
Ru(II)-polypyridyl surface functionalised gold nanoparticles as DNA targeting supramolecular structures and luminescent cellular imaging agents†
Miguel
Martínez-Calvo‡
a,
Kim N.
Orange‡
b,
Robert B. P.
Elmes
ac,
Bjørn
la Cour Poulsen
a,
D. Clive
Williams
*b and
Thorfinnur
Gunnlaugsson
*a
aSchool of Chemistry and Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie; martinm9@tcd.ie; Tel: +353 1 896 1947, +353 1 896 3459
bSchool of Biochemistry and Immunology and Trinity Biomedical Sciences Institute, Trinity College, Dublin 2, Ireland. E-mail: clive.williams@tcd.ie; Tel: +353 1 8962596
cDepartment of Chemistry, Maynooth University, National University of Ireland, Maynooth, Ireland
First published on 26th November 2015
Abstract
The development of Ru(II) functionalized gold nanoparticles 1–3·AuNP is described. These systems were found to be mono-disperse with a hydrodynamic radius of ca. 15 nm in water but gave rise to the formation of higher order structures in buffered solution. The interaction of 1–3·AuNP with DNA was also studied by spectroscopic and microscopic methods and suggested the formation of large self-assembly structures in solution. The uptake of 1–3·AuNP by cancer cells was studied using both confocal fluorescence as well as transmission electron microscopy (TEM), with the aim of investigating their potential as tools for cellular biology. These systems displaying a non-toxic profile with favourable photophysical properties may have application across various biological fields including diagnostics and therapeutics.
Introduction
Nanoparticles (NPs) are defined as solid materials with a nanometric particle size. Metallic nanoparticles, due to their tuneable size, shape and biocompatibility, have been shown to be effective materials for use in biomedical applications, as a vast variety of chemical species can be anchored to their surface.1 Nanoparticles offer great potential for diagnostic and therapeutic applications over that of single molecules,2 where intrinsic features such as optical, magnetic, thermal or mechanical properties permit vast potential applications. To date NPs have been capitalised on for use in applications such as in molecular recognition, cellular signalling, drug delivery and sensing/bio-sensing to name just a few key areas.3 Most recently, the use of NPs to deliver cargo into cells has shown particular promise and has been extensively demonstrated by the work of Mirkin et al.1a,d,2l,m,3a,d,j,n and Rotello et al.1a,b,2a,f,3b,g,q The small size of NPs and their large surface area allows anchoring of a large variety of ligands. Gold nanoparticles (AuNPs), in particular are highly attractive inorganic NPs that offer robust frameworks in which several components can be incorporated to give multifunctional capabilities.2,3 AuNPs are stable structures that can be easily synthesized and functionalized under ambient conditions.1 AuNPs are thought to be relatively non-cytotoxic as the bulk form of the metal is chemically inert, and any toxicity is usually associated with their surface modified groups/ligands rather than gold itself.2,3 Mirkin et al., have pioneered the use of AuNPs as drug carriers, photo-responsive therapeutics, imaging agents and gene regulating agents.3a Hence, these are clearly highly attractive platforms with great potential for use in medical applications.Ruthenium (Ru(II)) based polypyridyl complexes have also been intensively studied to date due to their rich photophysical properties, and they have found application in various fields of research. In particular, Ru(II) based polypyridyl complexes have been developed as sensitive and structure specific luminescent DNA probes and photoreagents.4 Here, the binding to DNA can occur either through intercalation5 or by groove binding6 causing modulation in the photophysical properties of the Ru-complexes; being dictated by the structural and physical nature of the polypyridyl ligands employed. Since targeting DNA with such complexes can prevent DNA replication within cancer cells and induce programmed cell death,7–9 it is of interest to develop new means of delivery into cells. Recently several examples of Ru(II) polypyridyl complexes have exhibited live cellular uptake with many being localized in both nuclei, mitochondria, lysosomes and endoplasmic reticulum.10 However, to date, examples of Ru(II) functionalised NPs, which provide possibility of delivering high concentrations of Ru(II) complexes into live cells, remain few.11 Pikramenou and co workers have recently studied the luminescent properties of several Ru(II) functionalised NPs,11j,k coated with a fluorinated surfactant, and shown that the emission properties (including lifetimes and quantum yields) of such conjugated systems is (AuNP) distance depended and can be enhanced over the individual complexes, which is highly desirable for application in biology. We have recently developed cell targeted probes capable of penetrating cells and inducing apoptosis using both organic, as well as metal ion complexes.12,13 In addition to these studies our group was the first to demonstrate the use of luminescent Ru(II)-polypyridyl conjugated to AuNP (Ru(II)-AuNPs). While we demonstrated that the photophysical properties of the Ru(II) complexes were preserved, the small size of AuNPs used, (average size of 4–5 nm), limited their applications, where it proved difficult to further investigate and characterize their uptake mechanism in cancer cells.11a As the size of the AuNP is known to be able to strongly influence the chemistry and the physical properties of the surface functionalised ligands themselves (e.g. due to stacking, self-assembly interactions, etc.), as well as the their biological properties (interactions with membrane bound proteins, etc.), we set out to investigate the use of larger AuNPs for cellular applications, in the hope of profiling the mechanism of cellular uptake in more detail and their subsequent distribution in the cytosol. We foresaw that our investigations would contribute to the general understanding of the behaviour of such nano-structures in biological environments and lead to their possible application in biological media; particularly in imaging and for the delivery of novel targeting therapeutics. To enable a direct comparison with our previous work, we have synthesized the polypyridyl complexes 1, 2 and 3, which have been tethered, via a phen spacer, to AuNPs with a larger diameter of ca. 30–40 nm (1·AuNP, 2·AuNP and 3·AuNP, Fig. 1). Herein, we present our results, where we have undertaken a detailed investigation of the physical and photophysical properties of these new nanostructures, their ability to interact with DNA and their biological activities in a cervical cancer cell line, using confocal fluorescence and transmission electron microscopy’, which demonstrates that while the increasing size of the AuNP allows for cellular uptakes studies to be profiled more accurately, the enhanced population of the complexes at the AuNP surface greatly affects their application due to increased inter- and intramolecular interactions.
