
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Virosome engineering of colloidal particles and surfaces: bioinspired fusion to supported lipid layers†
J.
Fleddermann
a,
E.
Diamanti
b,
S.
Azinas
cd,
M.
Košutić
b,
L.
Dähne
e,
I.
Estrela-Lopis
a,
M.
Amacker
f,
E.
Donath
a and
S. E.
Moya
*b
aInstitute of Medical Physics and Biophysics, Faculty of Medicine, University of Leipzig, 04107 Leipzig, Germany
bSoft Matter Nanotechnology Group, CIC biomaGUNE, Paseo Miramón 182 C, 20009 San Sebastián, Guipúzcoa, Spain. E-mail: smoya@cicbiomagune.es
cBiosurfaces Group, CIC biomaGUNE, Paseo Miramón 182 C, 20009 San Sebastián, Guipúzcoa, Spain
dStructural Biology Unit, CIC bioGUNE Technological Park, Bld 800 48160 Derio, Vizcaya, Spain
eSurflay Nanotec GmbH, Max Planck Str.3, 12489 Berlin, Germany
fMymetics SA, Route de la Corniche 4, 1066 Epalinges, Switzerland
First published on 7th March 2016
Abstract
Immunostimulating reconstituted influenza virosomes (IRIVs) are liposomes with functional viral envelope glycoproteins: influenza virus hemagglutinin (HA) and neuraminidase intercalated in the phospholipid bilayer. Here we address the fusion of IRIVs to artificial supported lipid membranes assembled on polyelectrolyte multilayers on both colloidal particles and planar substrates. The R18 assay is used to prove the IRIV fusion in dependence of pH, temperature and HA concentration. IRIVs display a pH-dependent fusion mechanism, fusing at low pH in analogy to the influenza virus. The pH dependence is confirmed by the Quartz Crystal Microbalance technique. Atomic Force Microscopy imaging shows that at low pH virosomes are integrated in the supported membrane displaying flattened features and a reduced vertical thickness. Virosome fusion offers a new strategy for transferring biological functions on artificial supported membranes with potential applications in targeted delivery and sensing.
Introduction
Immunostimulating reconstituted influenza virosomes (IRIVs) are virus like nanoparticles similar to influenza virus envelopes but lacking any viral genetic material.1 IRIVs are essentially spherical, unilamellar vesicles with a mean diameter of less than 200 nm.2–4 Their base is a liposome comprised of phosphatidylcholine (PC), phosphatidylethanolamine (PE) and lipids derived from the influenza virus.5 In contrast to liposomes, virosomes contain functional viral envelope glycoproteins: influenza virus hemagglutinin (HA) and neuraminidase (NA) intercalated in the phospholipid bilayer. Thus, reconstituted influenza virosomes retain the receptor binding and membrane fusion activity of the influenza virus.1 Virosomes have demonstrated to be a versatile and efficient carrier system for a variety of antigens, for example proteins, peptides, nucleic acids and carbohydrates.3,6Virosomes have been mainly developed as prospective adjuvants to potentiate an immune response, since antigens alone are often poor immunogens.1,7 While most adjuvants, including IRIVs, induce a humoral immune response, Kammer et al. developed a new generation of influenza virosomes (TIRIVs) that induced both cytotoxic T-cell and humoral responses.1 This new type of IRIV contains TC- or DC-cholesterol as stabilizers, which cause their possible lyophilization. After reconstitution, the immunogenic properties (e.g. membrane fusion activity) are maintained.1 Furthermore, virosomes have potential applications as drug carriers, like liposomes, with the advantage that the viral glycoprotein hemagglutinin promotes the fusion of the virosome with the cell membrane, which can in turn lead to the release of the virosome cargo directly in the cytoplasm of the cell.8 An unexplored domain where virosomes can be applied is their use to engineer colloids and surfaces in the way that liposomes have been often applied for the development of biocompatible surfaces, biosensors or drug delivery systems. Since virosomes retain the fusion properties of virus they could also be fused on artificial lipid membranes bringing the functionalities carried on their membranes to the supported lipids. Colloidal particles engineered with virosomes could have interesting applications in drug delivery besides their obvious importance in vaccine development. Virosome engineered particles could find applications as adjuvants, as influenza virus proteins will be present on the surface of the colloidal particles.9–11 An additional advantage of virosomes is that other proteins like antibodies or specific ligands can be integrated into the virosome or attached to the surface of the virosomes employing a lipid anchor.12,13 These biomolecules would be then transferred to the supported membranes.14–16 This represents an alternative to the direct functionalization of supported membranes, which is not always possible once the membrane is formed. Also the use of lipid vesicles entailing proteins and other biomolecules does not always result in the formation of a bilayer as they can affect the assembly, rupture and fusion process of liposomes. For the colloidal particles the presence of proteins in liposomes can also lead to major particle aggregation if the biomolecule prevents charge compensation during an electrostatically driven assembly. Therefore, virosomes could act as transfer agents of biological functionalities to supported membranes in a sequential way through a fusion mechanism without jeopardizing membrane formation and reducing particle aggregation in the case of colloids.
In this manuscript we will show that it is possible to fuse influenza virosomes on supported membranes, both assembled on colloidal particles and planar surfaces. We will prove that the fusion of the influenza virosomes is triggered by low pH, as occurring with the influenza virus.17 Thereby, the fusion of the virosomes on supported membranes can be used as a model for the virosome fusion on cell membranes.
Despite the fact that the fusion of virosomes has been proven in cell cultures,12,18,19 the fusion mechanism and the state of virosomes and glycoproteins after fusion have not been studied in detail. Our results can provide a better understanding of the fusion mechanism of virosomes with membranes that is relevant for drug delivery and vaccine applications.
Virosome fusion has been studied via flow cytometry, confocal laser scanning microscopy (CLSM), fluorescence spectroscopy, quartz crystal microbalance with dissipation (QCM-D), atomic force microscopy (AFM) and cryo-transmission electron microscopy (Cryo-TEM). By combining these techniques we have been able to address the influence of pH, time, temperature, and virosome concentration on their fusion into lipid bilayer supported membranes.
Supported membranes were fabricated on self-assembled polyelectrolyte multilayers (PEMs), which were chosen as model supports as they can be assembled on both planar and colloidal interfaces. PEMs were chosen as supports since membranes in cells are also supported by a polymer cushion of glycoproteins. The supported membranes were assembled from the mixed vesicles of DOPS and DOPC, under conditions that ensure the formation of a single lipid bilayer.20
Experimental
Materials and methods
Polyallylamine hydrochloride, (PAH, Mw 15 kDa), polystyrene sulfonate sodium salt, (PSS, Mw 70 kDa), and Rhodamine B octadecyl ester perchlorate were obtained from Sigma-Aldrich. PAH (Mw 15 kDa) labeled with Rhodamine B isothiocyanate (Rhd) and PAH (Mw 15 kDa) labeled with Cy5 were kindly offered from Surflay Nanotec GmbH. The phospholipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, 10 mg mL−1 in chloroform) and 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS, sodium salt, 10 mg mL−1 in chloroform) were purchased from Avanti Polar Lipids, Inc. SiO2 particles with a diameter of 3 μm were purchased from Attenbio. The immunostimulating reconstituted influenza virosomes (IRIVs); 7 mg per mL lipid [DOPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OPPE in a mass ratio 4:1] were prepared with 1.3 mg per mL hemagglutinin of the influenza strain A/Sing/6/86 H1N1. Phosphate buffered saline (PBS), sodium chloride (NaCl), citric acid, sodium phosphate dibasic and chloroform anhydrous (>99%) were purchased from Sigma-Aldrich. Acetic acid (99.8%) was obtained from Sigma Aldrich–Fluka. Absolute ethanol (99.9% HPLC) was obtained from Scharlau S.A.
:OPPE in a mass ratio 4:1] were prepared with 1.3 mg per mL hemagglutinin of the influenza strain A/Sing/6/86 H1N1. Phosphate buffered saline (PBS), sodium chloride (NaCl), citric acid, sodium phosphate dibasic and chloroform anhydrous (>99%) were purchased from Sigma-Aldrich. Acetic acid (99.8%) was obtained from Sigma Aldrich–Fluka. Absolute ethanol (99.9% HPLC) was obtained from Scharlau S.A.
Small unilamellar lipid vesicles (SUVs) composed of mixtures of 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) were prepared as follows: lipid stock solutions (10 mg mL−1 in chloroform) were mixed together at a molar ratio of 40:60 (DOPC:DOPS). The chloroform was evaporated under vacuum (1 mbar, room temperature) for at least 1 h. The lipid film was rehydrated immediately with PBS (10 mM, pH 7.4) and the resulting lipid solution was extruded through a polycarbonate membrane with 50 nm diameter pores to form SUVs.
For the assembly of a lipid bilayer, LbL-coated silica particles and planar surfaces were washed three times with PBS. After addition of SUVs (5 mg mL−1 in PBS) the sample was incubated for 15 min at room temperature under continuous shaking. Surplus SUVs were removed by washing three times with PBS.
SUVs and virosomes were characterized regarding the size and zeta-potential. The size was determined after 1:100 dilution (v/v) in buffer (50 mM phosphate, 83 mM NaCl; pH 7.4) by dynamic light scattering (DLS) using a Malvern ZetaSizer 3000HS and Nanotracking analysis (NTA) (NanoSight 2.3). The zeta potential was measured in 1 mM Tris (pH 7) with the Malvern ZetaSizer 3000HS.
:10 dilution in PBS from an ethanolic R18 stock solution (3 mg mL−1 in water containing 20% (v/v) ethanol) at room temperature for 1 h under gentle shaking. Due to the excess of R18 (5 mol% R18 of total lipid) and the resulting dye–dye interaction in the supported lipid membrane, the fluorescence of the probe is self-quenched.21,22 Surplus R18 was removed by Sephadex G-50 chromatography. When virosome fusion takes place into an unlabeled lipid bilayer the probe will dilute and the fluorescence increases accordingly.22 The fluorescence increase after fusion was measured by fluorescence spectroscopy and flow cytometry.
Virosome–vesicle fusion. R18 labeled virosomes (0.5 mg per mL lipid) were diluted in citric acid/phosphate buffer (pH 4.5 or pH 7.4). The fluorescence of the R18 probe was monitored immediately after virosome mixing with SUVs for 10 min using a Varian Cary50 spectrofluorimeter (λex: 532 nm, λem: 584 nm). After 1 min unlabeled SUVs (0.5 mg mL−1) were added to the virosome dispersion. Finally Triton X-100 (1% (v/v)) was added to derive the maximum R18 fluorescence value. Due to a small self-fluorescence of Triton X-100, control measurements in the absence of virosomes were performed in buffer and subtracted from the Triton value of the sample.
Virosome fusion with lipid coated colloids. Lipid coated LbL engineered beads (5 wt%) were diluted 1
:500 in citric acid/phosphate buffer (pH 4.5 or pH 7.4). Control measurements without virosomes were performed. After addition of R18-labeled virosomes (0.9 mg per mL lipid) the sample was incubated at 37 °C in a water bath for different times. The fluorescence of the sample was recorded using a Varian Cary50 Eclipse spectrofluorometer (λex: 532 nm, λem: 584 nm). Triton X-100 (1% (v/v)) was added, mixed and measured to obtain the maximal value of R18 fluorescence.
:PS (molar ratio 40:60) SUVs in PBS (0.1 mg mL−1). The suitable conditions for the formation of a lipid bilayer membrane from a mixture of DOPC and DOPS lipids on top of PAH/PSS PEM have been previously described.20,23 When the bilayer was formed and the frequency reached a plateau the membrane was rinsed with PBS to remove surplus vesicles. Subsequently, the chamber was pre-equilibrated with milli-Q water. The QCM-D chamber was filled with CIP buffer (pH 4.5) or PBS (pH 7.4) before injection of the virosomes. Virosomes were added at a concentration of 0.014 μg mL−1 and left in the chamber for at least 1 h. Finally, the chamber was rinsed with PBS and milli-Q water in order to remove non-fused virosomes from the membrane.
Results and discussion
Supported membrane assembly
SUVs were prepared as described in the experimental part and characterized regarding the size and zeta potential. The size of the SUVs was 127.2 ± 2.3 nm and 87 ± 5 nm as measured by DLS and NTA, respectively. The zeta potential of the SUVs was −23.7 ± 7.49 mV in 1 mM Tris (pH 7). The negative zeta potential value is consistent with the presence of DOPS in the vesicles. The SUVs were employed to deposit a lipid bilayer on silica particles covered with a PEM consisting of 11 layers with PAH being the top layer. Cryo-TEM showed that the LbL silica colloids have been successfully coated by a continuous lipid bilayer, which forms the basis for subsequent virosome fusion.20Fusion of virosomes with PEM supported lipid membranes
Hemagglutinin, the membrane fusion protein of the influenza virus, is activated and triggered by low pH.2,3,8,17 Upon exposure to low pH in endosomes, conformational changes of the hemagglutinin protein result in the exposure of the fusion peptide, the hydrophobic N-terminus of the HA2 polypeptide subunit.24,25 The hydrophobic peptide is inserted into the hydrocarbon part of the cell membrane.8,25,26 At pH 7.4 the virosomes remain attached to the surface of the membrane, since hemagglutinin is not triggered to initiate fusion with the lipid bilayer. Since the pH is important for the influenza virus for the fusion with lipid membranes, the fusion activity of the virosomes was tested on supported lipid membranes at acidic and neutral pH. Furthermore the best conditions for virosome fusion were established by evaluation of the temperature and virosome concentration.At first, IRIVs and free protein liposomes with the same composition and lipid content of the IRIVs, without hemagglutinin, were characterized in liquid solution regarding size and zeta potential (Fig. 1). The PDI of 0.096 (IRIVs) demonstrates that no aggregation or degradation takes place.
 | ||
| Fig. 1 Particle size, zeta-potential and Cryo-TEM image of IRIVs and protein free equivalent liposomes. | ||
The fusion of the virosomes was studied by means of the fluorescence self-quenching R18 assay. The assay is based on the incorporation of the lipophilic Rhodamin B- octadecylester-perchlorate (R18) into lipid membranes taking advantage of the alkyl chain of the dye conjugate. Virosomes were labeled by adding a R18 solution in ethanol to the virosome suspension at such a concentration that the R18 fluorescence becomes self-quenched as a result of the dye–dye interaction. The R18 quenching was about 50–70% (Fig. S3†).
The fusion of R18-labeled virosomes with SUVs was monitored by fluorescence spectroscopy measurements. Fig. 2 shows that the virosome–liposome fusion depended on the pH value. At pH 4.5 the fluorescence intensity increased after addition of SUVs reaching a plateau after approximately 3 min. At pH 7.4 the fluorescence intensity did not increase after addition of virosomes, which indicates that at neutral pH virosome–liposome fusion did not occur. Control studies with protein free liposomes with otherwise identical lipid composition and concentration to IRIVs showed no increase of fluorescence with time after their addition to the SUVs for both pH 4.5 and pH 7.4.
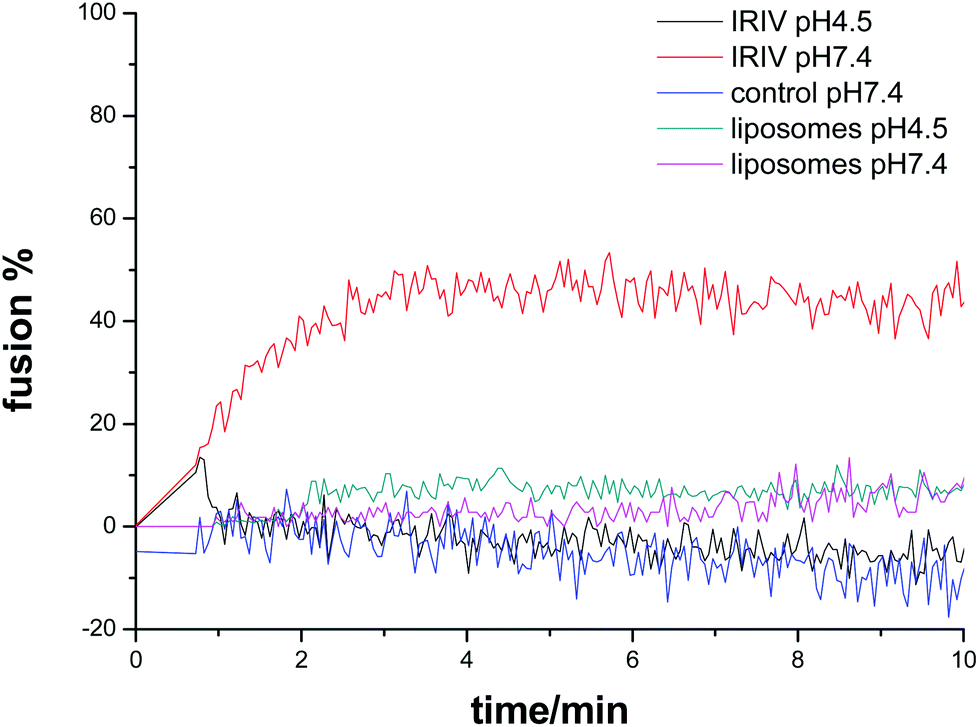 | ||
| Fig. 2 Kinetic analysis of the virosome fusion with liposomes. SUVs were incubated in citric acid/phosphate buffer with pH 4.5 or pH 7.4. After 1 min R18-labeled IRIVs or R18-labelled free protein liposomes analogous to IRIV (0.5 mg per mL of total lipid) were added. A sample without addition of R18-labeled virosomes was used as a control. The fluorescence intensity (λex: 532 nm, λem: 584 nm) was recorded every 0.05 min. Fusion% was calculated with (F − F0)/Fmax × 100. F is the fluorescence intensity after addition of virosomes or protein free liposomes or PBS (control). F0 is the fluorescence intensity before addition of virosomes. Fmax is the maximal fluorescence intensity after lysis with Triton X-100. | ||
The fusion of R18-labeled IRIVs with lipid-coated LbL particles was studied by flow cytometry. Fig. 3 shows that fusion was strongly dependent on pH. The mean fluorescence intensity was measured as a function of time and pH. At pH 4.5 the R18 fluorescence intensity increased over 20 min by more than a factor of two, while at pH 7.4 the fluorescence remained approximately constant over this period of time. Further experiments showed that the fusion of virosomes with lipid coated beads was completed within about 30 min at pH 4.5. The fusion of virosomes with the supported lipid layer on the beads proceeded much slower than the virosome–vesicle fusion. We attribute this difference to the restricted mobility and flexibility of the supported membrane compared with the free membrane of the vesicles. The different lipid surface ratios of liposomes and lipid-coated LbL-colloids may also play a role. Control measurements with protein free liposomes at both pH values showed also no increase of fluorescence with time, confirming that the presence of HA is responsible for the virosome fusion.
 | ||
| Fig. 3 Kinetic analysis of the virosome fusion with lipid-coated LbL-silica particles. Lipid-coated LbL-colloids were incubated in citric acid/phosphate buffer with pH 4.5 or pH 7.4 before addition of R18-labeled IRIVs or R18-labeled liposomes of IRIV. The mean fluorescence intensity distribution relative to the mean fluorescence intensity of the start value (0 min) is plotted as a function of the time. The fluorescence intensity (λex: 532 nm, λem in FL4) was measured after different times (0, 1, 2, 5, 10, 15, 20 min) by flow cytometry (n = 200–500 events). | ||
Virosome fusion with lipid-coated LbL-colloids was studied as a function of temperature. Fig. 4 shows that after 10 min the fluorescence intensity at 37 °C was only slightly larger than at 21 °C. The fluorescence intensity increases from 0 min (virosome addition) to 10 min by 2 times for 21 °C and 1.7 times for 37 °C. However, after lysis with Triton X-100 the R18 fluorescence intensity was much larger at 21 °C than at 37 °C. This indicates that a comparatively large amount of virosomes at 21 °C was still adsorbed on the lipid coated particles rather than fused within the lipid bilayer. This finding demonstrates that the temperature increase facilitates fusion as demonstrated by Haywood et al. for the fusion of the influenza virus with liposomes.27
 | ||
| Fig. 4 Fusion of IRIVs with lipid-coated LbL-silica particles in dependence of the temperature measured as the R18 fluorescence intensity. Lipid-coated LbL-silica particles were incubated with R18-labeled IRIVs (0.5 μg hemagglutinin) in citric acid/phosphate buffer with pH 4.5 at room temperature (21 °C) or 37 °C for 10 min. The fluorescence intensity (λex: 532 nm, λem: 584 nm) before and after lysis by addition of Triton X-100 was measured. | ||
Furthermore, the effect of virosome concentration on the membrane fusion was studied. In Fig. 5 two different IRIV concentrations are compared. The virosome concentration, given in the final amount of hemagglutinin per ml, was 0.5 μg and 2 μg HA. At pH 4.5 the fluorescence increases 2 times for 0.5 μg HA and one time for 2 μg HA after 10 min incubation to virosome addition at 0 min. This represents a more efficient fusion for the lower virosome concentration to lipid-coated LbL silica particles. At pH 4.5, after lysis with Triton X-100 the R18-fluorescence intensity increased 14 times for 2 μg hemagglutinin, a significantly larger increase than the observed approximately 4 times increase for 0.5 μg hemagglutinin (Fig. 5a and b). The pronounced difference in the fusion efficiency between 0.5 and 2 μg hemagglutinin suggests that in the latter case a large amount of virosomes remained attached to the supported membrane instead of fusing. It may also be that being the concentration of R18 significantly higher for the case with the larger concentration of virosomes their fusion does not cause a complete dequenching of the R18. However, the confocal fluorescence microscopy image (Fig. 5d) shows a rather inhomogeneous fluorescence distribution thus confirming the adsorption of virosomes to the supported membrane. It may be that if the lipid membrane is densely covered with virosomes, fusion becomes inhibited as a consequence of topological constraints. In contrast for 0.5 μg hemagglutinin the fluorescence distribution is homogenous indicating the virosome fusion to the supported membrane (Fig. 5c).
 | ||
| Fig. 5 Fusion of IRIVs with lipid-coated LbL-silica particles in dependence of hemagglutinin concentration. R18 fluorescence intensity (λex: 532 nm, λem: 584 nm) was measured before and after lysis with Triton X-100 for 0.5 and 2 μg hemagglutinin at (a) pH 4.5 and (b) pH 7.4. Confocal images (c) and (d) corresponding to (a) and (b), respectively. Lipid-coated LbL-silica particles were incubated with R18-labeled IRIVs concentrations either 0.5 μg or 2 μg hemagglutinin in citric acid/phosphate buffer with pH 4.5 at 37 °C for 10 min. The color bar indicates the R18-fluorescence intensity from a value of 0 (black) to 255 (blue). | ||
Virosome fusion on planar supported membranes
Virosome adsorption and fusion was also studied by means of QCM-D on planar supported membranes. Fig. 6 shows the results from the real time monitoring of fusion of the IRIVs for the two different pH values 4.5 and 7.4. The QCM-D sensor is sensitive to surface mass; the frequency decrease is related to the increase of the mass deposited on the sensor. As referred to in the Experimental part, a lipid bilayer was deposited on top of the 11 layers of PAH/PSS cushion prior to virosome deposition. Fig. 6a shows the frequency and dissipation changes occurring upon interaction of the IRIVs diluted in CIP buffer with the supported membrane at pH 4.5 as a function of time. The frequency followed a steadily decreasing tendency for more than two hours reaching a total frequency shift of Δf = −99 Hz. This comparatively large frequency shift demonstrates an ongoing adsorption of IRIVs on the membrane indicating that at this pH the HA protein in the IRIVs is able to recognize the lipid membrane. The strong parallel increase in dissipation confirms adsorption. The increase in dissipation is also indicative of the increase of the surface roughness during the attachment of the virosomes to the membrane leading to an enhanced friction of the soft virosomes. At this stage the QCM-D data do not allow the separation of fusion and adsorption. However, after rinsing with PBS non-fused IRIVs were removed as seen by the decrease in dissipation and the increase in frequency reaching a Δf = −46 Hz. The frequency increased even further to Δf = −21 Hz when rinsing with water. So finally the total frequency shift for the fused IRIVs in the supported membrane at acidic pH was Δf = −32 Hz. We attribute the frequency change after rinsing with PBS and water to the net mass increase caused by virosome membrane fusion. At neutral pH the QCM-D data demonstrate a completely different behavior as shown in Fig. 6b. The frequency changes only slightly upon the deposition of the IRIVs. The small increase in dissipation, note the different scale in Fig. 6a and b, is consistent with the adsorption of a small number of virosomes, which do not subsequently fuse with the lipid membrane. This was followed by the small total change in frequency Δf = −8 Hz after rinsing. | ||
| Fig. 6 Frequency (blue) and dissipation (red) curves measured by QCMD for the formation of a phospholipid bilayer deposited on a PEM of 11 layer of PAH/PSS and later exposed to (a) IRIVs in acidic pH (CIP 4.5), (b) IRIVs in neutral pH (PBS 7.4) and (c) protein free liposomes with the same lipid composition and content as IRIVs in acidic pH (CIP 4.5). | ||
Liposomes analogous to the IRIV, without HA proteins, did not fuse at pH 4.5 on the supported membranes (Fig. 6c) confirming again that the effect of the pH is only related to the hemagglutinin.
AFM imaging was conducted to have visual proof of the virosome fusion to the lipid membrane. In Fig. 7a the fused IRIVs at pH 4.5 can be recognized as they form features of a varying size of 190–270 nm, which is about twice as much as the diameter of the IRIVs. These features were not present in the supported membrane before the exposure to the virosomes. The observed features have a height of 50–70 nm. These values are consistent with the measured size of the IRIVs of about 115 nm. From the geometrical considerations it is obvious that after fusion of the IRIVs with the lipid membrane the latter has to be lifted off in the regions of the fusion events, as fusion goes along with a net increase of the total membrane surface. From this it follows that the virosome membranes were embedded in the supported membrane, while the enclosed volume was released during fusion. The fused virosomes appear to be aggregated and randomly distributed on the surface. The control at neutral pH is more revealing. Fig. 7b shows features of a height of ∼100 nm and lateral dimensions of 150–350 nm, which correspond to non-fused IRIVs displaying a size comparable to the one obtained by DLS and Cryo-TEM measurements for the IRIVs in bulk. The IRIVs at this pH appear as if they remained attached on the supported membrane without fusing. AFM was also conducted for supported membranes exposed to protein free liposomes without HA, at pH 4.5. In this case the lipid surface displays the same features as the supported membranes prior to the exposure to the liposomes but with a few large liposome aggregates on top with a height of 120–130 nm and a width of ∼500 nm (Fig. 7c). The extended aggregation of the liposomes could be due to the lack of proteins. No hints of fused liposomes could be observed.
 | ||
| Fig. 7 AFM images (2 × 2 μm) acquired in 1.5 mM NaCl (liquid mode) of (PAH/PSS)5.5 PC:PS lipid bilayer membrane after addition of a) IRIVs in pH 4.5 or (b) IRIVs in pH 7.4 and (c) protein free liposomes in pH 4.5. Bottom panels display roughness profiles taken as cross sections of the above images. | ||
The virosome fusion is based on their interaction with the lipid membrane driven by the hemagglutinin (HA) protein present on the surface of the virosomes, which is responsible for the pH dependent fusion. Fig. 8a displays a scheme of the behaviour of the virosomes at neutral pH where they remain on the membrane without fusing, while at low pH they fuse (Fig. 8b and c). In the figure the change in the conformation of the HA with the hydrophobic peptide penetrating in the hydrocarbon part of the cell membrane has been sketched (Fig. 8b).8,25,26 In Fig. 8c the possible arrangement of the lipids and proteins after the fusion is shown. It is remarkable that the fusion of virosomes is taking place in the absence of a sialoglycan or any other receptor on the membrane that promotes the interaction of the virosome with the membrane. The fact that our supported membranes are formed by lipids only lacking additional components of cell membranes like the glycocalyx or proteins could explain that the virosomes can fuse directly on the membranes being the lipid layers easily accessible for the virosomes to fuse triggered by the low pH when they get in close vicinity with the supported membrane.
 | ||
| Fig. 8 Schematic illustration of the (a) virosome attached to a supported membrane at neutral pH without fusing; and (b) and (c) fusion of the virosomes at low pH on the supported membrane. The virosome makes contact with the lipid membrane and there is a rearrangement of the hemagglutinin protein at a low pH of 4.5 (b), the zoom out image shows in more detail the rearrangement of the hemagglutinin protein. The virosome is fused to the lipid bilayer membrane (c), the zoom out image shows how the proteins are arranged at the place where the fusion occurs. | ||
Conclusions
We have shown that influenza virosomes carrying hemagglutinin can fuse on supported membranes on colloids as well as on planar surfaces. The mechanism of fusion is pH dependent as has been confirmed independently by the R18 assay, QCM-D and AFM. Virosomes fuse at low pH 4.5, but not at neutral pH 7.4. AFM imaging proves that the virosomes are integrated in the supported membrane at low pH as they can be recognized in the membrane as flattened features and have a reduced vertical thickness. Virosome fusion on supported membranes offers a novel strategy for the functionalization of supported membranes that can be used for the design of complex colloidal systems which can have applications in drug delivery or sensing devices.Acknowledgements
This work was financed by the FP7 PEOPLE-IAPP Project VIROMA, Grant agreement: 612453. The authors also acknowledge the project MAT2013-48169-R from the Spanish Ministry of Economy (MINECO).Notes and references
- A. R. Kammer, M. Amacker, S. Rasi, N. Westerfeld, C. Gremion, D. Neuhaus and R. Zurbriggen, Vaccine, 2007, 25, 7065–7074 CrossRef CAS PubMed.
- R. Glück and I. C. Metcalfe, Vaccine, 2003, 21, 611–615 CrossRef.
- M. Amacker, O. Engler, A. R. Kammer, S. Vadrucci, D. Oberholzer, A. Cerny and R. Zurbriggen, Int. Immunol., 2005, 17, 695–704 CrossRef CAS PubMed.
- M. Amacker, S. Moese, A. Kammer, A. Helenius and R. Zurbriggen, Delivery Technologies for Biopharmaceuticals, ed. L. Jorgensen and H. N. Nielsen, John Wiley & Sons Ltd, London, 2009 Search PubMed.
- C. Moser, M. Müller, M. Käser, U. Weydemann and M. Amacker, Expert Rev. Vaccines, 2013, 12, 779–791 CrossRef CAS PubMed.
- D. Felnerova, J. F. Viret, R. Gluck and C. Moser, Curr. Opin. Biotechnol., 2004, 15, 518–529 CrossRef CAS PubMed.
- B. Gaurav, K. Manisha and K. Vilasrao, World J. Pharm. Pharm. Sci., 2014, 3, 437–447 Search PubMed.
- S. C. Harrison, Nat. Struct. Mol. Biol., 2008, 15, 690–698 CAS.
- I. A. de Bruijn, J. Nauta, L. Gerez and A. M. Palache, Vaccine, 2006, 24, 6629–6631 CrossRef CAS PubMed.
- T. Douglas and M. Young, Science, 2006, 312, 873–875 CrossRef CAS PubMed.
- C. Moser, M. Amacker and R. Zurbriggen, Expert Rev. Vaccines, 2011, 10, 437–446 CrossRef CAS PubMed.
- L. Bungener, K. Serre, L. Bijl, L. Leserman, J. Wilschut, T. Daemena and P. Machy, Vaccine, 2002, 20, 2287–2295 CrossRef CAS PubMed.
- L. A. Lee, Z. Niu and Q. Wang, Nano Res., 2010, 2, 349–364 CrossRef.
- M. Fischlechner, O. Zschornig, J. Hofmann and E. Donath, Angew. Chem., Int. Ed., 2005, 44, 2892–2895 CrossRef CAS PubMed.
- M. Fischlechner and E. Donath, Angew. Chem., Int. Ed., 2007, 46, 3184–3193 CrossRef CAS PubMed.
- L. Toellner, M. Fischlechner, B. Ferko, R. M. Grabherr and E. Donath, Clin. Chem., 2006, 52, 1575–1583 CAS.
- J. M. White, S. E. Delos, M. Brecher and K. Schornberg, Crit. Rev. Biochem. Mol. Biol., 2008, 43, 189–219 CrossRef CAS PubMed.
- H. S. Kim and Y. S. Park, J. Biochem. Mol. Biol., 2002, 35, 459–464 CrossRef CAS PubMed.
- D. C. Johnson, M. Wittels and P. G. Spear, J. Virol., 1984, 52, 238–247 CAS.
- E. Diamanti, L. Cuellar, D. Gregurec, S. E. Moya and E. Donath, Langmuir, 2015, 31, 8623–8632 CrossRef CAS PubMed.
- A. Domecq, E. A. Disalvo, D. L. Bernik, F. Florenzano and M. J. Politi, Drug Delivery, 2001, 8, 155–160 CrossRef CAS PubMed.
- T. Buranda, Y. Wu, D. Perez, A. Chigaev and L. A. Sklar, J. Phys. Chem. B, 2010, 114, 1336–1349 CrossRef CAS PubMed.
- M. Fischlechner, M. Zaulig, S. Meyer, I. Estrela-Lopis, L. Cuéllar, J. Irigoyen, P. Pescador, M. Brumen, P. Messner, S. Moya and E. Donath, Soft Matter, 2008, 4, 2245 RSC.
- M. Kielian, Annu. Rev. Virol., 2014, 1, 171–189 CrossRef PubMed.
- B. S. Hamilton, G. R. Whittaker and S. Daniel, Viruses, 2012, 4, 1144–1168 CrossRef CAS PubMed.
- X. Han, J. H. Bushweller, D. S. Cafiso and L. K. Tamm, Nat. Struct. Biol., 2001, 8, 715–720 CrossRef CAS PubMed.
- A. M. Haywood and B. P. Boyer, J. Gen. Virol., 1986, 67, 2813–2817 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr08169f |
| This journal is © The Royal Society of Chemistry 2016 |