
Prospecting for new bacterial metabolites: a glossary of approaches for inducing, activating and upregulating the biosynthesis of bacterial cryptic or silent natural products
Joseph Scott
Zarins-Tutt
a,
Tania Triscari
Barberi
a,
Hong
Gao
a,
Andrew
Mearns-Spragg
b,
Lixin
Zhang
c,
David J.
Newman
d and
Rebecca Jane Miriam
Goss
*a
aSchool of Chemistry, University of St Andrews, St Andrews, KY16 9ST Scotland, UK. E-mail: rjmg@st-andrews.ac.uk; Fax: +44 (0)1334 463 808; Tel: +44 (0)1334 463 856
bAquapharm Biodiscovery Ltd., European Centre for Marine Biotechnology, Dunstaffnage, Oban, Argyll, PA37 1QA UK. E-mail: andrew@jellagen.co.uk
cCAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, 100101, China
dFrederick National Laboratories for Cancer Research, Natural Products Branch, Frederick, MD 21702, USA
First published on 5th November 2015
Abstract
Covering: up to 2015
Over the centuries, microbial secondary metabolites have played a central role in the treatment of human diseases and have revolutionised the pharmaceutical industry. With the increasing number of sequenced microbial genomes revealing a plethora of novel biosynthetic genes, natural product drug discovery is entering an exciting second golden age. Here, we provide a concise overview as an introductory guide to the main methods employed to unlock or up-regulate these so called ‘cryptic’, ‘silent’ and ‘orphan’ gene clusters, and increase the production of the encoded natural product. With a predominant focus on bacterial natural products we will discuss the importance of the bioinformatics approach for genome mining, the use of first different and simple culturing techniques and then the application of genetic engineering to unlock the microbial treasure trove.
1 Introduction
For millennia, natural products have been the cornerstone of medicine: for example, the ancient Egyptians utilised the antibiotic properties of honey to aid wound healing.1 It was not until the beginning of the last century, however, that our knowledge and understanding of science and nature enabled us to look at these health conferring natural products at a molecular level and identify the bioactive compounds that reside within them. The epiphany of modern natural product chemistry is famously exemplified by Alexander Fleming's seminal discovery that Penicillium rubens was capable of producing a substance with antibiotic properties.2 The structure of this bioactive compound, penicillin (1), was theorised by Ernst Boris Chain3 and later confirmed by Dorothy Hodgkin.4 This notable observation resulted in an intense period of pursuit of bioactive microbial secondary metabolites. As a result, a wealth of medicinally-relevant compounds have been revealed with many notable examples including the antifungal agent nystatin (2), the antibiotics erythromycin (3) and vancomycin (4), the cholesterol lowering drug lovastatin (5) and the immunosuppressant rapamycin (6) (Fig. 1).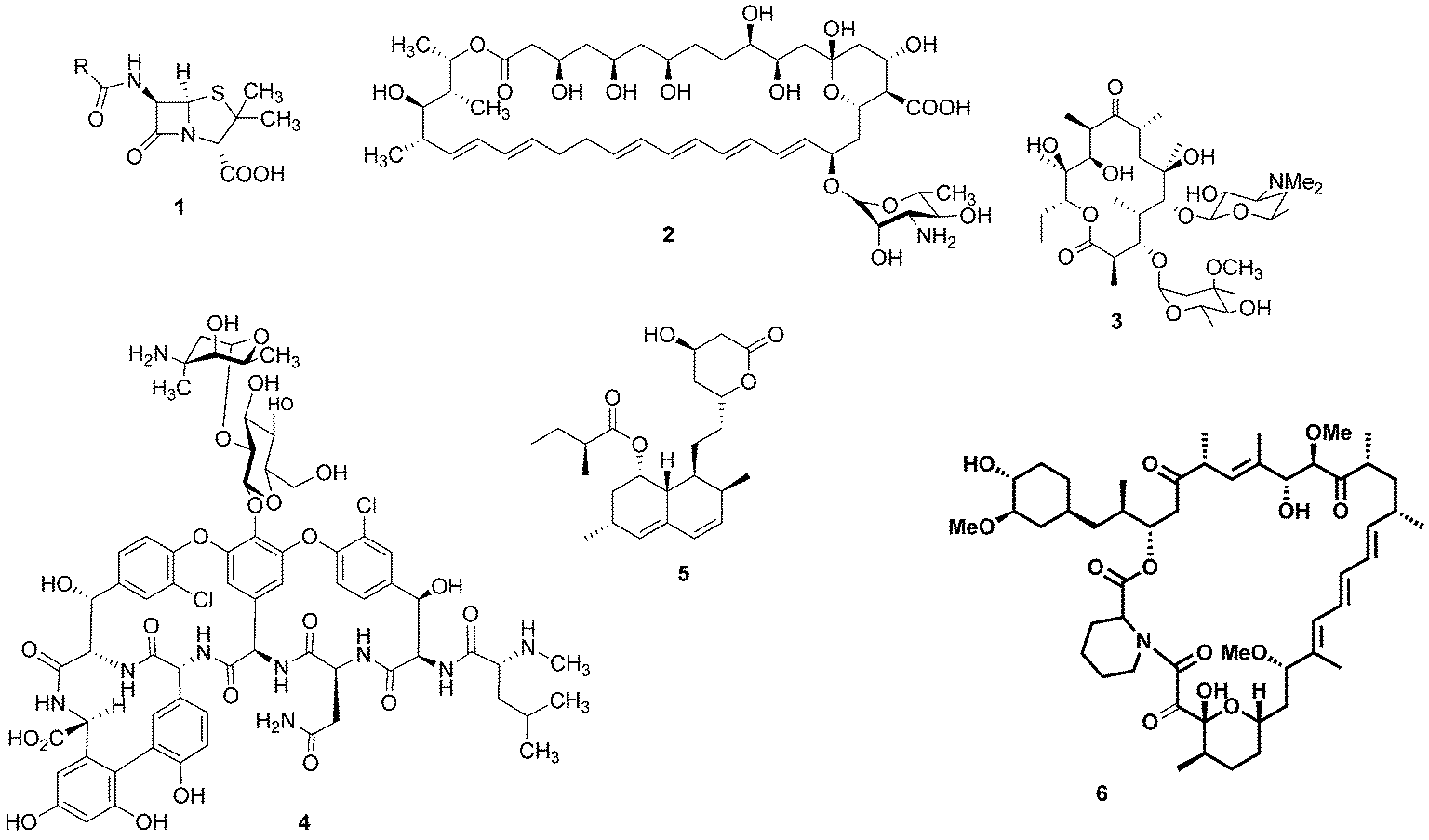 | ||
| Fig. 1 Penicillin (1), nystatin (2), erythromycin (3), vancomycin (4), lovastatin (5) and rapamycin (6). | ||
