
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A study on the AMACR catalysed elimination reaction and its application to inhibitor testing†
Maksims
Yevglevskis
a,
Guat L.
Lee
a,
Jenny
Sun
ab,
Shiyi
Zhou
ab,
Xiaolong
Sun
c,
Gabriele
Kociok-Köhn
c,
Tony D.
James
c,
Timothy J.
Woodman
a and
Matthew D.
Lloyd
*a
aMedicinal Chemistry, Department of Pharmacy & Pharmacology, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: M.D.Lloyd@bath.ac.uk; Fax: +44 (0)1225 386114
bDepartment of Pharmacy, Shandong University, People's Republic of China
cDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK
First published on 27th October 2015
Abstract
α-Methylacyl-CoA racemase (AMACR; P504S) catalyses a key step in the degradation of branched-chain fatty acids and is important for the pharmacological activation of Ibuprofen and related drugs. Levels of AMACR are increased in prostate and other cancers, and it is a drug target. Development of AMACR as a drug target is hampered by lack of a convenient assay. AMACR irreversibly catalyses the elimination of HF from 3-fluoro-2-methylacyl-CoA substrates, and this reaction was investigated for use as an assay. Several known inhibitors and alternative substrates reduced conversion of 3-fluoro-2-methyldecanoyl-CoA by AMACR, as determined by 1H NMR. The greatest reduction of activity was observed with known potent inhibitors. A series of novel acyl-CoA esters with aromatic side chains were synthesised for testing as chromophoric substrates. These acyl-CoA esters were converted to unsaturated products by AMACR, but their use was limited by non-enzymatic elimination. Fluoride sensors were also investigated as a method of quantifying released fluoride and thus AMACR activity. These sensors generally suffered from high background signal and lacked reproducibility under the assay conditions. In summary, the elimination reaction can be used to characterise inhibitors, but it was not possible to develop a convenient colorimetric or fluorescent assay using 3-fluoro-2-methylacyl-CoA substrates.
Introduction
α-Methylacyl-CoA racemase (AMACR, P504S; E.C. 5.1.99.4) catalyses a key step in the degradation of branched-chain fatty acids.1–3 The enzyme catalyses the conversion of either epimer of a 2-methylacyl-CoA ester into a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2R- and 2S-epimers.4,5 β-Oxidation of 2-methylacyl-CoAs requires 2S-configuration,6,7 but both R- and S-2-methylacyl-CoAs are produced in vivo and are derived from dietary fatty acids.3 Thus, AMACR enables metabolism of R-2-methylacyl-CoAs. It is also important in the pharmacological activation of R-Ibuprofen and related drugs.3,5,8 AMACR has also been proposed to be involved in the uni-directional chiral inversion of mandelic acid in mammals9 but this was recently disproved.10
:1 mixture of 2R- and 2S-epimers.4,5 β-Oxidation of 2-methylacyl-CoAs requires 2S-configuration,6,7 but both R- and S-2-methylacyl-CoAs are produced in vivo and are derived from dietary fatty acids.3 Thus, AMACR enables metabolism of R-2-methylacyl-CoAs. It is also important in the pharmacological activation of R-Ibuprofen and related drugs.3,5,8 AMACR has also been proposed to be involved in the uni-directional chiral inversion of mandelic acid in mammals9 but this was recently disproved.10
AMACR protein levels and enzyme activity are increased in prostate cancers,11,12 myxofibrosarcomas,13 a subset of colon cancers14 and various other cancers8 and it is widely recognised as a promising drug target.3,8,15–18 Genetic knock-down of AMACR reduces proliferation of cultured cancer cells13,15,16,19 and restores androgen-dependent growth in some prostate cancer cell lines.16 Relatively few chemical inhibitors of AMACR have been reported,17–20 largely due to the lack of a convenient, high-throughput assay. Current assay methods include wash-out of tritium from a labelled substrate followed by measurement of radioactive water1,2,19 or wash-in of deuterium from D2O followed by 1H NMR analyses.4,5,10 These assays are probably subject to a kinetic isotope effect, and are low-throughput and labour-intensive. In addition, the 1H NMR assay also suffers from signal overlap of the substrate/product 2-methyl group, thus making it more difficult to quantify activity. Characterisation of AMACR inhibitors using HPLC assays17,18 has also been reported, but these assays are also low throughput. Despite these difficulties, one of the identified inhibitor pro-drugs, trifluoroibuprofen, shows promising anti-prostate cancer effects in in vivo models.21
Other approaches have also been used to develop a convenient assay for AMACR activity. The use of acyl-CoA oxidase as a coupling enzyme enables a colorimetric assay to be performed.22 This enzyme is not commercially available and rationally designed acyl-CoA inhibitors of AMACR are also likely to inhibit the coupling enzyme, complicating the analysis. Coupled enzyme assays for other racemases/epimerases have also been reported,23–27 but these are not readily adaptable to measuring AMACR activity.
Direct measurement of racemisation by MCR (the bacterial homologue of AMACR from M. tuberculosis) using circular dichroism has been reported,28 but this was not used for inhibitor testing. Assays for several other racemases/epimerases using circular dichroism27,29–31 or polarimetry32–34 have been developed, but these are generally low-throughput. Moreover, acyl-CoA inhibitors with aromatic side-chains are likely to undergo racemisation and this will complicate the determination of inhibitor properties. Therefore, these assays have serious limitations when determining inhibitor potency.
It has recently been reported that AMACR performs an elimination reaction in which HF is eliminated from 3-fluoro-2-methylacyl-CoAs (such as 1) to give unsaturated acyl-CoAs (such as 2) (Scheme 1).35 This reaction is irreversible which is an advantage compared to assays using isotopic labels1,2,4,5,10,19 or ‘racemisation’17,18 as their reversibility makes them more difficult to interpret. In addition, the 2-methyl peaks of 1 and 2 are non-overlapping in the 1H NMR spectrum, simplifying the measurement of substrate conversion.
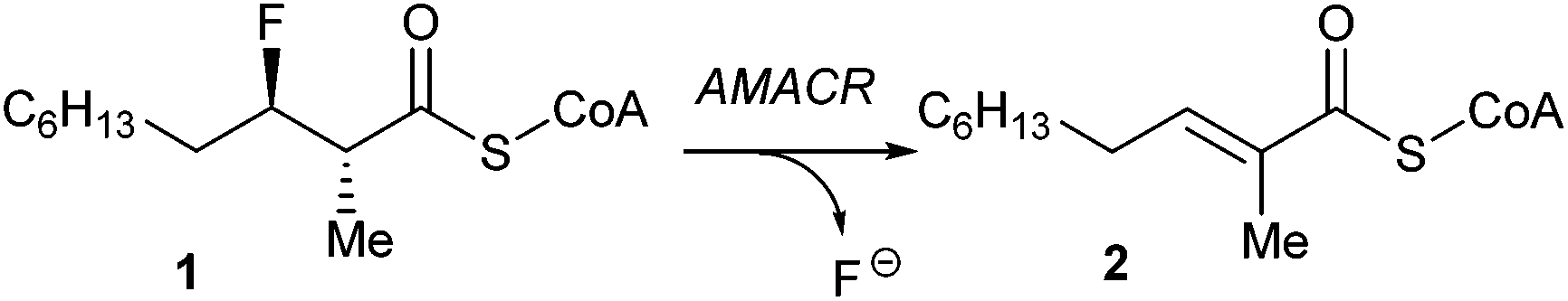 | ||
| Scheme 1 The elimination reaction catalysed by AMACR. | ||
The elimination reaction also offers the possibility of translation into a convenient, colorimetric or fluorometric assay by manipulation of the substrate side-chain or by the use of fluoride sensors. Assays using fluoride-specific electrodes to measure enzyme activity have also been reported,36–38 but these are generally low-throughput, require relatively large volumes and are not easy to adapt to a microtitre plate format.39 A number of highly sensitive molecular fluoride sensors have been reported in the literature, which give an increase in absorbance or fluorescence upon reaction with fluoride. However, there are relatively few that can be used in aqueous buffers.40–43 The development of a convenient high-throughput assay is essential for the development of AMACR as a drug target.
In this paper, the use of the AMACR-catalysed fluoride elimination reaction for the characterisation of inhibitors is investigated. Reduction in enzyme activity in the presence of other known AMACR substrates and inhibitors was observed by 1H NMR. The use of 3-fluoro-2-methylacyl-CoA substrates with aromatic side-chains and fluoride sensors in order to translate this reaction into a colorimetric or fluorescent assay format is also investigated.
Results and discussion
Use of the elimination assay for inhibitor characterisation was initially performed by incubation of recombinant human AMACR 1A4 with a series of known ‘inhibitors’ (Table 1) and substrate 1. The chosen ‘inhibitors’ included the known AMACR substrates5 Fenoprofenoyl-CoA 3, Flurbiprofenoyl-CoA 4, Ibuprofenoyl-CoA 5, Ketoprofenoyl-CoA 6, and Naproxenoyl-CoA 7. These are expected to behave as competitive inhibitors. Also chosen was N-dodecyl-N-methyl-carbamoyl-CoA 8, a transition state analogue and the most potent AMACR inhibitor described to date.18 Ebselen 9, Ebselen oxide 10 and Rose Bengal 11 were also chosen for study as these are reported to be good inhibitors of human AMACR 1A.19 Enzyme was pre-incubated with inhibitor for 10 min. to allow binding before addition of substrate at 100 μM final concentration. After 1 h the assay was terminated and the level of substrate conversion was determined by 1H NMR. Control experiments showed that the enzyme was fully active35 in the presence of 1 mM fluoride solution, indicating that any reduction in activity was due to the presence of the ‘inhibitor’.|
|
||
|---|---|---|
| Inhibitory compound | Relative conversion in presence of inhibitor | Relative reduction compared to no inhibitor |
| None | 100% | 0% |
| 3 | 74.9 ± 0.4% | 25.1% |
| 4 | 91.6 ± 2.0% | 8.4% |
| 5 | 74.1 ± 7.2% | 25.9% |
| 6 | 92.0 ± 8.6% | 8.0% |
| 7 | 88.8 ± 4.0% | 11.2% |
| 8 | 16.8 ± 1.8% | 83.2% |
| 9 | <5% | >95% |
| 10 | 88.2 ± 5.4% | 11.8% |
| 11 | <5% | >95% |
In the absence of inhibitor, ca. 50% of substrate 1 was converted into unsaturated product 2 by active AMACR. Negative controls containing heat-inactivated enzyme showed <5% conversion of 1 to 2, levels of which did not change over the incubation period. The presence of each ‘inhibitor’ (at 100 μM final concentration) resulted in a reduction in the level of conversion of 1 (Table 1). Compounds 3–7 showed moderate levels of inhibition in most cases, with the most significant reduction in activity occurring with Fenoprofenoyl-CoA 3 and Ibuprofenoyl-CoA 5. Modest levels of inhibition are expected with compounds 3–7, as the concentration of substrate 1 (100 μM) is significantly above its reported Km value (21 μM).35 These high substrate concentrations will reduce the apparent effect of competitive inhibitors, but relative high concentrations of 1 are required to perform the 1H NMR analyses. Incubation of the known highly potent AMACR inhibitor 818 resulted in a very significant reduction in activity (Fig. 1), consistent with it being a good inhibitor (reported IC50 value of 98 nM18). Ebselen 9 and Rose Bengal 11 were also highly potent under the assay conditions, with no detectable conversion of 1. In contrast, Ebselen oxide 10 was a modest inhibitor, having a similar potency to the alternative substrates 3–7. This result was surprising as Wilson et al.19 report that 10 was their most potent inhibitor (IC50 value of 790 nM), compared to 9 (IC50 value of 10 μM) and 11 (IC50 value of 2.8 μM). Ebselen 9 is reported to be an irreversible inhibitor of AMACR,19 and IC50 values are an inappropriate measure of potency as inhibition levels are dependent on the rate of inactivation. Rose Bengal 11 is a non-specific inhibitor of a number of enzymes, and inhibition appears to be related to generation of reactive oxygen species upon irradiation with UV light.44–46 The differences in observed behaviour between the two studies probably results from the different modes of action for 9, 10 and 11, meaning that the results of the two studies are not directly comparable.
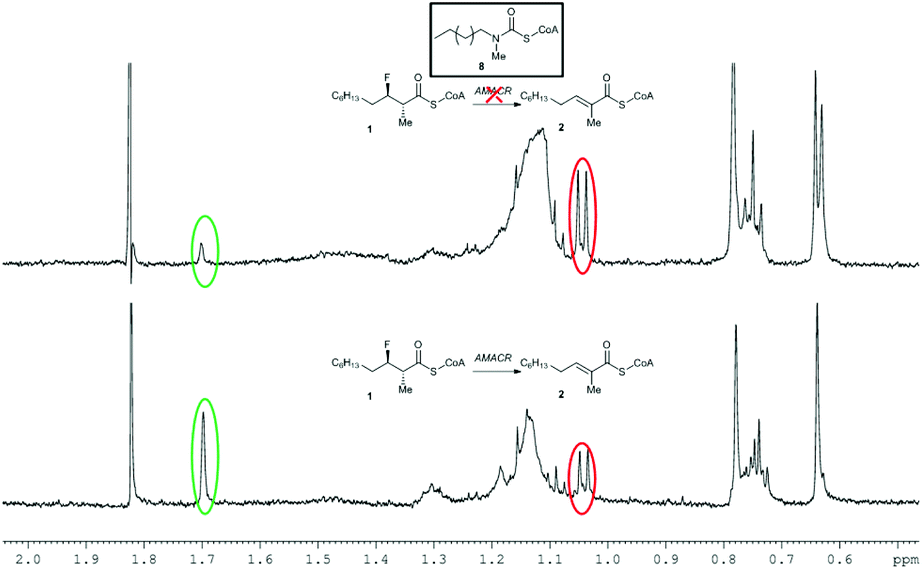 | ||
| Fig. 1 Reduction of conversion of 1 to 2 in the presence of inhibitor 8. Above, 1H NMR spectrum following incubation with active AMACR in the presence of 8; below, 1H NMR spectrum of positive control containing active AMACR in the absence of inhibitor. Red and green circles show methyl group signals for substrate 1 and unsaturated product 2, respectively. | ||
Although AMACR inhibitor testing using this method offers a number of advantages, including irreversible formation of the product and non-overlapping signals for substrate 1 and product 2, the method is still low-throughput as 1H NMR is used to quantify conversion levels. Translation of this method to a colorimetric or fluorescent assay is therefore desirable.
In order to do this, synthesis of acyl-CoA substrates with aromatic side-chains was investigated as it was anticipated that the unsaturated product would absorb in the visible spectrum. Synthesis of the anti-substrates was desired, as syn-substrates are prone to undergo non-enzymatic elimination.35 In the first synthesis, benzaldehyde 12 and the R-Evan's auxiliary protected propanoic acid 13 were condensed to give alcohol 14 (Scheme 2). However, treatment of 14 with DAST resulted in loss of stereochemistry upon introduction of the fluoride to give a mixture of diastereomers of 15. For aliphatic side-chains, the replacement of the 3-hydroxy group with fluoride proceeds via an SN2 mechanism with inversion of stereochemistry. This loss of stereochemistry is probably due to an SN1 reaction occurring, with consequent addition of fluoride to both faces of the stabilised benzylic carbocation. Conversion of 14 to the methyl ester 16 followed by treatment with DAST also resulted in significant loss of stereochemistry on conversion to 17, suggesting that steric hindrance by the chiral auxiliary was not the deciding factor.
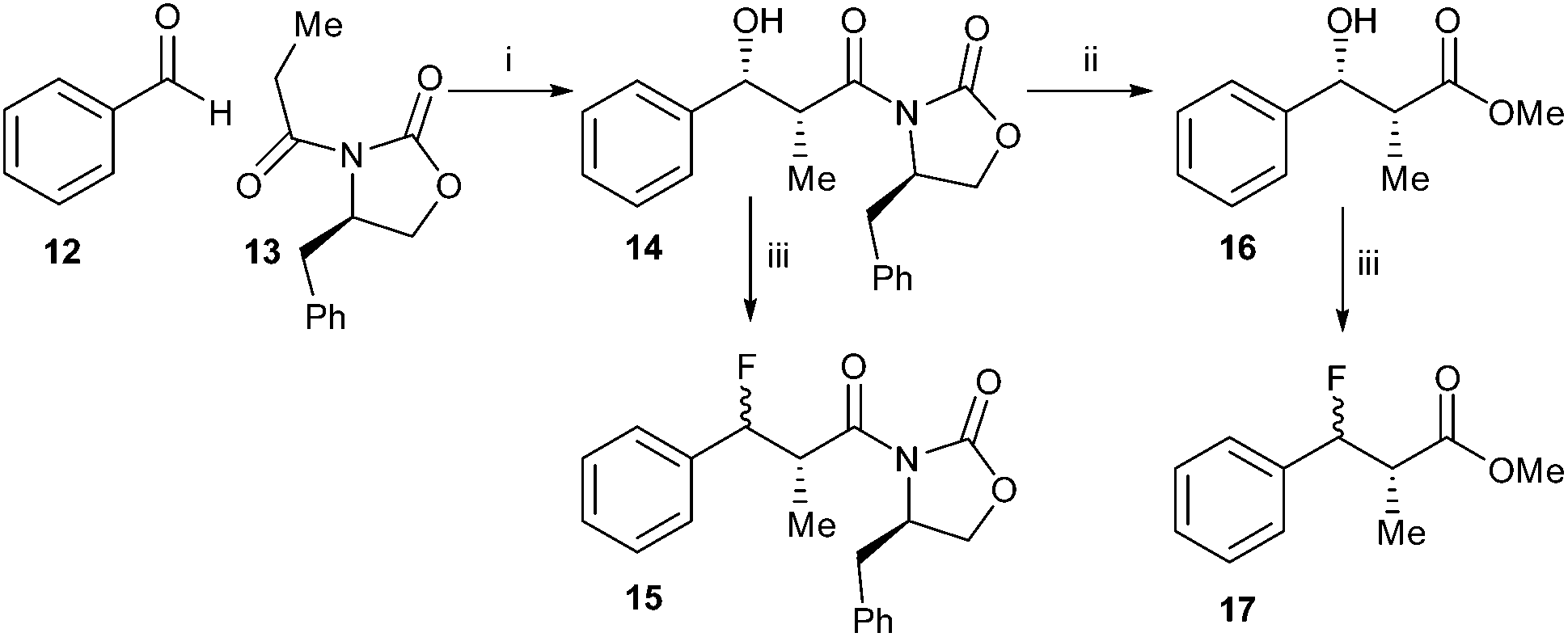 | ||
| Scheme 2 Synthesis of 15 and 17. Reagents and conditions: i: Bu2BOTf, i-Pr2EtN, DCM, −78 °C, 99%; ii: NaOMe, MeOH, 0 °C, 36%; iii. DAST, DCM, −78 °C. Stereochemical course of reaction iii: 14 to 15, 74%, 53% de; 16 to 17, 53%, 50% de. | ||
Synthesis of the 4-nitrophenyl- derivative was investigated (Scheme 3) in order to destabilise the carbocation intermediate and hence improve diastereoselectivity. Condensation of 4-nitrobenzaldehyde 18 with S-Evan's auxiliary protected propanoic acid 19 gave 20, which was converted to methyl ester 21. However, treatment with DAST still resulted in a mixture of syn- and anti-22 (68% de). The diastereomeric selectivity was somewhat improved compared to conversion of 16 to 17, suggesting that a carbocation intermediate had been destabilised and the SN2 reaction was now more favoured.
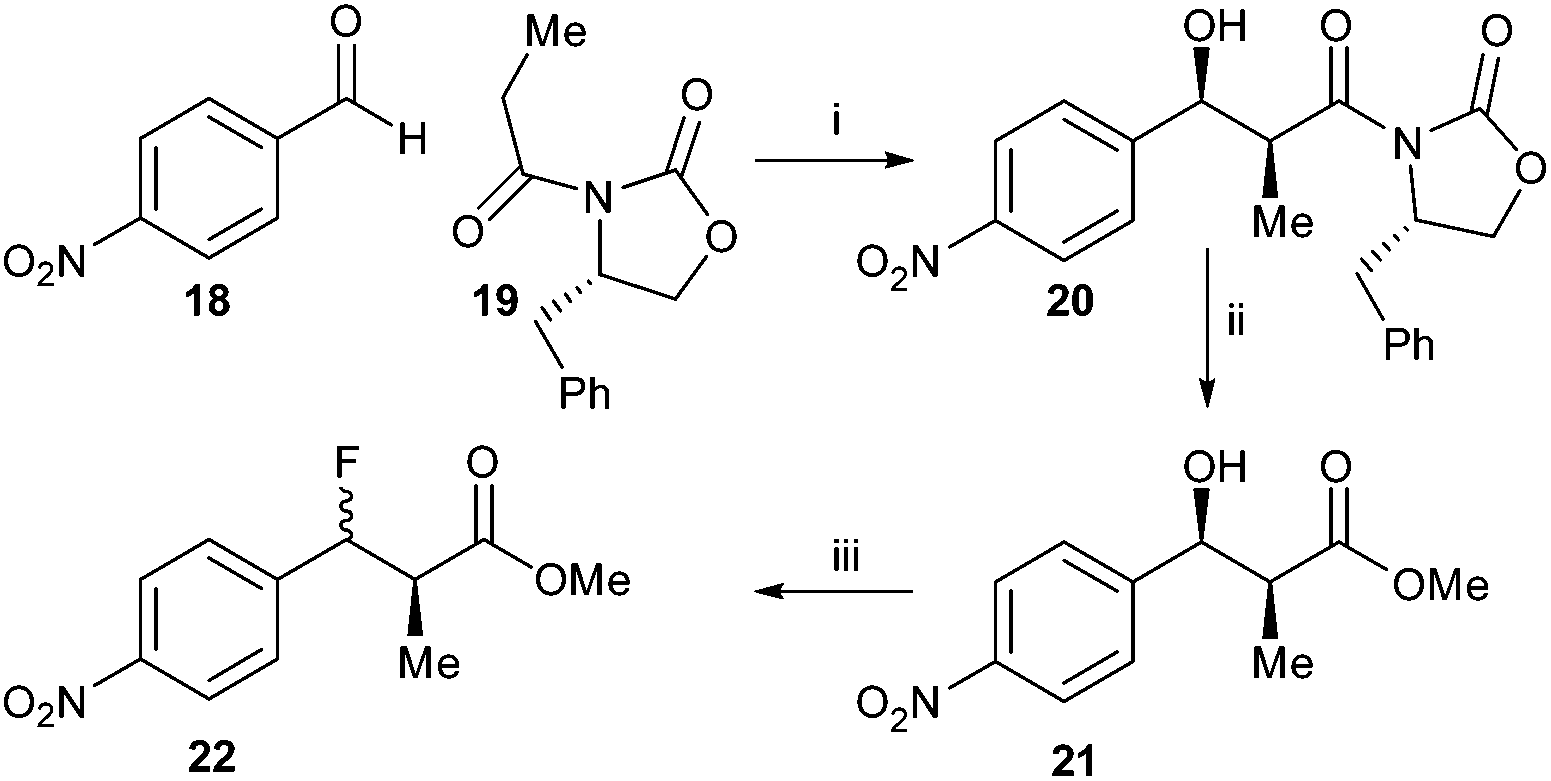 | ||
| Scheme 3 Synthesis of 22. Reagents and conditions: MgCl2, TMSCl, i-Pr2EtN, EtOAc, rt, then TFA:MeOH 1:9, rt, 71%; ii: NaOMe, MeOH, 0 °C, 53%; iii: DAST, DCM, −78 °C. 69% (68% de). | ||
In contrast, condensation of tert-butyl-protected propanoic acid 23 with 4-nitrobenzaldehyde 18 gave 24 as a pair of enantiomers. Treatment with DAST gave the desired ester 25 (Scheme 4).
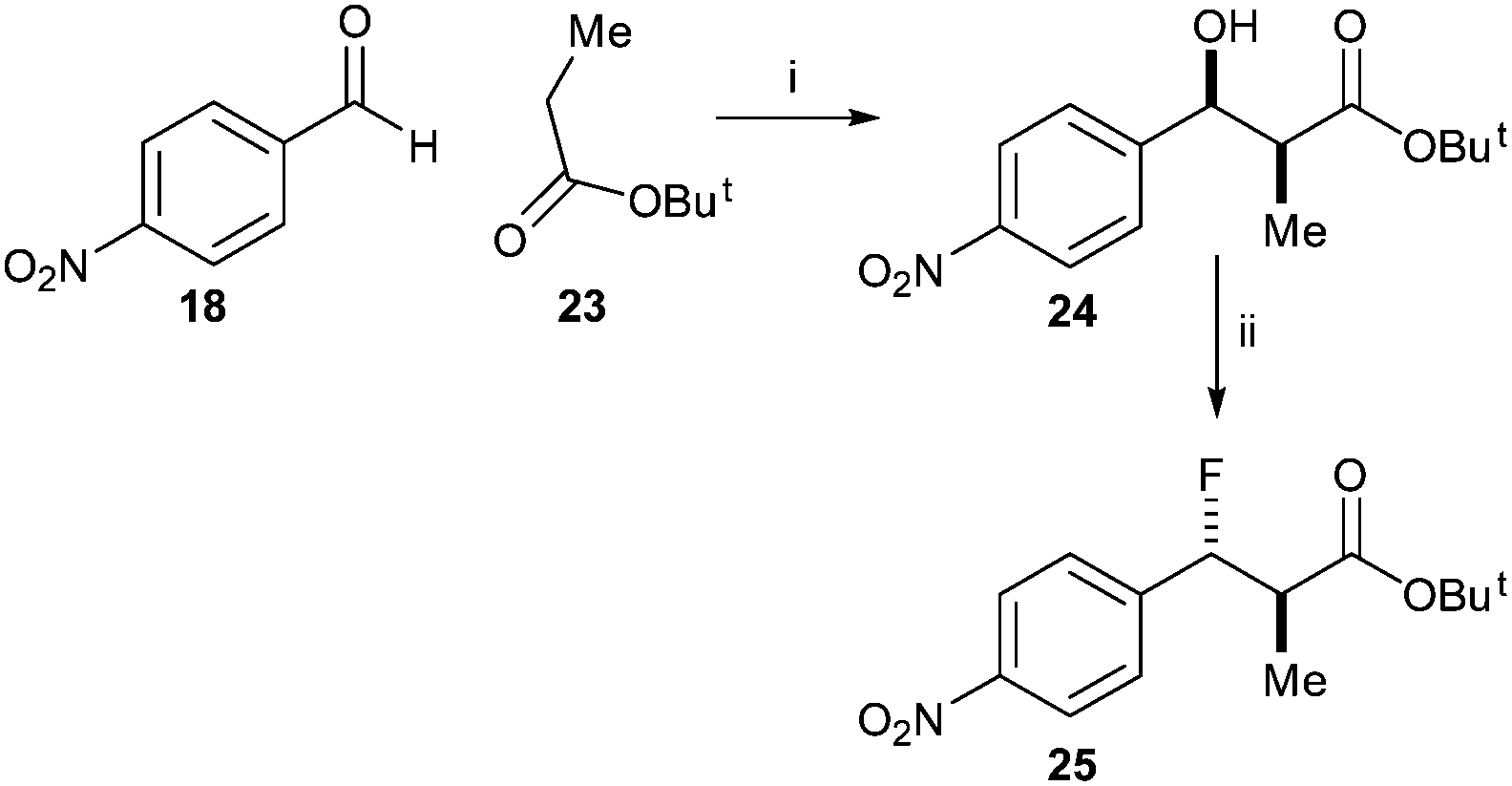 | ||
| Scheme 4 Synthesis of tert-butyl 3-fluoro-2-methyl-3-(4-nitrophenyl)propanoate 25 Reagents and conditions: i. LDA, THF, −78 °C, 52%; ii. DAST, DCM, −78 °C, 51%. | ||
Similarly, reaction of benzyl-protected propanoic acid 26 with 4-nitrobenzaldehyde 18 gave 27. Treatment with DAST gave the desired anti-product 28 (Scheme 5). Removal of the benzyl protecting group with TMSI gave acid 29. However, conversion of the acid to the acyl-CoA ester 30 using CDI resulted in formation of a significant amount of the eliminated acyl-CoA ester 31, the expected enzymatic product. This substrate and product mixture could not be easily separated. The elimination of 30 to give 31 is probably driven by the thermodynamic stability of the conjugated product.
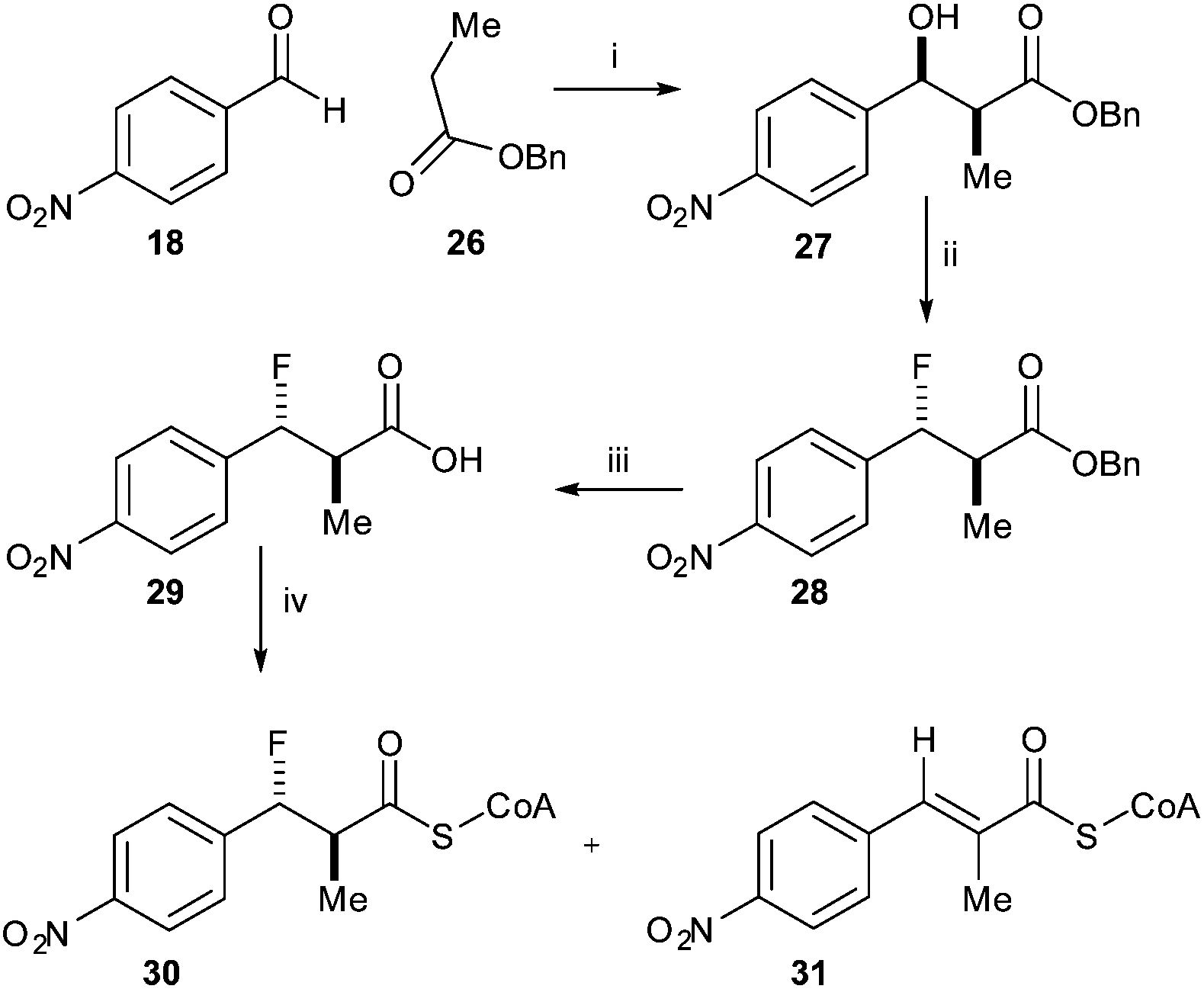 | ||
| Scheme 5 Synthesis of anti-3-fluoro-2-methyl-3-(4-nitrophenyl)propanoyl-CoA 30. Reagents and conditions: i: LDA, THF, −78 °C, separation of syn- and anti-isomers; 17% yield for syn-isomer; ii: DAST, DCM, −78 °C, 42%; iii: TMSI, CHCl3, 40 °C, 90%; iv: CDI, DCM, rt, then CoA-Li3, 0.1 M NaHCO3 aq., THF, rt. | ||
Incubation of a mixture of 30/31 with active AMACR confirmed that 30 was converted to 31, as judged by reduction of the doublet at 0.96 ppm and appearance of the Me-group singlet at 1.97 ppm (Fig. 2). A change in the ratio of the triplets at 2.32 and 2.37 ppm (CH2 groups in the CoA side-chain) was also observed. These changes were not observed when using heat-inactivated enzyme, showing that the elimination was enzyme-catalysed. Product 31 absorbs at a maximum wavelength of <340 nm and this is not ideally suited for use in a microtitre plate assay. It was anticipated that addition of further electron-withdrawing groups or extension of the aromatic system would increase the wavelength of the product chromophore to >340 nm, but this would result in higher levels of HF elimination from the acid when conversion of the acyl-CoA ester was attempted. Therefore, further development of this approach was not undertaken.
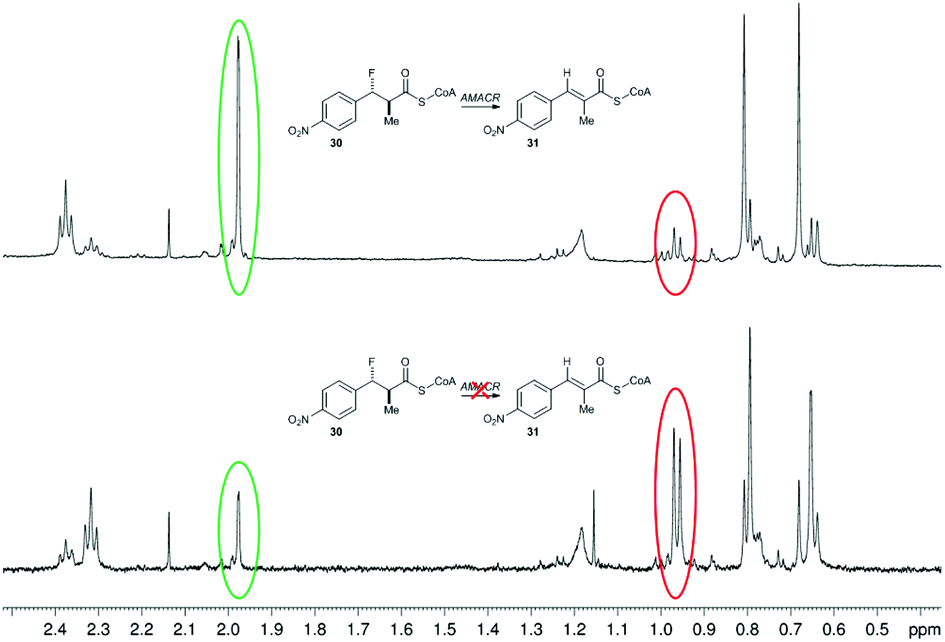 | ||
| Fig. 2 Conversion of 3-fluoro-2-methyl-3-(4-nitrophenyl)propanoyl-CoA 30 by human AMACR 1A. Above, 1H NMR spectrum following incubation with active AMACR; below, 1H NMR spectrum following incubation with heat-inactivated AMACR. Red and green circles show methyl group signals for substrate 30 and unsaturated product 31, respectively. | ||
A second approach to developing a colorimetric or fluorescent assay for AMACR is to utilise a molecular fluoride sensor in order to measure the fluoride released during the enzymatic reaction. An advantage of this approach is that it allows assaying of a wide variety of potential AMACR substrates, including those with alkyl side-chains.35 Although there are many fluoride sensors reported, few of them can be used in aqueous systems. Fluorescent sensors 32,4233,4034,47 and 3543 and the colorimetric sensor 3641 (Fig. 3) were selected for investigation due to their apparent sensitivity and compatibility with aqueous systems.
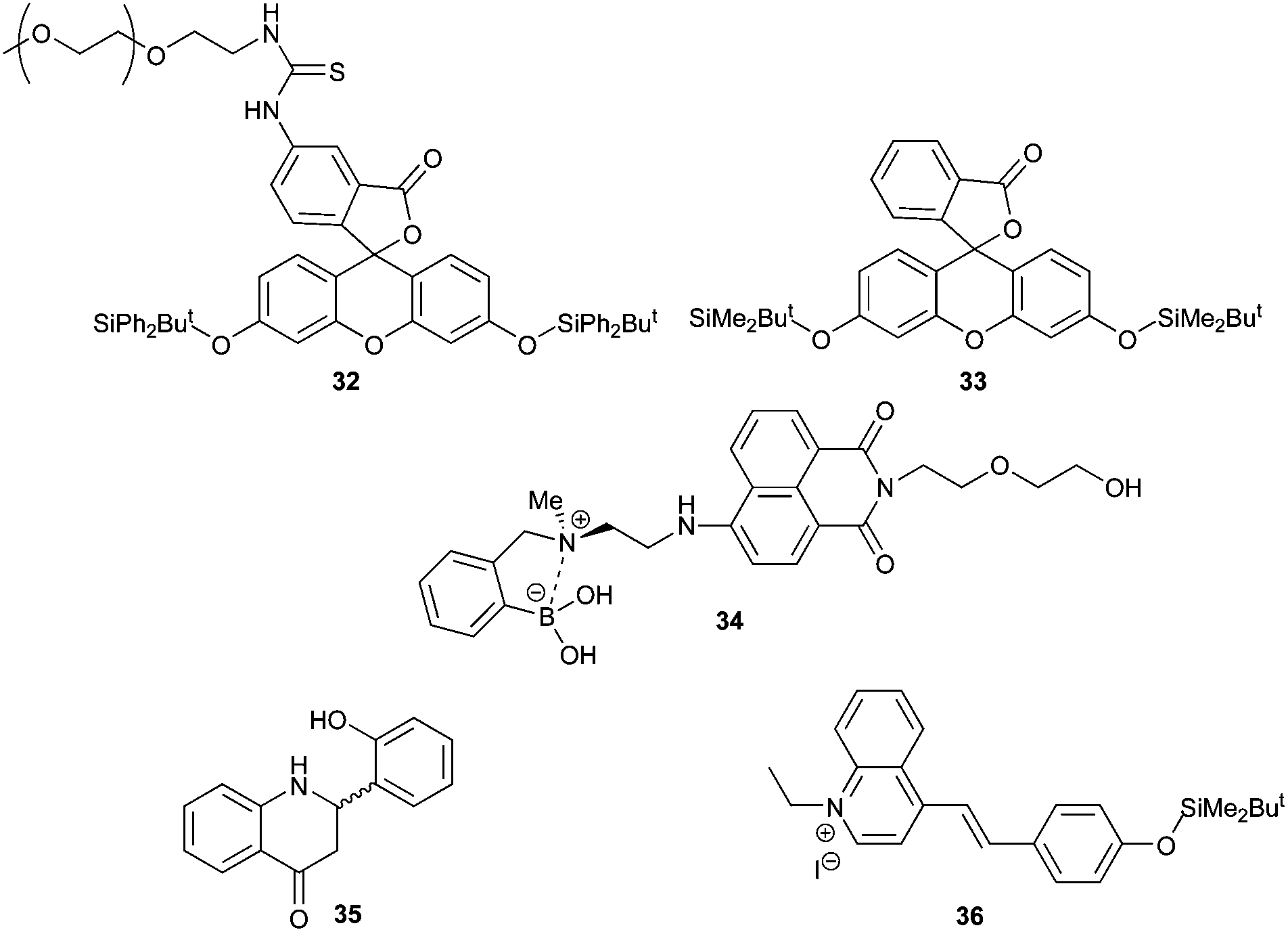 | ||
| Fig. 3 Fluoride sensors selected for study. | ||
Synthesis of these selected sensors and incubation with fluoride in buffered aqueous solution was undertaken to validate the method. Sensor 32 initially gave a low fluorescent signal which rapidly increased with time, regardless of whether fluoride was present or not. This was also true with solutions prepared using highly purified water. It was concluded that spontaneous loss of the silyl-protecting group occurred due to the formation of the highly stabilised aromatic fluorescein. Sensor 33, which has previously been used to assay γ-butyrobetaine hydroxylase activity,40 was therefore investigated. This sensor was more stable, but large variations in signal intensity at low aqueous fluoride concentrations were observed, limiting its use in enzymatic assays (ESI, Fig. S25†). Fluorescent detection of fluoride was also attempted using the ‘turn off’ sensor 34. Reaction of strong nucleophiles such as fluoride35 results in fluorescent quenching of 34 due to a weakening of the interaction between the nitrogen and boron. However, incubation of 34 with standard fluoride solutions resulted in highly variable readings in aqueous solutions even in the presence of high organic solvent concentrations (ESI, Fig. S26 and S27†).
Sensor 35 was also investigated, as it is reported to be highly sensitive and to work by a mechanism that does not involve silyl-protecting group removal.43 Synthesis was accomplished by modification of the literature procedure (Scheme 6).43 2-Hydroxybenzaldehyde was protected with MeI to give 37 and 2-aminoacetophenone was protected with acetyl chloride to give 38. Compounds 37 and 38 were condensed together under alkali conditions to give 39. Hydrolysis of the acetyl group from 39, followed by cyclisation gave 40. Removal of the O-methyl group with NaSEt gave 35, whose structure was confirmed by X-ray crystallography (see ESI† for details). However, no fluorescence was observed for 35 in the presence of fluoride. It was noted that the spectroscopic data for 35 did not match that reported in the literature.43
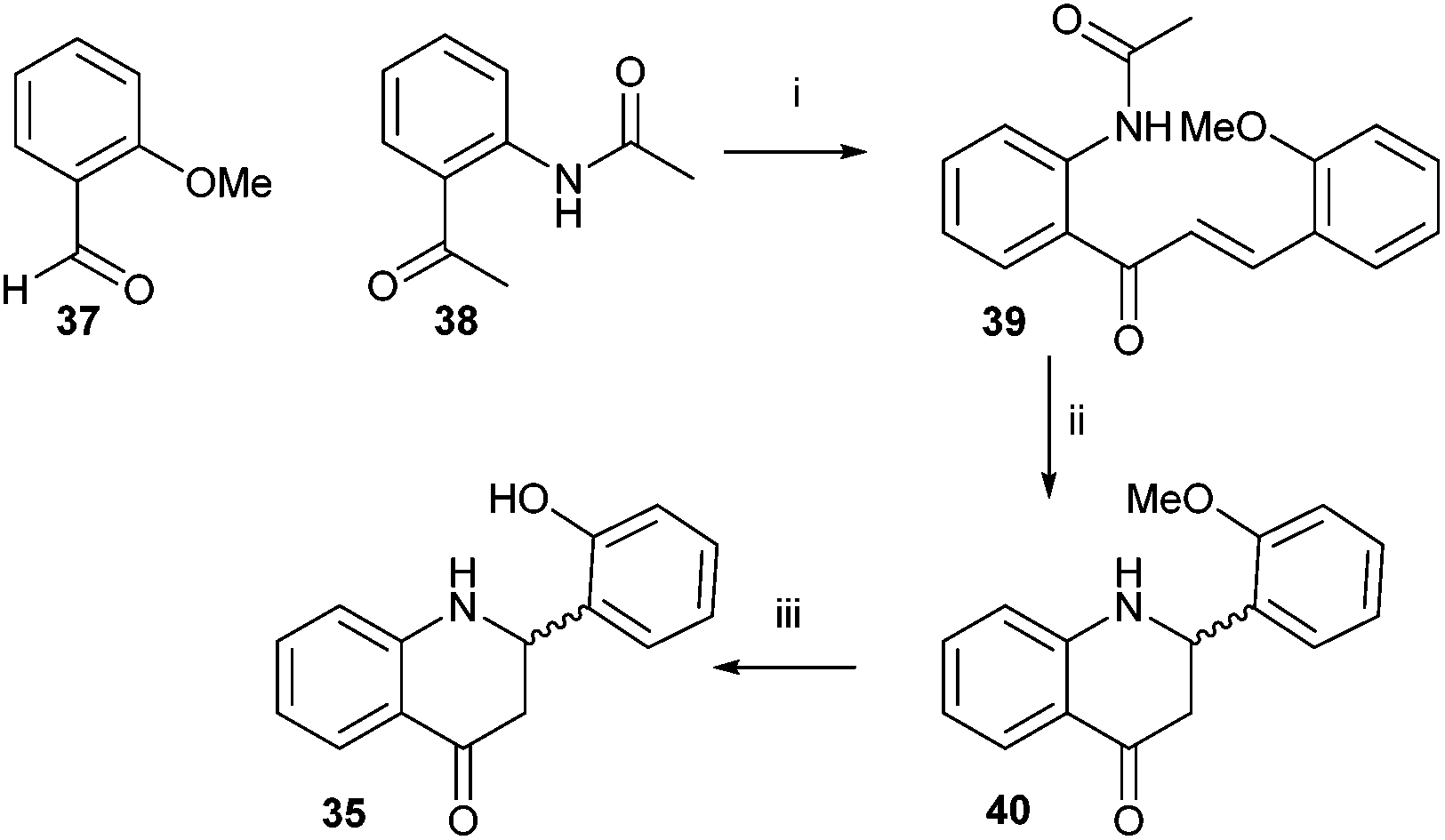 | ||
| Scheme 6 Synthesis of sensor 35. Reagents and conditions: i. 5% (w/v) NaOH aq., MeOH, rt, 72%; ii. 5% (v/v) HCl aq., reflux, 62%; iii. NaSEt, DMF, 140 °C, 80%. | ||
Finally, a colorimetric method for determining fluoride concentrations was investigated. The protected cyanine dye system 36 reported by Zhu et al.,41 was chosen since this is reported to be a highly sensitive system. The required dye was synthesised by reaction of lepidine 41 with ethyl iodide followed by coupling of the product 42 with 4-hydroxybenzaldehyde to give 43 (Scheme 7). However, protection of 43 with TBDMSiCl to give 36 could not be achieved using a number of different conditions, including those originally reported.41
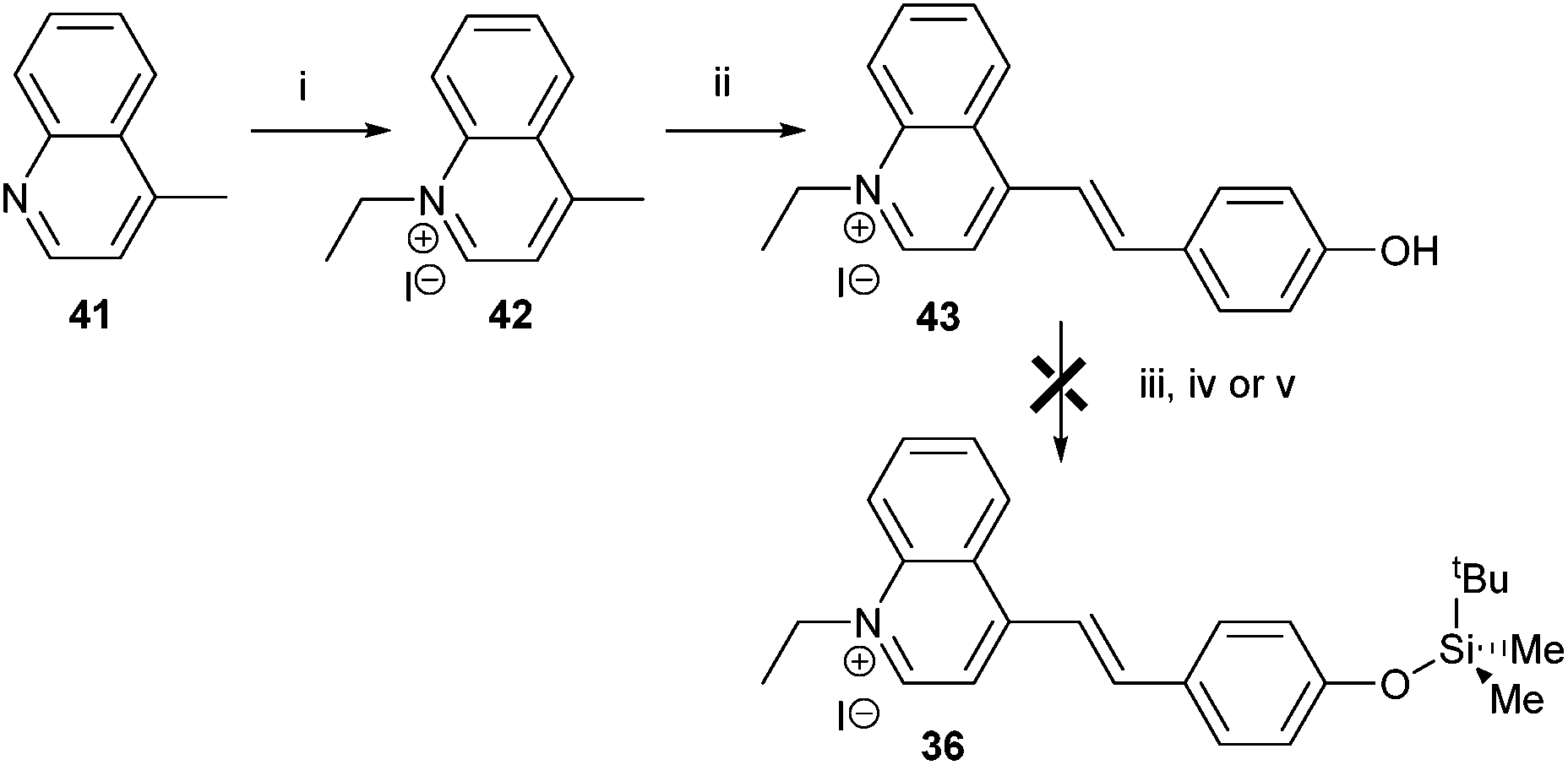 | ||
| Scheme 7 Attempted synthesis of fluoride sensor 36. Reagents and conditions: i: ethyl iodide, toluene, reflux, 89%; ii: 4-hydroxybenzaldehyde, piperidine, MeOH, reflux, 60%; iii: TBDMSiCl, pyridine, reflux, 0%; iv: TBDMSiCl, NEt3, DCM, rt, 0%; v: TBDMSiCl, NEt3, CHCl3, reflux, 0%. | ||
Conclusions
The results herein demonstrate that the elimination reaction catalysed by AMACR can be used to evaluate the potency of inhibitors. Conversion of substrate 1 to product 2 and HF was reduced in the presence of known AMACR substrates (acting as competitive inhibitors) and known inhibitors. N-Dodecyl-N-methyl-carbamoyl-CoA 8 gave the largest reduction in the conversion of 1 to 2 of all the acyl-CoA esters, consistent with previous reports18 that it is a potent inhibitor. Some of the inhibitors reported by Wilson et al.19 also potently inhibited the enzyme. The throughput of this assay is not sufficient for high-throughput screening purposes, but it may allow preliminary characterisation of inhibitors identified through other approaches and would be a useful secondary screen for inhibitors identified using other methods.Attempts to adapt the elimination assay to produce a colorimetric or fluorescent read-out were not very successful. Acyl-CoA substrates with aromatic side-chains were synthesised, but the presence of the aromatic side-chain resulted in loss of stereochemistry upon introduction of the fluoride leaving group. Loss of stereochemical integrity limits the application of these substrates as non-enzymatic elimination occurs in substrates in which the methyl group and fluorine atom are in a syn-arrangement. This approach is also limited by fluoride elimination upon conversion of the acid to the acyl-CoA. The results show that AMACR catalyses the elimination of fluoride from acyl-CoAs with aromatic side-chains and hence extends the range of known substrates.
The alternative approach of assaying AMACR activity by quantifying fluoride using sensors was also not very successful. Although there are several fluoride sensors reported for use in aqueous solutions, the high levels of hydration of the fluoride anion35 makes such methods slow and it can be difficult to achieve sufficient reproducibility.
Experimental
Sources of materials
All chemicals were obtained from the Sigma-Aldrich Chemical Co. or Fisher Scientific Ltd and were used without further purification, unless otherwise noted. Reagents were of analytical grade or equivalent (synthesis) or biochemical grade. Oasis HLB cartridges were obtained from Waters Corporation. Construction of the expression plasmid for human AMACR 1A has been previously described.4 The Rosetta2 (DE3) expression strain and auto-induction media system 1 were obtained from Novagen. (2R,3R)-3-Fluoro-2-methyldecanoyl-CoA 1 was synthesised as previously described.35 Fenoprofenoyl-CoA 3, Flurbiprofenoyl-CoA 4, Ibuprofenoyl-CoA 5, Ketoprofenoyl-CoA 6 and Naproxenoyl-CoA 7 were synthesised as previously described.5 Ebselen 9 and Ebselen oxide 10 were obtained from Cayman Chemical. Compounds 1448 and 1649 were synthesised by their reported methods. The PEGylated fluorescein derivative 32 was synthesised by the method of Zheng et al.42tert-Butyldimethylsilyl-protected fluorescein 33 was synthesised by the method of Rydzik et al.40 The boronic acid sensor 34 was synthesised by the method described by Sun et al.47 Intermediates 37,5038,5139,52 and 4052 for the synthesis of sensor 35 were produced by known methods. Intermediates 4253 and 4353 required for the synthesis of 36 were produced by known methods.General experimental procedures
Solvents were removed using Büchi rotary evaporators. Thin layer chromatography was performed on Merck silica aluminium plates 60 (F254) and UV light, potassium permanganate or phosphomolibdic acid were used for visualisation. Column chromatography was performed using Fisher silica gel (particle size 35–70 micron). Purifications of acyl-CoA esters were performed by solid phase extraction using Oasis HLB 6cc (200 mg) extraction cartridges. Phosphate buffer was prepared from monobasic and dibasic potassium phosphates at the required proportion for 0.1 M pH 7.0 buffer. Optical rotations were recorded on an Optical Activity AA-10 Automatic polarimeter instrument and are reported in 10−1 deg cm2 g−1. IR spectra were recorded on Perkin-Elmer RXI FTIR spectrometer instrument. NMR spectra were recorded on Bruker Avance III 400.04 MHz or 500.13 MHz spectrometers in D2O or CDCl3 and solvent was used as an internal standard. Shifts are given in ppm and J values reported to 0.1 Hz. Multiplicities are described as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Stock concentrations of acyl-CoA esters for assays were determined using 1H NMR.35 Mass spectra were recorded by ESI TOF at the University of Bath Mass Spectrometry Service. High resolution mass spectra were recorded in ES mode. Aqueous solutions for biological experiments were prepared in 18.2 MΩ cm−1 Nanopure water and pH-adjusted with aq. HCl or NaOH. Syntheses were carried out at ambient temperature, unless otherwise specified. Solutions in organic solvents were dried over anhydrous magnesium sulfate and evaporated under reduced pressure.Synthesis of N-dodecyl-N-methyl-carbamoyl-CoA (8)18
Compound 8 was synthesised by the method of Carnell et al.18 using 1-[dodecyl(methyl)carbamoyl]-3-methyl-1H-imidazol-3-ium iodide (40.0 mg, 0.092 mmol) and CoA-Li3 (17.0 mg, 0.020 mmol) in a mixture of dilute aqueous sodium hydrogen carbonate and THF. The crude aqueous product was freeze-dried and purified with solid phase extraction to obtain a white solid (7.1 mg). 1H NMR (500.13 MHz, CDCl3): δ 8.62 (1H, s), 8.35 (1H, s), 6.15 (1H, d, J = 6.0 Hz), 4.57–4.48 (1H, m), 4.23–4.13 (2H, m), 3.97 (1H, s), 3.84 (3H, s), 3.81–3.76 (1H, m), 3.53–3.47 (1H, m), 3.39 (2H, t, J = 6.5 Hz), 3.35–3.22 (3H, m), 2.95–2.82 (4H, m), 2.38 (2H, t, J = 6.5 Hz), 1.55–1.35 (2H, m), 1.25–1.05 (18H, m), 0.87 (3H, s), 0.76 (3H, t, J = 7.0 Hz), 0.72 (3H, s); HRMS (ES) [M + 2Na − 3H]− Calcd. For C35H60N8Na2O17P3S: 1035.2805, found 1035.3050.Attempted synthesis of (R)-4-benzyl-3-[(2S,3S)-3-fluoro-2-methyl-3-phenylpropanoyl]oxazolidin-2-one (15)
A solution of DAST (0.09 mL, 0.68 mmol) in anhydrous DCM (1 mL) was added dropwise to a solution of 14 (230 mg, 0.68 mmol) in anhydrous DCM (3 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 2 h, then allowed to reach ambient temperature. The reaction mixture was quenched by the slow addition of water (5 mL). The organic layer was washed with saturated NaHCO3 aq. and brine. The solution was dried over MgSO4, filtered and then the solvent was removed under reduced pressure. The product was purified by column chromatography (Pe:EtOAc 5:1) to give 15 as a mixture of diastereoisomers (200 mg, 87%, 50% de) as a colourless oil. 1H NMR (400.04 MHz, CDCl3): δ Major diastereomer (selected isolated peaks) 7.43–7.16 (10H, m), 5.63 (1H, dd, J = 46.4, 9.8 Hz), 4.80–4.72 (1H, m), 3.30 (1H, dd, J = 13.4, 3.4 Hz), 2.83 (1H, dd, J = 13.4, 9.5 Hz), 1.02 (3H, d, J = 7.0 Hz); minor diastereomer (selected isolated peaks) 7.43–7.16 (10H, m), 5.69 (1H, dd, J = 47.5, 6.6 Hz), 3.24 (1H, dd, J = 13.4, 3.3 Hz), 2.74 (1H, dd, J = 13.4, 9.7 Hz), 1.38 (3H, dd, J = 6.8, 0.8 Hz).
Attempted synthesis of (2S,3S)-methyl-3-fluoro-2-methyl-3-phenylpropanoate (17)
A solution of DAST (92 μL, 0.70 mmol) in anhydrous DCM (1 mL) was added dropwise to a solution of 16 (135 mg, 0.70 mmol) in anhydrous DCM (4 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 1 h, then allowed to reach ambient temperature. The reaction mixture was quenched by the slow addition of water (5 mL). The organic layer was washed with saturated NaHCO3 aq. and brine. The solution was dried over MgSO4, filtered and then the solvent was removed under reduced pressure. The product was purified by column chromatography (Pe:EtOAc 20:1) to give 17 (100 mg, 72%, 50% de) as a colourless oil. 1H NMR54 (400.04 MHz, CDCl3): δ Major diastereomer: 7.42–7.29 (5H, m), 5.54 (1H, dd, J = 46.1, 9.5 Hz), 3.77 (3H, s), 3.07–2.97 (1H, m), 0.96 (3H, d, J = 7.2 Hz); minor diastereomer: 7.42–7.29 (5H, m), 5.76 (1H, dd, J = 46.7, 6.1 Hz), 3.63 (3H, s), 2.97–2.87 (1H, m), 1.26 (3H, dd, J = 7.0, 0.8 Hz).
Synthesis of (S)-4-benzyl-3-[(2S,3S)-3-hydroxy-2-methyl-3-(4-nitrophenyl)propanoyl]-oxazolidin-2-one (20)
A solution of dibutylboron triflate in DCM (1.0 M, 4.72 mL, 4.72 mmol) and diisopropylethylamine (0.90 mL, 5.14 mmol) were added to a stirred solution of oxazolidinone 19 (1.000 g, 4.29 mmol) in 10 mL of DCM at −78 °C and the resulting solution was stirred for 30 min at this temperature. p-Nitrobenzaldehyde 18 (0.907 g, 6.00 mmol) in 3 mL of DCM was added dropwise and the reaction mixture was stirred at −78 °C for 30 min and then allowed to reach ambient temperature. The reaction was quenched by slow addition of phosphate buffer (0.1 M, pH = 7.0, 10 mL), the organic layer was then washed with 1 M HCl aq., then saturated NaHCO3 aq. and brine. The solution was dried over MgSO4, filtered and then the solvent was removed under reduced pressure. The product was purified by column chromatography (Pe:EtOAc 2:1) to give 20 (1.460 g, 89%) as a yellow solid. m.p. 136–138 °C; [α]21D = +59.0 (CHCl3, c = 0.43); IR (KBr disc, cm−1): 3525.3, 1775.2, 1683.4 1H NMR (400.04 MHz, CDCl3): δ 8.22 (2H, d, J = 8.7 Hz), 7.59 (2H, d, J = 8.7 Hz), 7.38–7.27 (3H, m), 7.23–7.17 (2H, m), 5.28–5.24 (1H, m), 4.78–4.70 (1H, m), 4.30–4.20 (2H, m), 4.05 (1H, dq, J = 7.0, 2.8 Hz), 3.54–3.48 (1H, m), 3.26 (1H, dd, J = 13.4, 3.4 Hz), 2.82 (1H, dd, J = 13.4, 9.4 Hz), 1.13 (3H, d, J = 7.0 Hz). 13C NMR (100.60 MHz, CDCl3) δ 176.67, 152.86, 148.34, 147.27, 134.66, 129.35, 129.01, 127.54, 126.93, 123.45, 72.19, 66.34, 55.01, 43.95, 37.71, 9.99. HRMS (ES) [M + H]+ Calcd. for C20H21N2O6: 385.1400, Found: 385.1421; [M + Na]+ Calcd. for C20H20N2NaO6: 407.1219, Found: 407.1216.
Synthesis of (2S,3S)-methyl 3-hydroxy-2-methyl-3-(4-nitrophenyl)propanoate (21)
Compound 21 was synthesised by a new procedure: sodium metal (49 mg, 2.13 mmol) was reacted with anhydrous MeOH (20 mL), cooled to 0 °C, then treated with a solution of compound 20 (511 mg, 1.33 mmol) in anhydrous MeOH (5 mL). The resulting reaction mixture was stirred at 0 °C for 15 min. The reaction was quenched by the slow addition of phosphate buffer (0.1 M, pH = 7.0, 20 mL). The reaction mixture was extracted with DCM (4 × 20 mL) and the combined organic extracts were washed with brine, dried over MgSO4, filtered and the solvents were removed under reduced pressure. The product was purified by column chromatography (Pe:EtOAc 3:1) to give 21 (170 mg, 53%) as a yellow oil. 1H NMR55 (400.04 MHz, CDCl3): δ 8.23–8.17 (2H, m), 7.55–7.50 (2H, m), 5.24 (1H, m), 3.72 (3H, s), 3.32 (1H, d, J = 3.0 Hz), 2.79 (1H, dq, J = 7.3, 3.4 Hz), 1.07 (3H, d, J = 7.3 Hz).
Synthesis of (2R,3R,S)-methyl-3-fluoro-2-methyl-3-(4-nitrophenyl)propanoate (22)
A solution of DAST (90 μL, 0.69 mmol) in anhydrous DCM (1 mL) was added dropwise to a solution of compound 21 (165 mg, 0.69 mmol) in anhydrous DCM (4 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 1 h, then allowed to reach ambient temperature. The reaction mixture was quenched by the slow addition of water (5 mL). The organic layer was washed with saturated NaHCO3 aq. and brine. The solution was dried over MgSO4, filtered and then the solvent was removed under reduced pressure. The product was purified by column chromatography (Pe:EtOAc 20:1) to give 22 (93 mg, 56%, 77% de) as a colourless oil. 1H NMR (400.04 MHz, CDCl3): δ Major diastereomer: 8.27–8.19 (2H, m), 7.53–7.47 (2H, m), 5.71 (1H, dd, J = 45.8, 8.3 Hz), 3.75 (3H, s), 3.08–3.00 (1H, m), 1.02 (3H, d, J = 7.2 Hz); minor diastereomer: 8.27–8.19 (2H, m), 7.53–7.47 (2H, m), 5.87 (1H, dd, J = 46.5, 5.8 Hz), 3.66 (3H, s), 3.00–2.85 (1H, m), 1.25 (3H, dd, J = 7.1, 0.9 Hz).
Synthesis of syn–tert-butyl 3-hydroxy-2-methyl-3-(4-nitrophenyl)propanoate (24)
tert-Butyl propionate 23 (1.00 mL, 865 mg, 6.64 mmol) was dissolved in anhydrous THF (20 mL), cooled to −78 °C, then lithium diisopropylamide in THF (2.0 M, 3.2 mL, 6.64 mmol) was added dropwise and the reaction mixture was stirred at this temperature for 30 min. 4-Nitrobenzaldehyde 18 (1004 mg, 6.64 mmol) in anhydrous THF (7 mL) was added to the reaction mixture, stirred for 2 h and then the reaction mixture was allowed to reach ambient temperature. The reaction mixture was quenched by slow addition of saturated NH4Cl aq. (20 mL), extracted with DCM. The organic layer was washed with water and brine, dried over MgSO4, filtered and the solvents were removed under reduced pressure. The residue was purified by column chromatography (Pe:EtOAc 10:1) to give compound 24 (970 mg, 52%) as a yellow oil. 1H NMR (400.04 MHz, CDCl3): δ 8.22–8.17 (2H, m), 7.55–7.50 (2H, m), 5.18 (1H, dd, J = 3.5, 2.8 Hz), 3.55 (1H, d, J = 2.8 Hz), 2.67 (1H, dq, J = 7.2, 3.5 Hz), 1.44 (9H, s), 1.03 (3H, d, J = 7.2 Hz). 13C NMR (100.59 MHz, CDCl3) δ 175.20, 148.80, 147.16, 126.87, 123.37, 81.81, 72.46, 46.27, 27.93, 10.33. HRMS (ES) [M + Na]+ Calcd. for C14H19NNaO5: 304.1161, Found: 304.1160.
Synthesis of anti–tert-butyl 3-fluoro-2-methyl-3-(4-nitrophenyl)propanoate (25)
A solution of DAST (94 μL, 0.71 mmol) in anhydrous DCM (1 mL) was added dropwise to a solution of compound 24 (200 mg, 0.71 mmol) in anhydrous DCM (5 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 2 h, then allowed to reach ambient temperature. The reaction mixture was quenched by the slow addition of water (10 mL). The organic layer was washed with saturated NaHCO3 aq. and brine. The solution was dried over MgSO4, filtered and then the solvent was removed under reduced pressure. The product was purified by column chromatography (Pe:EtOAc 30:1) to give 25 (102 mg, 51%) as white solid. 1H NMR (400.04 MHz, CDCl3): δ 8.26–8.21 (2H, m), 7.55–7.48 (2H, m), 5.78 (1H, dd, J = 46.8, 6.4 Hz), 2.89–2.74 (1H, m), 1.36 (9H, s), 1.23 (3H, dd, J = 7.1, 0.9 Hz). 13C NMR (125.76 MHz, CDCl3) δ 171.83 (d, J = 3.9 Hz), 148.15, 144.46 (d, J = 20.2 Hz), 127.35 (d, J = 7.0 Hz), 123.68, 94.25 (d, J = 176.6 Hz), 81.64, 46.99 (d, J = 24.6 Hz), 28.00, 12.92 (d, J = 6.7 Hz). 19F NMR (470.52 MHz, CDCl3) δ −175.39. HRMS (ES) [M + Na]+ Calcd. for C14H18FNNaO4: 306.1118, Found: 306.1106.
Synthesis of syn-benzyl 3-hydroxy-2-methyl-3-(4-nitrophenyl)propanoate (27)
Benzyl propionate 26 (1.00 mL, 1.04 g, 6.33 mmol) was dissolved in anhydrous THF (20 mL), cooled to −78 °C, then LDA in THF (2.0 M, 3.2 mL, 6.33 mmol) was added dropwise and the reaction mixture was stirred at this temperature for 30 min. 4-Nitrobenzaldehyde 18 (956 mg, 6.33 mmol) in anhydrous THF (7 mL) was added to the reaction mixture, stirred for 2 h and then the reaction mixture was allowed to reach ambient temperature. Reaction mixture was quenched by slow addition of saturated NH4Cl aq. (20 mL) and extracted with DCM. The organic layer was washed with water and brine, dried over MgSO4, filtered and the solvents were removed under reduced pressure. The residue was purified by column chromatography (Pe:EtOAc 10:1) to give compound 27 (519 mg, 26%) as a yellow oil. 1H NMR56 (400.04 MHz, CDCl3): δ 8.19–8.12 (2H, m), 7.52–7.45 (2H, m), 7.38–7.26 (5H, m), 5.21 (1H, dd, J = 3.9, 3.2 Hz), 5.18–5.09 (2H, m), 3.21 (1H, d, J = 3.2 Hz), 2.84 (1H, dq, J = 7.2, 3.9 Hz), 1.12 (3H, d, J = 7.2 Hz). HRMS (ES) [M + Na]+ Calcd. for C17H17NNaO5: 338.1004, Found: 338.1006.
Synthesis of anti-benzyl 3-fluoro-2-methyl-3-(4-nitrophenyl)propanoate (28)
A solution of DAST (0.46 mL, 3.45 mmol) in anhydrous DCM (10 mL) was added dropwise to a solution of compound 27 (989 mg, 3.14 mmol) in anhydrous DCM (20 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 2 h, then allowed to reach ambient temperature. The reaction mixture was quenched by the slow addition of water (20 mL). The organic layer was washed with saturated NaHCO3 aq. and brine. The solution was dried over MgSO4, filtered and then the solvent was removed under reduced pressure. The product was purified by column chromatography (Pe:EtOAc 10:1) to give 28 (410 mg, 42%) as a colourless oil. 1H NMR (500.13 MHz, CDCl3): δ 8.23–8.16 (2H, m), 7.48–7.41 (2H, m), 7.40–7.31 (5H, m), 5.74 (1H, dd, J = 45.7, 7.9 Hz), 5.23–5.14 (2H, m), 3.14–3.03 (1H, m), 1.06 (3H, d, J = 7.2 Hz). 13C NMR (125.76 MHz, CDCl3) δ 172.25 (d, J = 4.2 Hz), 148.09, 143.91 (d, J = 20.4 Hz), 135.34, 128.58, 128.45, 128.31, 127.14 (d, J = 7.1 Hz), 123.65, 93.78 (d, J = 177.6 Hz), 66.88, 46.03 (d, J = 24.5 Hz), 12.66 (d, J = 6.3 Hz). 19F NMR (470.52 MHz, CDCl3) δ −176.09. HRMS (ES) [M + Na]+ Calcd. for C17H16FNNaO4: 340.0961, Found: 340.0947.
Synthesis of anti-3-fluoro-2-methyl-3-(4-nitrophenyl)propanoic acid (29)
Compound 28 (216 mg, 0.68 mmol) was dissolved in anhydrous CHCl3 (10 mL), then TMSI (0.31 mL, 2.18 mmol) was added and the reaction mixture was stirred at 40 °C for 16 h. The reaction was quenched by slow addition of water (10 mL). The organic layer was washed with water and brine, dried over MgSO4, filtered and the solvent was removed under reduced pressure. The residue was purified by column chromatography (DCM:MeOH 10:1) to give 29 (139 mg, 90%) as a colourless oil. 1H NMR (500.13 MHz, CDCl3): δ 8.32–8.25 (2H, m), 7.58–7.51 (2H, m), 5.74 (1H, dd, J = 45.7, 8.6 Hz), 3.11–3.01 (1H, m), 1.08 (3H, d, J = 7.2 Hz). 13C NMR (125.76 MHz, CDCl3) δ 178.28, 148.32, 143.55 (d, J = 20.4 Hz), 127.40 (d, J = 6.9 Hz), 123.84, 93.68 (d, J = 177.1 Hz), 45.89 (d, J = 24.8 Hz), 12.81 (d, J = 6.5 Hz). 19F NMR (470.52 MHz, CDCl3) δ −173.92. HRMS (ES) [M + Na]+ Calcd. for C10H10FNNaO4: 250.0492, Found: 250.0477.
Attempted synthesis of anti-3-fluoro-2-methyl-3-(4-nitrophenyl)propanoyl-CoA (30)
Compound 30 was prepared from the acid 29 using CDI and CoA-Li3 according to the usual procedure5 and purified with solid phase extraction to give a white powder. 1H NMR analysis showed that the product was a mixture of 30 and the eliminated acyl-CoA ester 31 in an approximate 4:1 ratio. Full characterisation was not possible, however selected peaks from the 1H spectrum of 30 can be reported. 1H NMR (500.13 MHz, D2O): δ 8.60 (1H, m), 8.34 (1H, m), 8.17 (2H, m), 7.53 (2H, m), 6.13 (1H, dd, J = 6.0, 2.0 Hz), 5.77–5.65 (2H, two overlapping dd, J = 45.5, 7.5 Hz),), 4.53 (1H, m), 4.20–4.14 (2H, m), 3.98 (1H, s), 3.82–3.75 (1H, m), 3.55–3.48 (1H, m), 3.42–3.22 (5H, m), 2.97 (2H, t, J = 6.8 Hz), 2.37 (2H, t, J = 6.8 Hz), 1.01 (3H, d, J = 7.2 Hz), 0.87 (3H, s), 0.74 (3H, s).
Synthesis of 2-(2-hydroxyphenyl)-2,3-dihydroquinolin-4(1H)-one (35)57
NaSEt (2.602 g, 30.94 mmol) was added to a stirred solution of compound 40 (1.306 g, 5.16 mmol) in anhydrous DMF (46 mL) and the reaction mixture was stirred at 140 °C for 17 h. DMF was removed under reduced pressure, and the residue was dissolved in EtOAc, washed with saturated NH4Cl aq. and the organic layer was washed with water and brine, dried over MgSO4, filtered and the solvents were removed under reduced pressure. The residue was purified by column chromatography (Pe:EtOAc 5:1) to give compound 35 (990 mg, 80%) as a yellow solid. m.p. 179–181 °C, lit.57 165–167 °C. IR (KBr disk, cm−1): 3096.5, 1639.1, 1607.6. 1H NMR (400.04 MHz, CDCl3): δ 7.94 (1H, dd, J = 7.9, 1.5 Hz), 7.66 (1H, s), 7.41 (1H, ddd, J = 8.2, 7.2, 1.6 Hz), 7.24 (1H, ddd, J = 8.2, 7.5, 1.7 Hz), 7.16 (1H, dd, J = 7.5, 1.6 Hz), 6.98–6.87 (3H, m), 6.83 (1H, d, J = 8.2 Hz), 4.89 (1H, ddd, J = 14.1, 3.6, 0.8 Hz), 4.70 (1H, br s), 3.10 (1H, dd, J = 16.8, 14.1 Hz), 2.83 (1H, ddd, J = 16.8, 3.6, 1.8 Hz). 13C NMR (125.77 MHz, CDCl3) δ 193.70, 155.21, 150.67, 135.51, 129.84, 127.90, 127.80, 124.86, 120.6, 120.46, 117.34, 117.25, 57.42, 43.62. HRMS (ES) [M + H]+ Calcd. for C15H14NO2: 240.1025, Found: 240.1005; [M + Na]+ Calcd. for C15H13NNaO2: 262.0844, Found: 262.0828. Details of the crystal structure determination and parameters are reported in the ESI.†
Expression and purification of AMACR 1A
The plasmid for wild-type AMACR 1A4 was transformed into competent Rosetta2 (DE3) cells and plated onto Lennox LB media supplemented with 1% (w/v) agar, 30 μg mL−1 kanamycin sulfate and 32 μg mL−1 chloramphenicol. A single colony was picked into 10 mL Lennox LB media supplemented with 30 μg mL−1 kanamycin sulfate and 32 μg mL−1 chloramphenicol and grown overnight at 28 °C and 220 rpm. Growth of starter culture at 37 °C resulted in ‘leaky’ expression of wild-type AMACR. Starter culture was used to inoculate 500 mL of LB media supplemented with the same antibiotics and 1× auto-induction media and grown under the same conditions overnight. Cells were harvested (Beckman JA-10 rotor, 9000 rpm, 16000g, 30 min) and stored at −80 °C.
Cells (∼2 g) were re-suspended in 30 mL start buffer and AMACR was purified as previously described.35 Fractions containing AMACR were identified by SDS-PAGE analyses using 10% gels, pooled and dialysed into 10 mM NaH2PO4–NaOH, pH 7.4. Protein concentrations were determined by absorbance at 280 nm, and extinction coefficients and molecular weights for the His-tag protein calculated using Protparam (http://web.expasy.org/protparam/).
1H NMR assay of AMACR activity
Enzyme assays with inhibitor were performed using a similar method to previously reported.35 Enzyme (0.12 mg mL−1; 2.54 μM) was incubated with inhibitor (200 μM) in the presence of NaH2PO4–NaOH, pH 7.4 and ca. 88% (v/v) D2O (275 μL) for 10 min. An equal volume of (2R,3R)-3-fluoro-2-methyldecanoyl-CoA substrate 1 (200 μM) in buffer and D2O (275 μL) was added to the enzyme/inhibitor mixture, and the assay was incubated at 30 °C for 60 min. Enzyme was inactivated by heating at 60 °C for 10 min before 1H NMR analysis. Conversion of substrates was quantified by conversion of the 2-Me doublet at ca. 1.0 ppm into a singlet at ca. 1.75 ppm, and levels were corrected for non-enzymatic conversion in heat-inactivated negative controls (<5% conversion).4,5 Reported conversions are relative to positive controls lacking an inhibitor (100% activity). Approximately 50% of substrate 1 was converted to 2 after 1 h. Substrate conversion levels in the presence and absence of each inhibitor are given in the ESI (Table S1†) Concentrations of acyl-CoA substrate and inhibitor stock solutions were determined by 1H NMR.5Fluorescent detection of fluoride
Sensor 33 in DMSO (256 μM, 80 μL) was incubated with NaF (0–640 μM, 20 μL) in 50 mM Tris-HCl, pH 7.5 in a black microtitre plate at ambient temperature. After 1 h, 50 μL of 50 mM HEPES–NaOH, pH 7.0 was added. Fluorescence was determined using a FLUOstar Omega plate reader (BMG Labtech) with excitation wavelength 480 nm and emission wavelength 520 nm.40 The graph is shown in the ESI as Fig. S25.†Sensor 34 (final concentration 2 μM) was incubated with fluoride (final concentrations 0–300 μM) in a total volume of 200 μL for 3 min. Reactions were carried out in 100% acetonitrile (using tetra-n-butylammonium fluoride) or in 50 mM NaH2PO4–NaOH, pH 7.4 (using NaF) and acetonitrile [1:19 (v/v)]. Fluorescence was determined using a FLUOstar Omega plate reader (BMG Labtech) with excitation wavelength 350 nm and emission wavelength 520 nm.47 The graphs are shown in the ESI as Fig. S26 and S27,† respectively.
Abbreviations
| AMACR | α-Methylacyl-CoA racemase (P504S) |
| CoA | Coenzyme A |
| CDI | Carbonyldiimidazole |
| DAST | Diethylaminosulfur trifluoride |
| DCM | Dichloromethane |
| de | Diastereomeric excess |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| D2O | Deuterium oxide |
| EtOAc | Ethyl acetate |
| EtOH | Ethanol |
| ESI TOF | Electrospray ionisation time-of-flight |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethane sulfonic acid |
| HPLC | High performance liquid chromatography |
| HRMS | High resolution mass spectrometry |
| IR | Infra-red |
| LB | Luria-Bertani |
| MeI | Methyl iodide |
| MeOH | Methanol |
| m.p. | Melting point |
| NaSEt | Sodium ethanethiolate |
| NMR | Nuclear magnetic resonance |
| Pe | Petroleum ether |
| rpm | Revolutions per minute |
| ppm | Parts per million |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TBDPSi- | tert-Butyldiphenylsilyl- |
| THF | Tetrahydrofuran |
| TMSI | Trimethylsilyl iodide |
| Tris | Tris(hydroxymethyl)methylamine |
Acknowledgements
This work was funded by Prostate Cancer UK (S10-03 and PG14-009), a University of Bath Overseas Research Studentship, and Shandong-Bath undergraduate exchange studentships. We thank Mr N. Christodoulou and Ms Y. Petrova for assistance in synthesising compound 8. The authors are members of the Cancer Research @ Bath (CR@B) network.Notes and references
- W. Schmitz, C. Albers, R. Fingerhut and E. Conzelmann, Eur. J. Biochem., 1995, 231, 815–822 CrossRef CAS PubMed.
- W. Schmitz, R. Fingerhut and E. Conzelmann, Eur. J. Biochem., 1994, 222, 313–323 CrossRef CAS PubMed.
- M. D. Lloyd, M. Yevglevskis, G. L. Lee, P. J. Wood, M. D. Threadgill and T. J. Woodman, Prog. Lipid Res., 2013, 52, 220–230 CrossRef CAS PubMed.
- D. J. Darley, D. S. Butler, S. J. Prideaux, T. W. Thornton, A. D. Wilson, T. J. Woodman, M. D. Threadgill and M. D. Lloyd, Org. Biomol. Chem., 2009, 7, 543–552 CAS.
- T. J. Woodman, P. J. Wood, A. S. Thompson, T. J. Hutchings, G. R. Steel, P. Jiao, M. D. Threadgill and M. D. Lloyd, Chem. Commun., 2011, 47, 7332–7334 RSC.
- W. Schmitz and E. Conzelmann, Eur. J. Biochem., 1997, 244, 434–440 CAS.
- P. P. VanVeldhoven, K. Croes, S. Asselberghs, P. Herdewijn and G. P. Mannaerts, FEBS Lett., 1996, 388, 80–84 CrossRef CAS.
- M. D. Lloyd, D. J. Darley, A. S. Wierzbicki and M. D. Threadgill, FEBS J., 2008, 275, 1089–1102 CrossRef CAS PubMed.
- L.-B. Gao, J.-Z. Wang, T.-W. Yao and S. Zeng, Chirality, 2012, 24, 86–95 CrossRef CAS PubMed.
- M. Yevglevskis, C. R. Bowskill, C. C. Y. Chan, J. H.-J. Heng, M. D. Threadgill, T. J. Woodman and M. D. Lloyd, Org. Biomol. Chem., 2014, 12, 6737–6744 CAS.
- J. Luo, S. Zha, W. R. Gage, T. A. Dunn, J. L. Hicks, C. J. Bennett, C. N. Ewing, E. A. Platz, S. Ferdinandusse, R. J. Wanders, J. M. Trent, W. B. Isaacs and A. M. De Marzo, Cancer Res., 2002, 62, 2220–2226 CAS.
- C. Kumar-Sinha, R. B. Shah, B. Laxman, S. A. Tomlins, J. Harwood, W. Schmitz, E. Conzelmann, M. G. Sanda, J. T. Wei, M. A. Rubin and A. M. Chinnaiyan, Am. J. Pathol., 2004, 164, 787–793 CrossRef CAS PubMed.
- C.-F. Li, F.-M. Fang, J. Lan, J.-W. Wang, H.-J. Kung, L.-T. Chen, T.-J. Chen, S.-H. Li, Y.-H. Wang, H.-C. Tai, S.-C. Yu and H.-Y. Huang, Clin. Cancer Res., 2014, 20, 6141–6152 CrossRef CAS PubMed.
- Z. Jiang, G. R. Fanger, B. F. Banner, B. A. Woda, P. Algate, K. Dresser, J. C. Xu, S. G. Reed, K. L. Rock and P. G. Chu, Cancer Detect. Prev., 2003, 27, 422–426 CrossRef CAS PubMed.
- S. Zha, S. Ferdinandusse, S. Denis, R. J. Wanders, C. M. Ewing, J. Luo, A. M. De Marzo and W. B. Isaacs, Cancer Res., 2003, 63, 7365–7376 CAS.
- K. Takahara, H. Azuma, T. Sakamoto, S. Kiyama, T. Inamoto, N. Ibuki, T. Nishida, H. Nomi, T. Ubai, N. Segawa and Y. Katsuoka, Anticancer Res., 2009, 29, 2497–2505 CAS.
- A. J. Carnell, I. Hale, S. Denis, R. J. A. Wanders, W. B. Isaacs, B. A. Wilson and S. Ferdinandusse, J. Med. Chem., 2007, 50, 2700–2707 CrossRef CAS PubMed.
- A. J. Carnell, R. Kirk, M. Smith, S. McKenna, L.-Y. Lian and R. Gibson, ChemMedChem, 2013, 8, 1643–1647 CAS.
- B. A. P. Wilson, H. Wang, B. A. Nacev, R. C. Mease, J. O. Liu, M. G. Pomper and W. B. Isaacs, Mol. Cancer Ther., 2011, 10, 825–838 CrossRef CAS PubMed.
- A. Morgenroth, E. A. Urusova, C. Dinger, E. Al-Momani, T. Kull, G. Glatting, H. Frauendorf, O. Jahn, F. M. Mottaghy, S. N. Reske and B. D. Zlatopolskiy, Chem. – Eur. J., 2011, 17, 10144–10150 CrossRef CAS PubMed.
- C. Festuccia, G. L. Gravina, A. Mancini, P. Muzi, E. Di Cesare, R. Kirk, M. Smith, S. Hughes, R. Gibson, L.-Y. Lian, E. Ricevuto and A. J. Carnell, Anti-Cancer Agents Med. Chem., 2014, 14, 1031–1041 CrossRef PubMed.
- P. P. VanVeldhoven, K. Croes, M. Casteels and G. P. Mannaerts, Biochim. Biophys. Acta, Lipids Lipid Metab., 1997, 1347, 62–68 CrossRef CAS.
- P. F. Leadlay and J. Q. Fuller, Biochem. J., 1983, 213, 635–642 CrossRef CAS PubMed.
- G. Rudnick and R. H. Abeles, Biochemistry, 1975, 14, 4515–4522 CrossRef CAS PubMed.
- C. W. Koo and J. S. Blanchard, Biochemistry, 1999, 38, 4416–4422 CrossRef CAS PubMed.
- J. Q. Fuller and P. F. Leadlay, Biochem. J., 1983, 213, 643–650 CrossRef CAS PubMed.
- K. A. Gallo and J. R. Knowles, Biochemistry, 1993, 32, 3981–3990 CrossRef CAS PubMed.
- D. Ouazia and S. L. Bearne, Anal. Biochem., 2010, 398, 45–51 CrossRef CAS PubMed.
- T. R. Sharp, G. D. Hegeman and G. L. Kenyon, Anal. Biochem., 1979, 94, 329–334 CrossRef CAS PubMed.
- M. E. Tanner, K. A. Gallo and J. R. Knowles, Biochemistry, 1993, 32, 3998–4006 CrossRef CAS PubMed.
- M. Noda, Y. Matoba, T. Kumagai and M. Sugiyama, Biochem. J., 2005, 389, 491–496 CrossRef CAS PubMed.
- G. J. Cardinal and R. H. Abeles, Biochemistry, 1968, 7, 3970–3978 CrossRef.
- D. L. Schonfeld and U. T. Bornscheuer, Anal. Chem., 2004, 76, 1184–1188 CrossRef PubMed.
- H. Stecher, A. Hermetter and K. Faber, Biotechnol. Tech., 1998, 12, 257–261 CrossRef CAS.
- M. Yevglevskis, G. L. Lee, M. D. Threadgill, T. J. Woodman and M. D. Lloyd, Chem. Commun., 2014, 50, 14164–14166 RSC.
- K. L. Gorres and R. T. Raines, Anal. Biochem., 2009, 386, 181–185 CrossRef CAS PubMed.
- C. S. Rye and S. G. Withers, Anal. Biochem., 2002, 308, 77–82 CrossRef CAS PubMed.
- R. Koncki, D. Ogonczyk and S. Glab, Anal. Chim. Acta, 2005, 538, 257–261 CrossRef CAS.
- S. Abdellaoui, A. Noiriel, R. Henkens, C. Bonaventura, L. J. Blum and B. Doumeche, Anal. Chem., 2013, 85, 3690–3697 CrossRef CAS PubMed.
- A. M. Rydzik, I. K. H. Leung, G. T. Kochan, A. Thalhammer, U. Oppermann, T. D. W. Claridge and C. J. Schofield, ChemBioChem, 2012, 13, 1559–1563 CrossRef CAS PubMed.
- C. Q. Zhu, J. L. Chen, H. Zheng, Y. Q. Wu and J. G. Xu, Anal. Chim. Acta, 2005, 539, 311–316 CrossRef CAS.
- F. Zheng, F. Zeng, C. Yu, X. Hou and S. Wu, Chem. – Eur. J., 2013, 19, 936–942 CrossRef CAS PubMed.
- K. Kanagaraj and K. Pitchumani, Chem. – Asian J., 2014, 9, 146–152 CrossRef CAS PubMed.
- M. Khajehpour, T. Troxler and J. M. Vanderkool, Photochem. Photobiol., 2004, 80, 359–365 CrossRef CAS PubMed.
- S. Y. Kim, J. K. Tak and J. W. Park, Biochimie, 2004, 86, 501–507 CrossRef CAS PubMed.
- P. E. Morgan, R. T. Dean and M. J. Davies, Eur. J. Biochem., 2002, 269, 1916–1925 CrossRef CAS PubMed.
- X. Sun, Q. Xu, G. Kim, S. E. Flower, J. P. Lowe, J. Yoon, J. S. Fossey, X. Qian, S. D. Bull and T. D. James, Chem. Sci., 2014, 5, 3368–3373 RSC.
- N. Schlaeger and A. Kirschning, Org. Biomol. Chem., 2012, 10, 7721–7729 CAS.
- Y. Watanabe, N. Shimada, M. Anada and S. Hashimoto, Tetrahedron: Asymmetry, 2014, 25, 63–73 CrossRef CAS.
- K. Jyothish, Q. Wang and W. Zhang, Adv. Synth. Catal., 2012, 354, 2073–2078 CrossRef CAS.
- A. Furstner and D. N. Jumbam, Tetrahedron, 1992, 48, 5991–6010 CrossRef.
- R. Dhiman, S. Sharma, G. Singh, K. Nepali and P. M. S. Bedi, Arch. Pharm., 2013, 346, 7–16 CrossRef CAS PubMed.
- A. J. Winstead, G. Nyambura, R. Matthews, D. Toney and S. Oyaghire, Molecules, 2013, 18, 14306–14319 CrossRef PubMed.
- A. I. Ayi, M. Remli, R. Guedj and R. Pastor, J. Fluorine Chem., 1981, 17, 127–143 CrossRef CAS.
- P. V. Ramachandran and D. Pratihar, Org. Lett., 2009, 11, 1467–1470 CrossRef CAS PubMed.
- S.-I. Kiyooka, S. Matsumoto, T. Shibata and K.-I. Shinozaki, Tetrahedron, 2010, 66, 1806–1816 CrossRef CAS.
- H. Zheng, Q. Liu, S. Wen, H. Yang and Y. Luo, Tetrahedron: Asymmetry, 2013, 24, 875–882 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of synthesised compounds; details of X-ray crystal structure determination of compound 35; original data for Table 1; plots of fluorescence resulting from reaction of sensors 33 and 34 with fluoride solutions. CCDC 1408401. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob01541c |
| This journal is © The Royal Society of Chemistry 2016 |