
A method for investigating the stereochemical course of terpene cyclisations†
Patrick
Rabe
,
Jan
Rinkel
,
Tim A.
Klapschinski
,
Lena
Barra
and
Jeroen S.
Dickschat
*
Kekulé-Institut für Organische Chemie und Biochemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Straße 1, 53121 Bonn, Germany. E-mail: dickschat@uni-bonn.de
First published on 15th October 2015
Abstract
Three sesquiterpene cyclases from Streptomyces scabei 87.22, Streptomyces venezuelae ATCC 10712 and Streptomyces clavuligerus ATCC 27064 were characterised and their products were identified as (−)-neomeranol B, (+)-isodauc-8-en-11-ol and (+)-intermedeol, respectively. The stereochemical courses of the terpene cyclisations were investigated by use of various 13C-labelled FPP isotopomers. A quick and easy test was developed that allows to distinguish reprotonations of olefinic double bonds in neutral intermediates from the two stereoheterotopic faces. The method makes use of incubating 13C-FPP isotopomers labelled at the reprotonated carbon in deuterium oxide and subsequent HSQC analysis of the product. A 1,7-cyclisation towards (+)-isodauc-8-en-11-ol was followed by use of (1,7-13C2)FPP. Surprisingly, the (+)-isodauc-8-en-11-ol also accepted (2Z,6E)-FPP resulting in the same product profile as obtained from (2E,6E)-FPP.
Introduction
Terpenes are a structurally diverse class of natural products which are present in all kingdoms of life. Their biosynthesis proceeds via the linear precursors geranyl diphosphate (monoterpenes, C10), farnesyl diphosphate (sesquiterpenes, C15) or geranylgeranyl diphosphate (diterpenes, C20) that are converted into (poly)cyclic terpene hydrocarbons or alcohols by terpene cyclases. As crystallographic data reveal,1–8 class 1 enzymes exhibit, despite an overall low sequence conservation, a highly conserved α-helical fold, and in their active sites the highly conserved aspartate-rich motif (DDXX(X)(D,E)) near position 90 and approximately 130 residues downstream the NSE triad (NDXXSXX(R,K)(E,D)) for binding of a trinuclear Mg2+ cluster. The substrate binds in turn with its diphosphate moiety to the Mg2+ cations for formation of a highly reactive allyl cation. A reaction cascade involving cyclisations, carbon backbone rearrangements, hydride migrations and a terminal deprotonation or nucleophilic attack of water yields a terpene hydrocarbon or alcohol.9,10 The recently obtained crystal structure of selina-4(15),7(11)-diene synthase from Streptomyces pristinaespiralis in combination with site-specific mutations demonstrated the additional involvement of a highly conserved arginine (pyrophosphate sensor) that is part of an effector triad located at the helix G break, in most bacterial enzymes exactly 46 residues upstream of the NSE triad, and a RY dimer near the enzyme's C-terminus that are both important for substrate recognition.8 Cationic intermediates along the cyclisation cascade can be stabilised by cation–π-interactions with aromatic residues.4,5,7,8,11 To date, the products of nearly 50 bacterial type I terpene synthases have been characterized,11–31 and the functions of many enzymes can be delineated from their high sequence identity to these known enzymes. This work has resulted in the assignment of a function to nearly half of the ca. 600 presumptive terpene cyclases encoded in the genomes of sequenced bacteria, while the other half of these enzymes still awaits functional characterisation. Here we present the molecular cloning and expression of three bacterial terpene cyclases and structure elucidation of the products made by these enzymes. Furthermore, the enzyme mechanisms were investigated by isotopic labelling experiments.Results and discussion
The gene of an unidentified terpene synthase from Streptomyces scabiei 87.22 (WP_013004899) was cloned into the expression vector pYE-Express by homologous recombination in yeast.26 The His-tagged recombinant protein was expressed in E. coli, purified by Ni-NTA chromatography and incubated with oligoprenyl diphosphates for functional characterisation. While GPP and GGPP were not accepted as substrates, FPP conversion yielded a product with an EI mass spectrum that was not included in our mass spectral libraries (Fig. 1a of ESI†). EI-MS-QTOF analysis pointed to a sesquiterpene alcohol (m/z = 222.1970, calc. for C15H26O+: 222.1978). The compound was purified from an enzymatic conversion of 80 mg FPP, yielding 4.1 mg of the pure sesquiterpene alcohol, and its structure was determined by one- and two-dimensional NMR spectroscopy (Table 1).| 1 Ca | 1H (δ, m, J, int)b | 13C (δ)c | 3 Ca | 1H (δ, m, J, int)b | 13C (δ)c | 4 Ca | 1H (δ, m, J, int)b | 13C (δ)c |