Despite the proven success of natural products as drugs, many pharmaceutical companies abandoned their natural product drug discovery programmes due in part to high rates of re-discovery of known compounds and the difficulty of accessing synthetic analogues. They favoured instead the generation of large libraries of synthetic compounds that were more compatible with high-throughput screening.5 Bacterial-based natural product discovery programmes have traditionally employed the fermentation of such microbes in a series of media and then the screening of extracts for any promising bioactives that are produced. However, as we moved into the 21st century, the advent of genomic sequencing has initiated a second golden age of drug discovery as gene clusters encoding the assembly of previously undiscovered natural products could be mined out from the massive number of published genomes. Though considerable research has been devoted to unlocking cryptic pathways in fungi, as overviewed in a number of reviews,6–11 the unlocking of bacterial pathways is still in its infancy and represents an exciting opportunity. A growing number of different approaches, with the aim of providing a more global solution to unlocking bacterial cryptic pathways, have been published in recent years, including studies with a focus on unlocking the treasures of the megasynthases through increasing transcription levels and “reviving dead” genes.74,76 Streptomycetaceae are known to be a prolific source of bioactive compounds: so far over 200 drugs based on products isolated from these microorganisms are in clinical trials or FDA approved12 and have been used for treating infections, cancer circulation and immune system disorders and many other disease states. When the genome sequence of the archetypal streptomycetaceae Streptomyces coelicolor A3(2) was published in 2002,13 the sequence data revealed, along with the known secondary metabolite clusters, a plethora of genes encoding for as yet unobserved molecules, including nonribosomal peptides and polyketides.13,14 Secondary metabolite clusters that produce no noticeable quantities of metabolite have been termed “silent” and it is unknown what conditions trigger their biosynthesis; further, there are orphan clusters, with as yet undiscovered products.15 The emerging area of triggering the production of cryptic metabolites in microorganisms was overviewed by Hertweck in 2009.16 With more than 1800 bacterial genomes now sequenced17 there is the potential for the discovery of new structural classes by simply reading the genome, then selectively pursuing these previously uninvestigated compounds. Using this approach, new drug leads with novel modes of action may be revealed.
This highlight aims to provide an easy to utilise glossary of approaches that can be applied by natural product scientists to elicit the production of cryptic natural products and thereby enable their purification and identification. Although there is substantial overlap and complementarity between approaches, we group such methods into two main categories, ‘physiological triggers’ and ‘metabolic engineering’ via genetic manipulation. We start by looking at the prediction of cryptic gene clusters by in silico genome mining. Then we examine the traditional approaches of OSMAC (One Strain Many Compounds), where different culturing conditions are used to trigger and optimise compound production; co-culturing is then explored before looking at employment of chemical elicitors. Leading on from that, the genetic methods for unlocking or upregulating biosynthetic pathways, including promoter and regulator manipulation, heterologous expression and epigenetic modification will be discussed.