 | ||
| Fig. 1 The Ru(II)polypyridyl complexes-AuNP hybrid materials 1–3·AuNP on 15 nm gold nanoparticles developed for this study. These were formed by surface modification of freshly made AuNP using the thiol-terminal conjugated complexes 1–3 (see ESI† for detail). | ||
Results and discussion
Synthesis and characterisation of 1–3·AuNP
The synthesis of 1–3 was achieved according to Scheme S1 (see ESI†). This involved the formation of the Ru(II) precursors cis-[Ru(bpy)2Cl2], cis-[Ru(TAP)2Cl2], [RuCl2(η4-COD)] in accordance with established literature procedures, which were then reacted with phen derivative 4 (see ESI, Scheme 1†) 11-mercapto-N-(1,10-phenanthrolin-5-yl)-undecanamide in a single step using a peptide coupling reaction.11a All the complexes were analysed using conventional methods (see ESI†), the absorption spectra of each in water and buffered pH 7.4 solution are included in the ESI.† The AuNP employed, in the formation of 1–3·AuNP, were formed using the established sodium citrate method, which resulted in the formation of ca. 15–20 nm AuNP according to TEM analysis.14The formation of 1–3·AuNP was achieved by carrying out surface modification of the AuNPs in water (see Experimental). In the case of 1–3·AuNP, this involved the addition of the appropriate Ru complex (in water) to an aqueous solution of AuNPs. Adjustment of the pH of the reaction to pH 3, using HCl, followed by stirring at room temperature for 12 hours provided the desired Ru(II) functionalized AuNPs 1–3·AuNP. Purification was achieved by addition of a concentrated solution of ammonium hexafluorophosphate to an aqueous solution of 1–3·AuNP, followed by centrifugation, where the isolated solid was washed with MeOH. A second anion exchange process was achieved by firstly dissolving the isolated solid in CH3CN, before addition of a concentrated solution of tetrabutylammonium chloride. The resulting suspension was then centrifuged, and the isolated dark solid was washed with acetone. The full upload over the surface of the AuNP will compromise ca. 3000–4000 of the Ru(II) complexes 1–3. The resulting surface modified AuNPs were analysed using UV-Vis absorption spectroscopy, Dynamic Light Scattering (DLS) and Transmission Electron Microscopy (TEM) imaging in both water and in buffered solution at pH 7.4. Recording the Zeta potential, gave rise to a positive potential of 19 mV in the case of 3·AuNP, indicating the cationic nature of the AuNP after surface modification with 3. In the cases of 1–2·AuNP the value obtained for the Zeta potential was closer to be 0 mV, indicating that these conjugates were not colloidally stable. Nevertheless, we decided to study the photophysical properties and the uptake mechanism of these conjugates for comparative reasons with our previous 4–5 nm Ru-AuNP conjugates.13a
The UV-Visible absorption spectra of bare AuNPs and 1–3·AuNP are shown in Fig. 2, measured in water (Fig. 2a), and in 10 mM phosphate buffer at pH 7.4, Fig. 2b. In both solutions, the bare AuNP SPR band was centred at 524 nm, demonstrating that it was not dependent on the media. In contrast, the surface modification of AuNP by complexes 1–3 gave rise to a red shift in the absorption spectra of the SPR band (shown as normalized spectra in Fig. 2) for all three complexes, in both media. In both solvent media the MLCT band of the Ru(II) polypyridyl complexes was also visible for 3·AuNP, while the MLCT bands of 1·AuNP and 2·AuNP were masked by the SPR band of the AuNP itself. However, the higher energy Ru(II) polypyridyl band is clearly visible (see ESI,† and later discussion). In the case of 3·AuNP the MLCT transitions can be seen as broad bands, centred at 417 nm with a shoulder at 455 nm, structurally matching that of complex 3 itself (cf. the UV-Vis spectra of 1–3 in the ESI†). As can be seen from Fig. 2a, when recorded in water, the SPR band was centred for 1–3·AuNP at 535 nm, 547 nm and 525 nm, respectively. These solutions were found to be stable over a period of several months, as only very minor changes were observed in the UV-Vis absorption spectra.
 | ||
| Fig. 2 The UV-Visible absorption spectra of 1–3·AuNP in: (a) water and (b) in pH 7.4 phosphate buffered solutions. | ||
The stability in solution of 3·AuNP was further monitored by recording the UV-Vis absorption spectra over 16 hours. Whilst over the first 6 hours no changes were observed, a small reduction (5%) in absorbance was seen between 6–10 hours for the SPR band. In contrast to that seen in water, in pH 7.4 phosphate buffered solution, Fig. 2b, the absorption spectra of 1–3·AuNP were slightly red shifted. Here, both 1·AuNP and 2·AuNP appeared identical, being centred at 560 nm, whilst the SPR band for 3·AuNP was centred at 535 nm. Such a shift of ca. 15–20 nm is an indication of an agglomeration effect in the case of 1·AuNP and 2·AuNP. In the case of 3·AuNP, stability measurements were also carried out, by observing the changes in the absorption spectra over 16 hours. Unlike that seen in water, no changes (within experimental error) were seen in the SPR band of 3·AuNP over this time period, indicating that 3·AuNP was less susceptible to aggregation in pH 7.4 buffered solutions. We also carried out stability measurements in the presence of 160 mM NaCl, which again did not give rise to changes in the absorption spectra of 3·AuNP over period of 16 hours.
The luminescent properties of these surface modified AuNPs were also investigated. The excitation of 1–3·AuNPs (at the λmax for all the MLCT bands as determined for 1–3) gave rise to MLCT centered emission at long wavelengths (see discussion below) typical of that seen for such complexes. Moreover, the excitation spectra of these AuNPs also gave rise to spectra that were structurally similar to those observed for the absorption spectra of the complexes alone (see ESI†). However, as previously observed, 1–3·AuNP displayed reduced emission in comparison to their parent complexes 1–3, despite the fact that the complexes are separated from the gold by a covalent spacer.15
To investigate the structural nature of these surface modified AuNPs further, both TEM and DLS analysis were carried out. The DLS analysis (see ESI†) of these AuNPs in deionized water revealed that the hydrodynamic radii of 1·AuNP and 3·AuNP were in the expected region of ca. 15 nm, while 2·AuNP gave rise to larger structures in solution, being ca. twice of that obtained for 1·AuNP and 3·AuNP. However when these measurements were repeated in 10 mM phosphate buffer at pH 7.4 the hydrodynamic radius increased for 1–3·AuNP compared to that seen in deionized water (see ESI†) indicating some degree of agglomeration. Of these, the hydrodynamic radius of 3·AuNP increased the least, with an ionic radius of ca. 15 nm, while both 1·AuNP and 2·AuNP showed the formation of much larger structures with hydrodynamic radii of ca. 100 nm in solution. These results indicate that 1–3·AuNP are susceptible to some degree of aggregation in buffered solution. TEM images obtained for 1–3·AuNP in water are shown in Fig. 3, and clearly show monodisperse spherical nanoparticles. The measured average sizes of the core of the AuNPs were determined as 15.3 ± 3.0, 15.4 ± 3.3 and 15.8 ± 3.8 nm for 1·AuNP, 2·AuNP and 3·AuNP respectively.
 | ||
| Fig. 3 TEM images obtained using functionalized nanoparticles: (a) 1·AuNP, (b) 2·AuNP, (c) 3·AuNP. | ||
In contrast, the TEM imaging of 1–3·AuNP in 10 mM phosphate buffer solutions are shown in Fig. 4 and confirm the formation of agglomerates of 1·AuNP and 2·AuNP, where aggregation of 2·AuNP was particularly apparent. In contrast 3·AuNP did not give rise to any obvious aggregate formation, supporting the results observed through DLS analysis. Such behavior may be due to the known ability of the TAP ligand and related Ru(II) TAP based complexes to participate in hydrogen bonding with protic solvents thereby reducing their tendency to aggregate. In fact, the hydrogen bonding capability of the TAP ligands has been shown to play an important role in their interaction with the DNA and this may be an important factor when considering their potential in biological applications.16 To further investigate these effects we proceeded to study the binding behaviour of all three systems with double stranded DNA in solution.
 | ||
| Fig. 4 TEM images obtained using functionalized nanoparticles: (a) 1·AuNP, (b) 2·AuNP, (c) 3·AuNP in 10 mM phosphate buffer. | ||
Photophysical analysis of 1·AuNP–3·AuNP with st-DNA
Having carried out the above photophysical evaluations of 1–3·AuNP in water and buffered solutions, we conducted photophysical studies to establish the interaction of these compounds with double stranded DNA. This was achieved using UV-vis absorption, fluorescence emission and circular dichroism spectroscopy, in 10 mM phosphate buffered solution at pH 7.4 in the presence of salmon testes (st) DNA. The changes upon binding of complexes 1–3 to DNA has previously been investigated in our laboratory and showed that all three complexes to have high affinity for st-DNA, with binding constants of 5.5–6.5 × 105 M−1 in this phosphate buffer at pH 7.4.11a All measurements were repeated in triplicate using various batches of 1–3·AuNP to ensure reproducibility.The changes observed in the absorbance spectrum of 3·AuNP upon titration of these AuNPs with st-DNA, are shown in Fig. 5a. Each absorption band was clearly affected upon addition of aliquots of DNA; 282 nm (due to the transitions of the ancillary ligands, π–π*; ε = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 M−1 cm−1), MLCT band centred at 418 nm (ε = 10700 M−1 cm−1) a shoulder at 462 nm and in the SPR band at 535 nm. Abrupt changes were observed in the higher energy transitions, as well as in the MLCT band, which underwent a 29% hypochromic displacement, with a similar (26%) effect being observed for the SPR band centred at 535 nm within the addition of 0 → 1 P/D (Phosphate/Dye ratio); with further changes occurring up to the addition of 2.5 P/D, as demonstrated in Fig. 5b. These are similar changes to those observed for the complex alone upon titration with st-DNA;11a which are consistent with strong association of 3·AuNP with DNA. The intrinsic binding constant Kb, was also determined using the model of Bard et al. using these changes.17 A representative plot of (εa − εf)/(εb − εf) vs. [base pairs] and the corresponding best fit of the data to the Bard equation (R = 0.98) is shown if Fig. 5c. From these changes, a binding constant Kb of 2.4 × 106 M−1 was determined. This is a comparable binding affinity to that seen for other Ru(II) complexes, under such experimental conditions.18 The value obtained for the binding site for these titrations is unity and an indication that 3·AuNP interacts with the DNA by groove binding, or electrostatic binding, since this value is much lower than the expected value of two commonly seen for interaction by intercalation.17 This is not surprising giving that the conjugation of these complexes to the AuNPs will have an effect on the degree of freedom for the binding of these to the biomolecule.
