
Par-4 secretion: stoichiometry of 3-arylquinoline binding to vimentin
Vitaliy M.
Sviripa
ab,
Ravshan
Burikhanov
cd,
Josiah M.
Obiero
e,
Yaxia
Yuan
bef,
Justin R.
Nickell
e,
Linda P.
Dwoskin
e,
Chang-Guo
Zhan
bef,
Chunming
Liu
ad,
Oleg V.
Tsodikov
*e,
Vivek M.
Rangnekar
*cdg and
David S.
Watt
*abde
aDepartment of Molecular and Cellular Biochemistry, College of Medicine, University of Kentucky, Lexington, KY 40536-0509, USA. E-mail: dwatt@uky.edu
bCenter for Pharmaceutical Research and Innovation, College of Pharmacy, University of Kentucky, Lexington, KY 40536-0596, USA
cDepartment of Radiation Medicine, College of Medicine, University of Kentucky, Lexington, KY 40506-9983, USA. E-mail: vmrang01@uky.edu
dLucille Parker Markey Cancer Center, College of Medicine, University of Kentucky, Lexington, KY 40536-0096, USA
eDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, Lexington, KY 40536-0596, USA. E-mail: oleg.tsodikov@uky.edu
fMolecular Modeling and Biopharmaceutical Center, College of Pharmacy, University of Kentucky, Lexington, KY 40536-0596, USA
gGraduate Center for Toxicology, College of Medicine, University of Kentucky, Lexington, KY 40536-0305, USA
First published on 3rd November 2015
Abstract
Advanced prostate tumors usually metastasize to the lung, bone, and other vital tissues and are resistant to conventional therapy. Prostate apoptosis response-4 protein (Par-4) is a tumor suppressor that causes apoptosis in therapy-resistant prostate cancer cells by binding specifically to a receptor, Glucose-regulated protein-78 (GRP78), found only on the surface of cancer cells. 3-Arylquinolines or “arylquins” induce normal cells to release Par-4 from the intermediate filament protein, vimentin and promote Par-4 secretion that targets cancer cells in a paracrine manner. A structure–activity study identified arylquins that promote Par-4 secretion, and an evaluation of arylquin binding to the hERG potassium ion channel using a [3H]-dofetilide binding assay permitted the identification of structural features that separated this undesired activity from the desired Par-4 secretory activity. A binding study that relied on the natural fluorescence of arylquins and that used the purified rod domain of vimentin (residues 99-411) suggested that the mechanism behind Par-4 release involved arylquin binding to multiple sites in the rod domain.
Introduction
Advanced prostate tumors resist androgen-deprivation therapy, and the metastasis of these tumor cells to bone, lung or liver ultimately produces fatal outcomes.1–3 Although therapy-resistant cancer cells are susceptible to apoptosis by the pro-apoptotic, tumor suppressor called Prostate Apoptosis Response-4 (Par-4),4–6 cancer cells possess several mechanisms, including sequestration and phosphorylation, to elude the effects of endogenous, self-generated Par-4. Overexpression, for example, of the intermediate filament protein, vimentin, in cancer cells7 serves to sequester Par-4 (Fig. 1). Normal cells also impound Par-4 using vimentin and only export it at low levels unless subjected to stress-related circumstances. Once in circulation, extracellular Par-4 selectively binds to Glucose-regulated Protein-788–10 (GRP78), a specific receptor that appears only on the surface of cancer cells.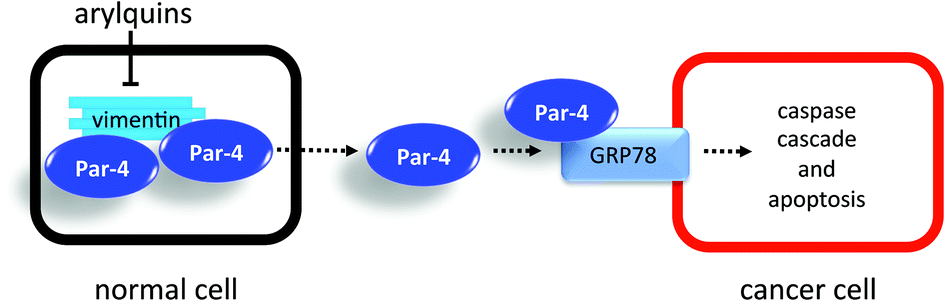 | ||
| Fig. 1 Cartoon depiction of Par-4 sequestration by vimentin and antineoplastic effect of Par-4 secretion. | ||
This binding event induces the caspase-driven apoptosis of cancer cells.5 Various cancers, including advanced prostate cancers, express GRP78, and small-molecule agents capable of promoting the enhanced secretion of Par-4 from normal cells, which vastly outnumber cancer cells, represent a new and attractive therapeutic approach.
We recently reported that substituted 3-arylquinolines 1 (Fig. 2), which we called “arylquins”, targeted vimentin and promoted a dose-dependent secretion of Par-4 from normal cells when administered at nanomolar concentrations.11 The most active arylquins 1 possessed amino groups at C-2 (i.e., 1 where X = H, Y = NH2), at C-7 (i.e., 1 where X = N(CH3)2, Y = H) or at both positions and possessed an aryl group at C-3 bearing an ortho-fluorine or ortho-chlorine substituent. We identified vimentin as the arylquin target using a pulldown experiment with a biologically active, biotinylated arylquin and streptavidin-sepharose beads.11
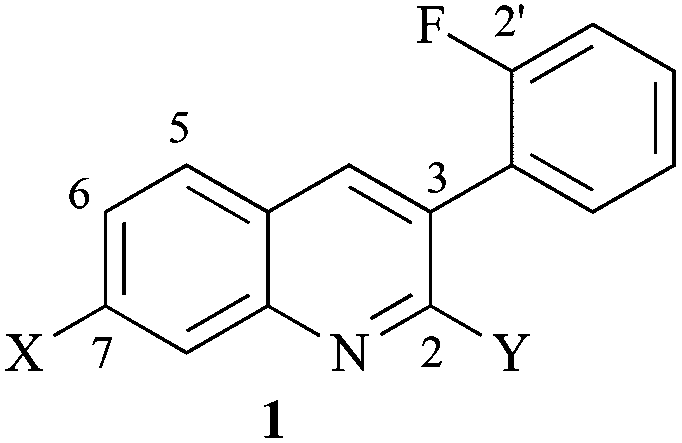 | ||
| Fig. 2 A representative arylquin, 3-(2′-fluorophenyl)-N7,N7-dimethylquinoline-2,7-diamine (1a) where X = N(CH3)2 and Y = NH2. | ||
Appropriate controls excluded the adventitious binding of vimentin to sepharose, and the absence of other, significant bands on gels of the pulldown experiment excluded the possible association of arylquins with a secondary protein that bound vimentin. Confocal microscopy established the co-localization of Par-4 and vimentin and the release of Par-4 induced by arylquin treatment. In addition, because the expression levels of vimentin were not altered by arylquins, we excluded the possible inhibition of a transcription factor that could reduce vimentin expression and thereby release Par-4 for secretion.
Targeting filament-forming proteins with compounds that inhibit filament growth or hyperstabilize filaments represents a successful strategy for the development of chemical probes and potential therapeutics. Among the prominent examples of such compounds are swinholide that inhibits actin filament growth;12 taxol that stabilizes tubulin filaments;13 vinblastine that inhibits tubulin filament growth;14,15 and withaferin A that covalently modifies Cys328 in the 2B subdomain of vimentin rod domain and causes aggregation.16 Both actin and tubulin are globular proteins as monomers, composed of both β sheets and α helices, and both possess cavities that are suitable for binding small molecules. On the other hand, a vimentin monomer is structurally distinct from actin and tubulin because of its linear rather than a globular structure. The N- and C-terminal regions of vimentin lack secondary structure, but its core, rod domain consists of α-helices connected by flexible linker regions.17 In contrast to the relatively rigid actin and tubulin monomers, a vimentin monomer is predicted to have a fairly dynamic structure, and consequently, it is not a classical, druggable target.
Monomers of vimentin associate into coiled-coil, dimeric structures that are more rigid than the flexible monomers. The dimers form antiparallel tetramers and, eventually, higher-order thick, elongated filament assemblies that are sixteen dimers thick.18 Although the exact structure of a fully-assembled vimentin filament remains unknown, this assembly possesses a well-featured surface topography. Its overexpression in various cancers, such as prostate cancer, makes it an attractive, albeit difficult, target for drug development.7 Among the unanswered questions regarding the effect of arylquins on Par-4 secretion is the stoichiometry of arylquin binding to vimentin. We now report structure–activity studies of arylquins and related compounds to identify potent arylquins for these binding studies, and we report binding experiments between arylquins and purified rod domain of vimentin (residues 99-411) that probe the issue of one-site versus multiple-binding-sites. We also successfully eliminated the undesired, human Ether-à-go-go-Related Gene (hERG) activity as a prelude to moving these compounds forward into preclinical development.
Results and discussion
Synthesis of arylquins 1 or arylquinolones 2 that were needed for the vimentin binding studies utilized the Friedländer condensation19 of substituted ortho-aminobenzaldehyde 3 and arylacetonitriles 4a or utilized the Knoevenagel condensation20 of the adduct between 3 and arylacetyl chlorides 4b, respectively (Scheme 1).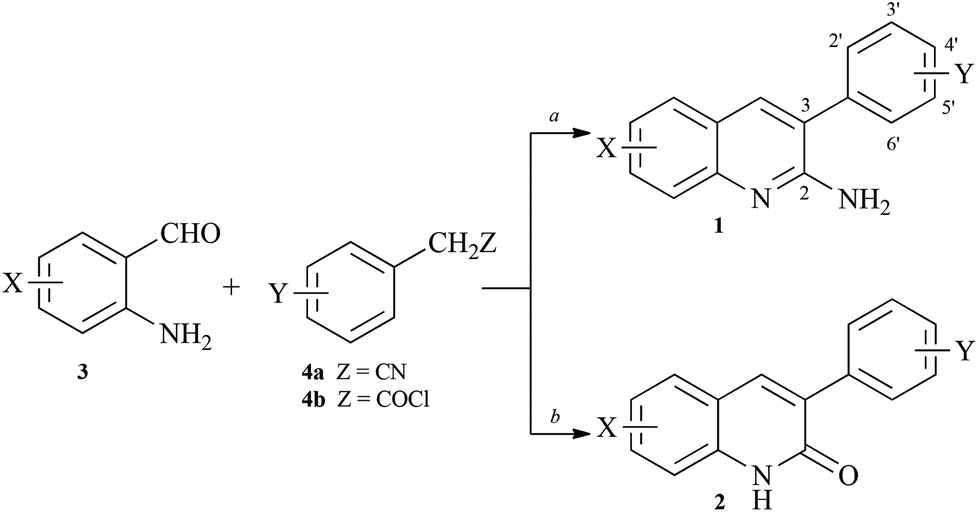 | ||
| Scheme 1 Synthesis of arylquins 1 or arylquinolones 2. Reagents: a, XC6H4CH2CN, tert-BuOK, DMF, 90 °C, 1–3 h; b, XC6H4CH2COCl, Et3N, reflux, 2 h and K2CO3, DMF, 90 °C, 4 h. | ||
Similar condensations of 4-(N,N-dimethylamino)-2-aminobenzaldehyde with 2-arylacetonitriles in which the aryl ring possessed either a ortho-cyano-or ortho-carboxylate-substituent led to the little-known but inactive, tetracyclic dibenzo[b,f][1,8]naphthyridine 5 or dibenzo[b,f][1,8]naphthyridin-5(6H)-one216, respectively (Scheme 2).
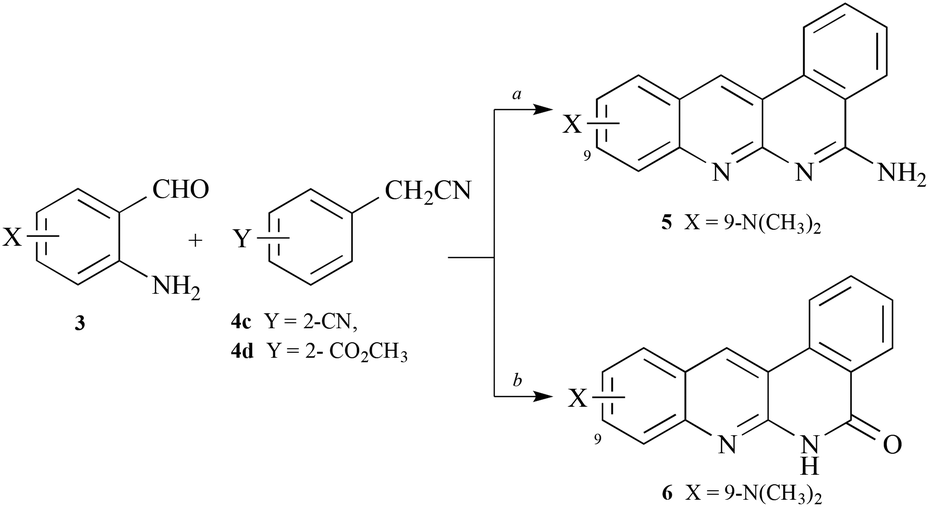 | ||
| Scheme 2 Synthesis of dibenzo[b,f][1,8]naphthyridine 5 and dibenzo[b,f][1,8]naphthyridin-5(6H)-one 6. Reagents: a, o-(NC)C6H4CH2CN (4c) or o-(CH3OCO)C6H4CH2CN (4d), tert-BuOK, DMF, 90 °C, 1–3 h. | ||
Finally, the arylquinolones 2 provided access to the thioquinolones 7, 2-thiomethoxyquinolines 8, 2-chloroquinolines 9, 2-(N-methylpiperazinyl)quinolines 10 and the 2-(morpholinyl)quinolines 11 in a straightforward series of reactions (Scheme 3). Friedländer condensation of other ortho-aminobenzaldehydes 3 with arylacetonitriles led to 12–19. These compounds (Table 1) were characterized by NMR and mass spectrometry, and their purity was established by combustion analysis.
 | ||
| Scheme 3 Synthesis of arylquins 8–19 modified at positions C-2, 5, 6 and 7. Reagents: a, 2,4-bis(4-methoxyphenyl)-2,4-dithioxo-1,3,2,4-dithiadiphosphetane (Lawesson's reagent), dioxane, reflux, 5 h; b, CH3I, K2CO3, DMF; c, POCl3, reflux 1 h; d, N-methylpiperazine or morpholine, reflux, 12 h; e, o-FC6H4CH2CN, tert-BuOK, DMF, 90 °C, 1–3 h. | ||
| Aqa | C-2 | C-3 | C-5 | C-6 | C-7 | Relative levels of Par-4 secretion by arylquins administered at 500 nM | Ratio of Par-4 secretion by arylquin relative to vehicle | Selected hERG IC50 (μM) values |
|---|---|---|---|---|---|---|---|---|
| Legend:a Ar = arylquin.b MOR = morpholino.c NMP = N-methylpiperazinyl. Numbering scheme is shown in Fig. 2. | ||||||||
| 1a 8 | NH2 | 2′-FC6H4 | H | H | N(CH3)2 | 104.7 ± 10.7 | 3.5 | 3.74 ± 1.17 |
| 1b | NH2 | 2′-ClC6H4 | H | H | N(CH3)2 | 88.7 ± 8.4 | 3.0 | 7.70 ± 2.07 |
| 1c | NH2 | 2′-BrC6H4 | H | H | N(CH3)2 | 33.6 ± 14.8 | 5.69 ± 2.56 | |
| 1d 11 | NH2 | 3′-FC6H4 | H | H | N(CH3)2 | 66.8 ± 10.9 | 1.59 ± 0.39 | |
| 1e | NH2 | 3′-ClC6H4 | H | H | N(CH3)2 | 78.3 ± 15.3 | 2.6 | 2.50 ± 0.59 |
| 1f 11 | NH2 | 4′-FC6H4 | H | H | N(CH3)2 | 12.3 ± 2.0 | 30.2 ± 10.3 | |
| 1g | NH2 | 4′-ClC6H4 | H | H | N(CH3)2 | 21.3 ± 4.6 | 8.19 ± 1.13 | |
| 1h | NH2 | 2′,3′-F2C6H3 | H | H | N(CH3)2 | 93.2 ± 10.6 | 3.1 | 5.31 ± 1.05 |
| 1i | NH2 | 2′,3′-Cl2C6H3 | H | H | N(CH3)2 | 88.4 ± 1.3 | 3.0 | 5.51 ± 0.67 |
| 1j | NH2 | 2′,4′-F2C6H3 | H | H | N(CH3)2 | 80.8 ± 3.0 | 2.7 | 1.44 ± 0.04 |
| 1k | NH2 | 2′,5′-F2C6H3 | H | H | N(CH3)2 | 86.2 ± 7.5 | 2.9 | 4.85 ± 1.11 |
| 1l | NH2 | 2′,5′-Cl2C6H3 | H | H | N(CH3)2 | 64.9 ± 13.1 | 2.2 | 6.91 ± 0.50 |
| 1m | NH2 | 2′,6′-F2C6H3 | H | H | N(CH3)2 | 44.6 ± 4.6 | 1.5 | 3.32 ± 1.91 |
| 1n | NH2 | 2′-F-6′-ClC6H3 | H | H | N(CH3)2 | 126.6 ± 1.4 | 2.4 | |
| 1o | NH2 | 2′,6′-Cl2C6H3 | H | H | N(CH3)2 | 122.7 ± 1.6 | 2.4 | |
| 1p | NH2 | 3′,4′-F2C6H3 | H | H | N(CH3)2 | 58.4 ± 14.1 | 2.0 | 2.79 ± 0.97 |
| 1q | NH2 | 3′,5′-F2C6H3 | H | H | N(CH3)2 | 47.4 ± 16.8 | 4.10 ± 0.49 | |
| 1r | NH2 | 2′-CH3OC6H4 | H | H | N(CH3)2 | 86.0 ± 12.2 | 1.7 | |
| 2 11 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
2′-FC6H4 | H | H | N(CH3)2 | 33.2 ± 0.9 | 1.1 | >100 |
| 7 11 | CS |
2′-FC6H4 | H | H | N(CH3)2 | 51.7 ± 4.9 | 1.7 | >100 |
| 8 | SCH3 | 2′-FC6H4 | H | H | N(CH3)2 | 75.4 ± 7.0 | 2.5 | ca. 100 |
| 9 | Cl | 2′-FC6H4 | H | H | N(CH3)2 | 104.5 ± 26.9 | 3.5 | ca. 100 |
| 10 | MORb | 2′-FC6H4 | H | H | N(CH3)2 | 54.4 ± 2.8 | 1.8 | 23.3 ± 3.05 |
| 11 | NMPc | 2′-FC6H4 | H | H | N(CH3)2 | 65.6 ± 3.6 | 2.2 | 9.41 ± 1.89 |
| 12 11 | NH2 | 2′-FC6H4 | H | H | H | 82.1 ± 21.2 | 2.8 | 14.6 ± 5.82 |
| 13 | NH2 | 2′-FC6H4 | F | H | H | 83.4 | 2.8 | |
| 14 | NH2 | 2′-FC6H4 | Cl | H | H | 63.2 ± 20.5 | 2.1 | ca. 100 |
| 15 | NH2 | 2′-FC6H4 | Br | H | H | 44.6 | 1.5 | |
| 16 | NH2 | 2′-FC6H4 | H | Cl | H | 81.9 ± 11.2 | 2.8 | 12.7 ± 6.83 |
| 17 | NH2 | 2′-FC6H4 | H | H | Cl | <50 | ||
| 18 | NH2 | 2′-FC6H4 | H | H | OCH3 | <50 | ||
| 19 | NH2 | 2′-FC6H4 | H | H | CO2H | <50 | ||
Prior studies indicated that arylquin 1a induced a robust secretion of Par-4 from normal cells when administered at nanomolar concentrations, and the amount of Par-4 secreted by 1a in cell culture-conditioned medium (CM) was adequate to induce apoptosis of diverse lung and prostate cancer cells.11 In the current study, mouse fibroblasts were treated with arylquins 1 at 500 nM concentrations using vehicle alone as a control. After 24 h exposure, the CM and lysates were subjected to quantitative Western blot analysis for Par-4 and collagen (Col1A1). Col1A1 secretion from the cells was not altered by exogenous application of arylquins, and therefore, Col1A1 served as a loading control to normalize Par-4 secretion in the CM. Arylquin 1a (500 nM) exposure increased Par-4 concentrations in the CM by 2-fold from basal levels of 430 pM with vehicle alone to 840 pM (Fig. 3). An indirect Western blot analysis of secreted Par-4 provided a suitable assay for examining the structure–activity relationships (SAR) for evaluating the arylquins and related compounds (Table 1). Attempts to use a commercial ELISA assay for the direct quantification of Par-4 in the Conditioned Medium (CM) were not effective due to a high background signal from the antibodies present in the residual serum, a component of the CM. Absent an effective ELISA assay, a Western blot analysis was used because previous studies indicated that quantification provided by this analysis correlated well with apoptosis induced by the CM in cancer cells.
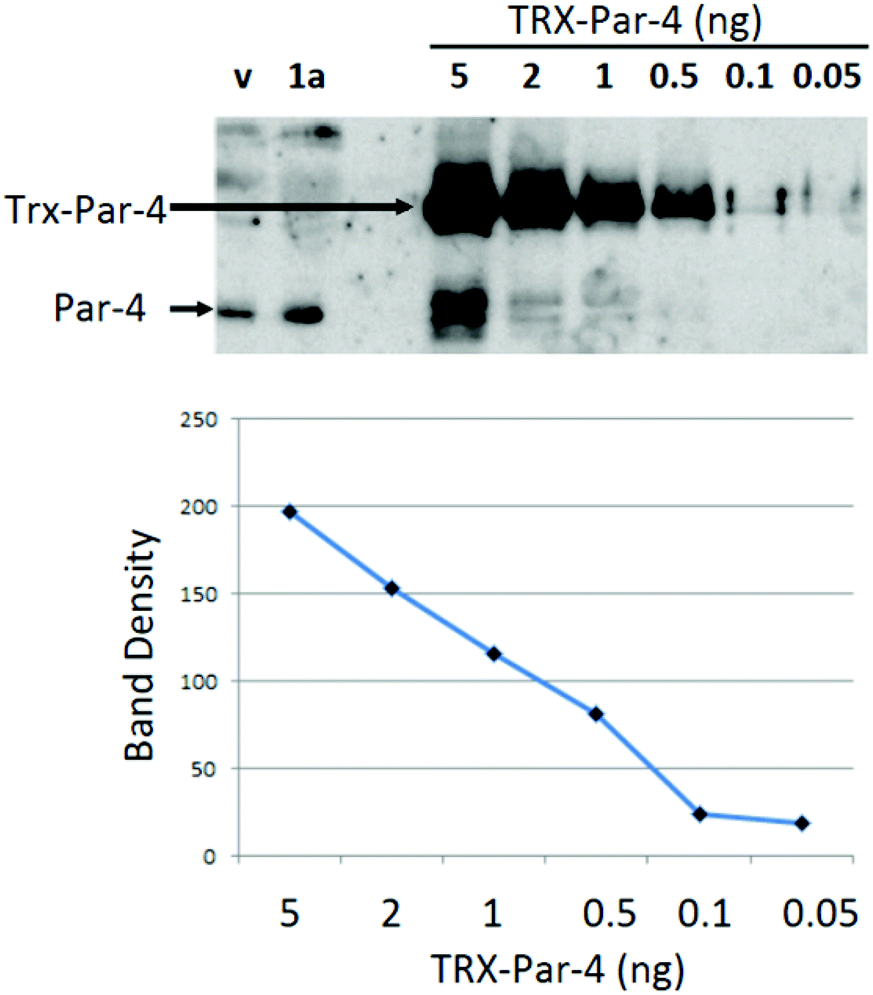 | ||
| Fig. 3 Quantitation of Par-4 secretion induced by 500 nM arylquin 1a. Calibration curve developed using fixed amounts (0.05–5 ng) of recombinant TRX-Par-4. Calculation for Par-4 concentration using arylquin 1a: band density of 40 μL of Par-4 containing solution (lane marked 1a) was 95, which corresponded to 0.42 ng of Par-4. Therefore the concentration was 0.82 ng/40 μL or 540 pM. In the same fashion, calculation for Par-4 concentration using vehicle (v) was 280 pM. | ||
It should also be emphasized that the departure point for the current SAR study was arylquin 1a that exhibited potency in the mid-nanomolar range, and consequently, only a relatively modest improvement in potency was necessary to achieve potency in the low nanomolar range needed for the binding studies described in this work and future drug development.
Computational modeling11 suggested that arylquins bound to a single, putative site on a fragment of the vimentin rod domain (residues 328-406) that was competitive with the binding site of withaferin A and presumably with a recently reported 3-azidowithaferin derivative.16,22,23 The “one-site model” suggested that the binding of arylquin 1a involved three non-covalent interactions: [1] van der Waals interactions between the C-7 N,N-dimethylamino in 1a and Leu326 and Val330; [2] a hydrogen bond between the C-2 amino group in 1a and Asp331; and [3] a hydrogen bond between the ortho-fluoro group in the C-3 aryl ring in 1a and Cys328. We probed these computational predictions by evaluating SAR modifications within arylquin 1a. As our SAR studies progressed, it became clear that some features of this “one-site model” were important. For example, arylquin 1a with an ortho-fluorophenyl group at C-3 was superior in potency as a Par-4 secretagogue to analogs lacking any substituents or to those with meta- and para-fluorophenyl groups as in 1d and 1f, respectively.11 However, not all SAR modifications were consistent with this one-site model. For example, replacement of the 7-N,N-dimethylamino group in arylquin 1a with either a 5-fluoro group, as in arylquin 13, or 6-chloro group, as in arylquin 16, produced analogs equipotent to 1a for reasons that were not consistent with the one-site model.
Suspecting that the one-site model for arylquin binding was inaccurate, we turned our attention to measuring directly the stoichiometry of arylquin binding to vimentin. Vimentin is a 53 kDa polypeptide comprised of 466 amino acids, with a highly conserved α-helical “rod” domain flanked by non-α-helical N-terminal head (77 residues) and C-terminal tail (61 residues) regions.24 Together, vimentin monomers associate in parallel and in-register to form a coiled-coil that forms the basic structural building block for the entire intermediate-filament family of proteins.25 We tested whether arylquins bound the purified rod domain of vimentin (residues 99-411), which is a much larger portion of vimentin than the segment used for the computational modeling. The rod domain consists of helices interrupted by short loops, and it has been demonstrated to form a parallel, coiled-coil dimer splayed at both termini. This dimer is a basic building block of an intermediate filament formed by the full-length vimentin.17 We titrated arylquin analogs 1a and 17 at constant concentrations with the vimentin rod domain and monitored the intrinsic fluorescence of the arylquins. Binding of the vimentin rod domain to arylquins was typically characterized by 2–3 fold enhancement in arylquin fluorescence. The intrinsic Trp fluorescence contribution to this fluorescence increase was insignificant. For example, arylquin 1a displayed activity in the Par-4 secretion assay bound to the vimentin rod domain with a Kd < 500 nM, but arylquin 17, which did not induce Par-4 secretion, did not bind the vimentin rod domain to a measurable extent.
We observed saturation of arylquin 1a at the stoichiometry of 3–4 molecules per monomer of the rod domain (Fig. 4), indicating that arylquin binding to the dimeric coiled-coil rod domain occurred at multiple sites and that the higher-order assembly of a full-length vimentin contained multiple, structurally similar arylquin binding sites. These experiments provided a direct arylquin-vimentin binding assay and demonstrated that arylquins binding to the rod domain correlated directly with ability to promote Par-4 secretion. This multi-site model was consistent with the narrow range of potencies found in the SAR study, since various arylquins could bind with differential affinities to the various sites in the rod domain. Moreover, the multiple binding site model has a structural rationale: the vimentin coiled-coil structure is quasi-repetitive because of the presence of a repetitive heptad and hendecad sequences.17
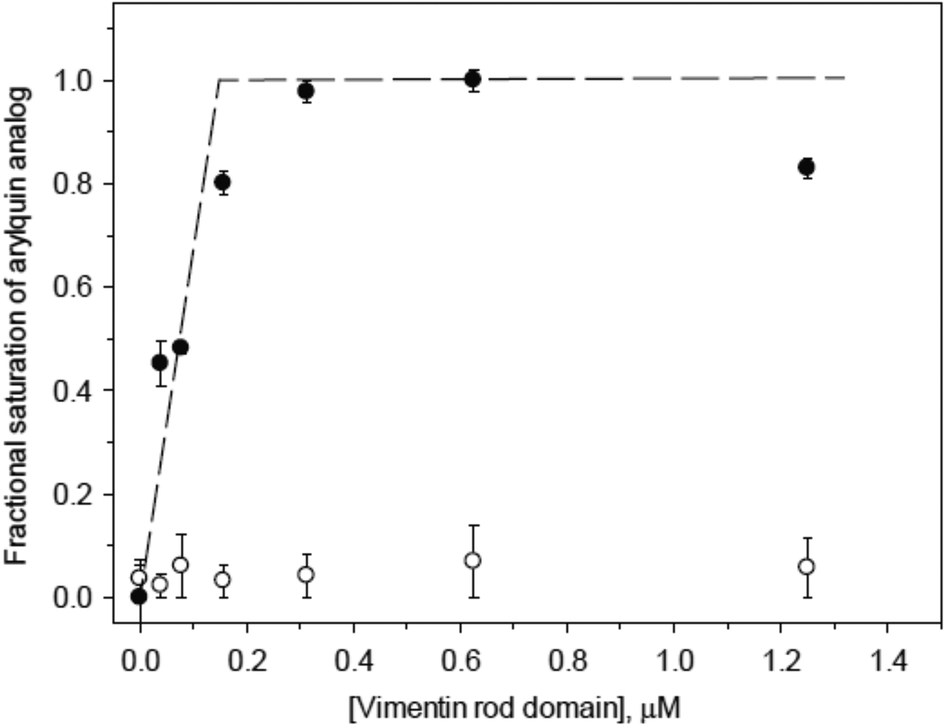 | ||
| Fig. 4 Equilibrium binding titrations of arylquins 1a (filled circles) and 17 (open circles), with vimentin rod domain. The arylquins were used at constant concentrations (500 nM for 1a and 1 μM for 17) throughout the titration. The fractional saturation for arylquin 1a was calculated assuming 100% saturation at 10 μM vimentin rod domain. Arylquin 17 fluorescence signal did not change in the titration. | ||
Potency alone, however, is no longer sufficient to guide translational drug development. We sought arylquins that minimized inhibition of the potassium channel produced from human Ether-à-go-go-Related Gene (hERG), which is associated with drug-induced, adverse cardiac events.26 We utilized a [3H]-dofetilide binding assay to evaluate the interaction of these arylquins with hERG and excluded for further development any arylquins interacting with hERG at IC50 values less than 1 μM.27
While the ideal candidate for development would exhibit no affinity for the hERG channel, such compounds are scarce, and most acceptable compounds exhibit IC50 values >5 μM, which is associated with moderate hERG inhibition.27 The arylquins reported here display a range of hERG inhibitory activities: sixteen exhibited IC50 values within the acceptable, moderately inhibiting range of 1–10 μM (Table 1); and nine were very acceptable, weak inhibitors of the hERG channel. Importantly, the structural modifications responsible for this reduction in hERG inhibition did not correlate with alterations in the ability of the compounds to facilitate Par-4 secretion of Par-4. Arylquins 11 and 12, for example, exhibited no affinity for the hERG channel and yet stimulated high levels of Par-4 secretion.
Conclusions
In summary, a structure–activity study of the arylquins using a Western blot analysis of Par-4 secretion produced compounds with potencies comparable to the originally reported11 arylquin 1a but with desired, reduced hERG inhibition. A binding study using arylquins and the purified rod domain of vimentin (residues 99-411) led to saturation at a stoichiometry of 3–4 arylquins per monomer of the rod domain. Future structural work will focus on understanding the details of arylquin binding to fragments of the rod domain of vimentin as well as the release of Par-4 for secretion.Experimental procedures
Chemicals and instruments
Chemicals were purchased from Sigma Aldrich or Fisher Scientific or were synthesized according to literature procedures. Solvents were used from commercial vendors without further purification unless otherwise noted. Nuclear magnetic resonance spectra were determined in dimethyl sulfoxide-d6 using a Varian instrument (1H, 400 MHz; 13C, 100 MHz). High resolution electrospray ionization mass spectra (HRMS ESI) were recorded on a LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The FT resolution was set at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (at 400 m/z). Samples were introduced through direct infusion using a syringe pump with a flow rate of 5 μL min−1. Purity of compounds was >95% as established by combustion analyses that was conducted by Atlantic Microlabs (Norcross, GA). Compounds were chromatographed on preparative layer Merck silica gel F254 purchased from Fisher Scientific (Pittsburgh, PA) unless otherwise indicated. Organic solutions were dried over anhydrous magnesium sulfate unless otherwise specified.
000 (at 400 m/z). Samples were introduced through direct infusion using a syringe pump with a flow rate of 5 μL min−1. Purity of compounds was >95% as established by combustion analyses that was conducted by Atlantic Microlabs (Norcross, GA). Compounds were chromatographed on preparative layer Merck silica gel F254 purchased from Fisher Scientific (Pittsburgh, PA) unless otherwise indicated. Organic solutions were dried over anhydrous magnesium sulfate unless otherwise specified.
General procedure for the synthesis of arylquins (1,5 and 6)
To a solution of 2.38 mmol (1.3 equiv.) of an appropriate arylacetonitrile 4a, 4c or 4d in 3 mL of anhydrous DMF at 0 °C was added 2.38 mmol (1.3 equiv.) of potassium tert-butoxide. The mixture was stirred for 15 min, and 1.83 mmol of a substituted ortho-aminobenzaldehyde 3 in 1 mL of anhydrous DMF was added dropwise at 0 °C. The mixture was allowed to warm to the room temperature and stirred for 1–3 h at 90 °C. After cooling, the mixture was quenched with water with vigorous stirring. The solution was adjusted to pH 7 only in the case of 2-amino-3-(2-fluorophenyl)quinoline-7-carboxylic acid (19). A precipitate was collected by filtration and purified by recrystallization and/or chromatography as noted for individual compounds described below.:10 methanol–dichloromethane (Rf = 0.74) to afford 120 mg (57%) of 8 as a pale yellow solid: mp 161–162 °C (recrystallized from ethanol). 1H NMR: δ 7.85 (s, 1H), 7.71 (d, 1H, J = 9.2 Hz), 7.53–7.47 (m, 1H), 7.42 (td, 1H, J = 7.2 Hz, 2.0 Hz), 7.35–7.28 (m, 2H), 7.18 (dd, 1H, J = 8.8 Hz, 2.6 Hz), 6.93 (d, 1H, J = 2.4 Hz), 3.08 (s, 6H), 2.53 (s, 3H). 13C NMR: δ 159.66 (d, J = 243.8 Hz), 157.95, 151.56, 148.86, 135.54, 132.31 (d, J = 2.7 Hz), 130.51 (d, J = 8.1 Hz), 128.51, 125.31 (d, J = 16.1 Hz), 124.45 (d, J = 3.3 Hz), 123.20, 117.16, 115.65 (d, J = 22.2 Hz), 114.88, 104.87, 40.02 (2C), 12.72. HRMS (ESI) Calcd for C18H18FN2S [MH+]: 313.11692. Found: 313.11790. Anal. Calcd for C18H17FN2S: C, 69.20; H, 5.48. Found: C, 68.93; H, 5.66.
:5 ethylacetate–hexane (Rf = 0.25) to afford 416 mg (78%) of 9 as a pale yellow solid: mp 170–172 °C. 1H NMR: δ 8.20 (s, 1H), 7.83 (d, 1H, J = 9.2 Hz), 7.54–7.46 (m, 2H), 7.36–7.29 (m, 3H), 6.95 (d, 1H, J = 2.4 Hz), 3.09 (s, 6H). 13C NMR: δ 159.36 (d, J = 243.6 Hz), 151.97, 148.83, 148.76, 139.50, 132.05 (d, J = 3.0 Hz), 130.50 (d, J = 8.4 Hz), 128.59, 125.54 (d, J = 16.0 Hz), 125.51 (d, J = 3.8 Hz), 122.92, 118.79, 116.87, 115.52 (d, J = 21.2 Hz), 104.18, 39.86 (2C). HRMS (ESI) Calcd for C17H14ClFN2 [MH+]: 301.09023. Found: 301.08961. Anal. Calcd for C17H14ClFN2: C, 67.89; H, 4.69. Found: C, 67.76; H, 4.52.
:20 methanol–dichloromethane (Rf = 0.60) to afford 52 mg (45%) of 10: mp 166–168 °C. 1H NMR: δ 7.87 (s, 1H), 7.64 (d, 1H, J = 9.2 Hz), 7.60 (td, 1H, J = 8.0 Hz, 1.6 Hz), 7.45–7.40 (m, 1H), 7.34–7.28 (m, 2H), 7.05 (dd, 1H, J = 8.6 Hz, 2.6 Hz), 6.79 (d, 1H, J = 2.0 Hz), 3.48 (t, 4H, J = 4.6 Hz), 3.04 (t, 4H, J = 4.6 Hz), 3.05 (s, 6H). 13C NMR: δ 158.76 (d, J = 244.4 Hz), 158.49, 151.51, 147.96, 139.23, 130.98 (d, J = 12.0 Hz), 129.42 (d, J = 8.4 Hz), 128.14, 127.26 (d, J = 14.4 Hz), 124.68 (d, J = 3.8 Hz), 117.15, 116.56, 115.97 (d, J = 22.0 Hz), 113.49, 104.90, 65.87 (2C), 49.21 (2C), 40.04 (2C). HRMS (ESI) Calcd for C21H23FN3O [MH+]: 352.1820. Found: 352.1820. Anal. Calcd for C21H22FN3O: C, 71.77; H, 6.31. Found: C, 71.59; H, 6.25.
:10 methanol–dichloromethane (Rf = 0.45) to afford 65 mg (54%) of 11: mp 177–179 °C. 1H NMR: δ 7.83 (s, 1H), 7.62 (d, 1H, J = 8.8 Hz), 7.56 (td, 1H, J = 7.6 Hz, 1.4 Hz), 7.44–7.39 (m, 1H), 7.33–7.28 (m, 2H), 7.03 (dd, 1H, J = 9.2 Hz, 2.8 Hz), 6.78 (d, 1H, J = 2.0 Hz), 3.06 (t, 4H, J = 4.6 Hz), 3.04 (s, 6H), 2.21 (t, 4H, J = 4.6 Hz), 2.12 (s, 3H). 13C NMR: δ 158.75 (d, J = 243.6 Hz), 158.66, 151.45, 148.00, 139.06, 130.97 (d, J = 3.1 Hz), 129.27 (d, J = 7.6 Hz), 128.08, 127.48 (d, J = 14.4 Hz), 124.63 (d, J = 3.1 Hz), 117.32, 116.47, 115.95 (d, J = 22.1 Hz), 113.29, 104.93, 54.37 (2C), 48.42 (2C), 45.63, 40.06 (2C). HRMS (ESI) Calcd for C22H26FN4 [MH+]: 365.2136. Found: 365.2136. Anal. Calcd for C22H25FN4: C, 72.50; H, 6.91. Found: C, 72.29; H, 6.68.
Cells and plasmids
Lung cancer cells H1299, HOP92, A549, H460, prostate cancer cells PC-3, and primary lung fibroblast cells HEL and epithelial cells HBEC and BEAS-2B were from ATCC, MD; normal human prostate epithelial cells PrEC were from Lonza Inc., Allendale, NJ. PC-3 derivatives PC-3 MM2 were from Sue-Hwa Lin, MD Anderson Cancer Center, Houston, TX. Par-4+/+ and Par-4−/− MEFs were derived from wild type and Par-4-null C57/B6 mice generated by Taconic. Vimentin-null and wild type MEFs, as well as vimentin-expressing and vimentin-deficient SW13cells were from Anthony Brown (The Ohio State University, Columbus, OH).Antibodies and siRNA duplexes
Par-4 (R334), Col1A1 (H-197), vimentin (H-84) for Western blot; vimentin (RV202) for ICC; GRP78, Col1A1 (H-197), and pan-cytokeratin (C11) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). The β-actin antibody was from Sigma Chemical Corp. (Saint Louis, MO).Co-immunoprecipitation and Western blot analysis for Par-4
Protein extracted from cell lysates was filtered, pre-cleared with 25 μL (bed volume) of protein G-Sepharose beads and immunoprecipitated with 1 μg of respective antibodies. The eluted proteins were resolved by SDS-PAGE, and subjected to Western blot analysis as described previously.11Apoptosis assays and detection of cell surface GRP78
Apoptotic nuclei were identified by ICC analysis for active caspase-3 or by DAPI staining.5,6 A total of three independent experiments were performed; and approximately 500 cells were scored in each experiment for apoptosis under a fluorescent microscope. Cell surface GRP78 expression on the cancer cell surface was quantified by FACS analysis in the Flow Cytometry Shared Resource Facility, Markey Cancer Center as previously described.5,6Computational modeling
Molecular modeling of vimentin binding with arylquin 1a and other analogs was performed using the AutoDock 4.2 program28 and the previously reported computational protocol.16,22 Briefly, a ligand was docked into the binding cavity and the resulting poses were refined by molecular dynamics (MD)-simulations. The most favorable binding mode (with the lowest binding free energy), which was identified in the docking procedure, was subjected to an MD simulation for 1 ns at 298 °K and used in binding free energy calculations.Statistical analyses
All experiments were performed in triplicate to verify the reproducibility of the findings. Data represent the mean ± standard deviation of at least three experiments. Statistical analyses were carried out with Statistical Analysis System software (SAS Institute, Cary, NC) and P values were calculated using the Student t test.hERG binding studies
To evaluate the concentration-dependent interaction of arylquins with the hERG channel, we utilized a [3H]-dofetilide binding assay employing hERG-HEK293 cell membranes.29 HEK293 cells stably expressing the hERG potassium channel (accession number U04270), referred to as hERG-HEK cells, were received at passage 11 (P11) from Millipore (CYL3006, lot 2, Billerica, MA). [3H]-Dofetilide (specific activity of 80 Ci mmol−1; labeled on the N-methyl group) was obtained from American Radiolabeled Chemicals (St. Louis, MO). Polyethylenimine (PEI) was obtained from Fluka/Sigma-Aldrich (St. Louis, MO). Essential Medium (MEM) with GlutaMAX™ and phenol red, MEM non-essential amino acids solution (NEAA, 100×), G418 disulfate salt solution, fetal bovine serum (FBS), 0.05% Trypsin-EDTA 1× with phenol red, and Hank's balanced salt solution (HBSS) were obtained from Life Technologies (Carlsbad, CA). All other chemicals and solvents were obtained from Sigma-Aldrich (Milwaukee, WI).hERG-HEK cell culture
hERG-HEK cells were cultured according to the protocol provided by Millipore. Specifically, cells were maintained in MEM (with glutamax and phenol red) supplemented with 10% FBS, 1% NEAA and 400 μg mL−1 of geneticin, and incubated at 37 °C in a humidified atmosphere with 5% CO2. Frozen aliquots of cells were transferred into T-75 cm2 flasks and allowed to adhere for 4–8 h. Culture medium was replaced every 2 days. Passages were carried out at least 3 times after thawing at 6 day intervals. Cells were dissociated with trypsin/EDTA and seeded into new 150 × 25 mm dishes at 2–3 × 106 cells per dish and placed at 30 °C, 5% CO2, for 40–48 h prior to membrane preparation. Membrane preparation occurred 6 days after the last passage (passage 20) and was based on previous methods.29–32 Cells were rinsed twice with HBSS at 37 °C and collected by scraping the dishes in ∼20 mL of ice-cold 0.32 M sucrose and homogenized on ice with a Teflon pestle using a Maximal Digital homogenizer (Fisher Scientific, Pittsburgh, PA) at ∼280 rpm for 30 s. Homogenates were centrifuged at 4 °C at 300g and 800g for 4 min each. Pellets were resuspended in 9 mL of ice-cold Milli-Q water and osmolarity restored by addition of 1 mL of 500 mM Tris buffer (pH 7.4) followed by suspension and centrifugation at 20000g for 30 min at 4 °C. Pellets were homogenized in 2 mL assay buffer (50 mM Tris, 10 mM KCl, and 1 mM MgCl2, 4 °C) and aliquots of cell membrane suspensions were stored at −80 °C and thawed the day of the [3H]-dofetilide binding assay. Prior to [3H]-dofetilide binding assay, protein content of cell membrane aliquots was determined prior to the assay using a Bradford protein assay with bovine serum albumin as the standard. Binding assays were initiated by adding cell membrane suspension (5 μg) to duplicate tubes containing assay buffer, 25 μL of a single concentration of arylquin (concentration range of 10 nM–100 μM for each experiment), and 25 μL of [3H]-dofetilide (5 nM, final concentration) for an assay volume of 250 μL, and incubating for 60 min at 25 °C. Assays were terminated by rapid filtration through Whatman GF/B filters, which were pre-soaked in 0.25% PEI overnight, using a Brandel cell/membrane harvester (M-48; Brandel Inc., Gaithersburg, MD). Filters were washed 3 times with ∼1 mL of ice-cold assay buffer. Radioactivity was determined by liquid scintillation spectrometry using the Tri-Carb 2100-TR Liquid Scintillation Analyzer (Perkin-Elmer Life and Analytical Sciences). Arylquin concentrations that produced 50% inhibition (IC50) in the biological studies were determined from the concentration–response curves via the non-linear regression one-site competition-fitting program (Prism 5.04; GraphPad Software Inc. San Diego, CA).
Cloning, expression and purification of the rod domain of vimentin
The human vimentin gene fragment encoding the rod domain (residues 99-411) was amplified by polymerase chain reaction (PCR) from a cDNA clone (clone ID: 2985712) purchased from Open Biosystems/GE Dharmacon (Lafayette, CO, USA) by using primers (5′-ATCAACCACATATGACCCGCACCAACGAGAAGGTGG-3′) and (5′-ATCGGATCCTCAAATCCTGCTCTCCTCGCCTTCC-3′). This insert was cloned between NdeI and BamHI sites of pET19b-pps vector33 encoding an N-terminal decahistidine tag cleavable by PreScission protease (GE Healthcare, Piscataway, NJ, USA). The sequence of the insert was verified by DNA sequencing (Eurofins, New Orleans, LA, USA). The plasmid was transformed into chemically-competent E. coli BL21(DE3). The transformation mixture was plated on Luria-Bertani (LB) agar containing 100 μg mL−1 of ampicillin and incubated overnight at 37 °C. A single colony from a fresh plate was inoculated into 2 mL of LB broth with 100 μg mL−1 ampicillin (LB/Amp). This culture was used to inoculate 2 L of LB/Amp and was incubated with shaking at 15 °C until the attenuance at 600 nm of 0.6. Protein overexpression was then induced by 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). The induced culture was then incubated with shaking for 16 h at 15 °C. The cells were and harvested by centrifugation (10min, 8000g, 4 °C). The cell pellet from the 2 L culture was resuspended in 40 mL of buffer A (40 mM Tris-HCl pH 8.0, 400 mM NaCl, 10% of glycerol, 2 mM β-mercaptoethanol). The cells were disrupted by sonication on ice and lysate was clarified by centrifugation at 38000g for 40 min. The supernatant was filtered through a 0.45 μm Millex-HV PVDF filter (Millipore, Billerica, MA, USA) and applied to a 5 mL Ni-IMAC HisTrap FF column (GE Healthcare) equilibrated with buffer A. The column was washed with 25 ml of buffer A containing 50 mM imidazole, and protein was eluted with 10 mL of buffer A containing 500 mM imidazole. The eluate was dialyzed against buffer B (40 mM Tris-HCl pH 8.0, 200 mM NaCl, 2 mM β-mercaptoethanol), concentrated, and loaded onto a size-exclusion S-200 column (GE Healthcare). Peak fractions (5 mL) were collected and the purity of the protein was checked on a Coomassie-stained SDS–PAGE gel. The fractions containing the rod domain of vimentin were pooled and concentrated to 5 mg mL−1.
Vimentin-arylquin binding assay
Arylquins displayed a natural fluorescence excitation maximum at 320 nm. The emission maxima were then identified for each compound by a fluorescence emission scan from 400 nm to 600 nm. The emission maxima were as follows: arylquin 1a was 450 nm, and arylquin 17 was 430 nm. Vimentin rod domain was serially diluted in the buffer B on ice. Binding assays were carried out in buffer B with 500 nM of arylquin 1a, or 1 μM of arylquin 17 with increasing protein concentration, as specified in the text. After the binding mix was incubated for 15 min at 22 °C, the fluorescence yield measurements were carried out on a SpectraMax M5 multimode plate reader (Sunnyvale, CA, USA) in flat-bottom 96-well black plates (Monroe, NC) at 22 °C. The fluorescence yield was measured at the excitation wavelength of 320 nm and the emission wavelength at the above emission maximum for each compound.Acknowledgements
CL and DSW were supported by R21 CA139359 and CA172379 from the NIH. VMR, C-GZ and DSW were supported by CA 187273 from the NIH. VMR was supported by R01 CA060872 and R21 CA179283 from the NIH. CL, DSW and VMR were supported by a University of Kentucky Center for Clinical and Translational Science Drug Discovery Pilot Grant. LPD and JRN were supported by U01 DA013519, UL1TR000117 and T32 DA016176 from the NIH. DSW was supported by the Office of the Dean of the College of Medicine and research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P30 GM110787. In accord with University of Kentucky policy, Chunming Liu, David S. Watt, Vivek Rangnekar, Ravshan Burikhanov, and Vitaliy M. Sviripa have disclosed this work to the University's Intellectual Property Committee that has sought patent protection. Chunming Liu, David S. Watt, and Vivek Rangnekar have partial ownership in a private venture, Curonc, LLC, to develop this technology. The manuscript's contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the NIGMS.References
- R. Siegel, D. Naishadham and A. Jemal, CA-Cancer J. Clin., 2013, 63, 1 CrossRef PubMed.
- T. Karantanos, P. G. Corn and T. C. Thompson, Oncogene, 2013, 32, 5501–5511 CrossRef CAS PubMed.
- L. Bubendorf, A. Schöpfer, U. Wagner, G. Sauter, H. Moch, N. Willi, T. C. Gasser and M. J. Mihatsch, Hum. Pathol., 2000, 31, 578 CrossRef CAS PubMed.
- N. Hebbar, C. Wang and V. M. Rangnekar, J. Cell Phys., 2012, 227, 3715 CrossRef CAS PubMed.
- R. Burikhanov, Y. Zhao, A. Goswami, S. Qiu, S. R. Schwarze and V. M. Rangnekar, Cell, 2009, 138, 377 CrossRef CAS PubMed.
- R. Burikhanov, T. Shrestha-Bhattarai, S. Qiu, N. Shukla, N. Hebbar, S. M. Lele, C. Horbinski and V. M. Rangnekar, Cancer Res., 2013, 73, 1011 CrossRef CAS PubMed.
- A. Satelli and S. Li, Cell. Mol. Life Sci., 2011, 68, 3033–3046 CrossRef CAS PubMed.
- J. Li and A. S. Lee, Curr. Mol. Med., 2006, 6, 45 CrossRef CAS PubMed.
- A. S. Lee, Trends Biochem. Sci., 2001, 26, 504 CrossRef CAS PubMed.
- A. S. Lee, Cancer Res., 2007, 67, 3496 CrossRef CAS PubMed.
- R. Burikhanov, V. M. Sviripa, N. Hebbar, W. Zhang, W. J. Layton, A. Hamza, C.-G. Zhan, D. S. Watt, C. Liu and V. M. Rangnekar, Nat. Chem. Biol., 2014, 10, 924 CrossRef CAS PubMed.
- M. R. Bubb, I. Spector, A. D. Bershadsky and E. Korn, J. Biol. Chem., 1995, 270, 3463 CrossRef CAS PubMed.
- P. B. Schiff, J. Fant and S. B. Horwitz, Nature, 1979, 277, 665 CrossRef CAS PubMed.
- K. G. Bensch and S. E. Malawista, Nature, 1968, 218, 1176 CrossRef CAS PubMed.
- K. G. Bensch, R. Marantz, H. Wisniewski and M. Shelanski, Science, 1969, 165, 495 CAS.
- P. Bargagna-Mohan, A. Hamza, Y. E. Kim, Y. Khuan Abby Ho, N. Mor-Vaknin, N. Wendschlag, J. Liu, R. M. Evans, D. M. Markovitz, C.-G. Zhan, K. B. Kim and R. Mohan, Chem. Biol., 2007, 14, 623 CrossRef CAS PubMed.
- A. A. Chernyatina, S. Nicolet, U. Aebi, H. Herrmann and S. V. Strelkov, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13620 CrossRef CAS PubMed.
- H. Herrmann, M. Haner, M. Brettel, S. A. Muller, K. N. Goldie, B. Fedtke, A. Lustig, W. W. Franke and U. Aebi, J. Mol. Biol., 1996, 264, 933 CrossRef CAS PubMed.
- J. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M. do Carmo Carreiras and E. Soriano, Chem. Rev., 2009, 109, 2652 CrossRef CAS PubMed.
- G. Jones, Org. React., 1967, 15, 204 CAS.
- A. Venkatesan, A. Agarwal, T. Abe, H. Ushirogochi, T. Takasaki, A. Mihira and T. S. Mansour, ChemMedChem, 2008, 3, 1658 CrossRef CAS PubMed.
- P. Bargagna-Mohan, R. R. Paranthan, A. Hamza, N. Dimova, B. Trucchi, C. Srinivasan, G. I. Elliott, C.-G. Zhan, D. L. Lau, H. Zhu, K. Kasahara, M. Inagaki, F. Cambi and R. Mohan, J. Biol. Chem., 2010, 285, 7657 CrossRef CAS PubMed.
- R. Rah, R. U. Rasool, D. Nayak, S. K. Yousuf, D. Mukherjee, L. D. Kumar and A. Goswami, Autophagy, 2015, 11, 314 CrossRef PubMed.
- K. N. Goldie, T. Wedig, A. K. Mitra, U. Aebi, H. Herrmann and A. Hoenger, J. Struct. Biol., 2007, 158, 378 CrossRef CAS PubMed.
- E. Fuchs and I. Hanukoglu, Cell, 1983, 34, 332 CrossRef CAS PubMed.
- W. S. Redfern, L. Carlsson, A. S. Davis, W. G. Lynch, I. MacKenzie, S. Palethorpe, P. K. Siegl, I. Strang, A. T. Sullivan, R. Wallis, A. J. Camm and T. G. Hammond, Cardiovasc. Res., 2003, 58, 32 CrossRef CAS PubMed.
- B. D. Guth and G. Rast, Brit. J. Pharmcol., 2010, 159, 22–24 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785 CrossRef CAS PubMed.
- T. Chen, A Practical Guide to Assay Development and High-throughput Screening in Drug Discovery, CRC Press Taylor and Francis Group, Boca Raton, FL, 2010 Search PubMed.
- S.-H. Jo, J. B. Youm, C. O. Lee, Y. E. Earm and W.-K. Ho, Br. J. Pharmacol., 2000, 129, 1474 CrossRef CAS PubMed.
- T. Biswas, E. Resto-Roldán, S. K. Sawyer, I. Artsimovitch and O. V. Tsodikov, Nucleic Acids Res., 2013, 41, e56 CrossRef CAS PubMed.
- P. M. Greengrass, M. Stewart and C. M. Wood, Patent WO2003021271 A2, 2003 Search PubMed.
- T. Biswas and O. V. Tsodikov, FEBS J., 2008, 275, 3064 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |