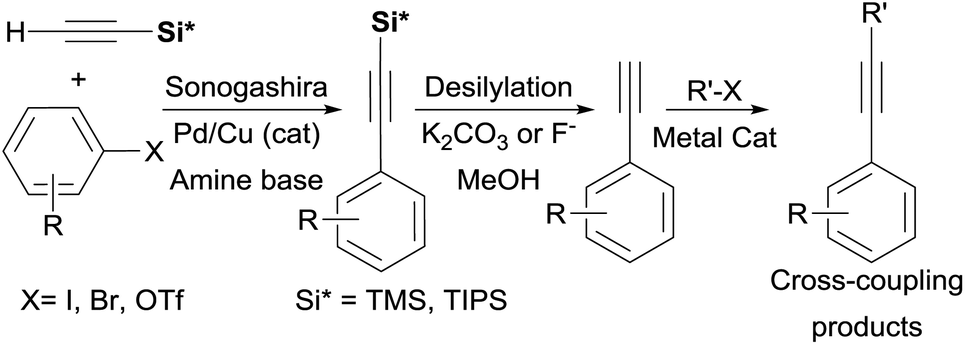 | ||
| Scheme 2 Iterative trialkylsilylalkyne synthesis, deprotection and cross-coupling. | ||
The use of alkynylsilanes 2 directly26 as coupling partners significantly simplifies this three-step procedure, dramatically enhances the reaction scope, as well as avoids dependence on commercial sources for terminal alkynes. Additionally, the use of alkynylsilanes could provide a fertile ground for the development of one-pot or multicomponent protocols (vide infra). Furthermore, alkynylation of several aryl electrophiles (Cl, OTf, OMs, OPO3Et2)27 with TMSA has been recently shown to proceed under metal-free conditions, thus, making the use of 2 considerably more attractive.
Results and discussion
Although direct transmetalation from silicon to copper to form Cu(I) acetylides has been shown to proceed without the need of an external activator,25e,28,29 our optimization studies were focused on the use of phenylacetylene 3a which would form upon in situ deprotection of the corresponding silyl derivative 2a in a protic solvent. Several combinations of solvent, copper source and bases were screened to identify suitable reaction conditions for the desired transformation, and the results are summarized in Table 1.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Alkyne | Solvent | Cat. (equiv.) | Ligand (equiv.) | Rxn time | Yield (%) |
| a Reaction conditions: 0.5 mmol of 1a, 1.5 equiv. of alkyne Cs2CO3 (2 equiv.), ligand and solvent (1 mL, degassed) were placed in a crimp top vial, sealed and heated at 130 °C for the indicated time. Isolated Yields are shown. Average of 2 consecutive runs. b Reaction was performed at 80 °C. c n-Bu4NBr (0.75 M) was added. d K2CO3 (2 equiv.) was used. e K3PO4 was used (2 equiv.). f n-Bu4NBr (0.5 M) was used. g Reaction was performed at 100 °C. h 3 equiv. of Cs2CO3 were used. | ||||||
| 1b | 3a | MeOH | CuCl2 (0.2) | L1 (0.2) | 24 h | 60 |
| 2 | 3a | H2Oc | CuCl2 (0.2) | L2 (0.2) | 30 min | 52 |
| 3 | 3a | H2Oc | Cu(OTf)2 (0.2) | L2 (0.2) | 30 min | 57 |
| 4 | 3a | H2Oc | CuI (0.1) | L2 (0.2) | 30 min | 47 |
| 5 | 3a | H2Oc | CuBr (0.1) | L2 (0.2) | 30 min | 52 |
| 6 | 3a | H2Oc | CuCI (0.1) | L1 (0.2) | 30 min | 52 |
| 7 | 3a | H2Oc | CuCI (0.1) | L2 (0.2) | 30 min | 65 |
| 8 | 3a | H2Oc | CuCI (0.1) | L3(0.2) | 30 min | 60 |
| 9 | 3a | H2Oc | CuCI (0.1) | L4 (0.2) | 30 min | 50 |
| 10 | 3a | H2Oc | CuCI (0.1) | L5 (0.2) | 30 min | 46 |
| 11 | 3a | H2Oc | CuCI (0.1) | L6 (0.2) | 30 min | 59 |
| 12 | 3a | H 2 O | CuCI (0.1) | L 2 (0.3) | 30 min | 70 |
| 13 | 3a | H2Oc | CuCI (0.1) | None | 30 min | 40 |
| 14d | 3a | H2Oc | CuCI (0.1) | L2 (0.3) | 30 min | 63 |
| 15e | 3a | H2Oc | CuCI (0.1) | L2 (0.3) | 30 min | 58 |
| 16f | 3a | H2Oc | CuCI (0.1) | L2 (0.3) | 30 min | n.d |
| 17g | 3a | H2Oc | CuCI (0.1) | L2 (0.3) | 30 min | n.d. |
| 18 | 2a | H 2 O | CuCI (0.1) | L 2 (0.3) | 30 min | 70 |
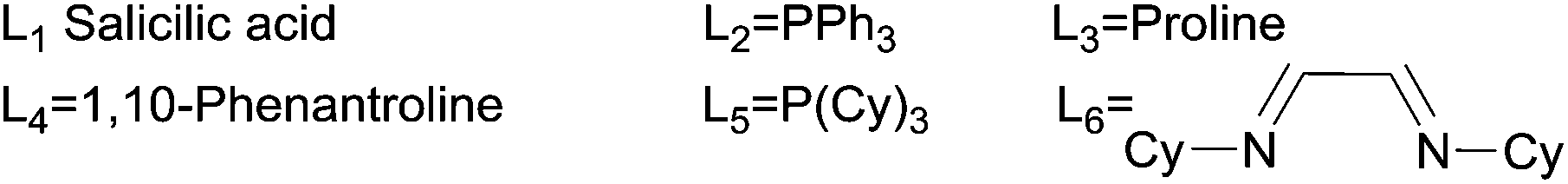
|
||||||
Initial screening was performed utilizing 10 mol% of CuCl2, salicylic acid (20 mol%) as a ligand and Cs2CO3 (2 equiv.) as a base. Since protodesilylation is known to proceed under basic, protic media, MeOH was first tried as a reaction medium and it was found to be a suitable solvent affording 4a in 60% yield after 24 h at 80 °C (Table 1, entry 1).30 To our delight, the use of water as a solvent significantly enhanced the kinetics of the reaction affording 4a in 52% yield in only 30 minutes at 130 °C, using n-tetrabutylammonium bromide (TBAB) as a phase-transfer agent (Table 1, entry 2). When Cu(OTf)2/PPh3 system was used, the yield of 4a was slightly enhanced to 57% (Table 1, entry 3). Further screening revealed that among the cuprous salts studied, CuCl was superior (Table 1, entries 4–6) and PPh3 was better than salicylic acid as a ligand affording 4a in a 65% isolated yield (Table 1, entry 7). Several other N–N, N–O and P ligands were also studied, but their use did not lead to any major improvements (Table 1, entries 8–11).