|---|---|---|---|---|---|---|---|---|
| a Carbon numbering as in Fig. 1. b Chemical shifts δ in ppm, multiplicity m (s = singlet, d = doublet, t = triplet, m = multiplet, br = broad), coupling constants J are given in Hertz (spectra of 1 and 3 recorded at 500 MHz, spectra of 4 recorded at 600 MHz). c Chemical shifts δ in ppm and assignment of carbons by 13C-DEPT135 spectroscopy. For NMR data of 3 in (2H)chloroform cf. Table 1 of ESI. | ||||||||
| 1a | 1.78 (dt, 3J = 9.4, 2J = 12.5, 1H) | 35.7 (CH2) | 1 | — | 42.5 (Cq) | 1a | 1.22 (m, 1H) | 41.7 (CH2) |
| 1b | 1.09 (m, 1H) | 1b | 0.98 (m, 1H) | |||||
| 2a | 1.63 (m, 1H) | 19.2 (CH2) | 2a | 1.33 (m, 1H) | 41.9 (CH2) | 2 | 1.33 (m, 2H) | 22.5 (CH2) |
| 2b | 1.65 (m, 1H) | 2b | 1.20 (m, 1H) | |||||
| 3a | 2.49 (m, 1H) | 34.9 (CH2) | 3a | 1.55 (m, 1H) | 27.5 (CH2) | 3a | 1.58 (m, 1H) | 43.8 (CH2) |
| 3b | 1.46 (m, 1H) | 3b | 1.50 (m, 1H) | 3b | 1.17 (m, 1H) | |||
| 4 | — | 52.3 (Cq) | 4 | 2.13 (dt, 3J = 11.4, 3J = 9.2, 1 H) | 53.55 (CH) | 4 | — | 71.3 (Cq) |
| 5 | 3.19 (d, 3J = 9.1, 1H) | 69.5 (CH) | 5 | 1.71 (dt, 3J = 12.5, 3J = 2.2, 1 H) | 57.4 (CH) | 5 | 1.38 (m, 1H) | 49.1 (CH) |
| 6 | 0.48 (dd, 3J = 9.5, 3J = 9.2, 1H) | 31.2 (CH) | 6a | 1.43 (ddt, 2J = 13.5, 3J = 11.8, 3J = 2.4, 1 H) | 23.9 (CH2) | 6a | 1.81 (m, 1H) | 23.9 (CH2) |
| 6b | 2.31 (ddt, 2J = 13.5, 3J = 5.6, 3J = 2.4, 1 H) | 6b | 1.63 (m, 1H) | |||||
| 7 | 0.52 (dt, 3J = 9.5, 3J = 6.2, 1H) | 27.9 (CH) | 7a | 2.03 (m, 1H) | 36.5 (CH2) | 7 | 2.34 (br, 1H) | 39.8 (CH) |
| 7b | 1.97 (m, 1H) | |||||||
| 8a | 1.03 (m, 1H) | 20.5 (CH2) | 8 | — | 139.4 (Cq) | 8a | 2.15 (dtt, 3J = 12.6, 3J = 1.8, 1H) | 23.1 (CH2) |
| 8b | 1.51 (m, 1H) | 8b | 1.27 (m, 1H) | |||||
| 9a | 1.37 (m, 1H) | 37.3 (CH2) | 9 | 5.48 (m, 1H) | 123.3 (CH) | 9a | 1.42 (m, 1H) | 40.7 (CH2) |
| 9b | 1.42 (m, 1H) | 9b | 1.05 (m, 1H) | |||||
| 10 | — | 46.1 (Cq) | 10a | 1.86 (m, 1H) | 42.9 (CH2) | 10 | — | 35.4 (Cq) |
| 10b | 2.07 (m, 1H) | |||||||
| 11 | — | 19.3 (Cq) | 11 | — | 73.7 (Cq) | 11 | — | 146.9 (Cq) |
| 12 | 0.95 (s, 3H) | 29.0 (CH3) | 12 | 1.06 (s, 3H) | 27.7 (CH3) | 12 | 1.78 (dt, 4J = 0.7, 4J = 0.6, 3H) | 23.0 (CH3) |
| 13 | 1.02 (s, 3H) | 15.2 (CH3) | 13 | 1.02 (s, 3H) | 32.4 (CH3) | 13 | 5.08 (dm, 2J = 9.4, 4J = 0.7, 2 H) | 111.4 (CH2) |
| 14 | 0.87 (d, 4J = 0.8, 3H) | 28.0 (CH3) | 14 | 1.75 (m, 3H) | 27.3 (CH3) | 14 | 0.79 (t, 4J = 0.8, 3H) | 18.6 (CH3) |
| 15 | 1.04 (s, 3H) | 15.2 (CH3) | 15 | 0.85 (s, 3H) | 19.4 (CH3) | 15 | 0.98 (d, 4J = 0.7, 3H) | 22.7 (CH3) |
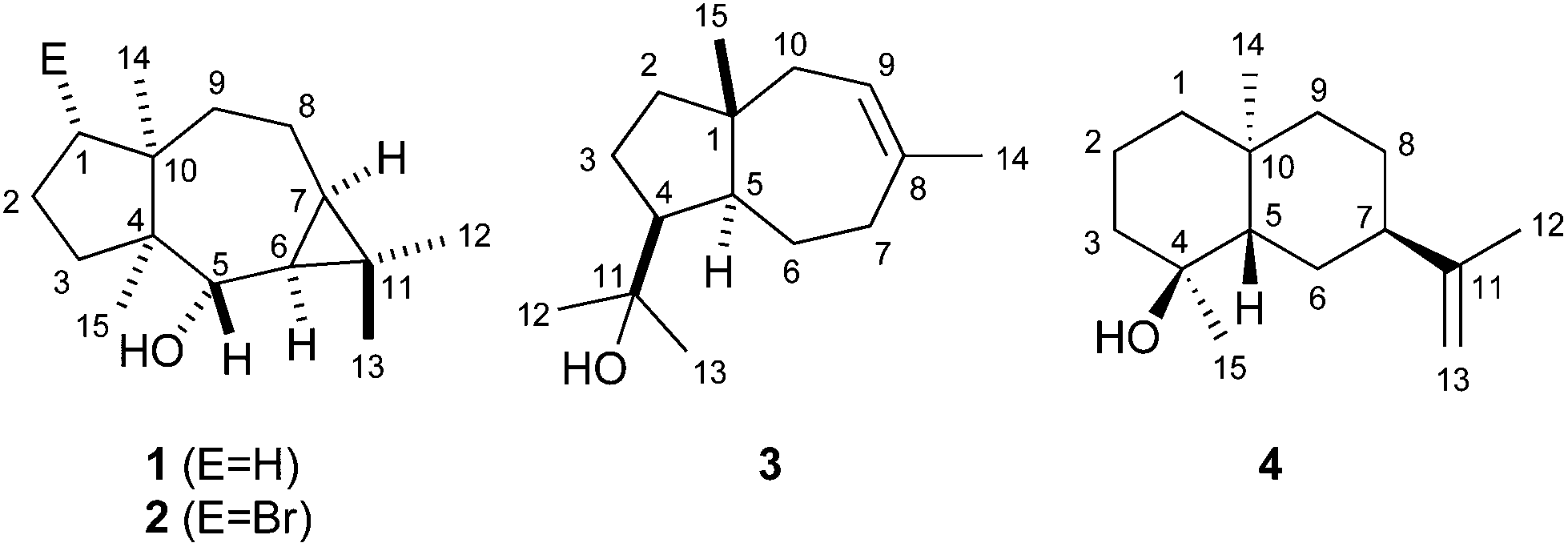
The 13C-NMR and 13C-DEPT135-NMR spectra of 1 exhibited fifteen carbon signals (four methyl, five methylene, two methine, one oxygenated methine and three quarternary carbons). The absence of signals for sp2 carbons suggested a tricyclic ring system. The signals for the corresponding protons for each carbon were assigned by heteronuclear single quantum correlation (HSQC) spectroscopy. 1H,1H-correlation spectroscopy (COSY) revealed two contiguous spin systems (C-5-6-7-8-9 and C-1-2-3, Fig. 2a). The 1H-NMR signals for all four methyl groups appeared as singlets revealing their attachment to quarternary carbons. The highfield shifts of the protons attached to C-6 and C-7 (δ = 0.48 and 0.52 ppm) suggested the presence of a cyclopropane ring. Heteronuclear multiple bond correlation (HMBC) spectroscopy showed cross peaks from C-12 and C-13 to C-6, C-7 and C-11, from C-14 to C-1, C-4, C-8, C-9 and C-10, and from C-15 to C-3, C-4, C-5 and C-10. These and all other HMBC correlations (including C-6 to C-5, C-7 and C-8; C-10 to C-2 and C-1 to C-3) were in agreement with the structure of 1. The relative configuration was determined by nuclear Overhauser spectroscopy (NOESY, Fig. 2a). Key correlations were observed between H-12, H-6 and H-7, indicating the cis-orientation of H-6 and H-7. Correlations between both bridgehead methyl groups H-14 and H-15 supported the cis-fused 7-5 ring system. Further diagnostic crosspeaks were observed between H-5 and H-13 and between H-6 and H-15, resulting in a fully assigned relative stereochemistry. The optical rotary power was determined as [α]24D = −10.2 (c = 0.082, CH2Cl2).
 | ||
| Fig. 1 Structures of neomeranol B (1), neomeranol (2), isodauc-8-en-11-ol (3) and (+)-intermedeol (4). | ||
 | ||
| Fig. 2 Key HMBC (single headed arrows) and NOESY correlations (double headed arrows) for (a) neomeranol B (1) and (b) isodauc-8-en-11-ol (3). Contiguous 1H-spin systems are shown in bold. | ||
The sesquiterpene alcohol 1 (neomeranol B) is a new natural product with a neomerane-type carbon backbone as previously described for neomeranol (2), a compound that was isolated from the green alga Neomeris annulata and has cytotoxic activity against brine shrimp.32,33 A series of oxidised neomeranes was recently isolated from Valeriana officinalis.34 Compound 1 is also present in headspace extracts of Streptomyces scabiei 87.22 (Fig. 2 of ESI†), demonstrating that the neomeranol B synthase is expressed under laboratory culture conditions.
Using the same approach a terpene synthase from Streptomyces venezuelae ATCC 10712 (CCA53839) also yielded an unknown sesquiterpene alcohol (EI-MS-QTOF: m/z = 222.1973, the EI mass spectrum is shown in Fig. 1b of ESI†) from FPP, while GPP and GGPP were not accepted. The enzymatic conversion of 90 mg FPP followed by purification via column chromatography yielded 12 mg of the pure compound 3. Its 13C-NMR and 13C-DEPT135 spectra showed fifteen carbon signals for four methyl, five methylene, three methine and three quarternary carbons (Table 1). One of the quarternary carbons (δ = 73.7 ppm) carried the alcohol function, while the signals of two carbons appeared in the olefinic region, indicating a bicyclic system. HSQC allowed for a correlation of the 1H signals with the 13C signals, while the 1H,1H-COSY spectrum revealed two spin systems (C-9-10 and C-2-3-4-5-6-7, Fig. 2b). HMBC connectivities from C-4 to C-11, C-12 and C-13 indicated the 1-hydroxy-1-methylethyl moiety, while further key HMBC correlations from C-5 to C-3, C-4, C-6, C-7, C-11 and C-15, and from C-14 to C-7, C-8, C-9 and C-10 explained the bicyclo[5.3.0]decene system. The relative configuration was determined by NOESY (Fig. 2b) that showed diagnostic correlations between H-15 and H-12, and between H-5, H-4 and H-13, in agreement with a trans-fused ring system and the cis-orientation of H-4 and H-5. The optical rotary power was determined as [α]22D = +19.4 (c = 0.505, CH2Cl2). In summary, compound 3 was identified as the new natural product (+)-isodauc-8-en-11-ol.
Recently, Ikeda and coworkers reported on the production of (+)-dauca-8,11-diene by the same terpene cyclase during heterologous expression in Streptomyces avermitilis.29 This compound was observed as a minor product (3%) of the purified recombinant enzyme in the conversion of FPP (Fig. 3a of ESI†). In agreement with this finding only the sesquiterpene alcohol 3, but not dauca-8,11-diene is present in headspace extracts of S. venezuelae ATCC 10712 (Fig. 3b of ESI†).35 In conclusion, the function of the sesquiterpene cyclase from S. venezuelae must be reassigned as (+)-isodauc-8-en-11-ol synthase.
The incubation of FPP with a third terpene cyclase from S. clavuligerus ATCC 27064 (WP_003955204) yielded a sesquiterpene alcohol, whereas GPP and GGPP were not converted by the enzyme. Its EI mass spectrum (Fig. 1c of ESI†) and all NMR data were in agreement with literature data for intermedeol (4) (Tables 1 and 2 of ESI†).36,37 The absolute configuration was determined as (+)-(4S,5S,7R,10S)-4 based on the measured optical rotary power of [α]25D = +11.9 (c = 0.50, CH2Cl2) in comparison to reported data for (+)-4 of [α]22D = +18.0 (c = 2.80, CHCl3).36 The sesquiterpene alcohol 4 is also found in the volatiles fraction of S. clavuligerus ATCC 27064 (Fig. 4 of ESI†).
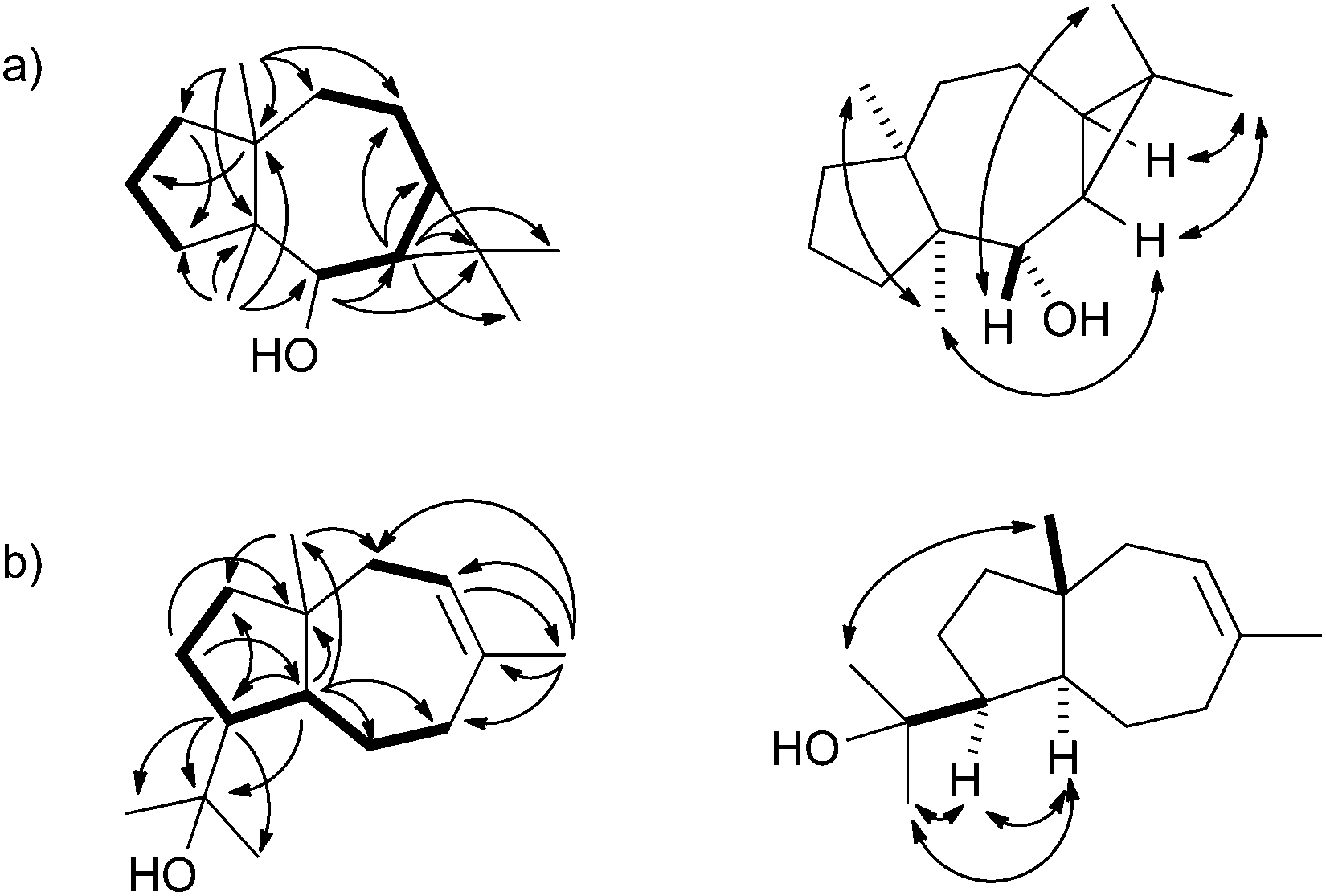
The biosynthetic mechanisms of the three characterised bacterial sesquiterpene cyclases were investigated by isotopic labelling experiments. For the biosynthesis of 2 a pathway via cyclisation of FPP to the (E,E)-germacradienyl cation (5) followed by deprotonation to bicyclogermacrene (6) was proposed (Scheme 1a).32 Several types of halogenases including vanadium-dependent chloroperoxidases or FADH2-dependent halogenases provide Hal+ equivalents in the biosynthesis of halogenated natural products.38,39 The halogenation of 6 with an electrophilic bromine species to the cationic intermediate 8 may initiate a second cyclisation with attack of water, either in a concerted process or via a cyclopropylcarbinyl cation as discussed for the biosynthesis of avermitilol,40 to yield 2. For the biosynthesis of 1 a very similar cyclisation cascade can be assumed in which the electrophile “Br+” is substituted by a proton. Such a cyclisation cascade via the neutral intermediate 6 can be catalysed by a single terpene cyclase, while the biosynthesis of 2 likely requires two enzymes: a terpene cyclase for the formation of 6 from FPP and a halogenase for the conversion of 6 into 2. The pathway for the formation of 1 by the terpene cyclase from S. scabiei via the neutral intermediate 6 is supported by its occurrence as a trace compound in enzyme incubations of FPP (Fig. 2a of ESI†).
 | ||
| Scheme 1 Proposed biosynthesis of (a) neomeranols 1 and 2, (b) (+)-(4S,5S,7R,10S)-intermedeol (4) and isodauc-8-en-11-ol (3). Red circles indicate 13C-labelled carbons. | ||
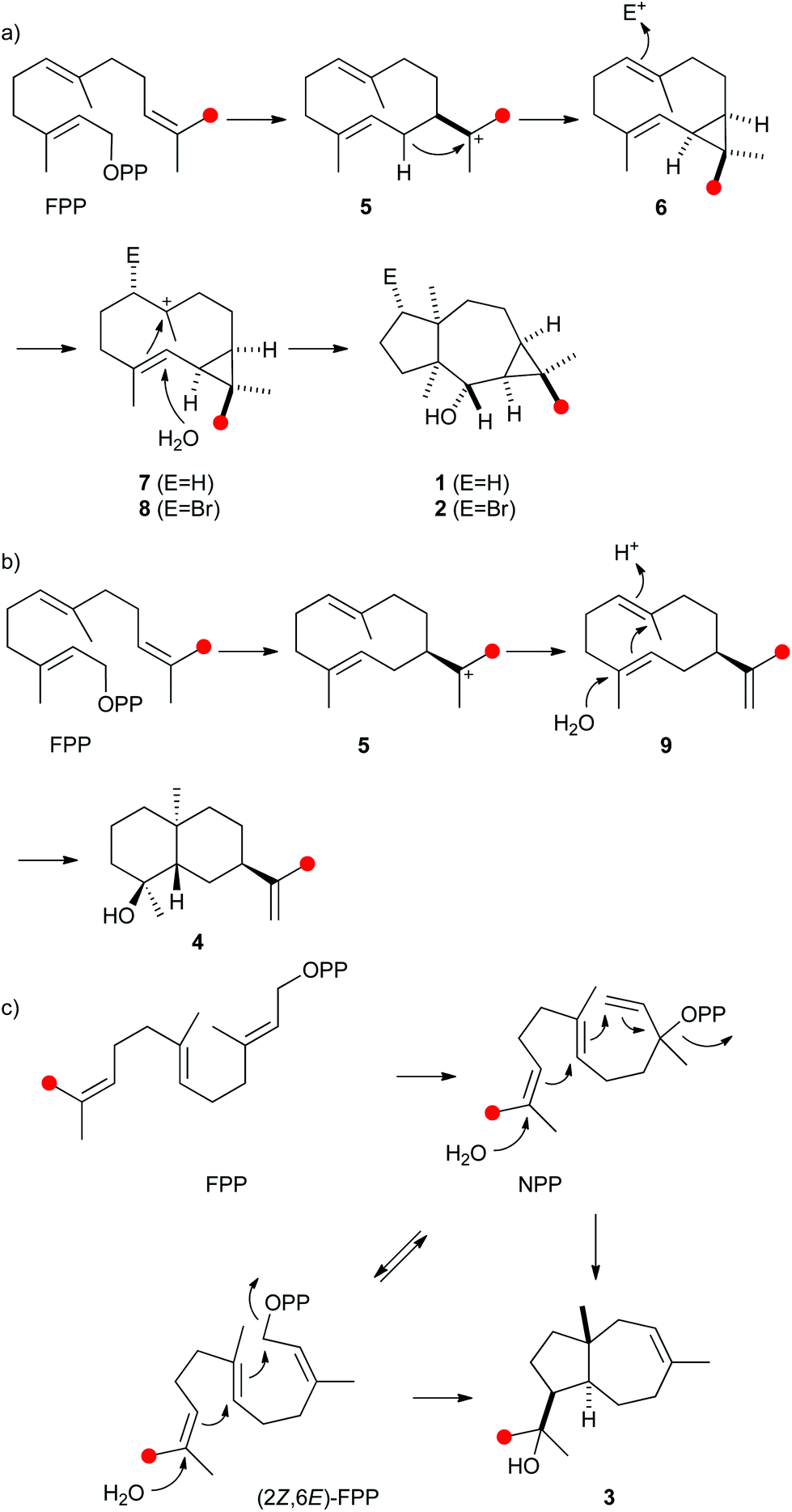
The protonation of 6 at C-1 was evident from incubations of (6-13C)FPP31 in water and in deuterium oxide followed by direct 13C-NMR analysis of the product that was simply extracted with (2H6)benzene, resulting in a strongly enhanced singlet for C-1 of 1 in the incubation experiment in water and a triplet due to 2H,13C-coupling in the experiment in deuterium oxide (Fig. 3a and b). The stereochemical course of the protonation was followed by HSQC analysis of the two obtained samples. While (1-13C)-1 gave two distinct crosspeaks for coupling of C-1 with the two directly bound diastereotopic protons at δH = 1.09 and 1.78 ppm, only one crosspeak at δH = 1.78 ppm was observed for (1-13C,1-2H)-1 obtained from the incubation of FPP in deuterium oxide (Fig. 3c). Thus, the proton at δH = 1.09 was substituted by deuterium, which is cis-oriented to Me-14, as was established by NOESY with the unlabelled compound (this hydrogen shows a crosspeak with the neighbouring methyl group (H-14), while the second proton at C-1 shows a crosspeak to H-5, next to the hydroxy function; Fig. 12 of ESI†). In summary, this experiment revealed a strict stereochemical course for the reprotonation of the neutral intermediate 6 to yield 1 with a cis-orientation of the introduced hydrogen and Me-14. Because of the unknown absolute configuration of 1 it remains unknown whether this corresponds to a Re or a Si face attack to intermediate 6.
 | ||
| Fig. 3 Incubation experiments with (6-13C)FPP and the neomeranol B synthase in water and in deuterium oxide. Red circles indicate 13C-labelled carbons. | ||
A model for the biosynthesis of (4S,5S,7R,10S)-4 starts with a 1,10-cyclisation of FPP to the germacradienyl cation (5) that forms germacrene A (9) upon loss of a proton (Scheme 1b). Reprotonation at C-1 initiates a second cyclisation and subsequent attack of water to yield 4. This suggested mechanism was studied using the same approach as described above for the neomeranol B synthase, i.e. via the incubation of (6-13C)FPP with the intermedeol synthase in water and in deuterium oxide. NMR analysis of the extracted product resulted in a strongly enhanced singlet (incubation in water) or triplet for the 13C-labelled carbon due to 2H,13C-coupling (incubation in deuterium oxide), thereby proving the reprotonation at C-1 (Fig. 4a and b). The stereochemistry of the protonation step was again evident from HSQC analysis of both samples, revealing that the crosspeak at δH = 0.98 ppm (pro-R proton as evident from the NOESY spectrum, Fig. 26 of ESI†) is retained in the deuterated sample, while the crosspeak at δH = 1.22 ppm (pro-S proton) is lost (Fig. 4c). Here, reprotonation of the neutral intermediate 9 from the Si face can be concluded, since the absolute configuration of 4 is known.
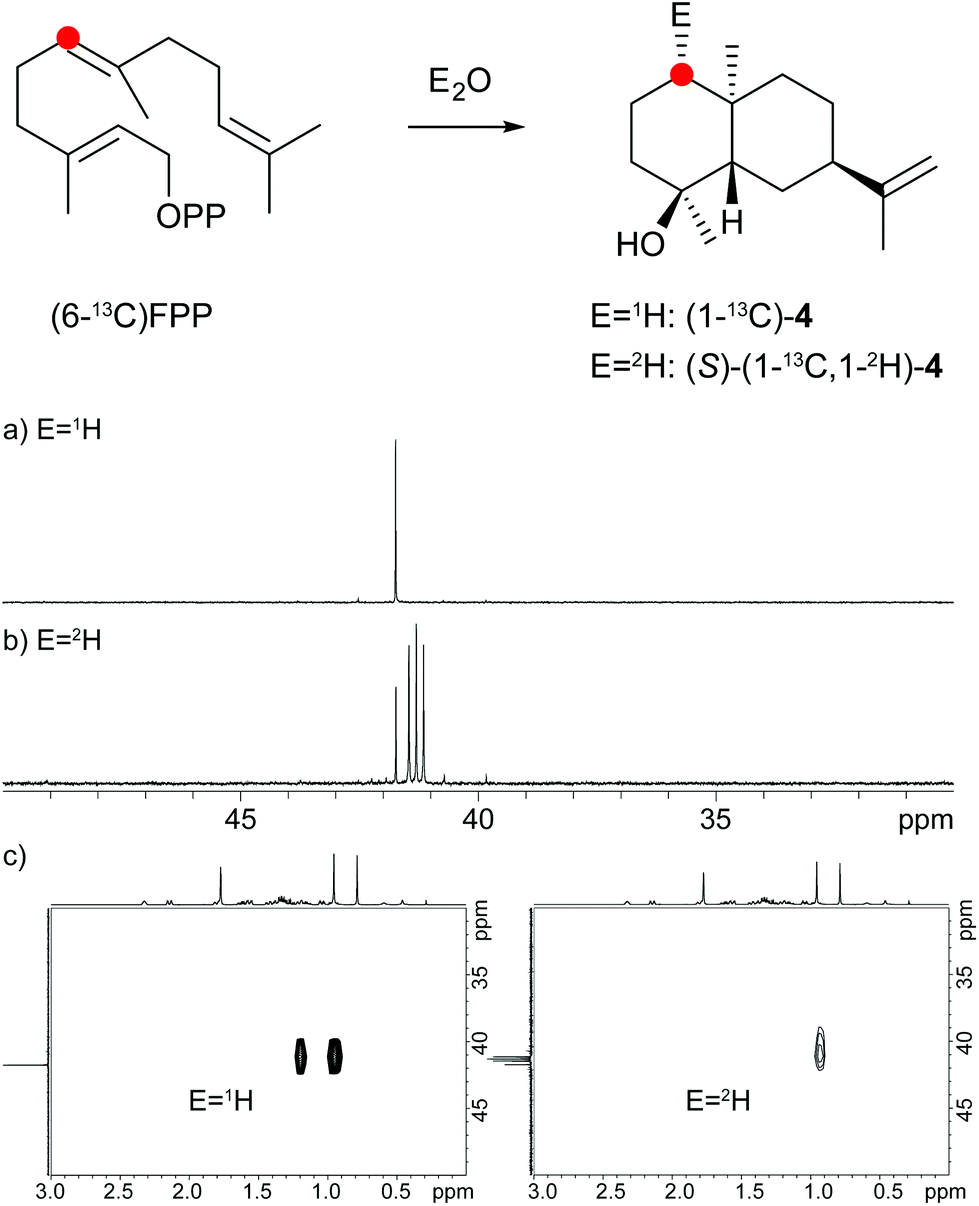 | ||
| Fig. 4 Incubation experiments with (6-13C)FPP and the intermedeol synthase in water and in deuterium oxide. Red circles indicate 13C-labelled carbons. | ||
A biosynthetic proposal for the formation of 3 includes the initial isomerisation of FPP to NPP that may react in a zipper mechanism in two concerted 1,7- and 6,10-cyclisations with concomittant attack of water at C-11 (Scheme 1c). Experimental evidence for this mechanism was obtained by incubation of (1,7-13C2)FPP (for synthesis cf. Fig. 5 of ESI†) with the purified recombinant isodauc-8-en-11-ol synthase that gave two doublets for 1JC,C-coupling in the 13C-NMR spectrum with a coupling constant of 35.0 Hz and a strong roof effect (Fig. 5). The biosynthesis of related sesquiterpenoids from Ferula jaeshkeana was suggested by Dev et al. in 1973 to proceed via (2Z,6E)-FPP.41 Although the generally accepted mechanism for the biosynthesis of sesquiterpenes with an initial 1,6- or 1,7-cyclisation proceeds via NPP, the possibility of an enzymatic conversion of (2Z,6E)-FPP by the isodauc-8-en-11-ol synthase was tested. (2Z,6E)-FPP7 was indeed accepted as substrate and converted into the same main product 3 and all minor side products as observed from (2E,6E)-FPP (Fig. 3c of ESI†). In previous work the conversion of (2Z,6E)-FPP was also demonstrated for tobacco 5-epi-aristolochene synthase42,43 and δ-cadinene synthase from cotton,44 but in contrast to our findings for the isodauc-8-en-11-ol synthase these experiments yielded different product spectra as obtained from (2E,6E)-FPP. Whether the results presented here mean that (2Z,6E)-FPP is a true intermediate in the biosynthesis of 3 that may itself be formed from (2E,6E)-FPP via NPP (Scheme 1c),45 or just fits with a similar conformation as NPP into the enzyme's active site and is thus transformed, is a question that will be difficult to address experimentally.
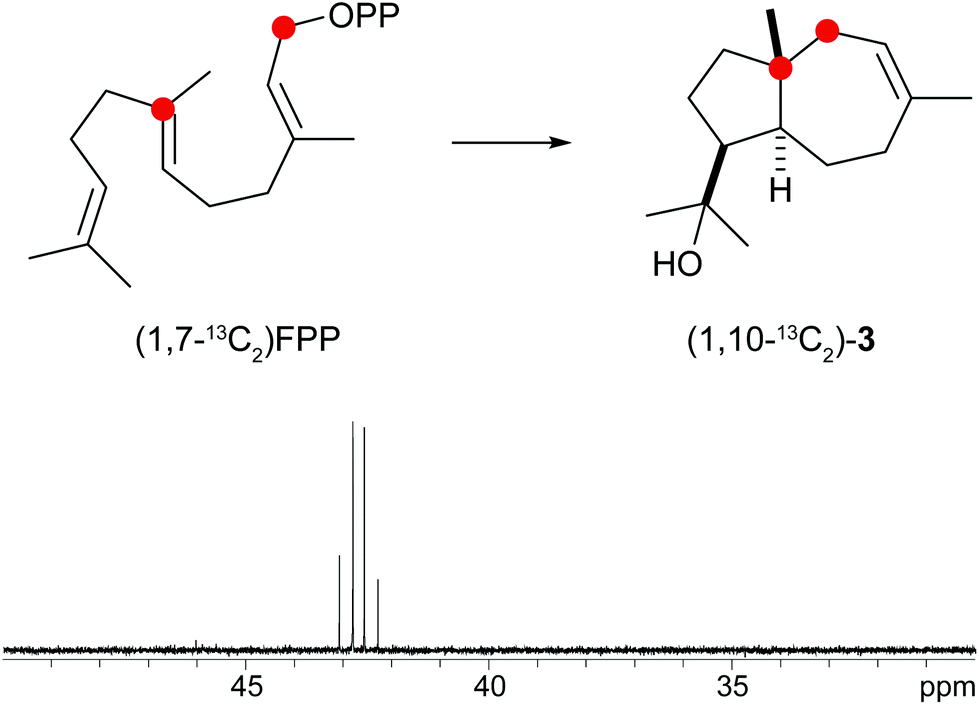 | ||
| Fig. 5 Incubation of (1,7-13C2)FPP with isodauc-8-en-11-ol synthase. The two doublets in the 13C-NMR spectrum show a strong roof effect. Red circles indicate 13C-labelled carbons. | ||
Finally, the stereochemical course of all three bacterial sesquiterpene cyclases in terms of the fate of the terminal E- and Z methyl groups (C-12 and C-13) of FPP was investigated by incubation experiments with (13-13C)FPP (red circles in Scheme 1). As was followed by 13C-NMR spectroscopy (Fig. 6) the isotopic labelling from (13-13C)FPP showed up at C-13 (δ = 15.2 ppm), but not at C-12 of 1 (δ = 29.0 ppm) in the incubation experiment with neomeranol B synthase, while the enzymatic conversion of (13-13C)FPP with intermedeol synthase produced 4 with specific introduction of labelling into C-12 (δ = 23.0 ppm) and not C-13 (δ = 111.4 ppm; a very small peak is visible here that likely originates from a small contamination (<1%) of synthetic (13-13C)FPP with (12-13C)FPP). Finally, the incubation of (13-13C)FPP with the isodauc-8-en-11-ol synthase resulted in a specific labelling at C-13 (δ = 32.4 ppm), but not at C-12 of 3 (δ = 27.7 ppm). In conclusion, all three enzymes revealed a very strict stereochemical course.
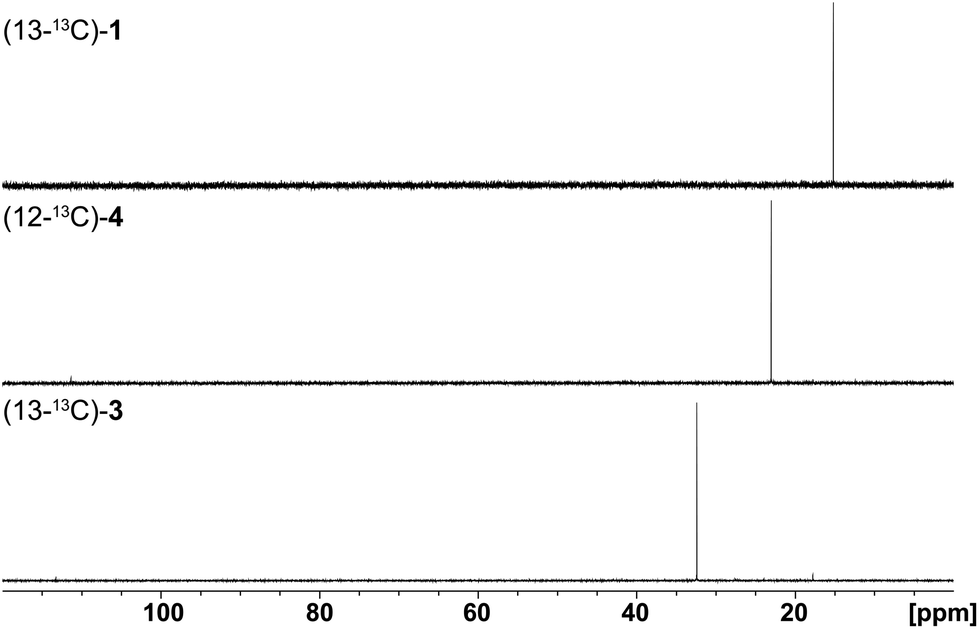 | ||
| Fig. 6 Results (13C-NMR spectra) obtained from incubation experiments with (13-13C)FPP. The corresponding isotopic labellings are shown in Scheme 1. | ||
Conclusions
We have characterised the enzyme products of three bacterial terpene cyclases as (−)-neomeranol B (1), (+)-isodauc-8-en-11-ol (3) and (+)-intermedeol (4). The compounds 1 and 3 have been isolated from natural sources for the first time. The terpene cyclisations of FPP to 1 and 4 proceed via the neutral intermediates bicyclogermacrene and germacrene A, that are reprotonated for further conversion into the final products. We provide here a fast and easy method to investigate the stereochemical courses, i.e. to distinguish reprotonations from the two stereochemically different faces of an olefinic double bond, by using a 13C-FPP isotopomer with labelling at the reprotonated carbon for incubations in deuterium oxide. A simple HSQC spectrum in combination with NOESY analysis of the unlabelled enzyme product gives then information about the stereochemical course. Furthermore, we have investigated the stereochemical courses of terpene cyclisations with respect to the fate of the stereochemically different E- and Z methyl groups of FPP (C-12 and C-13) by usage of (13-13C)FPP. A similar experiment using deuterium labels has previously been performed, inter alea, with the epi-isozizaene synthase from Streptomyces coelicolor A3(2).46 As demonstrated here and in previous work from our and other groups isotopic labellings are very useful for structure elucidations and mechanistic biosynthetic investigations.28,31,47–52 We will continue to investigate interesting aspects of terpene cyclisations via this approach.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft DFG (DI1536/7-1) and by the Beilstein-Institut zur Förderung der Chemischen Wissenschaften with a PhD scholarship to PR. We thank Dr Senada Nozinovic (Bonn) for recording the NMR spectra.Notes and references
- C. M. Starks, K. Back, J. Chappell and J. P. Noel, Science, 1997, 277, 1815–1820 CrossRef CAS PubMed.
- C. A. Lesburg, G. Zhai, D. E. Cane and D. W. Christianson, Science, 1997, 277, 1820–1824 CrossRef CAS PubMed.
- M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13543–13548 CrossRef CAS PubMed.
- E. Y. Shishova, L. Di Constanzo, D. E. Cane and D. W. Christianson, Biochemistry, 2007, 46, 1941–1951 CrossRef CAS PubMed.
- J. A. Aaron, X. Lin, D. E. Cane and D. W. Christianson, Biochemistry, 2010, 49, 1787–1797 CrossRef CAS PubMed.
- M. Köksal, Y. Jin, R. M. Coates, R. Croteau and D. W. Christianson, Nature, 2011, 469, 116–120 CrossRef PubMed.
- P. Baer, P. Rabe, C. A. Citron, C. C. de Oliveira Mann, N. Kaufmann, M. Groll and J. S. Dickschat, ChemBioChem, 2014, 15, 213–216 CrossRef CAS PubMed.
- P. Baer, P. Rabe, K. Fischer, C. A. Citron, T. A. Klapschinski, M. Groll and J. S. Dickschat, Angew. Chem., Int. Ed., 2014, 53, 7652–7656 CrossRef CAS PubMed.
- J. S. Dickschat, Nat. Prod. Rep., 2011, 28, 1917–1936 RSC.
- N. L. Brock and J. S. Dickschat, Biosynthesis of Terpenoids, in Handbook of Natural Products, ed. K. G. Ramawat and J.-M. Mérillon, Springer, 2013, pp. 2693–2732 Search PubMed.
- M. Seemann, G. Zhai, J.-W. de Kraker, C. M. Paschell, D. W. Christianson and D. E. Cane, J. Am. Chem. Soc., 2002, 124, 7861–7869 CrossRef.
- D. E. Cane, J. K. Sohng, C. R. Lamberson, S. M. Rudnicki, Z. Wu, M. D. Lloyd, J. S. Oliver and B. R. Hubbard, Biochemistry, 1994, 33, 5846–5857 CrossRef CAS PubMed.
- X. Lin, R. Hopson and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 6022–6023 CrossRef CAS PubMed.
- J. Jiang, X. He and D. E. Cane, Nat. Chem. Biol., 2007, 3, 711–715 CrossRef CAS PubMed.
- C.-M. Wang and D. E. Cane, J. Am. Chem. Soc., 2008, 130, 8908–8909 CrossRef CAS PubMed.
- S. A. Agger, F. Lopez-Gallego, T. R. Hoye and C. Schmidt-Dannert, J. Bacteriol., 2008, 190, 6084–6096 CrossRef CAS PubMed.
- M. Komatsu, M. Tsuda, S. Omura, H. Oikawa and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7422–7427 CrossRef CAS PubMed.
- W. K. W. Chou, I. Fanizza, T. Uchiyama, M. Komatsu, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2010, 132, 8850–8851 CrossRef CAS PubMed.
- Y. Hu, W. K. W. Chou, R. Hopson and D. E. Cane, Chem. Biol., 2011, 18, 32–37 CrossRef CAS PubMed.
- C. Nakano, M. H.-K. Kim and Y. Ohnishi, ChemBioChem, 2011, 12, 1988–1991 CrossRef CAS PubMed.
