
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A protocol for amide bond formation with electron deficient amines and sterically hindered substrates†
Maria E.
Due-Hansen
,
Sunil K.
Pandey
,
Elisabeth
Christiansen
,
Rikke
Andersen
,
Steffen V. F.
Hansen
and
Trond
Ulven
*
Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, DK-5230 Odense M, Denmark. E-mail: ulven@sdu.dk; Fax: +45 6615 8780; Tel: +45 6550 2568
First published on 5th November 2015
Abstract
A protocol for amide coupling by in situ formation of acyl fluorides and reaction with amines at elevated temperature has been developed and found to be efficient for coupling of sterically hindered substrates and electron deficient amines where standard methods failed.
Amide coupling reactions are common in organic synthesis and the most frequent reaction used in medicinal chemistry.1 The reaction is typically performed by combining an activated carboxylic acid with an amine and a substantial number of highly efficient amide coupling protocols have been developed, especially for peptide synthesis.2 Although often considered to be a solved problem in organic synthesis, amide couplings that perform poorly with established protocols are relatively frequently encountered, in particular with sterically hindered substrates and electron deficient amines, and alternative strategies for synthesis of such amides have been developed.3 We here report a protocol for coupling of carboxylic acids with amines that has proven efficient where other methods have failed.
In our project directed towards the free fatty acid receptor 2 (FFA2/GPR43), we wished to synthesize a compound disclosed in the patent literature via intermediate 1 (Table 1).4 The literature indicated that the central coupling between carboxylic acid 1a and the N-cyclopropyl-2-aminothiazole 1b had been performed with HATU and DIPEA in anhydrous DMF over four days.4b However, this method produced unsatisfactory results in our hands (Table 1, entry 1), especially in light of the laborious multistep sequences required for the production of both 1a and 1b. We proceeded by exploring other conventional amide coupling methods, such as, EDC/HOBt, DCC/DMAP, coupling via the acid chloride using Ghosez’s reagent,5 and by Ag(I)-promoted activation of the acid chloride,6 however, none of these provided the desired product in significant amounts (Table 1, entries 2–5).

The difficulties observed with this coupling were linked to the electron poor and sterically hindered amine in combination with a hindered carboxylic acid containing a tert-butyl ester that is sensitive to forcing conditions.10 We reasoned that a coupling strategy that minimized steric hindrance between the coupling partners would be more likely to succeed. Acyl fluorides are ideal in this respect and are well established as relatively stable and easily handled substrates that nevertheless exhibit high reactivity towards amines in amide couplings of for example sterically