Increasing the loading of PPh3 to 30 mol% significantly improved the yield of 4a (70%), while its absence resulted in a noticeable decrease in yield (40%), emphasizing the importance of PPh3 in the present reaction (Table 1, entries 12–13). Efforts to further optimize the conditions by changing the base were met with rather limited success (Table 1, entries 14–15). Additionally, the use of TBAB as a phase-transfer agent was found an essential component of the present protocol.
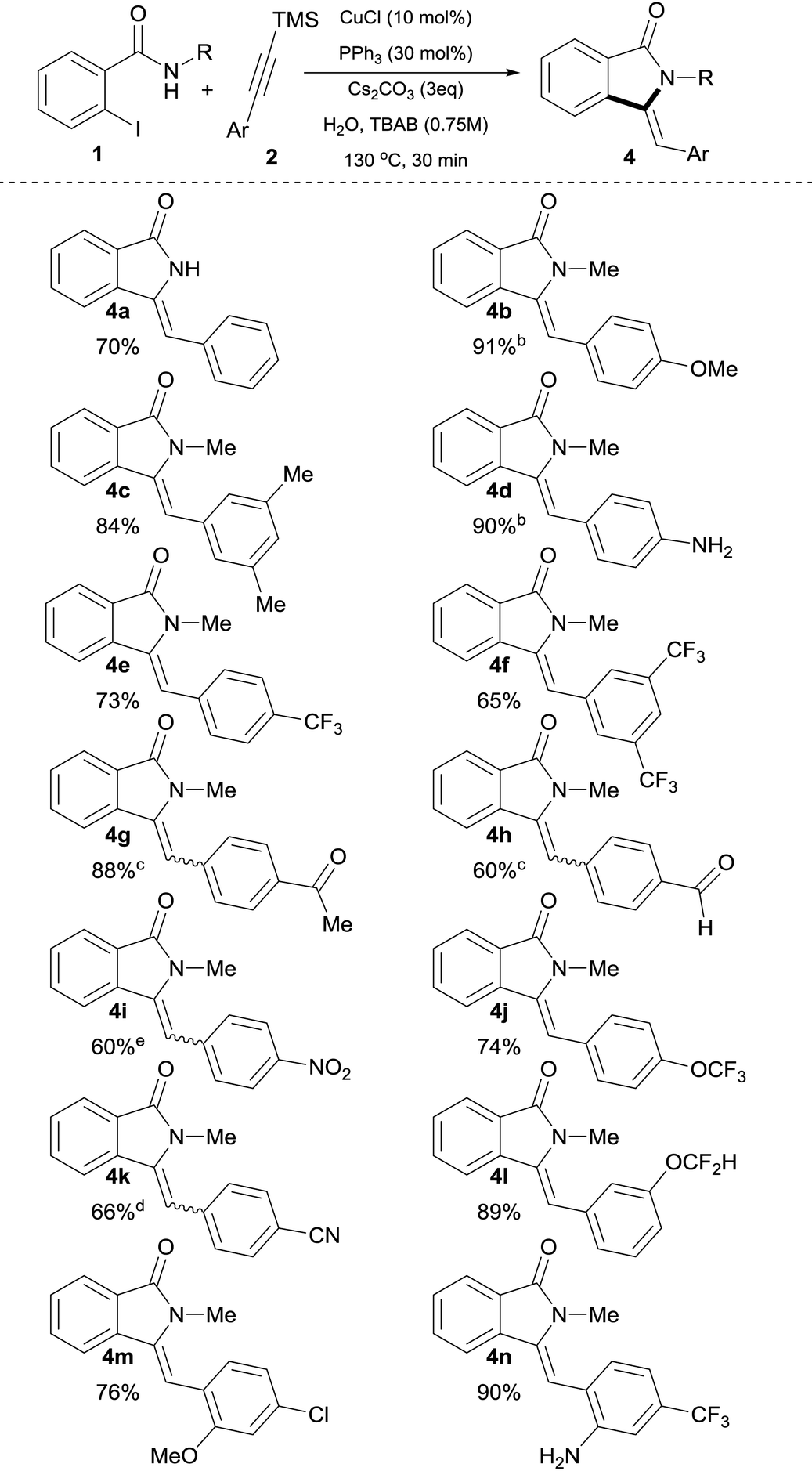
Lower concentrations of TBAB or lower reaction temperature led to incomplete conversion of starting materials (Table 1, entries 16–17). As expected, when 2a was used in the presence of 3 equiv. of Cs2CO3 under otherwise identical conditions, it underwent in situ desilylation31 followed by a cross-coupling and hydroamidation sequence without compromising the yield (Table 1, entry 18).32 With these optimized conditions in hand, parallel studies were performed using both trimethylsilyl alkynes 2 (Table 2) as well as commercially available terminal alkynes 3 (Table 3) under aqueous phase-transfer conditions.