900 M−1 cm−1), MLCT band centred at 418 nm (ε = 10700 M−1 cm−1) a shoulder at 462 nm and in the SPR band at 535 nm. Abrupt changes were observed in the higher energy transitions, as well as in the MLCT band, which underwent a 29% hypochromic displacement, with a similar (26%) effect being observed for the SPR band centred at 535 nm within the addition of 0 → 1 P/D (Phosphate/Dye ratio); with further changes occurring up to the addition of 2.5 P/D, as demonstrated in Fig. 5b. These are similar changes to those observed for the complex alone upon titration with st-DNA;11a which are consistent with strong association of 3·AuNP with DNA. The intrinsic binding constant Kb, was also determined using the model of Bard et al. using these changes.17 A representative plot of (εa − εf)/(εb − εf) vs. [base pairs] and the corresponding best fit of the data to the Bard equation (R = 0.98) is shown if Fig. 5c. From these changes, a binding constant Kb of 2.4 × 106 M−1 was determined. This is a comparable binding affinity to that seen for other Ru(II) complexes, under such experimental conditions.18 The value obtained for the binding site for these titrations is unity and an indication that 3·AuNP interacts with the DNA by groove binding, or electrostatic binding, since this value is much lower than the expected value of two commonly seen for interaction by intercalation.17 This is not surprising giving that the conjugation of these complexes to the AuNPs will have an effect on the degree of freedom for the binding of these to the biomolecule.
 | ||
| Fig. 5 (A) Representation of the UV-Vis titration of 3·AuNP with st-DNA (inset, zoom of the region from 350 to 800 nm); (B) binding isotherms of the MLCT band at 440 nm and the SPR band 535 nm and (C) Bard fitting plot of the MLCT band cantered at 440 nm. | ||
The high affinity of 3·AuNP and the strong interaction observed in the absorption spectra was further confirmed by observing the changes in the fluorescence emission spectra upon excitation at 440 nm. The overall changes in MLCT emission (centred at 637 nm) are shown in Fig. 6, demonstrating that the MLCT emission was quenched by ca. 70%, after the addition of 0.5 P/D of st DNA (as shown as inset in Fig. 6); being blue-shifted by ca. 15 nm. Once more, spectroscopic changes of this nature are in agreement with results obtained previously by our group11a and by others for similar Ru(II)-polypyridyl complexes containing two TAP ligands which have been shown to photo-oxidise guanine containing nucleotides through a proton coupled photoinduced electron transfer (PCET) process.19
 | ||
| Fig. 6 Emission spectra of 3·AuNP during its titration with st-DNA (inset, the binding isotherm at 646 nm). | ||
For comparison with our previously reported Ru-AuNPs, we also carried out the titration of 1–2·AuNP with st-DNA. Prior to the titration, the absorbance spectrum of 1·AuNP (cf. ESI,† and discussion above) possess two bands, one centred at 262 nm (ε = 49500 M−1 cm−1) corresponding to the π–π* transitions of the ancillary ligands of the Ru(II) which were monitored upon titration with DNA as the MLCT band was masked by the SPR band of the AuNP. These transitions were blue shifted by ca. 10 nm compared to the free complex (possibly due to the π–π stacking interactions between the aromatic rings of neighbouring molecules of 1).20 Upon increasing addition of st-DNA the π–π* transition initially underwent a hypochromic shift up to 1–1.5 P/D followed by a hyperchromic shift thereafter. A slight red hypochromic shift was also observed in the SPR band upon addition of DNA up to 2 P/D (Fig. S9A†). A very similar effect was observed in the absorption spectrum of 2·AuNP upon carrying out such DNA titrations (monitoring both the 266 nm ε = 72700 M−1 cm−1 and the MLCT transitions, see ESI†). Excitation of 1–2·AuNP at 420 nm, corresponding to the MLCT band, gave rise to an MLCT emission centred at 600 nm, which was structurally similar to that of the free complex (excitation spectra confirmed this as well). However, upon addition of st-DNA, no significant changes were observed in the MLCT centred emission spectra. These results confirmed that the agglomeration of 1–2·AuNP showed by the TEM imaging is hindering these conjugates to interact with the st-DNA. In our previous study with 4–5 nm Ru-AuNP conjugates all of them were able to interact with the stDNA whereas in our new conjugates it was possible to study in detail the 3·AuNP derivative. This could be due to the different stabilization of the AuNP. In our previous work the surfactant TOAB was employed to prevent aggregation of the AuNP. However, in the present work sodium citrate was employed, making that the coating of the gold surface implicated the removal of the citrate and the consequent aggregation of the Ru-AuNP with the exception of 3·AuNP where the availability of the TAP ligands to form H-Bonds prevents their aggregation and allows their interaction with the stDNA in a similar manner than the previous 4–5 Ru-AuNP.
To further study the interaction of these modified AuNP conjugates with DNA, CD-titrations were also carried out (see ESI†), by adding various concentrations of 1–3·AuNP to a solution of st-DNA and monitoring the changes in the DNA CD spectra between 200–300 nm. Significant induced changes were observed for the CD-spectra in the presence of 1–3·AuNP. The DNA transitions,21 seen as a positive CD-band at 280 nm and a negative CD-band at 250 nm were affected, but no long wavelength transitions were observed. For 1–3·AuNP, the long wavelength DNA-transition was reduced in intensity, while lesser changes were seen in the higher energy CD-band. Of these, the order of changes were largest in the case of 2·AuNP and 3·AuNP, where the 280 nm band was almost fully reduced to zero, in the case of 3·AuNP, or with a slight cross over to a negative signal in the case of 2·AuNP. While these titrations did not induce changes in the MLCT band the loss of the right handed helical structure in these CD measurements serve to confirm the above results where it is clear that 1–3·AuNPs are causing some changes to the double helical structure.