2 Reading the genome
The massive acceleration in the development of sequencing technologies over the past decade has resulted in easy and affordable access to bacterial genomes. Streptomycetaceae, a large family of soil dwelling bacteria, are responsible for producing over 80% of all known antibiotics.18 The first Streptomyces genome (that of S. coelicolor A3(2)) was published in 200213 and the availability of its complete annotated sequence allowed the prediction and characterization of pathways involved in the production of at least 17 chemically distinct classes of bioactive compounds as reviewed by Challis.19 Completely assembled and annotated genome sequences for over 65 streptomycetes and hundreds of bacterial species demonstrated that more than 50% are likely to have one or more gene cluster involved in the biosynthesis of polyketides or non-ribosomal peptides.20 Additionally, a recent review by Hertweck highlights the biosynthetic potential showcased in the sequence data of anaerobic microbes.21 Across bacteria, there clearly exists a huge treasure trove of as yet undiscovered medicines that remains to be unlocked. The rise of metagenomic sequencing projects has further expanded the dataset. Considering the vast number of data available for mining, advanced gene analysis tools such as antiSMASH,22 CLUSTSCAN,23 SBSPKS24 and NP searcher25 have made genome reading more accessible to increasing numbers of researchers with an interest in natural products science. Frequently in microbial systems, genes encoding a particular biosynthetic pathway cluster together oftentimes with specific regulatory elements and genes encoding resistance mechanisms present in the same cluster. Nonetheless, it is worth to consider genes that cluster in an unusual way as they can suggest novel structural types of natural products.20 Much about the structure of the product encoded by the biosynthetic cluster (cryptic or otherwise) may be deduced by reading the genome (Fig. 2). By comparing the genes of interest to characterized homologues it is often possible to accurately predict the biogenesis of the product, for example whether it contains a polyketide component and what the length, oxidation state and dehydration level of the nascent polyketide chain is. Furthermore, it is often possible to speculate upon substrate specificity and stereoselectivity, particularly for non-ribosomal peptide synthetases.26,27 | ||
| Fig. 2 Genome mining. gDNA is sequenced and read (1). The raw sequence data is annotated based upon sequence homology to existing data, identifying ORF (2). Clustered genes can be extracted and identified from the genome annotation and analysed further by specialised bioinformatics software (3) which allows prediction of substrate (4). The often linear nature of their assembly then allows for prediction of the final chemical structure (5). If the structure is accurately predicted, substrate specificity may be deduced enabling informed media preparation and fermentation (6). | ||
