
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced protein internalization and efficient endosomal escape using polyampholyte-modified liposomes and freeze concentration†
Sana
Ahmed
a,
Satoshi
Fujita
b and
Kazuaki
Matsumura
*a
aSchool of Materials Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: mkazuaki@jaist.ac.jp
bDepartment of Fiber technology and Science, Graduate School of Engineering, University of Fukui, Fukui 910-8507, Japan
First published on 11th July 2016
Abstract
Here we show a new strategy for efficient freeze concentration-mediated cytoplasmic delivery of proteins, obtained via the endosomal escape property of polyampholyte-modified liposomes. The freeze concentration method successfully induces the efficient internalization of proteins simply by freezing cells with protein and nanocarrier complexes. However, the mechanism of protein internalization remains unclear. Here, we designed a novel protein delivery carrier by modifying liposomes through incorporating hydrophobic polyampholytes therein. These complexes were characterized for particle size, encapsulation efficiency, and cytotoxicity. Flow cytometry and microscopic analysis showed that the adsorption and internalization of protein-loaded polyampholyte-modified liposomes after freezing were enhanced compared with that observed in unfrozen complexes. Inhibition studies demonstrated that the internalization mechanism differs between unmodified and polyampholyte-modified liposomes. Furthermore, polyampholyte-modified liposomes exhibited high efficacy in facilitating endosomal escape to enhance protein delivery to the cytoplasm with low toxicity. These results strongly suggest that the freeze concentration-based strategy could be widely utilised for efficient cargo delivery into the cytoplasm in vitro not only in cancer treatment but also for gene therapy as well.
Introduction
The targeted intracellular delivery of drugs has received considerable attention over the last few decades for improvement of biological activity.1 Various biomaterials are available, and their use has already been shown to be a milestone in the treatment of deadly diseases.2 Protein-based therapeutic materials play a prominent role in the medical field for the treatment of various disorders such as cancer,3 diabetes,4 and inflammatory diseases.5 However, peptide-based drugs have several inherent problems as they show difficulty in entering the plasma membrane. Currently, several methodologies are being tested to improve their targeted delivery. Of these, controlled release has been reported for only a few methods, including electroporation,6 microinjection,7 and ultrasonication.8 The most actively investigated approach, electroporation, induces cell death when the permeabilizing electric field is applied because of the associated loss of cell homeostasis.9 Therefore, site-specific and efficient delivery systems still pose difficulties. Consequently, suitable nanocarriers have been studied for improving the safe and controlled release of peptides to ensure that they reach their targets to a greater extent.10 To date, various nanocarriers including liposomes, polymeric micelles, and nanoparticles have been studied for the delivery of therapeutic materials;11,12 however, many have shown limitations such as cytotoxicity, low stability, and low efficacy.13,14 Among the nanocarriers, liposomes have attracted much attention as a desirable protein-based drug carrier system since they possess the advantages of being feasible under mild conditions, biocompatible with low toxicity, and exhibiting high affinity toward the cell membrane.15 Furthermore, additional properties such as ease of size control and the ability to modify their surfaces enhances their suitability as a vehicle system.16,17More recently, liposomes modified by polymers have been developed to improve their targeting ability. For example, many groups such as Kono et al. have successfully developed pH sensitive liposomes by modifying their surface using polyglycidol derivatives; these liposomes efficiently delivered antigenic molecules to the cytosol of dendritic cells in vitro.18 There are applications for the transport and intracellular delivery of proteins using pH-sensitive liposomes in cancer therapy and also in gene therapy.
To address these issues, we developed an approach called freeze concentration, as previously described.19 Freeze concentration is recognized as a physicochemical phenomenon wherein water molecules crystallize to form ice, leading to increased solute concentrations in the remaining unfrozen solution forming a phase separation during freezing.20 Specifically, spontaneous ice nucleation occurs and ice grows in all directions when a solution is supercooled at −5 to −45 °C. A high solute concentration remains in the unfrozen solution leading to a concentrated solute around the cells located in the residual solution.21,22 Previously, we calculated the sodium ion concentration during freezing in the presence of a cryoprotectant by measuring the amount of residual water by using 1H-NMR.19 When we used DMSO as a cryoprotectant, at −40 °C the sodium ion concentration was approximately 7 times higher than it was before freezing. When we used polyampholyte as a cryoprotectant, the sodium ion concentrated >10 times higher than that at room temperature. This finding indicates that the extracellular concentration of certain materials increases because they are ejected from ice crystals during freezing. This phenomenon might be one of the best strategies identified so far to enhance adsorption of the protein/carrier complex applied to cells, owing to the increase in the peripheral cell concentration. Within this strategy, the interaction between the cell membrane and protein/carrier complex is quite important because after thawing, the adsorbed complex should be internalized instead of diffusing back into the solution. This suggests that we can reduce the quantity of valuable materials that is internalized into cells. Additionally, freeze concentration strategies have several advantages in that they are simple, cost-effective, and highly reliable, and they are characterized by a lack of toxicity, high cell viability, and enhanced interaction between drugs and the cell membrane.
In our previous research we had designed novel polyampholyte nanoparticles as a carrier system by modification with dodecylsuccinic anhydride (DDSA) as a hydrophobic moiety that showed self-assembly, forming intermolecular hydrophobic and electrostatic interactions. However, these nanoparticles became cytotoxic at certain concentration levels.19 We have shown that these protein–nanocarrier complexes were highly internalized using the freeze concentration methodology, although the endosomal escape and uptake mechanism of the complexes that had obtained internalization by passing through the plasma membrane was not elucidated.
The endosomal escape process is crucial for the functionality of internalized proteins. Most particles enter cells through endocytosis and subsequently reach vesicles known as endosomes with pH 5.5 via the endosomal pathway.23 However, numerous nanocarrier/protein complexes are entrapped within the endosome and are then destroyed after fusion with lysosomes, which are the primary sites of enzymatic degradation.24,25 Various pathways exist for the internalization of vesicles including caveolae-, clathrin-, or receptor-mediated endocytosis, phagocytosis, or macropinocytosis. The phagocytosis process is regulated by specialized cells such as macrophages and monocytes. In contrast, clathrin- and caveolae-mediated endocytosis and macropinocytosis are important processes of pinocytosis that include receptor ligand interactions based on particle size and surface chemistry.26,27 Among the strategies utilised, membrane disruptive carriers have great potential to facilitate antigen escape from the endosomes.28 This approach is beneficial for delivering anticancer drugs, genes, or vaccines into cancerous cells. In this study, we present the development of a new carrier system composed of liposomes modified by hydrophobic polyampholytes in which the liposomes themselves function as a low toxicity and biocompatible material. Lysozyme was used as a model protein in this study because it is positively charged under physiological conditions and has a high affinity for liposomes under aqueous conditions. We confirmed the efficient uptake of the protein/liposome complexes by endocytosis following freeze concentration and furthermore, we describe the mechanism underlying the enhanced cellular uptake pathways for internalization. In addition, the endosomal escape ability of unmodified and polyampholyte-modified liposomes was also determined. These findings will provide a mechanistic understanding of the use of the novel freeze concentration approach for cell cargo delivery purposes. This methodology will likely be useful for in vitro gene delivery applications in future.
Results and discussion
Preparation of polyampholytes
A polyampholyte cryoprotectant was synthesized by succinylation with succinic anhydride (SA) to ε-poly-L-lysine (PLL) (PLL-SA, Scheme S1, ESI†). From the 1H-NMR chart, it was found that 65% of the amino groups were succinylated (Fig. S1, ESI†) and this compound was shown to have highly cryoprotective properties in 10% aqueous solution.22 A hydrophobic-modified polyampholyte was synthesized by the reaction of PLL, DDSA, and SA (Scheme S2†). The degree of substitution of SA obtained approximated 63.8% and of DDSA was 4.6% as determined by 1H NMR (Fig. S1†). We denoted the polyampholyte cryoprotectant and hydrophobic polyampholyte as PLL-SA and PLL-DDSA-SA, respectively.Preparation of protein-encapsulating liposomes
We prepared 2 types of liposomes, the first consisting of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of the zwitterionic lipids, 1,2 dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2, dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and the other being a PLL-DDSA-SA-modified liposome. To encapsulate the lysozyme protein into the liposomes, we used the lipid film hydration method. Briefly, a DOPC/DOPE solution in chloroform was dried under vacuum to obtain a dry lipid layer including lipid film hydration with protein containing solutions with or without PLL-DDSA-SA that was subsequently extruded. The surfaces of lipid membranes composed of DOPC/DOPE with or without PLL-DDSA-SA were negatively charged. We investigated the amount of protein encapsulation into the liposomes and its efficiency using a Bradford assay. For protein encapsulation into liposomes, 1 mL phosphate buffer saline without calcium and magnesium (PBS (−)) containing various concentrations of lysozyme (2 to 30 mg mL−1) was mixed with dried lipids and extruded to produce a liposome suspension, with or without PLL-DDSA-SA. A schematic illustration of unmodified and polyampholyte-modified liposomes is shown in Fig. 1A. The encapsulation efficiency was compared among various liposomes that constituted different concentrations of the lysozyme protein. Fig. 1B illustrates that the encapsulation efficiency of the lysozyme protein into unmodified liposomes decreases with increasing amounts of loaded protein concentration. Fig. 1C shows the amount of the protein encapsulated at different loading concentrations under physiological conditions. Fig. 1C depicts the sharp increment that was observed upon increasing the amount of the encapsulated lysozyme protein but then decreases owing to the high concentration of the lysozyme protein and to a certain extent its hydrophilic nature. The same trend was observed for PLL-DDSA-SA-modified liposomes; however, the values of encapsulation efficiency were higher than those in unmodified liposomes at each lysozyme protein concentration. Introduction of polyampholyte molecules into the liposomes increased the minus value of the zeta-potential from −5.04 to −11.25 mV. This might explain the higher encapsulation efficiency of polyampholyte-modified liposomes due to the electrostatic interactions between the liposomes and lysozyme.
:1 molar ratio of the zwitterionic lipids, 1,2 dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2, dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and the other being a PLL-DDSA-SA-modified liposome. To encapsulate the lysozyme protein into the liposomes, we used the lipid film hydration method. Briefly, a DOPC/DOPE solution in chloroform was dried under vacuum to obtain a dry lipid layer including lipid film hydration with protein containing solutions with or without PLL-DDSA-SA that was subsequently extruded. The surfaces of lipid membranes composed of DOPC/DOPE with or without PLL-DDSA-SA were negatively charged. We investigated the amount of protein encapsulation into the liposomes and its efficiency using a Bradford assay. For protein encapsulation into liposomes, 1 mL phosphate buffer saline without calcium and magnesium (PBS (−)) containing various concentrations of lysozyme (2 to 30 mg mL−1) was mixed with dried lipids and extruded to produce a liposome suspension, with or without PLL-DDSA-SA. A schematic illustration of unmodified and polyampholyte-modified liposomes is shown in Fig. 1A. The encapsulation efficiency was compared among various liposomes that constituted different concentrations of the lysozyme protein. Fig. 1B illustrates that the encapsulation efficiency of the lysozyme protein into unmodified liposomes decreases with increasing amounts of loaded protein concentration. Fig. 1C shows the amount of the protein encapsulated at different loading concentrations under physiological conditions. Fig. 1C depicts the sharp increment that was observed upon increasing the amount of the encapsulated lysozyme protein but then decreases owing to the high concentration of the lysozyme protein and to a certain extent its hydrophilic nature. The same trend was observed for PLL-DDSA-SA-modified liposomes; however, the values of encapsulation efficiency were higher than those in unmodified liposomes at each lysozyme protein concentration. Introduction of polyampholyte molecules into the liposomes increased the minus value of the zeta-potential from −5.04 to −11.25 mV. This might explain the higher encapsulation efficiency of polyampholyte-modified liposomes due to the electrostatic interactions between the liposomes and lysozyme.
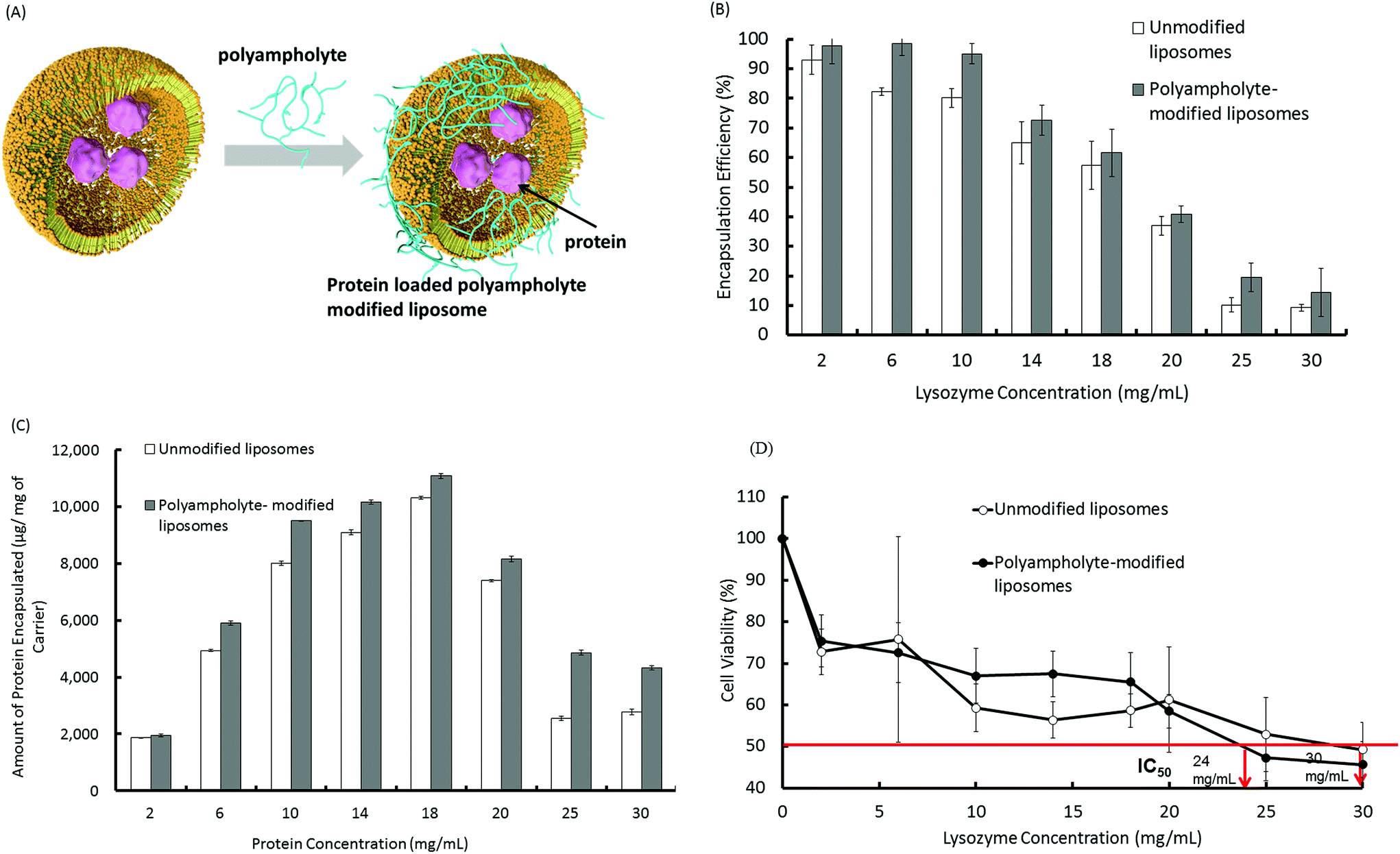 | ||
| Fig. 1 Protein encapsulation into unmodified and polyampholyte-modified liposomes with different protein concentrations (2 to 30 mg mL−1). (A) Schematic illustrations of the preparation of protein loaded liposomes and polyampholyte-modified liposomes. (B) Encapsulation efficiency (C) amount of protein encapsulated, and (D) cytotoxicity of liposome-encapsulated proteins. L929 cells were incubated with liposomes loaded with different protein concentrations for 48 h, followed by MTT assay analysis. IC50 represents the concentration of proteins that caused a 50% reduction in MTT uptake by a treated cell culture compared with the untreated control culture, data are expressed as the mean ± standard deviation (SD). | ||
To optimize the conditions for the preparation of protein-loaded liposomes, cytotoxic behaviour was evaluated. Surface charge is an important factor that can be responsible for inducing cytotoxicity.29 In this regard, cationic surface-charged liposomes showed a greater extent of cytotoxicity.30 Therefore, we selected the negatively charged liposomes for use as a carrier in subsequent experiments.31Fig. 1B depicts that as the amount of lysozyme protein increases, the encapsulation efficiency considerably decreases, which results in the unencapsulated protein remaining in the solution. Therefore, the negative surface charges decrease their magnitude and ultimately show a positive charge that could potentially show toxic behaviour. Fig. 1D describes the high cell viability of L929 cells shifting downward as the loading amount of protein increases, which might be explained by the zeta potential. We therefore determined the zeta potential of unmodified and PLL-DDSA-SA-modified liposomes with different lysozyme protein loading amounts. The surface charges were shown to drastically change to a positive potential (Fig. S2†). We then investigated the cytotoxic effects after the addition of PLL-DDSA-SA to the liposomes. A small amount of PLL-DDSA-SA did not show any toxic activity. However after crossing the threshold ratio of 6:4 (lipids:polyampholytes), the cell viability decreased drastically (Fig. S3†). This behaviour could possibly be due to the cytotoxicity of the polyampholyte at higher concentrations. Hence, we have chosen the liposome and polyampholyte composition (7:3) and loading lysozyme protein concentration (10 mg mL−1) accordingly for further investigations.
Lysozyme-encapsulating liposomes showed increments in their particle size over 7 days as the potential significantly changed from −4.91 to −23.3 mV (Fig. S4A and B†). On the other hand, the zeta potential of PLL-DDSA-SA-modified liposome complexes did not change even after 7 days (Fig. S4B†).
Furthermore, we investigated the particle size stability of unmodified or polyampholyte-modified liposomes under an ultra-cold temperature at −80 °C. Protein-encapsulated unmodified and polyampholyte-modified liposomes were frozen at −80 °C for 1 day with or without the use of any cryoprotectant. The solutions were thawed at 37 °C, and changes in particle sizes were investigated using dynamic light scattering (DLS). Without a cryoprotectant, the particle sizes were extremely large, showing the destabilization of protein molecules, which led to particle aggregation. However, the particle sizes of protein-encapsulated unmodified and polyampholyte-modified liposomes did not change when they were frozen in the presence of a polymeric cryoprotectant (Fig. S5†). These results clearly indicated that protein-encapsulated liposomes exhibit stability after treatment with a polymeric cryoprotectant. Based on these results, we successfully prepared stable liposomes loaded with a low-toxicity lysozyme and protein, both with and without PLL-DDSA-SA. The liposomes had a suitable size to serve as protein-delivery vesicles.
Adsorption of protein-encapsulating liposomes onto cells via freeze concentration
 | ||
| Fig. 2 Confocal microphotographs of L929 cells before and after freezing along with protein-encapsulating carriers with 10% PLL-SA as a cryoprotectant. Liposomes were stained with FITC-PE and lysozymes were labelled by TR red. (A) Unmodified liposomes. (B) Polyampholyte-modified liposomes. Scale bars: 30 μm. | ||
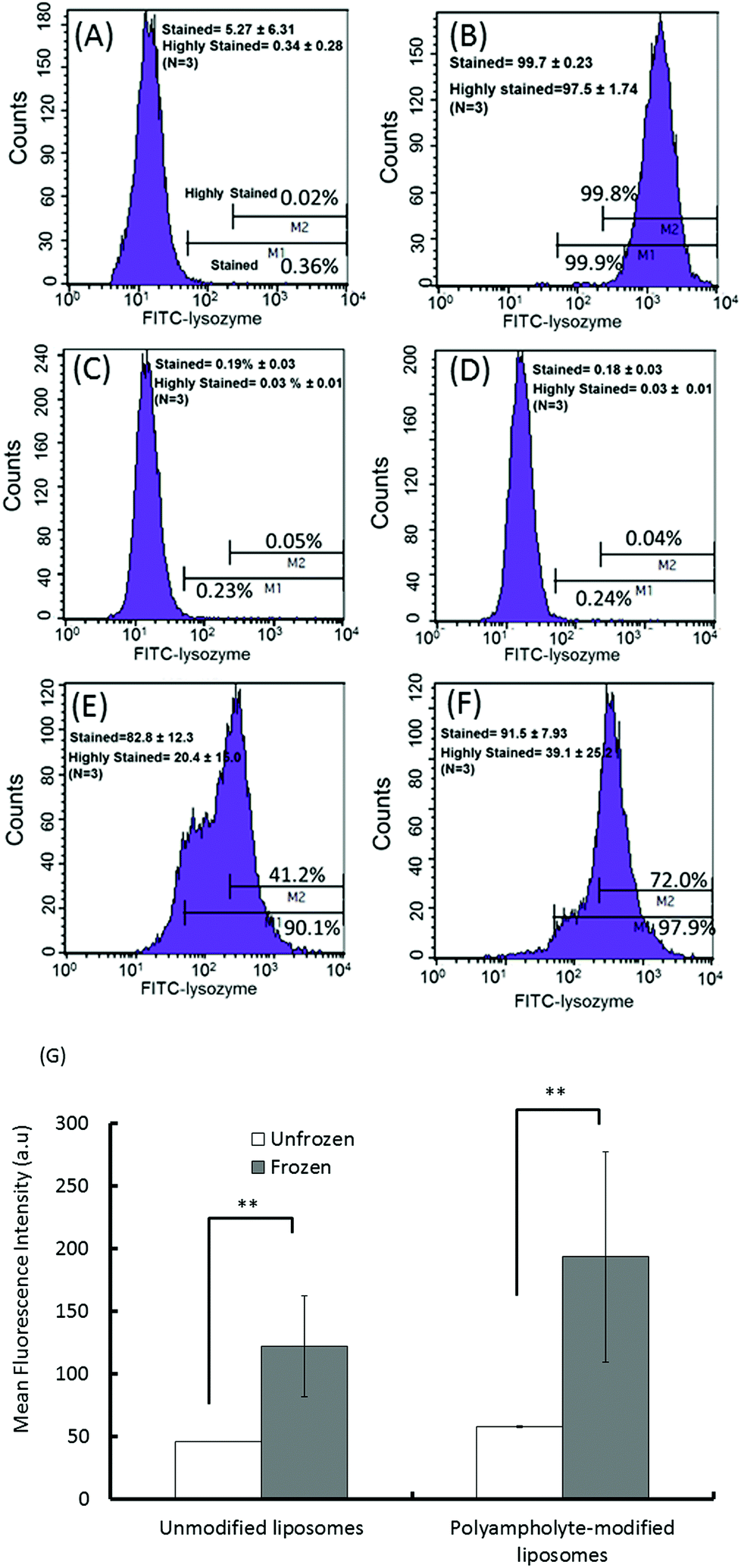 | ||
| Fig. 3 Flow cytometric analysis of frozen and unfrozen cells with various protein-loaded (5 mg mL−1) liposomes. (A) Negative control (cells only); (B) positive control (cells cryopreserved without a cryoprotectant). Unfrozen cells with (C) unmodified liposomes or (D) polyampholyte-modified liposomes. Frozen cells with (E) unmodified liposomes or (F) polyampholyte-modified liposomes. (G) Mean fluorescence intensity of frozen and unfrozen unmodified liposomes and polyampholyte-modified liposomes by flow cytometric analysis. Data are expressed as mean ± SD. **P < 0.01. | ||
As quantified in Fig. 3G, the cells treated with lysozyme protein loaded polyampholyte-modified liposomes showed enhanced fluorescence as compared to those treated with unmodified liposomes. This could be explained by the strong interaction of polyampholyte-modified liposomes with cells, which are likely to have more association owing to charge-inducing factors or hydrophobicity. The hydrophobicity of the liposomal membranes might be enhanced by the presence of polyampholyte nanoparticles that strongly favour the enhanced interactions between the liposomal membranes and cells. On the other hand, the effect was much more prominent upon increasing the dose of lysozyme protein-encapsulated in liposomes. The same observations for unmodified and polyampholyte-modified liposomes were noted even after low (1 mg mL−1) and medium doses (3 mg mL−1) were applied (Fig. S8†), wherein the gated numbers of stained and highly stained cells were higher than those obtained for unfrozen cells (5 mg mL−1). For the low dose concentration (1 mg mL−1) corresponding to unmodified to polyampholyte modified liposomes, the proportion of stained cells increased from 34.2 ± 12.0% to 48.7 ± 9.50% and the highly stained cells similarly increased from 5.61 ± 2.93% to 5.99 ± 3.25%. From these results, it is suggested that the efficiency is highly dependent on dose concentration, which resulted in a highly effective adsorption of the lysozyme protein (Fig. S8A–D† and Fig. 3E, F). In addition, the geometric means, which were calculated from flow cytometric analyses, showed the enhancement of fluorescence intensity upon increasing the dose of the liposome-encapsulated lysozyme protein (Fig. S8E†). Therefore, for our further investigations, we have chosen an optimum dose (final concentration 5 mg mL−1, 500 μL) of the liposome-encapsulated lysozyme protein for efficacious delivery into cells. Based on the above findings, it is expected that this strategy would promote high internalization efficacy through an endocytic pathway. Thus, we evaluated protein internalization after its accumulation onto the membrane.
Internalization of lysozyme protein-loaded liposomes after seeding
It is generally believed that membrane fusion is important for cytoplasmic delivery through endocytosis. Liposome uptake studies were performed on L929 fibroblast cells, wherein unmodified liposomes and polyampholyte-modified liposomes were labelled by FITC-phosphatidylethanolamine (FITC-PE) whereas the lysozyme protein was labelled by Texas Red (TR) dye. Cells mixed with lysozyme protein-loaded unmodified or polyampholyte-modified liposomes were cryopreserved at −80 °C in medium without FBS, replaced by fresh growth medium and seeded after thawing following incubation for 24 h at 37 °C to allow internalization. Cells were then washed with PBS and observed using CLSM. Both unmodified and polyampholyte-modified liposomes showed a tendency to be internalized using the freeze concentration methodology (Fig. 4A and B). Fluorescence was measured by confocal microscopy. The intensity of the red fluorescence of the internalized TR-labelled lysozyme protein showed that polyampholyte-modified liposomes exhibited a significantly greater capacity for protein intake in comparison with unmodified liposomes (Fig. 4C). These results demonstrate that the freeze concentration technique can enhance the cytosolic delivery of proteins. Because it is important to compare our system with other current systems, control experiments were performed using a commercially available PULSin™ protein-delivery kit. This reagent contains cationic, amphiphilic molecules that enhance adsorption on the cell membrane, but the cationic charge causes cytotoxicity. We followed the delivery protocol for suspension cells to compare the results obtained with the freeze concentration methodology. Because a significant decrease in cell viability (<60% viability) was observed using the protein/PULSin™ complexes after a 4 h incubation (due to the cationic charge), we decreased the incubation time to 0.5 h at 37 °C, according to the protocol. The medium without FBS containing protein/PULSin™ complexes was replaced with fresh growth medium to allow protein internalization after seeding to the glass-bottom dish.35 The fluorescence intensity of internalization of FITC-lysozyme/PULSin™ was lower compared with freeze concentration-mediated internalization (Fig. S9 [A–F]†). These results strongly suggested that the freeze concentration methodology is more efficient and less toxic than the current method, based on cationic amphiphiles used to deliver proteins to the cytosolic compartment of cells.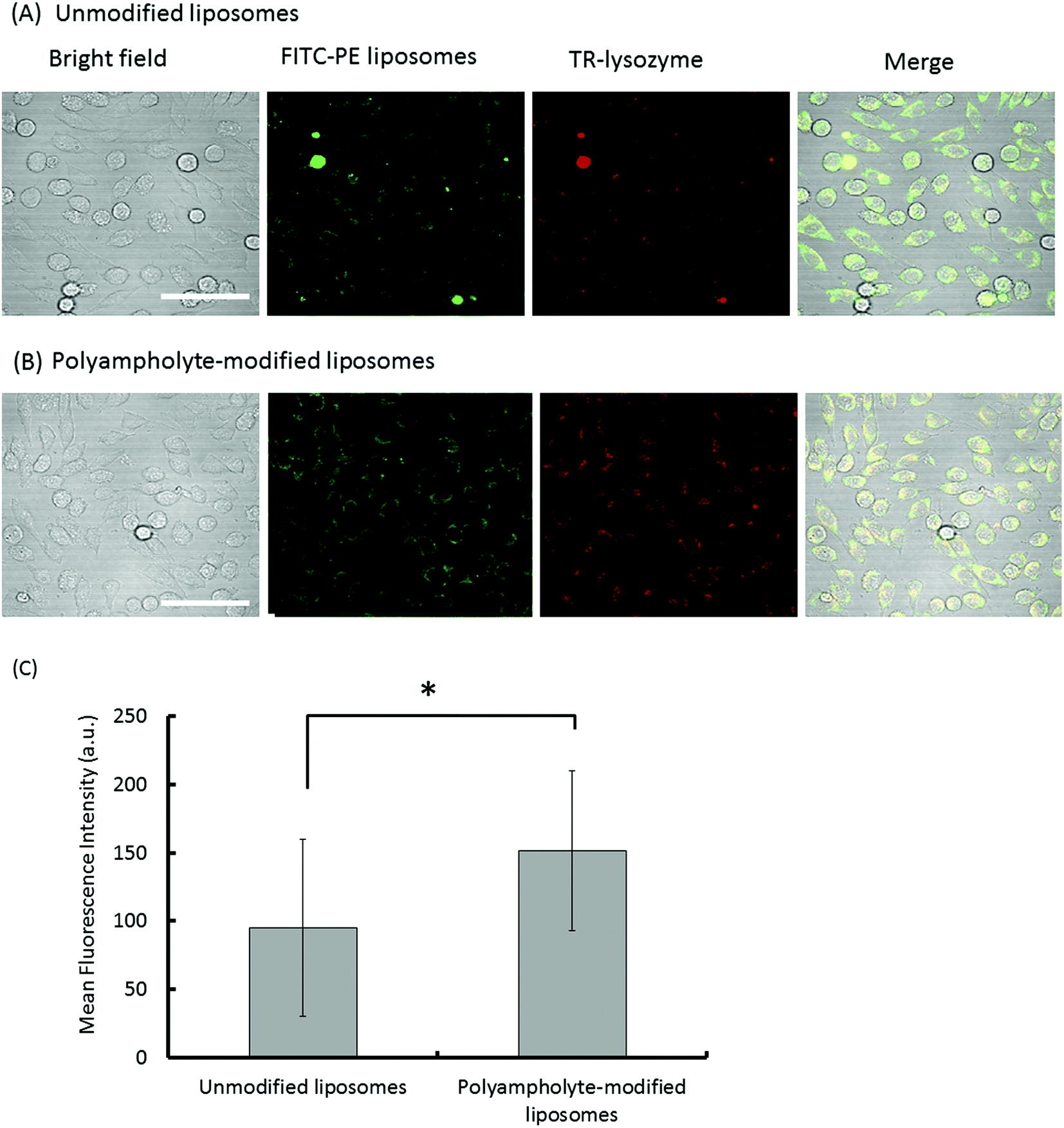 | ||
| Fig. 4 Confocal microphotographs of L929 cells. The images show that lysozyme protein internalization occurs via endocytosis during culture after being frozen with lysozyme-loaded modified liposomes using 10% PLL-SA as a cryoprotectant. (A) Unmodified liposomes; (B) polyampholyte-modified liposomes. Scale bars: 50 μm. (C) Mean fluorescence intensity of unmodified and polyampholyte-modified liposomes after internalization as determined by confocal microscopy. Data are expressed as mean ± SD. *P < 0.05. | ||
Recent studies have shown that unmodified liposomal delivery efficiency is very low whereas liposomes modified with a polymer are more capable of inducing the selective release of materials from endosomes into the cytoplasm. Kono et al. reported that liposomes modified by a polymer had a higher efficiency of entering the cells, ultimately resulting in internalization of the lysozyme protein.36 Furthermore, they also indicated that the addition of a polymer to liposomes effected the enhancement of intracellular delivery.18Fig. 4A and B show that the intensity of red-stained protein localized in the cytosol with polyampholyte-modified liposomes was greater than that observed with unmodified liposomes. This finding suggested that polyampholyte-modified liposomes transferred more lysozyme protein to the cytosol of treated cells. This result is in good agreement with our previous report.19 This enhanced internalization might be due to the efficient release of the lysozyme protein into the cytoplasm via endosomal escape. Next, we investigated the internalization pathway to study endosomal escape using our polyampholyte-modified liposomes.
Internalization mechanism of FITC-lysozyme-loaded polyampholyte-modified liposomes
Most particles cannot readily cross into cells because their large size and charge make it difficult to pass through the plasma membrane; however, lipoparticles have the tendency to be transported by one of the several modes. Therefore, it is important to elucidate the mechanism for the particles that actually obtain entry into the plasma membrane of the cells. The endocytosis pathway is a specialized pathway that mediates the active transportation of nanomedicines and targets them to such regions as the mitochondria, endosome, nucleus, or other specific organelles.37To investigate the mechanism associated with unmodified or polyampholyte-modified liposomes’ entry into the plasma membrane after freezing, a variety of inhibitors were selected to block specific endocytic pathways. We chose 3 inhibitors of the endocytotic pathway: chloropromazine (for clathrin-mediated endocytosis), 5(-N-ethyl-N-isopropyl)amiloride (EIPA) (macropinocytosis), and filipin (caveolae-mediated endocytosis).38 L929 cells were treated with different concentrations of each inhibitor followed by the addition of FITC-lysozyme-encapsulating liposomes in the presence of a polymeric cryoprotectant, and measured using a fluorescence microplate reader and by CLSM. The cell viability was determined to optimize the inhibitor concentration to select concentrations not associated with cytotoxicity (Fig. S10†). For confirmation of the endocytosis mechanism, confocal microscope observation (Fig. 5) and fluorescence microplate reader analysis (Fig. 6) were conducted. These results showed an agreement between the confocal images and the quantification of fluorescence for the determination of uptake. In these images, considerable fluorescence was observed in the L929 cells used as a positive control without any inhibitor for the unmodified and polyampholyte-modified liposomes; however, when specific inhibitors were used to block the pathway, a decrease in the fluorescence was observed that affected the uptake and did not allow the lysozyme protein to diffuse into the membrane. For unmodified liposomes, the fluorescence intensity after adding filipin declined considerably, whereas treatment with the other inhibitors had no such effect (Fig. 5A–D). The significant decrease in intensity following filipin treatment suggested that unmodified liposomes were internalized into the fibroblast L929 cells by caveolae-mediated endocytosis (Fig. 6A). Caveolae contain a hydrophobic domain that is rich in cholesterol and glycosphingolipids. When particles are internalized by caveolae, caveosomes are formed, which can directly transport the particles to specific organelles.39 Many reports have indicated that charged particles were likely to adopt caveolae-dependent endocytosis.40 However, in our investigation, using PLL-DDSA-SA-modified liposomes, the finding that both EIPA and filipin inhibitor significantly resulted in a decline of the fluorescence intensity suggest that these liposomes, unlike unmodified liposomes, tend to adopt 1 of the 2 pathways: caveolae-mediated endocytosis or macropinocytotic endocytosis (Fig. 5E–H and 6B). Consequently, it can be concluded that the hydrophobic polyampholyte in liposomes is responsible for promoting the macropinocytotic endocytic route. In macropinocytosis, the internalization of the particles occurs into large vacuoles called macropinosomes with a diameter of 0.5–1.0 μm.41 Macropinocytosis has received much attention in gene delivery as well as in cancer therapy fields. The most advantageous feature of macropinocytosis is that it allows for endosomal escape, which can avoid the lysosomal degradation of antigens and genes. One report has demonstrated that an octaarginine peptide-mediated gene expression system that showed high transfection efficacy ultimately adopted the macropinocytotic pathway.42 Thus, a notable finding in our study is that polyampholyte-modified liposomes adopted 2 methods of internalization. Various factors such as size43 and surface charges40 are associated with internalization, based on physicochemical characterization.44 However, no common factor has been elucidated yet to explain the associated mechanism involving entry into endocytic pathways.
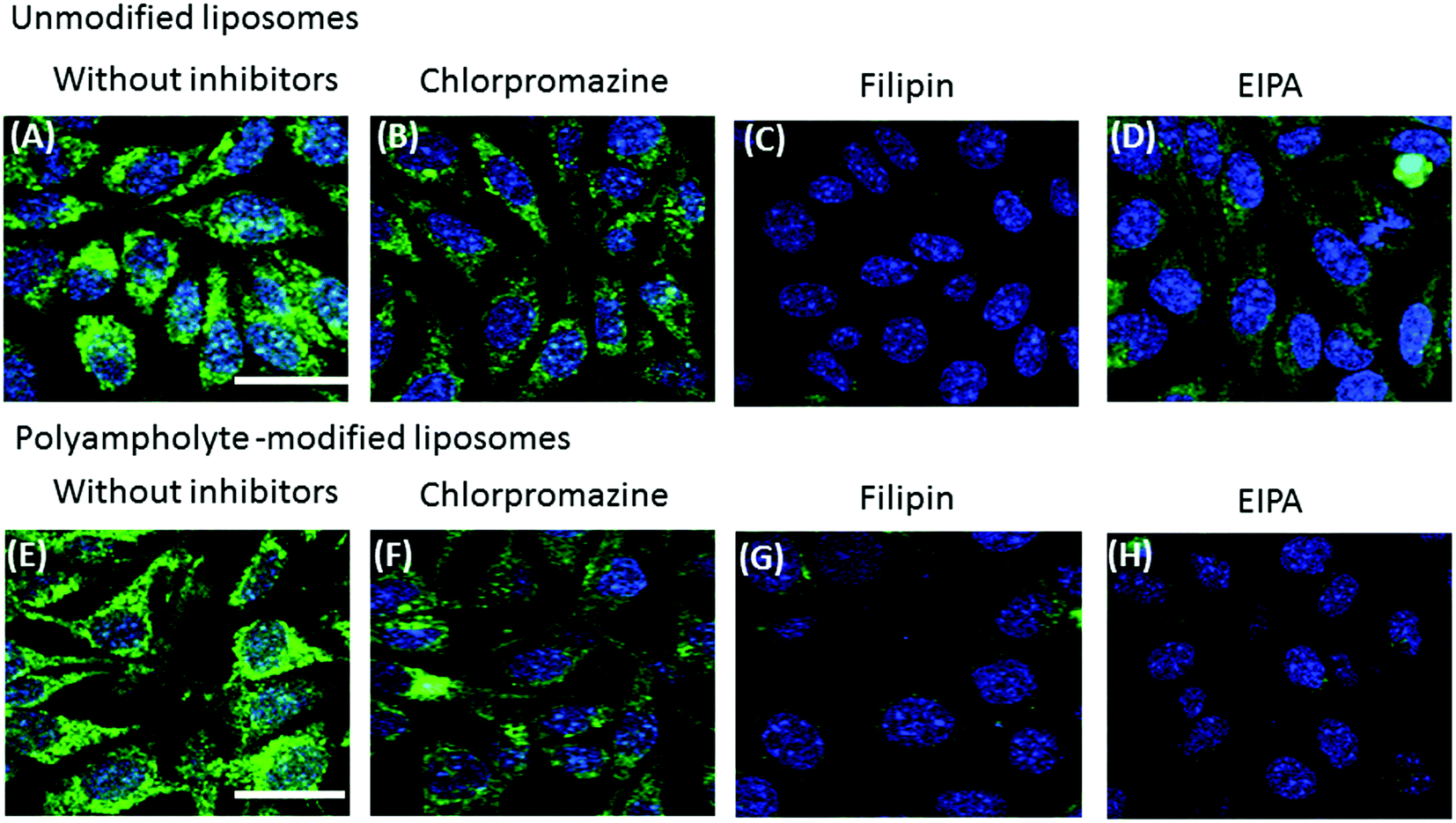 | ||
| Fig. 5 Effects of endocytic uptake of unmodified liposomes (A–D) or polyampholyte-modified liposomes (E–H). Encapsulated FITC-labelled lysozymes were pre-incubated with different inhibitors (chloropromazine, filipin, or EIPA) in the presence of a polymeric cryoprotectant at −80 °C. After thawing, the cells were seeded and incubated for at least 8 h. Confocal microscopy analysis without inhibitors (A, E), chloropromazine (B, F), filipin (C, G), or EIPA (D, H). Scale bars: 20 μm. | ||
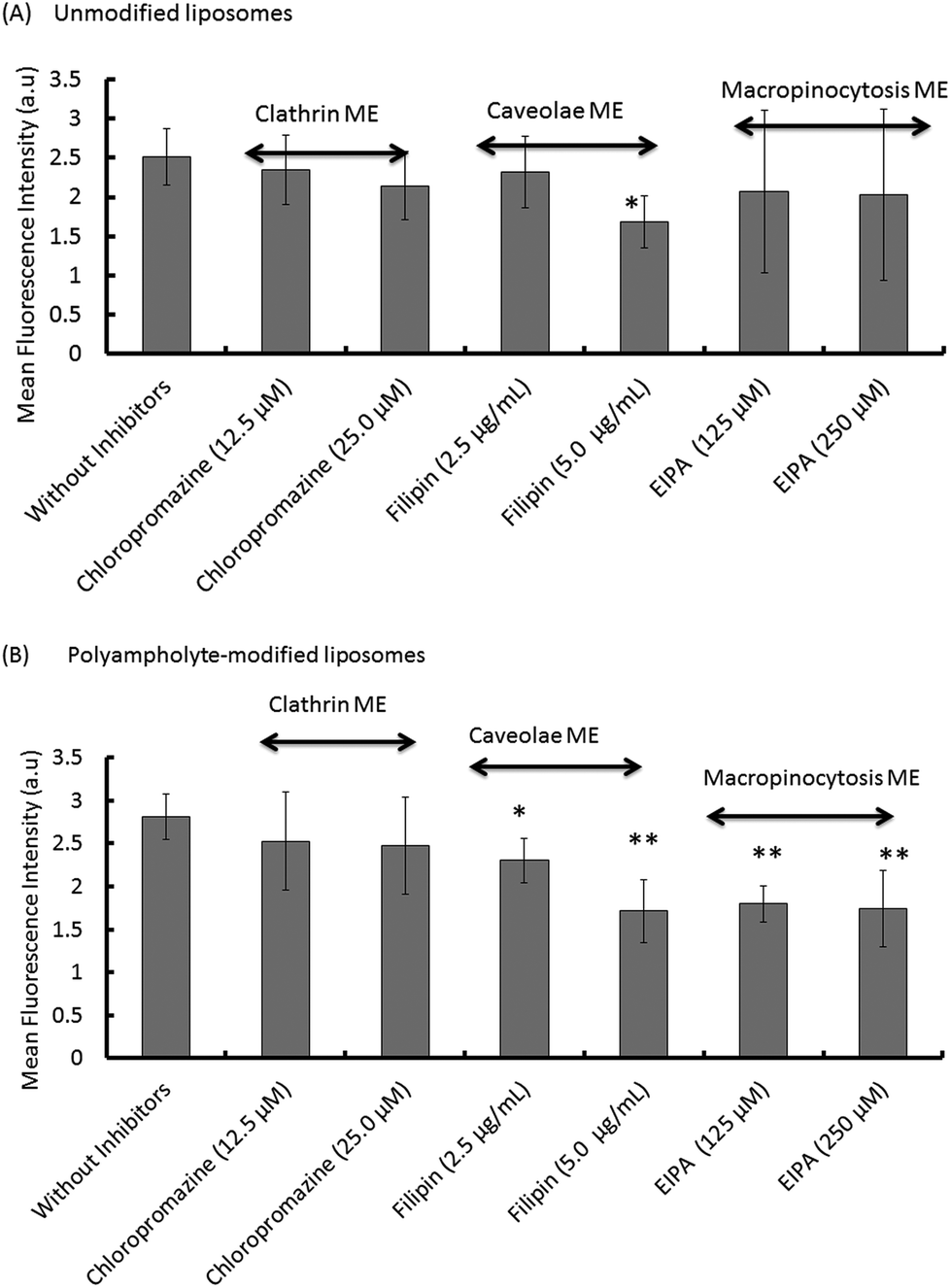 | ||
| Fig. 6 Quantitative analysis with a fluorescent microplate reader of the fluorescence intensity observed during endocytic uptake via clathrin-mediated endocytosis (ME), caveolae ME, and macropinocytosis ME, following treatment with different inhibitors. (A) Unmodified liposomes. (B) Polyampholyte-modified liposomes. Data are expressed as mean ± SD. **P < 0.01, *P < 0.05 vs. without inhibition. | ||
Taken together, these results not only suggested that both unmodified liposomes and polyampholyte-modified liposomes utilise caveolae-mediated endocytosis, but that polyampholyte triggers the adoption of an additional specific pathway, macropinocytosis, with a different uptake mechanism. Therefore, we suggest that polyampholyte-modified liposomes facilitate intracellular delivery through a different mechanism from that which occurs with unmodified liposomes.
Intracellular localization and endosomal escape of lysozyme proteins
To be effective for therapeutic purposes, it is required that delivered proteins must escape lysosomal degradation; in addition, they should be delivered into the cytosol of the cells for high efficiency. Thus, our investigation of the ability of materials to effect intracellular delivery also included an examination of the capability to release their contents into the cell cytoplasm.To confirm the entry of a lysozyme protein into endosomes and to determine whether or not the protein was subsequently trafficked to lysosomes wherein a large variety of macromolecules can be degraded, an investigation of intracellular trafficking is required for eventual successful design in protein delivery schemes. We examined the potential for endosomal protein escape using a combination of unmodified or polyampholyte-modified liposomes and freeze concentration-based internalization. To observe the intracellular distribution of cargo proteins in L929 cells, TR-labelled lysozyme-encapsulating liposomes were prepared. The endosomes and lysosomes were stained with LysoTracker Green and nuclei were stained with Hoechst 33258 for 30 min respectively prior to observations. As shown in Fig. 7A, the colocalization of lysozyme proteins and lysosomes were represented by yellow fluorescent regions indicating the presence of the proteins in the endosomes, which is consistent with the internalization of unmodified liposomes. This shows that unmodified liposomes remain intact even after their release from the endosome, suggesting that the associated encapsulated lysozyme proteins might have difficulty in being released from the vesicles. In contrast, the green fluorescence was partially separated from red fluorescence over time for polyampholyte-modified liposomes, indicating the successful release of their protein cargo (Fig. 7B). The images shown in Fig. 7 suggest a triggered release of the lysozyme protein from endosomes for engineered liposomes incorporating hydrophobic polyampholytes. The possible mechanism behind endosomal release might be that polyampholytes could be adsorbed onto the endosome membrane thereby destabilizing it, which leads to the release of the lysozyme protein; accordingly, we identified an enhanced in vitro endosomal escape efficacy with very low associated toxicity.
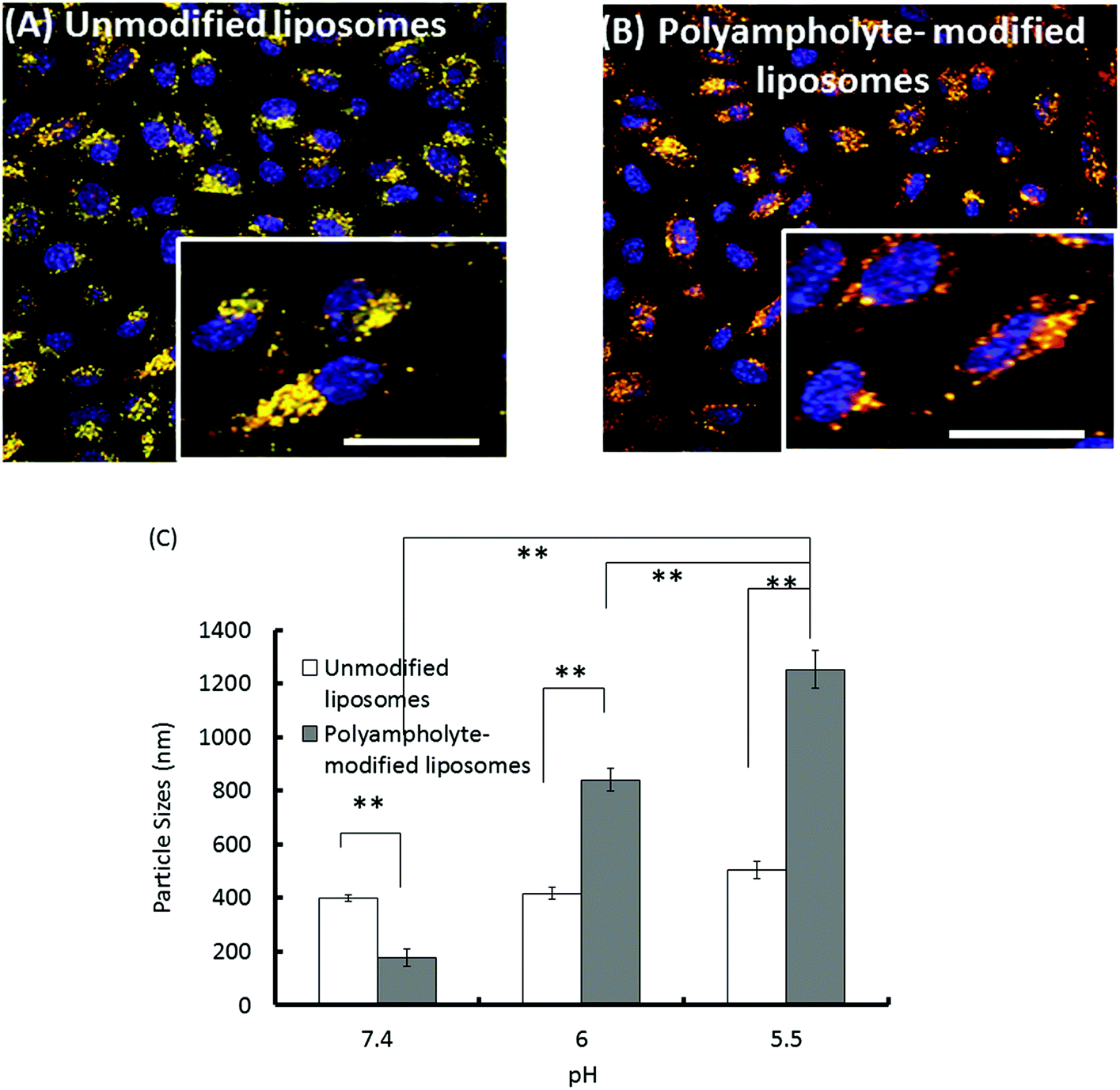 | ||
| Fig. 7 Intracellular delivery of TR-labelled lysozymes in L929 cells. We cryopreserved 1 × 106 cells with the polymeric cryoprotectant PLL-SA and protein containing liposomes. The cells were thawed and seeded for 12 h at 37 °C. After incubation, the endosomes/lysosomes and nuclei were stained by LysoTracker Green and Hoechst blue 33258, respectively. (A) Unmodified liposomes. (B) Polyampholyte-modified liposomes. Scale bar: 10 μm. (C) Mean diameter of unmodified or polyampholyte-modified liposomes after overnight incubation at various pH values. Data are expressed as mean ± SD. **P < 0.01. | ||
Therefore, we found that the fluorescence of TR-labelled lysozyme proteins did not increase and that they were effectively internalized with unmodified liposomes (Fig. 4A), whereas polyampholyte-modified liposomes led to efficient protein release and higher fluorescence due to the endosomal escape of proteins (Fig. 4B and C).
We have also evaluated the size variation of liposomes with or without polyampholyte-modification caused by changing their pH using a DLS technique. Unmodified liposomes did not show any remarkable change even at different pH values. However, polyampholyte-modified liposomes showed a size increase, indicating aggregation at pH 5.5 (Fig. 7C). This factor might induce destabilization of the liposomal membrane because polyampholyte-containing carboxyl groups are protonate,45 and these carboxylate ions can lose their negative charge causing destabilization of liposomal membranes.46–50 Upon endocytosis, the low pH in the endosomes induces fusion of the liposomal membrane with the endosomal membrane, causing the release of the contents of the liposomes into the cytoplasm of the cells. One reason for this might be that pH-sensitive liposomes undergo acidification responsible for disruption of the liposomal bilayer, with change of its configuration causing the release of the encapsulated material through the endosomal pathway. Thus, the polyampholyte-modified liposomes were capable of enhancing the endosomal escape efficiency. In addition, after escaping from endosomes, it is extremely important to investigate the drug delivery to a disease site in future.
Conclusion
In this study, we prepared a novel form of hydrophobic polyampholyte-modified liposomes using a combination of PLL-DDSA-SA. These polyampholyte-modified liposomes can successfully escape the endocytic pathway and can introduce lysozyme proteins into the cell cytosol through the use of a simplistic strategy involving freeze-thawing of cells with the encapsulated lysozyme protein complexes. The present study focused on providing a mechanistic overview of lysozyme protein delivery by using unmodified and polyampholyte-modified liposomes in conjunction with the freeze concentration method. These hydrophobic polyampholyte-modified liposomes are stable at physiological pH 7.0, and exhibit low cytotoxicity and high protein encapsulation efficiency. The results of flow cytometry analysis show that by using the freeze concentration method, the uptake and adsorption of lysozyme proteins was enhanced by 4-fold in comparison with that obtained using unfrozen cells. In addition, we found that the unmodified and polyampholyte-modified liposomes adopted different pathways for the cytoplasmic delivery of proteins, with the latter preferentially bypassing lysosomal degradation. Therefore, although further investigation in vitro using immune cells and in vivo using model systems should be performed, these positive results including the protein endosomal escape property suggest that the intracellular delivery of lysozyme proteins by hydrophobic polyampholyte-modified liposomes and the freeze concentration methodology might be very beneficial for in vitro applications in cancer treatment or gene therapy in future.Experimental
Reagents
DOPC, DOPE, Rh-PE, FITC-PE were purchased from Avanti Polar Lipids (Alabaster, AL, USA), and LysoTracker Green DND-26 and Hoechst 33342 were purchased from Molecular Probes Inc. (Eugene, OR, USA). Filipin, EIPA, and lysozyme were obtained from Sigma Aldrich (St Louis, MO, USA). Chloropromazine was obtained from Nacalai Tesque (Kyoto, Japan). Bradford Ultra was purchased from Expedeon Ltd (Cambridge, UK), and Sephadex G25 was obtained from GE Healthcare Bioscience Corp. (Piscataway, NJ, USA).Preparation of FITC-labelled lysozyme
Lysozyme (10 mg) and FITC (1 mg mL−1; Dojindo Laboratory, Kumamoto, Japan) solution was dissolved in sodium bicarbonate buffer solution (1 mL; 0.5 M, pH 9.0) with gentle stirring and incubated at 4 °C overnight with subsequent dialysis (molecular weight cut off: 3 kDa, Spectra/Por, Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA) for 3 days against water and freeze dried.18Preparation of polyampholyte cryoprotective agent and hydrophobic polyampholyte
A polyampholyte cryoprotectant was synthesized by succinylation of the polymer (PLL). To obtain the PLL-SA cryoprotective agent, an aqueous solution of 25% (w/w) PLL (10 mL, JNC Corp., Tokyo, Japan) and SA (1.3 g; Wako Pure Chem. Ind. Ltd, Osaka, Japan) were mixed at 50 °C for 2 h to convert 65% of the amino groups to the carboxyl groups (Scheme S1†).19,22 Polyampholyte nanoparticles were synthesized according to a previous report.19 Briefly, an aqueous solution of ε-PLL (10 mL; 25% w/w, JNC Co. Ltd, Yokohama, Japan) was added to 5% molar ratio DDSA (Wako Pure Chem. Ind. Ltd, Osaka, Japan) at 100 °C and allowed to mix for 2 h to obtain hydrophobically modified PLL (Scheme S2A†). Subsequently, SA was added at 65% molar ratio (COOH/NH2) and was allowed to react for 2 h at 50 °C (Scheme S2B†). The degrees of substitution of SA and DDSA were obtained by 1H-NMR. The spectra were obtained at 25 °C on a Bruker AVANCE III 400 spectrometer (Bruker BioSpin Inc., Fällanden, Switzerland) in D2O.Preparation of liposomes
Liposomes were composed of a DOPC and DOPE mixture at a molar ratio of 1:1. We used DOPE because it tends to form a hexagonal inverted phase leading to the formation of a non-lamellar structure that can facilitate aggregation, which in turn favours destabilization.51 Briefly, appropriate amounts of lipid DOPC (10 mg) and DOPE (9.46 mg) were dissolved in chloroform (1 mL). Chloroform was allowed to evaporate under a steady stream of nitrogen gas, following which the tubes were dried under vacuum to facilitate complete evaporation of the residual solvent. The dried lipids were dispersed in 1 mL of PBS (−) and extruded through a 100 nm polycarbonate membrane. When preparing lysozyme-encapsulating liposomes, lysozyme (10 mg) was dissolved in 1 mL PBS (−). Hydrophobic polyampholyte-modified liposomes were also prepared using the same method with the dry membrane of a lipid/polymer mixture (7:3 w/w). Liposomes were then purified on a Sephadex G25 column to remove unreacted polyampholytes.18
Zeta potential and particle size measurements
The mean particle sizes, size distribution, and the surface charge measurements of the zeta potential of unmodified and polyampholyte modified liposomes were analysed by DLS analysis using a Zetasizer 3000 (Malvern Instruments, Worcestershire, UK) with a scattering angle of 135°. The colloidal suspension of liposomes was diluted with PBS and the particle size analysis was carried out at a scattering angle of 135° and a temperature of 25 °C. The liposomes were dispersed in PBS (−) and the zeta potential values were measured at the default parameters of a dielectric constant at 78.5 and a refractive index at 1.6. Data were obtained as an average of more than 3 measurements on different samples.Determination of encapsulation efficiency
After preparing lysozyme-modified liposomes or polyampholyte-modified liposomes, a working dispersion (1 mL) was made using a liposome suspension (20 μL) in PBS (−). Then the working dispersion (500 μL) was mixed with 6% (v/v) Triton X-100 (100 μL) and this solution was maintained at 65 °C for 5 min to disrupt all the vesicles. The solution (400 μL) was transferred into an ultra-0.5 centrifugal device for the removal of unencapsulated lysozyme (molecular weight cut off 50 kDa, Amicon® ultra (0.5 mL), Merck Millipore, Darmstadt, Germany) and centrifuged at 19515g for 10 min.52 PBS solution (400 μL) was again added, and the same procedure was repeated. The amount of un-encapsulated lysozyme was quantified by the Bradford assay using a Bradford Ultra reagent and the efficiency was determined by ultraviolet spectroscopy at 595 nm using lysozyme as a standard as follows: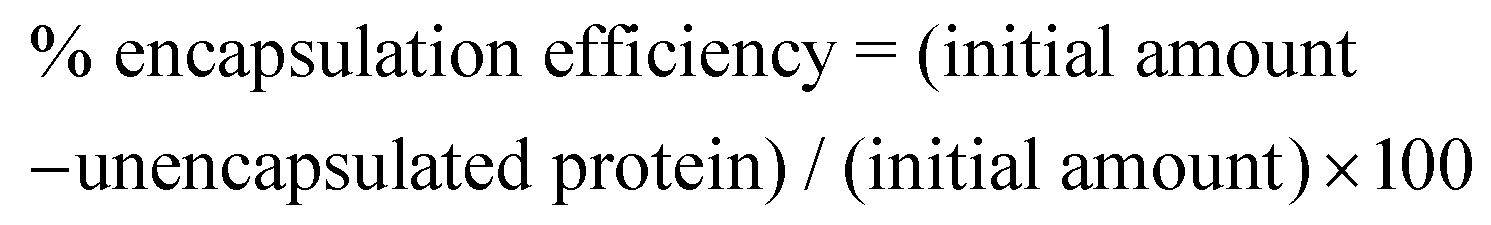
Cell culture
Mouse fibroblast L929 cells (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma Aldrich) supplemented with 10% FBS at 37 °C under 5% CO2 in a humidified atmosphere. When the cells reached 80% confluence, they were removed by 0.25% (w/v) trypsin containing 0.02% (w/v) ethylenediamine tetra-acetic acid in PBS (−) and were seeded on a new tissue culture plate for subculture.Cytotoxicity assay of unmodified and polyampholyte-modified liposomes
Cytotoxicity was determined using an MTT assay. In a 96 well plate, L929 cells at a density of 1 × 103 cells per mL were cultured in each well and incubated under saturated humid conditions at 37 °C and 5% CO2. After 24 h of incubation, unmodified liposomes and polyampholyte-modified liposome-containing medium were added and incubated for 48 h. Then, 3-(4,5-dimethyl thial-2-yl)-2,5-diphenyltetrazalium bromide (MTT) solution (0.1 mL, 300 μg mL−1 in medium) was added to the cultured cells. The cells were incubated for 4 h at 37 °C. The solutions were removed and subsequently replaced by DMSO (100 μL) and allowed to stand for 15 min to allow a complete reaction. The resulting colour intensity measured using a microplate reader (Versa max, Molecular Devices Co., Sunnyvale, CA, USA) at 540 nm was proportional to the number of viable cells. The concentration of the liposomes leading to 50% cell killing (IC50) was calculated from a concentration-dependent cell viability curve.22Cell freezing with lysozyme-encapsulated liposomes
To prepare FITC-labelled lysozyme-encapsulating liposomes, Rh-PE-labelled liposomes (1.27 × 10−5 mol) and FITC-labelled lysozyme (10 mg mL−1) were prepared as described previously.18 The solution was applied to a Sepharose 4B column to remove unencapsulated proteins. L929 cells were counted and resuspended in 10% PLL-SA cryoprotectant (1 mL) to avoid freezing damage along with the unmodified or polyampholyte-modified liposome encapsulated lysozyme protein (5 mg mL−1, 500 μL) without FBS at 4 °C at a density of 1 × 106 cells per mL in 1.9 mL cryovials (Nalgene, Rochester, NY, USA) and were stored in a −80 °C freezer overnight. These vials were thawed at 37 °C, diluted with DMEM, and the cells were washed 3 times with DMEM with 10% FBS.19,22 The cell viability was analysed by trypan blue dye exclusion using a hemocytometer. The total number of cells stained with trypan blue was recorded. Cell viability was determined as the number of viable cells divided by the total number of cells. Each condition was analysed in triplicate. The adsorption of unfrozen and frozen protein-encapsulating liposomes was observed using a CLSM (FV-1000-D; Olympus, Tokyo, Japan).Quantification of the adsorption of lysozyme protein onto cells by freeze concentration of unfrozen and frozen cells using flow cytometry
To determine the lysozyme protein uptake efficiency between unfrozen and frozen L929 cells, flow cytometric analysis was conducted. Cell freezing with the polyampholyte cryoprotectant solution incorporating lysozyme protein-encapsulating unmodified liposomes and polyampholyte-modified liposomes was discussed above. We used 1 × 106 cells for sample preparation and analysis by flow cytometry. The cells were then thawed, the old medium was removed, and the cells were washed 3 times with PBS (−).53 Data acquisition and analysis were performed using a FACSCalibur instrument (BD Biosciences, Franklin Lakes, NJ, USA). The region of live cells was determined by FSC-SSC gating to exclude dead cells and debris noise. The flow cytometry analysis plot showed the gating strategy for identifying stained and highly stained populations referring to non-stained cells (negative control) and cells cryopreserved in the absence of a cryoprotectant (positive control), respectively. A minimum of 20000 cells were collected for each histogram.
Internalization
After thawing, the cells were washed with the medium and seeded in a glass bottom dish. The cells were incubated for 1 day. Then the attached cells were washed with PBS and the internalization of the protein/liposome complexes was observed using CLSM.Determination of the internalization pathway via inhibition assay
Cells were pretreated with different concentrations of specific endocytotic inhibitors such as chloropromazine (for clathrin-mediated endocytosis), EIPA (macropinocytosis), or filipin (caveolae-mediated endocytosis) to determine their optimal concentration using a trypan blue exclusion assay and then were cryopreserved with 10% polymeric cryoprotectant carrying unmodified or polyampholyte-modified liposomes in the cell culture medium at −80 °C. Solutions were thawed and the cells were counted to select the concentration producing the highest viability after inhibitor treatment. After addition of the optimized inhibitor concentration, cells at a density of 1 × 103 cells per mL were seeded into 96-well plates for at least 8 h to determine the uptake of particles from the extracellular solution. At the start of the experiment, the cell culture medium was aspirated and washed with PBS (−) 3 times to remove any traces of inhibitors. The mean fluorescence intensity was evaluated using a fluorescence microplate reader (Varioskan flash, Thermo Fisher Scientific, Inc., Waltham, MA, USA).38 The endocytic uptake was also confirmed using CLSM.Intracellular localization of lysozyme proteins in L929 cells
A thawed solution of L929 cells at a density of 1 × 103 cells per mL containing 10% cryoprotectant comprising lysozyme encapsulating unmodified or polyampholyte-modified liposomes was seeded onto a glass bottom dish. The cells were incubated for 12 h under a 37 °C humidified atmosphere with 5% CO2. LysoTracker Green® DND-26 and Hoechst dye were added and incubated for 30 min prior to investigation. Samples were rinsed with PBS buffer and counterstained prior to imaging. The cells were analysed using CLSM.54Internalization of the lysozyme protein using a currently available cationic amphiphile-based protein delivery reagent
A control experiment for protein delivery was performed using the PULSin™ protein delivery reagent (Polyplus transfection SA, Illkirch, France), according to the manufacturer's protocol for suspension cells. Briefly, 1 × 106 L929 cells were suspended in 1 mL serum-free medium in a sterile 15 mL centrifuge tube. In a separate tube, 7 μg of the lysozyme protein was mixed gently in 200 μL of HEPES buffer ((4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (20 mM, pH-7.4)), after which 28 μL of the PULSin™ reagent was added immediately. Next, both solutions were incubated for 0.5 h or 4 h at 37 °C under a humidified atmosphere containing 5% CO2. After incubation at 37 °C, the cells were centrifuged at 1000 rpm for 4 min and suspended in 1 mL cell growth medium. Cell viability was assessed by trypan blue exclusion. The cells were seeded in a glass-bottom dish to allow the internalization of proteins, as described previously.35Statistical analysis
All data are expressed as means ± standard deviation (SD). All experiments were conducted in triplicate. To compare data among more than 3 groups, a 1-way analysis of variance with a post-hoc Fischer's protected least significant difference test was used. To compare data between 2 groups, the Student's t-test was used. The differences were considered statistically significant at a P value of <0.05.Acknowledgements
This study was supported in part by a Grant-in-Aid, KAKENHI (15K12538 and 16K12895), for scientific research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- U. H. Weidle, B. Schneider, G. Georges and U. Brinkmann, Cancer Genomics Proteomics, 2012, 9, 357–372 CAS.
- O. Veiseh, B. C. Tang, K. A. Whitehead, D. G. Anderson and R. Langer, Nat. Rev. Drug Discovery, 2015, 14, 45–57 CrossRef CAS PubMed.
- H. Patterson, R. Nibbs, I. McInnes and S. Siebert, Clin. Exp. Immunol., 2014, 176, 1–10 CrossRef CAS PubMed.
- N. A. Charoo, Z. Rahman, M. A. Repka and S. N. Murthy, Curr. Drug Delivery, 2010, 7(2), 125–136 CrossRef CAS.
- D. V. McAllister, M. G. Allen and M. R. Prausnitz, Annu. Rev. Biomed. Eng., 2000, 2, 289–313 CrossRef CAS PubMed.
- G. A. Husseini and W. G. Pitt, J. Pharm. Sci., 2009, 98(3), 795–811 CrossRef CAS PubMed.
- S. J. Beebe, N. M. Sain and W. Ren, Cells, 2013, 2, 136–162 CrossRef CAS PubMed.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic- Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- Z. Gu, A. Biswas, M. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638–3655 RSC.
- S. Arayachukiat, J. Seemork, P. Pan-In, K. Amornwachirabodee, N. Sangphech, T. Sansureerungsikul, K. Sathornsantikun, C. Vilaivan, K. Shigyou, P. Pienpinijtham, T. Vilaivan, T. Palaga, W. Banlunara, T. Hamada and S. Wanichwecharungruang, Nano Lett., 2015, 15, 3370–3376 CrossRef CAS PubMed.
- D. S. Pisal, M. P. Kosloski and S. V. Balu-Iyer, J. Pharm. Sci., 2010, 99(6), 2557–2575 CrossRef CAS PubMed.
- S. J. Soenen, W. J. Parak, J. Rejman and B. Manshian, Chem. Rev., 2015, 11, 2109–2135 CrossRef PubMed.
- V. P. Torchillin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef PubMed.
- M. L. Immordino, F. Dosio and L. Cattel, Int. J. Nanomed., 2006, 1(3), 297–315 CrossRef CAS PubMed.
- S. Martins, B. Sarmento, D. C. Ferreira and E. B. Souto, Int. J. Nanomed., 2007, 2(4), 595–607 CAS.
- E. Yuba, A. Harada, Y. Sakanishi, S. Watarai and K. Kono, Biomaterials, 2013, 34, 3042–3052 CrossRef CAS PubMed.
- S. Ahmed, F. Hayashi, T. Nagashima and K. Matsumura, Biomaterials, 2014, 35, 6508–6518 CrossRef CAS PubMed.
- B. S. Bhatnagar, M. J. Pikal and R. H. Bogner, J. Pharm. Sci., 2008, 97(2), 798–814 CrossRef CAS PubMed.
- P. Mazur and N. Rigopoulos, Cryobiology, 1983, 20(3), 274–289 CrossRef CAS PubMed.
- K. Matsumura and S. H. Hyon, Biomaterials, 2009, 30, 4842–4849 CrossRef CAS PubMed.
- J. Gruenberg and F. G. Vandergoot, Nat. Rev. Mol. Cell Biol., 2006, 7, 495–504 CrossRef CAS PubMed.
- H. Raagel, P. Saalik and M. Pooga, Biochim. Biophys. Acta, 2010, 1798, 2240–2248 CrossRef PubMed.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145(3), 182–195 CrossRef CAS PubMed.
- S. R. Sarker, R. Hokama and S. Takeoka, Mol. Pharm., 2014, 11, 164–174 CrossRef CAS PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- T. Akagi, H. Kim and M. Akashi, J. Biomater. Sci., Polym. Ed., 2010, 21, 315–328 CrossRef CAS PubMed.
- E. Frohlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- S. Takano, Y. Aramaki and S. Tsuchiya, Pharm. Res., 2003, 20(7), 962–968 CrossRef CAS.
- M. R. Mozafari, C. J. Reed, C. Rostron, C. Kocum and E. Piskin, Cell. Mol. Biol. Lett., 2002, 7(2), 243–244 Search PubMed.
- N. K. Jain and I. Roy, Protein Sci., 2009, 18(1), 24–36 CAS.
- P. Fonte, S. Sousa, A. Costa, V. Seabra, S. Reis and B. Sarmento, Biomacromolecules, 2014, 15(10), 3753–3765 CrossRef CAS PubMed.
- Y. Liu, Y. Yin, L. Wang, W. Zhang, X. Chen, X. Yang, J. Xu and G. Ma, J. Mater. Chem. B, 2013, 1, 3888–3896 RSC.
- C. O. Weill, S. Biri, A. Adib and P. Erbacher, Cytotechnology, 2008, 56, 41–48 CrossRef CAS PubMed.
- E. Yuba, A. Harada, Y. Sakanishi and K. Kono, J. Controlled Release, 2011, 149, 72–80 CrossRef CAS PubMed.
- L. M. Bareford and P. W. Swaan, Adv. Drug Delivery, 2007, 59(8), 748–758 CrossRef CAS PubMed.
- T. Akagi, F. Shima and M. Akashi, Biomaterials, 2011, 32, 4959–4967 CrossRef CAS PubMed.
- A. L. Kiss and E. Botos, J. Cell. Mol. Med., 2009, 13, 1228–1237 CrossRef PubMed.
- O. P. Perumal, R. Inapagolla, S. Kannan and R. M. Kannan, Biomaterials, 2008, 29, 3469–3476 CrossRef CAS PubMed.
- F. Cardarelli, D. Pozzi, A. Bifone, C. Marchini and G. Caracciolo, Mol. Pharmaceutics, 2012, 9, 334–340 CrossRef CAS PubMed.
- I. A. Khalil, K. Kogure, S. Futaki and H. Harashima, J. Biol. Chem., 2006, 281, 3544–3551 CrossRef CAS PubMed.
- S. Zhang, J. L. G. Lykotrafitis, G. Bao and S. Suresh, Adv. Mater., 2009, 21, 419–424 CrossRef CAS PubMed.
- L. Kou, J. Sun, Y. Zhai and Z. He, Asian J. Pharm., 2013, 8, 1–10 CAS.
- N. Sakaguchi, C. Kojima, A. Harada and K. Kono, Bioconjugate Chem., 2008, 19, 1040–1048 CrossRef CAS PubMed.
- A. E. Felber, M. H. Dufresne and J. C. Leroux, Adv. Drug Delivery Rev., 2012, 64, 979–992 CrossRef CAS PubMed.
- O. Meyer, D. Papahadjopoulos and J. C. Leroux, FEBS Lett., 1998, 421, 61–64 CrossRef CAS PubMed.
- J. C. Leroux, E. Roux, D. L. Garrec, K. Hong and D. C. Drummond, J. Controlled Release, 2001, 72, 71–84 CrossRef CAS PubMed.
- D. C. Drummond, M. Zignani and J. C. Leroux, Prog. Lipid Res., 2000, 39, 409–460 CrossRef CAS PubMed.
- T. Chen, D. McIntosh, H. Yuehua, J. Kim, D. A. Tirrell, P. Scherrer, D. B. Fenske, A. P. Sandhu and P. R. Cullis, Mol. Membr. Biol., 2004, 21, 385–393 CrossRef CAS PubMed.
- D. A. Balazs and W. T. Godbey, J. Drug Delivery, 2011, 2011, 1–12 CrossRef PubMed.
- X. Xu, A. Costa and D. J. Burgess, Pharm. Res., 2012, 29, 1919–1931 CrossRef CAS PubMed.
- U. S. Huth, R. Schubert and R. Peschka-Su, J. Controlled Release, 2006, 110, 490–504 CrossRef CAS PubMed.
- M. M. Fretza, G. A. Koning, E. Mastrobattista, W. Jiskoot and G. Storm, Biochim. Biophys. Acta, 2004, 1665, 48–56 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed scheme of hydrophobic polyampholyte synthesis and its characterization by 1H NMR, cytotoxicity, zeta potential, and zeta size characterization of unmodified and polyampholyte-modified liposome-encapsulated protein. Comparison of the existing and freeze concentration method, and particle size stability at different temperatures. Cell viability of L929 cells after freeze thawing, quantification of dose dependency using flow cytometry and determination of cell viability after treating with different inhibitors. See DOI: 10.1039/c6nr03940e |
| This journal is © The Royal Society of Chemistry 2016 |