hindered amino acids.2c–e One-pot amide coupling reactions via acyl fluorides have been developed with the fluorination reagents DAST and Deoxo-Fluor, but these may give rise to side products from coupling with the released diethylamine or bis(methoxyethyl)amine.2d,11 XtalFluor-E is a related reagent that also efficiently promote amide coupling under mild conditions, but release of diethylamine and side product formation is a concern also for this reagent.12
We decided to explore fluorouronium reagents such as TFFH13 and BTFFH,14 that have given excellent results in solid-phase coupling of sterically hindered amino acids.13–15 Using conditions inspired from solid-phase peptide synthesis,13 a conversion of only 8% was obtained after 24 hours (Table 2, entry 1). Despite the modest success, the reaction was repeated with heating to 80 °C in a microwave reactor, resulting in an encouraging 43% conversion and 20% isolated yield of 1 (entry 2). By HPLC monitoring, formation of the acid fluoride in CH2Cl2 was found to be significantly faster and less prone to hydrolysis than in DMF. CH2Cl2 as solvent gave a conversion of 23% after 24 h at room temperature (entry 3). As expected, TFFH gave a similar conversion (entry 4). BTFFH is preferred over TFFH as the latter is reported to form toxic by-products.2b,16 A conversion of 85% and an isolated yield of 61% after 4 h at 80 °C was observed with BTFFH in the microwave reactor (entry 5). In contrast, only 50% conversion was observed in refluxing 1,2-dichloroethane (entry 6). DMAP has been reported to catalyse the coupling of acyl fluorides with alcohols and thiols.17 In our system, the addition of 0.2 equiv. DMAP rather had a detrimental effect on the yield (entry 7), possibly due to the higher steric demand of the N-acyl DMAP intermediate. Increasing the temperature above 80 °C did not result in significant improvements (entries 8–10), whereas extending the reaction time to 24 hours using conventional heating in a sealed vial eventually resulted in complete conversion and 85% isolated yield of the desired product (entry 11). By increasing the concentration of the reaction mixture, the reaction time could be reduced to 12 hours (Table 3). The challenging nature of this reaction is illustrated by the unusual instability of the amide of 1, for which prolonged exposure to even weak acids such as acetic acid leads to clean hydrolysis back to 1a and 1b. Deprotection of the tert-butyl ester by treatment with TFA under anhydrous conditions is tolerated, but the carboxylic acid product must be stored in solution or on salt form to avoid self-catalysed hydrolysis.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent (cat.) | Temp. (°C) | Time (h) | Conv.b (%) | Yieldc (%) |
| a Reaction conditions: 1a (1.3 equiv.), BTFFH (1.5 equiv.), DIPEA (4.5 equiv.) in 4 mL mmol−1 solvent, 30 min at rt; then 1b (0.25 mmol, 1 equiv.). b Determined by HPLC. c Isolated yields. d 1a (1 equiv.), BTFFH (1.2 equiv.). e Not determined. f As entry 3 but with TFFH. g 1a (1.1 equiv.) h Microwave heating. i Reflux. j 1a (1.2 equiv.) k Conventional heating in sealed vial. | |||||