In 1999, Stachelhaus et al. demonstrated a correlation between the essential amino acid residues in adenylation domains of non-ribosomal peptide synthetases (NRPSs) and substrate specificity.28 Almost at the same time, Challis et al. established a method for predicting the substrate specificity of adenylation domains, responsible for selecting and activating specific amino acids based on the genome sequence of NRPSs.29 Polyketide and non-ribosomal peptide natural products, particularly those of canonical construction, provide the best exemplifications of prediction of these biosynthetic systems. The modular nature of higher polyketide synthases (PKSs) and NRPSs combined with the current understanding of their substrate specificity allows for the logical deduction as to which chemical building blocks are incorporated and in which order. Enzymatic tailoring of the assembly line, such as glycosylation methylation and oxidation, may also be postulated from the genome sequence. Analysis of the genome of the strict anaerobe, Clostridium beijerinckii, (NCIMB 8052, JGI project ID 3634512), used extensively in the industrial manufacture of solvents including ethanol, acetone and butanol indicated the presence of a series of biosynthetic clusters although no secondary metabolites had been reported from this organism. Hertweck and coworkers sought to investigate this further, and as the industrially utilised strain, whose genome sequence they had analysed, was not readily obtainable, instead they investigated a related C. beijerinckii strain (HKI0724) from the HKI strain collection. In contrast to other C. beijerinckii strains reported in the literature, fermentation of this organism resulted in a purple-red broth. Isolation and characterisation of the pigment revealed it to be a potent polyphenolic antibiotic of polyketide origin, but with a non-canonical folding pattern. This first polyketide to be reported to be derived from an anaerobic bacterium has been named clostrubin (7) (Fig. 3).30
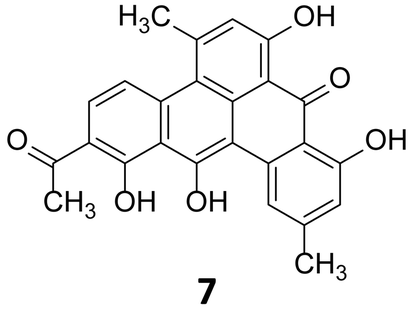 | ||
| Fig. 3 Structure of clostrubin (7).30 | ||
Recent new advances have been pioneered by Nguyen and colleagues who have developed approaches to link LC-MS/MS metabolomic profiles with gene clusters responsible for the production of secondary metabolites;31 the advantage of this approach is that it can be extended to organisms with unsequenced genomes based on the fact that “gene cluster families” involved in natural products biosynthesis can be directly linked to known “molecular families” by simply reading the mass spectral fragmentation pattern. The in silico prediction of metabolites and gene clusters responsible for their production represents a very powerful tool for the identification of pathways encoding secondary metabolites and can enable the chemist to prioritize and focus resource toward the isolation and identification of novel natural products.