Thermal denaturation and cleavage ability of 1–3·AuNP with st-DNA and plasmid DNA, respectively
Having investigated the interactions of 1–3·AuNP with double stranded DNA using various titration experiments, thermal denaturation studies were also carried out on st-DNA in the presence of 1–3·AuNP; in the absence of these nano-structures, a Tm value of 69.8 °C was determined for the st-DNA under the experimental conditions used. Complexes 1–3 were all shown to stabilize double stranded DNA prior to their conjugation to the AuNPs (see ESI†). The melting profiles of st-DNA upon interaction with 1–3·AuNP (see ESI†) were measured at P/D = 10, and all showed only minor changes in the Tm values; these being most pronounced with 2·AuNP and 3·AuNP, both of which showed a slight increase in the Tm (70.2 °C). These results are similar to that seen for smaller AuNPs previously developed in our laboratory11a and indicate that the conjugation of Ru(II) complexes to the AuNP surface seems to alter their ability to stabilize double stranded DNA.The DNA photocleavage efficiencies of 1–3·AuNP were also compared to the known DNA photocleavage agent [Ru(bpy)3]2+ by treating pBR322 plasmid DNA (1 mg mL−1) (see Figure and Table S1 in ESI†) with each of the Ru(II) AuNPs.22 As above, the complexes 1–3, all gave rise to nicking of plasmid DNA, determined using horizontal agarose gel electrophoresis. However, 3·AuNP showed lower cleavage ability compared to the free complex 3, with a 2.1%, 4.6% and 5.4% conversion to open form DNA at P/D ratios of 20, 10 and 5, respectively; and this small capability was removed in the presence of NaN3. It is clear that whilst the above spectroscopic titration results, clearly demonstrate high affinity of 3·AuNP for DNA, the thermal denaturation measurements do not seem to lead to increased stability in double stranded DNA. Moreover, it is clear that 3·AuNP does cleave DNA to some small extent in the presence of light activation, as had been seen for 3. These results clearly demonstrate that surface modification of AuNP with 3, has a major effect on the ability of the Ru(II) complex to either stabilize or cleave DNA; commonly seen properties for Ru(II)-polypyridine complexes. Hence, the results above suggest that these systems would not function as therapeutic candidates, but their potential as luminescent biological imaging agents or as carriers for other drug substrates (or other type of cargo, such as in gene-delivery, cf. discussion in introduction section) is feasible. Therefore the potential as luminescent biological imaging agents was investigated further and will be discussed below. However, the cause for the lack of these nano-structures to carry out functions (i.e. stabilise or cleave DNA) seen for the complexes 1–3 alone, seem to stem from their functionalisation to AuNP. Above TEM and DLS results strongly suggested media dependent agglomeration of 1·AuNP and 2·AuNP with the effect being less pronounced for 3·AuNP. In order to investigate these results further, we explored the binding of these nanostructures to st-DNA using both DLS and TEM analysis.
DLS and TEM studies of 1–3·AuNP with DNA
The DLS studies of 1–3·AuNP with st-DNA were carried out in 10 mM phosphate buffered solution at different P/D ratios. These studies (see Fig. 7 and ESI†) confirmed the results of the spectroscopic studies above, where upon interaction with DNA, binding or some sort of self-assembly formation occurred which led to significant agglomeration in the case of 1·AuNP and 2·AuNP, with an increase in the hydrodynamic radius of these systems. Given that Ru(II)-polypyridyl complexes are well known to either groove bind or intercalate with DNA, it is not unreasonable to expect that such a phenomenon might be occurring. In the case of 3·AuNP, these particles have a diameter in solution of about 36 nm that increased regularly with the addition of st-DNA up to 0.23 P/D. Such behaviour clearly demonstrates a gradual self-assembly formation between these particles and DNA, where the self-assembly reached an average diameter of 500 nm. Subsequent additions of st-DNA, from 1 → 100 P/D, did not produce any further changes in the measured diameter of these DNA aggregated particles. This was further demonstrated by TEM analysis as observed in Fig. 8 showing the TEM images of 3·AuNP in the presence of DNA. In the case of conjugates 1·AuNP and 2·AuNP (Fig. S16 and S17†) there was a similar pattern for values of P/D (∼0.20), however, at higher values of P/D the particle diameter varied randomly, which could indicate a weaker interaction than in the case of 3·AuNP. To confirm these results further, TEM studies were carried out using samples of 1–3·AuNP treated with different ratios of st-DNA (P/D of 0, 0.23, 1.00 and 8.00, respectively) in 10 mM phosphate buffer solutions. The results for 3.AuNP are shown in Fig. 8 (see ESI† for 1·AuNP and 2·AuNP). Comparing Fig. 8a with that of Fig. 8b, it is clear that initially the nanoparticles are more monodisperse while at a P/D-ratio = 0.23, clusters are beginning to form due to interaction with the st-DNA. Upon further increasing the P/D ratio, the self-assembly formation between 3·AuNP and st-DNA, is even more apparent as seen in Fig. 8c and d, respectively.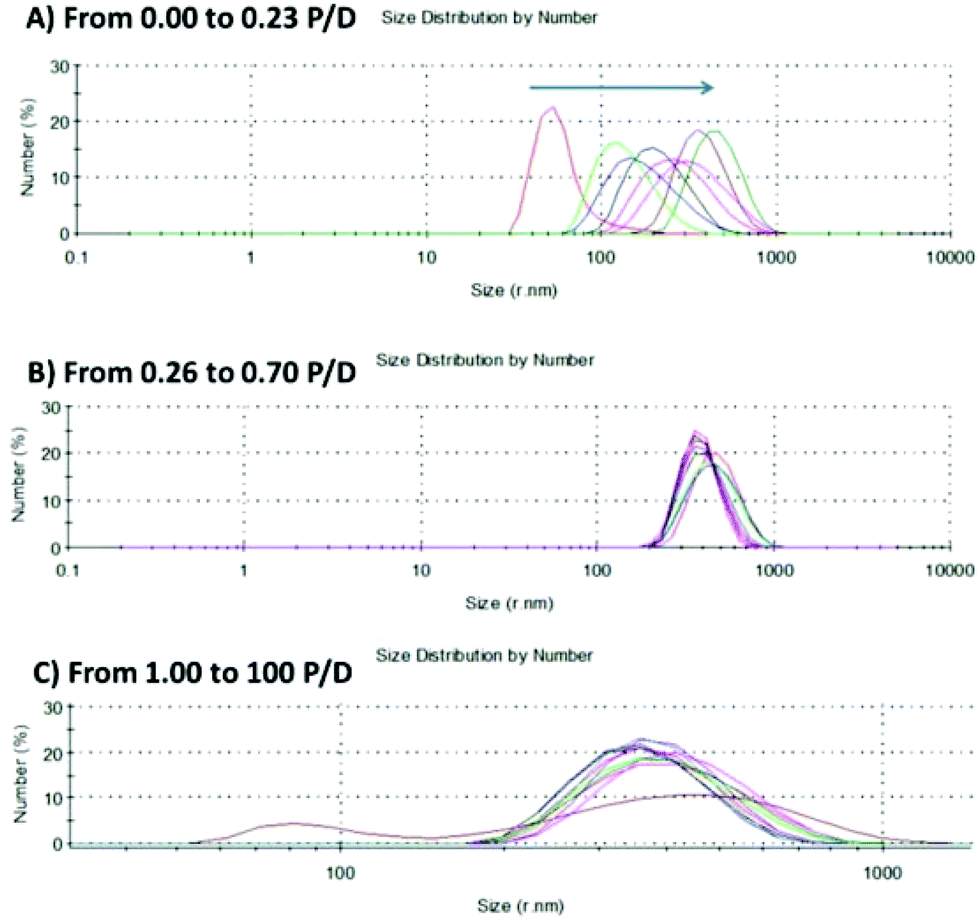 | ||
| Fig. 7 Changes in the DLS in 10 mM buffered solution of the size distribution of 3·AuNPs upon increasing concentration of st-DNA (expressed as P/D values): (a) The gradual changes occurring between 0 (shown in red) to 0.23 P/D (fare right green curve). (b) The changes between 0.23 → 0.70 P/D. (c) Zoomed in onto the larger DNA agglomerated 3·AuNPs changes occurring from 1.00 → 100 P/D. These measurements were repeated three times using freshly made solutions. | ||
 | ||
| Fig. 8 Images obtained of 3·AuNP by TEM at different P/D ratios: (a) in absence of any DNA; (b) P/D = 0.23, (c) P/D = 1.00, (d) P/D = 8.00. | ||
In the case of 1·AuNP continuous increments of the size of the AuNPs aggregates were observed with the addition of the increasing concentrations of st-DNA (cf. ESI†). While in the case of 2·AuNP fast agglomeration of these nanoparticles (cf. ESI†) occurred instantly upon addition of st-DNA. These TEM results confirm a strong self-assembly interaction with st-DNA where 3·AuNP seems to form the most ordered assemblies. In order to probe the effect that this self-assembly process may have in biological systems we next set about investigating both the cytotoxic and cellular uptake abilities of 1–3·AuNP.
Cytotoxic studies of 1–3·AuNPin vitro
To assess the cytotoxic effects of 1–3·AuNP, studies were carried out in vitro using the cervical cancer cell line HeLa, and Alamar blue cell viability assay. The cytotoxicity was investigated both with and without light irradiation; the latter being carried out in order to establish, if these systems were phototoxic, particularly in the case of 3·AuNP, which is known to form adducts with DNA upon irradiation. The HeLa cells were initially treated in the dark and either remained incubated in the dark or exposed to mild irradiation of 4 J m−2 of light. Hela cells treated with 3·AuNP and incubated in the dark did not show a reduction in cell viability when compared to the untreated control cells, indicating that these AuNPs were not cytotoxic in the dark. Upon light irradiation there was a small reduction in HeLa cell viability when compared to the untreated control cells (Fig. 9). Similar results were observed for 1·AuNP (see ESI†). However in the case of 2·AuNP, a slight reduction in cell viability was observed both in the dark and upon light activation (see ESI†). | ||
| Fig. 9 HeLa cell viability upon incubation with 3·AuNP (20μM) both with and with irradiation (4 J m−2). Cell viability was assessed using Alamar blue assay. These results represent three independent experiments carried out in triplicate. | ||
Fluorescence confocal microscopy studies in HeLa cells
Fluorescence confocal microscopy was used for real time observation of the complexes within live HeLa cells, the results of which are shown in Fig. 10. The HeLa cells were incubated with the compounds in the dark for various time points and then imaged using fluorescence confocal microscopy. The bright field images (BF), Fig. 10, show the morphologies of the treated cells remained similar to the untreated control cells (Fig. S21 and S22†). From the BF images 3·AuNP can be seen as dense aggregates within the cytoplasm of the cells, which increase or become more dense/darker overtime.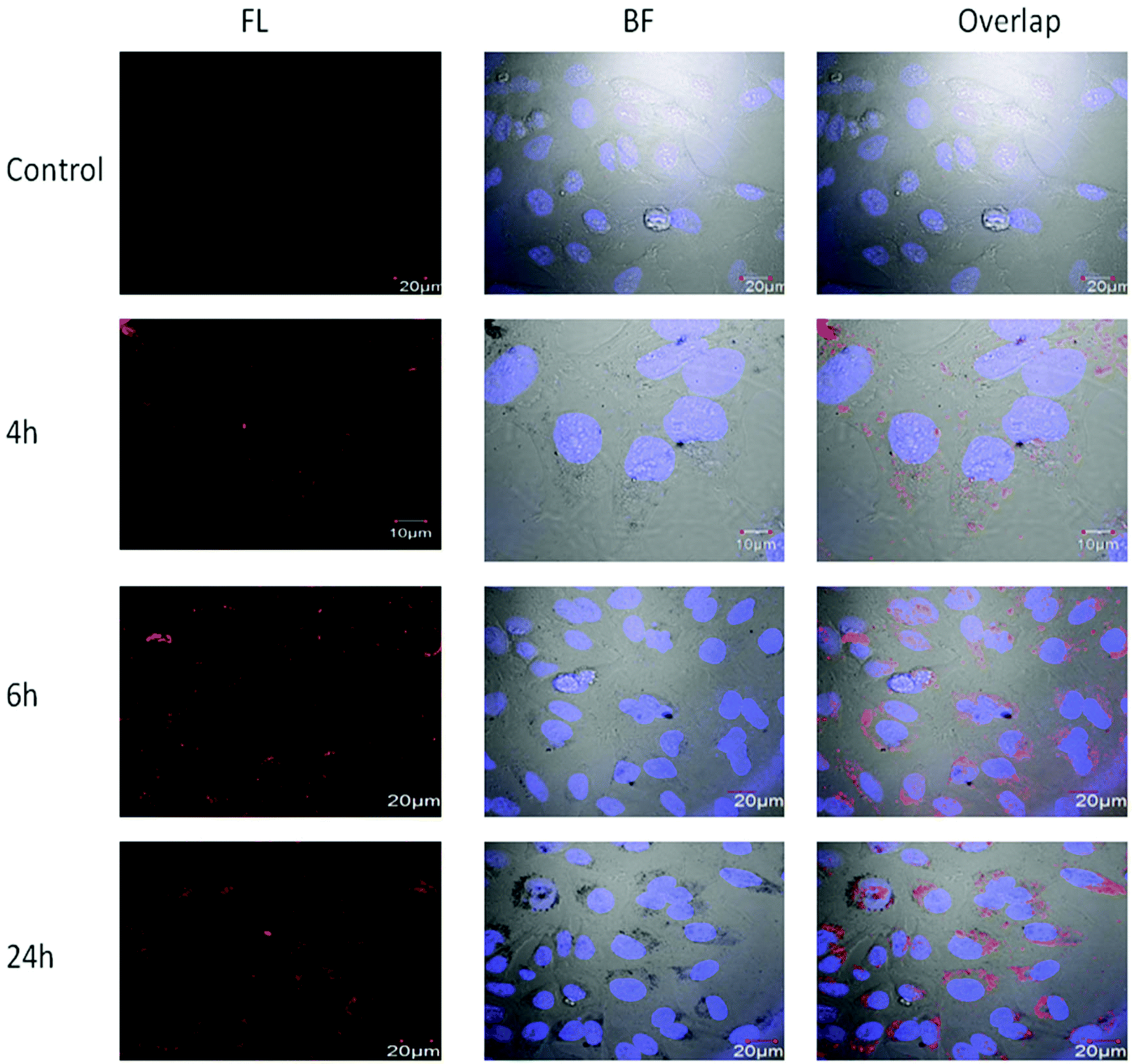 | ||
| Fig. 10 Confocal fluorescence microscopy images of HeLa cells treated with 3·AuNP for 2–24 h. Fluorescence images (FL) where nuclear stain DAPI is blue and 3·AuNP in red and bright field images (BF). | ||
The nucleus of the cells was stained with the nuclear stain DAPI that emits blue fluorescence upon excitation with the 405 nm laser. By exciting the AuNP at the MLCT absorption using a 488 nm laser and measuring the MLCT based emission between 600–700 nm they produce red fluorescence within the live Hela cells (FL channel). In Fig. 10 this red fluorescence can be seen to overlap directly with the dense patches observed in the bright field images, confirming the successful uptake of 3·AuNP into these cells. Despite 1–3·AuNP not being as luminescent as the free complexes 1–3, their emission can be clearly seen within the cells. Almost all cells in the field of view contained either the dense patches in the bright field images or red fluorescence due to 3·AuNP. Moreover, it can be seen that 3·AuNP was not distributed throughout the cell uniformly, but rather appeared as spots, which over time became brighter and moved closer to the nucleus. By varying the incubation times we concluded that the uptake of the AuNP complexes increased over time and was therefore time-dependent, Fig. 10. We also noted that over time the compound moved closer towards the nucleus but there was no evidence of the compounds entering the nucleus of the cells. These results are representative for all compounds (the same results were observed for both 1·AuNP and 2·AuNP, Fig. S19 and S20†). We next imaged the HeLa cells that had been treated with 3·AuNP for 24 h, Fig. 11. From these images it was clear that 3·AuNP was not uniformly distributed throughout the cell but accumulated at one side of the nucleus as seen in the images as black spots or red fluorescence after a period. The emission arising from 3·AuNP was notably brighter than at the earlier time points as demonstrated in Fig. 10, an observation that may suggest either increased fluorescence due to aggregation or an increase in the concentration of 3·AuNP within the cell over time. These images also confirmed that the 3·AuNP was not localizing within the nucleus, but instead the results suggest that it may be in vesicle like structures due to the spherical shape of both the ‘emission spots’ and the dense patches observed in Fig. 11b. Hence, these results clearly demonstrate that 3·AuNP is rapidly taken up by cells and that, whilst the precise localization of the NPs is not clear, accumulation close to the nuclear membrane is observed in HeLa cells by using TEM imaging.
 | ||
| Fig. 11 Fluorescence confocal microscopy images of HeLa cells treated with 20 μM 3·AuNP for 24 h. Fluorescence image (A → C) where nuclear stain DAPI (B) is blue and 3·AuNP in red and brightfield images (C). | ||
TEM time dependent uptake and localisation studies
The time dependent uptake of 3·AuNP by HeLa cells was also measured using TEM, Fig. 12. The results clearly demonstrate that the AuNPs are taken up by HeLa cells over time. Due to the AuNPs favorable size of 15 nm 3·AuNP, uptake could be easily imaged and observed as dark spots within the HeLa cells, even after short incubation times. Moreover, Fig. 12b, c, e and f, all demonstrated the presence of 3·AuNP within the HeLa cells in what appear to be endocytic vesicles. The imaging also shows that cells treated with 3·AuNP have the same morphology as that observed for the untreated HeLa cells, Fig. 12A, confirming that the AuNPs did not exhibit any toxicity towards the cells at that concentration (20 μM) or exposure time (1–24 hours). These results also confirm our previous observations that 3·AuNP is not toxic towards HeLa cells in the dark. The high magnification of the TEM images and the larger diameter of 3·AuNP allowed individual particles to be observed and measured within vesicles, confirming that the AuNPs maintain their original morphology once inside the cells (Fig. S23–S25†). These results also demonstrate the ease with which such Ru(II) polypyridyl modified AuNPs can be internalized by cells and suggest that 1–3·AuNP show significant potential for application in cellular biology as either cargo delivery agents or as diagnostic fluorescence imaging probes. Such factors are further emphasized by the potential to easily functionalize the surfaces of such AuNPs by other Transition metal complexes/ligands, etc. The TEM time dependent imaging demonstrated that 3·AuNP was taken up by the cells within 1–2 hours, and localized within what appear to be single membrane vesicles located in the cytoplasm of the cell. Moreover, after 4–6 hours, 3·AuNP was seen to be located at the nuclear edge (Fig. 11) as seen previously in the confocal results in Fig. 10. After 24 hours incubation, 3·AuNP remained in vesicles within the HeLa cell explaining why the AuNPs appeared as spherical aggregates in the brightfield images of our previous confocal images, Fig. 10 and 11. There was no evidence from the TEM images of 3·AuNP free in the cytoplasm or in the nucleus of the HeLa cells. These results would confirm that as anticipated the uptake and delivery of 3·AuNP is by an endocytotic mechanism. | ||
| Fig. 12 TEM images of HeLa cells incubated with 3·AuNP (20 μM) for (B) 1 h (C) 2 h (D) 4 h (E) 6 h (F) 24 h or left untreated (A). | ||
We also investigated the uptake and localization of 3·AuNP in the Murine macrophage cell line; RAW 264.7, using TEM, Fig. 13. After 24 h incubation 3·AuNP was located both inside the RAW cells within vesicle like structures (Fig. 13A and B) and at the outer edge of the cells plasma membrane (Fig. 13C and D). Similar to our studies in HeLa cells individual particles could be visualized within the vesicle but not free in the cytoplasm or in the nuclei of the cells. These results confirm the possibility of endocytosis as the uptake mechanism involved with these large AuNPs. The RAW cells displayed intact membranes and similar morphology to untreated RAW cells (see ESI†) further emphasizing 3·AuNPs lack of dark cytotoxicity.
 | ||
| Fig. 13 TEM images of RAW 264.7 cells incubated with 20 μM 3·AuNP for, 24 h. Cells were treated then fixed and processed for TEM imaging. | ||
Conclusion
In this present work we have synthesized three Ru(II) polypyridyl functionalized AuNPs 1–3·AuNP with an average size of ca. 15 nm, and a hydrodynamic radius of ca. 30 nm. We have demonstrated that these structures are stable in aqueous buffered solution, and that all give rise to absorption spectra consisting of the MLCT absorption contribution from the Ru(II) polypyridyl complexes and from the gold SPR bands. Moreover, upon excitation of the MLCT absorption a red MLCT centered emission is observed, despite quenching by the gold surface. We further show, using both TEM and DLS techniques that of the three systems 3·AuNP is the most highly stable remaining unchanged in solution over a period of 16 hours, even in the presence of buffer, high ionic strength or in the presence of DNA. In the case of 1·AuNP and 2·AuNP larger aggregates are formed over time, a phenomenon that becomes more apparent in the presence of DNA. We further demonstrate that these AuNPs are rapidly taken up into live human cancer cells in vitro; the mechanism of which appears to be endocytosis as demonstrated by both TEM and confocal fluorescence microscopic imaging. Upon excitation of the MLCT transition, these AuNPs display red emission within cells with no observed cytotoxicity. We show that these AuNPs are localized within the cytosol, and over time (24 hours) migrate towards the nuclear membrane. Using TEM imaging, we further show that these AuNPs are taken into cells and accumulate within vesicles. Cytotoxic analysis using cell viability assays confirmed that 1–3·AuNP are relatively non-toxic either in the dark or upon light excitation. Overall, the results from this investigation clearly demonstrate, the potential use of 15 nm Ru(II) functionalized AuNPs as valuable tools for chemical biology. The results obtained from our investigation shed further light on the behavior of 1–3·AuNP in a biological setting and overwhelmingly support their possible use in cellular applications. We are currently pursuing these and related areas and results will be published in due course.Experimental
Synthesis of the AuNPs and functionalization with the Ru(II)-polypyridyl complexes
For the preparation of the AuNP we chose the method of reducing gold with sodium citrate, which resulted in the formation of 15–20 nm AuNP.23 A solution of HAuCl4 (0.040 g) in Millipore water (300 mL) was boiled at 100 °C for 15 minutes. To this solution 10 mL of 4.04 × 10−2 M sodium citrate solution was added and the solution was left to reflux for another 15 minutes. During this time the solution changed its color from yellow to dark purple to finally turn into a wine color solution, and it was left to cool down to room temperature before using. For the functionalization of the AuNPs with the Ru(II)-polypyridyl complexes a solution was prepared by adding 1 mL of a 0.7 × 10−3 M solution of the corresponding Ru(II) polypyridyl complex to 1 mL of the AuNPs solution (3.38 × 10−4 M) in water. The mixture was left stirring overnight at pH 3 to improve the loading of the complexes onto the surface of the AuNPs adapting the method of J. Liu et al. in literature.24 The addition of a concentrated aqueous solution of NH4PF6 (0.5 mL) afforded flocculation of a dark solid which was collected by centrifugation and washed with H2O (3 × 10 mL). The solid was redissolved in MeCN (10 mL) before addition of a conc. solution of TBACl resulted in the formation of a flocculate which was collected by centrifugation and washed with MeCN (3 × 10 mL) before being dried under high vacuum.Acknowledgements
We thank Science Foundation Ireland (SFI RFP 2009 and SFI PI Awards 10/IN.1/B2999 and 13/IA/1865), the Ministerio of Economía y Competitividad of Spain (MMC Postdoctoral Fellowship), The Irish Research Council for Science, Engineering and Technology (IRCSET Postgraduate Studentship to RPBE), HEA PRTLI Cycle 4 and TCD for financial support. TEM images were obtained with the help from the CMA facility in TCD and funded by a CMA TEM bursary awarded to Miss Kim Orange in 2013. We like to thank Dr Sandra Bright for her help and support during this work.Notes and references
- (a) S. Comby, E. M. Surender, O. Kotova, L. K. Truman, J. M. Molloy and T. Gunnlaugsson, Inorg. Chem., 2014, 53, 1867 CrossRef CAS PubMed (and references therein). (b) O. R. Miranda, B. Creran and V. M. Rotello, Curr. Opin. Chem. Biol., 2010, 14, 728 CrossRef CAS PubMed; (c) D. J. Lewis and Z. Pikramenou, Coord. Chem. Rev., 2014, 273–274, 213 CrossRef CAS; (d) D. A. Giljohann and C. A. Mirkin, Nature, 2009, 462, 461 CrossRef CAS PubMed; (e) E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759 RSC; (f) S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209 RSC; (g) J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J. Y. Kim, J. Y. Park, J.-H. Hwang and N.-M. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS PubMed; (h) H. Hiramatsu and F. E. A. Osterloh, Chem. Mater., 2004, 16, 2509 CrossRef CAS; (i) C. M. Niemeyer, Angew. Chem., Int. Ed., 2001, 40, 4128 CrossRef CAS; (j) Z. Medarova, W. Pham, C. Farrer, V. Petkova and A. Moore, Nat. Med., 2007, 13, 37 CrossRef PubMed; (k) J. Massue, S. J. Quinn and T. Gunnlaugsson, J. Am. Chem. Soc., 2008, 130, 6900 CrossRef CAS PubMed; (l) A. J. Hallett, M. Broomfield, P. Christian and S. J. P. Pope, Transition Met. Chem., 2014, 39, 195 CrossRef CAS; (m) A. J. Hallett, P. Christian, J. E. Jones and S. J. A. Pope, Chem. Commun., 2009, 4278 RSC.
- (a) K. Saha, S. S. Agasti, C. Kim, X. N. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739 CrossRef CAS PubMed; (b) J. J. Li, D. Hartono, C. N. Ong, B. H. Bay and L. Y. Yung, Biomaterials, 2010, 31, 5996 CrossRef CAS PubMed; (c) L. K. Truman, S. Comby and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 9624 CrossRef CAS PubMed; (d) S. Comby and T. Gunnlaugsson, ACS Nano, 2011, 5, 7184 CrossRef CAS PubMed; (e) F. Marano, S. Hussain, F. Rodrigues-Lima, A. Baeza-Squiban and S. Boland, Arch. Toxicol., 2011, 85, 733 CrossRef CAS PubMed; (f) B. Kim, G. Han, B. J. Toley, C. K. Kim, V. M. Rotello and N. S. Forbes, Nat. Nanotechnol., 2010, 5, 465 CrossRef CAS PubMed; (g) J. Drbohlavova, R. Hrdy, V. Adam, R. Kizek, O. Schneeweiss and J. Hubalek, Sensors, 2009, 9, 2352 CrossRef CAS PubMed; (h) G. K. Kouassi and J. Irudayaraj, Anal. Chem., 2006, 78, 3234 CrossRef CAS PubMed; (i) S. C. Erwin, L. J. Zu, M. I. Haftel, A. L. Efros, T. A. Kennedy and D. J. Norris, Nature, 2005, 436, 91 CrossRef CAS PubMed; (j) X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS PubMed; (k) V. Biju, T. Itoh, A. Anas, A. Sujith and M. Ishikawa, Anal. Bioanal. Chem., 2008, 391, 2469 CrossRef CAS PubMed; (l) D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., In. Ed., 2010, 49, 3280 CrossRef CAS PubMed; (m) N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS PubMed; (n) K. Huan, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681 CrossRef PubMed.
- (a) D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280 CrossRef CAS PubMed; (b) S. Rana, A. Bajaj, R. Mout and V. M. Rotello, Adv. Drug Delivery Rev., 2012, 64, 200 CrossRef CAS PubMed; (c) S. D. Brown, P. Nativo, J. A. Smith, D. Stirling, P. R. Edwards, B. Vengopal, D. J. Flint, J. A. Plumb, D. Graham and N. J. Wheate, J. Am. Chem. Soc., 2010, 132, 4678 CrossRef CAS PubMed; (d) S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652 CrossRef CAS PubMed; (e) N. Lewinski, V. Colvin, R. Drezek and E. E. Connor, Small, 2008, 4, 26 CrossRef CAS PubMed; (f) J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325 CrossRef PubMed; (g) C. M. Goodman, C. D. McCusker, T. Yil-maz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897 CrossRef CAS PubMed; (h) P. Nativo, I. A. Prior and M. Brust, ACS Nano, 2008, 2, 1639 CrossRef CAS PubMed; (i) Y. Cheng, A. C. Samia, J. D. Meyers, I. Panagopoulos, B. Fei and C. J. Burda, Am. Chem. Soc., 2008, 130, 10643 CrossRef CAS PubMed; (j) T. A. Taton, C. A. Mirkin and R. L. Letsinger, Science, 2000, 289, 1757 CrossRef CAS PubMed; (k) N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2011, 44, 925 CrossRef CAS PubMed; (l) C. C. Huang, Z. Yang, K. H. Lee and H. T. Chang, Angew. Chem., Int. Ed., 2007, 46, 6824 CrossRef CAS PubMed; (m) P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nano Today, 2007, 2, 18 CrossRef; (n) A. E. Prigodich, D. S. Seferos, M. D. Massich, D. A. Giljohann, B. C. Lane and C. A. Mirkin, ACS Nano, 2009, 3, 2147 CrossRef CAS PubMed; (o) C. C. You, A. R. Miranda, B. Gider, P. S. Ghosh, I.-B. Kim, B. Erdogan, S. A. Krovi, U. H. F. Buenz and V. M. Rotello, Nat. Nanotechnol., 2007, 2, 318 CrossRef CAS PubMed; (p) M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Neil, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889 CrossRef CAS PubMed; (q) M. De, S. Rana, H. Akpinar, O. R. Miranda, R. R. Arvizo, U. H. F. Bunz and V. M. Rotello, Nat. Chem., 2009, 1, 461 CrossRef CAS PubMed.
- (a) N. Hadjiliadis and E. Sletten, Metal Complex - DNA Interactions, Wiley-Blackwell, 2009 Search PubMed; (b) E. Alessio, Medicinal Chemistry, Ed. Bioinorganic, Wiley-VCH Verlag GmbH & Co., Germany, 2011 Search PubMed; (c) K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777 CrossRef CAS PubMed; (d) R. Zhao, R. Hammitt, R. P. Thummel, Y. Liu, C. Turro and R. M. Snapka, Dalton Trans., 2009, 10926 RSC; (e) Q. Yu, Y. Liu, L. Xu, C. Zheng, F. Le, X. Qin, Y. Liu and J. Liu, Eur. J. Med. Chem., 2014, 82, 82 CrossRef CAS PubMed; (f) C. A. Puckett and J. K. Barton, Biochemistry, 2008, 47, 11711 CrossRef CAS PubMed; (g) J. G. Vos and J. M. Kelly, Dalton Trans., 2006, 4869 RSC; (h) B. Elias and A. K.-D. Mesmaeker, Coord. Chem. Rev., 2006, 250, 1627 CrossRef CAS; (i) A. Ghosh, P. Das, M. R. Gill, P. Kar, M. G. Walker, J. A. Thomas and A. Das, Chem. – Eur. J., 2011, 17, 2089 CrossRef CAS PubMed; (j) I. Ortmans, S. Content, N. Boutonnet, A. K.-D. Mesmaeker, W. Bannwarth, J. F. Constant, E. Defrancq and J. Lhomme, Chem. – Eur. J., 1999, 5, 2712 CrossRef CAS; (k) G. J. Ryan, S. Quinn and T. Gunnlaugsson, Inorg. Chem., 2008, 47, 401 CrossRef CAS PubMed; (l) A. M. Nonat, S. Quinn and T. Gunnlaugsson, Inorg. Chem., 2009, 48, 4646 CrossRef CAS PubMed; (m) P. M. Keane, F. E. Poynton, J. P. Hall, I. P. Clark, I. V. Sazanovich, M. Towrie, T. Gunnlaugsson, S. Quinn, C. J. Cardin and J. M. Kelly, J. Phys. Chem. Lett., 2015, 6, 734–738 CrossRef CAS PubMed.
- (a) J. P. Hall, D. Cook, S. Ruiz-Morte, P. McIntyre, K. Buchner, H. Beer, D. J. Cardin, J. A. Brazier, G. Winter, J. M. Kelly and C. J. Cardin, J. Am. Chem. Soc., 2013, 135, 12652 CrossRef CAS PubMed; (b) P. Nordell and P. Lincoln, J. Am. Chem. Soc., 2005, 127, 9670 CrossRef CAS PubMed.
- (a) D. A. Lutterman, A. Chouai, Y. Liu, Y. Sun, C. D. Stewart, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2008, 130, 1163 CrossRef CAS PubMed; (b) A. Ghosh, P. Das, M. R. Gill, P. Kar, M. G. Walker, J. A. Thomas and A. Das, Chem. – Eur. J., 2011, 17, 2089 CrossRef CAS PubMed; (c) I. Ortmans, B. Elias, J. M. Kelly, C. Moucheron and A. K.-D. Mesmaeker, Dalton Trans., 2004, 668 RSC; (d) B. Elias, C. Creely, G. W. Doorley, M. M. Feeney, C. Moucheron, A. K.-D. Mesmaeker, J. Dyer, D. C. Grills, M. W. George, P. Matousek, A. W. Parker, M. Towrie and J. M. Kelly, Chem. – Eur. J., 2008, 14, 369 CrossRef CAS PubMed.
- (a) S. Neidle and M. Waring, Molecular Aspects of Anti-cancer Drug DNA Interactions, Taylor & Francis, Bosa Roca, 1994 Search PubMed; (b) I. Kostova, Curr. Med. Chem., 2006, 13, 1085 CrossRef CAS PubMed; (c) J.-Q. Wang, P.-Y. Zhang, C. Qian, X.-J. Hou, L.-N. Ji and H. J. Chao, Biol. Inorg. Chem., 2014, 19, 335 CrossRef CAS PubMed.
- (a) M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662 CrossRef CAS PubMed; (b) M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179 RSC; (c) S. J. Turrell, M. H. Filby, A. Whitehouse and A. J. Wilson, Bioorg. Med. Chem. Lett., 2012, 22, 985 CrossRef CAS PubMed.
- R. B. P. Elmes, J. A. Kitchen, D. C. Williams and T. Gunnlaugsson, Dalton Trans., 2012, 41, 6607 RSC.
- (a) C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC; (b) C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2009, 131, 8738 CrossRef CAS PubMed.
- (a) R. B. P. Elmes, K. N. Orange, S. M. Cloonan, D. C. Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2011, 133, 15862 CrossRef CAS PubMed. Other recent examples of Ru(II) based nano-material includes: (b) J. Moreau, F. Lux, M. Four, J. Olesi-ak-Banska, K. Matczyszyn, P. Perriat, C. Frochot, P. Ar-noux, O. Tillement, M. Samoc, G. Ponterini, S. Roux and G. Lemercier, Phys. Chem. Chem. Phys., 2014, 16, 14826 RSC; (c) L. He, Y. Huang, H. Zhu, G. Pang, W. Zheng, Y.-S. Wong and T. Chen, Adv. Mater., 2014, 24, 2754 CAS; (d) F. C.-M. Leung, A. Y.-Y. Tam, V. K.-M. Au, M.-J. Li and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2014, 6, 6644 CrossRef CAS PubMed; (e) S. Pramod, P. K. Sudeep, K. G. Thomas and P. V. Kamat, J. Phys. Chem. B, 2006, 110, 20737 CrossRef PubMed; (f) L. Zedler, F. Theil, A. Csáki, W. Fritzsche, S. Rau, M. Schmitt, J. Popp and B. Dietzek, RSC Adv., 2012, 2, 4463 RSC; (g) C. R. Mayer, E. Dumas and F. J. Sécheresse, Colloid Interface Sci., 2008, 328, 452 CrossRef CAS PubMed; (h) C. R. Mayer, E. Dumas, F. Miomandre, R. Meallet-Renault, F. Warmont, J. Vigneron, R. Pansu, A. Etcheberry and F. Sécheresse, New J. Chem., 2006, 30, 1628 RSC; (i) T. Huang and R. W. Murray, Langmuir, 2002, 18, 7077 CrossRef CAS; (j) N. J. Rogers, S. Claire, R. M. Harris, S. Farabi, G. Zikeli, I. B. Styles, N. J. Hodges and Z. Pikramenou, Chem. Commun., 2014, 50, 617 RSC; (k) S. A. M. Osborne and Z. Pikramenou, Faraday Discuss., 2015 10.1039/C5FD00108K.
- (a) E. B. Veale, D. O. Frimannsson, M. Lawler and T. Gunnlaugsson, Org. Lett., 2009, 11, 4040 CrossRef CAS PubMed; (b) E. B. Veale and T. Gunnlaugsson, J. Org. Chem., 2010, 75, 5513 CrossRef CAS PubMed; (c) S. Murphy, S. A. Bright, F. E. Poynton, T. McCabe, J. A. Kitchen, E. B. Veale, D. C. Williams and T. Gunnlaugsson, Org. Biomol. Chem., 2014, 12, 6610 RSC; (d) S. Banerjee, S. A. Bright, J. A. Smith, J. Burgeat, M. Martinez-Calvo, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, J. Org. Chem., 2014, 79, 9272 CrossRef CAS PubMed.
- (a) S. M. Cloonan, R. B. P. Elmes, M. Erby, S. A. Bright, F. E. Poynton, D. E. Nolan, S. Quinn, T. Gunnlaugsson and D. C. Williams, J. Med. Chem., 2015, 58, 4494–4505 CrossRef CAS PubMed; (b) R. B. P. Elmes, M. Erby, S. M. Cloonan, S. Quinn, D. C. Williams and T. Gunnlaugsson, Chem. Commun., 2011, 47, 686 RSC.
- J. W. Liu and Y. Lu, Nat. Protoc., 2006, 1, 246 CrossRef CAS PubMed.
- (a) T. Huang and R. W. Murray, Langmuir, 2002, 18, 7077 CrossRef CAS; (b) P. Reineck, D. Gómez, S. H. Ng, M. Karg, T. Bell, P. Mulvaney and U. Bach, ACS Nano, 2013, 7, 6636 CrossRef CAS PubMed.
- W. Vanderlinden, M. Blunt, C. C. David, C. Moucheron, A. Kirsch-De Mesmaeker and S. De Feyter, J. Am. Chem. Soc., 2012, 134, 10214 CrossRef CAS PubMed.
- M. T. Carter, M. Rodriguez and A. J. Bard, J. Am. Chem. Soc., 1989, 111, 8901 CrossRef CAS.
- R. B. Nair, E. S. Teng, S. L. Kirkland and C. J. Murphy, Inorg. Chem., 1998, 37, 139 CrossRef CAS PubMed.
- (a) J.-P. Lecomte, A. K.-D. Mesmaeker, M. M. Feeney and J. M. Kelly, Inorg. Chem., 1995, 34, 6481 CrossRef CAS; (b) B. Elias, C. Creely, G. W. Doorley, M. M. Feeney, C. Moucheron, A. Kirsch-Demesmaeker, J. Dyer, D. C. Grills, M. W. George, P. Matousek, A. W. Parker, M. Towrie and J. M. Kelly, Chem. – Eur. J., 2007, 14, 369 CrossRef PubMed.
- (a) A. C. Templeton, J. J. Pietron, R. W. Murray and P. Mulvaney, J. Phys. Chem. B, 2000, 104, 564 CrossRef CAS; (b) C. J. Murphy, A. M. Gole, S. E. Hunyadi, J. W. Stone, P. N. Sisco, A. Alkilany, B. E. Kinard and P. Hankins, Chem. Commun., 2008, 544 RSC.
- M. Eriksson, M. Leijon, C. Hiort, B. Norden and A. Grae-slund, Biochemistry, 1994, 33, 5031 CrossRef CAS PubMed.
- S. Sun, Y. He, Z. Yang, Y. Pang, F. Liu, J. Fan, L. Sund and X. Peng, Dalton Trans., 2010, 39, 4411 RSC.
- J. W. Liu and Y. Lu, Nat. Protoc., 2006, 1, 246 CrossRef CAS PubMed.
- X. Zhang, M. R. Servos and J. Liu, J. Am. Chem. Soc., 2012, 134, 7266 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr05598a |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |