
Highly sensitive self-complementary DNA nanoswitches triggered by polyelectrolytes†
Jincai
Wu‡
a,
Feng
Yu‡
a,
Zheng
Zhang
a,
Yong
Chen
a,
Jie
Du
*a and
Atsushi
Maruyama
*b
aCollege of Materials and Chemistry Engineering, Hainan University, Haikou 570228, China. E-mail: dujie@hainu.edu.cn
bDepartment of Biomolecular Engineering, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, 4259-B57 Nagatsuta, Midori, Yokohama, 226-8501, Japan. E-mail: amaruyama@bio.titech.ac.jp
First published on 19th November 2015
Abstract
Dimerization of two homologous strands of genomic DNA/RNA is an essential feature of retroviral replication. Herein we show that a cationic comb-type copolymer (CCC), poly(L-lysine)-graft-dextran, accelerates the dimerization of self-complementary stem–loop DNA, frequently found in functional DNA/RNA molecules, such as aptamers. Furthermore, an anionic polymer poly(sodium vinylsulfonate) (PVS) dissociates CCC from the duplex shortly within a few seconds. Then single stem–loop DNA spontaneously transforms from its dimer. Thus we can easily control the dimer and stem–loop DNA by switching on/off CCC activity. Both polyelectrolytes and DNA concentrations are in the nanomole per liter range. The polyelectrolyte-assisted transconformation and sequences design strategy ensures the reversible state control with rapid response and effective switching under physiologically relevant conditions. A further application of this sensitive assembly is to construct an aptamer-type drug delivery system, bind or release functional molecules responding to its transconformation.
Introduction
Nanometer-scaled DNA devices based on programmed assembly of DNA helices has become a powerful tool in DNA nanotechnology fields beyond its biological importance.1–4 Numerous endeavors have been reported on the use of light irradiation,5–8 changes in environmental pH9–14or ion,15,16 and the addition of DNA oligonucleotides17–20 or enzymes21–23 as external stimuli for DNA nanomachines.For most of the DNA nanomachines constructed so far, oligonucleotides have been generally used as fuel. The common operating principles employed in this DNA-fueled nanomachine involve sequence-specific hybridizations and strand exchange reactions between the machine body and the sequential addition of DNA fuels. However, the operating conditions of the DNA-fueled nanomachines are limited. For example, the switch frequency is limited by the hybridization kinetics of complementary strands; it needs a high concentration of DNA oligonucleotide to retain the machine's dynamic action; a DNA duplex waste is produced in each cycle, and accumulation of the waste products leads to a progressive loss of operating efficiency.24,25 To overcome these weaknesses, a new strategy is therefore required for the development of DNA nanotechnology.
Our group developed a cationic copolymer-assisted strategy to solve this problem using G-quadruplex and DNA tweezer nanomachines as models.26,27 This strategy is partially an extended work of our previous studies on interactions between cationic comb-type copolymers (CCCs) and triplex or duplex DNA.28–30 In those studies, we demonstrated that CCCs composed of a polycation backbone and abundant hydrophilic graft chains (>90 wt%) influence the kinetics and thermodynamics of nucleic acid hybridization under physiologically relevant conditions. Detailedly, we found that the poly(L-lysine)-graft-dextran (PLL-g-Dex) copolymer significantly accelerates DNA hybridization.31 Moreover, the copolymer markedly accelerates the strand-exchange reaction (SER) and increases the stability of dsDNA relative to that in buffer alone.32–34 The SER acceleration of the copolymer has been applied for refining the DNA detection method35–37 and DNA nanodevices with improved response even at 200 times lower strand concentration (nanomole per liter range) than those in other reports.20,27 The foundation of our research is the interaction mechanism of CCCs and nucleic acid. The CCCs reduce an entropically unfavourable counterion condensation effect that accompanies a nucleation process of SER, lowering the energy barrier associated with breakage and reassociation of the nucleic acid base pairs.32
Nucleic acid chaperones catalyze the folding of nucleic acid into the most stable conformation. As artificial nucleic acid chaperones, a unique aspect of CCCs is that they accelerate the SER while stabilizing DNA hybrids. A naturally occurring nucleic acid chaperone, HIV-1 NCp7, also has the nucleic acid chaperone activity and plays a crucial role in the proviral life cycle.38,39 Dimerization of two homologous strands of genomic RNA is an essential feature of retroviral replication. In HIV-1, a conserved stem–loop sequence, the dimerization initiation site (DIS), has been identified as the domain primarily responsible for the initiation of this aspect of viral assembly.40,41 The DIS loop contains a self-complementary sequence and can form a homodimer through a loop–loop kissing interaction. NCp7 acts catalytically to induce the structural isomerization by accelerating strand exchange between the two stem–loops, and converts the DIS dimer into an extended duplex isoform.
Herein we examined the activity of CCCs to trigger the dimerization of a self-complementary stem–loop DNA, DIS25, similar to DIS in HIV-1 genome. The purpose of this study was to test whether CCCs facilitate the dimerization of DIS like in NCp7. Further, we assessed whether the anionic polymer could switch off CCC activity, soon dissociating a single stem–loop DIS from its dimer. If this strategy is viable, we can easily control DIS dimer and single stem–loop by switching on/off CCC activity. A biomedical application of this programmable assembly is to construct a smart aptamer-type nanoswitch that can bind or release functional molecules responding to its transconformation. Beyond this objective, the reversible state control with rapid response and effective switching is in great need, which is the main work in this paper.
Experimental
Materials
All oligonucleotides were supplied by FASMAC Co., Ltd and purified by reverse-phase high performance liquid chromatography. The sequence of DIS25, DIS25-2a, DIS25-3a; Tm of stem–loop DNA and the dimer are listed in Fig. S1 (ESI†). The cationic comb-type copolymer PLL-g-Dex (Mn = 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was prepared by a reductive amination reaction of PLL·HBr (Mn = 20000, BACHEM) with dextran (Mn = 5900, Dextran T-10, Amersham Pharmacia Biotech) as described previously.28 The dextran content of the copolymer was 91 wt% as determined by 1H NMR (Fig. S2, ESI†). DNA samples were treated by annealing (heating to 90 °C for 5 min and quick cooling on ice). PLL-g-Dex/DNA complexes were constructed at various N/P ratios (the ratios of moles of the amine groups of cationic polymers to those of the phosphate ones of DNA).
000) was prepared by a reductive amination reaction of PLL·HBr (Mn = 20000, BACHEM) with dextran (Mn = 5900, Dextran T-10, Amersham Pharmacia Biotech) as described previously.28 The dextran content of the copolymer was 91 wt% as determined by 1H NMR (Fig. S2, ESI†). DNA samples were treated by annealing (heating to 90 °C for 5 min and quick cooling on ice). PLL-g-Dex/DNA complexes were constructed at various N/P ratios (the ratios of moles of the amine groups of cationic polymers to those of the phosphate ones of DNA).
Native polyacrylamide gel electrophoresis
DNA solutions were prepared in 10 mM sodium phosphate buffer (pH 7.2) containing 0.5 mM EDTA and 150 mM NaCl with or without PLL-g-Dex. The DNA samples were incubated at 37 °C for 1 h or 3 h. Native PAGE (13%) was performed in TBE buffer at 25 °C for 2 h at 100 V. After electrophoresis, the gel was stained with 0.01% EtBr.Fluorescence spectroscopy
The DNA solution (4:1 mixture of non-labeled DIS25 and TAMRA and DABCYL double-labeled DIS25, T-DIS25-D) was dissolved in sodium phosphate buffer (10 mM sodium phosphate, 0.5 mM EDTA, 150 mM NaCl, pH 7.2). Baseline emission values were first recorded for about 5 minutes, and then PLL-g-Dex and PVS were successively added to the DNA solution with a syringe. The change in fluorescence intensity of the mixture (total volume 2 ml) in a 10 mm-square quartz cuvette was recorded on a JASCO FP-6500 fluorescence spectrometer (JASCO) with a Peltier thermostatically controlled cell holder at excitation and emission wavelengths of 540 nm and 570 nm, respectively. The TAMRA emission of T-DIS25-D is quenched by DABCYL when T-DIS25-D folds into the stem–loop structure. The TAMRA emission recovers when T-DIS25-D forms a dimer with non-labeled DIS25.
UV-melting point (Tm) measurement
The concentration of DIS25 in sodium phosphate buffer (10 mM sodium phosphate, 0.5 mM EDTA, 150 mM NaCl, pH 7.2) is 2 μM. The mixture of DNA solution was heated at 95 °C for 3 min and gradually cooled to r.t. UV spectra at 260 nm was recorded by a Shimadzu UV-1650 PC spectrometer equipped with a TMSPC-8 temperature controller (Shimadzu, Kyoto, Japan). Melting curves were obtained at a heating rate of 0.5 °C min−1 and a cooling rate of 0.5 °C min−1.Results and discussion
Firstly, the effect of CCC, PLL-g-Dex on DIS25 dimerization was evaluated by native polyacrylamide gel electrophoresis (PAGE) assay using a 4:1 mixture of non-labeled DIS25 and TAMRA and DABCYL double-labeled DIS25, T-DIS25-D. DIS25 and T-DIS25-D mixture (2 μM) were incubated at 25 °C in 10 mM sodium phosphate buffer (pH 7.2) containing 150 mM NaCl and 0.5 mM EDTA in the absence or presence of PLL-g-Dex (N/P = 1) for 1 h or 3 h.
After the incubation, 0.2 wt% poly(sodium vinylsulfonate) (PVS) was added to dissociate each copolymer from DNA. Gel electrophoresis was then carried out on a 13% polyacrylamide gel at 4 °C in TBE buffer. The TAMRA emission of T-DIS25-D is quenched by DABCYL when T-DIS25-D folds into the stem–loop structure. The TAMRA emission recovers when T-DIS25-D forms a dimer with non-labeled DIS25. Fig. 1A shows the PAGE images acquired by TAMRA emission or EtBr staining. The band with higher electrophoretic mobility is identified as the stem–loop monomer. After the gel is stained with EtBr, both the monomer and dimer bands can be captured. The gel images showed that the dimerization of DIS25/T-DIS25-D in the absence of PLL-g-Dex hardly occurred even after 3 h incubation. On the other hand, the hybridization was accelerated by PLL-g-Dex, which revealed that PLL-g-Dex thermodynamically stabilizes the intermolecular dimer preferentially over the stem–loop intramolecular structure. Another study with more PLL-g-Dex (N/P = 2, N/P = 6) and longer incubation (3 h, 10 h, 24 h) evaluated by PAGE is shown in Fig. S3 (ESI†). The results further testified that PLL-g-Dex markedly facilitates dimer formation. Note that upper bands of DNA in the presence of PLL-g-Dex are in the same level with the band of DNA after the renaturation process (heating to 90 °C for 5 min and slowly cooling to room temperature), which confirms that the upper band is for DNA duplex, not for the single strand coil with two ends far away from each other.
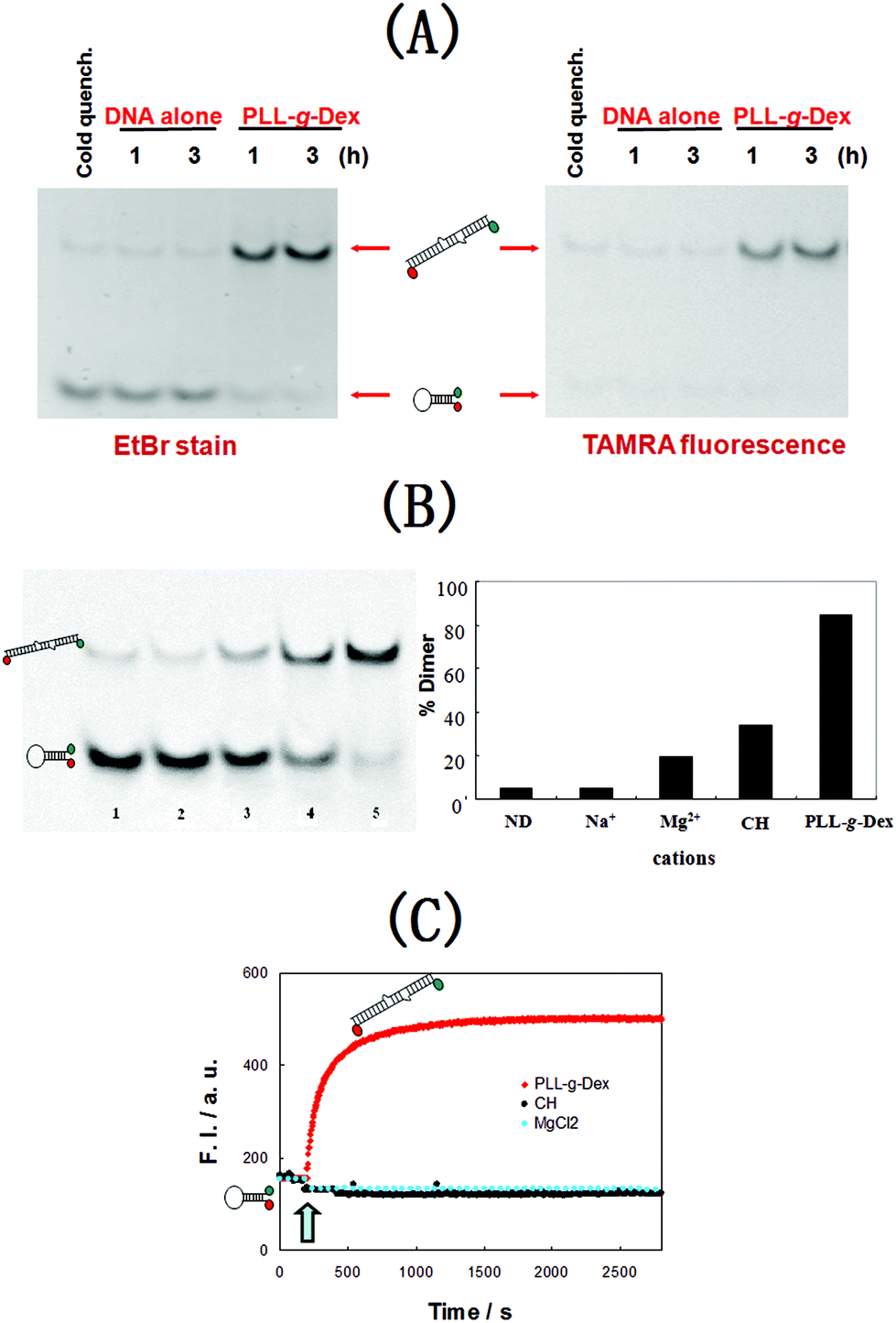 | ||
| Fig. 1 (A) Gel electrophoretic analysis of dimerization of DIS25 with or without PLL-g-Dex. [DIS25] = 1.6 μM, [T-DIS25-D] = 0.4 μM, N/P = 1. (B) Gel electrophoretic analysis of dependency of dimerization of DIS25 on the cationic substance. [DIS25] = 1.6 μM, [T-DIS25-D] = 0.4 μM, in 10 mM sodium phosphate buffer (pH 7.2, 0.5 mM EDTA, 150 mM NaCl), 37 °C for 2 h. Lane 1, no additive (quench); lane 2, no additive; lane 3, 5 mM MgCl2; lane 4, 500 μM [Co(NH3)6]3+; lane 5, 100 μM PLL-g-Dex. The gel image was captured with EtBr staining. (C) Time course of dimerization of DIS25 in the presence of different cationic substances. A stirred DNA solution ([DIS25] = 80 nM, [T-DIS25-D] = 20 nM) in 10 mM sodium phosphate buffer (pH 7.2, 0.5 mM EDTA, 150 mM NaCl) was mixed at 37 °C with 5 mM Mg2+ (cyan), 500 μM [Co(NH3)6]3+ (black), and 100 μM PLL-g-Dex (red). | ||
We then compared the activity of the copolymer (100 μM PLL-g-Dex) with those of different cationic substances, 5 mM Mg2+ and 500 μM [Co(NH3)6]3+ under physiologically relevant conditions, pH 7.2, containing 150 mM NaCl at 37 °C. As shown in Fig. 1B, 5 mM Mg2+ and 500 μM [Co(NH3)6]3+ resulted in 18% and 42%, respectively, conversion to DIS25 dimer, while PLL-g-Dex at lower concentration (100 μM) induced almost full conversion (>90%) to the dimer state from the stem–loop state. The result implied an effective stablilization effect of CCC on the DIS25 dimer.
In order to observe the kinetic effect of the cationic substances on the dimerization reaction in real time with a resolution of seconds, we employed the fluorescence resonance energy transfer (FRET) assay. Fig. 1C shows that when we added PLL-g-Dex into the DNA solution, the fluorescence intensity increased to the maximum value within 900 s. The fluorescence intensity changes were not obvious in other cationic substances. These results showed that PLL-g-Dex significantly accelerated dimerization to stabilize the linear dimer structure even at 100 nM DNA concentration.
During the dimerization formation, CCCs spontaneously interact with DNA to form inter-polyelectrolyte complexes. The fluorescence polarization can be used to measure the apparent volume (or molecular weight) of DNA duplex. This measurement is possible because a larger duplex rotates more slowly. Hence, if CCC binds to the DNA duplex, the rotational rate decreases, and the polarity increases. According to this theory, the fluorescence polarity of the DIS25 dimer should be increased after CCC–DNA complex formation. Thus, we can investigate the changes of fluorescence polarity of the DNA duplex to examine CCCs’ binding. As shown in Fig. S4 (ESI†), the fluorescence polarity increased after PLL-g-Dex (N/P = 1) was added into the DNA solution. To confirm that the increase in the polarity was due to CCC–DNA complex formation, we subsequently added 1.2 times charge concentrated anionic polymer, PVS, to the mixture, and the fluorescence was polarity recovered, which shows that the anionic polymer can dissociate the cationic copolymer from the duplex. That means CCC activity can be switched off by a polyanion. Without CCC chaperoning, could the dimer stably maintain the conformation? To answer this question, the transconformation between the single stem–loop DIS25 and extended dimer induced by switching on/off CCC activity was evaluated by the FRET assay. The kinetic effects of PLL-g-Dex/PVS on dimerization of DIS25 or dissociation of the dimer at different temperatures were also investigated (Fig. 2 and 3).
 | ||
| Fig. 2 Kinetic dimerization of DIS25 triggered by PLL-g-Dex (N/P = 1). A stirred DNA solution ([DIS25] = 90 nM, [T-DIS25-D] = 10 nM) in 10 mM sodium phosphate buffer (pH 7.2, 0.5 mM EDTA, 150 mM NaCl) was mixed (A) at 37 °C, (B) at 45 °C, (C) at 50 °C, (D) at 60 °C. The value of fluorescence intensity was calculated with the following equation to yield a rate constant of the second-order reaction. kt = ln[(I∞ − It)/(I∞ − I0)], where I0 is the initial fluorescence intensity of TAMRA-labeled DNA, It is that at time t, and I∞ is that after the reaction reached equilibrium. | ||
 | ||
| Fig. 3 Kinetic dissociation of DIS25 dimer triggered by PVS (1.2 times excess added after the course in Fig. 3). (A) at 37 °C, (B) at 45 °C, (C) at 50 °C, (D) at 60 °C. The value of fluorescence intensity was calculated with the following equation to yield a rate constant of the second-order reaction. kt = ln[(It− I∞)/(I0 − I∞)], where I0 is the initial fluorescence intensity of TAMRA-labeled DNA before adding PVS, It is that at time t, and I∞ is that after the reaction reached equilibrium. | ||
After the addition of PLL-g-Dex, fluorescence intensity increased, indicating the transformation of the stem–loop to the extended dimer form. As shown in Fig. 2, with increasing temperature, the dimerization rate slightly accelerated, while the variation range of the fluorescence intensity decreased above 50 °C. Note that the dimer was formed even at 60 °C in the presence of PLL-g-Dex. The copolymer increased Tm of the dimer from 54.5 °C (estimated by mFold) to 66.7 °C as evaluated by UV-Tm analysis (Fig. S5, ESI†).
Subsequent addition of PVS resulted in a relatively slow decrease in the fluorescence intensity, especially at 37 °C, as shown in Fig. 3. Since PVS-induced dissociation of DNA from CCC occurred shortly within a few seconds both at 37 °C and 60 °C (Fig. S4, ESI†), PVS easily divests the PLL-g-Dex–DNA complex of CCC chaperoning even at a low temperature. Thus the slow decrease in fluorescence intensity (Fig. 3) indicated that without CCC chaperoning the dimer was not still stable and spontaneously transformed into the stem–loop DNA. Further, the spontaneous dissociation of the extended dimer from the stem–loop DNA was considerably slower than the CCC-assisted dimerization. Since the stem–loop (Tm = 62.0 °C estimated by mFold) is thermodynamically more stable than the extended dimer (Tm = 54.5 °C estimated by mFold) in the absence of the CCC, transconformation from the dimer to the stem–loop is kinetically restricted by the high energy barrier between dissociation and reassociation of base pairs. As expected, the dissociation rate was accelerated at higher temperature close to Tm. Above 50 °C, the transition was very rapid (Fig. 3). The Ea value estimated for spontaneous dimer dissociation was 366.2 kJ mol−1, while Ea of CCC-assisted dimerization was estimated to be 113.2 kJ mol−1 (Fig. S6, ESI†), and also testified that spontaneous dissociation of the extended dimer to the stem–loop DNA was considerably slower than the CCC-assisted dimerization.
As mentioned above, at a relatively low temperature such as 37 °C, the slower dissociation rate resulted in a weaker transconformation response (Fig. 3), although the dimerization occurred rapidly in the presence of CCC, which is an obstacle to its further application in biological fields. To overcome this limitation, we found that modification of a DNA oligonucleotide sequence was a pathway to accelerate the transformation from dimer to stem–loop DNA. We slightly changed the loop sequence of DIS25, and changed G/C to A/T. The diverse stem–loop DNAs were marked as DIS25-2a and DIS25-3a and the sequences are listed in Fig. S1 (ESI†). Estimated by mFold at 2 μM and 150 mM NaCl, the melting temperatures of stem–loop DIS25, DIS25-2a and DIS25-3a are the same (62.0 °C), while the melting temperatures of the dimer DIS25-2a (45.3 °C) and dimer DIS25-3a (42.3 °C), are lower than dimer DIS25 (54.5 °C). That means the stabilities of dimers DIS25-2a and DIS25-3a are lower compared to DIS25, while the stabilities of stem–loop are maintained. In response to the added polyelectrolyte, the transitions of DIS25-2a and DIS25-3a were rapid and reversible and occurred at lower temperatures, 46 °C and 42 °C, respectively, as shown in Fig. 4.
 | ||
| Fig. 4 Dimerization of stem–loop DNA and its dissociation triggered by PLL-g-Dex (N/P = 1) and PVS (1.2 times charge excess). Final concentration of DNA was 50 nM. FRET assay was performed at indicated temperatures in 10 mM sodium phosphate buffer (pH 7.2, 0.5 mM EDTA, 150 mM NaCl). (A) DIS25-2a at 46 °C. (B) DIS25-2a at 42 °C. (C) DIS25-3a at 42 °C. (D) DIS25-3a at 37 °C. | ||
On the basis of the results above, we can construct a kind of a DNA mechanical device that allows easy state control of the reversible extended dimer and single stem–loop DNA by switching on/off CCC activity. The operating principle employed in this system is shown in Fig. 5. This switch can be operated at very low concentrations of both DNA and fuels owing to strong interaction between polycation and polyanion or DNA.
 | ||
| Fig. 5 Switching between stem–loop DNA and extended dimer drived by PLL-g-Dex and PVS. | ||
We carried out the FRET assay for the real-time observation of the transformations of the nanoswitches. The operating temperature was raised to near Tm of the dimer to obtain quick responses. Based on this principle, self-complementary DNA nanoswitches (DIS25, DIS25-2a, and DIS25-3a series) with high sensitivity driven by a polyelectrolyte were constructed respectively at 60 °C, 46 °C and 42 °C, as shown in Fig. 6A, C, and D. PLL-g-Dex and PVS were added successively, which induced the transformation between the dimer and stem–loop DNA. The gel electrophoresis assay testified DIS25 nanoswitch's transformation of the first two cycles shown in Fig. 6B.
 | ||
| Fig. 6 Transconformation switching of DIS derivatives triggered by polyelectrolytes. Final concentration of DNA was 50 nM. FRET assay was performed at indicated temperatures in 10 mM sodium phosphate buffer (pH 7.2, 0.5 mM EDTA, 150 mM NaCl). PLL-g-Dex and PVS (1.2 times excess) were added successively. (A) [DIS25]:[T-DIS25-D] = 4:1, at 60 °C. (B) [DIS25-2a]:[T-DIS25-2a-D] = 39:1, at 46 °C. (C) [DIS25-3a]:[T-DIS25-3a-D] = 39:1, at 42 °C. (D) Gel electrophoretic analysis to confirm transconformation between dimer and single stem–loop DIS25 in response to successive addition of PLL-g-Dex and PVS. Gel images were captured with EtBr staining. Lane 1: DNA alone, Lane 2: 1st PLL-g-Dex addition, Lane 3: 1st PVS addition, Lane 4: 2nd PLL-g-Dex addition, Lane 5: 2nd PVS addition. | ||
Unlike many other DNA-fueled nanomachines, it can operate without a supply of additional DNA strands, and there is no DNA duplex waste produced in each cycle. Therefore, at very low concentrations of both DNA and polymer fuels, switching should be highly reversible with negligible changes in efficiency. To testify this hypothesis, we changed the N/P value in DIS25-3a nanoswitch system at 42 °C (Fig. 7). The switching efficiency (N/P = 2, Fig. 7A) was decreased almost by half in the second cycle. N/P = 1.2 (Fig. 7B) and N/P = 1 (Fig. 7C) also showed an obvious decrease in switching efficiency after 4 and 6 cycles, respectively. When the N/P value was reduced to 0.5, the switching was well reversible and decreased only slightly in efficiency even after 13 cycles (Fig. 7E). Since a polycation–polyanion complex is produced in each operating cycle, high concentration of this waste polyelectrolyte complex induces loss of switching efficiency after several cycles. Therefore, the switching efficiency was increased with the N/P value decreasing. Using this programmable strategy, we further designed a double stem–loop DNA (DIS42), and constructed a more complicated nanoswitch system at 37 °C (Fig. S7).†
 | ||
| Fig. 7 Transconformation switching of DIS25-3a triggered by polyelectrolytes. Final concentration of DNA was 100 nM ([DIS25-3a]:[T-DIS25-3a-D] = 4:1). FRET assay was performed at indicated temperatures in 10 mM sodium phosphate buffer (pH 7.2, 0.5 mM EDTA, 150 mM NaCl) at 42 °C. PLL-g-Dex and PVS (1.2 times excess) were added successively. (A) N/P = 2 (B) N/P = 1.2 (C) N/P = 1 (D) N/P = 0.8 (E) N/P = 0.5. | ||
So far, a series of polyelectrolyte-assisted DNA mechanical nanodevices with rapid response were offered, achieving the original aim of this study. As we know, self-complementary stem–loop structures are frequently found in functional DNA and RNA molecules, such as aptamers. Aptamers provide additional functional information about a target protein, which makes them useful for therapeutic applications. Our study in progress is constructing a polyelectrolyte-assisted aptamer-type DNA nanoswitch system that can be instructed repeatedly to bind or release functional proteins, in rapid response to transconformation between the single stem–loop and extended dimer. This nanodevice has potential application as an intelligent drug delivery system.
Conclusions
In summary, a cationic comb-type copolymer, PLL-g-Dex, considerably promoted dimer formation of a self-complementary stem–loop DNA. Dimerization of the stem–loop DNA assisted by PLL-g-Dex was significantly faster than spontaneous dissociation of the dimer. Quick responses were obtained near Tm of the dimer. Stem–loop structures are found naturally in some functional DNA/RNA, such as aptamer. Based on the strategy we have developed here, nanodevices with biological function would permit us to use them in combination with DNA-acting enzymes or other small biomolecules, such as ligase, polymerase, and restrictive proteins. We expect that this strategy might extend the applicability of DNA nanodevices in biotechnology and nanotechnology.Acknowledgements
This work was supported by the National Science Foundation of China (Grant No. 21404028, 51362009), “Nanomedicine Molecular Science” (Japan, no. 2306), and Grant-in-Aid for Scientific Research (Japan, no. 23240074). J. D. was supported by the JSPS postdoctoral fellowship. J. C. W. also thanks the Graduate Students Innovation Research Project of Hainan Province.Notes and references
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795 CrossRef CAS PubMed.
- X. Liu, C. H. Lu and I. Willner, Acc. Chem. Res., 2014, 47, 1673 CrossRef CAS PubMed.
- A. E. Marras, L. Zhou, H. J. Su and C. E. Castro, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 713 CrossRef CAS PubMed.
- P. Yin, H. M. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318 CrossRef CAS PubMed.
- T. Goldau, K. Murayama, C. Brieke, S. Steinwand, P. Mondal, M. Biswas, I. Burghardt, J. Wachtveitl, H. Asanuma and A. Heckel, Chem. – Eur. J., 2015, 21, 2845 CrossRef CAS PubMed.
- Y. Kamiya and H. Asanuma, Acc. Chem. Res., 2014, 47, 1663 CrossRef CAS PubMed.
- X. Liang, H. Nishioka, N. Takenaka and H. Asanuma, ChemBioChem, 2008, 9, 702 CrossRef CAS PubMed.
- H. Liu, Y. Xu, F. Li, Y. Yang, W. Wang, Y. Song and D. Liu, Angew. Chem., Int. Ed., 2007, 46, 2515 CrossRef CAS PubMed.
- Y. Dong, Z. Yang and D. Liu, Acc. Chem. Res., 2014, 47, 1853 CrossRef CAS PubMed.
- A. Idili, A. Vallée-Bélisle and F. Ricci, J. Am. Chem. Soc., 2014, 136, 5836 CrossRef CAS PubMed.
- Q. Ke, Y. Zheng, F. Yang, H. Zhang and X. Yang, Talanta, 2014, 129, 539 CrossRef CAS PubMed.
- D. Liu and S. Balasubramanian, Angew. Chem., Int. Ed., 2003, 42, 5734 CrossRef CAS PubMed.
- S. Modi, M. Swetha, D. Goswami, G. D. Gupta, S. Mayor and Y. Krishnan, Nat. Nanotechnol., 2009, 4, 325 CrossRef CAS PubMed.
- I. V. Nesterova and E. E. Nesterov, J. Am. Chem. Soc., 2014, 136, 8843 CrossRef CAS PubMed.
- C. Mao, W. Sun, Z. Shen and N. C. Seeman, Nature, 1999, 397, 144 CrossRef CAS PubMed.
- C. M. Niemeyer, M. Adler, S. Lenhert, S. Gao, H. Fuchs and L. Chi, ChemBioChem, 2001, 2, 260 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield, A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605 CrossRef CAS PubMed.
- H. Yan, X. Zhang, Z. Shen and N. C. Seeman, Nature, 2002, 415, 62 CrossRef CAS PubMed.
- W. U. Dittmer, A. Reuter and F. C. Simmel, Angew. Chem., Int. Ed., 2004, 43, 3550 CrossRef CAS PubMed.
- P. Alberti and J. L. Mergny, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1569 CrossRef CAS PubMed.
- Y. Chen, M. Wang and C. Mao, Angew. Chem., Int. Ed., 2004, 43, 3554 CrossRef CAS PubMed.
- P. Yin, H. Yan, X. G. Daniell, A. J. Turberfield and J. H. Reif, Angew. Chem., Int. Ed., 2004, 43, 4906 CrossRef CAS PubMed.
- L. Hu, C. H. Lu and I. Willner, Nano Lett., 2015, 15, 2099 CrossRef CAS PubMed.
- J. Bath and A. J. Turberfield, Nat. Nanotechnol., 2007, 2, 275 CrossRef CAS PubMed.
- P. Alberti, A. Bourdoncle, B. Saccà, L. Lacroix and J. L. Mergny, Org. Biomol. Chem., 2006, 4, 3383 CAS.
- N. Makita, S. Inoue, T. Akaike and A. Maruyama, Nucleic Acids Symp. Ser., 2004, 48, 173 CrossRef PubMed.
- S. W. Choi, N. Makita, S. Inoue, C. Lesoil, A. Yamayoshi, A. Kano, T. Akaike and A. Maruyama, Nano Lett., 2007, 7, 172 CrossRef CAS PubMed.
- A. Maruyama, M. Katoh, T. Ishihara and T. Akaike, Bioconjugate Chem., 1997, 8, 3 CrossRef CAS PubMed.
- A. Ferdous, H. Watanabe, T. Akaike and A. Maruyama, Nucleic Acids Res., 1998, 26, 3949 CrossRef CAS PubMed.
- A. Maruyama, H. Watanabe, A. Ferdous, M. Katoh, T. Ishihara and T. Akaike, Bioconjugate Chem., 1998, 9, 292 CrossRef CAS PubMed.
- L. Wu, N. Shimada, A. Kano and A. Maruyama, Soft Matter, 2008, 4, 744 RSC.
- W. J. Kim, T. Akaike and A. Maruyama, J. Am. Chem. Soc., 2002, 124, 12676 CrossRef CAS PubMed.
- W. J. Kim, Y. Sato, T. Akaike and A. Maruyama, Nat. Mater., 2003, 2, 815 CrossRef CAS PubMed.
- N. Shimada, W. Song and A. Maruyama, Biomater. Sci., 2014, 2, 1480 RSC.
- K. Yamana, Y. Fukunaga, Y. Ohtani, S. Sato, M. Nakamura, W. J. Kim, T. Akaike and A. Maruyama, Chem. Commun., 2005, 2509 RSC.
- H. Asanuma, T. Osawa, H. Kashida, T. Fujii, X. Liang, K. Niwa, Y. Yoshida, N. Shimada and A. Maruyama, Chem. Commun., 2012, 48, 1760 RSC.
- J. Michaelis, A. Maruyama and O. Seitz, Chem. Commun., 2013, 49, 618 RSC.
- K. M. Stewart-Maynard, M. Cruceanu, F. Wang, M.-N. Vo, R. J. Gorelick, M. C. Williams, I. Rouzina and K. Musier-Forsyth, J. Virol., 2008, 82, 10129 CrossRef CAS PubMed.
- G. Krishnamoorthy, B. Roques, J.-L. Darlix and Y. Mély, Nucleic Acids Res., 2003, 31, 5425 CrossRef CAS PubMed.
- C. Liang, L. Rong, E. Cherry, L. Kleiman, M. Laughrea and M. A. Wainberg, J. Virol., 1999, 73, 6147 CAS.
- M.-R. Mihailescu and J. P. Marino, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1189 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: I. Sequences of DIS25, DIS25-2a and DIS25-3a. II. Structural formula of poly(L-lysine)-graft-dextran (PLL-g-Dex). 1H-NMR spectra of PLL-g-Dex in D2O. III. Gel electrophoretic analysis of dimerization of DIS25 with various N/P ratios. IV. The effect of polyelectrolyte on the fluorescence polarity of TAMRA-labeled duplex. V. UV absorption/Tm profiles of DIS25. VI. Arrhenius plots for spontaneous dissociation of the DIS25 dimer and PLL-g-Dex-assisted dimerization of DIS25.VII. Switching between double stem–loop DIS42 and extended multiplex drived by PLL-g-Dex and PVS. See DOI: 10.1039/c5nr05193b |
| ‡ J. C. W. and F. Y. contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2016 |