
Uniform Au@Pt core–shell nanodendrites supported on molybdenum disulfide nanosheets for the methanol oxidation reaction†
Shao
Su
*a,
Chi
Zhang
a,
Lihui
Yuwen
a,
Xingfen
Liu
a,
Lihua
Wang
b,
Chunhai
Fan
ab and
Lianhui
Wang
*a
aKey Laboratory for Organic Electronics and Information Displays & Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing University of Posts & Telecommunications, 9 Wenyuan Road, Nanjing 210023, China
bDivision of Physical Biology, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China. E-mail: iamssu@njupt.edu.cn; iamlhwang@njupt.edu.cn
First published on 16th November 2015
Abstract
Herein, we presented a facile seeded growth method to prepare high-quality three-dimensional (3D) Au@Pt bimetallic nanodendrite-decorated molybdenum disulfide (MoS2) nanosheets (Au@Pt/MoS2). Transmission electron microscopy (TEM) and high-resolution TEM exhibited that Au@Pt core–shell nanostructures were dispersed onto the surface of MoS2 nanosheets. More importantly, the thickness of the Pt shell of the Au@Pt bimetallic nanodendrites on the surface of the MoS2 nanosheets could be easily tuned via simply changing the synthesis parameters, such as the concentration of H2PtCl6, reaction time and temperature, which greatly influence the catalytic ability of Au@Pt/MoS2 nanohybrids. Both cyclic voltammetry (CV) and chronoamperometry (CA) demonstrated that the as-prepared Au@Pt/MoS2 nanohybrids possessed much higher electrocatalytic activity and stability than Pt/MoS2 or commercial Pt/C catalyst. The peak current mass density of the selected Au@Pt/MoS2 was 6.24 A mg−1, which was 3389 and 20.3 times those of Pt/C (0.00184 A mg−1) and Pt/MoS2 (0.307 A mg−1), respectively. The presented method may be a facile approach for the synthesis of MoS2-supported bimetallic nanocomposites, which is significant for the development of high performance MoS2-based sensors and catalysts.
1. Introduction
The direct methanol fuel cell (DMFC) is regarded as one of the most promising energy sources due to low toxicity, high energy density of methanol, availability at a low price and biological renewability. As we all know, Pt-based nanostructures are considered as the most common and efficient catalysts for the methanol oxidation reaction (MOR).1–3 However, methanol oxidation on a Pt/C catalyst usually requires high Pt loading to achieve high efficiency. Moreover, Pt/C catalysts easily suffer from poisoning by CO, which limits the MOR kinetics and long-term stability. To improve the electrocatalytic performance of Pt and minimize the usage of Pt, Pt-based bimetallic nanohybrids have attracted more and more attention to replace pure Pt as electrocatalysts during the past decade.4–9 Among these bimetallic nanohybrids, such as Pd–Pt, Au–Pt, Fe–Pt, Ni–Pt, Ag–Pt and Ru–Pt, Pt-based core–shell nanostructures are particularly interesting because of their excellent catalytic activities.10–18 The Au@Pt core–shell bimetallic nanohybrid is one of the most studied Pt-based core–shell nanostructures.14,16,17 Unfortunately, such Au@Pt bimetallic nanostructures usually suffer from heavy aggregation during the synthetic process and reuse in catalytic reactions. Therefore, supporting materials have been introduced into constructing Au@Pt-substrate nanocomposites, which is not only to efficiently solve aggregation but also greatly enhance the performance of the catalyst. For example, Zeng's group successfully synthesized a Au–Pt nanoparticle-dispersed carbon (Au@Pt/C) catalyst, which exhibited excellent electrocatalytic activity toward methanol oxidation.18 The presence of Au underneath a very thin Pt shell could facilitate the removal of inhibiting CO-like reaction intermediates. Cui et al. employed an under-potential deposition redox replacement technique to synthesize a Au–Pt core–shell catalyst supported on a reduced graphene oxide surface. The prepared Au–Pt/RGO exhibited excellent catalytic activity towards the oxygen reduction reaction and the methanol oxidation reaction.19As a graphene analogue, MoS2 has attracted increasing interest in sensors, catalysis, capacitors, lithium batteries and energy harvesting due to its novel nanoelectronic and optoelectronic properties.20–31 Like graphene, MoS2 is also considered as a promising supporting material to stabilize metal nanoparticles (NPs), forming hierarchical nanocomposites. Therefore, it is expected that MoS2 nanosheets decorated with noble metal nanoparticles (NPs) could potentially be used as novel catalytic nanomaterials. Huang et al. developed the epitaxial growth of Pd, Pt, Au and Ag nanoparticles on the surface of single-layer MoS2 nanosheets. Among these hybrid nanomaterials, Pt–MoS2 nanocomposites showed higher catalytic activity toward the hydrogen evolution reaction compared with commercial Pt catalysts.32 Wang's group reported that PdNPs–MoS2 nanocomposites could significantly enhance catalytic activity toward the methanol oxidation reaction.33 Considering that single noble metal nanoparticle-decorated MoS2 had excellent electrocatalytic activity, noble bimetallic nanostructures grown on the surface of MoS2 nanosheets could possess better electrocatalytic performance. Up to now, there is no report of the synthesis of a Au@Pt/MoS2 hybrid nanocomposite and the performance of its electrocatalytic activity.
