
Atomistic modeling of BN nanofillers for mechanical and thermal properties: a review
Rajesh
Kumar
and
Avinash
Parashar
*
Department of Mechanical and Industrial Engineering, Indian Institute of Technology, Roorkee - 247667, India. E-mail: drap1fme@iitr.ac.in; Tel: +91-1332-284801
First published on 12th November 2015
Abstract
Due to their exceptional mechanical properties, thermal conductivity and a wide band gap (5–6 eV), boron nitride nanotubes and nanosheets have promising applications in the field of engineering and biomedical science. Accurate modeling of failure or fracture in a nanomaterial inherently involves coupling of atomic domains of cracks and voids as well as a deformation mechanism originating from grain boundaries. This review highlights the recent progress made in the atomistic modeling of boron nitride nanofillers. Continuous improvements in computational power have made it possible to study the structural properties of these nanofillers at the atomistic scale.
 Rajesh Kumar | Mr Rajesh Kumar is a PhD candidate in the department of mechanical and industrial engineering at the Indian Institute of Technology-Roorkee. His research area focuses on atomistic modelling of BN nanofillers and their hybrids. |
 Avinash Parashar | Dr Avinash Parashar received his PhD in mechanical engineering (2012) from the University of Alberta. He worked as an attached scientist with the Computational Reactor Physics laboratory in Atomic Energy of Canada Limited. Currently, he is working as an assistant professor in the department of Mechanical and Industrial Engineering at the Indian Institute of Technology-Roorkee. His research interest includes atomistic modeling of nanofillers and nanocomposites. |
1. Introduction
Boron nitride (BN) is a lab-grown binary compound consisting of an equal number of boron (B) and nitrogen (N) atoms. BN crystallizes either as a hexagonal layered structure or as a tetrahedral linked structure, similar to that of graphite and diamond respectively. Due to its white color and slippery properties hexagonal boron nitride (h-BN) is also known as white graphite or graphitic boron nitride.1,2 In 1995, BN was first synthesized as boron nitride nanotubes (BNNTs) and in continuation to that work, BN nanomeshes were further developed.3Similar to carbon based nanofillers e.g. graphene and carbon nanotubes; BN nanotubes (BNNTs) and nanosheets (BNNSs) also have a hexagonal closed packed atomic configuration.4–6 BN nanofillers (BNNT or BNNS) exist in the hexagonal form with alternatively placed boron (B) and nitrogen (N) atoms as illustrated in Fig. 1. Despite similar atomic configurations, and comparable mechanical and thermal properties,7,8 BN nanofillers are good electrical insulators1,9,10 as compared to the corresponding carbon based nanofillers.
 | ||
| Fig. 1 Atomic configuration of (a) single layer BNNS and (b) single-walled BNNT. Red and blue dots correspond to boron and nitrogen atoms, respectively. | ||
Due to its wide band gap, h-BN is preferred over graphene for specific applications such as electronic packaging.11–14 In addition to exceptional mechanical properties,15–26 thermal conductivity27 and electrical insulating properties,1,9,10 BN nanofillers are also highly stable against oxidation even at very high temperatures such as 800 °C.28 At such high working temperatures, BN nanofillers are considered chemically and structurally more stable as compared to the corresponding carbon based nanostructures.28,29 The thermal conductivity of BN nanofillers has been reported to be in the range of 1700–2000 W m−1 K−1.27 In addition to these exceptional properties, BN nanofillers have also shown deep ultraviolet photon emission,30 and a good amount of piezoelectricity.31–33
Due to the above mentioned exceptional properties, h-BN nanofillers have found applications in many areas such as nano-sensors,34–39 bio-sensors,34–37,39 biomaterials40–42 and in developing corrosion-resistant and thermally stable composites.43–49 The bio-compatibility, biosafety and high hemo/histocompatibility of BN nanofillers are encouraging researchers to use them as therapeutic agents to treat neuro-genetic disorders.50–55 The neutron absorption capacity of these nanofillers has also found applications in the field of cancer treatment such as cerebral glioblastoma multiforme.56 Adsorption of hydrogen on the surface of BNNTs and BNNSs57–59 also opens the door for hydrogen storage at ambient temperatures. Some other prominent applications of BN nanofillers include optoelectronics60–66 and tribology.67–69 Due to their excellent sorption capabilities for a wide range of oils, solvents and dyes, their application as a water purifier has also been proposed.70
Despite the availability of high yield synthesis techniques,71 commercial production of these nanofillers is still in an immature state, due to the limited availability of techniques for characterizing these nanofillers. Due to challenges associated with the characterization of these BN nanofillers, limited research has been published in this area as compared to that of carbon based nanofillers. A year wise comparative chart for the number of publications in the field of respective nanofillers is plotted in Fig. 2.
 | ||
| Fig. 2 A year wise graphical comparison of research papers published and containing respective phrases i.e. BNNT, BNNS, CNT & graphene in their title. Statistics based on Scopus database, as of June 15th, 2015. | ||
There are a number of methods that are currently being used for the production of h-BN nanofillers. Monolayer to few-layers of h-BN flakes can be exfoliated from bulk BN crystals either by mechanical cleavage/exfoliation72,73 or by a chemical-solution-derived method.74 Other synthesis methods for BN nanofillers include chemical vapor deposition (CVD),75–79 sonication,80,81 ball milling,81,82 high-energy electron beam irradiation,83,84 reaction of boric acid and urea,85,86 a metal-catalyst-free approach,87 chemical blowing (a mass production technique that relies on generating large bubbles of BNH or BNCH from a precursor ammonia borate compound),88,89 using substitution reaction,90,91via micro-fluidization92 and via laser ablation.93–96 The electrical insulating properties that single out BN nanofillers from other nanofillers have been extensively studied by researchers. In 1994, Blase et al.9 predicted a constant band gap for BN nanotubes and this was further explored by Watanabe et al.10 to establish the fact that BN nanotubes have a direct-band gap in the ultra-violet region. Zhang et al.97 predicted the stability and electronic structures of BNNTs with diameters below 0.4 nm. In their work on BNNTs numerical based techniques have been used to study the effect of nanotube diameter, chirality and multiwall BN structures on the band gap or electrical insulation. These exceptional properties, along with a diversified field of applications have aroused keen interest in the researchers to explore more about these nanofillers. Starting from 1994, researchers have been trying to study BN nanofillers using different techniques such as experiments, finite element based continuum models, molecular dynamics based atomistic models, and higher fidelity numerical simulations such as density functional theory (DFT).
So far, only a limited number of review papers have been published on BN nanofillers, and most of them are focused on the generalized topics. Atomistic modeling techniques are emerging as a focused area of research for researchers investigating the behavior of these nanofillers. The aim of the present article is to address the current state of the art techniques for simulating the mechanical as well as thermal behavior of BNNSs and BNNTs with the help of atomistic modeling based approaches. The authors have also attempted to illustrate the challenges associated with each kind of atomistic modeling technique.
2. Atomistic modeling techniques
During the last couple of years, several experimental and modeling techniques have been employed by the researchers to characterize or predict the mechanical and thermal behaviors of BN nanofillers. Despite the fact that experimental techniques are costly and time consuming, these experiments are still considered as a realistic tool for characterizing the behavior of materials. Transmission electron microscopy (TEM)15,16,23,24,85,98 and atomic force microscopy (AFM) based techniques15,16,73,99 are usually employed in the experimental characterization of nano materials. In addition to these common techniques, X-ray based scattering has also been employed for estimating the five elastic moduli of h-BN.100In addition to the cost and time involved, experimental techniques also have some limitations in estimating the localized mechanical or thermal behavior of these nanofillers at the atomistic level. Due to these limitations associated with experimental techniques, computational techniques based on finite element, DFT and MD are emerging as effective tools for predicting the mechanical as well as thermal behavior of these nanofillers. Atomistic simulations are not substituting experiments but are complementing them at the nanoscale.
2.1. Structural mechanics based approach
The theoretical approaches, mentioned above, can be further classified as atomistic and continuum based approaches. The atomistic approach includes classical molecular dynamics and density functional theory, whereas the continuum mechanics based approach mainly consists of classical continuum mechanics and shell modeling.101–103 The structural mechanics based approach developed and proposed by Li and Chou104 is considered to be a pioneer research in the field of atomistic modeling of nanofillers. Their model was based on the notion that the nanofillers (e.g. CNT) are geometrical space frame structures and can be studied with the help of structural mechanics. In the structural mechanics based approach atomic nuclei are treated as material points. The motion of atomic nuclei is regulated by a force field and this force field is generally expressed as the steric potential energy which depends on the relative position of atomic nuclei. In 2005, Tserpes and Papanikos105 implemented the concept of Li and Chou104 in the finite element method (FEM). In their FEM based approach, bonds are simulated as load carrying elements, and atoms as joints or nodes for connecting these elements as illustrated in Fig. 3. In FEM based approaches the material properties or the stiffness matrix is generated by establishing a linkage between molecular and continuum mechanics.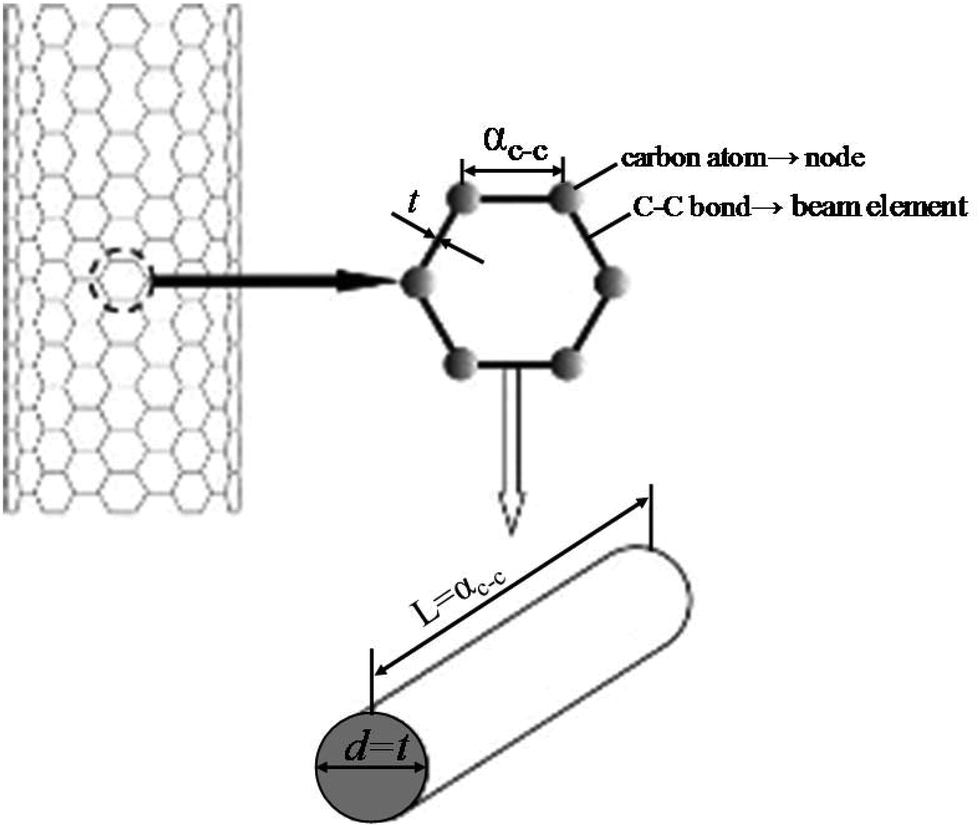 | ||
| Fig. 3 Schematic of the space frame model for CNT (reproduced with permission from ref. 105). | ||
Harmonic, Morse and Lennard Jones LJ 6–12 (van der Waals) like interatomic potentials are commonly employed in FEM based models for generating the stiffness matrix. These interatomic potentials account for all the bonded as well as non-bonded interactions. These structural mechanics based modeling techniques have also been extended by researchers for the modeling of BN nanofillers.106–108 In 2011, Boldrin et al.106 extended the space frame models for the simulation of h-BN nanosheets. They employed molecular mechanics in conjunction with finite element to study the effective mechanical properties of h-BN nanosheets. In addition to these space frame based atomistic models, the continuum based lattice approach has also been employed in conjunction with the Tersoff–Brenner potential for estimating the Young's modulus and Poisson's ratio of boron nitride crystals.109 Numerical techniques, based on the sub-hexagonal lattice-sums,110 have been further extended by Green et al.111 to study the elastic behavior of h-BN. Finite element based continuum models are considered suitable for predicting the average or the global properties of these nanofillers.
2.2. Density functional theory (DFT)
Density Functional Theory (DFT) is a quantum mechanics based higher fidelity simulation technique, and is common among the researchers for estimating the atomic level properties of materials in physics, materials science and chemistry. In DFT, atomic nuclei are usually treated as frozen while electrons are considered to be moving in the electrostatic field generated by the nuclei (Born–Oppenheimer approximation). In DFT based atomistic simulations, electron density is used as its central part to calculate the total energy of the system. This energy E[ρ] is a functional of the electron energy density ‘ρ’ and is expressed as:| E[ρ] = T[ρ] + EH[ρ] + Exc[ρ] + Eext[ρ] + Ezz | (1) |
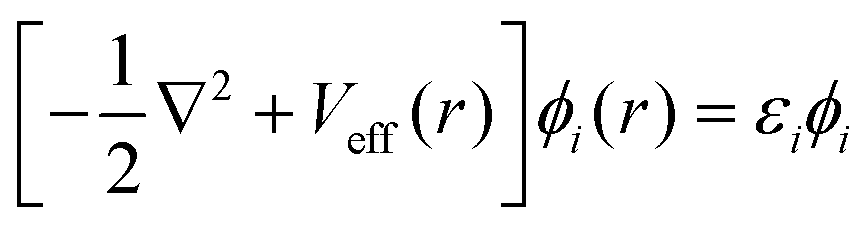 | (2) |
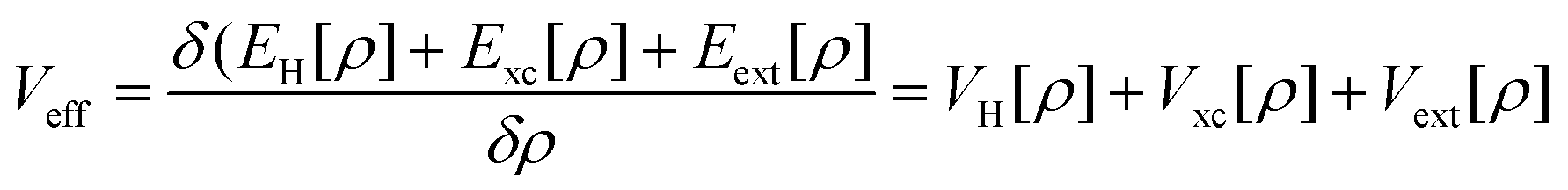 | (3) |
 | (4) |
Due to Kohn–Sham formulations, the multi-electron problem is reduced to one seeking a set of solutions to one electron problems. For solving the equations, an initial guess of electron density “ρ” is made and is used for further calculations. For solving the Kohn–Sham equations, two types of basis sets “atomic orbitals” or “plane wave” are usually used by researchers. In the first case, a linear combination of atomic orbitals (LCAO), such as Gaussian orbitals, is used. LCAO is especially suitable for a study of molecules or systems with strong directional bonding. But the non-orthogonal nature of these orbitals makes the calculations complex and creates practical inconvenience in a way that there is no systematic way that improves accuracy if the number of basis sets is increased. The other common basis set i.e. plane wave, is good for periodic systems (such as solids) but can also be adapted for finite size systems, such as atoms or molecules, with a supercell approach where large periodicity of the simulation box is ensured. The plane wave basis sets are usually used for valence spaces in conjunction with pseudopotentials.
Before solving Kohn–Sham equations, all the energy terms in the equations need to be evaluated. Except for exchange–correlation energy, all other energy terms can be estimated easily. The exchange–correlation energy is usually estimated by using some approximations such as local density approximation (LDA) [(ref. 112) (PW92)–(ref. 113) (PZ81)], generalized gradient approximation (GGA) [(ref. 114) (PBE)–(ref. 115) (LYP)] etc. LDA is the simplest one and neglects the gradients in electron density. Despite being simple, LDA gives quite good results even with this simple approach. Other approximations include meta-GGA [(ref. 116) (TPSS)], hybrid functional [(ref. 117 and 118) (PBE0)] and some meta-hybrid functionals and the choice of one out of them is application specific.
For solid state systems, a suitable method is to use the pseudopotential (PP) approach. In this method, interactions between core electrons and valence electrons are treated with a pseudopotential function. The pseudopotential approach is based on the consideration that core electrons do not participate significantly in the bonding process and can be neglected while solving for the electronic structure of the electrons. On the other hand, valence electrons, which are fewer in number, are treated with plane-wave basis sets. Although higher in number, plane-waves are used in solids because they are cheap to compute. Pseudopotentials may be an empirical pseudopotential or an ab initio pseudopotential. Ab initio pseudopotentials based on local solutions of wave functions near the atomic nuclei are commonly preferred over the empirical pseudopotentials. However, in more accurate DFT implementations, the non-local form of pseudopotentials is used.
The pseudopotential approach is much faster than all-electron calculations because tightly-bound core electrons are not directly taken into the account for calculations. After solving KS equations, the output electron density obtained is used to calculate the total ground state energy of the whole system. This procedure is repeated until a self-consistent solution is achieved. This optimized total ground state energy is further used to estimate the other structural properties of the material. An overview of the DFT process has been shown in Fig. 4. However, solving KS equations for construction of the density matrix from the Fock/Kohn–Sham matrix and then solving for density functional is an expensive process which makes DFT a computationally intensive technique. However, another approach based on DFT, which is faster with surprisingly good accuracy, is the tight-binding (TB) method in which valence electrons are treated by being ‘tightly bound’ to their nucleus and atomic orbitals are not altered during the bonding process. The tight-binding model is based on a set of approximations obtained from quantum mechanics and hence is less computationally intensive. Even though this method is less accurate as compared to ab initio DFT it is sufficiently accurate for the structures to which it is fitted. The method is usually applied to covalent systems such as semi-conductors (Si, Ge) and to carbon. In DFT calculations, mechanical properties are estimated using strain energy (U) obtained by the following equation:
| U = Uε − Uε=0 | (5) |
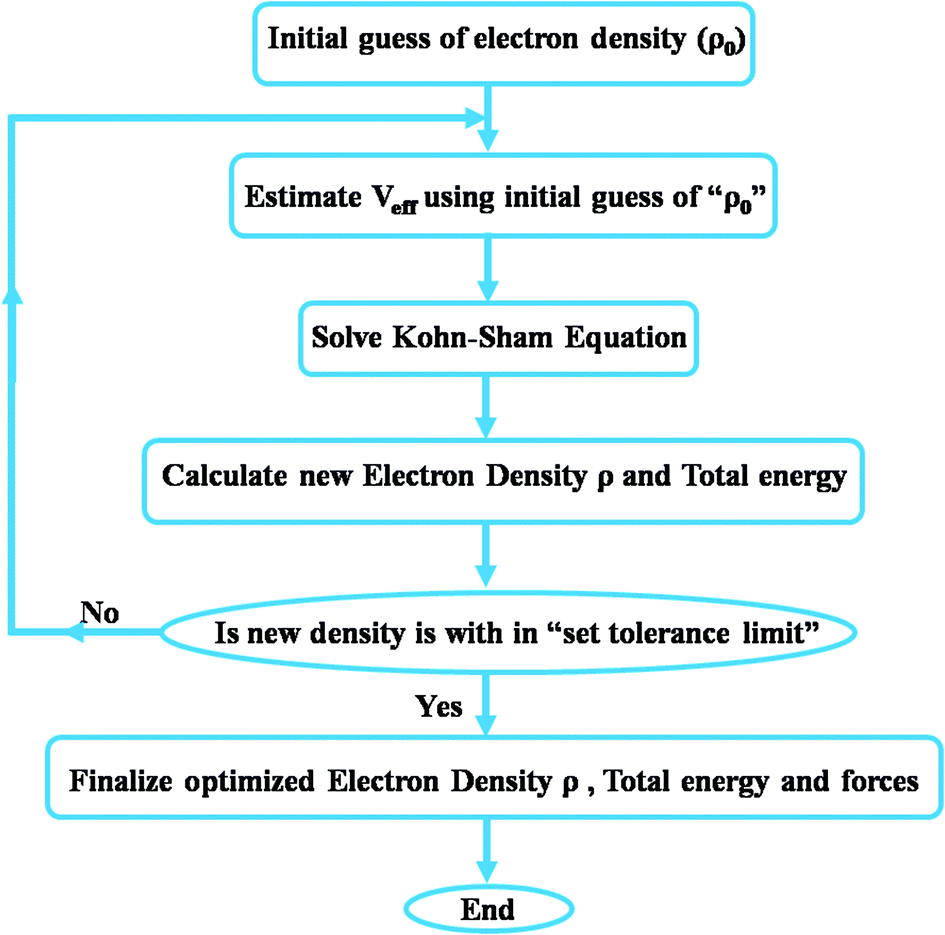 | ||
| Fig. 4 An overview chart of the iterative DFT process. | ||
The value of strained energy is further processed to calculate the Young's modulus (Y in N m−2) and 2D Young's modulus (YS in N m−1) as illustrated in eqn (6) and (7) respectively:
| Y = (∂2U/∂ε2)/V | (6) |
| YS = (∂2U/∂ε2)/S | (7) |
Here, V is the volume and S is the surface area of the system.
With BN nanofillers, DFT and other DFT based approaches have also been used by researchers to estimate the elastic as well as other structural properties.17–20,25,119–123 Kudin and Scuseria17 employed DFT with Gaussian type orbitals and PBE114 approximations to study the elastic behavior of BNNTs. They predicted an average value of nearly 810 GPa for the Young's modulus of the nanotubes which was in close agreement with tight-binding calculations of Hernández et al.124 Later on, Suryavanshi et al.24 also reported a value of nearly 722 GPa for the Young's modulus of BNNTs using experimental techniques. In 2009, Sahin et al.18 used the DFT framework to investigate the elastic and other properties of BN nanosheets. A combination of first-principles plane-wave calculations and projector augmented wave potentials125 was used to ensure accuracy and computation speed. They predicted a value of nearly 810 GPa (assuming a thickness of 0.33 nm for BNNS) for the Young's modulus which was close to the value reported by Bosak et al.100 using inelastic X-ray scattering measurements. Some more research studies19,20,126 performed in the DFT framework also predicted values of the Young's modulus to be nearly close to those reported in the studies mentioned previously in this paragraph. A combination of DFT and quasi-harmonic approximation (QHA)127 was successfully applied by researchers128 to evaluate the effect of temperature on the mechanical behavior of BN nanosheets. It was found that the Young's modulus of boron nanosheets and nanotubes increases with an increase in the temperature up to 800 °C. After this temperature, the rate of increase was slow and the temperature tended toward a constant value.
Although, the DFT based approach has been successfully applied for the atomistic modeling and predicting of properties of materials quite accurately yet it has certain limitations associated with it. As already mentioned generally it is considered computationally intensive and can only be performed with a limited number of atoms. Tight-binding methods are considered good from a computational point of view but these have issues of transferability to other systems and even have the issue of number of atoms. Moreover, DFT based approaches are very useful for developing new empirical interatomic potentials for empirical methods like molecular dynamics. These empirical potentials are considered as the back bone for the less computational intensive molecular dynamics based simulations.
2.3. Molecular dynamics (MD)
As discussed above, FEM and DFT based approaches have been applied pretty much successfully in atomistic modeling, but still have certain limitations. As compared to FEM and DFT based atomistic modeling techniques, the molecular dynamics (MD) based approach is emerging as yet another alternative for performing atomistic simulations.MD is a widely used computer based technique for the atomistic simulation of atoms/molecules in the context of N-body simulation or many body systems.129 In MD based simulations atoms are treated as classical particles. The atomic position and potential energy of the system help in computing the atomic forces that are further used in Newton's equation of motion. The relationship derived from the Newton's second law of motion is used for updating the position, velocity and acceleration of each atom in the system at an integrated time step. The fundamental equation solved in molecular dynamics based simulation is given as:
| mαaα = Fα = −(∂E/∂rα) | (8) |
Here, N is the total number of atoms; mα, rα and Fα are the mass, the position and the time dependent force acting on them due to external agents, respectively. The potential energy ‘E’ consists of an internal part (Eint) that accounts for the interaction between the atoms and an external part (Eext) that accounts for external fields and constraints. The potential energy (E) and hence force Fα are the functions of position and velocity vectors of the atoms. Statistical mechanics based approaches are commonly used over the time averages to derive macroscopic properties such as pressure, temperature, stresses etc.
In MD based simulations, position and velocity vectors are updated after each time step, usually by using any of the numerical integration algorithms such as Verlet algorithm, Velocity-Verlet algorithm,130 Leapfrog algorithm,131 and Beeman's algorithm.132 But the Velocity-Verlet algorithm (an extension of Verlet algorithm) is the most widely used integrating scheme in MD based simulations. The position vector ‘r’ at any time t and after an increment Δt, for any atom involved in the simulation can be expanded as a Taylor series expansion as given by eqn (9).
r(t + Δt) ≈ r(t) + ṙ(t)Δt + ![[( with combining umlaut]](https://www.rsc.org/images/entities/char_0028_0308.gif) (t)Δt2)/2 + … (t)Δt2)/2 + … | (9) |
The updated velocity vector after time ‘t + Δt’ can be easily obtained with the help of the Velocity-Verlet equation as:
| v(t + Δt) = v(t) + (F(t + Δt) + F(t)/2m)Δt | (10) |
MD simulations are carried out under different types of ‘Ensembles’. An ensemble is a collection of all possible different microstates of a system which have an identical macroscopic state. Micro-canonical (NVE), canonical (NVT) and isobaric–isothermal (NPT) ensembles are the three most commonly used ensembles in MD simulations.
Most of the interatomic potentials employed in MD based simulations are either derived or developed empirically with the help of experiments or higher fidelity numerical simulations. Interatomic potentials are mathematical expressions for estimating the potential energy (E) of the system of atoms. Commonly used interatomic potentials are Tersoff,133–136 LJ,137 the embedded atom method (EAM),138 REBO139 and AIREBO.140 Tersoff type133–136 interatomic potentials are commonly used for simulating the mechanical and thermal behaviors of BN nanofillers. The Tersoff potential can be expressed mathematically as given by eqn (11) and (12):
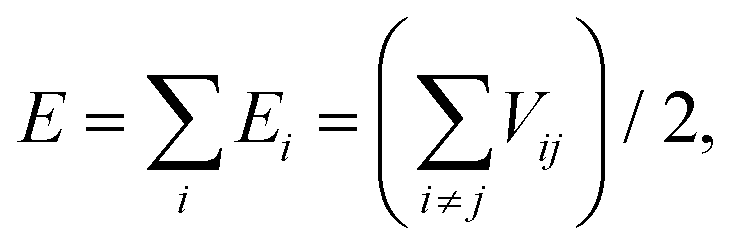 | (11) |
| Vij = fc(rij)[aijfR(rij) + bijfA(rij)]. | (12) |
Here, E is the total energy of the system, which is decomposed, for convenience, into site energy Ei and bond energy Vij. The indices i and j run over the atoms of the system, and rij is the distance between atoms i and j. The functions fR and fA represent the repulsive and attractive parts of the pair potential. The functions fc and bij are the cutoff function and the bond-order function respectively.
Significant progress has been made in the development of interatomic potentials for estimating the behavior of BN nanofillers with improved accuracy. For the modeling of BN nanofillers in a MD based environment, most of the researchers have employed either Tersoff or a modified form of the Tersoff potential.21,141,142 A harmonic force field has also been employed by a couple of researchers for managing the atomic interactions in BN nanofillers.22,143 The Tersoff potential that was initially introduced in ref. 133 has been further modified by Brenner144 to include additional terms for the inherent over-binding of radicals and for non-local effects. In addition to Brenner,144 various researchers have also proposed optimized Tersoff potential parameters for improving the accuracy of atomistic simulations. Sekkal et al.145 proposed Tersoff parameters for boron by fitting the bulk properties of cubic BN. In their simulations, BN was treated as a single entity, hence, an identical set of parameters was proposed for both boron and nitrogen. Tersoff potential parameters proposed by Sekkal et al.145 are limited to BN interactions only, and cannot be extended to study interactions of boron or nitrogen with other atoms. These potential parameters145 have been successfully employed by many researchers for predicting the structural and thermal properties of BN nanofillers. Sevik et al.146 proposed parameters for the Tersoff potential that effectively capture the experimental phonon dispersion in BN. Due to the capability of these parameters to effectively capture phonon dispersion, they are considered more suitable for estimating the lattice thermal conductivity of BN based nanostructures. Matsunaga et al.147–149 proposed separate sets of parameters for boron, nitrogen and carbons atoms. This set of potential parameters is considered more suitable for reproducing experimentally measured lattice constants, bulk modulus and the binding energy of cubic boron nitride. In addition to these parameters, Albe et al.150,151 also proposed Tersoff potential parameters for bulk BN materials to simulate ion-beam deposition of BN thin films. One big advantage with parameters provided by Albe et al.150,151 is that they account for the sp2 structure of BN bonding. The parameters proposed by Sekkal et al.,145 Sevik et al.,146 Matsunaga et al.,147–149 and Albe et al.150,151 have been summarized in Table 1.
| Parameter | Sekkal et al.145 | Sevik et al.146 | Matsunaga et al.147 (χi−j = 1.1593) | Albe et al.150 | |
|---|---|---|---|---|---|
| B–N | B–N | B–B | N–N | B–N | |
| m | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 |
| Γ | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| λ3 | 0.0 | 0.0 | 0.0 | 0.0 | 1.9925 |
| c | 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 049 049 |
25000 |
0.52629 | 79934 |
1092.9287 |
| d | 4.384 | 4.3484 | 0.001587 | 134.32 | 12.38 |
| h | −0.57058 | −0.89 | 0.5 | −0.9973 | −0.5413 |
| n | 0.72751 | 0.72751 | 3.9929 | 12.4498 | 0.364 53367 |
| β | 0.00000015724 | 0.0000001257 | 0.0000016 | 0.10562 | 0.000011134 |
| λ2 | 2.22 | 2.199 | 1.5856 | 2.5115 | 2.784247207 |
| B | 346.7 | 340 | 183.49 | 219.45 | 3613.431337 |
| R | 1.95 | 1.95 | 1.95 | 2.15 | 2.0 |
| D | 0.2 | 0.05 | 0.15 | 0.15 | 0.1 |
| λ1 | 3.468 | 3.568 | 1.9922 | 5.7708 | 2.998355817 |
| A | 1393.6 | 1380 | 277.02 | 11000 |
4460.833973 |
Due to accurate parameterization that helps in achieving accurate fit with the DFT calculations and experimental results, Tersoff type potentials have attracted more attention among the researchers for simulating the bonded interactions. On the other hand, reactive force field (ReaxFF) potentials are common among researchers for simulating the complex atomic structures of hydrocarbons.152–154 In addition to hydrocarbons, ReaxFF has also been applied for BN nanofillers and will be discussed further in the next section. For ReaxFF potentials, parameters are obtained from fittings to the results of quantum chemical analysis. Similar to other empirical non-reactive potentials, the total energy of the system is derived from various partial energy contributions (bonded as well as non-bonded) as expressed in eqn (13).
| Esystem = Ebond + Eover + Eunder + Eval + Epen + Etors + Econj + EvdWaals + EColumb | (13) |
ReaxFF type potentials are reported to be more suitable for simulating bond-breaking and bond-forming mechanisms. The fundamental difference between ReaxFF and most of the other empirical interatomic potentials is that ReaxFF does not employ any fixed connectivity for the bonded interaction. In ReaxFF type potentials the bond order is directly calculated from the instantaneous interatomic distance rij, which keeps on updating during the simulation. This bond order is responsible for the creation and dissociation of bonds during the simulation. The mathematical expression for calculating the bond order with respective details can be found in the papers.152–154
In MD based simulations, strain calculations are performed with the help of initial (Li) and final (Lf) lengths of the simulation box as expressed by eqn (14). The concept of stress in a material or a body is well known at the continuum level and is usually referred to as the Cauchy stress tensor. However, at the atomistic level the principles of continuum are not applicable, and hence an alternative stress referred to as virial stress is estimated at the atomistic level. Virial stress is the most commonly used definition of stress in discrete particle systems. Virial stress is the summation of two components. The first component depends upon the mass and velocity of the atoms in the MD simulations, whereas the second component depends upon the interatomic forces and atomic positions. The mathematical expression for virial stress as the summation of two components is given by eqn (15).
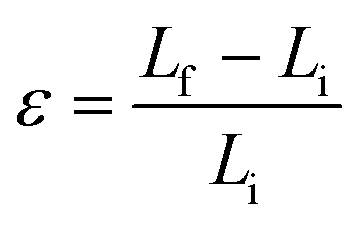 | (14) |
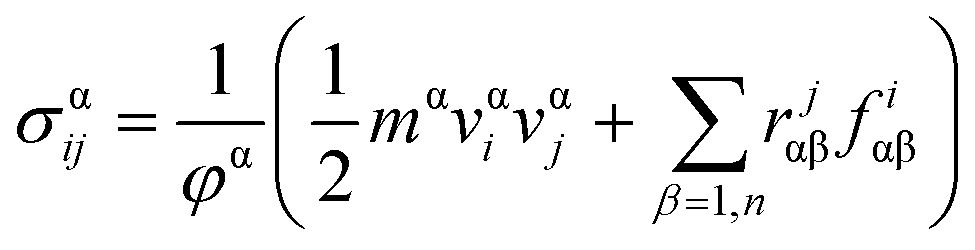 | (15) |
The strain energy based concepts, as discussed in the DFT section, are also applicable in MD simulations for estimating the Young's modulus and 2D Young's moduli values of the system as per eqn (5)–(7). In addition to predicting the mechanical behavior of an atomistic system, the MD based simulations have also been employed for predicting the thermal behavior of the system. Thermal conductivity (k) of a simulated atomistic configuration can be estimated by using the mathematical expressions given below:
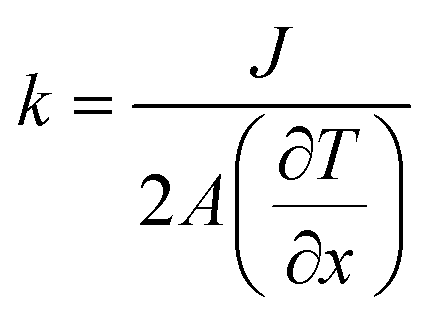 | (16) |
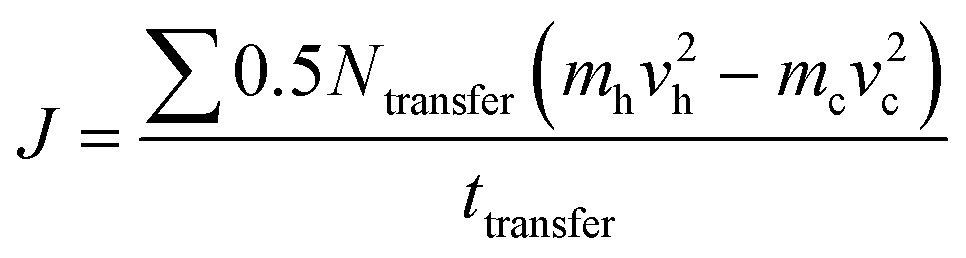 | (17) |
Here, in eqn (16) and (17), 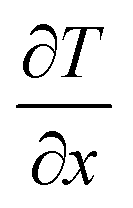 refers to the temperature gradient along the direction of heat flux (J), A is the cross sectional area, h and c refer to the hot and cold atoms, and Ntransfer and ttransfer are the summation time and the number of exchanges respectively.
refers to the temperature gradient along the direction of heat flux (J), A is the cross sectional area, h and c refer to the hot and cold atoms, and Ntransfer and ttransfer are the summation time and the number of exchanges respectively.
3. MD based simulations for BN nanofillers
As described in previous sections atomistic modeling techniques are emerging as a potential tool for predicting the mechanical and thermal behaviors of BN nanofillers. MD based simulations are considered sufficiently accurate and less computationally intensive as compared to FEM and DFT based approaches, respectively. In this part of the article, the authors have made an attempt to review the current state of art techniques for simulating the mechanical as well as thermal behavior of BN nanofillers with the help of molecular dynamics.3.1. Mechanical behavior
The mechanical behavior of BN nanofillers has been widely investigated by many researchers. Among various mechanical properties, Young's modulus is considered as the most widely predicted or estimated property of any material. Mechanical properties of BN nanofillers (BNNS or BNNT) predicted or estimated by using different means have been compiled in Tables 2 and 3. It can be inferred from these tables that researchers have simulated BN nanofillers both as an isotropic as well as an in-plane orthotropic material (arm-chair and zig-zag configurations). While treating BNNS as an isotropic material,18–20,75,100,106,109,111,155–157 researchers have predicted a range of Young's modulus starting from 0.79 TPa to 0.97 TPa (assuming 0.33 nm as the thickness of a BNNS). Similarly, for BNNTs, the Young's modulus values with isotropic material properties range from 0.77 TPa to 1.2 TPa.16,23,24,109,142,158 In addition to the Young's modulus, researchers have also predicted the 2D Young's modulus for BN nanofillers as tabulated in Table 3. At the atomistic level, these nanofillers are not isotropic in nature and usually have in-plane orthotropic properties that are dependent on the chirality vector. Based on the alignment of the bonds with the loading direction, BN nanofillers (BNNT or BNNS) can be further classified as zig-zag and armchair configurations. The Young's modulus (Y) as well as 2D Young's modulus (YS) for zig-zag and armchair directions has also been summarized in Tables 2 and 3, respectively.17,21,22,26,124,126,143,159–161 It can be inferred from these tables that the 2D Young’ modulus (YS) predicted through atomistic simulations and experimental techniques lies in the range of 200 N m−1 to 500 N m−1.75| Method | Reference | Nanofiller | Young's modulus (TPa) | Fracture stress (GPa) | Fracture strain (%) | Poisson's ratio |
|---|---|---|---|---|---|---|
| AC stands for Armchair and ZZ stands for Zigzag. | ||||||
| Experimental | Chopra et al.23 | BNNT | 1.22 ± 0.24 | — | — | — |
| Suryavanshi et al.24 | BNNT | 0.722 | — | — | — | |
| Wei et al.158 | BNNT | 0.895 | 33 | 3 | — | |
| Arenal et al.16 | BNNT | 1.11 ± 0.17 | — | — | — | |
| Bosak et al.100 | BNNS | 0.811 | — | — | — | |
| DFT | Hernández et al.124 | BNNT | 0.884ZZ | — | — | 0.23–0.27 |
| 0.912AC | ||||||
| Fakhrabad et al.25 | BNNT | 0.821AC | ||||
| 0.764AC | — | — | — | |||
| Ohba et al.155 | BNNS | 0.952 | — | — | — | |
| Mirnezhad et al.156 | BNNS | 0.829 | — | — | 0.21 | |
| Zhang et al.142 | BNNT | 0.850 | ||||
| MD | Verma et al.26 | BNNT | 0.982ZZ | — | — | 0.13ZZ |
| 1.111AC | 0.16AC | |||||
| BNNS | 1.004ZZ | — | — | — | ||
| 1.111AC | ||||||
| Mortazavi et al.159 | BNNS | 0.800ZZ | 165 | 30 | — | |
| 0.850AC | ||||||
| Zhao et al.160 | BNNS | 0.693ZZ | 120 | 28 | — | |
| 0.740AC | ||||||
| Han et al.157 | BNNS | 0.881 | 133 | 33 | — | |
| Structural mechanics & others | Eun-Suok Oh109 | BNNS | 0.977 | — | — | 0.17 |
| BNNT | 0.950 | |||||
| Boldrin et al.106 | BNNS | 0.797 | — | — | 0.21 | |
| Green et al.111 | BNNS | 0.803 | — | — | — | |
| Method | Reference | Nanofiller | Young's modulus (N m−1) | Fracture stress (N m−1) | Fracture strain (%) | Poisson's ratio |
|---|---|---|---|---|---|---|
| AC stands for Armchair and ZZ stands for Zigzag. | ||||||
| Experimental | Li et al.75 | BNNS | 200–500 | — | 22 | — |
| DFT | Kudin et al.17 | BNNT | 266ZZ | — | — | 0.21 |
| 267AC | ||||||
| Sahin et al.18 | BNNS | 267 | — | — | 0.21 | |
| Topsakal et al.126 | BNNS | 258AC | — | — | — | |
| Andrew et al.19 | BNNS | 276 | — | — | 0.22 | |
| Peng et al.20 | BNNS | 278 | — | — | — | |
| MD | Minh-Quy Le21 | BNNS | 263ZZ | 36ZZ | 23ZZ | — |
| 253AC | 30AC | 17AC | ||||
| Minh-Quy Le22 | BNNS | 284AC & ZZ | — | — | — | |
| BNNT | 283AC & ZZ | |||||
| Minh-Quy Le143 | BNNS | 296ZZ | — | — | — | |
| 286AC | ||||||
| Le and Nguyen161 | BNNS | 258ZZ | 38ZZ | 26ZZ | — | |
| 251AC | 36AC | 26AC | ||||
| 257ZZ | 31ZZ | 17ZZ | — | |||
| 249AC | 30AC | 18AC | ||||
| 257ZZ | 35ZZ | 21ZZ | — | |||
| 251AC | 32AC | 20AC | ||||
| 258ZZ | 31ZZ | 17ZZ | — | |||
| 251AC | 33AC | 22AC | ||||
| Structural mechanics | Minh-Quy Le143 | BNNS | 332 | — | — | — |
Le21 performed MD simulations using Tersoff-potential parameters provided by Sevik et al.146 to predict mechanical properties of BNNSs. In this study, the author extended the small cut-off distance to a large cut-off distance and obtained lower values of fracture stress and strains as compared with earlier studies.159 The fracture strain predicted by Le21 was in good agreement with DFT calculations performed by Topsakal et al.126 Molecular dynamics based simulations has been performed by Verma et al.26 to estimate the Young's modulus and shear modulus of BN nanofillers. In their computational work, interatomic interactions between boron and nitrogen atoms were estimated with the help of Tersoff-type potentials134,135 with parameters obtained from the work of Sekkal et al.145 The potential parameters provided by Sekkal et al.145 were slightly modified by Verma et al. to obtain the best fit for the bond length and cohesive energy of the h-BN sheet. Verma et al.26 predicted the values of the Young's modulus for BNNT as 1.11 TPa and 0.98 TPa in the armchair and zig-zag directions respectively. Nearly same values of Young's modulus were predicted for BNNSs i.e. 1.11 TPa and 1.0 TPa in the arm-chair and zig-zag directions, respectively. They further validated their computational model with the help of experimental results reported in the work of Chopra et al.23 It was concluded in the research work of Verma et al.26 that the Young's modulus of small diameter BNNTs increases with an increase in the diameter and attains a maximum value of 1.11 TPa at a radius of 0.95 nm, and then falls with a further increase in the diameter as illustrated in Fig. 5 (left). On the other hand, shear modulus decreases with an increase in the diameter of the BNNT and attains a constant value at a higher tube diameter as shown in Fig. 5 (right).
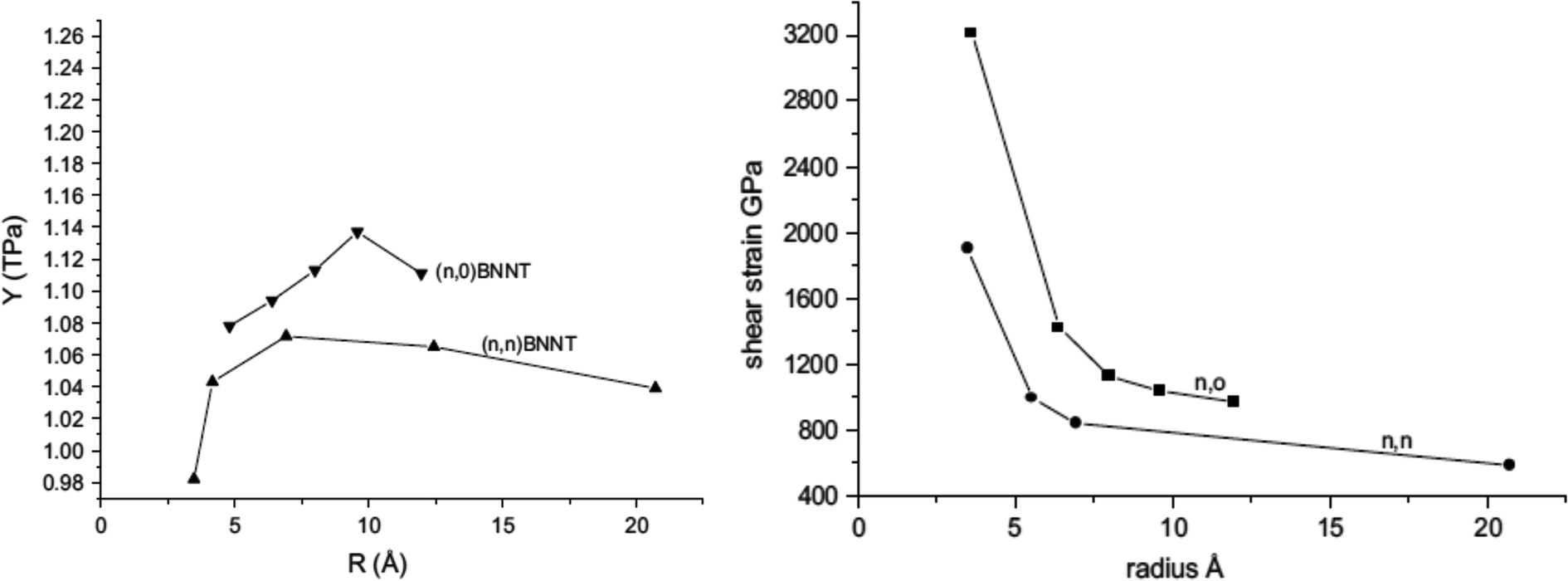 | ||
| Fig. 5 Variation of Young's modulus (left figure) and shear strain (right) with respect to the radius of BNNT (reproduced with permission from ref. 26). | ||
Zhang et al.142 studied the intrinsic twisting of helical BNNTs with the help of the tight-binding MD [TBMD] method, and estimated Young's and shear modulus values, as 0.850 and 0.370 TPa, respectively. In 2012, Mortazavia and his research team159 performed MD based simulations to study the tensile behavior of BNNSs. In their computational work, the Tersoff potential was used in conjunction with optimized parameters obtained from the work of Matsunaga et al.147,149 It was predicted in their computational work that BN nanostructures failed in a brittle manner, and had their Young's modulus in the range of 0.8–0.85 TPa.
MD based studies162,163 have also been performed to investigate the effect of grafted carboxyl groups on the elastic properties of BNNTs. Yuan and Liew162 studied the effects of grafting carboxyl groups on the elastic properties of BNNTs. It was predicted in their computational work that the Young's moduli of nanotubes started deteriorating with the attachment of a carboxyl group. The zig-zag configuration of BNNTs was reported to be affected the most as compared to the armchair configuration.
It was further concluded in the research of Yuan and Liew162 that the reduction in the elastic modulus for the zig-zag direction was least affected by the quantity of attached carboxyl groups, whereas the reduction in the armchair direction depends on the quantity of the chemical group. Recently in 2014, Ghazizadeh et al.163 conducted a MD based study on the mechanical properties of hydrogenated BNNTs. MD simulations were performed in conjunction with the universal force field (UFF) and Dreiding potentials.164–166a It was concluded in their research that the Young's modulus of BNNTs showed a decreasing trend when hydrogen atoms were bonded with all available nitrogen atoms.
3.2. Buckling behavior
In 2001, Srivastava et al.166b investigated the stiffness and plasticity of BNNTs. They studied the implications of rotated BN bonds on the buckling behavior of zig-zag nanotubes, and reported anisotropic strain release followed by anisotropic plastic buckling for axially compressed BNNTs. In their research paper,166b they also proposed a “skin-effect” model for smart nano composite materials that can localize structural damage towards the skin or the surface side of the material. Nejad et al.167 also investigated the buckling behavior of open-ended boron nitride nanotubes. They performed MD based simulations with the help of optimized Tersoff potential parameters obtained from the work of Liao et al.168 In their conclusion, it was reported that hydrogen physisorption decreased the buckling stability of BNNT by 10% in the temperature range of 300–3000° K as compared to pristine BNNTs. Hai-Jun Shen169 performed MD based simulations to compare the effect of radial compression on BNNTs and CNTs. The author predicted a comparable radial compressive stiffness for both CNTs and BNNTs. However, it was also observed in their simulations that BNNT had lower energy absorbing, load-support and deformation-support capabilities as compared to CNT. Nejad et al.170 examined the behavior of fixed length BNNTs with varying diameters. MD simulations were performed with the help of Lennard-Jones137 and harmonic potentials. It was concluded in their numerical simulations that critical buckling loads exhibited an increasing trend with an increase in the nanotube diameter.Slotman and Fasolino171 compared the bending rigidity of BNNTs with that of graphene. Simulations were performed with the help of the Tersoff potential136 in combination with optimized parameters obtained from the work of Albe et al.151 It was also predicted in their computational model that BNNSs had lower bending rigidity as compared to that of graphene sheets. In 2010, Liao et al.172 investigated the effects of temperature on the tensile and compressive behavior of BNNTs. They performed MD simulations in conjunction with the Tersoff potential parameters proposed by them after fitting MD simulation results to the one obtained from density functional theory. These potential parameters were suggested to be more suitable for the simulation of nano-sized BNNTs and also for simulating their bond breaking behavior. It was reported in their work that both failure as well as buckling strain decreased with an increase in temperature. It was further noted from their computational model that BNNTs failed in a chain like mode without any obvious yielding in tension, as shown in Fig. 6 whereas no chain formation was observed under compressive loading. Shokuhfar and his team173 investigated the effect of temperature (up to 3000° K) on the buckling behavior of BNNTs using potential parameters provided by Albe et al.151 It was predicted in their computational model that higher modeling temperatures reduced the value of critical buckling load as well as the critical buckling strain. A similar kind of study on BNNSs was performed by Yuan and Liew.174 They performed their investigation with the help of MD simulations in conjunction with the universal force field (UFF).164 A higher value of buckling strain was predicted in the MD simulations for the zig-zag configuration of BN nanofillers as compared to the armchair one.
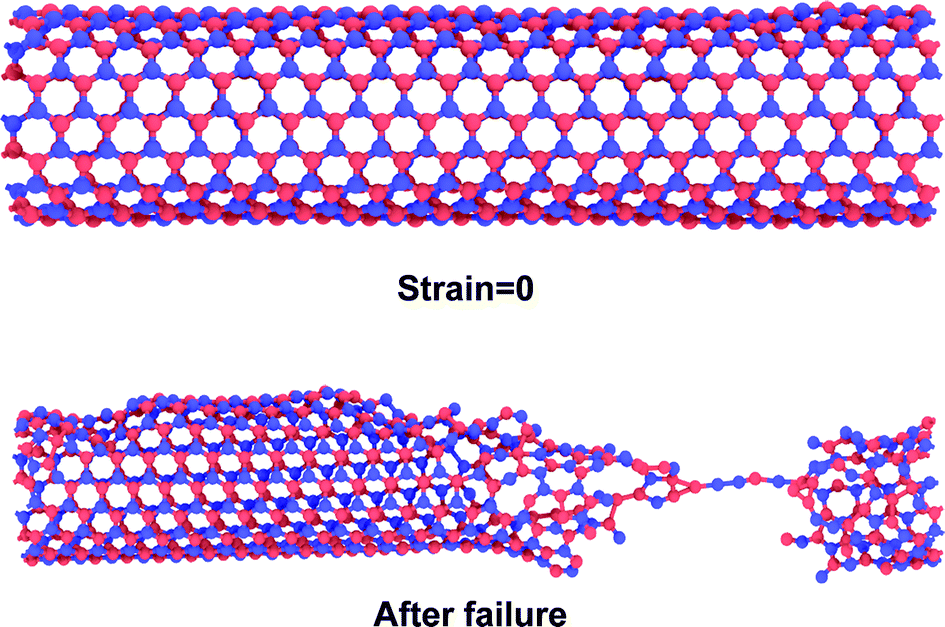 | ||
| Fig. 6 Deformation morphology of a BNNT subjected to tensile loading with a chain (adapted from ref. 172, simulations, with similar conditions as in ref. 172, were performed by authors of this paper to reproduce this image). | ||
3.3. Defects in BN nanofillers
BN nanofillers prepared experimentally are not flawless and usually contain some native defects such as vacancies, dislocation, Stone Wales defects etc. The existence of defects in BN nanofillers has already been established by experimental as well as theoretical studies.175,176 The following section reviews MD based studies conducted on defective BN nanofillers and the effects of these defects on their mechanical and thermal properties.Griebel et al.177 used MD based simulations in conjunction with the Parrinello-Rahman178 approach to study the impact of vacancy defects and functionalization on the Young's modulus of BNNTs. In their numerical work, the Tersoff potential in conjunction with parameters provided by Matsunaga et al.147 was used to estimate interactions between boron and nitrogen. In the original parameters proposed by Matsunaga et al.147 the possibility of bond formation between same atoms was excluded by only accounting for repulsive forces. But in the work of Griebel et al.177 the potential parameters were adapted to deal with B–B and N–N bonds as well. In order to account for homo-elemental bond formation the attraction term was also included in the potential function.
In their work, it was predicted that the Young's modulus of BNNTs starts deteriorating with an increase in concentration of vacancy defects. This reduction in the Young's modulus was found to be independent of the tube-chirality. In order to better visualize the mode of failure with single defects in BN nanotubes with different chiralities, snapshots from the simulation box at different levels of strain are shown in Fig. 7. Griebel and his team177 also investigated the effect of functionalization on the Young's modulus of BNNTs, and summarized the effect of functionalization as shown in Fig. 8. A general decrease in the value of Young's modulus up to 2/3rd of pristine BNNTs can be inferred from the plotted data in Fig. 8. No significant effect of functionalizing was observed in the range of 60% to full functionalization for the zig-zag configuration, whereas a slight increase in the Young's modulus was reported for the armchair configuration between the same ranges of functionalization.
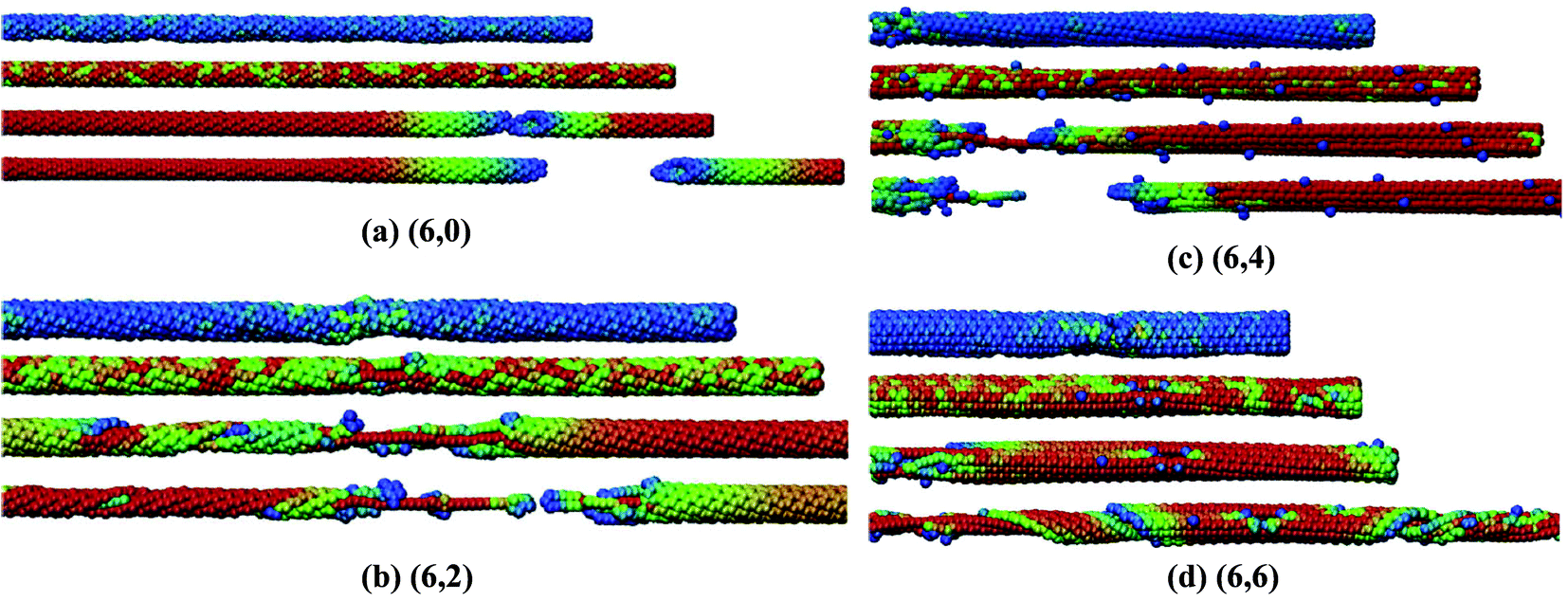 | ||
| Fig. 7 Stress induced deformation and failure of BNNT with different chiralities. Color scheme refers to the level of stress per atom values from blue over green to red. Four snapshots for each chiral nanotube refer to, no strain, strain before, at and after failure states (reproduced with permission from ref. 177). | ||
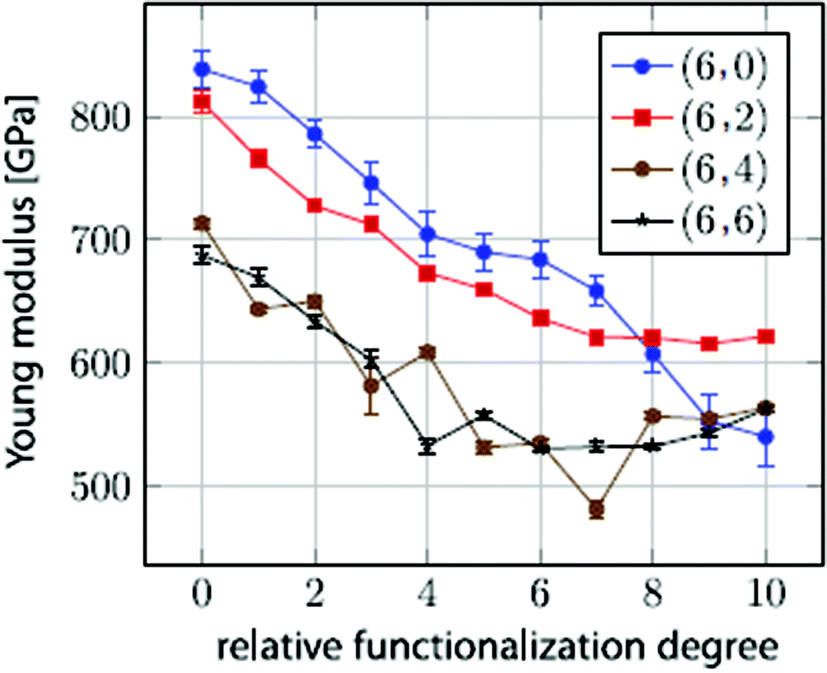 | ||
| Fig. 8 Young's modulus estimated as a function of the relative functionalization degree for (6, m) BN nanotubes (reproduced with permission from ref. 177). | ||
Shokuhfar et al.179 carried out MD simulations with GROMACS harmonic potential parameters for bonded interactions. In their MD based simulations the harmonic potential parameters were obtained by fitting the potential parameters of Liao et al.168 to the DFT calculations. In their simulations, the NVT ensemble was enforced to study the effect of vacancy on the critical buckling performance of BNNTs, and it was reported in the conclusion that the B–N-double vacancy was most detrimental to the structural strength, followed by B-vacancy and N-vacancy defects. It was also reported in their work that B-type vacancy was most detrimental to the fracture strain. In order to visualize the failure modes under compressive loading, snapshots from the simulations of BNNTs at the time of failure are shown in Fig. 9. It can be inferred from the snapshots that the failure morphology of BNNTs is chirality dependent.
 | ||
| Fig. 9 Defective BNNT after compressive buckling (a) (10,10), (b) (5,5), (c) (7,7) (reproduced with permission from ref. 179). | ||
Le and Nguyen161 examined the effects of vacancies and Stone–Wales (SW) defects on the tensile properties of h-BN nanosheets. The Tersoff potential in conjunction with parameters obtained from Sevik et al.146 was employed to simulate atomistic interactions. In order to avoid the overestimation of the maximum force to break the bond the cutoff function was removed from the potential function (fc(rij) = 1). The notion that overestimation of maximum forces can be avoided by removing the cutoff function has been proposed by the author with the help of Fig. 10. It can be inferred from their plotted data in Fig. 10 that the potential energy and force reduce smoothly to zero as the bond strain increases with the removed cutoff function. Le and Nguyen161 employed finite element in conjunction with the MD based simulations. The relaxed atomic positions of B and N atoms were determined with the help of MD simulations. It was concluded in their computational model that a centrally located single-defect noticeably reduced the fracture stress and strain values, as compared to those of pristine sheets. The Young's modulus of BNNSs was reported to be unaffected due to the presence of a single vacancy but a di-vacancy had detrimental effects on the values of fracture stress and strain. It was further observed that reduction in fracture properties was dependent on the applied loading direction and orientation of Stone–Wales defects.
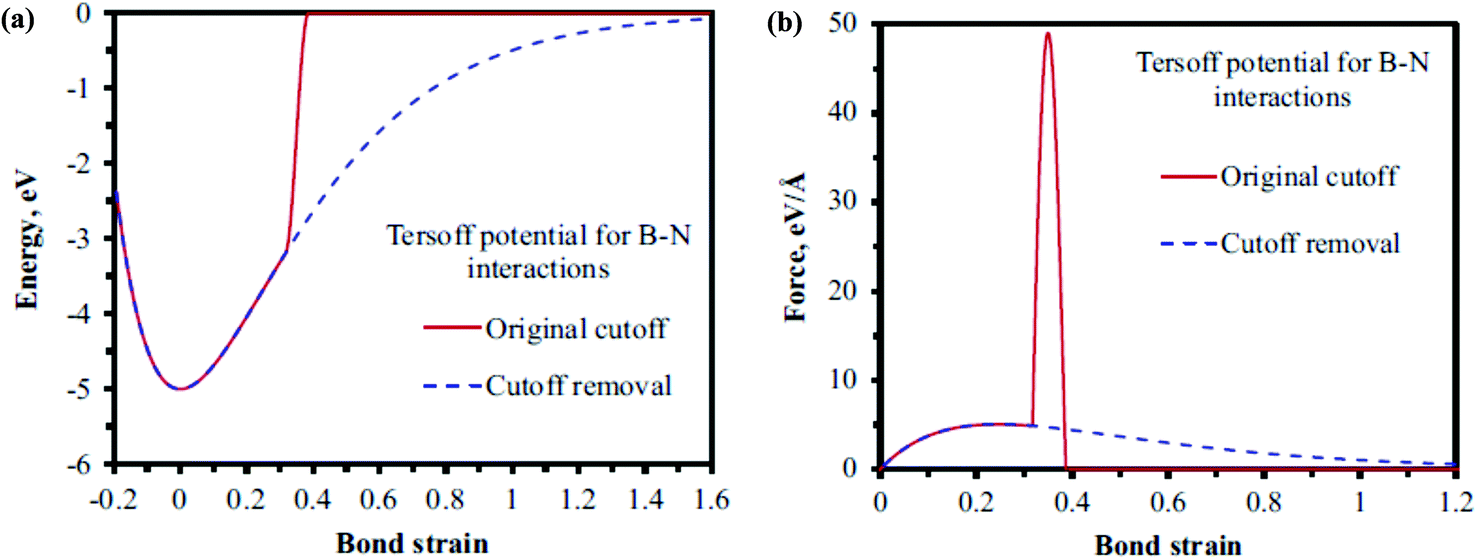 | ||
| Fig. 10 (a) Potential energy vs. bond strain. (b) Force vs. bond strain, on keeping the bond angle value constant according to the Tersoff potential (reproduced with permission from ref. 161). | ||
Recently in 2014, Sarma and his team180 used the MD based simulations in conjunction with reactive force field potentials (ReaxFF) with parameters taken from the work of Weismiller et al.153 It was predicted in their simulations that the point and line defects significantly reduced the tensile strength of BNNTs. Tube diameter and simulation temperature also had a significant effect on the tensile behavior of BNNTs. Similar to the work of Sarma et al.180 MD based simulations with the Tersoff–Brenner potential144 were also performed by Krishnan et al.181 to investigate the effects of vacancy and Stone–Wales (SW) defects on the failure behavior of BNNTs. The simulations were performed with the help of potential parameters proposed by Sevik et al.146 It was concluded in their work180 that BNNTs in the armchair configuration were highly sensitive to defects, whereas the zig-zag configurations were the least sensitive.
Pristine nanotubes (both armchair as well as zig-zag) failed in a brittle manner whereas in the case of defective nanotubes only the zig-zag configuration of nanotubes underwent brittle failure. The defect-induced plastic behavior was also observed in armchair BNNTs. The activation of different failure mechanisms in the zig-zag and arm-chair configurations of BNNTs resulted in plastic failure for the armchair configuration and brittle failure for the zig-zag configuration. In the armchair configuration of BNNTs, once the bond in pentagon breaks it leads to a failure of bonds oriented at an angle of 30° to the axis of BNNTs as shown in Fig. 11(a). This failure of bonds aligned at an angle leads to the formation of decagon-chains, as shown in Fig. 11(b). This sequence of events in the armchair configuration induces a plastic behavior in BNNTs. This plastic behavior due to SW defects had a strong radius-dependency in the armchair configuration of BNNTs, and turned into brittle behavior with radii greater than 2.5 nm as in the case of (20, 20) BNNTs. In the case of the zig-zag configuration of BNNTs, formation of a decagon due to the failure of the B–B bond is followed by the failure of the N–N bond which in turn is followed by the failure of the N–N bond and then rotation of the bond between B–N. This bond rotation further resulted in the formation of two new B–N bonds. The formation of these two new bonds brought back the pristine form and resulted in a brittle failure as compared to the armchair configuration.
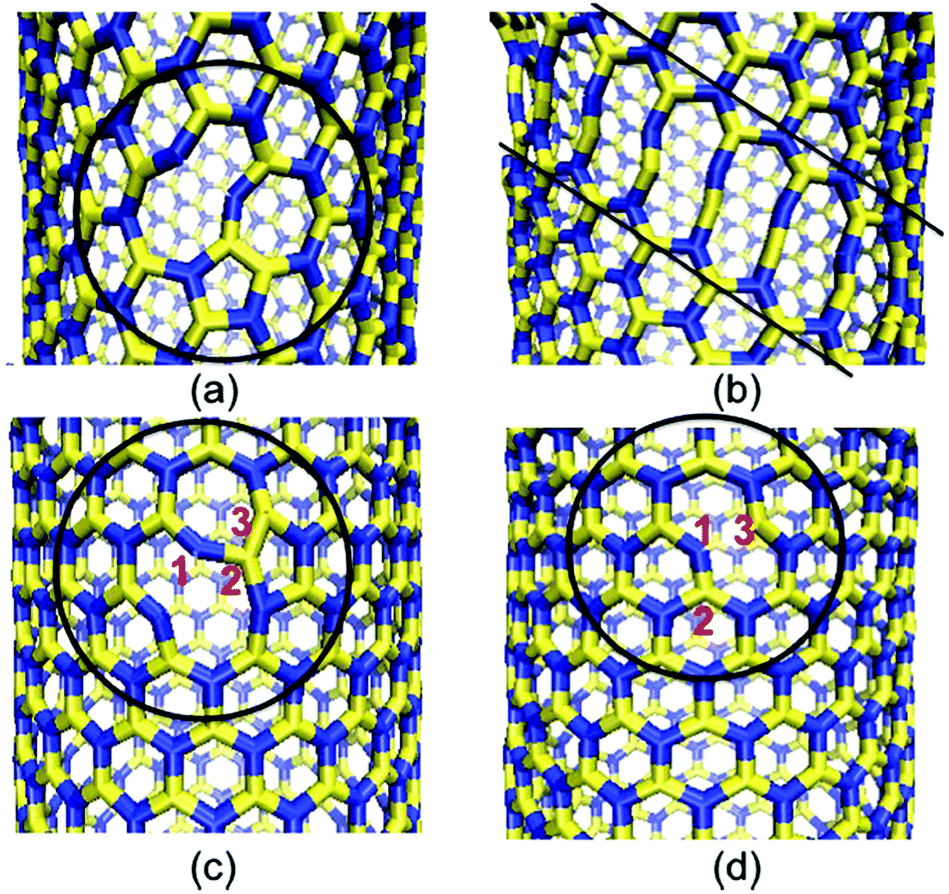 | ||
| Fig. 11 (a) Breaking of the B–B bond in the 5-7-7-5 defect in the arm chair configuration. (b) Formation of a decagon with the propagation of bond breaking. (c) Breaking of the B–B bond in the zig-zag configuration. (d) Rotation of defective B–N bonds to regain the pristine form (reproduced with permission from ref. 181). | ||
Mortazavi and Cuniberti (2014)182 examined the properties of polycrystalline BN nanosheets using the Tersoff-potential134 in conjunction with optimized parameters proposed by Matsunaga et al.149 They concluded in their work that a reduction in the grain size ultimately resulted in a gradual decrease in elastic moduli of polycrystalline films. They also suggested that experimentally fabricated polycrystalline BN nanosheets might possess remarkably high mechanical properties. Moon and Hwang183 performed MD based simulations using the Tersoff potential133–136 in combination with parameters proposed by Albe et al.150,151 to examine the structural and thermal behavior of BNNTs. It was reported in their computational work that the strain energy had a decreasing trend with an increase in the nanotube diameter. The strain energy was found to be proportional to the inverse of the square of the tube diameter. At higher temperatures, Stone–Wales SW defects were found in nanotubes during their thermal treatment. Also the formation energy of SW defects increased with an increase in the diameter of the nanotubes.
3.4. Failure/fracture
In addition to the elastic properties of BN nanofillers, their fracture behavior has also been investigated by many researchers with the help of a MD based platform. In 2011, Liao et al.168 investigated the deformation of armchair BNNTs on being subjected to axial tensile strains using a modified version of Tersoff potential parameters developed by Albe et al.150 The modified set of parameters predicted a better bond breaking behavior. A chain like failure mode (as discussed and explained earlier w.r.t. ref. 172), without any noticeable yielding, was reported in their computational work. Wei et al.184 examined and compared the behavior of CNTs and BNNTs subjected to tensile as well as compressive loading. In their atomistic simulation, it was concluded that under compression, the zig-zag configuration of CNTs as well as BNNTs had a higher Young's modulus as compared to the armchair configuration of the respective nanotubes. It was also concluded from the computational model of Wei et al.184 that the fracture behavior of CNTs or BNNTs is dependent on the chirality, and the nanotubes with the same chirality had similar deformation patterns regardless of the material.Tian et al.185 investigated the impact of shear displacement on the mechanical properties, wrinkling patterns and fracture behavior of h-BN nanosheets. In order to study the wrinkling in a single BN nanosheet, an analytical model was developed with geometrical parameters and boundary conditions as shown in Fig. 12. In Fig. 12, L, H and t are the length, width and thickness of the plane, respectively, E and ν are the Young's modulus and Poisson's ratio, respectively. The schematic of one wrinkle in a nanosheet is shown in Fig. 12, where σξ and ση are the applied stresses in ξ and η directions respectively. The mathematical expressions for shear stresses σξ and ση are provided in eqn (19), where γ is referred to as δ/H.
 | ||
| Fig. 12 Single wrinkle in a nanosheet (reproduced with permission from ref. 185). | ||
The out of plane displacement ω with respect to the mode shape is given mathematically by eqn (18). In this eqn (18), A and λ refer to the amplitude and wavelength of the wrinkle.
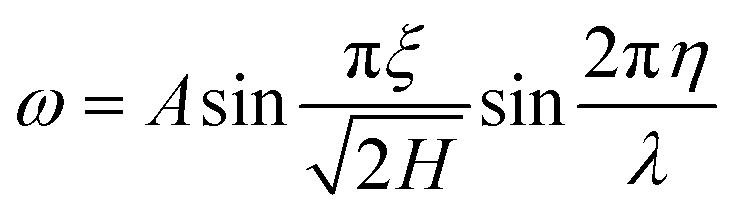 | (18) |
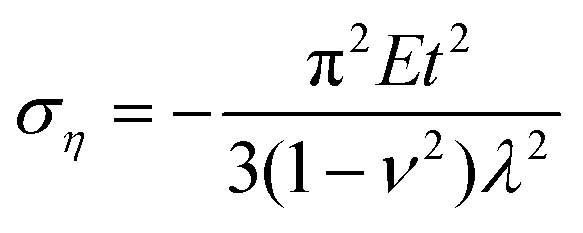 | (19a) |
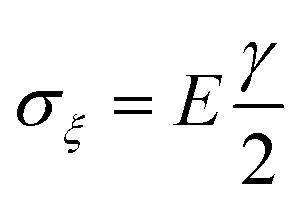 | (19b) |
| σηκη + σξκξ = 0 | (20) |
In the analytical model proposed by Tian et al.185 the stress state equilibrium equation is established as eqn (20), where κη and κξ are the curvatures along the directions η and ξ, respectively.
It can be inferred from the wrinkling pattern of monolayer BNNS, shown in Fig. 13(a), that wrinkling initiates at a point when in-plane shear displacement (δ) reaches a critical buckling value of 0.08 nm. At critical buckling the wrinkle initiates from the free edges and keeps the central part of the sheet wrinkle-free. Further increase in shear displacement results in a uniform distribution of wrinkle patterns with identical crest and trough as indicated in Fig. 13(c). It can be inferred from the results obtained from the simulations of Tian et al.185 that the average wrinkling amplitude of the nanosheets increases with an increase in shear displacement, whereas the wrinkling wavelength has a reverse effect as indicated in Fig. 14(a) and (b), respectively.
 | ||
| Fig. 13 Wrinkling pattern of nanosheets with respect to shear displacement δ (reproduced with permission from ref. 185). | ||
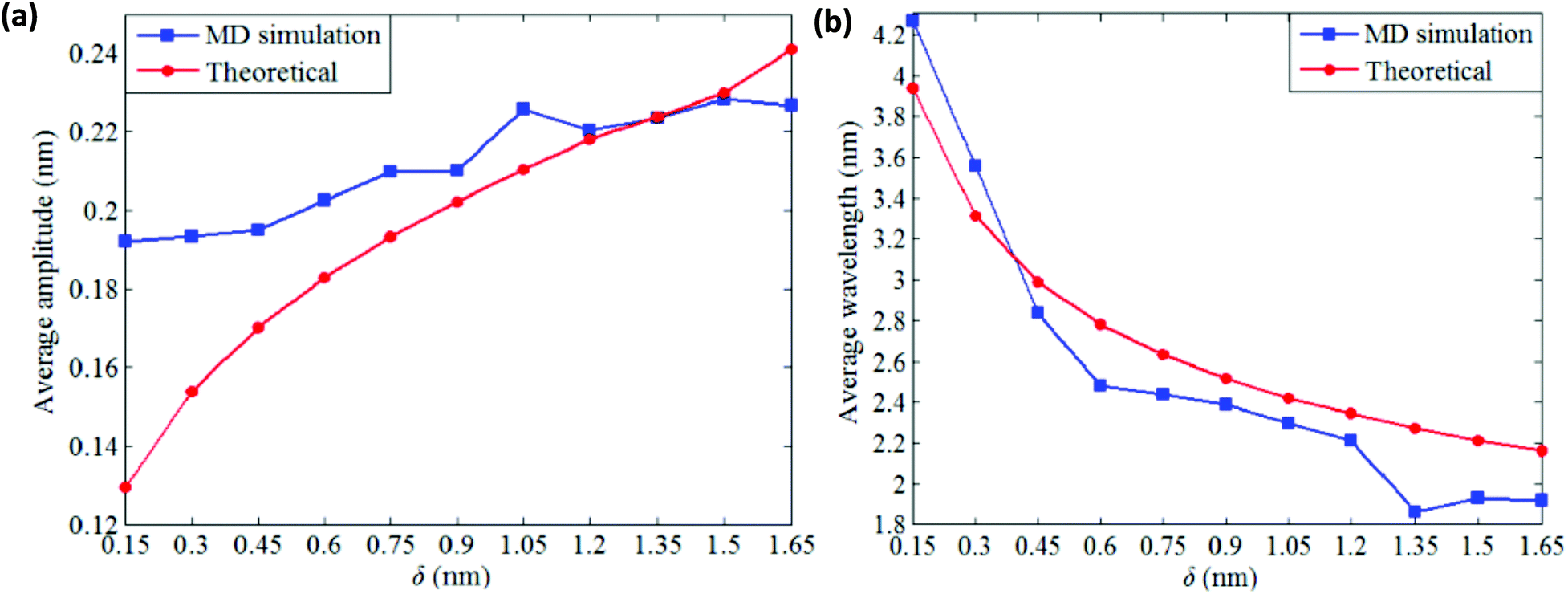 | ||
| Fig. 14 Influence of shear displacement on the wrinkle profile (reproduced with permission from ref. 185). | ||
An important contribution regarding MD based simulations on boron nitride came from Han et al.157 who studied the effects of temperature, strain rate and crystal orientations on the mechanical properties of BNNSs. The interactions between boron and nitrogen atoms were described by the parameters proposed by Albe et al.150,151 It was observed that the nanosheets were basically an anisotropic material and any increase in the temperature led to a decrease in the Young's modulus, fracture strength and fracture strain. A reduction in the Young's modulus was observed with an increase in the strain rate, while a reverse effect was observed on the fracture strength of BNNSs. Perim et al.186 appraised the fracture behavior of defective carbon and boron nitride nanotubes in accordance with MD based simulations in conjunction with the ReaxFF force field.152–154 The similarities and differences in their (CNT and BNNT) fracture patterns were outlined, under similar stress conditions.
3.5. Chirality & torsion
Krishnan and Ghosh187 studied the effect of chirality on the elastic properties of single-walled BNNTs subjected to tensile and torsional loading. Their simulations were performed with the help of the Tersoff-potential parameter set provided by Sevik et al.146 They concluded in their simulation results (as shown in Fig. 15) that with an increase in aspect ratios (length to diameter ratio of a nanotube) the elastic modulus also increases. It can also be inferred from their summarized data in Fig. 15 that at higher aspect ratios the variation in the Young's modulus is comparatively small. | ||
| Fig. 15 Elastic modulus of BNNT with respect to the aspect ratio (reproduced with permission from ref. 187). | ||
Recently in 2014, Ansari and Ajori188 studied the torsional vibration performance of double-wall BNNTs using Tersoff-potential parameters developed by Albe et al.150 They extended MD simulations in conjunction with continuum modeling to estimate the shear modulus of nanotubes. The torsional frequency was predicted to be varying with geometrical parameters such as length and boundary conditions. Moreover, the torsional frequency of BNNTs was observed to be higher than that of their carbon counterparts i.e. nanotubes (CNT).
Ajori and Ansari189 employed MD based simulations with a Tersoff-like potential and parameters reported by Albe and Möller150 to investigate the torsional buckling response of BN nanotubes. In their simulations, (6, 6) and (10, 10) armchair BNNTs with the length varying from 5.13 nm and 15.4 nm were chosen for investigation. They predicted size dependent values of the shear modulus which differed from 0.64 TPa at 5.13 nm and finally converged to a value of 0.75 TPa as the tube length was increased. Critical torque was found to be highly dependent upon the geometrical parameters such as length, chirality and boundary conditions. The critical shear strain was predicted to be independent of chirality.
Minh-Quy Le190 investigated the dependency of mechanical properties of BN nanoribbons on their size using potential parameters for boron and nitrogen from the work of Sevik et al.146 In the case of rectangular nanoribbons, tensile properties were found to be significantly affected by the length to width ratios. However, size effects were found to be low in the case of square boron nitride nanosheets as compared to those of rectangular ones. It was also observed that the Young's modulus, fracture stress and fracture strain had an increasing trend with decreasing width for the zigzag configuration of nanoribbons with a fixed length. In the case of armchair BN nanoribbons with a constant length these properties varied slightly as compared to the zig-zag configuration. BN nanoribbons showed higher mechanical strength in the zigzag direction as compared to the armchair direction. Tang et al.191 investigated the effect of the interfacial-structure on the mechanical properties of BN nano-bamboos (BNNB).192,193 In these simulations, interactions between boron and nitrogen atoms were modeled by the Tersoff-like potential134 with parameters taken from the work of Albe et al.151 and the van der Waals interactions at interfaces were modeled with the help of the Lennard-Jones (LJ) potential.137 BNNBs with interlocked joint interfacial structures with compressive interfacial stresses, showed higher tensile fracture strength and a Young's modulus up to 8.0 GPa and 225 GPa, respectively. Due to these interlocked joint interfacial structures a shift from the inter-planar sliding mode to the in-plane tensile elongation mode was observed in the deformation mechanism.
3.6. Thermal conductivity
BN nanofillers have exceptionally high thermal conductivities. Molecular dynamics based simulations have the capability to accurately predict the thermal conductivity of these nanofillers under different types of boundary and loading scenarios. Shen194 studied the variation in thermal conductivity of BNNTs with temperature and tube diameter with the help of Tersoff type potential parameters proposed by Albe et al.150 It was observed in his computational work that the thermal conductivity had a decreasing trend with an increase in the temperature and tube diameter. Sevik et al.146 studied the thermal transport properties of BN nanostructures using the parameterized Tersoff-potential under a (NVE) micro-canonical ensemble. Though, BN nanostructures were found to be possessing quite high thermal conductivities, they were lower as compared to their carbon counterparts. But qualitatively, both carbon and BN nanoribbons showed similar behavior with respect to the variation in the width and edge structure (zig-zag and armchair). It was further concluded that thermal conductivities were independent of chirality. Mortazavi and Rémond159 also evaluated the thermal conductivity of a single-layer of BN nanosheet through MD simulations with parameters for boron and nitrogen taken from the work of Matsunaga et al.147–149 and predicted a value of 80 W m−1 K−1 for thermal conductivity of the nanosheet.Kınacı et al.195 investigated the variation in thermal conductivity (κ) of hybrid graphene/h-BN nanostructures with stripe super-lattices (alternate stripes of graphene and BN) and BN dots embedded in graphene. The parameters for boron, nitrogen and carbon interactions were developed by the author and his team using B–N parameters from Sevik et al.146 and carbon interactions from the work of Lindsay and Broido.196 For stripe super-lattices, it was predicted that the interface in the zigzag configuration produced a higher κ in the direction parallel to the interface than in the armchair configuration, while the perpendicular conductivity was less prone to the details of the interface and was limited by the thermal conductivity of h-BN. However, the embedded dot structures having mixed zigzag and armchair interfaces, had a stronger effect on the thermal transport properties as compared to super-lattices. Recently in 2014, Chen et al.197 examined the thermal conductivity of BN nanoribbons in various transport directions (from 0° to full 30° chirality) using non-equilibrium molecular dynamics based simulations with the use of the Müller-Plathe198 method and Tersoff-type three-body potential.195 Edge specularity for boundary scattering (which is closely related to the edge roughness) was also studied with respect to different chiral angles. It was concluded that both thermal conductivities and edge specularity attained local maxima at a chiral angle of 19.11°. Thermal conductivity was predicted to be increasing with an increase in the ribbon length up to a certain critical length value depending upon the chiral angle.
Sevik et al.199 investigated the effect of boron isotopes on the thermal conductivity of BN nanotubes. It was concluded that thermal conductivity varied in the range of 450–500 W m−1 K−1 and 340–400 W m−1 K−1 for isotopically pure (100% 11B) and natural (80% 11B) BNNTs and h-BN nanosheets, respectively. In addition to these quantitative results, a temperature dependency of the thermal conductivity of BNNTs on isotopes was also studied with the same computational model. It was also concluded from their atomistic model that effects of isotopes were more significant at lower temperatures. The scattering of phonons from isotopes at lower temperatures was associated with the reduction in the thermal transport phenomenon. Sevik et al.199 further concluded that the effect of isotopes was more pronounced in 3D structures as compared to 1D and 2D structures. This dependency of thermal conductivity on structures was considered to be associated with average isotope–isotope separation, where smaller distances reduced the relaxation length, ultimately reducing the lattice thermal conductivity. Haijun Shen200 examined and compared the thermal conductivities of CNTs and BNNTs, under radial compression, with the help of the Tersoff-potential.135 It was predicted in his work that under local as well global compression, the thermal conductivity exhibited a decreasing trend. But global compression had a more profound effect than local compression. In addition to this an increase in temperature resulted in the lowering of thermal conductivity. Mashreghi201 also conducted a MD based study on thermal expansion/contraction of armchair BNNTs in various directions i.e. axial, radial and circumferential. Boron and nitrogen interactions were modeled using parameters provided by Albe et al.150 Coefficients of thermal expansion (CTE) were estimated along the axial direction with different diameters. The CTE in the radial direction were reported to be lower as compared to those in the circumferential direction.
3.7. Hybrids
So far, this review paper has exclusively discussed the current state of art techniques in MD based simulations of BN nanofillers either in the pristine or the defective form. But some progress has also been made in simulating the hybrids of BN nanofillers with polymers. Liew and Yuan202 investigated the structural performance of a double-walled nanotube consisting of a carbon nanotube inside a boron nitride nanotube. Liew and Yuan performed these simulations in the MD framework in combination with the universal force field.164,203 During their simulations, the nanotubes were subjected to axial compressive loading. It was reported in their simulations that at the annealing temperatures of 3500 K and 4000 K, distortion and critical buckling load of the inner CNTs were dependent on the chiral vector.It can be inferred from Fig. 16 that in the case of C(5, 5)@BN(10, 10) the outer BNNT starts showing instability at ε = 11.83% while the inner carbon nanotube remains stable till the higher values of strains (12.74%). But in the case of C(5, 5)@C(10, 10), both tubes became unstable simultaneously at lower strain values (9.10%) than in the previous case. Hence it can be concluded from the work of Liew and Yuan202 that BNNT acts as a better protection than a CNT when used as an outer protection tube.
 | ||
| Fig. 16 Coaxial nanotubes: (a) optimized C(5,5)@BN(10,10), (b) optimized C(5,5)@C(10,10) at different axial compressive strains (ε) at the temperature of 300° K (reproduced with permission from ref. 202). | ||
Yuan et al.204,205 investigated the structural stability and elastic properties of bilayer and trilayer graphene and boron nitride nanosheets by employing MD based simulations in conjunction with the universal force field.164 Their investigation on multiplayers of nanosheets was based on analyzing and comparing the binding energy, van der Waals interactions between layers and radial distribution function (RDF). In their computational model the full width half maximum (FWHM) of RDF was considered to be closely related to the integrity of the atomistic structure. The structures with sharp peaks and smaller values of FWHM for RDF were predicted to possess better integrity. They performed simulations with bilayers and trilayers of nanosheets, and compared the results with the monolayer of graphene and BNNS. FWHM values obtained from the first, second and third peaks (refer to Table 4) suggested a small deviation between the bilayer and single sheets of BNNS, and also these changes in values of FWHM for BNNS are comparatively lower as compared to graphene. These trends in FWHM between single and bilayers indicate that bilayer BNNS can be considered as a more integrated structure as compared to graphene.204
| Material | FWHM (1st peak) | FWHM (2nd peak) | FWHM (3rd peak) |
|---|---|---|---|
| Gr (monolayer) | 0.00258 nm | 0.00221 nm | 0.00257 nm |
| Gr–Gr (bilayer) | 0.00236 nm | 0.00225 nm | 0.00234 nm |
| Gr–Gr–Gr (trilayer) | 0.00231 nm | 0.00201 nm | 0.00255 |
| BN (monolayer) | 0.00301 nm | 0.00217 nm | 0.0301 nm |
| BN–BN (bilayer) | 0.00302 nm | 0.00219 nm | 0.00311 nm |
| BN–Gr–BN (sandwich) | 0.00221 nm | 0.00184 nm | 0.00255 nm |
Young's moduli of the combined bilayer nanosheets were found to be lower than those of the corresponding monolayers for graphene and BNNS. In the case of BN nanosheets, the reduction in the Young's modulus was less as compared to that of graphene nanosheets. Yuan and Liew205 also performed MD simulations to investigate the structural stability and high temperature distortion-resistance of a sandwich structure of graphene and BN nanosheets. The optimal interlayer equilibrium distances between graphene nanosheets (in the case of graphene sandwich) and graphene and BN nanosheets (in the case of the BNNS-graphene sandwich) were reported as 0.347 nm and 0.341 nm, respectively. Equilibrium distance was the spacing between nanosheets at which repulsive and attractive forces are zero. It was further predicted in their work that graphene sandwiched between BN nanosheets proved to be a more integrated structure as compared to monolayer graphene sandwiched between graphene nanosheets in a crystal structure on the basis of FWHM values obtained from the first peak in RDF as provided in Table 4 and illustrated in Fig. 17.
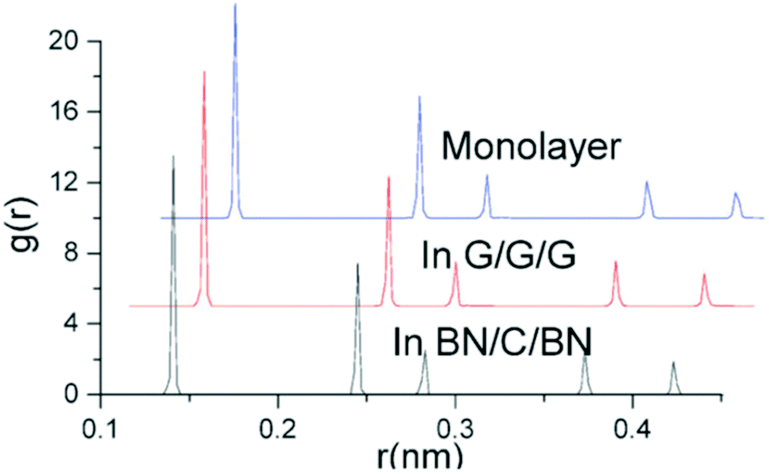 | ||
| Fig. 17 RDF for monolayer graphene, three layers of graphene and the sandwich structure of the graphene sheet between BNNS (reproduced with permission from ref. 205). | ||
Song and Medhekar206 also studied the hybrid nanostructures of graphene and h-BN nanoribbons. In their MD based simulations, interatomic interactions were modeled with the help of the Tersoff potential136 with optimized parameters taken from the work of Matsunaga et al.149 The thermal transport in the direction perpendicular to the interface was reported to be less affected by the composition. On the other hand, it was also concluded in their work that the thermal transport in these hybrid nanoribbons was significantly affected by the interface scattering and the presence of a low thermal conductivity component i.e. h-BN in the path of the thermal current. However, thermal transport parallel to the interface was limited by the graphene component. Shen Haijun207 investigated the compressive strength of single-walled BNNTs and the hybrid structure of BNNTs embedded into CNTs. The simulations were performed using B–N potential parameters from Albe et al.150 while non-bonded interactions were treated with the help of LJ potential parameters.164 It can be inferred from their conclusions that hybrid structures show higher compressive strength as well as melting temperatures as compared to single BNNTs. In 2013, Zhao and Xue160 examined the mechanical properties of hybrid structures of graphene and h-BN. While B–N, B–C and N–C interactions were modeled by mixing of potential parameters of Matsunaga et al.148 parameters provided by Lindsay and Broido196 were used to model carbon–carbon interactions. Hybrid sheets were predicted to show plastic behavior that was absent in pure h-BN and graphene nanosheets. The Young's modulus for hybrid nanosheets showed a decreasing trend with increasing concentrations of BN, irrespective of BN shapes and distribution. DFT based simulations have also been employed to investigate the hybrid structures of BN nanofillers. Zhang et al.208 investigated the electronic and transport properties of fullerene and vanadium based peapods of BNNTs using DFT calculations. Based on their simulation results, BNNT based peapods were predicted to be semiconducting with a narrow bandgap. Further details regarding this behavior of BNNTs can be found in their paper.208
3.8. Nanocomposites
So far, this review paper has exclusively discussed nanostructures such as BN nanotubes, BN nanosheets and hybrid structures of these BN nanofillers with graphene or CNT. But a decent amount of progress has also been made in simulating BN nanofiller based nanocomposites. This section focuses on the MD based simulations of BN nanocomposites. Nasrabadi and Foroutan209 used MD based simulations for investigating the interfacial binding of BN nanotubes with poly[m-phenylenevinylene-co-(2,5-dioctyloxy-p-phenylenevinylene)] PmPV, polystyrene, and polythiophene. In their simulations, MM3210 and Lennard Jones force fields have been employed for simulating the bonded and non-bonded interactions (interface), respectively. From their simulations, it can be inferred that the interaction energy of BNNT–polymer composites is strongly influenced by the specific monomer structure and nanotube radius. On the basis of higher values of the interaction energy of BNNT–polymer composites, they suggested that the BNNTs could be more efficient nano-fillers than the CNTs for nano-composite reinforcement applications. The effect of polarization of BNNTs on the interfacial strength was also studied in the same work209 and it was concluded that interfacial strength increased due to polarization of BNNTs.Fatemi and Foroutan211 investigated the dispersion of two BNNTs in an aqueous solution of triton X-100 as a surfactant, using MD based simulations in combination with the AMBER99 force field.212 The presence of the surfactant resulted in an increase in the surface angle between the two nanotubes and hence more gap was created between two BNNTs leading to the dispersion of the BNNTs. The presence of the surfactant shortened the bond distance between the nitrogen atom of the BNNTs and the hydrogen atom of water molecules. Moreover, the estimated values of the radius of gyration of the surfactant and the interfacial angle between two BNNTs shed more light on the arrangement of the surfactants around the BNNTs with and without the presence of water molecules.
Wang and his team213 applied axial compression on copper-filled BNNTs to study the buckling behavior of BNNTs using the MD framework. The universal force field, with parameters taken from the work of Jung et al.214 and Zhou et al.215 was used for simulating the atomic interactions between the atoms. It was observed that nanotubes with different chiralities exhibited different deformation patterns and energy loss. Side and top views for different tubes at the buckling stage have been shown in Fig. 18. Fig. 18(a) shows a 49 Cu@BNNT (7, 7) i.e. 49 copper atoms in BNNT (7, 7), Fig. 18(b) is for 49 Cu@BNNT (11, 11), both showing different deformation patterns. Fig. 18(c) and (d) depicted the same atomic configuration i.e. a 195 Cu@BNNT (11, 11) nanotube, but at different simulation steps. It is clear from these two figures that a regular hexahedron like shape evolved before buckling of the tube and at the time of buckling, this shape finally transformed into a square shape. In addition to these deformation patterns, BN nanotubes filled with a larger number of copper atoms were found to be more stable as compared to the nanotubes filled with a smaller number of copper atoms.
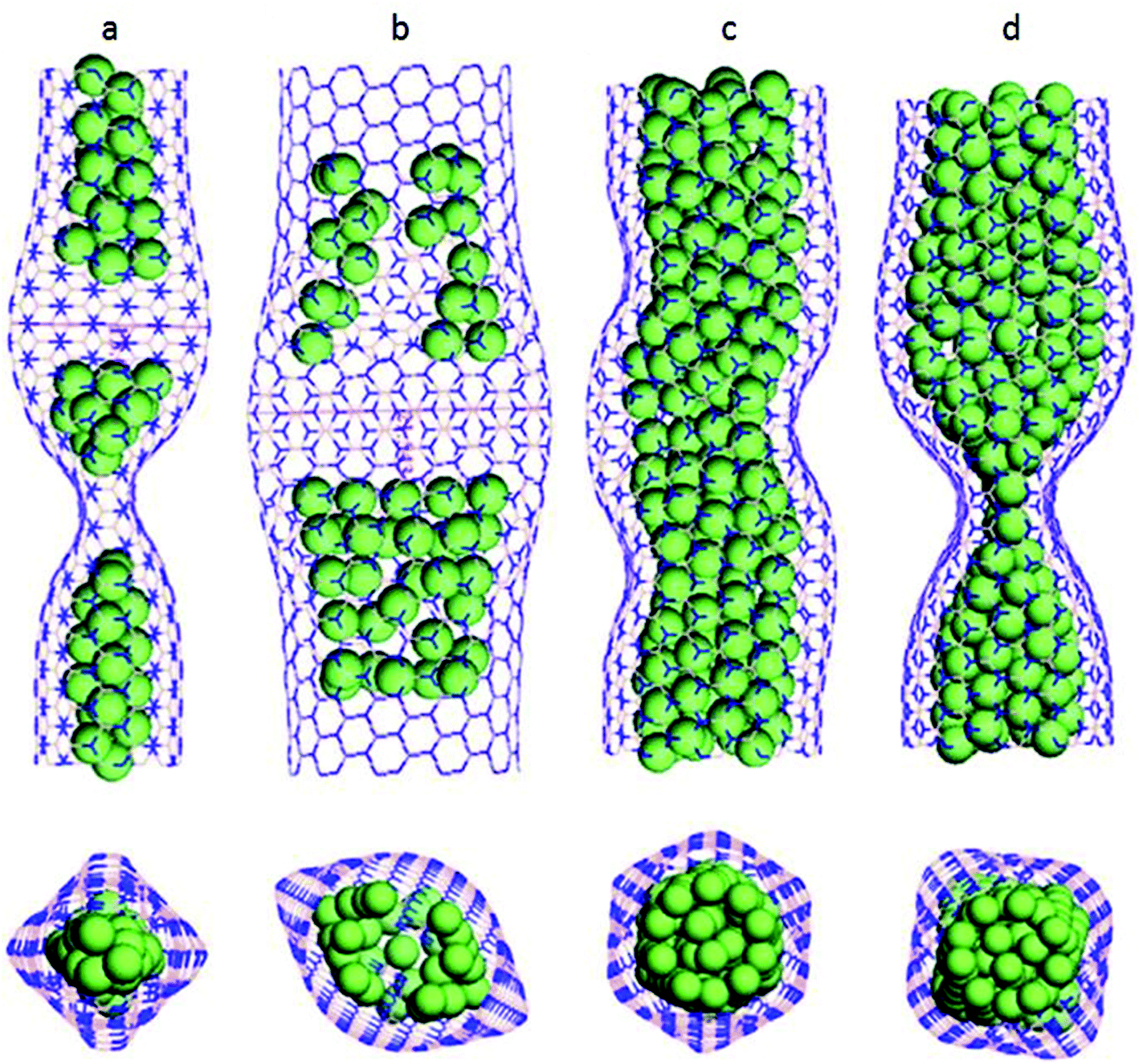 | ||
| Fig. 18 Deformation pattern of (a) 49 Cu@BNNT(7,7), (b) 49Cu@BNNT(11,11), (c) 195CuBNNT(11,11) at a simulation time t, (d) 195Cy@BNNT(11,11) after an increased simulation time t + Δt (reproduced with permission from Wang et al.213). | ||
Bari et al.216 carried out MD simulations to study the non-covalent interactions between polyvinyl pyrrolidone (PVP) and isolated BNNSs in a water medium. Interactions between the PVP molecule and water atoms were treated with a general AMBER force field217,218 while the LJ potential was used to take care of the interactions between the BNNSs and water with parameters from the work of Mayo et al.219 It was predicted in their research that in the initial stages of simulation, PVP chains showed a random thermal motion in water, and finally reached within an approachable distance to BNNS, so that, interactions could be possible between BNNS and PVP. It can be inferred from their conclusion that after the chain reaches within close proximity to BNNS, the distance between these two does not change any further. This emphasized that in the presence of water, there exists a strong affinity between the PVP chain and the BNNS. Griebel and Hamaekers220 examined the elastic properties of BNNT embedded into amorphous silicon–boron–nitride ceramics. The computational work was performed using Parrinello–Rahman's approach178,221 with potential parameters from the work of Matsunaga et al.147 The results obtained in their computational model further suggested that out of Si3BN5, Si3B2N6 and Si3B3N7 matrices, Si3B3N7 had the highest Young's modulus with the highest elastic range. Further examination of the properties of a ceramic composite material made of Si3B3N7 as the matrix and BN nanotubes as the filler was performed in the computational work of Griebel and Hamaekers.220 It was further suggested that the simple macroscopic rule-of-mixture, which just accounts for the volume fraction of the nanotube, could be used for composites with periodically infinitely long BN nanotubes.
Thomas and his team222 studied the interactions of BNNTs and CNTs embedded in lipid bilayers. They employed MD based simulations in conjunction with CHARMM36 force field parameters223,224 for investigating the interaction of BNNTs and CNTs with lipid bilayers. It was predicted in their simulations that the stability of BNNTs in cell membranes was comparable to that of functionalized CNTs. It was further observed in their simulations that a narrow BNNT might be blocked at one end by lipid molecules as indicated in Fig. 19(a), while a wider BNNT was only partially blocked by lipid molecules as shown in Fig. 19(b). On the other hand, CNT was reported to be completely occupied by lipid molecules as illustrated in Fig. 19(c). Yuan and Liew225 examined the strength of a coaxially placed silicon wire (SiNW) inside a BN nanotube. MD based studies were performed in conjunction with the universal force field147,199 to investigate the effect of the silicon nanowire on the structural strength of BN nanotubes. Two different types of silicon nanowires (SiNWs) were simulated by Yuan and Liew in their atomistic model as shown in Fig. 20. An axial monatomic chain with a helical shell of SiNWs, coaxially aligned inside a BNNT (10, 10) and only an axially aligned monatomic chain of SiNW inside the BNNT (5, 5) are shown in Fig. 20(a), (b) and (c), (d), respectively. It was reported in their work that BNNT and SiNW nanocomposites were more stable as compared to their respective pristine forms. It was also concluded in their research work that the deformation pattern and energy loss varied from form to form, whereas the buckling strain remained the same for the pristine BNNT and BNNT/Si NW composites.
 | ||
| Fig. 19 (a) Blocked lipid at the end of narrow BNNT, (b) partially blocked lipid along with a wider BNNT, (c) completely occupied CNT with the lipid molecules (reproduced with permission from Thomas et al.222). | ||
 | ||
| Fig. 20 (a) Axial monatomic and helical shell of silicon nanowires coaxially aligned inside the BNNT (10,10), (b) side view of the atomic configuration shown in part (a). (c) Only a monatomic chain of silicon nanowire inside BNNT (5,5), (d) side view of the atomic configuration shown in part (c) (reproduced with permission from Yuan and Liew225). | ||
It can be inferred from the preceding review that Tersoff is the most common among the potentials for simulating interatomic interactions in BN nanofillers. In addition to the Tersoff type potential, ReaxFF, Harmonic, LJ, MM3, AMBER and CHARM potentials were also employed by few researchers for the modeling of BN nanofillers. A summary of different types of potentials employed in various MD simulations for estimating the mechanical and thermal behaviors of BN nanofillers is provided in Table 5.
| Potential parameters | Any modification | Ref. | Work done |
|---|---|---|---|
| Albe et al.150 | — | 157 | Effect of temperature and strain rate on mechanical properties of BNNS |
| — | 169 | Radial compression of BNNTs | |
| — | 171 | Bending rigidity of BNNSs | |
| — | 183 | Structural and thermal behavior of BNNTs with SW defects | |
| — | 184 | Fracture and buckling behavior of BNNTs | |
| — | 188 | Torsional vibrational performance of BNNTs | |
| — | 189 | Torsional and buckling behavior of BNNTs | |
| — | 191 | Effect of interfacial structure on mechanical properties of BNNBs | |
| — | 194 | Effect of diameter and temperature on thermal conductivity of BNNTs, tensile properties of BNNTs | |
| — | 201 | Effect of temperature and diameter on coefficients of thermal (CTE) expansion of BNNTs | |
| — | 207 | Thermal-stability and compressive behavior of BNNT and BNNT@CNT | |
| Sevik et al.146 | — | 146 | Characterization of thermal transport in BN nanofillers |
| Cut-off function f(r) = 1 | 161 | Effects of vacancy and SW defects on mechanical properties of BNNSs | |
| Tersoff–Brenner potential | 181 | Effect of vacancy and SW defects on failure behavior of BNNTs under tension | |
| — | 187 | Effect of chirality on elastic properties of BNNTs under tension and torsion | |
| R ij = Sij | 190 | Size effects on mechanical properties of BNNRs | |
| — | 197 | Thermal conductivity of graphene–BNNS hybrids | |
| — | 199 | Impact of isotopes on thermal transport in BN nanostructures | |
| Matsunaga et al.147 | — | 159 | Investigation of tensile properties and thermal conductivity of BNNS |
| — | 160 | Mechanical properties of hybrid graphene and BNNS | |
| Inclusion of attractive potential term to deal with homo-elemental bonds | 177 | Effects of vacancies and functionalization on Young's modulus of BNNTs | |
| — | 182 | Mechanical properties of polycrystalline BNNSs | |
| — | 206 | Thermal transport in hybrid of graphene and h-BN | |
| — | 220 | Elastic properties of BNNTs embedded in amorphous Si–B–N | |
| Sekkal et al.145 | Slightly modified to get better fit to the bond length and bond energy | 26 | Elastic and shear moduli of BN nanofillers |
| Liao et al.168 | Modified version of parameters from Albe et al. to, better capture of properties of nanofillers, predict better bond breaking behavior | 168 | Deformation behaviors of BNNTs under axial tensile strains in armchair direction |
| — | 167 | Buckling behavior of pristine and hydrogenated BNNTs | |
| — | 172 | Effect of temperature on tensile and compressive behavior of BNNTs | |
| UFF & Dreiding | — | 163 | Mechanical properties of hydrogenated BNNTs |
| — | 174 | High-temperature thermal stability and in-plane compressive properties of a BNNT | |
| — | 202 | High-temperature thermal stability and axial compressive properties of a coaxial CNT@BNNT | |
| — | 205 | Structure stability and high-temperature distortion resistance of trilayer hybrid of graphene and BNNS | |
| — | 204 | Structural stability and elastic properties of bilayer hybrid of graphene and BNNS | |
| — | 213 | Axial compression of copper-filled BNNT | |
| — | 225 | Compressing behavior of coaxial silicon nanowires inside a BNNT | |
| Harmonic potential or LJ potential | Harmonic potential for bonded interactions and LJ potential non-bonded interactions | 170 | Buckling behavior of BNNTs under axial compression |
| Parameters from Liao et al. were used to obtain harmonic potential parameters for bonded interactions | 179 | Effects of vacancy defects on the compressive buckling of boron nitride nanotubes | |
| ReaxFF | — | 180 | Point and line defects on BNNTs |
| — | 186 | Fracture behavior of defective BNNTs | |
| MM3 + LJ | — | 209 | Interactions between polymers SWBNNT |
| AMBER force field + LJ | — | 211 | Dispersion of BNNTs inside water by triton X-100 surfactant |
| — | 216 | Liquid phase exfoliation and crumpling of BNNSs | |
| CHARMM36 force field | — | 222 | Stability of BNNTs into lipid bilayers |
4. Conclusion and future aspects
In this review, the authors have summarised recent progress made in the field of atomistic modeling of BN nanofillers. Due to their exceptional properties, BN nanofillers are emerging as potential candidates for a wide range of practical applications. On the basis of a wide band gap in the range of ∼5.5 eV, higher stability against oxidation even at higher temperatures and thermal stability, BN nanofillers have emerged as superior to carbon based nanofillers for specific applications and hence their further exploration has always drawn the attention of researchers.From the preceding review on atomistic modeling of BN nanofillers, it can be concluded that these approaches are capable of producing results that are in good agreement with experimental results. When compared to each other, each of these techniques has its own advantages along with certain limitations associated with it, as discussed in the review. In spite of the fact that DFT based simulations are more accurate, they have limitations of being computationally intensive and limit the overall system size. Although, tight binding calculations are faster they need further improvements, like in their scalability. Continuum based FEM techniques proved to be accurate for predicting the global properties, but have limitations while modeling the localised effects such as bond formation and rotation under an atomistic atmosphere. When it comes to MD, Tersoff type potentials have attracted more attention among the researchers for simulating the bonded interactions, due to accurate parameterizations that produce an accurate fit with the DFT calculations and experimental results. So far, different sets of Tersoff type potential parameters have been proposed by the researchers. Sekkal et al.,145 Sevik et al.,146 Matsunaga et al.,147 and Albe et al.150 are the prominent contributors for these parameters. Due to the empirical nature of these parameters, an overall scattered result in terms of mechanical, fracture and thermal properties has been reported in MD based simulations performed with these parameters. Some studies21,161 on mechanical properties of BN nanofillers with minor modifications in the cut-off function have predicted better results that are in good agreement with DFT and experiments. These studied were performed using the parameters from Sevik et al.146 However, no study with other sets of parameters, used in conjunction with a modified cut-off function, has been reported yet. So, a scope of further improvements, such as cut-off function optimisation, exists, as far as MD simulations of BN nanofillers are concerned.
Overall, atomistic modeling and simulation techniques are proving to be an excellent complement to the experimental techniques to explore further research directions. Properties which cannot be explored with experiments can be traceable using these techniques and hence, further improvements in these techniques will make them more versatile.
References
- A. Rubio, J. L. Corkill and M. L. Cohen, Theory of graphitic boron nitride nanotubes, Phys. Rev. B: Condens. Matter, 1994, 49, 5081–5084 CrossRef CAS.
- N. G. Chopra, R. J. Luyken, K. Cherrey, H. C. Vincent, L. C. Marvin, G. L. Steven and A. Zettl, Boron nitride nanotubes, Science, 1995, 269, 966–967 CrossRef CAS PubMed.
- M. Corso, W. Auwärter, M. Muntwiler, A. Tamai, T. Greber and J. Osterwalder, Boron Nitride Nanomesh, Science, 2004, 303, 217–220 CrossRef CAS PubMed.
- T. Sato, Studies on hexagonal and rhomobohedral layered boron nitrides: synthesis, crystal growth, and transformation under high pressure, Report of National Institute for Research in Inorganic Materials, Tsukuba, Japan, 1987 Search PubMed.
- J. J. Pouch and S. A. Alterovitz, Synthesis and properties of boron nitride, Mater. Manuf. Processes, 1990, 6, 373–374 CrossRef.
- S. Iijima, Helical microtubules of graphitic carbon, Nature, 1991, 354, 56–58 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang and S. V. Dubonos, Electric Field Effect in Atomically Thin Carbon Films, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich and S. V. Morozov, Two-dimensional atomic crystals, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- X. Blase, A. Rubio, S. G. Louie and M. L. Cohen, Stability and band gap constancy of boron nitride nanotubes, Europhys. Lett., 1994, 28, 335–340 CrossRef CAS.
- K. Watanabe, T. Taniguchi and H. Kanda, Direct. Bandgap Properties And Evidence For Ultraviolet Lasing of Hexagonal Boron Nitride Single Crystal, Nat. Mater., 2004, 3, 404–409 CrossRef CAS PubMed.
- Z. Lin, Y. Liu, K. Moon and C. Wong, Enhanced thermal transport of hexagonal boron nitride filled polymer composite by magnetic field-assisted alignment, Electronic Comp. and Tech. Conf. (ECTC), 2013 IEEE 63rd, 1692–96.
- K. C. Yung and H. Liem, Enhanced thermal conductivity of boron nitride epoxy-matrix composite through multi-modal particle size mixing, J. Appl. Polym. Sci., 2007, 106, 3587–3591 CrossRef CAS.
- X. Huang, S. Wang, M. Zhu, K. Yang, P. Jiang, Y. Bando, D. Golberg and C. Zhi, Thermally conductive, electrically insulating and melt-processable polystyrene/boron nitride nanocomposites prepared by in situ reversible addition fragmentation chain transfer polymerization, Nanotechnology, 2015, 26, 015705 CrossRef CAS PubMed.
- C. P. Wong and S. Raja, Bollampally. Comparative study of thermally conductive fillers for use in liquid encapsulants for electronic packaging, IEEE Trans. Adv. Packag., 1999, 22, 54–59 CrossRef CAS.
- D. Golberg, M. F. Pedro, J. Costa, O. Lourie, M. Masanori Mitome, X. Xuedong Bai, K. Kurashima, C. Zhi, C. Tang and T. Bando, Direct Force Measurements and Kinking under Elastic Deformation of Individual Multiwalled Boron Nitride Nanotubes, Nano Lett., 2007, 7, 2146–2151 CrossRef CAS.
- R. Arenal, M. Wang, Z. Xu, A. Loiseau and D. Golberg, Young Modulus. Mechanical and Electrical Properties of Isolated Individual and Bundled Single-Walled Boron Nitride Nanotubes, Nanotechnology, 2001, 22, 26570 Search PubMed.
- K. N. Kudin, G. E. Scuseria and B. I. Yakobson, C2F, BN, and C Nanoshell Elasticity from Ab Initio Computations, Phys. Rev. B: Condens. Matter, 2001, 64, 235406 CrossRef.
- H. Sahin, S. Cahangirov, M. Topsakal, E. Bekaroglu, E. Akturk, R. T. Senger and S. Ciraci, Monolayer Honeycomb Structures Of Group-IV Elements And III–V Binary Compounds: First-Principles Calculations, Phys. Rev. B: Condens. Matter, 2009, 80, 155453 CrossRef.
- R. C. Andrew, R. E. Mapasha, A. M. Ukpong and N. Chetty, Mechanical Properties of Graphene and Boronitrene, Phys. Rev. B: Condens. Matter, 2012, 85, 125428 CrossRef.
- Q. Peng, W. Ji and S. De, Mechanical properties of the hexagonal boron nitride monolayer: ab initio study, Comput. Mater. Sci., 2012, 56, 11–17 CrossRef CAS.
- M. Q. Le, Atomistic study on the tensile properties of hexagonal AlN, BN, GaN, InN and SiC sheets, J. Comput. Theor. Nanosci., 2014, 11, 1458–1464 CrossRef CAS.
- M. Q. Le, Young's Modulus Prediction of Hexagonal Nanosheets and Nanotubes Based on Dimensional Analysis and Atomistic Simulations, Meccanica, 2014, 49, 1709–1719 CrossRef.
- N. G. Chopra and A. Zettla, Measurement of the elastic modulus of a multi-wall boron nitride nanotube, Solid State Commun., 1998, 105, 297–300 CrossRef CAS.
- A. P. Suryavanshi, M. F. Yu, J. Wen, C. Tang and Y. Bando, Elastic modulus and resonance behavior of boron nitride nanotubes, Appl. Phys. Lett., 2004, 84, 2527 CrossRef CAS.
- D. V. Fakhrabada and N. Shahtahmassebia, First-principles calculations of the Young's modulus of double wall boron-nitride nanotubes, Mater. Chem. Phys., 2013, 138, 963–966 CrossRef.
- V. Verma, V. K. Jindal and K. Dharamvir, Elastic Moduli of A Boron Nitride Nanotube, Nanotechnology, 2007, 18, 435711 CrossRef.
- T. Ouyang, Y. Chen, Y. Xie, K. Yang, Z. Bao and J. Zhong, Thermal transport in hexagonal boron nitride nanoribbons, Nanotechnology, 2010, 21, 245701 CrossRef PubMed.
- Y. Chen, J. Zou, S. J. Campbell and G. L. Caer, Boron Nitride Nanotubes: Pronounced Resistance to Oxidation, Appl. Phys. Lett., 2004, 84, 2430 CrossRef CAS.
- F. P. Bundy and R. H. Wentorf, Direct Transformation of Hexagonal Boron Nitride to Denser Forms, J. Chem. Phys., 1963, 38, 1144 CrossRef CAS.
- Y. Kubota, K. Watanabe, O. Tsuda and T. Taniguchi, Deep ultraviolet light-emitting hexagonal boron nitride synthesized at atmospheric pressure, Science, 2007, 317, 932–934 CrossRef CAS PubMed.
- M. Noor-A-Alam, H. J. Kima and Y. H. Shin, Dipolar polarization and piezoelectricity of a hexagonal boron nitride sheet decorated with hydrogen and fluorine, Phys. Chem. Chem. Phys., 2014, 16, 6575–6582 RSC.
- K. H. Michel and B. Verberck, Theory of elastic and piezoelectric effects in two-dimensional hexagonal boron nitride, Phys. Rev. B: Condens. Matter, 2009, 80, 224301 CrossRef.
- J. Qi, X. Qian, L. Qi, J. Feng, D. Shi and J. Li, Strain-Engineering of Band Gaps in Piezoelectric Boron Nitride Nanoribbons, Nano Lett., 2012, 12, 1224–1228 CrossRef CAS PubMed.
- Q. Huang, Y. Bando, L. Zhao, C. Y. Zhi and D. Golberg, pH Sensor Based On Boron Nitride Nanotubes, Nanotechnology, 2009, 20, 415501 CrossRef CAS PubMed.
- M. B. Panchal and S. H. Upadhyay, Boron. Nitride Nanotube-Based Bio-sensing of Various Bacterium/Viruses: Continuum Modeling-Based Simulation Approach, IET Nanobiotechnol., 2014, 8, 143–148 CrossRef PubMed.
- R. Chowdhury and S. Adhikari, Boron-Nitride Nanotubes as Zeptogram-Scale Bio-nano-sensors: Theoretical Investigations, IEEE Trans. Nanotechnol., 2010, 10, 659–667 CrossRef.
- M. B. Panchal and S. H. Upadhyay, Boron nitride nanotube-based biosensor for acetone detection: molecular structural mechanics-based simulation, Mol. Simul., 2014, 40, 103542 CrossRef.
- M. B. Panchal, S. H. Upadhyay and S. P. Harsha, Mass Detection Using Single Walled Boron Nitride Nanotube as A Nanomechanical Resonator, Nano, 2012, 07, 1250029 CrossRef.
- M. B. Panchal and S. H. Upadhyay, Single walled boron nitride nanotube-based biosensor: an atomistic finite element modeling approach, IET Nanobiotechnol., 2014, 8, 149–156 CrossRef CAS PubMed.
- X. Chen, P. Wu, M. Rousseas, D. Okawa, Z. Gartner, A. Zettl and C. R. Bertozzi, Boron Nitride Nanotubes Are Noncytotoxic and Can Be Functionalized for Interaction with Proteins and Cells, J. Am. Chem. Soc., 2009, 13, 890–891 CrossRef PubMed.
- G. Ciofania, V. Raffaa, A. Menciassia and A. Cuschieria, Boron nitride nanotubes: An innovative tool for nanomedicine, Nanotoday, 2009, 4, 8–10 CrossRef.
- G. Ciofani, S. Danti, G. G. Genchi, B. Mazzolai and V. Mattoli, Boron nitride nanotubes: biocompatibility and potential spill-over in nanomedicine, Small, 2013, 9, 1672–1685 CrossRef CAS PubMed.
- K. B. Shelimov and M. Moskovits, Composite Nanostructures Based on Template-Grown Boron Nitride Nanotubules, Chem. Mater., 2000, 12, 250–254 CrossRef CAS.
- C. Zhi, Y. Bando, C. Tang, S. Honda, K. Sato, H. Kuwahara and D. Golberg, Characteristics of Boron Nitride Nanotube–Polyaniline Composites, Angew. Chem., Int. Ed., 2005, 44, 7929–7932 CrossRef CAS PubMed.
- W. Zhou, S. Qi, Q. An, H. Zhao and N. Liu, Thermal conductivity of boron nitride reinforced polyethylene composites, Mater. Res. Bull., 2007, 42, 1863–1873 CrossRef CAS.
- H. Ishida and S. Rimdusit, Very high thermal conductivity obtained by boron nitride-filled polybenzoxazine, Thermochim. Acta, 1998, 320, 177–186 CrossRef CAS.
- W. Zhou, S. Qi, H. Li and S. Shao, Study on insulating thermal conductive BN/HDPE composites, Thermochim. Acta, 2007, 452, 36–42 CrossRef CAS.
- M. Yi, Z. Shen, L. Lei Liua and S. Lianga, Size-selected boron nitride Nanosheets as oxygen-atom corrosion resistant fillers, RSC Adv., 2015, 5, 2983–2987 RSC.
- X. Wang, A. Pakdel, J. Zhang, Q. Weng, T. Zhai, C. Zhi, D. Golberg and Y. Bando, Large-surface-area BN nanosheets and their utilization in polymeric composites with improved thermal and dielectric properties, Nanoscale Res. Lett., 2012, 7, 662 CrossRef PubMed.
- G. Ciofani, S. Danti, L. Ricotti, D. D'Alessandro, S. Moscato, S. Berrettini, V. Mattoli and A. Menciassi, Boron Nitride Nanotubes: Production, Properties, Biological Interactions and Potential Applications as Therapeutic Agents in Brain Disease, Curr. Nanosci., 2011, 7, 94–109 CrossRef CAS.
- G. Ciofani, Potential Applications of Boron Nitride Nanotubes As Drug Delivery Systems, Expert Opin. Drug Delivery, 2010, 7, 889–893 CrossRef CAS PubMed.
- X. Li, C. Zhi, N. Hanagata, M. Yamaguchi, Y. Bandoa and D. Golberg, Boron Nitride Nanotubes Functionalized With Mesoporous Silica For Intracellular Delivery Of Chemotherapy Drugs, Chem. Commun., 2013, 49, 7337–7339 RSC.
- T. H. Ferreira, D. C. F. Soares, L. M. C. Moreira, P. R. O. da Silva, R. G. dos Santos and E. M. B. de Sousa, Boron nitride nanotubes coated with organic hydrophilic agents: stability and cytocompatibility studies, Mater. Sci. Eng., C, 2013, 33, 4616–4623 CrossRef CAS PubMed.
- S. Del Turco, G. Ciofani, V. Cappello, M. Gemmi, T. Cervelli, C. Saponaro and V. Mattioli, Cytocompatibility evaluation of glycol-chitosan coated boron nitride nanotubes in human endothelial cells, Colloids Surf., B, 2013, 111, 142–149 CrossRef CAS PubMed.
- T. H. Ferreira, L. M. Hollanda, M. Lancellotti and E. M. B. de Sousa, Boron Nitride Nanotubes Chemically Functionalized with Glycol Chitosan for Gene Transfection in Eukaryotic Cell Lines, J. Biomed. Mater. Res., Part A, 2015, 103, 2176–2185 CrossRef CAS PubMed.
- G. Ciofani, V. Raffa, A. Menciassi and A. Cuschieri, Folate Functionalized Boron Nitride Nanotubes and their Selective Uptake by Glioblastoma Multiforme Cells: Implications for their Use as Boron Carriers in Clinical Boron Neutron Capture Therapy, Nanoscale Res. Lett., 2009, 4, 113–121 CrossRef CAS PubMed.
- S. H. Jhi and Y. K. Kwon, Hydrogen adsorption on boron nitride nanotubes: A path to room-temperature hydrogen storage, Phys. Rev. B: Condens. Matter, 2004, 69, 245407 CrossRef.
- H. Zhang, C. J. Tong, Y. Zhang, Y. N. Zhang and L. M. Liu, Porous BN for hydrogen generation and storage, J. Mater. Chem. A, 2015, 3, 9632–9637 CAS.
- W. Lei, H. Zhang, Y. Wu, B. Zhang, D. Liu, S. Qin and Y. Chen, Oxygen-doped boron nitride nanosheets with excellent performance in hydrogen storage, Nano Energy, 2014, 6, 219–224 CrossRef CAS.
- C. Lee, Q. Li, W. Kalb, X. Z. Liu, H. Berger, R. W. Carpick and J. Hone, Frictional characteristics of atomically thin sheets, Science, 2010, 328, 76–80 CrossRef CAS PubMed.
- Y. Lin, C. E. Bunker, K. S. Fernando and J. W. Connell, Aqueously Dispersed Silver Nanoparticle-Decorated Boron Nitride Nanosheets for Reusable, Thermal Oxidation-Resistant Surface Enhanced Raman Spectroscopy (SERS) Devices, ACS Appl. Mater. Interfaces, 2012, 4, 1110–1117 CAS.
- K. Watanabe, T. Taniguchi, T. Niiyama, K. Miya and M. Taniguchi, Far-ultraviolet plane-emission handheld device based on hexagonal boron nitride, Nat. Photonics, 2009, 3, 591–594 CrossRef CAS.
- R. Haubner, M. Wilhelm, R. Weissenbacher and B. Lux, Boron Nitrides-Properties, Synthesis and Applications, in High Performance Non-Oxide Ceramics II, Struct. Bonding, 2002, 102, 1–45 CrossRef CAS.
- W. Lei, D. Portehault, R. Dimova and M. Antonietti, Boron Carbon Nitride Nanostructures from Salt Melts: Tunable-Soluble Phosphors, J. Am. Chem. Soc., 2011, 133, 7121–7127 CrossRef CAS PubMed.
- L. H. Li, Y. Chen, B. M. Cheng, M. Y. Lin, S. L. Chou and Y. C. Peng, Photoluminescence of boron nitride nanosheets exfoliated by ball milling, Appl. Phys. Lett., 2012, 100, 261108 CrossRef.
- G. Y. Guo and J. C. Lin, Systematic ab initio study of the optical properties of BN nanotubes, Phys. Rev. B: Condens. Matter, 2005, 71, 165402 CrossRef.
- K. Miyoshi, D. H. Buckley, J. J. Pouch, S. A. Alterovitz and H. E. Sliney, Mechanical strength and tribological behavior of ion-beam-deposited boron nitride films on non-metallic substrates, Surf. Coat. Technol., 1987, 33, 221–233 CrossRef CAS.
- Z. Pawlak, T. Kaldonski, R. Pai, E. Bayraktar and A. Oloyede, A comparative study on the tribological behaviour of hexagonal boron nitride (h-BN) as lubricating micro-particles-An additive in porous sliding bearings for a car clutch, Wear, 2009, 267, 1198–1202 CrossRef CAS.
- D. Lahiri, V. Singh, A. P. Benaduce, S. Seal, L. Kos and A. Agarwal, Boron nitride nanotube reinforced hydroxyapatite composite: Mechanical and tribological performance and in vitro biocompatibility to osteoblasts, J. Mech. Behav. Biomed. Mater., 2011, 4, 44–56 CrossRef CAS PubMed.
- W. Lei, D. Portehault, D. Liu, S. Qin and Y. Chen, Porous boron nitride nanosheets for effective water cleaning, Nat. Commun., 2013, 4, 1777 CrossRef PubMed.
- M. W. Smith, K. C. Jordan, C. Park, J. W. Kim, P. T. Lillehei, R. Crooks and J. S. Harrison, Very long single- and few-walled boron nitride nanotubes via the pressurized vapor/condenser method, Nanotechnology, 2009, 20, 505604 CrossRef PubMed.
- D. Pacile, J. C. Meyer, C. O. Girit and A. Zettl, The two-dimensional phase of boron nitride: Few-atomic-layer sheets and suspended membranes, Appl. Phys. Lett., 2008, 92, 133107 CrossRef.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Two-dimensional atomic crystals, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- W. Q. Han, L. Wu, Y. Zhu, K. Watanabe and T. Taniguchi, Structure of chemically derived mono- and few-atomic-layer boron nitride sheets, Appl. Phys. Lett., 2008, 93, 223103 CrossRef.
- S. Li, L. Ci, H. Lu, P. B. Sorokin, C. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Large scale growth and characterization of atomic hexagonal boron nitride layers, Nano Lett., 2010, 10, 3209–3215 CrossRef PubMed.
- L. Ci, L. Song, C. Jin, D. Jariwala, D. Wu, Y. Li, S. Arivastava, Z. F. Wang, K. Storr, L. Balicas, F. Liu and P. M. Ajayan, Atomic layers of hybridized boron nitride and graphene, Nat. Mater., 2010, 9, 430–435 CrossRef CAS PubMed.
- Y. Shi, C. Hamsen, X. Jia, K. K. Kim, A. Reina, M. Hofmann, A. L. Hsu, K. Zhang, H. Li, Z. Y. Juang, M. S. Dresselhaus, L. J. Li and J. Kong, Synthesis of few-layer hexagonal boron nitride thin film by chemical vapor deposition, Nano Lett., 2010, 10, 4134–4139 CrossRef CAS PubMed.
- R. Gao, L. W. Yin, C. X. Wang, Y. X. Qi, N. Lun, L. Zhang, Y. X. Liu, L. Kang and X. Wang, High Yield Synthesis of Boron Nitride Nanosheets with Strong Ultraviolet Cathodoluminescence Emission, J. Phys. Chem. C, 2009, 113, 15160–15165 CAS.
- K. K. Kim, A. Hsu, X. Jia, S. M. Kim, Y. Shi and M. Hofmann, et al., Synthesis of Monolayer Hexagonal Boron Nitride on Cu Foil Using Chemical Vapor Deposition, Nano Lett., 2012, 12, 161–166 CrossRef PubMed.
- C. Zhi, Y. Bando, C. Tang, H. Kuwahara and D. Golberg, Large-Scale Fabrication of Boron Nitride Nanosheets and Their Utilization in Polymeric Composites with Improved Thermal and Mechanical Properties, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS.
- Y. Yao, Z. Lin, Z. Li, X. Song, K. S. Moon and C. P. Wong, Large-scale production of two-dimensional nanosheets, J. Mater. Chem., 2012, 22, 13494–13499 RSC.
- L. H. Li, Y. Chen, G. Behan, H. Zhang, M. Petravic and A. M. Glushenkov, Large-scale mechanical peeling of boron nitride nanosheets by low-energy ball milling, J. Mater. Chem., 2011, 21, 11862–11866 RSC.
- C. Jin, F. Lin, K. Suenaga and S. Iijima, Fabrication of a freestanding boron nitride single layer and its defect assignments, Phys. Rev. Lett., 2009, 102, 195505 CrossRef PubMed.
- J. C. Meyer, A. Chuvilin, G. Algara-Siller, J. Biskupek and U. Kaiser, Selective sputtering and atomic resolution imaging of atomically thin boron nitride membranes, Nano Lett., 2009, 9, 2683–2689 CrossRef CAS PubMed.
- A. Nag, K. Raidongia, K. P. Hembram, R. Datta, U. V. Waghmare and C. N. R. Rao, Graphene Analogues of BN: Novel Synthesis and Properties, ACS Nano, 2010, 4, 1539–1544 CrossRef CAS PubMed.
- K. Raidongia, A. Nag, K. P. S. S. Hembram, U. V. Waghmare, R. Datta and C. N. R. Rao, BCN: A Graphene Analogue with Remarkable Adsorptive Properties, Chem. – Eur. J., 2010, 16, 149–157 CrossRef CAS PubMed.
- R. Y. Tay, S. H. Tsang, M. Loeblein, W. L. Chow, G. C. Loh and J. W. Toh, et al., Direct growth of nanocrystalline hexagonal boron nitride films on dielectric substrates, Appl. Phys. Lett., 2015, 106, 101901 CrossRef.
- X. Wang, C. Zhi, L. Li, H. Zeng, C. Li and M. Mitome, et al., “Chemical Blowing” of Thin-Walled Bubbles: High-Throughput Fabrication of Large-Area, Few-Layered BN and Cx-BN Nanosheets, Adv. Mater., 2011, 23, 4072–4076 CrossRef CAS PubMed.
- X. Wang, A. Pakdel, C. Zhi, K. Watanabe, T. Sekiguchi, D. Golberg and Y. Bando, High-yield boron nitride nanosheets from ‘chemical blowing’: towards practical applications in polymer composites, J. Phys.: Condens. Matter, 2012, 24, 314205 CrossRef PubMed.
- W. Han, Y. Bando, K. Kurashima and T. Sato, Synthesis of boron nitride nanotubes from carbon nanotubes by a substitution reaction, Appl. Phys. Lett., 1998, 73, 3085 CrossRef CAS.
- D. Golberg, Y. Bando, K. Kurashima and T. Sato, Ropes of BN multi-walled nanotubes, Solid State Commun., 2000, 116, 1–6 CrossRef CAS.
- H. Yurdakul, Y. Göncü, O. Durukan, A. Akay, A. T. Seyhan, N. Ay and S. Turan, Nanoscopic characterization of two-dimensional (2D) boron nitride nanosheets (BNNSs) produced by microfluidization, Ceram. Int., 2012, 38, 2187–2193 CrossRef CAS.
- D. Golberg, Y. Bando, M. Eremets, K. Takemura, K. Kurashima and H. Yusa, Nanotubes in boron nitride laser heated at high pressure, Appl. Phys. Lett., 1996, 69, 2045 CrossRef CAS.
- D. Goldberg, Y. Bando, M. Eremets, K. Takemura, K. Kurashima, K. Tamiya and H. Yusa, Boron nitride nanotube growth defects and their annealing-out under electron irradiation, Chem. Phys. Lett., 1997, 279, 191–196 CrossRef.
- D. P. Yu, X. S. Sun, C. S. Lee, I. Bello, S. T. Lee and H. D. Gu, et al., Synthesis of boron nitride nanotubes by means of excimer laser ablation at high temperature, Appl. Phys. Lett., 1998, 72, 1966 CrossRef CAS.
- R. S. Lee, J. Gavillet, M. L. de La Chapelle, A. Loiseau, J. L. Cochon and D. Pigache, et al., Catalyst-free synthesis of boron nitride single-wall nanotubes with a preferred zig-zag configuration, Phys. Rev. B: Condens. Matter, 2001, 64, 121405 CrossRef.
- Z. Zhang, W. Guo and Y. Dai, Stability and electronic properties of small boron nitride nanotubes, J. Appl. Phys., 2009, 105, 084312 CrossRef.
- D. Golberg, Y. Bando, K. Kurashima and T. Sato, Synthesis and characterization of ropes made of BN multiwalled nanotubes, Scr. Mater., 2001, 44, 1561–1564 CrossRef CAS.
- C. Li, Y. Bando, C. Zhi, Y. Huang and D. Golberg, Thickness-dependent bending modulus of hexagonal boron nitride nanosheets, Nanotechnology, 2009, 20, 5707 Search PubMed.
- A. Bosak, J. Serrano, M. Krisch, K. Watanabe, T. Taniguchi and H. Kanda, Elasticity of hexagonal boron nitride: Inelastic x-ray scattering measurements, Phys. Rev. B: Condens. Matter, 2006, 73, 041402 CrossRef.
- A. Parashar and P. Mertiny, Study of Mode I Fracture of Graphene Sheets Using Atomistic Based Finite Element Modeling and Virtual Crack Closure Technique, Int. J. Fract., 2012, 176, 119–126 CrossRef CAS.
- C. Li and T. W. Tsu-Wei Chou, A structural mechanics approach for the analysis of carbon nanotubes, Int. J. Solids Struct., 2003, 40, 2487–2499 CrossRef.
- K. I. Tserpes and P. Papanikos, Finite element modeling of single-walled carbon nanotubes, Composites, Part B, 2005, 36, 468–477 CrossRef.
- C. Li and T. W. Chou, A structural mechanics approach for the analysis of carbon nanotubes, nt. J. Solids Struct., 2003, 40, 2487–2499 CrossRef.
- K. I. Tserpes and P. Papanikos, Finite element modeling of single-walled carbon nanotubes, Composites, Part B, 2005, 36, 468–477 CrossRef.
- L. Boldrin, F. Scarpa, R. Chowdhury and S. Adhikari, Effective mechanical properties of hexagonal boron nitride nanosheets, Nanotechnology, 2011, 22, 505702 CrossRef CAS PubMed.
- S. Trivedi, S. C. Sharma and S. P. Harsha, Evaluations of Young's Modulus of Boron Nitride Nanotube Reinforced Nano-composites, Procedia Mater. Sci., 2014, 6, 1899–1905 CrossRef CAS.
- S. Trivedi, S. C. Sharma and S. P. Harsha, Dynamic Analysis of Single Walled Boron Nitride Nanotube Reinforced Composite Based Nanomechanical Resonator, J. Inst. Eng. (India): Ser. D, 2014, 95, 7–18 CrossRef CAS.
- E. S. Oh, Elastic properties of boron-nitride nanotubes through the continuum lattice approach, Mater. Lett., 2010, 64, 859–862 CrossRef CAS.
- J. F. Green, T. K. Bolland and J. W. Bolland, Lennard-Jones interaction for hexagonal layered crystals, J. Chem. Phys., 1974, 61, 1637 CrossRef CAS.
- J. F. Green, T. K. Bolland and J. W. Bolland, Theoretical elastic behavior for hexagonal boron nitride, J. Chem. Phys., 1976, 64, 656 CrossRef CAS.
- J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B: Condens. Matter, 1992, 45, 13244 CrossRef.
- J. P. Perdew and A. Zunger, Self-interaction correction to density-functional approximations for many-electron systems, Phys. Rev. B: Condens. Matter, 1981, 23, 5048 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- J. P. Perdew, Matthias Ernzerhof and Kieron Burke. Rationale for mixing exact exchange with density functional approximations, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- B. Baumeier, P. Krüger and J. Pollmann, Structural, elastic, and electronic properties of SiC, BN, and BeO nanotubes, Phys. Rev. B: Condens. Matter, 2007, 76, 085407 CrossRef.
- R. Ansari, S. Malakpour and S. Ajori, Structural and elastic properties of hybrid bilayer graphene/h-BN with different interlayer distances using DFT, Superlattices Microstruct., 2014, 72, 230–237 CrossRef CAS.
- M. Topsakal, E. Aktürk and S. Ciraci, First-principles study of two- and one-dimensional honeycomb structures of boron nitride, Phys. Rev. B: Condens. Matter, 2009, 79, 115442 CrossRef.
- R. Ansari, S. Malakpour, M. Faghihnasiri and S. Ajori, Influence of Electric Field on the Mechanical Properties of Hexagonal Boron-Nitride Sheets Using ab initio Calculations, NANO, 2015, 10, 1550047 CrossRef.
- Q. Peng, W. Ji and S. De, Mechanical properties of the hexagonal boron nitride monolayer: Ab initio study, Comput. Mater. Sci., 2012, 56, 11–17 CrossRef CAS.
- E. Hernández, C. Goze, P. Bernier and A. Rubio, Elastic properties of single-wall nanotubes, Appl. Phys. A, 1999, 68, 287–292 CrossRef.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- M. Topsakal and S. Ciraci, Elastic and plastic deformation of graphene, silicene, and boron nitride honeycomb nanoribbons under uniaxial tension: A first-principles density-functional theory study, Phys. Rev. B: Condens. Matter, 2010, 81, 024107 CrossRef.
- S. Baroni, P. Giannozzi and E. Isaev, Density-Functional Perturbation Theory for Quasi Harmonic Calculation, Rev. Mineral. Geochem., 2010, 71, 39 CrossRef CAS.
- M. Mirnezhad, R. Ansari and A. Shahabodini, Temperature Effect on Young's Modulus of Boron Nitride Sheets, J. Therm. Stresses, 2013, 36, 152–159 CrossRef.
- E. B. Tadmor and E. Miller, Modeling Materials: Continuum, Atomistic and Multiscale Techniques, Cambridge University Press, 2012 Search PubMed.
- W. C. Swope, H. C. Andersen, P. H. Berens and K. R. Wilson, A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters, J. Chem. Phys., 1982, 76, 637 CrossRef CAS.
- R. W. Hockney and J. W. Eastwood, Computer Simulation Using Particles, McGraw-Hill, New York, 1981 Search PubMed.
- D. Beeman, Some multistep methods for use in molecular dynamics calculations, J. Comput. Phys., 1976, 20, 130–139 CrossRef.
- J. Tersoff, New empirical model for the structural properties of silicon, Phys. Rev. Lett., 1986, 56, 632 CrossRef CAS PubMed.
- J. Tersoff, New empirical approach for the structure and energy of covalent systems, Phys. Rev. B: Condens. Matter, 1988, 37, 6991 CrossRef.
- J. Tersoff, Empirical Interatomic Potential for Carbon, with Applications to Amorphous Carbon, Phys. Rev. Lett., 1988, 61, 2869 CrossRef PubMed.
- J. Tersoff, Modeling solid-state chemistry: Interatomic potentials for multicomponent system, Phys. Rev. B: Condens. Matter, 1989, 39, 5566 CrossRef.
- J. E. Jones, On the Determination of Molecular Fields. II. From the Equation of State of a Gas, Proc. R. Soc. London, Ser. A, 1924, 106, 463–477 CrossRef CAS.
- M. S. Daw and M. I. Basks, Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals, Phys. Rev. B: Condens. Matter, 1984, 29, 6443 CrossRef CAS.
- B. W. Jeong, J. K. Lim and S. B. Sinnott, Tensile mechanical behavior of hollow and filled carbon nanotubes under tension or combined tension-torsion, Appl. Phys. Lett., 2007, 90, 023102 CrossRef.
- S. J. Stuart, A. B. Tutein and J. A. Harrison, A reactive potential for hydrocarbons with intermolecular interactions, J. Chem. Phys., 2000, 112, 6472 CrossRef CAS.
- S. K. Singh, M. Neek-Amal, S. Costamagna and F. M. Peeters, Thermomechanical properties of a single hexagonal boron nitride sheets, Phys. Rev. B: Condens. Matter, 2013, 87, 184106 CrossRef.
- D. B. Zhang, E. Akatyeva and T. Dumitrică, Helical BN and ZnO nanotubes with intrinsic twisting: An objective molecular dynamics study, Phys. Rev. B: Condens. Matter, 2011, 84, 115431 CrossRef.
- M. Q. Le, Prediction of Young's modulus of hexagonal monolayer sheets based on molecular mechanics, Int. J. Mech. Mater. Des., 2015, 11, 15–24 CrossRef CAS.
- D. W. Brenner, Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films, Phys. Rev. B: Condens. Matter, 1990, 42, 9458 CrossRef CAS.
- W. Sekkal, B. Bouhafs, H. Aourag and M. Certier, Molecular-dynamics simulation of structural and thermodynamic properties of boron nitride, J. Phys.: Condens. Matter, 1998, 10, 4975 CrossRef CAS.
- C. Sevik, A. Kinaci, J. B. Haskins and T. Çağın, Characterization of thermal transport in low-dimensional boron nitride nanostructures, Phys. Rev. B: Condens. Matter, 2011, 84, 085409 CrossRef.
- K. Matsunaga and Y. Iwamoto, Molecular Dynamics Study of Atomic Structure and Diffusion Behavior in Amorphous Silicon Nitride Containing Boron, J. Am. Ceram. Soc., 2001, 84, 2213–2219 CrossRef CAS.
- K. Matsunaga, Indra, H. Osamu, I. Shoji and O. Yasushi, The new 5-HT 2A and analysis by receptor binding experiments and molecular modeling of the interaction with the receptor antagonist Nantenin the receptor, Jpn. Pharmacol. Mag., 2000, 116, 48–52 Search PubMed.
- K. Matsunaga, C. Fisher and H. Matsubara, Tersoff Potential Parameters for Simulating Cubic Boron Carbonitrides, Jpn. J. Appl. Phys., 2000, 39, 48 CrossRef.
- K. Albe and W. Möller, Modeling of boron nitride: Atomic scale simulations on thin film growth, Comput. Mater. Sci., 1998, 10, 111–115 CrossRef CAS.
- K. Albe, W. Möller and K. H. Heinig, Computer simulation and boron nitride, Radiat. Eff. Defects Solids, 1997, 141, 85–97 CrossRef CAS.
- A. C. Van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, ReaxFF: A Reactive Force Field for Hydrocarbons, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- M. R. Weismiller, A. C. V. Duin, J. Lee and R. A. Yetter, ReaxFF Reactive Force Field Development and Applications for Molecular Dynamics Simulations of Ammonia Borane Dehydrogenation and Combustion, J. Phys. Chem. A, 2010, 114, 5485–5492 CrossRef CAS PubMed.
- A. C. Van Duin, A. Strachan, S. Stewman, Q. Zhang, X. Xu and W. Goddard, ReaxFFSiO Reactive Force Field for Silicon and Silicon Oxide Systems, J. Phys. Chem. A, 2003, 107, 3803–3811 CrossRef CAS.
- N. Ohba, K. Miwa, N. Nagasako and A. Fukumoto, First-principles study on structural, dielectric, and dynamical properties for three BN polytypes, Phys. Rev. B: Condens. Matter, 2001, 63, 115207 CrossRef.
- M. Mirnezhad, R. Ansari and H. Rouhi, Mechanical properties of multilayer boron nitride with different stacking orders Super-lattices, Microstruct., 2013, 53, 223–231 CrossRef CAS.
- T. Han, Y. Luo and C. Wang, Effects of temperature and strain rate on the mechanical properties of hexagonal boron nitride nanosheets, J. Phys. D: Appl. Phys., 2014, 47, 025303 CrossRef.
- X. Wei, M. S. Wang, Y. Bando and D. Goldberg, Tensile Tests on Individual Multi-Walled Boron Nitride Nanotubes, Adv. Mater., 2010, 22, 4895–4899 CrossRef CAS PubMed.
- B. Mortazavi and Y. Rémond, Investigation of tensile response and thermal conductivity of boron-nitride nanosheets using molecular dynamics simulations, Physica E, 2012, 44, 1846–1852 CrossRef CAS.
- S. Zhao and J. Xue, Mechanical properties of hybrid graphene and hexagonal boron nitride sheets as revealed by molecular dynamic simulations, J. Phys. D: Appl. Phys., 2013, 46, 135303 CrossRef.
- M. Q. Le and D. T. Nguyen, Atomistic simulations of pristine and defective hexagonal BN and SiC sheets under uniaxial tension, Mater. Sci. Eng., A, 2014, 615, 481–488 CrossRef CAS.
- J. Yuan and K. M. Liew, The Effects of Grafted Carboxyl Groups on the Elastic Properties of Single-Walled Boron Nitride Nanotubes, J. Comput. Theor. Nanosci., 2010, 7, 1878–1884 CrossRef CAS.
- M. Ghazizadeh, J. E. Estevez, A. D. Kelkar and J. G. Ryan, Mechanical Properties Prediction of Hydrogenated Boron Nitride Nanotube's using Molecular Dynamic Simulations, JSM Nanotechnol. Nanomed., 2014, 2, 1030 Search PubMed.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- T. A. Hilder, R. Yang, V. Ganesh, D. Gordon, A. Bliznyuk, A. P. Rendell and S. H. Chung, Validity of current force fields for simulations on boron nitride nanotubes, Micro Nano Lett., 2010, 5, 150–156 CAS.
- (a) S. S. Han, J. K. Kang and H. M. Leea, The theoretical study on interaction of hydrogen with single-walled boron nitride nanotubes. I. The reactive force field ReaxFFHBN development, J. Chem. Phys., 2005, 123, 114703 CrossRef PubMed; (b) D. Srivastava, M. Menon and K. Cho, Anisotropic nanomechanics of boron nitride nanotubes: Nanostructured “skin” effect, Phys. Rev. B: Condens. Matter, 2001, 63, 195413 CrossRef.
- S. Ebrahimi-Nejad and A. Shokuhfar, Compressive buckling of open-ended boron nitride nanotubes in hydrogen storage applications, Physica E, 2013, 50, 29–36 CrossRef CAS.
- M. L. Liao, Y. C. Wang, S. P. Ju, T. W. Lien and L. F. Huang, Deformation behaviors of an armchair boron-nitride nanotube under axial tensile strain, J. Appl. Phys., 2011, 110, 054310 CrossRef.
- H. J. Shen, Mechanical properties and electronic structures of one BN nanotube under radial compression, Front. Mater. Sci. China, 2009, 3, 201–204 CrossRef.
- S. Ebrahimi-Nejad, A. Shokuhfar and A. Zare-Shahabadi, Molecular Dynamics Simulation of the Buckling Behavior of Boron Nitride Nanotubes under Uniaxial Compressive Loading, Defect Diffus. Forum, 2010, 297, 984–989 CrossRef.
- G. J. Slotman and A. Fasolino, Structure, stability and defects of single layer hexagonal BN in comparison to graphene, J. Phys.: Condens. Matter, 2013, 25, 045009 CrossRef CAS PubMed.
- M. L. Liao, T. W. Lian and S. P. Ju, Tensile and Compressive Behaviors of a Boron Nitride Nanotube: Temperature Effects, Mater. Sci. Forum, 2012, 700, 125–128 CrossRef CAS.
- A. Shokuhfar, S. Ebrahimi-Nejad, A. Hosseini-Sadegh and A. Zare-Shahabadi, The effect of temperature on the compressive buckling of boron nitride nanotubes, Phys. Status Solidi A, 2012, 209, 1266–1273 CrossRef CAS.
- J. Yuan and K. M. Liew, High-Temperature Thermal Stability and In-Plane Compressive Properties of a Graphene and a Boron-Nitride Nanosheet, J. Nanosci. Nanotechnol., 2012, 12, 2617–2624 CrossRef CAS PubMed.
- A. Zobelli, C. P. Ewels, A. Gloter, G. Seifert, O. Stephan, S. Csillag and C. Colliex, Defective Structure of BN Nanotubes: From Single Vacancies to Dislocation Lines, Nano Lett., 2006, 6, 1955–1960 CrossRef CAS PubMed.
- A. J. Stone and D. J. Wales, Theoretical studies of icosahedral C60 and some related species, Chem. Phys. Lett., 1986, 128, 501–503 CrossRef CAS.
- M. Griebel, J. Hamaekers and F. Heber, A molecular dynamics study on the impact of defects and functionalization on the Young modulus of boron–nitride nanotubes, Comput. Mater. Sci., 2009, 45, 1097–1103 CrossRef CAS.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys., 1981, 52, 7182 CrossRef CAS.
- A. Shokuhfar and S. Ebrahimi-Nejad, Effects of structural defects on the compressive buckling of boron nitride nanotubes, Physica E, 2013, 48, 53–60 CrossRef CAS.
- J. V. N. Sarma, R. Chowdhury, R. Jayaganthan and F. Scarpa, Atomistic Studies on Tensile Mechanics of BN Nanotubes in the Presence of Defects, Int. J. Nanosci., 2014, 13, 1450005 CrossRef.
- N. M. A. Krishnan and D. Debraj Ghosh, Defect induced plasticity and failure mechanism of boron nitride nanotubes under tension, J. Appl. Phys., 2014, 116, 044313 CrossRef.
- B. Mortazavi and G. Cuniberti, Mechanical properties of polycrystalline boron-nitride nanosheets, RSC Adv., 2014, 4, 19137–19143 RSC.
- W. H. Moon and H. J. Hwang, Molecular-dynamics simulation of structure and thermal behaviour of boron nitride nanotubes, Nanotechnology, 2004, 15, 431 CrossRef CAS.
- R. Wei, Y. Tian, V. Eichhorn and S. Fatikow, Compressive and tensile behaviors of carbon and boron nitride nanotubes, in Manipulation, Manufacturing and Measurement on the Nanoscale (3M-NANO), 2012, pp. 301–304 Search PubMed.
- Y. Tian, Z. Li, W. Gao, K. Cai, F. Wang and D. Zhang, et al., Mechanical properties investigation of monolayer h-BN sheet under in-plane shear displacement using molecular dynamics simulations, J. Appl. Phys., 2014, 115, 014308 CrossRef.
- E. Perim, R. P. Santos, P. A. Autreto and D. S. Galvao, Fracture Patterns of Boron Nitride Nanotubes, MRS Proc., 2013, 1526 Search PubMed.
- N. M. A. Krishnan and D. Ghosh, Chirality dependent elastic properties of single-walled boron nitride nanotubes under uniaxial and torsional loading, J. Appl. Phys., 2014, 115, 064303 CrossRef.
- A. Ansari and S. Ajori, Molecular dynamics study of the torsional vibration characteristics of boron-nitride nanotubes, Phys. Lett. A, 2014, 378, 2876–2880 CrossRef.
- S. Ajori and R. Ansari, Torsional buckling behavior of boron-nitride nanotubes using molecular dynamics simulations, Curr. Appl. Phys., 2014, 14, 1072–1077 CrossRef.
- M. Q. Le, Size effects in mechanical properties of boron nitride nanoribbons, J. Mech. Sci. Technol., 2014, 28, 4173–4178 CrossRef.
- D. M. Tang, C. L. Ren, X. Wei, M. S. Wang, C. Liu, Y. Bando and D. Golberg, Mechanical Properties of Bamboo-like Boron Nitride Nanotubes by In Situ TEM and MD Simulations: Strengthening Effect of Interlocked Joint Interfaces, ACS Nano, 2011, 5, 7362–7368 CrossRef CAS PubMed.
- L. T. Chaddertona and Y. Chenb, A model for the growth of bamboo and skeletal nanotubes: catalytic capillarity, J. Cryst. Growth, 2002, 240, 164–169 CrossRef.
- D. M. Tang, C. Liu and H. M. Cheng, Controlled synthesis of quasi-one-dimensional boron nitride nanostructures, J. Mater. Res., 2007, 22, 2809–2816 CrossRef CAS.
- H. J. Shen, Thermal-conductivity and tensile-properties of BN, SiC and Ge nanotubes, Comput. Mater. Sci., 2009, 47, 220–224 CrossRef CAS.
- A. Kınacı, J. B. Haskins, C. Sevik and T. Çağın, Thermal conductivity of BN-C nanostructures, Phys. Rev. B: Condens. Matter, 2012, 86, 115410 CrossRef.
- L. Lindsay and D. A. Broido, Optimized Tersoff and Brenner empirical potential parameters for lattice dynamics and phonon thermal transport in carbon nanotubes and graphene, Phys. Rev. B: Condens. Matter, 2010, 81, 205441 CrossRef.
- Y. C. Chen, S. C. Lee, T. H. Liu and C. C. Chang, Thermal conductivity of boron nitride nanoribbons: Anisotropic effects and boundary scattering, Int. J. Therm. Sci., 2015, 94, 72–78 CrossRef CAS.
- F. Müller-Plathe, A simple nonequilibrium molecular dynamics method for calculating the thermal conductivity, J. Chem. Phys., 1997, 106, 6082 CrossRef.
- C. Sevik, A. Kinaci, J. B. Haskins and T. Çağın, Influence of disorder on thermal transport properties of boron nitride nanostructures, Phys. Rev. B: Condens. Matter, 2012, 86, 075403 CrossRef.
- H. J. Shen, The Effects of Radial Compression on Thermal Conductivity of Carbon and Boron Nitride Nanotubes, J. Nanomater., 2012, 756791 Search PubMed.
- A. Mashreghi, Thermal expansion/contraction of boron nitride nanotubes in axial, radial and circumferential directions, Comput. Mater. Sci., 2012, 65, 356–364 CrossRef CAS.
- K. M. Liew and J. Yuan, High-temperature thermal stability and axial compressive properties of a coaxial carbon nanotube inside a boron nitride nanotube, Nanotechnology, 2011, 22, 085701 CrossRef CAS PubMed.
- A. K. Rappe, K. S. Colwell and C. J. Casewit, Application of a universal force field to metal complexes, Inorg. Chem., 1993, 32, 3438–3450 CrossRef CAS.
- J. Yuan, J. Liao, C. Yang and X. Shi, Structural Stability and Elastic Properties of Bilayer Graphenes and Boron Nitride Nanosheets, Curr. Nanosci., 2013, 9, 324–329 CrossRef CAS.
- J. Yuan and K. M. Liew, Structure stability and high-temperature distortion resistance of trilayer complexes formed from graphenes and boron nitride nanosheets, Phys. Chem. Chem. Phys., 2014, 16, 88–94 RSC.
- J. Song, V. Nikhil and N. V. Medhekar, Thermal transport in lattice-constrained 2D hybrid graphene heterostructures, J. Phys.: Condens. Matter, 2013, 25, 445007 CrossRef PubMed.
- H. J. Shen, Thermal-stability and compressive properties of one boron nitride nanotube embedded in another carbon tube, Micro Nano Lett., 2011, 6, 444–447 CAS.
- G. Zhang, R. Zhou and X. C. Zeng, Carbon nanotube and boron nitride nanotube hosted C60–V nanopeapods, J. Mater. Chem. C, 2013, 1, 4518–4526 RSC.
- A. T. Nasrabadi and M. Foroutan, Interactions between Polymers and Single-Walled Boron Nitride Nanotubes: A Molecular Dynamics Simulation Approach, J. Phys. Chem. B, 2010, 114, 15429–15436 CrossRef CAS PubMed.
- N. L. Allinger, Y. H. Yuh and J. H. Lii, Molecular mechanics. The MM3 force field for hydrocarbons, J. Am. Chem. Soc., 1989, 111, 8551–8566 CrossRef CAS.
- S. M. Fatemi and M. Foroutan, Study of dispersion of boron nitride nanotubes by triton X-100 surfactant using molecular dynamics simulations, J. Theor. Comput. Chem., 2014, 13, 1450063 CrossRef CAS.
- M. J. Frisch, G. W. Trudes and H. B. Schlegel, Gaussian 03, Revision C.02, Gaussian Inc., Wallingford, CT, 2004 Search PubMed.
- J. Wang, H. Li, Y. Li, H. Yu, Y. He and X. Song, Deformation of copper-filled single-walled boron-nitride nanotubes under axial compression, Physica E, 2011, 44, 286–289 CrossRef CAS.
- D. H. Jung, D. Kim, T. B. Lee, S. B. Choi, J. H. Yoon and J. Kim, et al., Grand Canonical Monte Carlo Simulation Study on the Catenation Effect on Hydrogen Adsorption onto the Interpenetrating Metal–Organic Frameworks, J. Phys. Chem. B, 2006, 110, 22987–22990 CrossRef CAS PubMed.
- Y. Zhou, S. Adams, R. P. Rao, D. D. Edwards, A. Neiman and N. Pestereva, Charge Transport by Polyatomic Anion Diffusion in Sc2(WO4)3, Chem. Mater., 2008, 20, 6335–6345 CrossRef CAS.
- R. Bari, D. Parviz, F. Khabaz, C. D. Klaassen, S. D. Metzler and M. J. Hansen, et al., Liquid phase exfoliation and crumpling of inorganic nanosheets, Phys. Chem. Chem. Phys., 2015, 17, 9383–9393 RSC.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and A. David, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and A. David, Automatic atom type and bond type perception in molecular mechanical calculations, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, DREIDING: a generic force field for molecular simulations, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- M. Griebel and J. Hamaekers, Molecular dynamics simulations of boron-nitride nanotubes embedded in amorphous Si-B-N, Comput. Mater. Sci., 2007, 39, 502–517 CrossRef CAS.
- M. Parrinello and A. Rahman, Crystal Structure and Pair Potentials: A Molecular-Dynamics Study, Phys. Rev. Lett., 1980, 45, 1196 CrossRef CAS.
- M. Thomas, M. Enciso and T. A. Hilder, Insertion Mechanism and Stability of Boron Nitride Nanotubes in Lipid Bilayers, J. Phys. Chem. B, 2015, 119, 4929–4936 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. L. D. R. Bellott, R. L. Dunbrack, J. D. Evanseck and M. J. Field, All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins, J. Phys. Chem. B, 1998, 102, 3586–3516 CrossRef CAS PubMed.
- A. D. MacKerell, M. Feig and C. L. Brooks, Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef CAS PubMed.
- J. Yuan and K. M. Liew, Formation and Compressing Behavior of Coaxial Silicon Nanowires inside a Boron Nitride Nanotube, J. Phys. Chem. C, 2011, 115, 431–435 CAS.
| This journal is © The Royal Society of Chemistry 2016 |