
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase control during the synthesis of nickel sulfide nanoparticles from dithiocarbamate precursors†
Anna
Roffey
a,
Nathan
Hollingsworth
*a,
Husn-Ubayda
Islam
a,
Maxime
Mercy
a,
Gopinathan
Sankar
a,
C. Richard A.
Catlow
a,
Graeme
Hogarth
*ab and
Nora H.
de Leeuw
ac
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: n.hollingsworth@ucl.ac.uk; graeme.hogarth@kcl.ac.uk
bDepartment of Chemistry, King's College London, Britannia House, 7 Trinity Street, London SE1 1DB, UK
cSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3XQ, UK
First published on 28th April 2016
Abstract
Square-planar nickel bis(dithiocarbamate) complexes, [Ni(S2CNR2)2], have been prepared and utilised as single source precursors to nanoparticulate nickel sulfides. While they are stable in the solid-state to around 300 °C, heating in oleylamine at 230 °C, 5 mM solutions afford pure α-NiS, where the outcome is independent of the substituents. DFT calculations show an electronic effect rather than steric hindrance influences the resulting particle size. Decomposition of the iso-butyl derivative, [Ni(S2CNiBu2)2], has been studied in detail. There is a temperature-dependence of the phase of the nickel sulfide formed. At low temperatures (150 °C), pure α-NiS is formed. Upon raising the temperature, increasing amounts of β-NiS are produced and at 280 °C this is formed in pure form. A range of concentrations (from 5–50 mM) was also investigated at 180 °C and while in all cases pure α-NiS was formed, particle sizes varied significantly. Thus at low concentrations average particle sizes were ca. 100 nm, but at higher concentrations they increased to ca. 150 nm. The addition of two equivalents of tetra-iso-butyl thiuram disulfide, (iBu2NCS2)2, to the decomposition mixture was found to influence the material formed. At 230 °C and above, α-NiS was generated, in contrast to the results found without added thiuram disulfide, suggesting that addition of (iBu2NCS2)2 stabilises the metastable α-NiS phase. At low temperatures (150–180 °C) and concentrations (5 mM), mixtures of α-NiS and Ni3S4, result. A growing proportion of Ni3S4 is noted upon increasing precursor concentration to 10 mM. At 20 mM a metastable phase of nickel sulfide, NiS2 is formed and as the concentration is increased, α-NiS appears alongside NiS2. Reasons for these variations are discussed.
Introduction
Nickel sulfides are an interesting and important group of materials exhibiting a plethora of phases, including α-NiS, β-NiS, NiS2, Ni3S2, Ni3S4, Ni7S6, Ni9S8, which find use in alternative energy applications. For example, Ni3S2 has potential as a low cost counter electrode material in dye-sensitised solar cells,1 while α-NiS has been used as a cathode material in lithium-ion batteries.2 Nickel sulfides have also been investigated as potential competitors to silicon thin film photovoltaic cells,3 as well as being used in the photocatalytic generation of hydrogen from water and other protic solvents.4,5Solvothermal decompositions of precursors are generally preferred for nanoparticle synthesis, where the solvent often acts as a heat sink and capping agent. Solvothermal decomposition of single-source precursors (SSP) such as thiazole,6 polysulfide,7 thiobiuret,8 imidodiphosphinate,9 thiobenzoate10 and xanthate11 nickel complexes have been reported to afford various nickel sulfides. Decomposition conditions such as solvent, precursor concentration and temperature have been varied, resulting in the ability to form up to two nickel sulfide phases, often as a mixture.
Reports detailing the decomposition of nickel dithiocarbamate complexes are limited,12–14 which is surprising given that nickel dithiocarbamate complexes, first reported by Delépine in 1907,15 are well studied.16 In addition, their ease of synthesis from cheap readily available starting materials has led to metal dithiocarbamates in general being widely employed as SSP for the synthesis of metal sulfide materials.17–26
We have recently reported the solvothermal decomposition of a nickel dithiocarbamate in primary amines.27 This work focussed on mechanistic aspects of the decomposition, giving a general mechanism for metal dithiocarbamate decomposition. We showed that the decomposition of nickel dithiocarbamates is dependent on the formation of amide exchange intermediates (Fig. 1a and b). The extent of amide exchange dictates the phase of the resultant nanoparticle. Ni dithiocarbamates that decompose through a single amide exchange (Fig. 1a) were shown to form NiS and those that decompose through a double amide-exchange (Fig. 1b) were shown to favour formation of Ni3S4 nanoparticles.
 | ||
| Fig. 1 Decomposition of nickel dithiocarbamate in primary amines via 1 amide exchange [a] and 2 amide exchange [b] intermediates. | ||
Here we report how changes to experimental conditions, the identity of the dithiocarbamate ligand and the addition of additives can affect the solvothermal synthesis of nickel sulfide nanoparticles. Our work reveals the remarkable versatility of this approach in accessing a range of phase-pure nickel sulfides upon simple changes to reaction conditions; an approach that is potentially applicable to the wide range of metal sulfide materials accessible through dithiocarbamate precursors.
Results and discussion
Synthesis and solid-state stability of nickel bis(dithiocarbamate) complexes
Complexes 1–4 (Fig. 2) were synthesised according to established literature methods28via the addition of two equivalents of NaS2CNR2 to an aqueous solution of nickel chloride. All were easily isolated and purified, although [Ni(S2CNMe2)2] (1) was only sparingly soluble in chlorinated solvents making purification on a large scale more difficult. | ||
| Fig. 2 Dithiocarbamate complexes employed in this study. | ||
1–4 are green, air and moisture stable solids, which were readily characterised by standard analytical and spectroscopic methods (see ESI†). A large number of crystallographic studies have been carried out on nickel bis(dithiocarbamate) complexes: all report nickel in a square-planar coordination environment, with intra-ligand bite-angles at nickel ranging from 78–80° and Ni–S bond distances between 2.16 and 2.23 Å.29–33 The square planar geometry of these complexes allows for efficient packing in the solid-state which accounts for the low solubility of 1, since here all thirteen atoms of complex 1 lie in a plane and thus can pack efficiently.
The relative stability of nickel bis(dithiocarbamate) complexes was first investigated in the solid-state by thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA for 2–4 (ESI Fig. S1†) are all are similar, exhibiting thermal stability until ca. 300 °C, whereupon they lose ca. 95% of their mass in one sharp step. The DSC graphs (ESI Fig. S1†) for 2–4 show melting peaks at 235, 177 and 118 °C respectively, consistent with literature values.34–36 The dimethyl derivative, 1 shows an unusual trace being different from those for 2–4. Thus, loss of a mass equivalent to S(SCNMe2)2 occurs gradually, beginning at 106 °C and continuing to 367 °C, suggesting that this complex decomposes rather than evaporates or sublimes. At this point the remaining mass is equal to NiS, but the sample continues to lose mass, leaving a percentage equal to less than that of elemental nickel. The DSC trace for 1 shows no melting peak, consistent with previous literature reports.34
Decomposition of nickel bis(dithiocarbamate) complexes
Complexes 1–4 were decomposed in solution at 230 °C with a single-source precursor concentration of 5 mM in oleylamine. All formed green solutions at room temperature with the exception of 1 which dissolved only upon heating to ca. 30 °C. As the solutions were heated, they slowly became brown at ca. 60 °C, becoming darker as the temperature was raised until around 130–140 °C, whereupon the clear brown solution became opaque and black (with the exception of 1, which started to become brown at 90 °C, and black at 140–150 °C).Powder XRD revealed the crystalline phase of the decomposed black powders to be pure α-NiS (ESI Fig. S2†) in all cases, showing that substituent variations do not significantly affect the phase of nickel sulfide formed under these conditions. Previous studies27 have shown that the decomposition of 3 in oleylamine proceeds through an intermediate with a single amide exchange, akin to Fig. 1a, resulting in an NiS product. The observation that 1–4 decompose to the same product suggests that the variation in dithiocarbamate does not affect the decomposition route through this single amide exchange in oleylamine. Thus the decomposition route is more likely to be affected by the choice of amine.
TEM analysis reveals nanoparticles that are primarily hexagonal in shape for all decompositions. Particles from the decomposition product of 3 (Fig. 3 right) are typical. HRTEM of these particles show d-spacings of 2.95 Å, consistent with the [100] plane of α-NiS (2.98 Å). The average particle diameter varies with substituents, such that as the steric bulk of the nickel bis-(dithiocarbamate) substituent increases, the average diameter of the nanoparticles increases.
 | ||
| Fig. 3 (Left) Average particle diameter of samples prepared from 1–4 with standard error, (Right) TEM image of the sample prepared from 3 with HRTEM insert. | ||
Computational modelling of nucleophilic attack on 1–4
In order to discover relationships between changes in dithiocarbamate moiety and resultant particle size, in silico studies have been performed with DFT (B3PW91/6-31G(d,p)). To reduce computational costs 1–4 were modelled with ethylamine as solvent, the linear un-branched nature of oleylamine affords this to be a sensible substitute and should have negligible effect on the resulting transition states. Metal dithiocarbamates of the type 1–4 have previously been shown to be stable to decomposition in the absence of primary amines, whereas addition of primary amines was shown to promote amide exchange intermediates of the type shown in Fig. 1a and b which are key to enabling precursor decomposition. Furthermore, the rate-determining transition state during the decomposition was identified as the substitution of the secondary amino group by a primary amine via a proton transfer (namely the amide exchange).Steric bulk effects during the initial amide exchange were explored first. The corresponding transition states (proton transfer between the two amines) for complexes 1–4 were computed (Fig. 4). Modelling showed the geometry of the four-membered transition state ring is the same whatever the size of the substituent considered in this study. Therefore steric hindrance by bulky substituents did not affect the stability of the transition state.
 | ||
| Fig. 4 Modelled transition states of the amide-exchange of ethylamine with 1–4. Bond distance (Å) and angle (°). Gibbs free energy (kJ mol−1) with the separate reactants as reference. | ||
However, there is a correlation between the calculated activation barrier to form this transition state and the particle size, such that, the lower the activation barrier, the smaller the particle observed as shown in Fig. 5. Generally, when keeping the amount of precursor constant, small particles will result when the number of nucleation sites is high, due to Ostwald ripening taking place on multiple sites, until all decomposed material is consumed. In contrast, larger particles can result when the number of nucleation sites is low, thereby resulting in fewer sites for the same amount of material to grow upon. Decomposition of nickel dithiocarbamates and so formation of nucleation sites is plausibly dependent on the rate of formation of amide-exchange intermediates by nucleophilic attack of a primary amine on the CS2 carbon of the dithiocarbamate moiety, which we suggest is the origin of the correlation between activation energy and particle size.
 | ||
| Fig. 5 (a) Correlation between particle size and the activation barrier to the amide exchange transition state at 368.15 K of 1–4. (b) Correlation between particle size and NAC of the sum of the first alkyl carbons of 1–4. | ||
It is likely that the electron withdrawing nature of the nickel dithiocarbamate will also play a role in stabilising S2Cδ+ and so provide an additional complication. In an attempt to address this suggestion we studied correlations between particle size and the natural atomic charges (NAC) of the N (δN) on the dithiocarbamate of complexes 1–4. No trend is observed with δN being ca. −0.41 in all cases. A trend is seen, however, when looking at ∑δCN, the sum of the NAC of the first two carbons of the alkyl chain connected to the N of the dithiocarbamate (NR2) (Fig. 5). The more negative the potential the smaller the nanoparticle formed. As such, a negative potential on the alkyl carbons may also affect amide exchange.
Temperature-dependent decomposition of 3
Complex 3, is especially easy to synthesise and purify, shows good solubility in oleylamine and produced well-faceted nanoparticles at 230 °C, and it was thus selected for temperature-dependent decomposition studies. 5 mM solutions were decomposed at 150, 180, 230, 260 and 280 °C but no visible differences were noted as the temperature was varied.XRD analysis of the resultant nanoparticles, however, reveals a relationship between decomposition temperature and the crystalline nickel sulfide phase (Fig. 6). At low temperature (150 °C) α-NiS is formed; however, at 260 °C, the majority of the crystalline material is β-NiS, with a small α-NiS impurity. At 280 °C α-NiS is no longer present and pure β-NiS is produced.
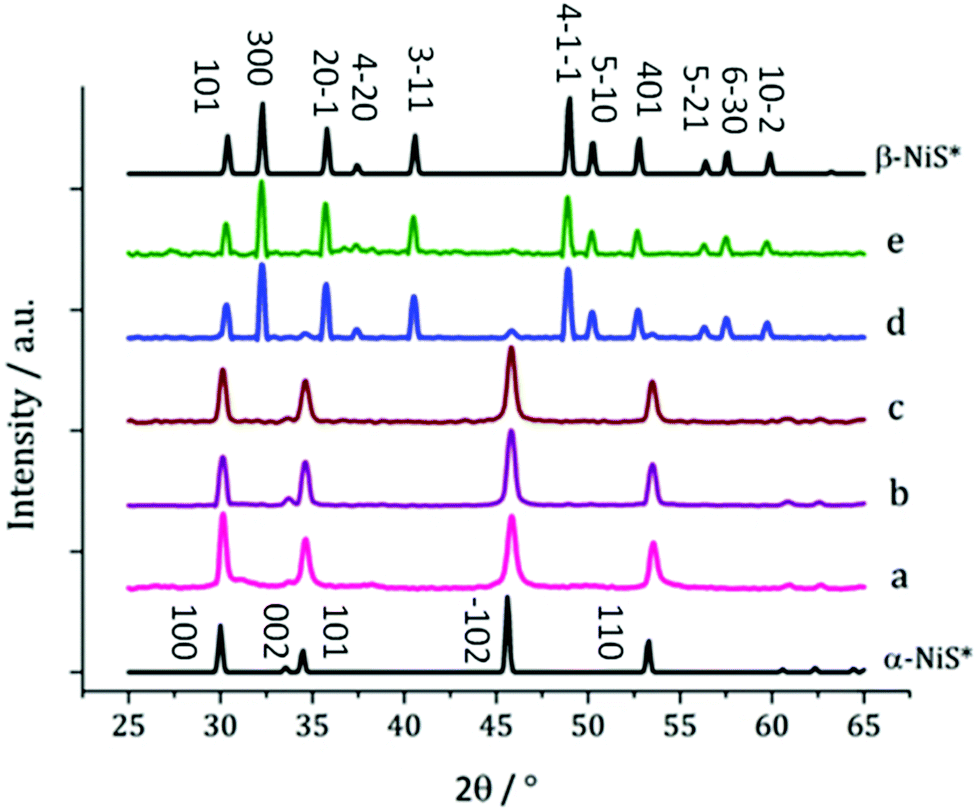 | ||
| Fig. 6 XRD patterns for samples prepared from 3 at (a) 150 °C, (b) 180 °C, (c) 230 °C, (d) 260 °C and (e) 280 °C, with reference patterns for bulk α-NiS (ICDD card no. 02-1273) and β-NiS (ICDD card no. 86-2281), major peaks have been indexed. | ||
This result contradicts the generally accepted behaviour of the α and β phases of NiS, being the high and low temperature phases, respectively.37 There is some precedent for this reversal of behaviour. Chen et al. reported the synthesis of β-NiS nanoparticles with some α-NiS impurity which was eliminated by increasing the reaction time,38 indicating that the β phase is actually the thermodynamically favoured phase in the solvothermal system.
The β-NiS nanoparticles formed at 260 and 280 °C exhibit larger average diameters than the α-NiS formed at lower temperatures (150–230 °C). In both samples of β-NiS, the nanoparticles appear agglomerated with a large size distribution (ESI Fig. S3†). HRTEM of particles produced at 280 °C shows d-spacings of 2.54 and 2.98 Å, consistent with the [−111] and [010] planes of β-NiS (2.51 and 2.95 Å respectively).
When 3 was decomposed at 150 and 230 °C, α-NiS nanoparticles of 38.0 and 37.9 nm respectively, were formed, while at 180 °C, much larger α-NiS nanoparticles were produced (105.3 nm).
Concentration-dependent decomposition of 3
A temperature of 180 °C was chosen for a detailed study of the effect of concentration on the system. Concentrations of 10, 20, 40 and 50 mM were used and XRD analysis of the products reveals that the precursor concentration has no effect on the phase of the resulting nickel sulfide nanoparticles generated, α-NiS being formed in all instances (Fig. S4†). TEM analysis of the resultant samples reveals no change to the morphology of the nanoparticles upon increasing precursor concentration, all being roughly hexagonal in accordance with the sample produced at 5 mM (Fig. S5†). The average size of the nanoparticles, however, was affected by varying precursor concentration, such that generally the higher the concentration of 3, the greater the average nanoparticle diameter (Fig. 7). | ||
| Fig. 7 Graph of average particle diameter with standard error against concentration of 3. | ||
Effect(s) of adding tetra-iso-butyl thiuram disulfide (5) to the decomposition mixture
Thiuram disulfides are particularly interesting as additives because they can act as a S source and a redox active complex when thermally decomposed within primary amines.39 The thiuram disulfides employed in this study, 5 and 6 are shown in Fig. 2.Mixtures of 3 (5 mM) and 5 (10 mM) were decomposed in oleylamine at 150, 180, 230, 260 and 280 °C. XRD of the black powders obtained are summarised in Fig. 8. Samples synthesised at 230 °C and above match the reference pattern for α-NiS, which is contrary to the results found without added 5, where β-NiS was formed, which suggests that the addition of 5 stabilises the metastable α-NiS phase. XRD patterns for samples prepared at 150 and 180 °C show mixtures of both α-NiS and Ni3S4, though the pattern at 150 °C is of poor quality being indicative of low crystallinity. Polydymite (Ni3S4) is a thiospinel and is a metastable phase. We have previously reported that the synthesis of Ni3S4 is likely to arise from the decomposition of a double amide-exchange intermediate, validating this claim with the decomposition of Ni(S2CN(H)Hexyl)2 at 120 °C to Ni3S4.27 At 180 °C there appears to be a mixture of a large component of α-NiS and smaller amounts of polydymite (Ni3S4).
 | ||
| Fig. 8 XRD patterns for samples prepared from 3 with tetra-iso-butyl-thiruam disulfide (5) at (a) 150 °C, (b) 180 °C, (c) 230 °C, (d) 260 °C and (e) 280 °C, with reference patterns for bulk Ni3S4 (ICDD card no. 43-1469) and α-NiS (ICDD card no. 02-1273), major reference peaks indexed. | ||
TEM images for all samples are shown in Fig. 9. The image of the sample at 150 °C (Fig. 9a) supports the XRD analysis, showing some very small particulates and amorphous areas which is consistent with the sample produced in the absence of thiuram disulfide at 150 °C, which was also found to be partially amorphous.
 | ||
| Fig. 9 TEM images of samples prepared from 3 with 5 at (a) 150, (b) 180, (c) 230, (d) 260 and (e) 280 °C. (f) Graph of average particle size against temperature of decomposition with standard error. | ||
TEM analysis of the other samples shows a progression from small, partially amorphous materials, to large well-faceted particles (resembling the α-NiS samples previously prepared) when the decomposition temperature is increased from 150 to 180 °C. This increase in size could be due to increased Ostwald ripening at the higher temperature of 180 °C. The average particle size drops, however, on increasing decomposition temperature from 180 to 230 °C, which could be due to an increased rate of decomposition and thus an increase in the number of nucleation sites at the higher temperature. Samples prepared at 230 °C and above increase in size again as the decomposition temperatures increase. The sample at 230 °C is made up of many small particles that have agglomerated, while the sample at 280 °C may be the result of Ostwald ripening of these smaller particles. As with the samples prepared without added thiuram disulfide, particles prepared at 180 °C are larger than those at other temperatures. This temperature appears to be significant in the nickel sulfide decomposition system.
Varying the concentration of thiuram disulfide additive (5) and 3
Mixtures of 3 and tetra-isobutyl thiuram disulfide (5) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio) were decomposed at 180 °C in increasing concentrations (10:20, 20:40, 40:80 and 50:100 mM) and various phases of nickel sulfide were formed as shown by XRD (Fig. 10). At low precursor concentration, a mixture of α-NiS and Ni3S4 was formed, with a growing proportion of Ni3S4 as the concentration of precursor is increased to 10 mM. The sample prepared using 20 mM of 3 (and 40 mM of 5) matches well to the pattern for NiS2 (ICDD card no. 89-3058). As the concentration is increased, peaks for α-NiS appear and grow alongside the NiS2 peaks. NiS2 is a metastable phase of nickel sulfide, only accessible in this system by the use of the thiuram disulfide additive 5, and at high precursor concentrations. All XRD patterns show broad peaks indicative of small crystallite size, which is confirmed by TEM images of the particles for the NiS2 containing samples, which all have very small particles (Fig. 10). The particles prepared from 5 and 10 mM solutions of 3 are larger (ca. 50 nm). The sample prepared from 10 mM of 3 contains a higher proportion of Ni3S4 which was imaged via HRTEM, showing d-spacings of 5.50 Å, matching well to literature values for the [111] plane (5.48 Å, ICDD card 43-1469). The morphology of this phase shows interlocking irregularly shaped sheets, quite unlike the morphology of the particles prepared at higher precursor concentration.
:2 ratio) were decomposed at 180 °C in increasing concentrations (10:20, 20:40, 40:80 and 50:100 mM) and various phases of nickel sulfide were formed as shown by XRD (Fig. 10). At low precursor concentration, a mixture of α-NiS and Ni3S4 was formed, with a growing proportion of Ni3S4 as the concentration of precursor is increased to 10 mM. The sample prepared using 20 mM of 3 (and 40 mM of 5) matches well to the pattern for NiS2 (ICDD card no. 89-3058). As the concentration is increased, peaks for α-NiS appear and grow alongside the NiS2 peaks. NiS2 is a metastable phase of nickel sulfide, only accessible in this system by the use of the thiuram disulfide additive 5, and at high precursor concentrations. All XRD patterns show broad peaks indicative of small crystallite size, which is confirmed by TEM images of the particles for the NiS2 containing samples, which all have very small particles (Fig. 10). The particles prepared from 5 and 10 mM solutions of 3 are larger (ca. 50 nm). The sample prepared from 10 mM of 3 contains a higher proportion of Ni3S4 which was imaged via HRTEM, showing d-spacings of 5.50 Å, matching well to literature values for the [111] plane (5.48 Å, ICDD card 43-1469). The morphology of this phase shows interlocking irregularly shaped sheets, quite unlike the morphology of the particles prepared at higher precursor concentration.
 | ||
| Fig. 10 XRD patterns for samples prepared from 3 with 5 at (a) 5, (b) 10, (c) 20, (d) 40 and (e) 50 mM concentration, with reference patterns for bulk α-NiS ICDD card no. 02-1273), Ni3S4 (ICDD card no. 43-1469) and NiS2 (ICDD card no. 89-3058). | ||
The particles formed from the decomposition of 3 at concentrations of 20–50 mM are very small (ca. 5 nm) and do not appear to aggregate in the same way (Fig. 11). HRTEM analysis reveals a d-spacing of 2.81 Å, which matches the [200] plane of NiS2 (2.81 Å, ICDD card no. 89-3058). Particles formed from 50 mM of 3 have a larger average diameter (7.60 nm), due to the presence of other large particles, which are probably the α-NiS impurity.
 | ||
| Fig. 11 TEM images of samples prepared from 3 with 5 at 180 °C at 10 (a, b) and 20 mM (c, d). | ||
Role of thiuram disulfide
It has been reported that during the photoreduction of the Ni(IV) dithiocarbamate species, thiuram disulfides are produced. The reversible nature of this reaction suggests that the addition of a thiuram disulfide to a Ni(II) dithiocarbamate would result in the formation of the Ni(IV) species (Fig. 12).40,41 | ||
| Fig. 12 Formation of Ni(IV) species on addition of thiuram disulfide. | ||
1H NMR has been used to probe this reaction. In order to make the interpretation of the spectra simpler, the tetramethyl thiuram disulfide (6) has been used in place of 5. Monitoring the addition of two equivalents of 6 to 3 shows the peaks associated with both 1, 5 and a new diamagnetic species, presumably due to the formation of the Ni(IV) tris(dimethyldithiocarbamate) cation, [Ni(S2CNMe2)3]+ (see ESI Fig. S6†).
XANES analysis of the addition of 5 to 3 reveals only the presence of 3 in a 2+ oxidation state (ESI Fig. S7–8†), which may seem surprising given the 1H NMR analysis showing a NiIV species. However, it has been reported42 that X-rays reduce NiIV species to NiII. Increasing the temperature of the mixture should push the equilibrium toward the NiIV species. However, given the intensity of the X-rays used, the NiIV was not observed during in situ monitoring of the decomposition, rendering monitoring this reaction via XAS unfeasible. The monitoring does reveal that the decomposition follows the same decomposition pathway previously reported for 3 in oleylamine. Thus the decomposition pathway in this case may be directed by the presence of X-rays.
Eckstein and Hoyer have studied the reduction of Ni(IV) dithiocarbamates,41 here they show the reduction takes place in three stages (Fig. 13). Relating back to our decomposition system, we have two processes taking place: first, the reduction of the as formed Ni(IV) species and, second, the amide exchange process essential for decomposition to nickel sulfide nanoparticles (Fig. 1). It follows plausibly that the extent of the reduction of the Ni(IV) species, (i.e. the position of the equilibrium in EQ1 through to EQ3, Fig. 13), prior to the point at which amide exchange takes place and thus decomposition, is temperature-dependent. Therefore, the formation of Ni3S4 (a thiospinel containing Ni2+ and Ni3+ cations) at low temperatures upon the addition of 5 to 3, is plausibly explained by the amide exchange process proceeding after partial reduction. The position of the equilibrium lies in favour of the products of EQ1 and EQ2 (Fig. 13) prior to amide exchange and hence decomposition, providing NiII and NiIII for the formation of Ni3S4 nanoparticles. On increasing the temperature, we suggest that the position of the equilibrium will be in favour of the products of EQ3 (Fig. 13) prior to amide exchange. Thus, we find the products of decomposition of 3 with 5 at high temperatures is α-NiS (a purely NiII mineral).
 | ||
| Fig. 13 Reduction of Ni(IV) dithiocarbamates, dtc corresponds to a dithiocarbamate ligand and (dtc)2 corresponds to a thiuram disulphide.41 | ||
Changing concentration, but fixing the temperature appears to form the expected Ni3S4 at 180 °C at low concentration. Surprisingly, at high concentration, a purely NiII cation-containing mineral is formed, suggesting that the concentration also affects the equilibrium position (Fig. 13) prior to amide exchange. In addition, the high temperature mineral formed is NiS2. The increase in sulfur is attributed to the decomposition of excess 5, providing a sulfur source for the resultant particles – a phenomenon that only appears to happen at high concentration.
Experimental
General
All reagents were procured commercially from Aldrich and used without further purification.All 1H and 13C NMR29 spectra were obtained on either a Bruker Avance III 400 or Avance 600 spectrometer, the latter being equipped with a cryoprobe. All spectra were recorded using CDCl3 which was dried and degassed over molecular sieves prior to use; 1H and 13C{1H} chemical shifts are reported relative to SiMe4. The mass spectra were obtained using either Micromass 70-SE spectrometer using Electron Ionisation (EI) or a Thermo Finnigan MAT900xp spectrometer using Fast Atom Bombardment (FAB). Elemental analysis was carried out using Elemental Analyser (CE-440) (Exeter Analytical Inc). Thermogravimetric analysis (TGA) was performed using a Netzsch STA 449C TGA system. Data was recorded from 25 to 600 °C with a constant heating rate of 10 °C min−1. XRD patterns were measured on a Bruker AXS D4 diffractometer using CuKα1 radiation. The diffraction patterns obtained were compared to database standards. For TEM characterisation a 4 μl droplet of nanoparticle suspension (chloroform) was placed on a holey carbon-coated copper TEM grid and allowed to evaporate in air under ambient laboratory conditions for several minutes. TEM images were obtained using a JEOL-1010 microscope at 100 kV equipped with a Gatan digital camera. HRTEM measurements were collected using a Jeol 2100 TEM with a LaB6 source operating at an acceleration voltage of 200 kV. Micrographs were taken on a Gatan Orius Charge-coupled device (CCD).
Precursor synthesis and characterisation
See ESI.†Decomposition studies
In a typical decomposition, 1 (5 mM) was added to oleylamine (20 mL) in a three-neck round-bottom flask attached to a condenser and evacuated and refilled with nitrogen repeatedly for 15 minutes. The solution was heated to 230 °C and held there for 1 h. The mixture was allowed to cool to RT slowly, whereupon methanol (80 mL) was added with stirring. The mixture was centrifuged and then the solution decanted leaving behind the resultant nanoparticles. This procedure was repeated three times and finally the material was allowed to dry in air. A number of parameters were varied including; (i) decompositions were carried out at 150, 180, 260 and 280 °C; (ii) precursors concentrations were varied between 10–50 mM; (iii) tetra-iso-butyl thiuram disulfide (5) was added in varying amounts.Computational details
All the geometry optimisations and energy calculation were performed according to the density functional theory level of theory employing the B3PW91 hybrid functional43 with a Pople basis set 6-31G(d,p).44 For all the geometries optimized, the nature of the steady states (minimum and transition state) were verified by analytical frequency calculations. The intrinsic reaction coordinate (IRC) was followed for the transition state to confirm the connection between the reactant and the product. The suite of programs Gaussian 09 Rev C was used for all the calculations.45 Natural bond orbital (NBO) analysis were performed using NBO 3.1 as implemented in Gaussian.46Conclusions
Through varying decomposition temperature, precursor concentration and the use of an additive, nickel sulfide nanoparticles of various mineral phases have been produced. These conditions can be tuned to obtain α-NiS, β-NiS, Ni3S4 and NiS2. Previous reports detailing the synthesis of a range of nickel sulfides47–53 have often employed metal salts combined with a sulfur source or employed a relatively long autoclave based synthesis procedure. The work presented here offers the advantage of short solvothermal decompositions from a cheap SSP.The decomposition study on precursor 3 (without additive) showed that in the solvothermal system, α-NiS is formed at low temperature, while β-NiS at high – which might suggest that the former is a kinetically favoured, metastable phase compared to the latter, being a more thermodynamically stable phase of nickel sulfide. This finding is in contrast to their relationship in the nickel sulfide phase diagram, where α-NiS is regarded as the high temperature phase compared to β-NiS. The reasons for this apparent contradiction are unclear, although they may be attributed to solvent interactions and suggest that the solvent has an important role in the decomposition mechanism.
The addition of two equivalent amounts of thiuram disulfide (5) to the decomposition system allows access to the metastable phase Ni3S4. Both nickel(II) and nickel(III) ions are present in Ni3S4, indicating that nickel(II) in the [Ni(S2CNiBu2)2] precursor has been partially oxidised.
At high precursor concentrations, with two equivalents of 5 present, NiS2 was produced. Nickel is in the 2+ oxidation state in this phase with an S22− dianion, providing further support for the hypothesis that the role of 5 in the decomposition mechanism is not to change the oxidation state of the metal, but to stabilise metastable phases of nickel sulfide in the system. The addition of 5 to the synthesis of other metal sulfides may have a similar stabilising effect, enabling the formation of metastable phases that are not easily prepared otherwise. Work is currently underway applying this approach to Fe, Co, In and Zn systems.
Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) for supporting this work financially (grant numbers; EP/H046313/1, EP/K035355/2 and EP/K001329/1). H-UI thanks; DUBBLE, ESRF for funding and beam time on BM26A DUBBLE EXAFS beamline, and also Wim Bras, Dipanjan Banerjee, Alessandro Longo for support on the beamline. The UK Catalysis Hub is thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (EP/K014706/1, EP/K014668/1, EP/K014854/1 and EP/K014714/1).Notes and references
- H. K. Mulmudi, S. K. Batabyal, M. Rao, R. R. Prabhakar, N. Mathews, Y. M. Lam and S. G. Mhaisalkar, Phys. Chem. Chem. Phys., 2011, 13, 19307–19309 RSC.
- S.-C. Han, K.-W. Kim, H.-J. Ahn, J.-H. Ahn and J.-Y. Lee, J. Alloys Compd., 2003, 361, 247–251 CrossRef CAS.
- C. Wadia, A. P. Alivisatos and D. M. Kammen, Environ. Sci. Technol., 2009, 43, 2072–2077 CrossRef CAS PubMed.
- W. Zhang, Y. Wang, Z. Wang, Z. Zhong and R. Xu, Chem. Commun., 2010, 46, 7631–7633 RSC.
- L. Zhang, B. Tian, F. Chen and J. Zhang, Int. J. Hydrogen Energy, 2012, 37, 17060–17067 CrossRef CAS.
- B. Geng, X. Liu, J. Ma and Q. Du, Mater. Sci. Eng., B, 2007, 145, 17–22 CrossRef CAS.
- J. H. L. Beal, P. G. Etchegoin and R. D. Tilley, J. Phys. Chem. C, 2010, 114, 3817–3821 CAS.
- A. L. Abdelhady, M. A. Malik, P. O'Brien and F. Tuna, J. Phys. Chem. C, 2011, 116, 2253–2259 Search PubMed.
- M. G. Babashkina, D. A. Safin and Y. Garcia, Dalton Trans., 2012, 41, 2234–2236 RSC.
- L. Tian, L. Y. Yep, T. T. Ong, J. Yi, J. Ding and J. J. Vittal, Cryst. Growth Des., 2008, 9, 352–357 Search PubMed.
- N. Pradhan, B. Katz and S. Efrima, J. Phys. Chem. B, 2003, 107, 13843–13854 CrossRef CAS.
- H. Cui, R. D. Pike, R. Kershaw, K. Dwight and A. Wold, J. Solid State Chem., 1992, 101, 115–118 CrossRef CAS.
- G. H. Singhal, R. I. Botto, L. D. Brown and K. S. Colle, J. Solid State Chem., 1994, 109, 166–171 CrossRef CAS.
- X. Chen, Z. Wang, X. Wang, J. Wan, J. Liu and Y. Qian, Chem. Lett., 2004, 33, 1294–1295 CrossRef CAS.
- M. Delépine, C. R. Hebd. Seances Acad. Sci., 1907, 144, 1125 Search PubMed.
- G. Hogarth, Prog. Inorg. Chem., 2005, 53, 71–561 CrossRef CAS.
- J. C. Bear, N. Hollingsworth, P. D. McNaughter, A. G. Mayes, M. B. Ward, T. Nann, G. Hogarth and I. P. Parkin, Angew. Chem., Int. Ed., 2014, 53, 1598–1601 CrossRef CAS PubMed.
- A. Roldan, N. Hollingsworth, A. Roffey, H. U. Islam, J. B. M. Goodall, C. R. A. Catlow, J. A. Darr, W. Bras, G. Sankar, K. B. Holt, G. Hogarth and N. H. de Leeuw, Chem. Commun., 2015, 51, 7501–7504 RSC.
- H.-U. Islam, A. Roffey, N. Hollingsworth, R. Catlow, M. Wolthers, N. De Leeuw, W. Bras, G. Sankar and G. Hogarth, J. Phys.: Conf. Ser., 2013, 430, 012050 CrossRef.
- J. C. Bear, N. Hollingsworth, A. Roffey, P. D. McNaughter, A. G. Mayes, T. J. Macdonald, T. Nann, W. H. Ng, A. J. Kenyon, G. Hogarth and I. P. Parkin, Adv. Opt. Mater., 2015, 3, 704–712 CrossRef CAS.
- E. A. Lewis, P. D. McNaughter, Z. Yin, Y. Chen, J. R. Brent, S. A. Saah, J. Raftery, J. A. M. Awudza, M. A. Malik, P. O'Brien and S. J. Haigh, Chem. Mater., 2015, 27, 2127–2136 CrossRef CAS.
- W. J. Peveler, A. Roldan, N. Hollingsworth, M. J. Porter and I. P. Parkin, ACS Nano, 2015, 10(1), 1139–1146 CrossRef PubMed.
- E. Sathiyaraj, G. Gurumoorthy and S. Thirumaran, New J. Chem., 2015, 39, 5336–5349 RSC.
- B. Arul Prakasam, M. Lahtinen, A. Peuronen, M. Muruganandham, E. Kolehmainen, E. Haapaniemi and M. Sillanpää, Polyhedron, 2014, 81, 588–596 CrossRef CAS.
- B. Arul Prakasam, M. Lahtinen, A. Peuronen, M. Muruganandham, E. Kolehmainen, E. Haapaniemi and M. Sillanpää, Inorg. Chim. Acta, 2015, 425, 239–246 CrossRef CAS.
- R. Chauhan, M. Trivedi, J. Singh, K. C. Molloy, G. Kociok-Köhn, U. P. Mulik, D. P. Amalnerkar and A. Kumar, Inorg. Chim. Acta, 2014, 415, 69–74 CrossRef CAS.
- N. Hollingsworth, A. Roffey, H.-U. Islam, M. Mercy, A. Roldan, W. Bras, M. Wolthers, C. R. A. Catlow, G. Sankar, G. Hogarth and N. H. de Leeuw, Chem. Mater., 2014, 26, 6281–6292 CrossRef CAS.
- F. A. Cotton and J. A. McCleverty, Inorg. Chem., 1964, 3, 1398–1402 CrossRef CAS.
- G. Rajput, V. Singh, A. N. Gupta, M. K. Yadav, V. Kumar, S. K. Singh, A. Prasad, M. G. B. Drew and N. Singh, CrystEngComm, 2013, 15, 4676–4683 RSC.
- G. J. Kettmann Viktor and K. Stefan, Collect. Czech. Chem. Commun., 1978, 43, 1204 CrossRef.
- J. Lokaj, F. Pavelčik, V. Kettmann, J. Masaryk, V. Vrábel and J. Garaj, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 926–928 CrossRef.
- J. Kamenfček, R. Pastorek, B. Cvek and J. Taraba, Z. Kristallogr. - New Cryst. Struct., 2013, 218, 205–206 Search PubMed.
- C. Raston and A. White, Aust. J. Chem., 1976, 29, 523–529 CrossRef CAS.
- J. Krupčík, J. Garaj, Š. Holotík, D. Oktavec and M. Košík, J. Chromatogr., A, 1975, 112, 189–196 CrossRef.
- M. L. Riekkola and O. Mäkitie, J. Therm. Anal., 1982, 25, 89–94 CrossRef CAS.
- G. D'Ascenzo and W. W. Wendlandt, J. Therm. Anal., 1969, 1, 423–434 CrossRef.
- G. Kullerud and R. A. Yund, J. Petrol., 1962, 3, 126–175 CrossRef CAS.
- S. Chen, K. Zeng, H. Li and F. Li, J. Solid State Chem., 2011, 184, 1989–1996 CrossRef CAS.
- L. Van Boi, Russ. Chem. Bull., 2000, 49, 335–343 CrossRef CAS.
- J. P. Fackler, A. Avdeef and R. G. Fischer, J. Am. Chem. Soc., 1973, 95, 774–782 CrossRef CAS.
- P. Eckstein and E. Hover, Z. Anorg. Allg. Chem., 1982, 487, 33–43 CrossRef CAS.
- D. Collison, C. David Garner, C. M. McGrath, J. Frederick, W. Mosselmans, E. Pidcock, M. D. Roper, B. G. Searle, J. M. W. Seddon, E. Sinn and N. A. Young, J. Chem. Soc., Dalton Trans., 1998, 4179–4186 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- R. W. Ditchfield, J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09 (Revision C 01), Pittsburgh, PA, 2009 Search PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO version 3.1, TCI, University of Wisconsin, Madison, 1998 Search PubMed.
- B. Yuan and W. Luan, Funct. Mater. Lett., 2014, 7, 1450003 CrossRef.
- X. Yang, L. Zhou, A. Feng, H. Tang, H. Zhang, Z. Ding, Y. Ma, M. Wu, S. Jin and G. Li, J. Mater. Res., 2014, 29, 935–941 CrossRef CAS.
- C. Sun, M. Ma, J. Yang, Y. Zhang, P. Chen, W. Huang and X. Dong, Sci. Rep., 2014, 4, 7054 CrossRef CAS PubMed.
- Z. Meng, Y. Peng, W. Yu and Y. Qian, Mater. Chem. Phys., 2002, 74, 230–233 CrossRef CAS.
- N. Mahmood, C. Zhang and Y. Hou, Small, 2013, 9, 1321–1328 CrossRef CAS PubMed.
- R. Karthikeyan, D. Thangaraju, N. Prakash and Y. Hayakawa, CrystEngComm, 2015, 17, 5431–5439 RSC.
- S. Chen, K. Zeng, H. Li and F. Li, J. Solid State Chem., 2011, 184, 1989–1996 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr00053c |
| This journal is © The Royal Society of Chemistry 2016 |