| 1d | DMF | rt | 24 | 8 | —e |
| 2 | DMF | 80 | 2.5 | 43 | 20 |
| 3d | CH2Cl2 | rt | 24 | 23 | —e |
| 4f | CH2Cl2 | rt | 24 | 21 | —e |
| 5g | CH2Cl2 | 80h | 4 | 85 | 61 |
| 6g | DCE | 84i | 24 | 50 | —e |
| 7g | CH2Cl2 (DMAP) | 80h | 4 | 63 | 35 |
| 8j | CH2Cl2 | 80h | 7 | 94 | —e |
| 9j | CH2Cl2 | 100h | 7 | 96 | 78 |
| 10j | CH2Cl2 | 140h | 3 | 87 | 75 |
| 11 | CH2Cl2 | 80k | 24 | 100 | 85 |

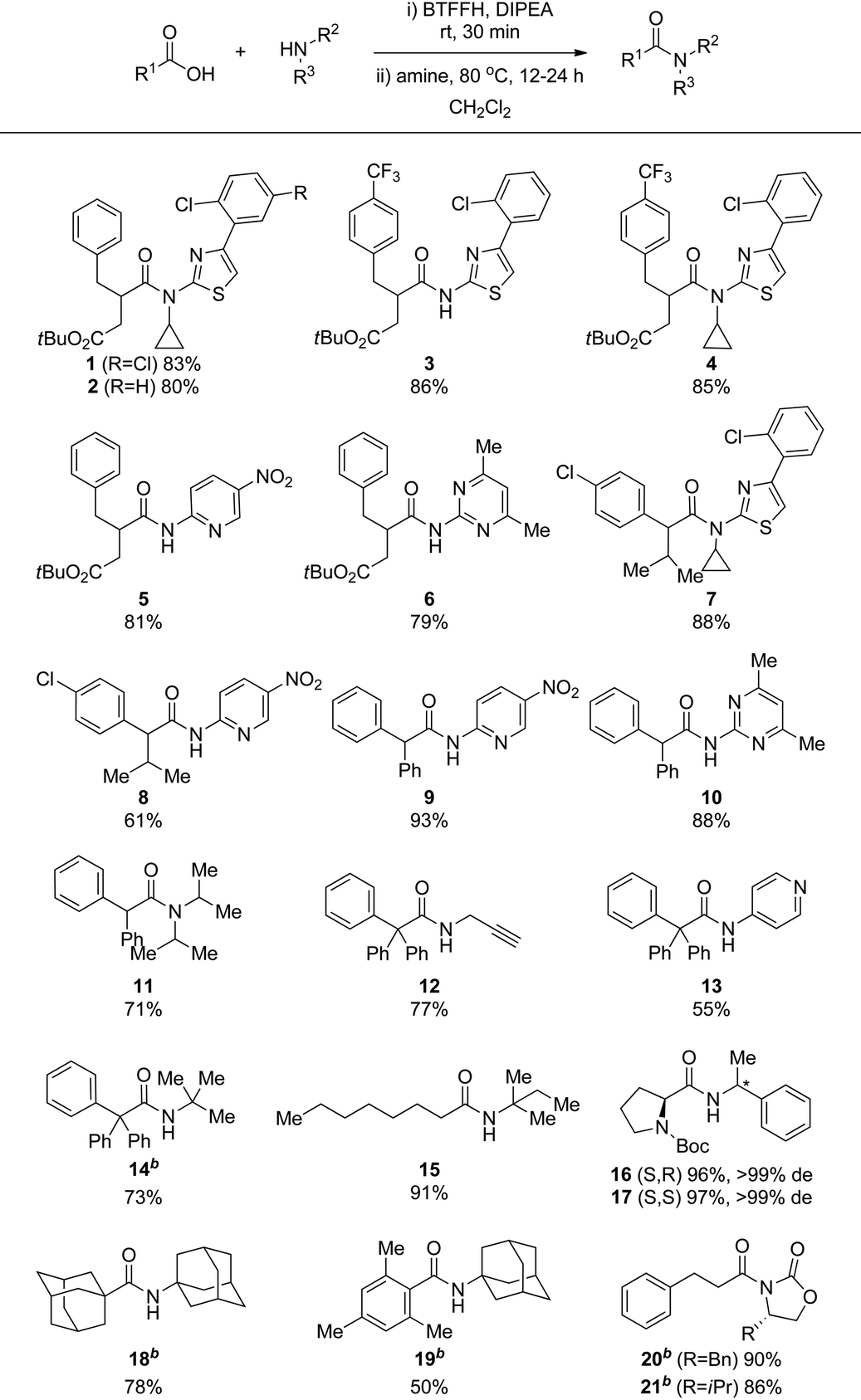
Pleased with the results from the optimized BTFFH promoted coupling of 1a with 1b, we wished to explore the scope of the reaction for coupling of other sterically hindered substrates and electron deficient amines (Table 3). Thus, a selection of analogous substrates all provided the desired amide in good to excellent yield (2–4). 1a was coupled with poor nucleophiles such as 2-amino-5-nitropyridine and 2-amino-4,6-dimethylpyrimidine, resulting in complete conversion, preserved tert-butyl esters, and good isolated yields of 5 and 6. The same electron deficient amines were also coupled with other hindered carboxylic acids such as 2-(4-chlorophenyl)-3-methylbutanoic acid and diphenylacetic acid, giving rise to moderate to high isolated yields of 7–10. To estimate the degree of challenge represented by these amide coupling reactions, they were also attempted using a standard EDC/HOBt method, resulting in 38% conversion for 7 and only trace of products 8–10 by HPLC.
The method was further tested on other amide coupling reactions reported with low yields in the literature. The coupling of diphenylacetic acid with diisopropylamine has been reported in 20% yield by a TaCl5-based method especially developed for coupling of hindered substrates, whereas no product was observed by DCC-mediated coupling.18 Since the diphenylacetyl fluoride intermediate and the product co-eluted on silica, an excess of the amine was used in this case, giving full conversion to 11 and 71% isolated yield. Coupling of the hindered triphenylacetic acid with propargylamine, 4-aminopyridine and tert-butylamine, by DCC or TaCl5 or via the acid chloride catalysed by DMAP are reported with low to moderate yields in the literature.18,19 In all cases, coupling by the optimized BTFFH method resulted in significantly improved yields of 12–14. Coupling of octanoic acid with tert-pentylamine by XtalFluor-E was reported in only 8% yield due to reaction with the released diethylamine.12 Using the optimized BTFFH method, 15 was isolated in 91% yield. Acyl fluorides are reported to be less prone to α-racemization than acyl chlorides.13,16 To investigate our method in this respect, we coupled N-Boc-L-proline with both enantiomers of 1-phenethylamine, giving 16 and 17 in excellent yields and with no sign of epimerization as determined after deprotection due to rotameric forms of the Boc group.
To access some of the most challenging extremely hindered amides, Bode and co-workers have devised a method involving addition of Grignard reagents to isocyanates.3b For example, the very hindered 18, previously synthesized in 19% via adamantanecarbonyl chloride,20 was obtained in 75% by addition of adamantylmagnesium bromide to adamantyl isocyanate.3b The BTFFH protocol provided 18 in a highly satisfactory 78% isolated yield. The sterically hindered amide 19, synthesized in 87% yield by addition of mesityl magnesium bromide to adamantyl isocyanate,3b was subjected to the BTFFH protocol, resulting in a satisfactory 50% isolated yield. Thus, the BTFFH protocol can also give access to some of the extremely hindered amides that has previously required Grignard addition to isocyanates for efficient synthesis. The advantage of the BTFFH protocol in this respect is a wider general scope that also includes tertiary amides and sensitive functional groups, although it is notable that methyl esters and ketones also can be accommodated in the isocyanate fragment with Grignard addition at low temperature.3b
N-Acyloxazolidinones are important intermediates, e.g. in Evans’ asymmetric aldol reaction, that are usually prepared by nBuLi promoted N-deprotonation followed by reaction with an acyl chloride.21 Carreira and co-workers recently found acyl fluorides to efficiently couple directly with oxazolidinones.22 To evaluate the suitability of our method for this reaction, 3-phenylpropionic acid was reacted with oxazolidinones, resulting in 20 and 21 in excellent isolated yield. The BTFFH protocol thus represents a more practical one-pot route to these structures.
Although the BTFFH protocol proved successful in the synthesis of a wide variety of challenging amide bonds, the method generally failed with arylacetic acid substrates lacking a second α-substituent such as phenylacetic acid, indole-3-acetic acid and benzothiophene-3-acetic acid. The cause for this has not been investigated but might be related to decomposition via ketene formation. Also, coupling partners with further increased steric hindrance, such as 2,2,6,6-tetramethylpiperidine or coupling of the hindered and electron deficient 2,5-dichloroaniline with mesitylcarboxylic acid, failed to provide the desired amide product and generally resulted in recovery of the acyl fluoride. The product from the latter coupling can be accessed by Bode's Grignard procedure,3b demonstrating that the two methods have complementary scopes.
Conclusions
We have developed a method for efficient coupling of sterically hindered carboxylic acids with hindered or electron deficient amines and oxazolidinones via the acyl fluoride at elevated temperature. No α-racemization was observed in the coupling of Boc-proline and the method was found to be efficient for a diverse variety of sterically hindered substrates and electron deficient amines, frequently providing the desired product in good to excellent isolated yield where other methods have failed to give satisfactory results.Acknowledgements
We thank Assoc. Prof. Paul C. Stein for NMR assistance, Lone Overgaard Storm for technical support and the Danish Council for Independent Research | Technology and Production Sciences (grant 09-070364) and the Danish Council for Strategic Research (grant 11-116196) for financial support.Notes and references
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed; (b) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS PubMed; (c) M. M. Joullié and K. M. Lassen, ARKIVOC, 2010, 8, 61 Search PubMed; (d) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC; (e) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS.
- (a) G. Schafer and J. W. Bode, Chimia, 2014, 68, 252 CrossRef CAS PubMed; (b) G. Schäfer, C. Matthey and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9173 CrossRef PubMed; (c) B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027 CrossRef CAS PubMed; (d) G. Schäfer and J. W. Bode, Org. Lett., 2014, 16, 1526 CrossRef PubMed.
- (a) H. Hoveyda, C. E. Brantis, G. Dutheuil, L. Zoute, D. Schils and G. Fraser, Compounds, pharmaceutical composition and methods for use in treating gastrointestinal disorders, WO2011076732A1, 2011 Search PubMed; (b) H. Hoveyda, C. E. Brantis, G. Dutheuil, L. Zoute, D. Schils and J. Bernard, Compounds, pharmaceutical composition and methods for use in treating metabolic disorders, WO2010066682A1, 2010 Search PubMed; (c) B. D. Hudson, M. E. Due-Hansen, E. Christiansen, A. M. Hansen, A. E. Mackenzie, H. Murdoch, S. K. Pandey, R. J. Ward, R. Marquez, I. G. Tikhonova, T. Ulven and G. Milligan, J. Biol. Chem., 2013, 288, 17296 CrossRef CAS PubMed.
- A. Devos, J. Remion, A.-M. Frisque-Hesbain, A. Colens and L. Ghosez, J. Chem. Soc., Chem. Commun., 1979, 1180 RSC.
- A. C. Spivey, J. McKendrick, R. Srikaran and B. A. Helm, J. Org. Chem., 2003, 68, 1843 CrossRef CAS PubMed.
- A. F. Larsen and T. Ulven, Org. Lett., 2011, 13, 3546 CrossRef CAS PubMed.
- E. Tsandi, C. G. Kokotos, S. Kousidou, V. Ragoussis and G. Kokotos, Tetrahedron, 2009, 65, 1444 CrossRef CAS.
- W.-C. Shieh, Z. Du, H. Kim, Y. Liu and M. Prashad, Org. Process Res. Dev., 2014, 18, 1339 CrossRef CAS.
- L. A. Carpino, D. Sadat-Aalaee, H. G. Chao and R. H. DeSelms, J. Am. Chem. Soc., 1990, 112, 9651 CrossRef CAS.
- J. M. White, A. R. Tunoori, B. J. Turunen and G. I. Georg, J. Org. Chem., 2004, 69, 2573 CrossRef CAS PubMed.
- A. Orliac, D. Gomez Pardo, A. Bombrun and J. Cossy, Org. Lett., 2013, 15, 902 CrossRef CAS PubMed.
- L. A. Carpino and A. El-Faham, J. Am. Chem. Soc., 1995, 117, 5401 CrossRef CAS.
- A. El-Faham, Chem. Lett., 1998, 27, 671 CrossRef.
- (a) K. Holland-Nell, M. I. Fernandez-Bachiller, Ahsanullah and J. Rademann, Org. Lett., 2014, 16, 4428 CrossRef CAS PubMed; (b) A. El-Faham, S. N. Khattab, M. Abdul-Ghani and F. Albericio, Eur. J. Org. Chem., 2006, 1563 CrossRef CAS; (c) Y.-A. Kim, H.-N. Shin, M.-S. Park, S.-H. Cho and S.-Y. Han, Tetrahedron Lett., 2003, 44, 2557 CrossRef CAS.
- J. W. Lippert, ARKIVOC, 2005, 14, 87 Search PubMed.
- M. Pittelkow, F. S. Kamounah, U. Boas, B. Pedersen and J. B. Christensen, Synthesis, 2004, 2485 CAS.
- J. B. Fang, R. Sanghi, J. Kohn and A. S. Goldman, Inorg. Chim. Acta, 2004, 357, 2415 CrossRef CAS.
- (a) K. Kizjakina, J. M. Bryson, G. Grandinetti and T. M. Reineke, Biomaterials, 2012, 33, 1851 CrossRef CAS PubMed; (b) R. S. Dothager, K. S. Putt, B. J. Allen, B. J. Leslie, V. Nesterenko and P. J. Hergenrother, J. Am. Chem. Soc., 2005, 127, 8686 CrossRef CAS PubMed.
- C. E. Wagner, M. L. Mohler, G. S. Kang, D. D. Miller, E. E. Geisert, Y.-A. Chang, E. B. Fleischer and K. J. Shea, J. Med. Chem., 2003, 46, 2823 CrossRef CAS PubMed.
- D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127 CrossRef CAS.
- C. S. Schindler, P. M. Forster and E. M. Carreira, Org. Lett., 2010, 12, 4102 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, compound characterization, NMR spectra. See DOI: 10.1039/c5ob02129d |
| This journal is © The Royal Society of Chemistry 2016 |