
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From porphyrins to pyrphyrins: adsorption study and metalation of a molecular catalyst on Au(111)†
Gerson
Mette‡
 *a,
Denys
Sutter
a,
Yeliz
Gurdal
b,
Stephan
Schnidrig
b,
Benjamin
Probst
b,
Marcella
Iannuzzi
b,
Jürg
Hutter
b,
Roger
Alberto
b and
Jürg
Osterwalder
*a
*a,
Denys
Sutter
a,
Yeliz
Gurdal
b,
Stephan
Schnidrig
b,
Benjamin
Probst
b,
Marcella
Iannuzzi
b,
Jürg
Hutter
b,
Roger
Alberto
b and
Jürg
Osterwalder
*a
aPhysik-Institut, Universität Zürich, CH-8057 Zürich, Switzerland. E-mail: gerson.mette@physik.uni-marburg.de; osterwal@physik.uzh.ch; Tel: +41 (0)44 635 5827
bInstitut für Chemie, Universität Zürich, CH-8057 Zürich, Switzerland
First published on 8th March 2016
Abstract
The molecular ligand pyrphyrin, a tetradentate bipyridine based macrocycle, represents an interesting but widely unexplored class of molecules. It resembles the well-known porphyrin, but consists of pyridyl subunits instead of pyrroles. Metal complexes based on pyrphyrin ligands have recently shown promise as water reduction catalysts in homogeneous photochemical water splitting reactions. In this study, the adsorption and metalation of pyrphyrin on a single crystalline Au(111) surface is investigated in an ultrahigh vacuum by means of scanning tunneling microscopy, low-energy electron diffraction, X-ray photoelectron spectroscopy and density functional theory. Pyrphyrin coverages of approximately one monolayer and less are obtained by sublimation of the molecules on the substrate kept at room temperature. The molecules self-assemble in two distinct phases of long-range molecular ordering depending on the surface coverage. The deposition of cobalt metal and subsequent annealing lead to the formation of Co-ligated pyrphyrin molecules accompanied by a pronounced change of the molecular self-assembly. Electronic structure calculations taking the herringbone reconstruction of Au(111) into account show that the molecules are physisorbed, but preferred adsorption sites are identified where Co and the N atoms of the two terminal cyano groups are optimally coordinated to the surface Au atoms. An intermediate state of the metalation reaction is observed and the reaction steps for the Co metalation of pyrphyrin molecules on Au(111) are established in a joint experimental and computational effort.
1. Introduction
The solution for how to satisfy the world's increasing energy consumption is still pending. However, there is an overall agreement that solar energy conversion has to be established on a grand scale. Within the search for new methods and materials for harvesting solar energy, photocatalytic water splitting plays a prominent role.1 In this framework, the tetradentate, bipyridine-based macrocycle pyrphyrin (Pyr) has been investigated recently for its applicability as a water reduction catalyst (WRC).2 The macrocycle as shown in Fig. 1(a) had already been synthesized by Ogawa et al. in 1984 but, until now, it has been only superficially examined.3,4 It shares some similarities with the far better known porphyrins, but in place of the pyrroles it is constituted by pyridyl subunits. A high stability of the pyrphyrin ligand and corresponding complexes is suggested owing to its conjugation, planarity and cyclic nature. Thus, complexes based on pyrphyrin may appear as promising as their porphyrin siblings for various applications, e.g. as photosensitizers or catalysts in photochemical reactions or in organic photovoltaics,5–8 or as molecular magnets in spin-based electronic devices.9–11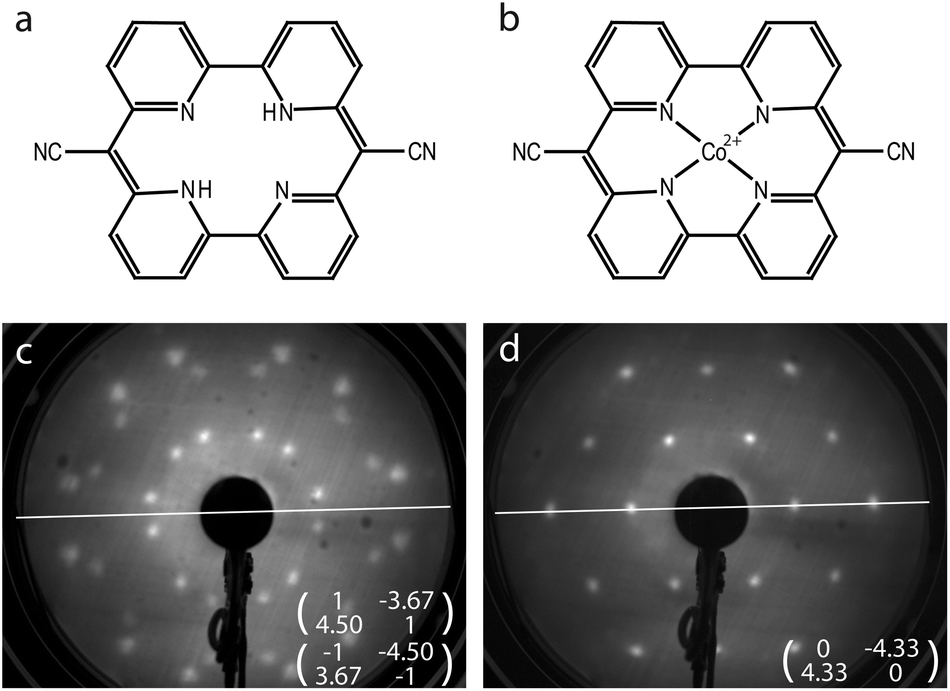 | ||
Fig. 1 Structural formulae of (a) pyrphyrin and (b) Co-pyrphyrin. LEED patterns at E = 14.1 eV of (c) the initial pyrphyrin covered Au surface and (d) after subsequent Co evaporation and annealing at 523 K for 12 hours, as well as the corresponding superstructure matrices. White lines indicate a [11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ] high-symmetry axis of the Au(111) surface. ] high-symmetry axis of the Au(111) surface. | ||
Indeed, metal complexes based on pyrphyrin ligands have recently shown promise as WRCs in homogeneous photochemical water splitting reactions, providing very high turnover numbers.2 However, in a practical water splitting device the WRC molecules will likely have to be immobilized on a solid surface as part of a heterogeneous photocatalytic reactor. It is therefore of interest to study the adsorption behavior of these metal complexes on well defined surfaces, and to explore whether their catalytic activity and stability are preserved in the adsorbed state. Examples of such studies can be found for surface-supported metallo-porphyrins. Hulsken et al.12 performed a mechanistic study of oxidation catalysis by monitoring individual manganese porphyrin catalysts on a Au(111) surface by scanning tunneling microscopy (STM) at the solid–liquid interface. Another example is the measurement of the adsorption isotherms of molecular oxygen on cobalt octaethylporphyrin molecules immobilized on highly-oriented pyrolytic graphite,13 again at the solid–liquid interface. Well defined monolayers of molecular catalysts therefore appear to lend themselves as model systems for heterogeneous catalysis. Furthermore, recent density functional theory (DFT) calculations of Co-ligated pyrphyrin (CoPyr) on TiO2 indicate that the pyrphyrin molecules also might be applied as a molecular photosensitizer.14
In this work, we studied the adsorption of pyrphyrin on a single crystalline surface and examined its controlled metalation in ultrahigh vacuum (UHV). Pyrphyrin coverages up to one monolayer were obtained by sublimation of the molecules on a Au(111) surface kept at room temperature. Deposition of Co metal at the level of 5% of a monolayer and subsequent annealing then led to the formation of 90% of CoPyr molecules. Similar recipes involving in situ two-step preparation in UHV had been successfully applied for the metalation of porphyrins and phthalocyanines.15–19 Our preparations of CoPyr metal complexes were characterized by means of low-energy electron diffraction (LEED), scanning tunneling microscopy, X-ray photoelectron spectroscopy (XPS) and density functional theory.
2. Experimental
All measurements were performed at room temperature in an UHV system based on a user-modified Vacuum Generator ESCALAB 220. For XPS studies, an unmonochromatised Mg Kα X-ray source was used, providing photons with an energy of hν = 1253.6 eV. All binding energies were referenced to Au 4d5/2 (335.1 eV).20 For quantitative analysis of the Shirley-background corrected XPS spectra, curve fitting was performed using convolutions of Doniach–Sunjic (DS) and Gaussian functions21 to account for the asymmetric line shape of the Au 4d, N 1s and C 1s spectra. For N 1s and C 1s, this asymmetry is based most likely on a complex satellite structure22,23 which is below the resolution limit of our measurements. In some cases symmetric functions were used approximated by setting the asymmetry parameter of the DS function to α = 0.01. The STM experiments were carried out in a Park Scientific VPII instrument, attached to the same UHV system, using etched W-tips. Specified tunneling voltages represent sample bias values.The Au(111) single crystal was cleaned by repetitive cycles of Ar sputtering and annealing up to 760 K resulting in the well-known herringbone reconstruction.24,25 Pyrphyrin was synthesized according to Ogawa et al.4 and purified by sublimation at 623 K and 5 × 10−5 mbar to obtain a pure dark wine red solid.2 The molecules were evaporated from a Knudsen cell at a temperature of 575 K and at a rate of 0.02 monolayers (ML) per minute and with the sample kept at room temperature. The deposition of Pyr was terminated early enough not to exceed one monolayer of Pyr. The cobalt source consists of a cobalt tube of high purity (99.998%) fitted on an Al2O3 ceramic tube that can be resistively heated by a tungsten filament threaded along its axis. During Co evaporation at a rate of 0.015 ML per minute the sample was held either at room temperature or at slightly elevated temperature of 323 K and afterwards annealed at temperatures up to 723 K.
Electronic structure calculations were performed at the Kohn–Sham DFT level, employing the Gaussian and plane wave (GPW) formalism as implemented in the CP2K package.26 Double-zeta plus polarization basis sets, optimized on molecular geometries,27 are assigned to all atomic types. The energy cutoff for the auxiliary PW basis is set at 500 Ry. The interaction between the valence electrons and the atomic cores is described through norm-conserving pseudo potentials.28 The valence shells contain 11, 17, 4, 1, and 5 electrons for Au, Co, C, H, and N, respectively. Periodic boundary conditions are always applied. As exchange correlation functional the spin polarized general gradient approximation by Perdew–Burke–Ernzerhof29 is used, augmented by the revised Vydrov–Voorhis van der Waals density functional,30 which accounts for the weaker dispersion interactions.
The CoPyr monomer in vacuum turns out to be a flat molecule in the doublet spin state. The adsorbed molecules on the reconstructed Au(111) surface are simulated employing a slab model, where the metal is represented by four atomic layers, stacked along the [111] direction, plus 20 Å of vacuum to avoid interactions with the periodic images in this direction. In order to reproduce the herringbone reconstruction, a minimum of 22 × 2 Au(111) hexagonal unit cells are needed in the lateral dimensions of the slab, where only in the topmost layer 23 atoms instead of 22 are positioned along the x-axis.31 The calculations, both for the herringbone reconstruction and the monomer adsorption, were carried out using a 22 × 6 Au(111) slab with four layers, where the three topmost layers of the slab are relaxed, whereas the fourth layer is kept fixed in the bulk positions. As already demonstrated in previous studies,32 once optimized, this model accurately reproduces the reconstruction. The adsorbed molecules are always placed flat on the already reconstructed Au(111) and their geometry is then further optimized. Geometry optimizations of the Pyr and CoPyr monomers and of two dimensional molecular networks in the gas phase serve as reference calculations. For the adsorbed molecular networks, the intermolecular distances in the starting geometry are chosen according to the experimental observation. However, several possible relative rotations of the molecules were tested in order to simultaneously optimize the intermolecular and the molecule–substrate interactions. The adsorption energy Eads is calculated as
| Eads = [Ecomplex − (Eslab + Emols)]/nmol | (1) |
3. Results and discussion
3.1. Experiment
First, the coverage dependent adsorption of pyrphyrin on Au(111) for coverages up to one monolayer was explored. The Pyr molecules self-assemble on the Au substrate, and two phases of different coverage and long-range molecular ordering were identified and characterized by LEED and STM.34 The present study uses the denser phase with a nominal coverage of 1 ML as a starting material before Co deposition. Every molecule covers an area associated with 17.5 Au atoms of the reconstructed Au(111) top layer. Fig. 1(c) shows the corresponding LEED pattern and the superstructure matrices deduced for the two observed rotational domains. Fig. 2(a) represents an STM image of one of these rotational domains with hexagonally-ordered Pyr molecules on top of the preserved herringbone reconstruction of the Au(111) surface. | ||
| Fig. 2 STM images of (a) a monolayer of Pyr molecules on Au(111) and (b, c) after the additional deposition of Co and a subsequent annealing at 523 K. (a) The Pyr molecules self-assemble in a hexagonally-ordered molecular layer on top of the preserved herringbone reconstruction of the Au(111) surface. The free-base molecules can be resolved as donut-shaped configurations (A) with a weak indication of two-fold symmetry as shown in the inset of (a). (b) The molecular contrast is changed clearly by the deposition of Co and the subsequent annealing. Approximately 90% of the molecules are resolved as bright grey dots and interpreted as CoPyr complexes (B). Non-metalated Pyr molecules appear darker and with a central depression (A). Defects appearing as black and white dots are assigned as missing Pyr molecules (C) and metallic cobalt clusters (D), respectively. (c) The preparation procedure and the metalation of the Pyr molecules obviously does not affect the underlying Au(111) herringbone reconstruction. Tunneling parameters: (a) 45 × 45 nm2, −1.1 V, 0.6 nA; inset 4 × 4 nm2, +0.8 V, 0.5 nA; (b) 45 × 45 nm2, −0.8 V, 0.5 nA; inset 4 × 4 nm2, −0.8 V, 0.5 nA; (c) 115 × 115 nm2, −0.8 V, 0.5 nA. | ||
The subsequent evaporation of Co with the sample held at 293 K was terminated at a Co coverage of 5% of 1 ML, where 1 ML is defined as one Co atom per surface Au atom. Finally the surface was annealed at 523 K for 12 hours. After this procedure a pronounced change of the long-range molecular ordering in the LEED pattern [Fig. 1(d)] occurs. Instead of the former two rotational domains, a single hexagonal molecular unit cell is observed, the vectors of which are well aligned with the Au high-symmetry axes. The area per molecule corresponds to 18.7 Au atoms, i.e., slightly larger than the starting Pyr superstructure. The change of the long-range molecular ordering is related to the evaporated Co: annealing the pure Pyr ligand in the same way does not induce any changes.
Fig. 2(b) and (c) show STM images of the annealed surface. On the large scale image [Fig. 2(c)] one still observes a hexagonally-ordered molecular layer on top of the preserved herringbone reconstruction of the Au(111) surface. The predominant molecular species and three kinds of defects in the molecular layer can be distinguished more easily in the smaller scale image of Fig. 2(b). While the majority of molecules are imaged as spots of relatively uniform and intermediate brightness (labelled as B), dark spots (C) mark point defects within the molecular lattice. Very bright spots (D) appear to be small remnant Co clusters resulting from the metal deposition (see below). Finally, there are darker grey spots (A) that are significantly less dark than the point defects (C). We identify them as non-metalated Pyr molecules. In a pristine, non-metalated monolayer the Pyr molecules can be imaged with submolecular resolution even at room temperature, where they appear donut-shaped with a weak indication of two-fold symmetry as shown in the inset of Fig. 2(a). This shape can just barely be discerned in the inset of Fig. 2(b) where the two darker grey spots (A) show a slight depression in their center, while the brighter molecules (B) do not. The interpretation of the images in Fig. 2(b) and (c) is thus that the predominant brighter spots, representing ≈90% of the molecular lattice, are CoPyr complexes, with Co atoms occupying the central, four-fold nitrogen-coordinated site. This assignment is further corroborated below by means of XPS measurements and DFT calculations.
The annealing step is important for reaching an almost complete metalation of the molecular layer, as it is illustrated in the sequence of STM images shown in Fig. 3. After the metal deposition at a sample temperature of 323 K one observes a number of irregular large islands (E) that can be attributed to clusters of metallic cobalt [Fig. 3(a) and (b)]. The clusters exhibit fuzzy boundaries perpendicular to the scanning direction which indicates either a high mobility of the Co atoms along the cluster boundaries or a strong tip–cluster interaction. Moreover, they appear to align on a lattice that reflects the periodicity of the elbows of the Au(111) herringbone reconstruction. This is reminiscent of the formation of ordered cluster arrays on the clean Au(111) surface, where the dislocations in the surface lattice at the elbows serve as nucleation sites.35–37 On the basis of these images it cannot be established whether the Co clusters are situated on top of the molecular lattice or underneath, but the periodic order of the clusters makes the latter scenario more likely.
 | ||
| Fig. 3 STM images covering (a) 50 × 50 nm2, (b)–(d) 25 × 25 nm2 and (insets) 4 × 4 nm2, recorded after Co evaporation on a monolayer Pyr/Au(111) surface at a sample temperature of 323 K (a, b), and after successive annealing steps to 373 K for 1 hour (c), and to 423 K for 14 hours (d). Labels A, B and D are defined as in Fig. 2; E marks Co clusters found after evaporation at 323 K, while features F represent molecules in an intermediate metalated state that is observed prior to the annealing step. Tunneling parameters: (a) −1.0 V, 1.0 nA; (b) +0.8 V, 0.5 nA; inset −1.0 V, 0.5 nA; (c) −0.8 V, 0.5 nA; inset −0.8 V, 0.5 nA; (d) −0.8 V, 1.0 nA. | ||
In between the clusters, the Pyr molecules can be imaged quite nicely [Fig. 3(b)]. At this stage most of them have maintained their donut shape (A), but some of them appear brighter exhibiting two distinct lobes (F). Successive annealing steps to 373 K and to 423 K increase the number of brighter molecules, but their contrast has changed from the two lobes to a single central protrusion (B). Concomitantly, the irregular Co clusters disappear [Fig. 3(c) and (d)]. The metalation thus appears to be temperature driven. After the Co evaporation, one part of the Co atoms assembles in the observed clusters whereas another part is already ligated to the cores of Pyr molecules. Due to the annealing, on the one hand Co atoms dissociate from the initially formed clusters and diffuse along the surface, before they are captured by Pyr molecules. On the other hand, the changing submolecular contrast of the Co-ligated Pyr species [cf. insets of Fig. 3(b) and (c)] indicates the existence of an intermediate state (F) prior to the annealing.
This scenario is well supported by the XPS data. Fig. 4(a) shows the Co 2p3/2 spectrum of the metalated pyrphyrin monolayer, obtained after annealing at 523 K, corresponding to the STM images of Fig. 2(b) and (c). One observes a broad structure of peaks with two maxima at binding energies of EB = 780.3 eV and 777.7 eV. While the latter component was also found after evaporation of cobalt onto the clean Au(111) surface [cf. Fig. S1(c) in the ESI†] and therefore can be attributed to metallic Co, the peak structure at higher binding energies has to be related to Co atoms ligated to the cores of Pyr molecules. However, the metallic contribution appears rather high in view of the STM images of Fig. 2(b) and (c) which were measured on the very same preparation showing only very little remnant Co on top of the molecular layer. This observation can be reconciled by referring to a recent study of the related systems Co-octaethylporphyrin (CoOEP) and Co-tetraphenylporphyrin (CoTPP) on Au(111),38 where a substantial metallic-like component was observed in the monolayer case, even though the Co-complexed ligands had been deposited directly onto the surface by evaporation. It was argued that the reconstructed Au(111) surface has transition regions between the fcc and hcp stacked parts of the herringbone unit cell which are more reactive. These regions therefore might interact more strongly with the Co atoms in the adsorbed porphyrin derivatives, leading to this metallic-like component shifted by about 1.5 eV to lower binding energies in the Co 2p3/2 spectrum.
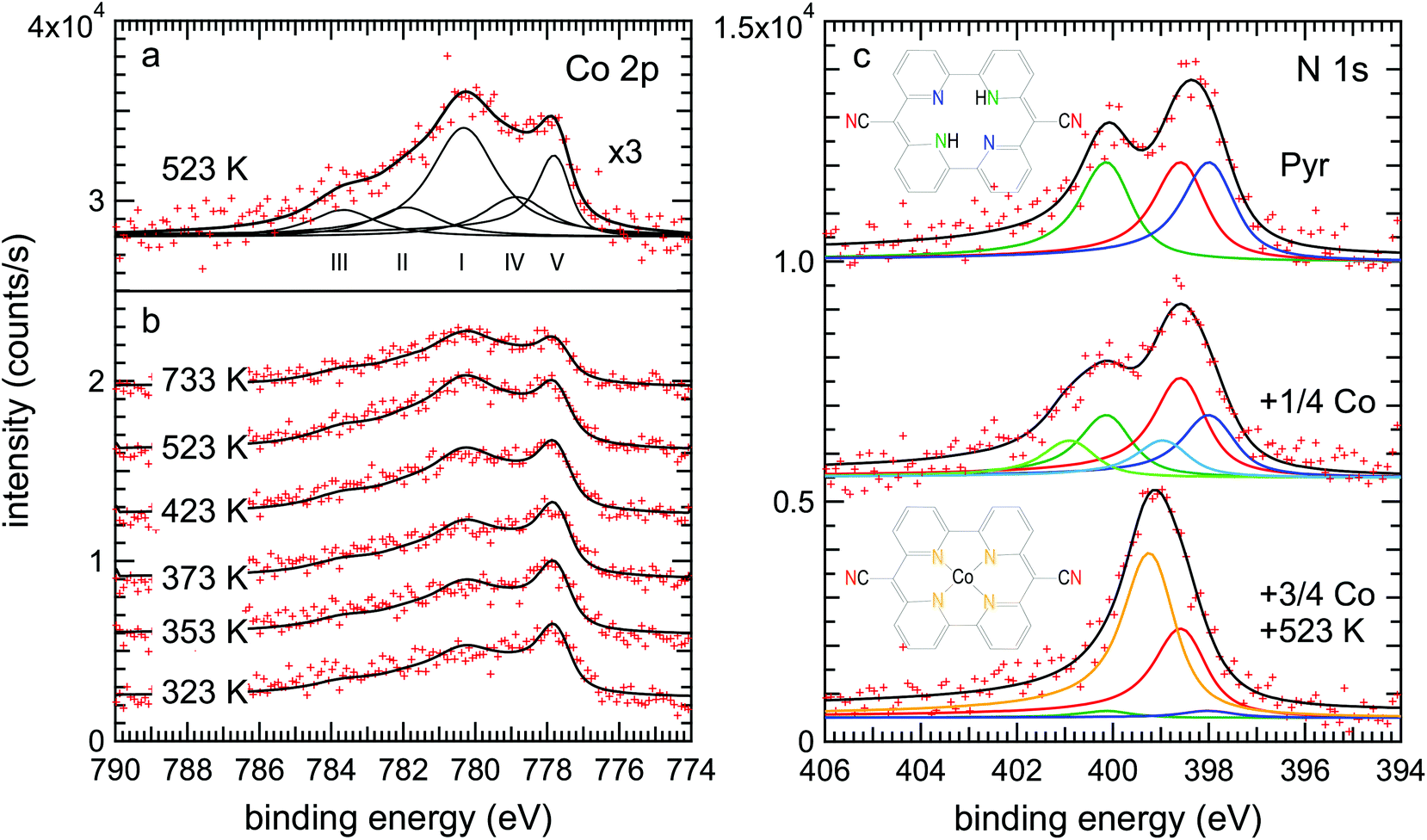 | ||
| Fig. 4 XPS data and corresponding fits of Co 2p3/2 and N 1s spectra before and after Co evaporation on a monolayer Pyr/Au(111) surface and after subsequent annealing. (a) Co 2p3/2 spectrum measured after a 12 hour annealing at 523 K fitted with three main components at binding energies of EB = 780.3 eV (I), 778.9 eV (IV) and 777.7 eV (V) and two additional satellites at 781.9 eV (II) and 783.7 eV (III). (b) Evolution of the Co 2p3/2 spectra as a function of the annealing temperature fitted with the same five components. (c) N 1s spectra before the Co evaporation (top), after partial Co evaporation (a quarter of the total deposition) without annealing (middle) and after the complemented Co evaporation (resulting in a total coverage of 0.043 ML) with subsequent annealing to 523 K (bottom). The top spectrum has been fitted with three peaks representing the three different species of nitrogen atoms in the pyrphyrin molecule: the cyano–N (398.5 eV, red), the pyridinic–N (400.1 eV, green) and the iminic–N (397.9 eV, blue). After Co metalation in the bottom spectrum, the pyridinic–N and iminic–N peaks are both greatly reduced and replaced by a new Co–N peak (399.2 eV, orange). Co evaporation without annealing produces a distinct shift of the middle spectrum to higher binding energies that can be accounted for by adding two additional components at 400.8 eV (light-green) and 398.9 eV (light-blue). | ||
In an attempt to further quantify the relative contributions from metallic Co clusters and CoPyr, we have fitted the spectrum from the 523 K annealed sample [Fig. 4(a)] with five components. The three components (I–III) represent the emission from Co2+ with its associated satellite structure38 resulting in particular from multiplet splitting.39 Following the assignment of Massimi et al., peaks (I) and (II) can be associated with the multiplet, while peak (III) is an additional satellite most likely due to shakeup processes.39,40 The multiplet has been attributed to the open-shell character of the Co2+ ion.38,39,41 Several approaches for the deconvolution of complex multiplet lines have been used according to different conceivable coupling mechanisms.39,40,42 However, the comparison of different evaluation methods for the multiplet structure observed for Fe2+ tetrapyrrole complexes on Au(111) revealed that an explicit attribution is ambiguous.40
For pragmatic reasons, the deconvolution of our Co 2p3/2 spectra was therefore aligned to the evaluation of Bai et al.38 In addition to the components (I–III), a fourth component (IV) is introduced which is shifted by ≈1.5 eV from the main Co2+ peak to lower binding energy due to a stronger interaction of the CoPyr with the substrate.38 Any satellite or multiplet structure of the component (IV) is neglected as in ref. 38. It has been argued that the stronger substrate interaction of the respective molecular complexes reduces the total spin at their metal centers which would also reduce the multiplet splitting (cf. ESI of ref. 42). While the four components (I–IV) represent the Co species incorporated within Pyr molecules adsorbed at different surface sites,38 an additional fifth component (V) is related to the metallic Co species present either in the remaining Co clusters (feature D) or incorporated into the Au(111) surface layer beneath the molecules. According to Bai et al.38 the four components attributed to the Co atoms ligated to the cores of Pyr molecules were fitted by symmetric functions while for the metallic component of the Co clusters an asymmetric DS function was used (cf. Experimental). The ratio of the components (IV) to (I–III) was fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.2 according to the reported values of 1:4.7 for CoTPP and 1:3.8 for CoOEP monolayers.38 With these assumptions the ratio of incorporated Co (I–IV) to free metallic Co (V) is quantified to 4.4:1. Note that due to the broad peak structure and the overall noise, the error bars of the individual Co peak intensities are about ±5% of the total Co 2p3/2 intensity. Accordingly, the uncertainty of the discussed (I–IV) to (V) ratio is quite large and values might vary between 6.4:1 and 3.3:1.
:4.2 according to the reported values of 1:4.7 for CoTPP and 1:3.8 for CoOEP monolayers.38 With these assumptions the ratio of incorporated Co (I–IV) to free metallic Co (V) is quantified to 4.4:1. Note that due to the broad peak structure and the overall noise, the error bars of the individual Co peak intensities are about ±5% of the total Co 2p3/2 intensity. Accordingly, the uncertainty of the discussed (I–IV) to (V) ratio is quite large and values might vary between 6.4:1 and 3.3:1.
Our assignment of the peaks (I–V) is in agreement with the evolution of the Co 2p3/2 spectrum as a function of the annealing temperature shown in Fig. 4(b) (see the ESI and Fig. S1 and S2† for additional information). The Co 2p3/2 spectrum obtained right after the Co deposition at 323 K exhibits a dominant metallic component (V). With increasing annealing temperature, the intensity of this metallic component is decreasing, in particular for temperatures above 373 K, thus reflecting the disappearance of the Co clusters observed by STM (Fig. 3). Simultaneously, the intensities of the components (I–IV) attributed to Co ligated to the Pyr centers are increasing continuously up to annealing temperatures of 523 K. The observed concomitant increase of the total intensity of the Co 2p3/2 spectra (cf. Fig. S2 in the ESI†) is most likely related to the decreasing attenuation of Co atoms dissolving from the clusters and/or ascending from below the molecular layer during metalation. After annealing at 723 K the ratio of the peaks (I–IV) to the peak (V) remains almost constant, but the overall Co 2p3/2 intensity decreases considerably. This decrease indicates the decomposition of some CoPyr molecules and the partial disappearance of Co.
The N 1s spectra shown in Fig. 4(c) represent direct probes of the local chemistry within the pyrphyrin molecules, and they allow a more accurate evaluation of the relative number of metalated molecules. The spectrum of Pyr/Au(111) (top curve) shows two peaks. In order to account for the three types of chemical environments of N atoms within the molecule, pyridinic (–NH–), iminic (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) and cyano (–C
N–) and cyano (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) nitrogen, curve fitting was done with three identical DS functions convoluted with Gaussians. The spectrum can be well fitted with these three components. We assign the high binding energy component at 400.1 eV to the pyridinic nitrogen, following the interpretation by Gottfried et al. for the case of tetraphenylporphyrin (TPP) on Ag(111), where the related pyrrolic nitrogen was also found at 400.1 eV.15 The two low binding energy peaks at 398.5 eV and 397.9 eV are assigned to cyano and iminic nitrogen, respectively.
N) nitrogen, curve fitting was done with three identical DS functions convoluted with Gaussians. The spectrum can be well fitted with these three components. We assign the high binding energy component at 400.1 eV to the pyridinic nitrogen, following the interpretation by Gottfried et al. for the case of tetraphenylporphyrin (TPP) on Ag(111), where the related pyrrolic nitrogen was also found at 400.1 eV.15 The two low binding energy peaks at 398.5 eV and 397.9 eV are assigned to cyano and iminic nitrogen, respectively.
After Co evaporation and subsequent annealing of the sample (bottom curve), the spectrum seemingly collapses into a single peak with a slight shoulder on the low binding energy side. It can be well fitted with four peaks, a major component at 399.2 eV and a smaller one at the cyano nitrogen position forming the shoulder, as well as two small remnant pyridinic and iminic components, reflecting a few unmetalated molecules. The binding energy of the major Co–N component compares well with the binding energy of 398.8 eV reported for CoTPP on Ag(111).15 Some constraints were made in order to sensibly fit a single broad maximum by four contributions. While the binding energy of the new Co-coordinated nitrogen component was left free, its intensity was constrained to be equal to the intensity loss of the decreasing pyridinic and iminic components. The cyano nitrogen intensity and all other parameters were fixed at the fit results of the unmetalated pyrphyrin. The resulting intensity ratios of the pyridinic, cyano, iminic and the Co-coordinated nitrogen are 0.08:1:0.08:1.84. This corresponds to a ratio of metalated to unmetalated Pyr molecules of 0.92:0.08 consistent with the STM measurements of Fig. 2(b). Note that the error bars of the N 1s peak intensities are about ±6% of their intensity in the case of the pristine Pyr. Thus, the error bars are of the same order as the intensities observed for the pyridinic and iminic peaks after the Co-metalation and one should not over interpret their absolute values. Overall, the observed changes in the N 1s spectrum after the annealing at 523 K are in excellent agreement with the incorporation of Co into the molecular cores. Only upon further annealing at 723 K we observed some rectangular network-like arrangements of molecules by STM, which might indicate the formation of Co coordination networks via cyano groups of neighboring molecules.43–45
Co deposition without annealing yields a distinct shift of the N 1s spectra to higher binding energies (middle spectrum). However, a deconvolution into the four already discussed components does not lead to a satisfactory agreement of the fit and the experimental data. As the STM experiments also show a clear difference between Co related species before and after the annealing, the existence of an intermediate state is strongly indicated. This intermediate might be a complex in which the Co atom is already coordinated by the Pyr molecule while the two hydrogen atoms have not yet desorbed (CoPyr-2H). Similar intermediate states were reported in studies on Zn and Fe metalation of TPP on Ag(111) and Ag(100) by means of XPS and DFT.46–48 Two main activation barriers were identified: one is the initial formation of a metal–porphyrin complex, which involves the breaking of a metal-to-substrate bond when the metal atom is incorporated into the fourfold N-coordinated site of the molecule.47 This step is activated when the sample is warmed up to 300 K after initial Zn deposition at 115 K. The second barrier is due to an isomerization reaction where one hydrogen atom is transferred from a pyrrolic nitrogen to the metal.47 It is this second barrier that is rate limiting for the release of hydrogen molecules, and which is higher in the case of Zn compared to the transition metals Fe, Co, Ni and Cu. Therefore, an annealing step to 550 K was found necessary to form the Zn–TPP complex.47 More recently, an intermediate state was identified for Cu metalation of tetrapyridylporphyrin (TPyP) on Au(111) by STM in combination with XPS and DFT.45 Similar to our findings, the reported intermediate state was imaged much brighter than the final configuration by STM.
Thus, in order to account for this probable CoPyr-2H intermediate, two additional nitrogen components (represented by the light-blue and light-green curves) were introduced in the N 1s fitting model. It was assumed again that the Co incorporation influences only the two core nitrogen species while the cyano nitrogens are unaffected. Furthermore, it was assumed that the above-named Co–N component is not yet existent as only a single type of Co-related species is found after Co deposition by STM [Fig. 3(b)]. The resulting fit can describe the experimental data very well. The intensities of the pyridinic, cyano, iminic and the two intermediate-state nitrogen components are 0.64:1:0.64:0.36:0.36. This corresponds to a ratio of intermediate species to pristine Pyr molecules of 0.36:0.64. This ratio is slightly higher than expected, as the Co deposition for this special preparation was interrupted after 25% of the deposition time used for the complete Co metalation. However, this deviation might be explained by possible rate fluctuations of the Co evaporator and the overall error of the XPS evaluation.
The C 1s spectra (cf. Fig. S3 in the ESI†) show two peaks at binding energies of 286.1 eV and 284.7 eV. From the observed intensity ratios of about 10:14, the two peaks can be attributed to C atoms with and without a neighboring N atom, respectively.22,23 As Co is not coordinated to any C atom, consistently, no pronounced changes are observed during metalation upon annealing up to temperatures of 523 K. Annealing at 723 K leads to spectral changes that suggest some rearrangements within the molecules in accordance with the observed decrease of the Co 2p intensity at high temperature.
Quantitatively, the pyrphyrin coverage corresponding to the preparation shown in Fig. 2 was 0.73 ± 0.04 ML as evaluated from C 1s and N 1s relative to the Au 4d5/2 peak intensities (including also inelastic attenuation of the photoelectrons of the substrate by the molecular layer). This value indicates a co-existence of the denser molecular phase and uncovered clean Au areas which is consistent with the observation of clean areas by STM. The total Co coverage for this preparation was 4.3 ± 0.4% of a ML as calculated from the Co 2p3/2 and Au 4d5/2 peak areas. By taking into account the total Pyr coverage of 0.73 ML, the evaluated ratio of metalated to unmetalated Pyr molecules of 0.92:0.08 as well as the density of Pyr molecules relative to the Au substrate atoms, one can calculate the coverage of Co atoms which are coordinated in the Pyr molecules to 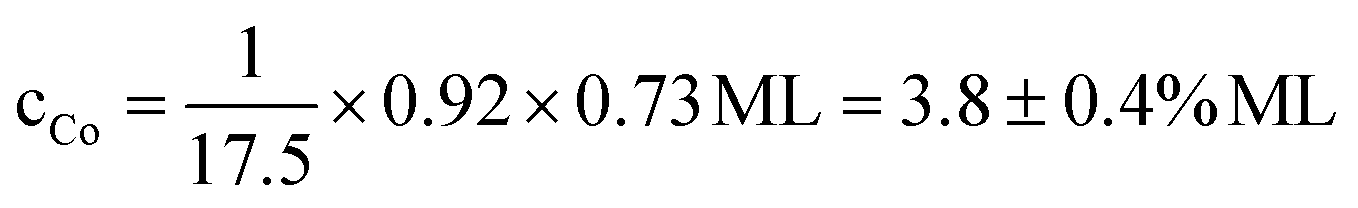 . That means that the exceeding Co leading to additional metallic clusters should be around 0.5% of a ML corresponding to about 12% of the total Co coverage. This is in good agreement with the results of the Co 2p3/2 fitting model which yields the free metallic Co at about 18 ± 5% of the total Co coverage.
. That means that the exceeding Co leading to additional metallic clusters should be around 0.5% of a ML corresponding to about 12% of the total Co coverage. This is in good agreement with the results of the Co 2p3/2 fitting model which yields the free metallic Co at about 18 ± 5% of the total Co coverage.
Calculation
The interaction between CoPyr and the reconstructed Au(111) surface has been studied in detail by means of DFT simulations. Most of the recent computational studies related to the adsorption of organic molecules on Au(111) use simpler ideal surfaces to limit the system size.49,50 However, modeling the reconstructed Au(111) with simulation cells containing more than 600 atoms is essential to take into account the modulation of the electronic structure over the different domains (fcc, transition, hcp).Different possible adsorption modes have been tested by rotating and displacing the molecule over the reconstructed surface. The results show that the interaction is maximized when both the cyano N (NCN) atoms and Co are close to the surface Au atoms. This condition is satisfied when the molecular cyano axis is rotated by 60° with respect to the [110] direction of the Au lattice, and NCN and Co are close to atop sites, as depicted in Fig. 5(a) and (b). The molecule is slightly bent along the cyano axis and the distance between NCN and the closest Au atom is 2.48 Å. The Au atom right underneath the Co atom is displaced outward by ≈0.2 Å, such that the Au–Co distance becomes 2.74 Å. This configuration corresponds to an adsorption energy of −4.16 eV when the molecule is placed above the fcc area of the herringbone reconstruction. Minimal differences are observed placing the molecule on the hcp or transition region. These are determined by slight variations in the interatomic distances among the surface Au atoms. Also pure Pyr adsorbs preferentially in the same orientation, being slightly bent to favor the NCN–Au interaction. However, the adsorption strength is more than half an eV weaker, indicating that Co contributes significantly to the bonding.
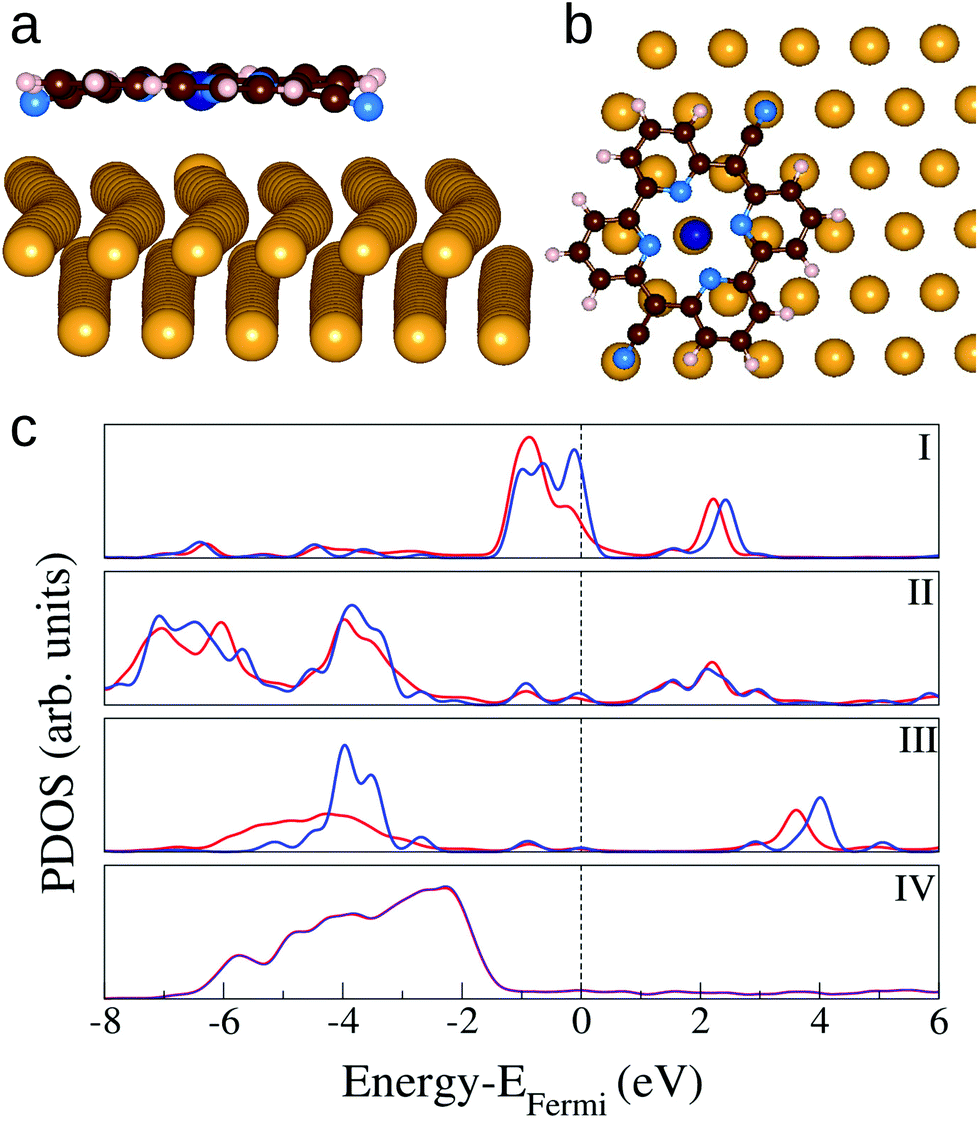 | ||
| Fig. 5 Side view (a) and top view (b) of the optimized structure of CoPyr on the reconstructed Au(111) surface. The molecular axis is at an angle of 60° with respect to [110] Au axis. For clarity only two Au layers and only the surface layer are shown in (a) and (b), respectively. Color code: dark blue: Co, blue: N, brown: C, pink: H, and yellow: Au. (c) PDOS on CoPyr atoms and Au surface atoms. (I) Co 3d; (II) 2p states of central nitrogen atoms; (III) 2p states of cyano nitrogen atoms; (IV) 5d states of Au surface atoms. Red and blue solid lines correspond to adsorbed CoPyr on Au and isolated CoPyr and bare Au, respectively. | ||
Fig. 5(c) shows the density of states projected on the different species of the complex in comparison with the same PDOS calculated for the isolated molecule and the bare surface. The main effects of the adsorption are the rearrangement of the charge distribution on Co involving the highest occupied molecular states. The spin polarisation (not shown) is lifted, some states near the Fermi energy are shifted to lower energy in the complex. The molecular states on the cyano N atoms are hybridized with the surface d-band. On the other hand, the charge transfer to the metal appears to be minimal and it affects only the Au atoms beneath Co and NCN. The corresponding changes in the PDOS of Au are marginal and become visible only by projection on the single atoms (not shown). These relatively small electronic structure changes confirm that the adsorption is dominated by dispersion forces. When CoPyr is adsorbed on different regions of the reconstructed surface the electronic structure rearrangements do not change significantly.51 The calculated PDOS obtained for CoPyr adsorbed on the hcp and transition regions are reported in the ESI.†
Fig. 6 displays the simulated STM images obtained for Pyr, CoPyr-2H, and CoPyr adsorbed on the reconstructed Au(111). The CoPyr-2H complex is considered as an intermediate state, where Co is partially inserted at the molecular center, and the two H atoms are still bound to the N atoms, but pointing out of the molecular plane (see the ESI†). We verified that this adsorbed complex represents also a minimum on the potential energy surface. We compared the three competitive complexes on Au(111), i.e., co-adsorbed Pyr and Co atom, CoPyr-2H, and adsorbed CoPyr with molecular hydrogen in the gas phase. It turns out that the adsorbed CoPyr-2H complex is more stable than co-adsorbed but separated Co and Pyr by more than 1.3 eV. Moreover, the final state, where CoPyr is adsorbed and H2 is released, is even more stable by 2.2 eV. These results thus confirm the experimentally observed progression of metalation by annealing. The simulated STM image of the CoPyr-2H complex shown in Fig. 6(b) exhibits higher electron density at both the molecular ligand and the center compared to the CoPyr complex [Fig. 6(c)]. Thus, it seems reasonable that the brighter features (F) discussed in Fig. 3(b) are associated with the CoPyr-2H intermediate state.
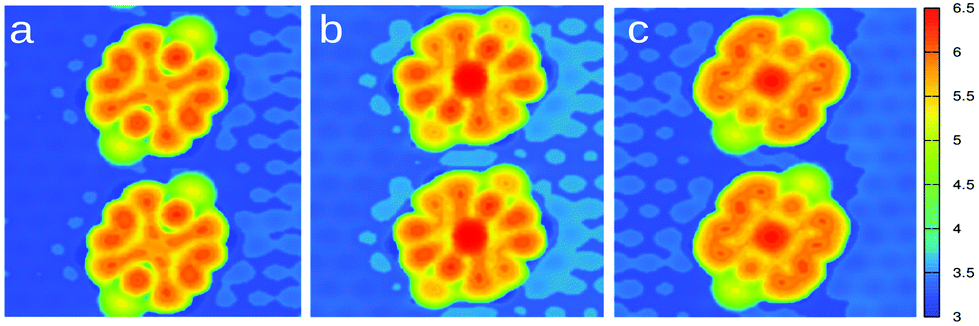 | ||
| Fig. 6 Simulated STM images of Pyr (a), CoPyr-2H (b), and CoPyr (c) adsorbed on the reconstructed Au surface. The image has been generated with a bias potential of −1.0 eV, selecting the iso-density surface corresponding to 10−5 e Å−3. | ||
As discussed above, upon Pyr metalation the molecular lattice undergoes some rearrangements. Our results suggest that the molecule–substrate interaction favors molecules that have Co and NCN at atop sites. In order to better understand this process, the molecule–molecule interactions need to be investigated and compared to the molecule–substrate ones. From the LEED measurements we know the lattice vectors of the molecular superstructure, but we have no clear indication on the relative orientation of the molecules. We selected a few trial structures for the molecular network, formed by four molecules arranged according to the hexagonal lattice determined by LEED. The models differ by the relative positions and orientations of the molecules. They are positioned in a way that avoids too close a contact with neighboring molecules, keeping the cyano groups at sufficient distance, but favoring the hydrogen bonding between the NCN and the neighboring H atoms. Obviously these conditions prevent that all molecules are at the optimal adsorption geometry. These simulations were performed with an unreconstructed Au(111) model, which can be obtained with smaller simulation cells, a 9 × 9 Au(111) slab with four layers, thus reducing the computational effort. By optimizing the structure, some adjustments are observed, where the molecules slightly shift and rotate, until, over all, the molecule–substrate binding is also maximized. Among those tested, the best model is the one represented in Fig. 7. In this geometry, the molecules rotate and shift to maximize attractive interactions between Co–Au and NCN–Au pairs as well as to increase hydrogen-bonding type interactions between the individual molecules. The NCN are still slightly bent towards the Au and the distances among the molecular centers are slightly changed due to the shifts. The adsorption energy per molecule in the monolayer geometry on the ideal Au(111) surface amounts to −3.98 eV. This energy is given by two contributions: the molecule–metal interaction which is −3.56 eV per molecule, and the molecule–molecule interaction, −0.42 eV per molecule, which is obtained by subtracting the energy of the single molecule from the total energy of the same network computed as free standing. We conclude that the molecule–substrate interactions also play a role in determining the monolayer structure.
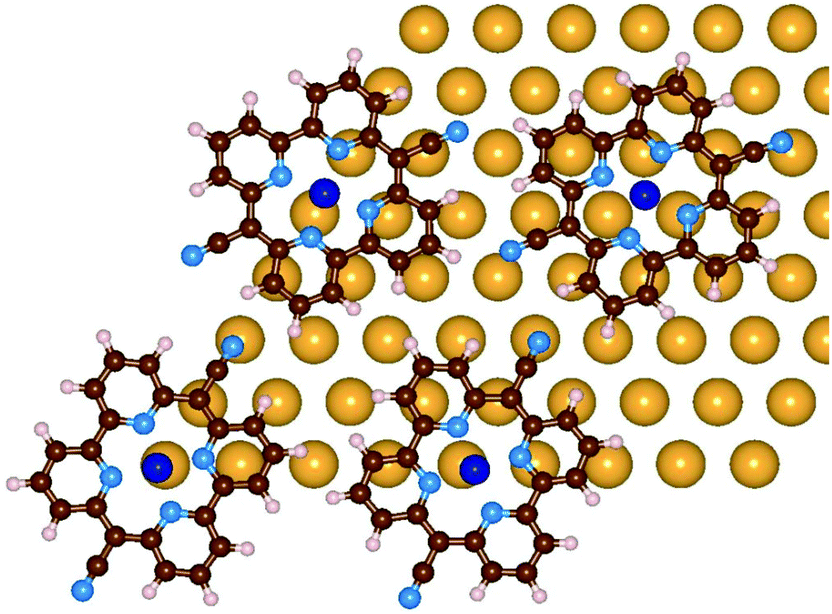 | ||
| Fig. 7 Optimized configuration of the CoPyr monolayer on the unreconstructed Au(111) surface. Only the surface Au layer is shown for clarity. Color code: see the previous figure. | ||
Finally, we have carried out a few simulations to shed light on the metalation process. We consider, in this case, the Co atom and the Pyr monomer simultaneously adsorbed on Au(111) at different distances, and we want to address the diffusion of Co towards the molecule and the substitution at the molecular center. In order to simplify the problem, we have moved the Co atom to different sites and investigated the properties of the intermediate geometries. A Co adatom is first adsorbed alone at different sites on the reconstructed surface, showing that the most stable state (by ≈0.1 eV) is at a hollow site of the fcc domain. Then, starting from the optimized Pyr adsorbed on the fcc region, we place Co once above the molecule and once in the intermediate space between Pyr and Au(111). It turns out that when Co is initially placed above Pyr, it relaxes and coordinates with the Pyr core, leading to a spontaneous insertion. In contrast, when Co is placed underneath the molecule, it relaxes on the surface, without coordinating to the Pyr center. These results suggest that metalation of Pyr from the top is a downhill process, whereas, since Co adatoms on the Au(111) surface form also a stable state, the process from underneath needs to be activated, since it occurs over an energy barrier.
In many related porphyrin monolayer systems the metalation reaction occurs rapidly and spontaneously upon metal deposition at 300 K. Cases in point are Co and Fe metalation of TPP,15,52 and Fe in TPyP,16 all on Ag(111), as well as Ni incorporation in TPP on Au(111).53 Interestingly, the peripheral substituents (e.g. phenyl- and pyridyl-groups) at the porphyrin core and with this the core's distance to the substrate do not have a significant influence on the metalation reaction.19 Instead, it seems to be the metal atoms that determine the activation barriers.47 Pyrphyrin seems to be different from these porphyrins: while porphyrin exhibits a rapid metalation by Co at 300 K,15 pyrphyrin metalation seems to have a higher activation barrier. On the one hand, one might presume an influence of the smaller ligand core size on the higher activation barrier of the pyrphyrins as the distance of the nitrogen atoms is 3.77 Å and 3.94 Å for Co-pyrphyrin and Co-porphyrin, respectively.14,47 On the other hand, recent results for the metalation of TPyP on Au(111) indicate an increased activation barrier compared to Ag(111).54 Thus, an influence of the substrate seems to be reasonable as well. In order to decode the mechanisms, systematic metalation studies for pyrphyrin using different metals and substrates are indicated. In particular the changing long-range molecular ordering induced by the metalation has to be explored in more detail as – to the best of our knowledge – it has not been observed in the case of the metalation of porphyrins and phthalocyanines.
4. Conclusions
From these systematic experimental and theoretical studies the following reaction steps for the Co metalation of a monolayer of pyrphyrin molecules on Au(111) were established.(i) Metal deposition onto the Pyr layer at temperatures below 323 K leads on the one hand to an intermediate, Co-related Pyr complex which might be associated with a CoPyr-2H complex. This metalation of Pyr from the top is a downhill process with the CoPyr-2H complex representing a minimum on the potential energy surface. On the other hand, Co deposition leads to the formation of Co clusters on the surface. Thus, some of the Co atoms arriving from the vapor phase diffuse along the surface, either on top of the molecular layer or underneath. Due to the observed nucleation at the elbow sites of the Au(111) herringbone reconstruction it is likely that the clusters are located underneath the pyrphyrin layer, directly on the Au(111) surface.
(ii) Upon annealing, Co atoms dissolve from the clusters and diffuse along the surface, reacting along the way with Pyr molecules. The diffusion likely takes place directly on the Au(111) surface with metalation occurring from underneath the molecules. Our calculations indicate an energy barrier for the metalation process from underneath consistent with our experimental findings. A similar behavior has recently been observed for the copper metalation of 5,15-diphenylporphyrin on Au(111).55 Moreover, in the self-metalation of porphyrin on Cu(111),56 incorporation of Cu atoms from the substrate into the molecules was observed at temperature starting from 373 K, while at room temperature the molecular layer consisted of unmetalated ligands.
(iii) In addition to the Co cluster dissolution at elevated temperatures, the complexation reaction proceeds from the CoPyr-2H intermediate to the final CoPyr configuration by the release of molecular hydrogen.
In conclusion, the adsorption and the in situ metalation of pyrphyrin on a metal substrate exhibit an overall astonishing resemblance to the reported porphyrin and phthalocyanine systems.19 However, there are distinct differences like the changing molecular self-assembly during the metalation or the activation barrier for the metalation process from underneath. Finally, one has to point out that the cyano groups can be exchanged to other (functional) substituents or anchor groups. Thus, the pyrphyrins provide a completely new class of molecules and the exploration of their properties and their adsorption behavior on solid substrates has just begun.
Acknowledgements
This project has been financed under the University Research Priority Program LightChEC of the University of Zurich, the FP7 Marie Curie COFUND scheme of the European Commission, and the National Centres of Competence in Research–Materials Revolution: Computational Design and Discovery of Novel Materials (NCCR–MARVEL). We acknowledge computing resources from the Swiss National Supercomputer Centre (CSCS) under the project ID s425 and expert technical support by Thomas Kälin.References
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- E. Joliat, S. Schnidrig, B. Probst, C. Bachmann, B. Spingler, K. K. Baldridge, F. von Rohr, A. Schilling and R. Alberto, Dalton Trans., 2016, 45, 1737–1745 RSC.
- S. Ogawa, R. Narushima and Y. Arai, J. Am. Chem. Soc., 1984, 106, 5760–5762 CrossRef CAS.
- S. Ogawa, T. Uchida, T. Uchiya, T. Hirano, M. Saburi and Y. Uchida, J. Chem. Soc., Perkin Trans. 1, 1990, 1649–1653 RSC.
- M. V. Martinez-Diaz, G. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090–7108 RSC.
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759–792 CrossRef CAS.
- C. Wang, J. Li, G. Mele, M.-Y. Duan, X.-F. Lü, L. Palmisano, G. Vasapollo and F.-X. Zhang, Dyes Pigm., 2010, 84, 183–189 CrossRef CAS.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- A. Scheybal, T. Ramsvik, R. Bertschinger, M. Putero, F. Nolting and T. A. Jung, Chem. Phys. Lett., 2005, 411, 214–220 CrossRef CAS.
- H. Wende, M. Bernien, J. Luo, C. Sorg, N. Ponpandian, J. Kurde, J. Miguel, M. Piantek, X. Xu, P. Eckhold, W. Kuch, K. Baberschke, P. M. Panchmatia, B. Sanyal, P. M. Oppeneer and O. Eriksson, Nat. Mater., 2007, 6, 516–520 CrossRef CAS PubMed.
- C. Iacovita, M. V. Rastei, B. W. Heinrich, T. Brumme, J. Kortus, L. Limot and J. P. Bucher, Phys. Rev. Lett., 2008, 101, 116602 CrossRef CAS PubMed.
- B. Hulsken, R. Van Hameren, J. W. Gerritsen, T. Khoury, P. Thordarson, M. J. Crossley, A. E. Rowan, R. J. M. Nolte, J. A. A. W. Elemans and S. Speller, Nat. Nanotechnol., 2007, 2, 285–289 CrossRef CAS PubMed.
- B. A. Friesen, A. Bhattarai, U. Mazur and K. W. Hipps, J. Am. Chem. Soc., 2012, 134, 14897–14904 CrossRef CAS PubMed.
- Y. Gurdal, S. Luber, J. Hutter and M. Iannuzzi, Phys. Chem. Chem. Phys., 2015, 17, 22846–22854 RSC.
- J. M. Gottfried, K. Flechtner, A. Kretschmann, T. Lukasczyk and H.-P. Steinrück, J. Am. Chem. Soc., 2006, 128, 5644–5645 CrossRef CAS PubMed.
- W. Auwärter, A. Weber-Bargioni, S. Brink, A. Riemann, A. Schiffrin, M. Ruben and J. V. Barth, ChemPhysChem, 2007, 8, 250–254 CrossRef PubMed.
- Y. Bai, F. Buchner, M. T. Wendahl, I. Kellner, A. Bayer, H.-P. Steinrück, H. Marbach and J. M. Gottfried, J. Phys. Chem. C, 2008, 112, 6087–6092 CAS.
- G. Di Santo, C. Castellarin-Cudia, M. Fanetti, B. Taleatu, P. Borghetti, L. Sangaletti, L. Floreano, E. Magnano, F. Bondino and A. Goldoni, J. Phys. Chem. C, 2011, 115, 4155–4162 CAS.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- W.-D. Schneider and C. Laubschat, Phys. Rev. B: Condens. Matter, 1981, 23, 997–1005 CrossRef CAS.
- S. Doniach and M. Šunjić, J. Phys. C: Solid State Phys., 1970, 3, 285–291 CrossRef CAS.
- H. Peisert, M. Knupfer and J. Fink, Surf. Sci., 2002, 515, 491–498 CrossRef CAS.
- Y. Alfredsson, B. Brena, K. Nilson, J. Åhlund, L. Kjeldgaard, M. Nyberg, Y. Luo, N. Mårtensson, A. Sandell, C. Puglia and H. Siegbahn, J. Chem. Phys., 2005, 122, 214723 CrossRef CAS PubMed.
- C. Wöll, S. Chiang, R. J. Wilson and P. H. Lippel, Phys. Rev. B: Condens. Matter, 1989, 39, 7988–7991 CrossRef.
- J. V. Barth, H. Brune, G. Ertl and R. J. Behm, Phys. Rev. B: Condens. Matter, 1990, 42, 9307–9318 CrossRef CAS.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CrossRef CAS.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter, 1996, 54, 1703–1710 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- O. A. Vydrov and T. Van Voorhis, Phys. Rev. Lett., 2009, 103, 063004 CrossRef PubMed.
- R. Gaspari, C. A. Pignedoli, R. Fasel, M. Treier and D. Passerone, Phys. Rev. B: Condens. Matter, 2010, 82, 041408 CrossRef.
- F. Hanke and J. Björk, Phys. Rev. B: Condens. Matter, 2013, 87, 235422 CrossRef.
- J. Tersoff and D. R. Hamann, Phys. Rev. B: Condens. Matter, 1985, 31, 805–813 CrossRef CAS.
- G. Mette, Y. Gurdal, S. Schnidrig, B. Probst, M. Iannuzzi, J. Hutter, R. Alberto and J. Osterwalder, unpublished work.
- B. Voigtländer, G. Meyer and N. M. Amer, Phys. Rev. B: Condens. Matter, 1991, 44, 10354–10357 CrossRef.
- S. Padovani, F. Scheurer and J. P. Bucher, Europhys. Lett., 1999, 45, 327–333 CrossRef CAS.
- F. Grillo, H. Früchtl, S. M. Francis and N. V. Richardson, New J. Phys., 2011, 13, 013044 CrossRef.
- Y. Bai, M. Sekita, M. Schmid, T. Bischof, H.-P. Steinrück and J. M. Gottfried, Phys. Chem. Chem. Phys., 2010, 12, 4336–4344 RSC.
- L. Massimi, M. Angelucci, P. Gargiani, M. G. Betti, S. Montoro and C. Mariani, J. Chem. Phys., 2014, 140, 244704 CrossRef PubMed.
- M. Schmid, J. Zirzlmeier, H.-P. Steinrück and J. M. Gottfried, J. Phys. Chem. C, 2011, 115, 17028–17035 CAS.
- T. Lukasczyk, K. Flechtner, L. R. Merte, N. Jux, F. Maier, J. M. Gottfried and H.-P. Steinrück, J. Phys. Chem. C, 2007, 111, 3090–3098 CAS.
- C. Isvoranu, B. Wang, K. Schulte, E. Ataman, J. Knudsen, J. N. Andersen, M. L. Bocquet and J. Schnadt, J. Phys.: Condens. Matter, 2010, 22, 472002 CrossRef CAS PubMed.
- L.-A. Fendt, M. Stöhr, N. Wintjes, M. Enache, T. A. Jung and F. Diederich, Chem. – Eur. J., 2009, 15, 11139–11150 CrossRef CAS PubMed.
- N. Henningsen, R. Rurali, C. Limbach, R. Drost, J. I. Pascual and K. J. Franke, J. Phys. Chem. Lett., 2011, 2, 55–61 CrossRef CAS PubMed.
- Y. Li, J. Xiao, T. E. Shubina, M. Chen, Z. Shi, M. Schmid, H.-P. Steinrück, J. M. Gottfried and N. Lin, J. Am. Chem. Soc., 2012, 134, 6401–6408 CrossRef CAS PubMed.
- A. Kretschmann, M.-M. Walz, K. Flechtner, H.-P. Steinrück and J. M. Gottfried, Chem. Commun., 2007, 568–570 RSC.
- T. E. Shubina, H. Marbach, K. Flechtner, A. Kretschmann, N. Jux, F. Buchner, H.-P. Steinrück, T. Clark and J. M. Gottfried, J. Am. Chem. Soc., 2007, 129, 9476–9483 CrossRef CAS PubMed.
- M. Röckert, M. Franke, Q. Tariq, H.-P. Steinrück and O. Lytken, Chem. Phys. Lett., 2015, 635, 60–62 CrossRef.
- K. Leung, S. B. Rempe, P. A. Schultz, E. M. Sproviero, V. S. Batista, M. E. Chandross and C. J. Medforth, J. Am. Chem. Soc., 2006, 128, 3659–3668 CrossRef CAS PubMed.
- J. Kučera and A. Gross, Langmuir, 2008, 24, 13985–13992 CrossRef.
- Y. Gurdal, J. Hutter and M. Iannuzzi, unpublished work.
- F. Buchner, V. Schwald, K. Comanici, H.-P. Steinrück and H. Marbach, ChemPhysChem, 2007, 8, 241–243 CrossRef CAS PubMed.
- M. Chen, X. Feng, L. Zhang, H. Ju, Q. Xu, J. Zhu, J. M. Gottfried, K. Ibrahim, H. Qian and J. Wang, J. Phys. Chem. C, 2010, 114, 9908–9916 CAS.
- T. Lin, G. Kuang, W. Wang and N. Lin, ACS Nano, 2014, 8, 8310–8316 CrossRef CAS PubMed.
- F. Xiang, C. Li, Z. Wang, X. Liu, D. Jiang, X. Leng, J. Ling and L. Wang, Surf. Sci., 2015, 633, 46–52 CrossRef CAS.
- K. Diller, F. Klappenberger, F. Allegretti, A. C. Papageorgiou, S. Fischer, A. Wiengarten, S. Joshi, K. Seufert, D. Écija, W. Auwärter and J. V. Barth, J. Chem. Phys., 2013, 138, 154710 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: More details and results of the XPS experiments and the DFT calculation including also the coordinates of the calculated configurations. See DOI: 10.1039/C5NR08953K |
| ‡ Present address: Fachbereich Physik, Philipps-Universität Marburg, D-35037 Marburg, Germany. |
| This journal is © The Royal Society of Chemistry 2016 |