In this paper, we demonstrate for the first time a wet-chemical approach to synthesize high-quality 3D Au@Pt bimetallic nanodendrites supported on MoS2 nanosheets (Au@Pt/MoS2). As shown in Fig. 1, gold nanoparticle-decorated MoS2 nanocomposites were used as nanoseeds to prepare Au@Pt/MoS2 hybrid nanocomposites. The morphology, structure and composition of the as-prepared Au@Pt/MoS2 were characterized by TEM, HRTEM, powder X-ray diffraction (XRD) and X-ray photoelectron spectroscopy. Interestingly, the thickness of the Pt shell and the catalytic performance of Au@Pt/MoS2 were easily tuned by altering the concentration of H2PtCl6, reaction time and temperature. The as-prepared MoS2-based substrates possessed synergistic effects of the intrinsic properties of the Au@Pt and MoS2, making the Au@Pt/MoS2 nanocomposite exhibit attractive electrocatalytic activity. More importantly, the thicker Pt shell (Au@Pt/MoS2-2) generated a larger surface area, which exhibited higher electrocatalytic activity toward the MOR than the thinner nanocomposites (Au@Pt/MoS2-1).
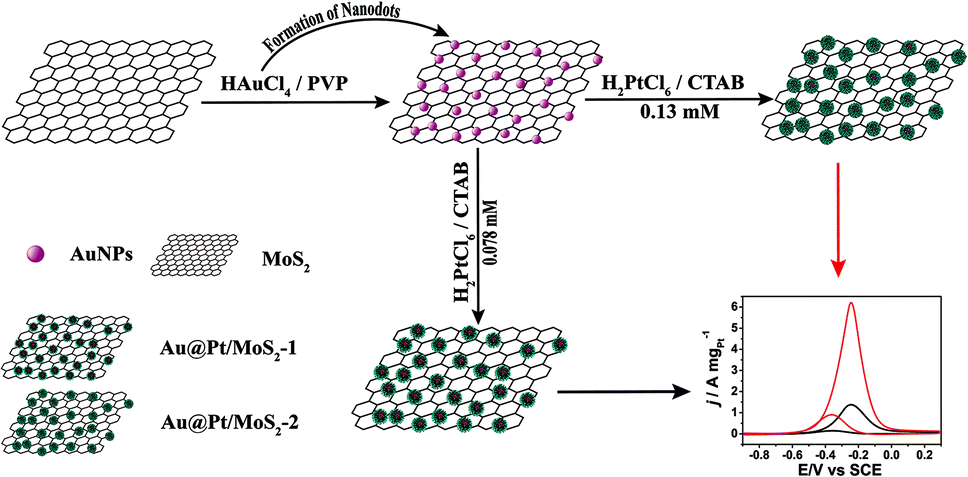 | ||
| Fig. 1 Schematic illustration for synthesizing Au@Pt/MoS2 nanocomposites and their application in methanol oxidation reaction. | ||
2. Experimental section
2.1. Reagents and chemicals
N-Butyllithium (n-BuLi, 2.4 M hexane solution) was bought from Amethyst. Gold(III) tetrachloride trihydrate (HAuCl4·3H2O, ≥49.0%), hydrogen hexachloroplatinate(IV) (H2PtCl6·xH2O, ≥99.9%), cetyltrimethylammonium bromide (CTAB, 96%), poly(N-vinyl-2-pyrrolidone) (PVP·K30, molecular weight = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–40000), sodium carboxymethylcellulose (CMC, 800–1200 mPa s), Pt/C (10 wt%) and molybdenum(IV) sulfide powder (<2 μm, 99%) were purchased from Sigma. L-Ascorbic acid (AA, ≥99.7%) and methanol (≥99.7%) were provided by Sinopharm Chemical Reagent Co., Ltd. All chemicals and reagents were directly used without further purification. All solutions were prepared with ultrapure millipore water (18.2 MΩ cm) from a Milli-pore system.
000–40000), sodium carboxymethylcellulose (CMC, 800–1200 mPa s), Pt/C (10 wt%) and molybdenum(IV) sulfide powder (<2 μm, 99%) were purchased from Sigma. L-Ascorbic acid (AA, ≥99.7%) and methanol (≥99.7%) were provided by Sinopharm Chemical Reagent Co., Ltd. All chemicals and reagents were directly used without further purification. All solutions were prepared with ultrapure millipore water (18.2 MΩ cm) from a Milli-pore system.
2.2. Apparatus
UV-vis-NIR adsorption spectra were measured on a Shimadzu UV-3600 spectrophotometer. Transmission electron microscopy (TEM) images were taken on a Hitachi H-7500 electron microscope (120 kV) and high-resolution TEM (HRTEM) characterization was performed on a Tecnai G2F20 S-Twin electron microscope (200 kV). Structural analysis of the selected Au@Pt/MoS2-2 nanocomposite was performed in scanning TEM (STEM) mode on a Philips CM 200 electron microscope (200 kV) equipped with an energy dispersive X-ray spectrometer (EDS). The STEM image was recorded with a high-angle annular dark-field (HAADF) detector. Powder X-ray diffraction (XRD) was performed using a D/max-γB diffractometer. X-ray photoelectron spectroscopy (XPS) was performed using a PHI 5000 Versa Probe with Al Kα as the excitation source. Surface morphology of MoS2 nanosheets was examined with an atomic force microscope (AFM, Bruker). The loading amounts of dendritic Pt were determined by using inductively coupled plasma mass spectroscopy (ICP-MS, Optima 5300DV, PE).Electrochemical measurements were carried out by using a CHI-660E Electrochemical Workstation with a three-electrode system (CHI Instruments, USA). The nanocomposite-modified carbon glass electrode (GCE) served as a working electrode. A Pt wire and saturated calomel electrode (SCE) were used as the counter and reference electrode, respectively. Details for preparation of the modified electrodes were provided in the ESI.† All the potentials were reported with respect to the SCE and all electrochemical data were obtained at room temperature.
2.3. Preparation of the MoS2, Au/MoS2 and Au@Pt/MoS2 nanocomposites
MoS2 nanosheets were prepared according to our previous works.33–35 The detailed process was listed in the ESI.† The morphology of MoS2 nanosheets was characterized by TEM. Fig. S1A† showed that the as-prepared MoS2 nanosheets exhibited a typical layered material with little bending and folding. The thickness of MoS2 nanosheets was determined by AFM. As shown in Fig. S2,† the average height of the MoS2 nanosheets was about 1.09 nm, suggesting that the exfoliated MoS2 nanosheet was a single layer.36,37The gold nanoparticle (AuNP)-decorated MoS2 nanocomposite (Au/MoS2) was synthesized by an in situ reduction growth method. In a typical synthesis, 4 mL (0.025 mg mL−1) of the MoS2 nanosheet dispersion was mixed with 400 μL of PVP (5%) under vigorous stirring for 2 min to obtain a stable mixture. Then, 110 μL 10 mM HAuCl4·3H2O was dropped into the mixture slowly and stirred vigorously for 20 min. The color of the mixture slowly turned from brown to wine. Finally, the product Au/MoS2 nanocomposite was stored at 4 °C without further purification. The morphology of Au/MoS2 was characterized by TEM. Uniform, high-density AuNPs were decorated on the surface of MoS2 with an average diameter of 15 nm (Fig. S1B†).
In a typical synthesis of the Au@Pt/MoS2 nanocomposite, 0.3 mL aqueous Au/MoS2 seed solution was added to 2 mL ultrapure water. Then 0.5 mL 10 mM CTAB solution was mixed with the seed solution for 5 min under vigorous stirring. After that, 1 mL AA (100 mM) was injected quickly into the mixed solution. At intervals of 5 s, 100 μL 5 mM H2PtCl6 was added to the solution under stirring. Then, the mixture was heated on a hotplate at approximately 100 °C for 6 min to grow dendritic Pt nanoshells. Finally, the product of Au@Pt/MoS2 nanocomposite was purified by washing and centrifugation (5000 rpm for 10 min) to remove excess reagent three times with acetone and water.
UV-vis absorption spectra and the color change of the solution have been employed to prove the whole synthesis process. Fig. S3† shows the UV-vis absorption spectra of MoS2, Au/MoS2 and Au@Pt/MoS2 dispersions. Three obvious absorption peaks of MoS2 were obtained at 255 nm, 304 nm and 400 nm, which were ascribed to exfoliated MoS2 nanosheets (black line). After the AuNPs grew on the MoS2 nanosheets, a new absorption peak corresponding to the Au plasma band at around 520 nm emerged (red line).38 For Au@Pt/MoS2, no obvious new peaks were obtained but the absorbance from 300 nm to 1000 nm noticeably increased, since Au@Pt nanodendrites have a wide absorption range with no distinct absorption peaks in the UV-vis-NIR range (blue line).33,39,40 Moreover, an obvious color change was observed during the Au/MoS2 and Au@Pt/MoS2 synthesis process. The color of the solution changed from brown to wine after HAuCl4 was added to the MoS2 solution. During the synthesis of the Au@Pt/MoS2 nanocomposite, the color of the solution changed from wine to a dark color (Fig. S3,† inset).
3. Results and discussion
3.1. Preparation and characterization of the Au@Pt/MoS2 nanocomposite
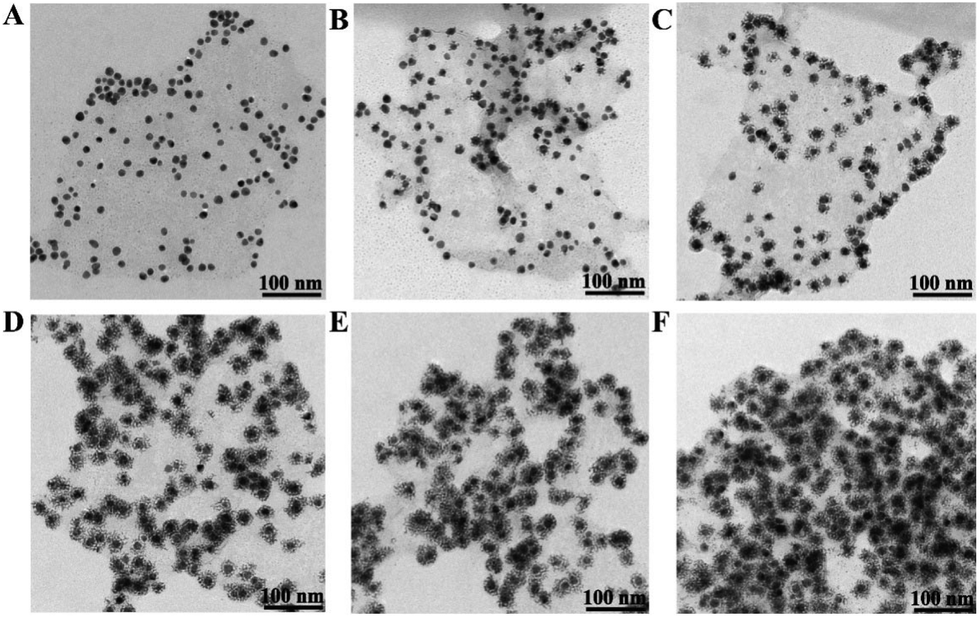
The synthesis process of the Au@Pt/MoS2 nanocomposite was monitored by TEM. First, the MoS2 nanosheets decorated with Au@Pt nanodendrites were examined by changing the concentration of H2PtCl6 and keeping other conditions unaltered. Interestingly, the morphology of the Au@Pt/MoS2 nanocomposite could be tuned by varying the concentration of H2PtCl6. As shown in Fig. 2A–F, the size of the Au@Pt nanodendrites gradually grew bigger with the concentration of H2PtCl6 changing from 0.026 to 0.21 mM. Only few Pt shells were dispersed on the AuNPs with addition of 0.026 mM H2PtCl6 (Fig. 2A). When the concentration of H2PtCl6 was increased, the Au@Pt grew larger (Fig. 2B and C). When the concentration of H2PtCl6 was 0.13 mM, a uniform, high-density Au@Pt/MoS2 nanocomposite was obtained (Fig. 2D). If the concentration of H2PtCl6 increased up to 0.17 mM and 0.21 mM, the thickness of the Pt shell increased a little (Fig. 2E and F). The corresponding particle size distribution of Au@Pt nanodendrites was listed in Fig. S4.† The data also proved that the particle size increased with the concentration of H2PtCl6. In order to save the usage of Pt, 0.13 mM H2PtCl6 was selected as the optimal concentration. | ||
| Fig. 2 TEM images of Au@Pt/MoS2 synthesized under different concentrations of H2PtCl6 (from A to F: 0.026 mM, 0.078 mM, 0.11 mM, 0.13 mM, 0.17 mM and 0.21 mM). | ||
As we know, reaction time usually plays a vital role in nanomaterials’ morphology.14,16 We further explored the influence of reaction time on the morphologies of Au@Pt/MoS2 nanohybrids by using 0.13 mM H2PtCl6 at 100 °C. As shown in Fig. 3, the thickness of the Pt shell increased with the reaction time in the range from 0 min to 6 min (Fig. 3A–D). When the reaction time was prolonged to 10–15 min, the thickness of the Pt shell was almost unchanged (Fig. 3E and F). These results indicated that the reaction process was fast, and only needed 6 min to form Au@Pt nanodendrites under the appropriate conditions.
 | ||
| Fig. 3 TEM images of Au@Pt/MoS2 formed under different times (from A to F: 0 min, 2 min, 4 min, 6 min, 10 min and 15 min). | ||
Based on the optimal concentration and time, the reaction temperature was also studied. Fig. 4 shows the TEM images of Au@Pt/MoS2 nanohybrids at different temperatures. The Pt nanoshell gradually grew thicker as the temperature was increased from 30 °C to 100 °C (Fig. 4A–D). When the temperature was 100 °C, a uniform and well-dispersed Pt nanoshell grew on the surface of the Au nanoparticles (Fig. 4D). If the temperature was over 100 °C, the thickness of the Pt nanoshell still increased (Fig. 4E and F). But it should be pointed out that the stability and dispersibility of the Au@Pt/MoS2 nanohybrids were worse than that prepared at 100 °C. As shown in Fig. S5,† the Au@Pt/MoS2 nanocomposite gradually settled to the bottom of the tube after 8 h when it was synthesized at 160 °C. Meanwhile, the as-prepared Au@Pt/MoS2 at 100 °C was still stable and dispersible. In this work, we chose the best temperature of 100 °C.
 | ||
| Fig. 4 TEM images of Au@Pt/MoS2 synthesized under different temperatures (from A to F: 30 °C, 60 °C, 80 °C, 100 °C, 120 °C and 160 °C). | ||
The morphology of the Au@Pt/MoS2 nanocomposite was further characterized by HRTEM. The magnified image revealed that the lattice distances for Au cores and Pt shells were 0.240 nm (image b) and 0.225 nm (image a), respectively, corresponding to the planes of face-centered cubic (fcc) Au and Pt, respectively, which suggested that bimetallic Au@Pt core–shell nanodendrites were successfully synthesized (Fig. 5A).16,33,41 The HAADF-STEM image further revealed the dendritic structure and the elemental distribution of the nanohybrid (Fig. 5B). The elemental mapping patterns (image a–c) and the line profiles of a single bimetallic nanodendrite (inset image d) further demonstrated the core–shell feature of the Au@Pt nanodendrites. Pt appears to be distributed over the entire surface of the Au core and does not overlap extensively, which agreed with previous reports.14,17,42
 | ||
| Fig. 5 (A) HRTEM images of the Au@Pt/MoS2 nanocomposite. (B) HAADF-STEM-EDS elemental mapping images (a–c) and cross-sectional compositional line profiles (d) taken from Au@Pt nanodendrites. The inset shows the corresponding HAADF-STEM image. | ||
Powder X-ray diffraction (XRD) was used to characterize the structure of MoS2, Au/MoS2 and Au@Pt/MoS2. As shown in Fig. S6,† a diffraction peak was located at about 14.40° for the MoS2 nanosheets, which matched the (001) index of 1T-MoS2.25,43 After modification, strong diffraction peaks emerged at 38.5° for the Pt@MoS2 and 40.0° for the Au@MoS2 nanocomposite, which can be assigned to the (111) planes of cubic Pt (JCPDS no. 04-0802) and Au (JCPDS no. 04-0784), respectively.16,17 All strong diffraction peaks of Pt and Au were obtained in the Au@Pt/MoS2 nanocomposite. The XRD data indicated the successful formation of Au/MoS2, Pt/MoS2 and Au@Pt/MoS2 nanocomposites. XPS was employed to investigate the structure and composition of the Au@Pt/MoS2 nanocomposite. It had been reported that a trigonal orismatic (2H) and an octahedral (1T) configuration of MoS2 could coexist during the lithium intercalation–exfoliation process.33,43–47 As shown in Fig. 6A, the binding energy peaks of the MoS2 nanosheets were fitted into three groups: (1) two larger peaks located near 232 eV and 229 eV, which correspond to Mo4+ 3d3/2 and Mo4+ 3d5/2 of 2H-MoS2, respectively; (2) the doublet peaks located around 231 eV and 228 eV could be assigned to Mo4+ 3d3/2 and Mo4+ 3d5/2 of 1T-MoS2, respectively; (3) the peaks of Mo 3d peaks with a higher oxidation state (Mo6+) located at 235 eV and 231 eV, which may originate from the partial oxidation of Mo atoms at the edges or defects of the MoS2 nanosheets during chemical exfoliation and preparation of the Au@MoS2 nanoseeds.33,48 The binding energies for S 2p3/2 and S 2p1/2 were 162.3 and 163.5 eV, respectively (Fig. 5B). Two peaks located at about 87.2 eV and 83.5 eV can be ascribed to Au 4f5/2 and Au 4f7/2, respectively (Fig. 6C).25 Peaks at 74.3 eV and 71.0 eV originated from Pt 4f5/2 and Pt 4f7/2, respectively (Fig. 6D).15Fig. 6C and D also suggested the successful synthesis of Au@Pt core–shell nanodendrites. The XPS results further confirmed the coexistence of Au, Pt and MoS2 in the Au@Pt/MoS2 nanocomposite, which agreed well with the XRD results.
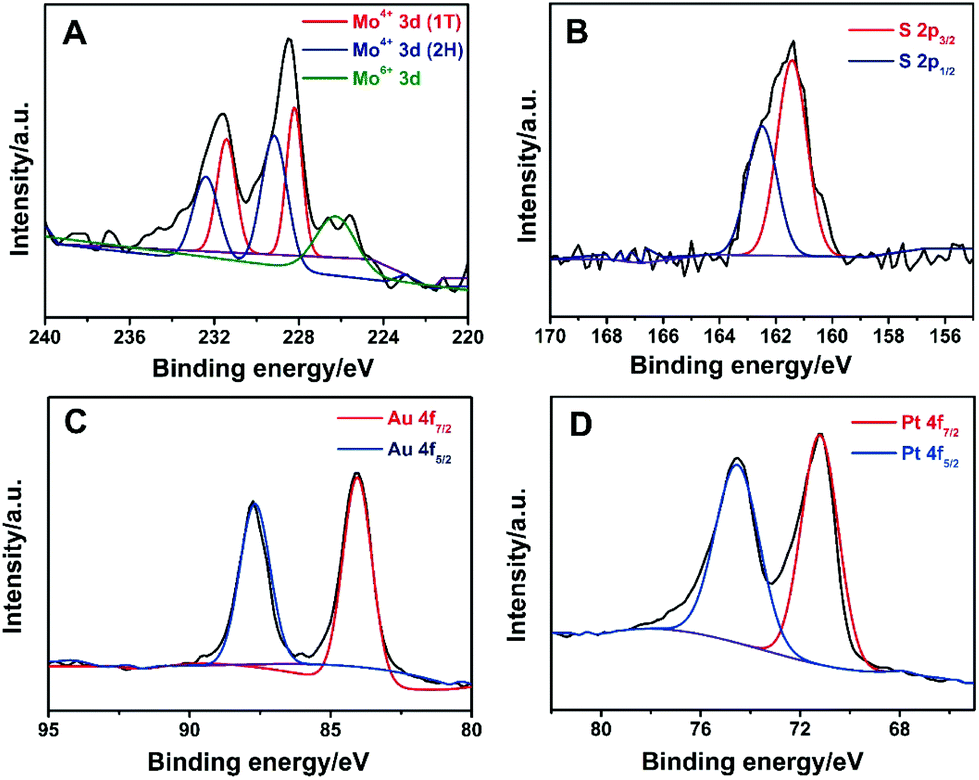 | ||
| Fig. 6 High-resolution (A) Mo 3d, (B) S 2s, (C) Au 4f and (D) Pt 3d XPS spectra of the Au@Pt/MoS2 nanocomposite. | ||
3.2. Electrochemical oxidation of methanol
To select the best catalyst, we explored the activity toward the methanol oxidation reaction of different thicknesses of the Pt shell for Pt@Au/MoS2 nanocomposites. We selected two typical nanocomposites to evaluate the catalytic performance, a Au@Pt/MoS2 nanocomposite with a thin Pt shell (Au@Pt/MoS2-1, Fig. 2B) and a Au@Pt/MoS2 nanocomposite with a thick Pt shell (Au@Pt/MoS2-2, Fig. 2D). For comparison, Pt/C (10%) and Pt/MoS2 were employed to assess the catalytic activity. The synthesis process of Pt/MoS2 is described in the ESI.† The TEM image of Pt/MoS2 showed that it possessed a similar structure to Au@Pt/MoS2 (Fig. S1C†). Fig. 7A shows the CV curves of Pt/C (10%) (black line), Pt/MoS2 (red line), Au@Pt/MoS2-1 (blue line) and Au@Pt/MoS2-2 (green line) in N2-saturated 0.5 M NaOH solution at 100 mV s−1. Clearly, the hydrogen adsorption–desorption peaks and the Pt oxidation/reduction peaks were well observed for all the catalysts, indicating that the surface composition of the catalysts was Pt. Obviously, the hydrogen adsorption–desorption peaks of Au@Pt/MoS2-2 were much larger than those of Pt/C, Pt/MoS2 and Au@Pt/MoS2-1, indicating that the Au@Pt/MoS2-2 catalyst had a larger electrochemical surface area (ECSA) and higher catalytic activity than the others. The improved catalytic activity could be ascribed to the 3D Au@Pt nanodendrites and the synergistic effects of Au@Pt and MoS2 nanosheets.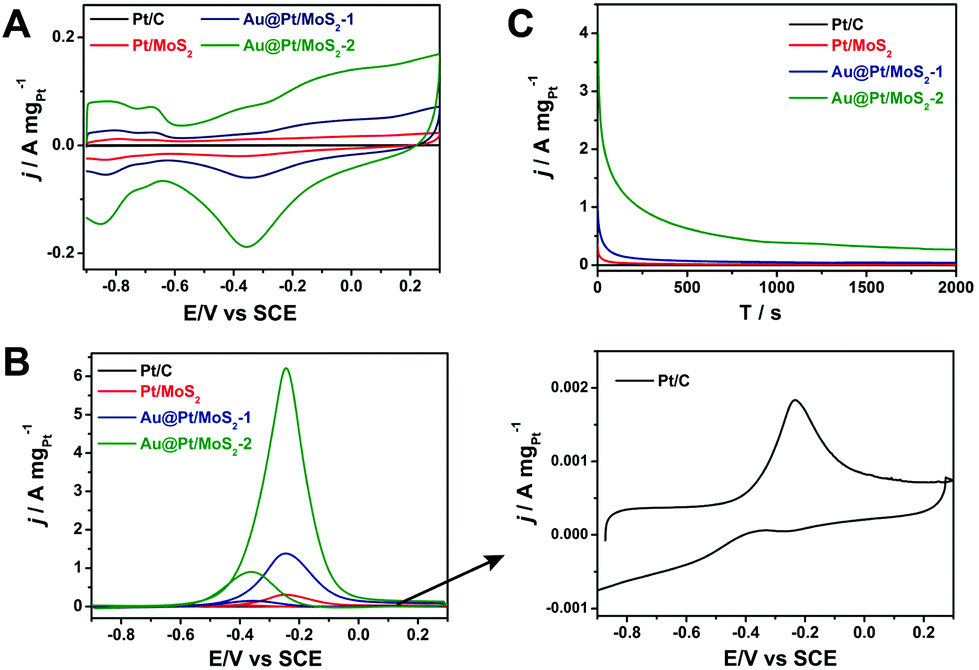 | ||
| Fig. 7 (A) Cyclic voltammograms of four catalysts in N2-saturated 0.5 M NaOH solution. (B) Cyclic voltammograms of four catalysts in 0.5 M NaOH + 1.0 M CH3OH. Scan rate: 100 mV s−1. (C) Chronoamperometric responses of four catalysts in 0.5 M NaOH + 1.0 M CH3OH. | ||
Fig. 7B exhibits the electrocatalytic activities of Pt/C (black line), Pt/MoS2 (red line), Au@Pt/MoS2-1 (blue line) and Au@Pt/MoS2-2 (green line) modified GCE in 0.5 M NaOH containing 1.0 M methanol. Clearly, the four catalysts displayed similar electrochemical performances. An oxidation peak was located at about −0.23 V in the forward scan, which was attributed to the direct oxidation of methanol molecules adsorbed on the electrode surface. And the other peak was located at about −0.38 V in the reverse scan, which could be assigned to the removal of carbonaceous species that were not completely oxidized in the forward scan.49 The Au/MoS2 nanocomposite showed no measurable MOR activity under the same experimental conditions (Fig. S7†). Au@Pt/MoS2-2 showed higher electrocatalytic activity toward methanol, the corresponding mass activity was 6.24 A mg−1, which is 3389, 20.3, and 4.5 times those of Pt/C (0.00184 A mg−1), Pt/MoS2 (0.307 A mg−1), and Au@Pt/MoS2-1 (1.39 A mg−1), respectively (the mass concentrations of Pt in the Pt/MoS2, Au@Pt/MoS2-1, and Au@Pt/MoS2-2 were determined by ICP-MS, Table S1†). The mass activity of Au@Pt/MoS2-2 was comparable to or better than several previous works.50–52
The onset peak potential of Au@Pt/MoS2-2 (−0.24 V) was more negative than that of Pt/C (−0.23 V), indicating that Au@Pt/MoS2-2 had a better tolerance towards CO poisoning. The durability of the four catalysts was also studied through the chronoamperometric measurements at −0.23 V (Fig. 7C). It can be seen that the Au@Pt/MoS2 catalyst had a much higher initial current than other Pt catalysts. After a sharp drop in the initial period of around 15 s, the currents decay at a much slower speed and then approach a flat line. Both Au@Pt/MoS2-1 and Au@Pt/MoS2-2 maintained higher currents within 2000 s of the test than the rest of the catalysts tested. More importantly, the catalyst Au@Pt/MoS2-2 showed a higher current than that of Au@Pt/MoS2-1, suggesting that a thicker Pt shell possessed better electrocatalytic activity. The stability of the catalysts was also proved by TEM investigation. Fig. S8† shows that the structures of Pt/MoS2, Au@Pt-MoS2-1 and Au@Pt-MoS2-2 had almost no change after the electrocatalytic process. These results were consistent with the CV measurements, further confirming that the Au@Pt/MoS2-2 catalyst possessed higher catalytic activity and long-term stability towards the methanol oxidation reaction.
The interfacial processes and kinetics of electrode reactions were studied by the electrochemical impedance spectroscopy (EIS) technique. Fig. S9† exhibits the Nyquist plots for methanol oxidation at different modified electrodes in 0.5 M NaOH containing 1.0 M methanol. The diameter of the charge transfer resistance decreased in the order Pt/C > Pt/MoS2 > Au@Pt/MoS2-1 > Au@Pt/MoS2-2. The lower charge transfer resistance of Au@Pt/MoS2-2 suggested that this catalyst possessed faster oxidation kinetics, which could be attributed to the prevention of the intermediate adsorption on the catalyst surface.53
4. Conclusions
In summary, dendritic Pt shell and Au core bimetallic heterostructures supported on MoS2 nanosheets could be easily synthesized by a seeded growth method. The Au@Pt/MoS2 nanocomposites exhibited higher electrocatalytic activity and long-term durability for methanol oxidation than those of the monometallic Pt catalyst. The improved catalytic performance could be attributed to the large surface area of MoS2, the unique porous nanostructure of Au@Pt, the high electrocatalytic activity of Pt nanoshells and their excellent synergistic effects. These MoS2-based nanocomposites possessed high electrocatalytic activity and excellent stability, which could be further employed as a potential nanomaterial in catalytic and fuel cell applications.Acknowledgements
This work was financially supported by the National Basic Research Program of China (2012CB933301), the National Natural Science Foundation of China (21305070, 21475064), the Natural Science Foundation of Jiangsu Province (BK20130861), the Sci-tech Support Plan of Jiangsu Province (BE2014719), the Ministry of Education of China (20133223120013), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- A. S. Arico, S. Srinivasan and V. Antonucci, Fuel Cells, 2001, 1, 133–161 CrossRef CAS.
- L. Brandao, D. M. Gattia, R. Marazzi, M. V. Antisari, S. Licoccia, A. D'Epifanio, E. Traversa and A. Mendes, Mater. Sci. Forum, 2010, 638, 1106–1111 CrossRef.
- M. S. Whittingham and T. Zawodzinski, Chem. Rev., 2004, 104, 4243–4244 CrossRef CAS PubMed.
- J. Y. Cao, M. W. Guo, J. Y. Wu, J. Xu, W. C. Wang and Z. D. Chen, J. Power Sources, 2015, 277, 155–160 CrossRef CAS.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- C. M. Sánchez-Sánchez, J. Solla-Gullón, F. J. Vidal-Iglesias, A. Aldaz, V. Montiel and E. Herrero, J. Am. Chem. Soc., 2010, 132, 5622–5624 CrossRef PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- Y. Yang, Phys. Chem. Chem. Phys., 2009, 11, 9759–9765 RSC.
- D. Zhao and B. Q. Xu, Angew. Chem., Int. Ed., 2006, 118, 5077–5081 CrossRef.
- S. Alayoglu and B. Eichhorn, J. Am. Chem. Soc., 2008, 130, 17479–17486 CrossRef CAS PubMed.
- E. Lee, J. H. Jang, M. A. Matin and Y. U. Kwon, Ultrason. Sonochem., 2014, 21, 317–323 CrossRef CAS PubMed.
- C. Li and Y. Yamauchi, Phys. Chem. Chem. Phys., 2013, 15, 3490–3496 RSC.
- J. B. Zhu, M. L. Xiao, K. Li, C. P. Liu and W. Xing, Chem. Commun., 2015, 51, 3215–3218 RSC.
- H. Ataee-Esfahani, L. Wang, Y. Nemoto and Y. Yamauchi, Chem. Mater., 2010, 22, 6310–6318 CrossRef CAS.
- S. J. Guo, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed.
- S. J. Guo, J. Li, S. J. Dong and E. K. Wang, J. Phys. Chem. C, 2010, 114, 15337–15342 CAS.
- Y. Kim, J. W. Hong, Y. W. Lee, M. Kim, D. Kim, W. S. Yun and S. W. Han, Angew. Chem., Int. Ed., 2010, 49, 10197–10201 CrossRef CAS PubMed.
- J. H. Zeng, J. Yang, J. Y. Lee and W. J. Zhou, J. Phys. Chem. B, 2006, 110, 24606–24611 CrossRef CAS PubMed.
- X. Cui, S. Wu, S. Jungwirth, Z. Chen, Z. Wang, L. Wang and Y. Li, Nanotechnology, 2013, 24, 295402 CrossRef PubMed.
- G. S. Bang, K. W. Nam, J. Y. Kim, J. Shin, J. W. Choi and S.-Y. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 7084–7089 CAS.
- R. Ganatra and Q. Zhang, ACS Nano, 2014, 8, 4074–4099 CrossRef CAS PubMed.
- H. Li, J. Wu, Z. Yin and H. Zhang, Acc. Chem. Res., 2014, 47, 1067–1075 CrossRef CAS PubMed.
- H. F. Sun, J. Chao, X. L. Zuo, S. Su, X. F. Liu, L. H. Yuwen, C. H. Fan and L. H. Wang, RSC Adv., 2014, 4, 27625–27629 RSC.
- H. Zhang, ACS Nano, 2015, 9, 9451–9469 CrossRef CAS PubMed.
- C. Tan and H. Zhang, Chem. Soc. Rev., 2015, 44, 2713–2731 RSC.
- X. Huang, C. Tan, Z. Yin and H. Zhang, Adv. Mater., 2014, 26, 2185–2204 CrossRef CAS PubMed.
- X. Huang, Z. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934–1946 RSC.
- X. Cao, Y. Shi, W. Shi, X. Rui, Q. Yan, J. Kong and H. Zhang, Small, 2013, 9, 3433–3438 CrossRef CAS PubMed.
- Z. Yin, B. Chen, M. Bosman, X. Cao, J. Chen, B. Zheng and H. Zhang, Small, 2014, 10, 3537–3543 CrossRef CAS PubMed.
- J. Chen, X. J. Wu, L. Yin, B. Li, X. Hong, Z. Fan, B. Chen, X. Can and H. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1210–1214 CrossRef CAS PubMed.
- S. Su, M. Zou, H. Zhao, C. Yuan, Y. Xu, C. Zhang, L. H. Wang, C. H. Fan and L. H. Wang, Nanoscale, 2015, 7, 19129–19135 RSC.
- X. Huang, Z. Zeng, S. Bao, M. Wang, X. Qi, Z. Fan and H. Zhang, Nat. Commun., 2013, 4, 1444 CrossRef PubMed.
- L. Yuwen, F. Xu, B. Xue, Z. Luo, Q. Zhang, B. Bao, S. Su, L. Weng, W. Huang and L. Wang, Nanoscale, 2014, 6, 5762–5769 RSC.
- S. Su, H. F. Sun, F. Xu, L. H. Yuwen and L. H. Wang, Electroanalysis, 2013, 25, 2523–2529 CrossRef CAS.
- S. Su, C. Zhang, L. Yuwen, J. Chao, X. Zuo, X. Liu, C. Song, C. Fan and L. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 18735–18741 CAS.
- S. S. Chou, M. De, J. Kim, S. Byun, C. Dykstra, J. Yu, J. Huang and V. P. Dravid, J. Am. Chem. Soc., 2013, 135, 4584–4587 CrossRef CAS PubMed.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- T. Teranishi, M. Hosoe, T. Tanaka and M. Miyake, J. Phys. Chem. B, 1999, 103, 3818–3827 CrossRef CAS.
- S. Y. Zhao, S. H. Chen, S. Y. Wang, D. G. Li and H. Y. Ma, Langmuir, 2002, 18, 3315–3318 CrossRef CAS.
- G. Zhang, C. Huang, R. Qin, Z. Shao, D. An, W. Zhang and Y. X. Wang, J. Mater. Chem. A, 2015, 3, 5204–5211 CAS.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2010, 132(39), 13636–13638 CrossRef CAS PubMed.
- W. Zhou, Z. Yin, Y. Du, X. Huang, Z. Zeng, Z. Fan, H. Liu, J. Wang and H. Zhang, Small, 2013, 9, 140–147 CrossRef CAS PubMed.
- S. Ji, Z. Yang, C. Zhang, Y.-E. Miao, W. W. Tjiu, J. Pan and T. Liu, Microchim. Acta, 2013, 180, 1127–1134 CrossRef CAS.
- J. Kim, S. Byun, A. J. Smith, J. Yu and J. Huang, J. Phys. Chem. Lett., 2013, 4, 1227–1232 CrossRef CAS PubMed.
- Y. Liu, Y.-X. Yu and W.-D. Zhang, J. Phys. Chem. C, 2013, 117, 12949–12957 CAS.
- Y. Shi, J. K. Huang, L. Jin, Y. T. Hsu, S. F. Yu, L. J. Li and H. Y. Yang, Sci. Rep., 2013, 3, 1839 Search PubMed.
- J. Kim, S. Byun, A. J. Smith, J. Yu and J. Huang, J. Phys. Chem. Lett., 2013, 4, 1227–1232 CrossRef CAS PubMed.
- Y. Chen, J. Yang, Y. Yang, Z. Peng, J. Li, T. Mei, J. Wang, M. Hao, Y. Chen, W. Xiong, L. Zhang and X. Wang, Chem. Commun., 2015, 10, 1039 Search PubMed.
- A. Akinpelu, B. Merzougui, S. Bukola, A.-M. Azad, R. A. Basheer, G. M. Swain, Q. Chang and M. Shao, Electrochim. Acta, 2015, 161, 305–311 CrossRef CAS.
- C.-S. Chen, F.-M. Pan and H.-J. Yu, Appl. Catal., B, 2011, 104, 382–389 CrossRef CAS.
- A. Maksic, Z. Rakocevic, M. Smiljanic, M. Nenadovic and S. Strbac, J. Power Sources, 2015, 273, 724–734 CrossRef CAS.
- A. Dutta and J. Ouyang, ACS Catal., 2015, 5, 1371–1380 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr06077j |
| This journal is © The Royal Society of Chemistry 2016 |