| a Reaction conditions: 0.5 mmol of 1b, silylalkyne 4 (1.5 equiv.), Cs2CO3 (3 equiv.), n-tetrabutylammonium bromide (TBAB, 0.75 M), CuCl (10 mol%), PPh3 (30 mol%) in 1 mL of H2O (degassed) were heated at 130 °C for 30 min in a crimp top vial under N2. b Reaction time = 40 min. c Z/E ratio = 2. d Z/E ratio = 1.2. e Z/E ratio = 0.2. |
|---|
|
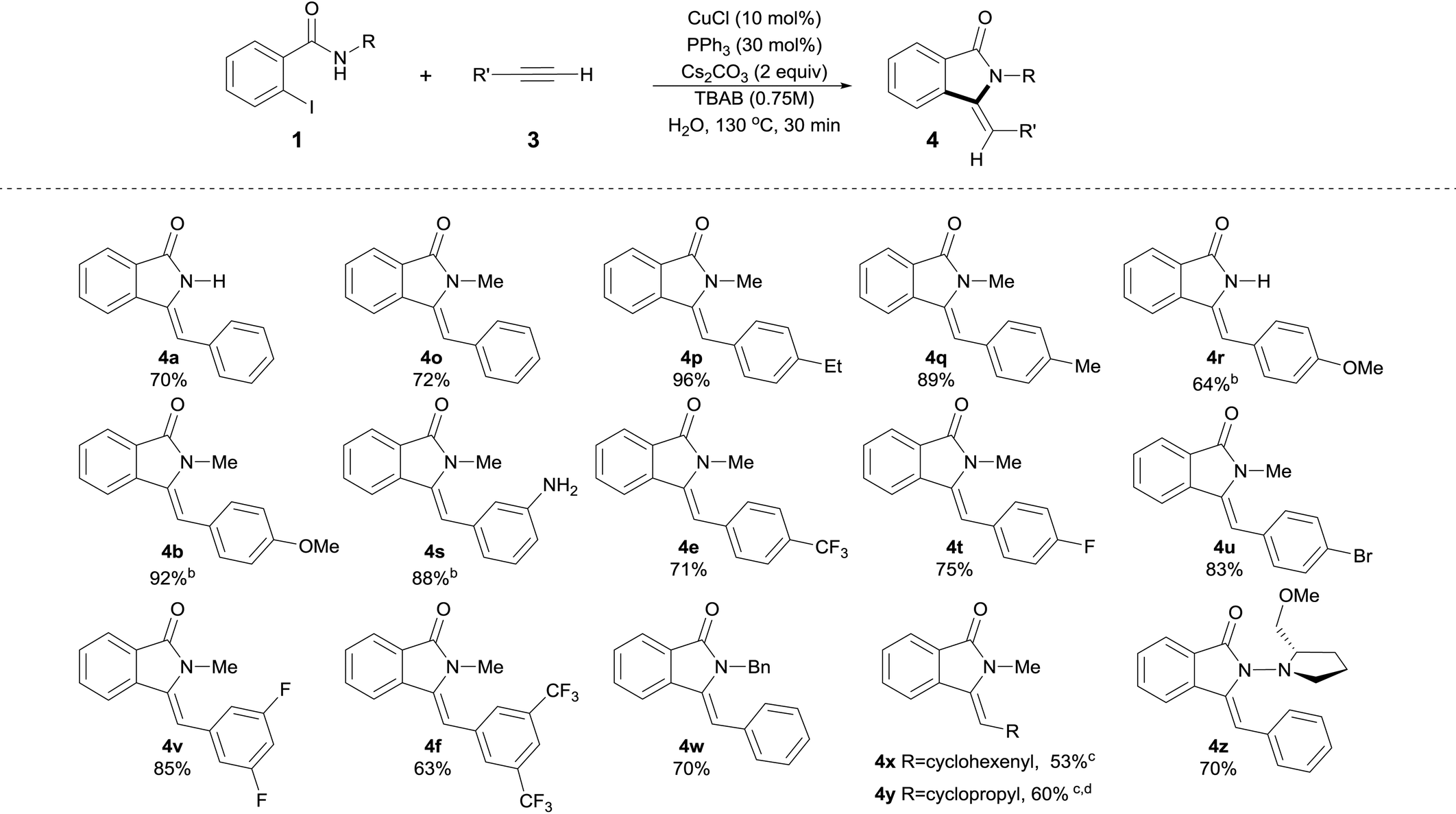
| a Isolated yields, average of two runs are shown. Reactions conditions: 0.5 mmol 2-Iodobenzamide derivatives 1, 1.5 equiv. of terminal alkyne 3, CuCl (10 mol%), PPh3 (30 mol%), Cs2CO3 (2 equiv.), n-tetrabutylammonium bromide (TBAB, 0.75 M) in 1 mL of H2O (degassed) were heated at 130 °C for 30 min. b Reaction time = 40 min. c 3 equiv. of alkyne were used. d Reaction time was 90 min. |
|---|
|
The scope of the process with respect to (silyl)alkynes was found to be broad and allowed access to the target molecules with a varied range of functionalities in good to excellent isolated yields (60–96%). Alkynylsilanes possessing electron-donating as well as electron-withdrawing groups were found suitable substrates, with electron donating groups giving generally higher yields. Accordingly, 4-MeO and 4-NH2 derivatives 4b and 4d (Table 2), were obtained in high yields (91% and 90% respectively), albeit under slightly longer reaction times (40 min). These results are in accordance with the effect of reduced electrophilicity of the corresponding o-alkynylbenzamide intermediate possessing electron-donating moieties.33
With the exception of 4g, 4h, 4i and 4k, which possess strong electron-withdrawing groups (–COMe, –CHO, –NO2 and –CN respectively) exclusive Z-configuration around the exocyclic double bond was revealed by NOESY experiments in all products obtained. Furthermore, Z-configuration was unambiguously determined by X-ray crystallography of 4a and 4y.34 It is worth mentioning that medicinally attractive CF3, OCF3 and OCF2H-substituted isoindolin-1-ones 4e, 4f, 4j and 4l were obtained in good yields and as single Z-isomers.
For products 4a, 4b, 4e and 4f, as can be seen by comparison of Tables 2 and 3, the use of either silylalkyne or terminal alkynes afforded the products in similar isolated yields, with silylalkynes requiring 3 equiv. of base. Noticeably, unprotected N–H functionality was readily incorporated, providing amino-substituted isoindolin-1-ones in a very high yield without competitive C–N bond formation (Table 2, 4d, 4n and Table 3, 4s). Silylalkynes with ortho substituents were also smoothly converted into the corresponding products in good yields (Table 2, 4m and 4n). Remarkably, for the ortho-NH2 substituted silylalkyne 2n, the competitive cyclization to form the corresponding indole16b,35 was completely suppressed and the corresponding isoindolinone 4n was isolated in 90% yield with exclusive Z-selectivity.
The use of halo-substituted (F, Cl and Br) substrates is notable as it allows the potential for further product functionalization by subsequent transition metal catalyzed coupling processes (Table 2, 4m and Table 3, 4t, 4u and 4v). The acetyl and formyl functionalities were also readily incorporated in good yields without competitive 1,2-addition, albeit with a decrease in Z-selectivity (Z/E ratio = 2, Table 2, 4g, and 4h). In the case of 4–cyano substituted silylalkyne 2k, the corresponding product was obtained in 66% isolated yield also with a decreased Z-selectivity (Z/E ratio = 1.2, Table 2, 4k). Similarly, NO2-substituted silylalkyne 2i was smoothly converted into product in 60% yield, however with a reverted stereoselectivity (Z/E ratio = 0.2).36 Aliphatic alkynes 1-ethynylcyclohex-1-ene 3x and ethynylcyclopropane 3y were utilized successfully in this process, providing products 4x and 4y in slightly diminished yields of 53% and 60%, respectively, after 90 min (Table 3). Although the decreased yields obtained with aliphatic alkynes represent a drawback in the present protocol, these results clearly highlight the 5-exo-selectivity of the process as evidenced by X-ray crystallographic studies of 4y.34 Investigation of N-substituted amide coupling partner showed the high tolerance of N–Me, N–Bn as well as free N–H functionalities, albeit the latter delivers slightly reduced yields of product (4a and 4r in 70% and 64% yield, respectively). Furthermore, (S)-1-amino-2-methoxymethylpyrrolidine, SAMP-derived benzamide 1z, provided the corresponding enantiomerically enriched isoindolinone 4z in 70% isolated yield (Table 3), which could potentially allow for subsequent asymmetric transformations.
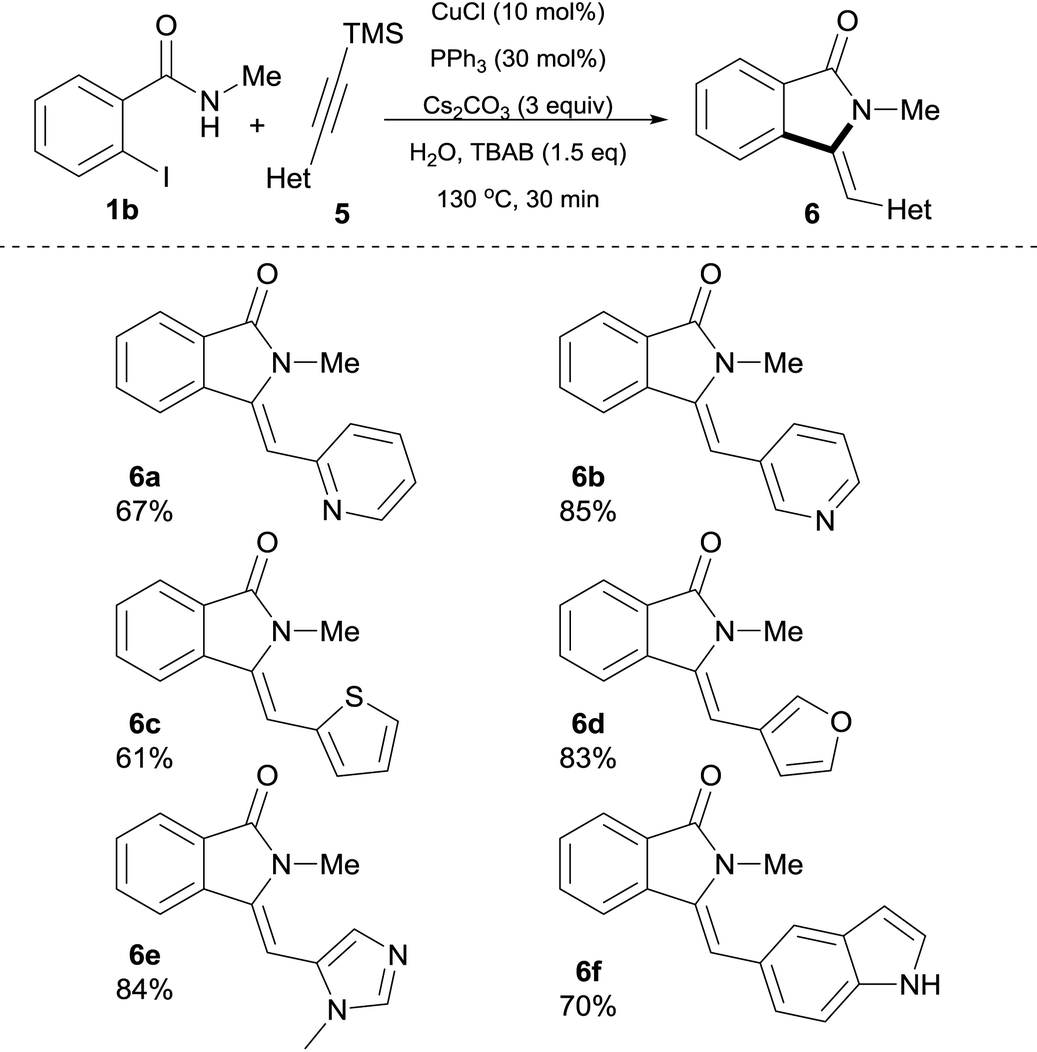
In pursuit of medicinally relevant isoindolin-1-ones scaffolds, we decided to explore the possibility of employing heteroaryl silylalkyne substrates 5 and the results are shown in Table 4. Gratifyingly, the present protocol successfully enabled the preparation of a series of heterocyclic alkyne-derived isoindolin-1-ones 6 in good yields. For example, 2-pyridyl as well as 3-pyridylsilylalkynes afforded 6a and 6b in 67% and 85% yield, respectively. Furthermore, 2-thienyl- and 3-furanyl-derived trimethylsilylacetylenes were smoothly converted into products affording isoindolinones 6c and 6d in good yields (61% and 83%, respectively) with exclusive Z-configuration. Other nitrogen heterocycles such as N-Me-imidazole and 1H-indole were also well tolerated and afforded 6e and 6f in 84% and 70% isolated yields, respectively. In this case, it is important to point out that competitive N-arylation37 of the indole moiety, a process known to be catalyzed by copper, was completely absent under our reaction conditions, thus, further emphasizing the tolerance of our method for this type of N–H heterocycles.
| a Reaction conditions: 0.5 mmol of 1b, heteroaryl silylalkyne 5 (1.5 equiv.), Cs2CO3 (3 equiv.), n-tetrabutylammonium bromide (0.75 M), CuCl (10 mol%), PPh3 (30 mol%) in 1 mL of H2O (degassed) were heated at 130 °C for 30 min in a crimp top vial under N2. |
|---|
|
As mentioned earlier, the direct use of alkynylsilanes as coupling partners could enable the development of one-pot or multicomponent processes. Accordingly, we investigated the possibility to access the target molecules by this protocol (Schemes 3 and 4). Towards this end, we performed the reaction of TMSA, 2-iodo-N-methylbenzamide 1b and 4-(trifluoromethoxy)iodobenzene 7j in a multicomponent fashion using Pd(PPh3)4 (3 mol%) and CuI (10 mol%) as co-catalysts in DMF/Et2NH system.38 After heating this mixture at 120 °C for 15 min, simple addition of aqueous Cs2CO3, TBAB and further heating for 30 min, led to the isolation of 4j in a 72% yield (Scheme 3, Conditions A). In the case of electron rich aryl iodides such as 4-iodoanisole 7b, best results were obtained by adding 1b in the second step, along with aqueous carbonate and TBAB39 (Conditions B). These preliminary results (although not yet optimized) clearly emphasize the importance of using alkynylsilanes to facilitate this and other consecutive cross-coupling processes, thus, enabling the creation of molecular complexity in a single operational step from readily available aryl electrophiles and TMSA as a convenient acetylene surrogate.
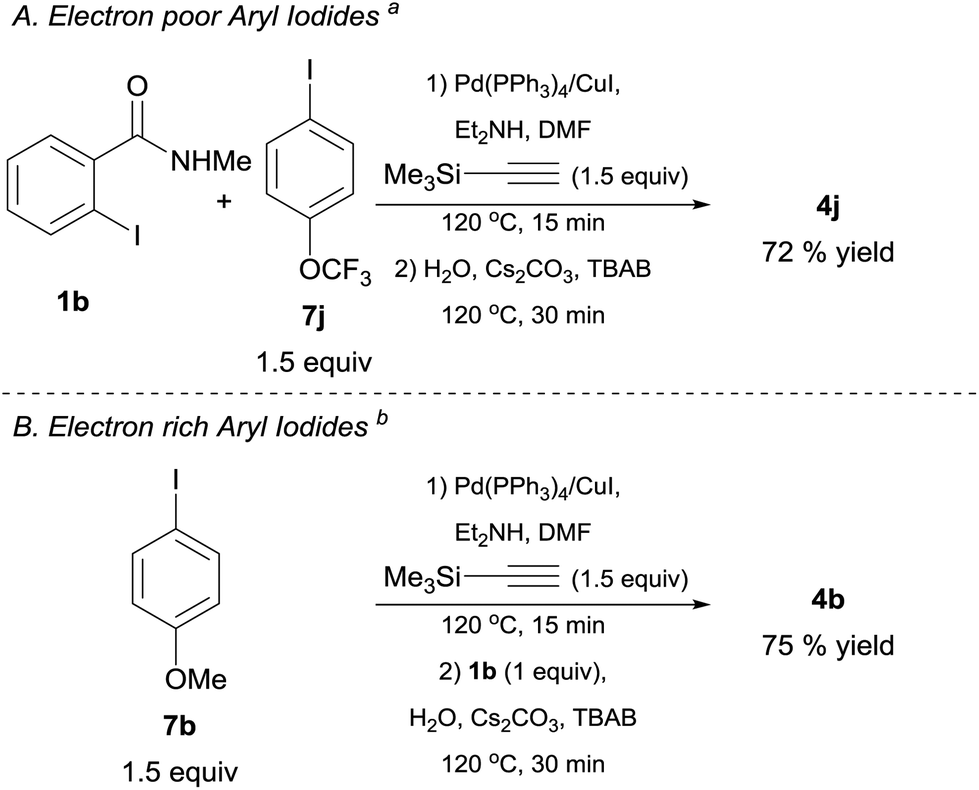 | ||
| Scheme 3 One-pot preparation of isoindolin-1-ones from 1b, TMSA and aryl iodides. Reaction conditions. Conditions A: 1b 0.5 mmol (1 equiv.), aryl iodide derivative 7 (1.5 equiv.), TMSA (1.5 equiv.), Pd(PPh3)4 3 mol%, CuI 10 mol%, DMF (1 mL), Et2NH (1 mL), were placed in a crimp-top vial, sealed under argon and heated at 120 °C for 15 min. Then 1 mL of an aqueous solution (degassed) of Cs2CO3 (3 equiv.) and TBAB (0.75 M) was added and heating was continued for 30 min. Conditions B: for electron rich aryl iodides, same as above but 1b 0.5 mmol (1 equiv.) is added after the first step in 1 mL of H2O (degassed), along with Cs2CO3 (3 equiv.) and TBAB (0.75 M). TMSA = trimethylsilylacetylene TBAB = n-tetrabutylammonium bromide. | ||
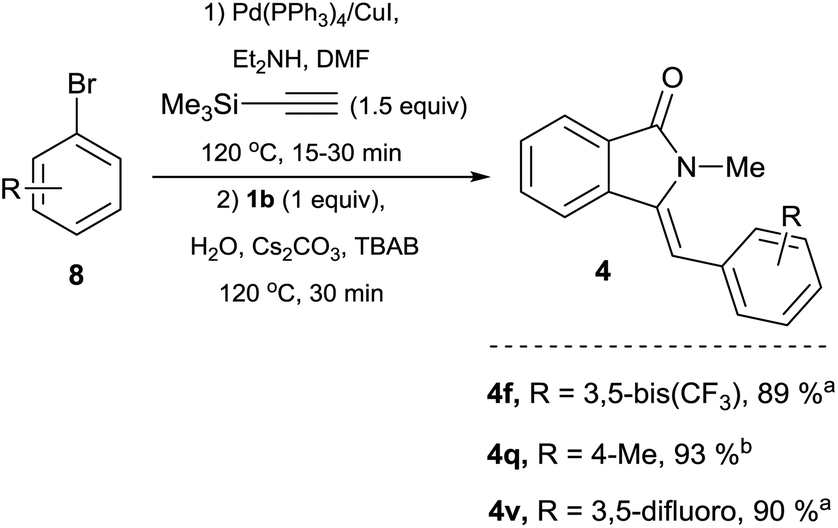 | ||
| Scheme 4 One-pot preparation of isoindolin-1-ones from 1b, TMSA and aryl bromides. Reaction conditions: aryl bromide derivative 8 (1.5 equiv.), TMSA (1.5 equiv.), Pd(PPh3)4 3 mol%, CuI 10 mol%, DMF (1 mL), Et2NH (1 mL), were placed in a crimp-top vial, sealed under argon and heated at 120 °C for (a) 15 min or (b) 30 min. Then 1b 0.5 mmol (1 equiv.), 1 ml of an aqueous solution (degassed) of Cs2CO3 (3 equiv.) and TBAB (0.75 M) was added and heating was continued for 30 min. Catalyst loadings are based on limiting reagent 1b. TMSA = trimethysilylacetylene TBAB = n-tetrabutylammonium bromide. See ESI† for full experimental and characterization details. | ||
Because of the inexpensive nature and greater atom-economy exhibited by aryl bromides 8 when compared to their aryl iodide counterparts, we explored the possibility of utilizing this type of coupling partner in a one-pot protocol. Gratifyingly, under conditions analogous to the ones used for electron rich aryl iodides, the reaction of 8f, 8q or 8v with TMSA under Pd/Cu catalysis, followed by addition of 1b, led to the clean formation of the corresponding isoindolin-1-ones 4f, 4q and 4v in yields superior to the ones obtained with the preformed alkynes. The simple and straightforward nature of this protocol clearly highlights the advantages of utilizing TMSA in a one-pot process (Scheme 4).
Conclusions
In summary, we have developed an inexpensive, fast and efficient CuCl/PPh3-catalyzed method for the synthesis of 3-methylene-isoindolin-1-ones 4 from 2-iodobenzamides 1 and silyl 2 or terminal 3 alkynes. Salient features of this protocol include the utilization of aqueous phase-transfer conditions and very short reaction times (30 min), affording the products with exclusive Z-configuration, with few exceptions. In addition, Br– Cl–, COMe–, CHO–, NH2–, CN– and NO2– substituents were well tolerated, thus, enabling further synthetic transformations. Furthermore, heteroaryl silylalkynes 5 are suitable substrates including the ones bearing unprotected N–H functionality. Preliminary results demonstrated the feasibility of a one-pot process with the use of Pd(PPh3)4/CuI co-catalyst system starting from 1b, TMSA and electron-rich as well as electron-poor aryl iodides 7 and aryl bromides 8. These results represent a successful example of a process combining the elements of pot, atom and step economy40 (PASE); characteristics which significantly simplify the synthesis of 3-methylene-isoindolin-1-ones and other heterocyclic frameworks while significantly contributing to the principles of green chemistry. The extension of this one-pot protocol to other halo-benzamides or other electrophiles such as Ar-OTf should also be possible and such studies are currently underway in our laboratories.Acknowledgements
Support by the Loker Hydrocarbon Research Institute and Mexico's Consejo Nacional de Ciencia y Tecnologia, CONACYT is gratefully acknowledged.Notes and references
- G. Blasko, D. J. Gula and M. Shamma, J. Nat. Prod., 1982, 45, 105 CrossRef CAS.
- T. R. Belliotti, W. A. Brink, S. R. Kesten, J. R. Rubin, D. J. Wustrow, K. T. Zoski, S. Z. Whetzel, A. E. Corbin, T. A. Pugsley, T. G. Heffner and L. D. Wise, Bioorg. Med. Chem. Lett., 1998, 8, 1499 CrossRef CAS PubMed.
- Y. Kato, M. Takemoto and K. Achiwa, Chem. Pharm. Bull., 1993, 41, 2003 CrossRef CAS PubMed.
- (a) M. Linden, D. Hadler and S. Hofmann, Hum. Psychopharmacol., 1997, 12, 445 CrossRef CAS; (b) Z.-P. Zhuang, M.-P. Kung, M. Mu and H. F. Kung, J. Med. Chem., 1998, 41, 157 CrossRef CAS PubMed; (c) M. H. Norman, D. J. Minick and G. C. Rigdon, J. Med. Chem., 1998, 39, 149 CrossRef PubMed.
- A. Murashiiri, Japanese Patent, 59046268, 1984 Search PubMed; Chem. Abstr. 1984 101 54922 Search PubMed.
- (a) I. Pendrak, S. Barney, R. Wittrock, D. M. Lambert and W. D. Kingsbury, J. Org. Chem., 1994, 59, 2623 CrossRef CAS; (b) E. De Clercq, J. Med. Chem., 1995, 38, 2491 CrossRef CAS PubMed.
- (a) E. C. Taylor, P. Zhou, L. D. Jennings, Z. Mao, B. Hu and J.-G. Jun, Tetrahedron Lett., 1997, 38, 521 CrossRef CAS; (b) C. Riedinger, J. A. Endicott, S. J. Kemp, L. A. Smyth, A. Watson, E. Valeur, B. T. Golding, R. J. Griffin, I. R. Hardcastle, M. E. Noble and J. M. McDonnell, J. Am. Chem. Soc., 2008, 130, 16038 CrossRef CAS PubMed.
- H. Peters and W. Flitsch, Tetrahedron Lett., 1969, 15, 1161 Search PubMed.
- (a) E.-C. Wang, H.-F. Chen, P.-K. Feng, Y.-L. Lin and M.-K. Hsu, Tetrahedron Lett., 2002, 43, 9163 CrossRef CAS; (b) Y. Kato, M. Takemoto and K. Achiwa, Chem. Pharm. Bull., 1993, 41, 2003 CrossRef CAS PubMed.
- M. Freccero, E. Fasani and A. Albini, J. Org. Chem., 1998, 58, 1740 CrossRef.
- A. Couture, E. Deniau and P. Grandclaudon, Tetrahedron, 1997, 53, 10313 CrossRef CAS.
- C. Sun and B. Xu, J. Org. Chem., 2008, 73, 7361 CrossRef CAS PubMed.
- Y. Kondo, F. Shiga, N. Murata, T. Sakamoto and H. Yamanaka, Tetrahedron, 1994, 50, 11803 CrossRef CAS.
- (a) P. Barrio, I. Ibáñez, L. Herrera, R. Román, S. Catalán and S. Fustero, Chem. – Eur. J., 2015, 21, 11579 CrossRef CAS PubMed. For other aminocarbonylation protocols see: (b) D. Marosvölgyi-Haskó, A. Takács, Z. Riedl and L. Kollár, Tetrahedron, 2011, 67, 1036 CrossRef; (c) M. Fujioka, T. Morimoto, T. Tsumagari, H. Tanimoto, Y. Nishiyama and K. Kakiuchi, J. Org. Chem., 2012, 77, 2911 CrossRef CAS PubMed; (d) R. Mancuso, I. Ziccarelli, D. Armentano, N. Marino, S. V. Giofre and B. Gabriele, J. Org. Chem., 2014, 79, 3506 CrossRef CAS PubMed; (e) Y. Xu, W. Hu, X. Tang, J. Zhao, W. Wu and H. Jiang, Chem. Commun., 2015, 51, 6843 RSC.
- H. Cao, L. Mcnamee and H. Alper, Org. Lett., 2008, 10, 5281 CrossRef CAS PubMed.
- (a) Y. Koseki, S. Kusano and T. Nagasaka, Tetrahedron Lett., 1998, 39, 3517 CrossRef CAS; (b) C. Koradin, W. Dohle, A. L. Rodriguez, B. Schmid and P. Knochel, Tetrahedron, 2003, 59, 1571 CrossRef CAS; (c) C. Kanazawa and M. Terada, Chem. – Asian J., 2009, 4, 1668 CrossRef CAS PubMed; (d) G. Bianchi, M. Chiarini, F. Marinelli, L. Rossi and A. Arcadi, Adv. Synth. Catal., 2010, 352, 136 CrossRef CAS. For a phase-transfer promoted cyclization see: (e) J. Hu, L. Liu, X. Wang, Y. Hu, S. Yang and Y. Liang, Green Sustainable Chem., 2011, 01, 165 CrossRef CAS; (f) A. Bubar, P. Estey, M. Lawson and S. Eisler, J. Org. Chem., 2012, 77, 1572 CrossRef CAS PubMed; (g) D. Y. Li, K. J. Shi, X. F. Mao, Z. Le Zhao, X. Y. Wu and P. N. Liu, Tetrahedron, 2014, 70, 7022 CrossRef CAS.
- (a) M. W. Khan and N. G. Kundu, Synlett, 1997, 1435 CrossRef; (b) N. G. Kundu and M. W. Khan, Tetrahedron, 2000, 56, 4777 CrossRef CAS.
- (a) M. Hellal and G. D. Cuny, Tetrahedron Lett., 2011, 52, 5508 CrossRef CAS; (b) S. Sarkar, S. Dutta, R. Dey and S. Naskar, Tetrahedron Lett., 2012, 53, 6789 CrossRef CAS.
- (a) L. Li, M. Wang, X. Zhang, Y. Jiang and D. Ma, Org. Lett., 2009, 11, 1309 CrossRef CAS PubMed; (b) J. Pan, Z. Xu, R. Zeng and J. Zou, Chin. J. Chem., 2013, 31, 1022 CrossRef CAS; (c) J. Dong, F. Wang and J. You, Org. Lett., 2014, 16, 2884 CrossRef CAS PubMed. For a Cu-catalyzed decarboxylative protocol starting from alkynyl carboxylic acids as alkyne sources see: (d) A. Gogoi, S. Guin, S. K. Rout, G. Majji and B. K. Patel, RSC Adv., 2014, 4, 59902 RSC.
- (a) Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef CAS; (b) Y. Hatanaka, K. Matsui and T. Hiyama, Tetrahedron Lett., 1989, 30, 2403 CrossRef CAS.
- S. E. Denmark and D. Wehrli, Org. Lett., 2000, 2, 565 CrossRef CAS PubMed.
- (a) S. E. Denmark and S. A. Tymonko, J. Org. Chem., 2003, 68, 9151 CrossRef CAS PubMed; (b) S. Chang, S. H. Yang and P. H. Lee, Tetrahedron Lett., 2001, 42, 4833 CrossRef CAS; (c) S. E. Denmark, L. Neuville, M. E. L. Christy and S. A. Tymonko, J. Org. Chem., 2006, 71, 8500 CrossRef CAS PubMed; (d) E. Alacid and C. Najera, J. Org. Chem., 2008, 73, 2315 CrossRef CAS PubMed.
- For accounts and review articles see: (a) Y. Hatanaka and T. Hiyama, Synlett, 1991, 845 CrossRef CAS; (b) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC; (c) S. E. Denmark and R. F. Sweis, Acc. Chem. Res., 2002, 35, 8359 CrossRef; (d) S. E. Denmark and J. D. Baird, Chem. – Eur. J., 2006, 12, 4954 CrossRef CAS PubMed.
- Although not uniformly used in the literature, the term “Sonogashira reaction” strictly refers to the Pd/Cu co-catalyzed C(sp)–C(sp2) coupling. Other types of alkynylation protocols are also available, in fact, a decade prior to the discovery of Sonogashira reaction, Stephens and Castro developed a protocol using stoichiometric amounts of copper acetylides. Reactions catalyzed by copper only should be referred to as catalytic “Castro–Stephens reactions”, whereas the Pd only protocols should be referred to as “Heck–Cassar coupling”. For a brief review on alkynylation catalyzed by different metals see: H. Plenio, Angew. Chem., Int. Ed., 2008, 47, 6954 CrossRef CAS PubMed.
- (a) S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627 CrossRef CAS; (b) M. Erdelyi and A. Gogoll, J. Org. Chem., 2001, 66, 4165 CrossRef CAS PubMed; (c) M. S. Mohamed Ahmed, A. Sekiguchi, K. Masui and A. Mori, Bull. Chem. Soc. Jpn., 2005, 78, 160 CrossRef; (d) G. Zengin, Asian J. Chem., 2007, 19, 3063 CAS; (e) Y. Nishihara, K. Ikegashira, A. Mori and T. Hiyama, Chem. Lett., 1997, 1233 CrossRef CAS. For reviews see: (f) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed; (g) E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979 CrossRef CAS PubMed.
- Y. Nishihara, S. Noyori, T. Okamoto, M. Suetsugu and M. Iwasaki, Chem. Lett., 2011, 40, 972 CrossRef CAS.
- S. Protti, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2005, 44, 5675 CrossRef CAS PubMed.
- Y. Nishihara, K. Ikegashira, A. Mori and T. Hiyama, Tetrahedron Lett., 1998, 39, 4075 CrossRef CAS.
- Y. Nishihara, K. Ikegashira, K. Hirabayashi, J. Ando, A. Mori and T. Hiyama, J. Org. Chem., 2000, 65, 1780 CrossRef CAS PubMed.
- Reaction was conducted in a crimp top vial. Due to methanol's high volatility, further enhancement of the reaction rate by increasing the reaction temperature was not pursued.
- The similar reaction rates for silyl and terminal alkynes, suggests that in situ deprotection is the dominant pathway, although direct transmetalation from silicon to copper cannot be ruled out.
- Only 2 equivalents of base were insufficient to achieve higher yields.
- Shorter reaction times led to the isolation of a mixture of isoindolinone and the uncyclized o-alkynylbenzamide derivative.
- See ESI† for details. Crystallographic information files have been deposited in the Cambridge Crystallographic Data Centre. CCDC numbers of products are 4a: 1427402 and 4y: 1426924.
- G. W. Kabalka, L. Wang and R. M. Pagni, Tetrahedron, 2001, 57, 8017 CrossRef CAS.
- The results of the Z/E ratio in this series are in perfect alignment with the electron withdrawing ability of the corresponding functional groups as judged by their σ(para) values. However, the p-CF3 substituted product 4e deviates from this trend. We attribute this observation to the decreased coordination of –CF3 to the copper Lewis acid. Coordination of moieties containing stronger Lewis basic sites most likely enhances the electron withdrawing ability of such functional groups.
- J. C. Antilla, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 11684 CrossRef CAS PubMed.
- Catalyst loadings mentioned are based on the limiting reagent 1b.
- If 1b is present in the first step, the product 4b could also be obtained, albeit in a decreased yield of 52% along with significant amounts of 1,2-bis(4-methoxyphenyl)ethyne, resulting from the competitive cross-coupling of 2b with 7b.
- P. a. Clarke, S. Santos and W. H. C. Martin, Green Chem., 2007, 9, 438 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1427402 and 1426924. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02187a |
| This journal is © The Royal Society of Chemistry 2016 |