- C. Nakano, F. Kudo, T. Eguchi and Y. Ohnishi, ChemBioChem, 2011, 12, 2271–2275 CrossRef CAS PubMed.
- C. Nakano, M. H.-K. Kim and Y. Ohnishi, ChemBioChem, 2011, 12, 2403–2407 CrossRef CAS PubMed.
- C. Nakano, S. Horinouchi and Y. Ohnishi, J. Biol. Chem., 2011, 286, 27980–27987 CrossRef CAS PubMed.
- C. Nakano, T. Tezuka, S. Horinouchi and Y. Ohnishi, J. Antibiot., 2012, 65, 551–558 CrossRef CAS PubMed.
- P. Rabe and J. S. Dickschat, Angew. Chem., Int. Ed., 2013, 51, 1810–1812 CrossRef PubMed.
- J. S. Dickschat, K. A. K. Pahirulzaman, P. Rabe and T. Klapschinski, ChemBioChem, 2014, 15, 810–814 CrossRef CAS PubMed.
- A. Schifrin, T. T. B. Ly, N. Günnewich, J. Zapp, V. Thiel, S. Schulz, F. Hannemann, Y. Khatri and R. Bernhardt, ChemBioChem, 2015, 16, 337–344 CrossRef CAS PubMed.
- P. Rabe, K. A. K. Pahirulzaman and J. S. Dickschat, Angew. Chem., Int. Ed., 2015, 54, 6041–6045 CrossRef CAS PubMed.
- Y. Yamada, T. Kuzuyama, M. Komatsu, K. Shin-ya, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 857–862 CrossRef CAS PubMed.
- J.-Y. Chow, B.-X. Tian, G. Ramamoorthy, B. S. Hillerich, R. D. Seidel, S. C. Almo, M. P. Jacobson and C. D. Poulter, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5661–5666 CrossRef CAS PubMed.
- P. Rabe, L. Barra, J. Rinkel, R. Riclea, C. A. Citron, T. A. Klapschinski, A. Janusko and J. S. Dickschat, Angew. Chem., Int. Ed., 2015, 53 DOI:10.1002/anie.201507615R1.
- D. E. Barnekow, J. H. Cardellina, A. S. Zektzer and G. E. Martin, J. Am. Chem. Soc., 1989, 111, 3511–3517 CrossRef CAS.
- V. J. Paul, J. M. Cronan and J. H. Cardellina, J. Chem. Ecol., 1993, 19, 1847–1860 CrossRef CAS PubMed.
- Z.-Z. Han, X.-P. Zu, J.-X. Wang, H.-L. Li, B.-Y. Chen, Q.-X. Liu, X.-Q. Hu, Z.-H. Yan and W.-D. Zhang, Tetrahedron, 2014, 70, 962–966 CrossRef CAS.
- C. A. Citron, J. Gleitzmann, G. Laurenzano, R. Pukall and J. S. Dickschat, ChemBioChem, 2012, 13, 202–214 CrossRef CAS PubMed.
- A. San Feliciano, M. Medarde, B. Del Rey, J. M. M. Del Corral and A. F. Barrero, Phytochemistry, 1990, 29, 3207–3211 CrossRef CAS.
- R. P. W. Kesselmans, J. B. P. A. Wijnberg, A. J. Minnaard, R. E. Walinga and A. de Groot, J. Org. Chem., 1991, 56, 7237–7244 CrossRef CAS.
- P. Bernhardt, T. Okino, J. M. Winter, A. Miyanaga and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 4268 CrossRef CAS PubMed.
- C. Wagner, M. El Omari and G. M. König, J. Nat. Prod., 2009, 72, 540 CrossRef CAS PubMed.
- Y. J. Hong and D. J. Tantillo, Org. Lett., 2011, 13, 1294–1297 CrossRef CAS PubMed.
- M. C. Sriraman, B. A. Nagasampagi, R. C. Pandey and S. Dev, Tetrahedron, 1973, 29, 985–991 CrossRef CAS.
- J. A. Faraldos, P. E. O'Maille, N. Dellas, J. P. Noel and R. M. Coates, J. Am. Chem. Soc., 2010, 132, 4281–4289 CrossRef CAS PubMed.
- J. P. Noel, N. Dallas, J. A. Faraldos, M. Zhao, B. A. Hess Jr., L. Smentek, R. M. Coates and P. E. O'Maille, ACS Chem. Biol., 2010, 5, 377–392 CrossRef CAS PubMed.
- J. A. Faraldos, D. J. Miller, V. Gonzalez, Z. Yoosuf-Aly, O. Cascon, A. Li and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 5900–5908 CrossRef CAS PubMed.
- D. Arigoni, Pure Appl. Chem., 1975, 41, 219–245 CrossRef CAS.
- X. Lin and D. E. Cane, J. Am. Chem. Soc., 2009, 131, 6332–6333 CrossRef CAS PubMed.
- L. Barra, K. Ibrom and J. S. Dickschat, Angew. Chem., Int. Ed., 2015, 54, 6637–6640 CrossRef CAS PubMed.
- R. Riclea and J. S. Dickschat, Angew. Chem., Int. Ed., 2015, 54, 12167–12170 CrossRef CAS PubMed.
- H. B. Bode, D. Reimer, S. W. Fuchs, F. Kirchner, C. Dauth, C. Kegler, W. Lorenzen, A. O. Brachmann and P. Grin, Chem. – Eur. J., 2012, 18, 2342–2348 CrossRef CAS PubMed.
- H. B. Bode, A. O. Brachmann, K. B. Jadhav, L. Seyfarth, C. Dauth, S. W. Fuchs, M. Kaiser, N. R. Waterfield, H. Sack, S. H. Heinemann and H.-D. Arndt, Angew. Chem., Int. Ed., 2015, 54, 10352–10355 CrossRef CAS PubMed.
- A. Meguro, Y. Motoyoshi, K. Teramoto, S. Ueda, Y. Totsuka, Y. Ando, T. Tomita, S.-Y. Kim, T. Kimura, M. Igarashi, R. Sawa, T. Shinada, M. Nishiyama and T. Kuzuyama, Angew. Chem., Int. Ed., 2015, 54, 4353–4356 CrossRef CAS PubMed.
- A. Ear, S. Amand, F. Blanchard, A. Blond, L. Dubost, D. Buisson and B. Nay, Org. Biomol. Chem., 2015, 13, 3662–3666 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Strains, culture conditions, gene cloning and expression conditions, details of incubation experiments and synthesis of (1,7-13C2)FPP including spectroscopic data, and gas chromatograms, mass spectra and NMR spectra of terpene cyclase products. See DOI: 10.1039/c5ob01998b |
| This journal is © The Royal Society of Chemistry 2016 |