3 Altering of chemical and physical conditions
3.1 OSMAC
Varying culturing conditions is a frequently used approach to elevate titres of compound production and to encourage the production of a wider range of the natural products from a microorganism. The approach termed OSMAC (One Strain Many Compounds)32 by Bode and co-workers (and initially explored by Hans Zahner),33 refers to the fact that a single bacterial strain is capable of producing a diverse array of structurally different compounds. A microorganism never produces the entire arsenal of compounds at the same time under one set of environmental conditions, this would be energetically and metabolically costly; rather, the biosynthesis of such metabolites is strictly controlled and depends on various internal and external cues. Each compound is produced only when needed, ensuring a competitive advantage when environmental conditions change. Consequently, by varying culturing conditions, it can be possible to induce or optimise the production of secondary metabolites. There have been many successful examples of the application of the OSMAC approach to enable the production of previously unknown natural products from microbial strains. Using such an approach, Knappe et al. found a new lasso peptide, capistruin (8), from the Burkholderia thailandensis E264 strain (Fig. 4).34 Initially, culturing in M9 minimal medium at 37 °C, trace amounts of the compound were observed. Different culturing conditions were explored and employed demonstrating that the production of capistruin could be increased approximately 300-fold when culturing in M20 medium at 42 °C. In 2011, Rateb et al. explored the potential of natural products biosynthesis of the Streptomyces strain C34 isolated from a hyper-arid desert soil.35 When C34 was cultured in ISP2 medium, two novel ansamycin compounds, chaxamycins A (9) and B (10), were found; however when the glucose in ISP2 was replaced with glycerol, two other novel compounds, chaxamycins C (11) and D (12) were produced instead. From 3 L culture broth, 75 mg chaxamycin A, 28 mg chaxamycin B, 2 mg chaxamycin C and 7 mg chaxamycin D could be purified. Following on from these results, they explored eight further media obtaining a series of diverse metabolic profiles. Three new compounds from the rare class of 22-membered macrolactone polyketides, chaxalactins A–C (13–15) were detected from a defined medium which also contained glycerol as the main carbon source, and 19 mg (chaxalactin A), 7 mg (chaxalactin B) and 11 mg (chaxalactin C) pure compounds were obtained from a 3 L culture (Fig. 5).36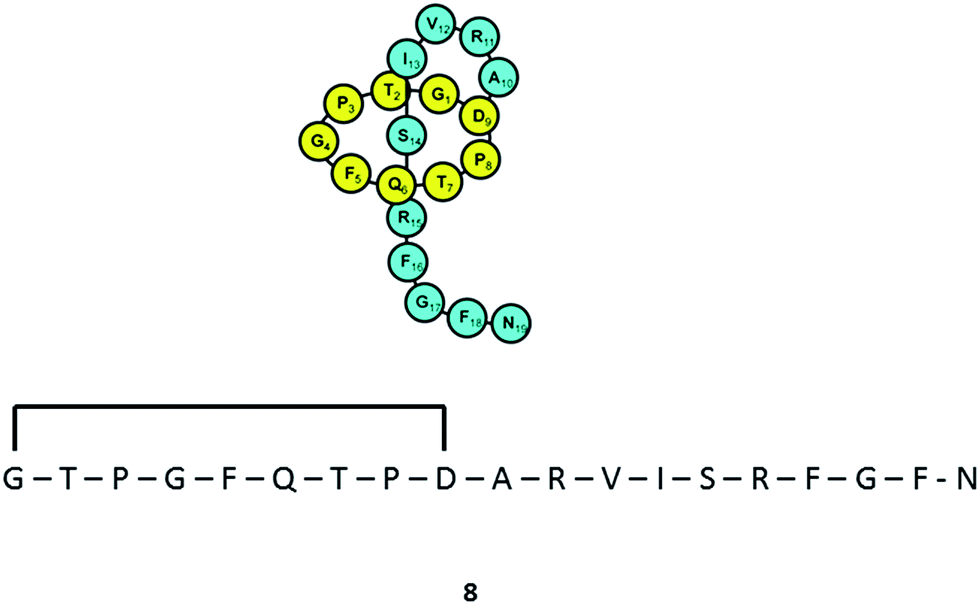 | ||
| Fig. 4 Lasso peptide, capistruin (8). | ||
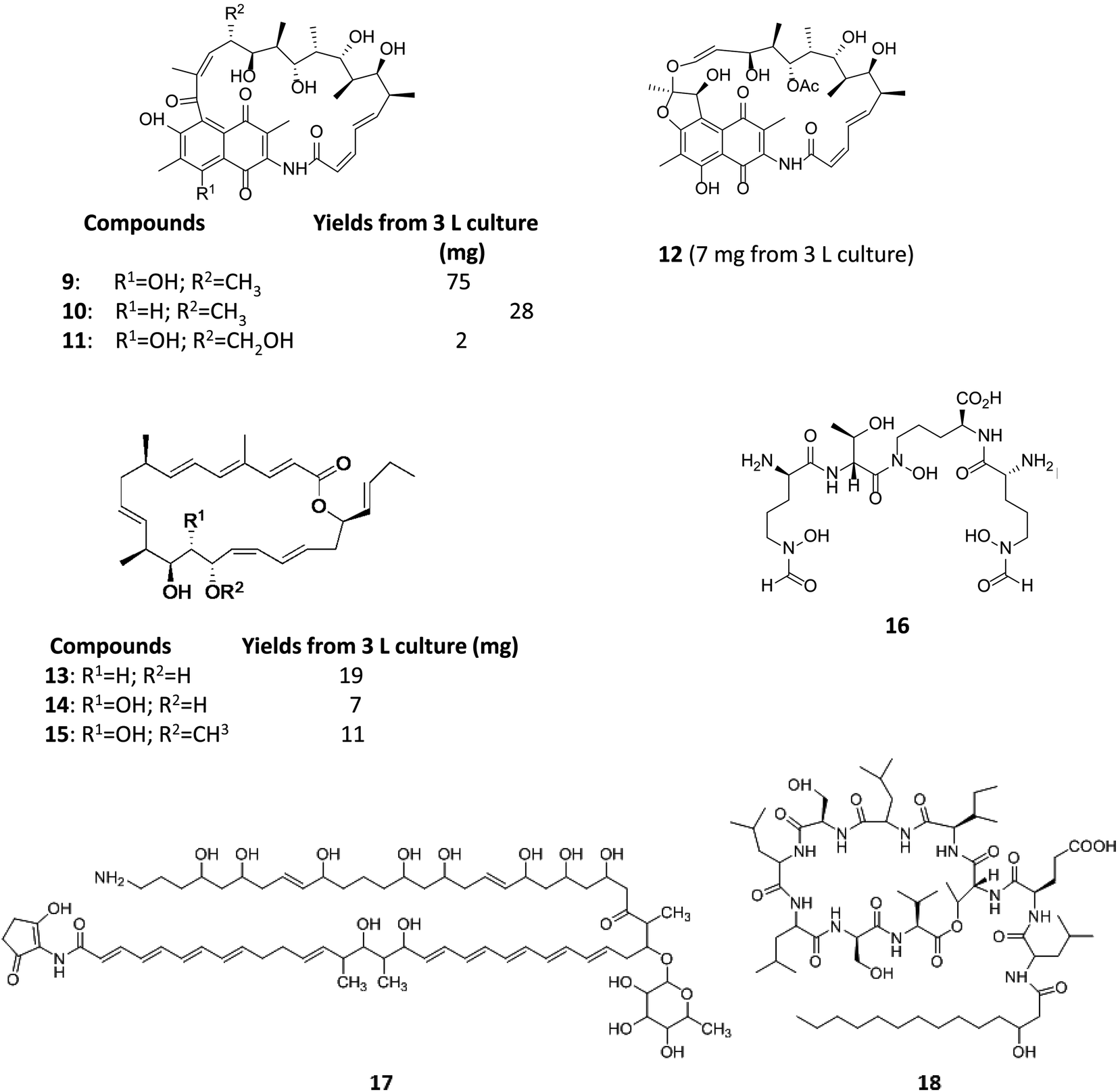 | ||
| Fig. 5 Chaxamycins A, B, C, D (9–12)35 chaxalactins A–C (13–15)36 coelichelin (16),37 ECO-02301 (17)39 and orfamide A (18).40 | ||
The OSMAC approach is not targeted to the activation of a specific cryptic gene cluster, but to the systematic alteration of readily accessible culturing parameters including medium components (salts, amino acids, carbon source), pH, culture aeration (including the type of culture vessel used) and temperature of growth. This makes the OSMAC approach an accessible, versatile, inexpensive and relatively simple tool to use. Variations in the entire metabolic profile can be observed when a chemical component is added/depleted or a physical parameter changed within the culturing system. In a similar vein, increasing precursor/substrate supply can have a profound impact upon production levels. The examples that we describe in this paragraph demonstrate that different carbon sources, nutrient availability and growth temperature are capable of influencing the onset of secondary metabolites production in bacteria.
3.2 Genome-informed culturing to coerce the production of selected natural products
Insights into the structural and physico-chemical features of novel cryptic secondary metabolites can lead to the informed exploration of specific culturing conditions in order to enable production of compounds predicted through genome reading. Genome scanning or sequencing followed by in silico gene mining is a powerful tool for the identification of genes encoding enzymes involved in the biosynthesis of natural product. Genome analysis enables canonical biosynthetic assembly lines to be read and their products to be postulated with a degree of confidence. By reading the genome of S. coelicolor Challis and Ravel were able to propose a tripeptide product for one of the 14 remaining cryptic secondary metabolite clusters of S. coelicolor.37,38 Their postulation as to the structure of the natural product led them to speculate that the compound might be an iron siderophore. Based on this hypothesis, an appropriate growth medium depleted in iron, was rationally selected to induce the compound's biosynthesis. This informed approach enabled sufficiently enhanced levels of production of coelichelin (16) for the compound to be isolated and characterised. The final structure was, however, found to be a tetrapeptide, illustrating existing flaws in the prediction of compound structure from sequence data when the biosynthetic machinery is used iteratively or skips modules.37 By analysing the genome of Streptomyces aizunensis NRRL B-11277, McAlpine et al. were able to identify 11 gene clusters encoding natural products. Detailed bioinformatics analysis of one of these clusters enabled prediction that the strain could be capable of the production of a linear polyene polyketide with a predicted mass of approximately 1297 Da; culturing media were screened and optimised for the production of this particular polyene (now known to be antifungal agent ECO-02301 (17)), and an understanding of the anticipated lipophilicity of the predicted molecule informed steps toward its isolation.39A similar approach may be employed with NRPs; as discussed above the amino acid composition of the compound can be predicted from the nucleotide sequence of the cluster encoding for the NRPSs.28,29 Using this approach, Gross et al. were able to predict and isolate orfamide A (18) from Pseudomonas fluorescens Pf-5.40 Amino acidic sequence alignment showed that the compound was related to lipopeptides of the amphisin and viscosin classes, in particular with a side chain connected to a leucine residue (Leu1). Based on the Gerwick's genomisotopic approach, a tool developed to assist genome mining, the labeled amino acid predicted as precursor was used thus enabling the identification and isolation of orfamide A (18) (Fig. 5).40
3.3 Co-culturing
Co-culturing provides a further straight-forward yet effective approach to elicit natural product generation by simply growing a microorganism together with one or more additional microbial strains. Interspecies cross talk can often trigger the production of specific bioactive compounds for defensive purposes (Fig. 6). In nature, bacteria live in complex, multi-species communities; unleashing the full biosynthetic potential of a bacterium in the laboratory is likely to rely upon mimicking the dynamic natural environment. There are many published examples in which a specific natural product's generation has been induced or increased with the aid of co-culturing techniques. For example a series of co-culturing experiments with 16 marine-derived bacterial strains, each co-cultured with one of the three terrestrial challengers, Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa, was reported by Mearns-Spragg et al.41 The challenger strain was added to the fermentation, constrained within dialysis tubing, allowing interspecies cross talk through the diffusion of small molecules. In these experiments, 12 of the 16 marine derived strains demonstrated enhanced antibiotic activity against either E. coli, S. aureus or P. aeruginosa, with some of the most striking results arising from co-culture with S. aureus 6871. Six of the marine derived strains, co-cultured with this challenger, produced supernatants with increased antibiotic activity. One marine strain (Mbbc1122) showed not only increased levels of activity, but also increased spectral activity. The supernatant of Mbbc1122 co-cultured with S. aureus 6871 was active against both the Gram-negative E. coli and the Gram-positive S. aureus whereas the control was only active against E. coli. This indicates that the co-culturing approach positively impacted upon both natural product titre as well as chemical diversity produced by the strain. The study also demonstrated that in many cases a living challenger was not required, with heat killed cells being as effective in eliciting antibiotic production. A later study also demonstrated that the supernatant of an unspecified bacterial fermentation could be used to induce antibiotic production in a number of marine derived microbes.42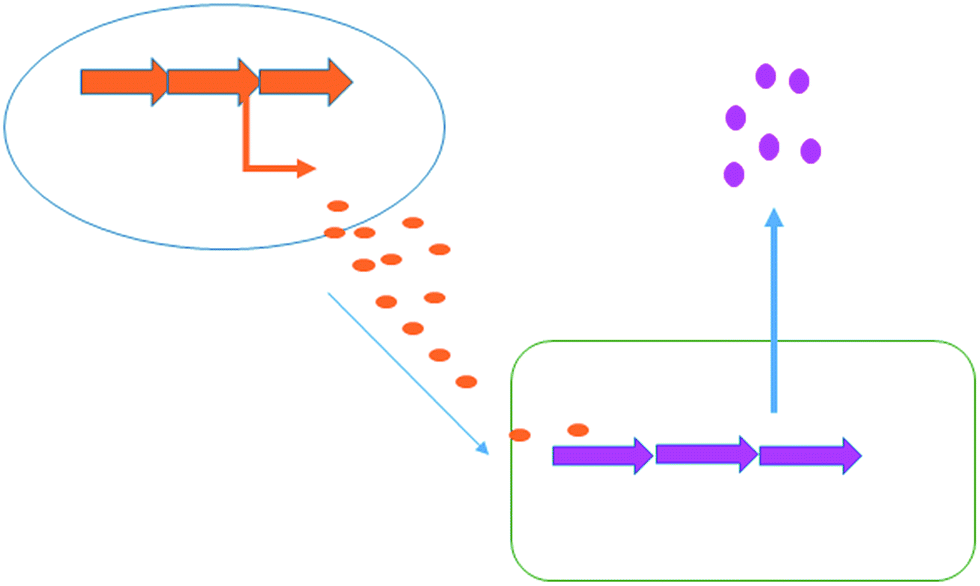 | ||
| Fig. 6 Co-culturing. Chemical signals produced by one bacterium (orange) are capable of inducing the biosynthesis of chemicals in a second bacterium (pink). | ||
Bacterial co-culturing has enabled the production and discovery of a number of new biologically active compounds. In 2001, growth of an unidentified Gram-negative bacterium CNJ-328 with a marine derived fungus, belonging to the Pestalotia spp., yielded a new chlorinated benzophenone, pestalone (19, Fig. 7).43 In 2003, co-culture of marine microbes C-148 and CF-20 with Vibrio anguillarum resulted in the production of a number of diketopiperazine (20) natural products with antibiotic properties, four of which had not been previously identified in nature.44Streptomyces endus S-522 is another example of a strain that, when cultured in the presence of the mycolic acid producer Tsukamurella pulmonis, was found to generate the novel secondary metabolite alchivemycin A (21).45 In the same study, T. pulmonis was also shown to induce red pigment production in Streptomyces lividans.45
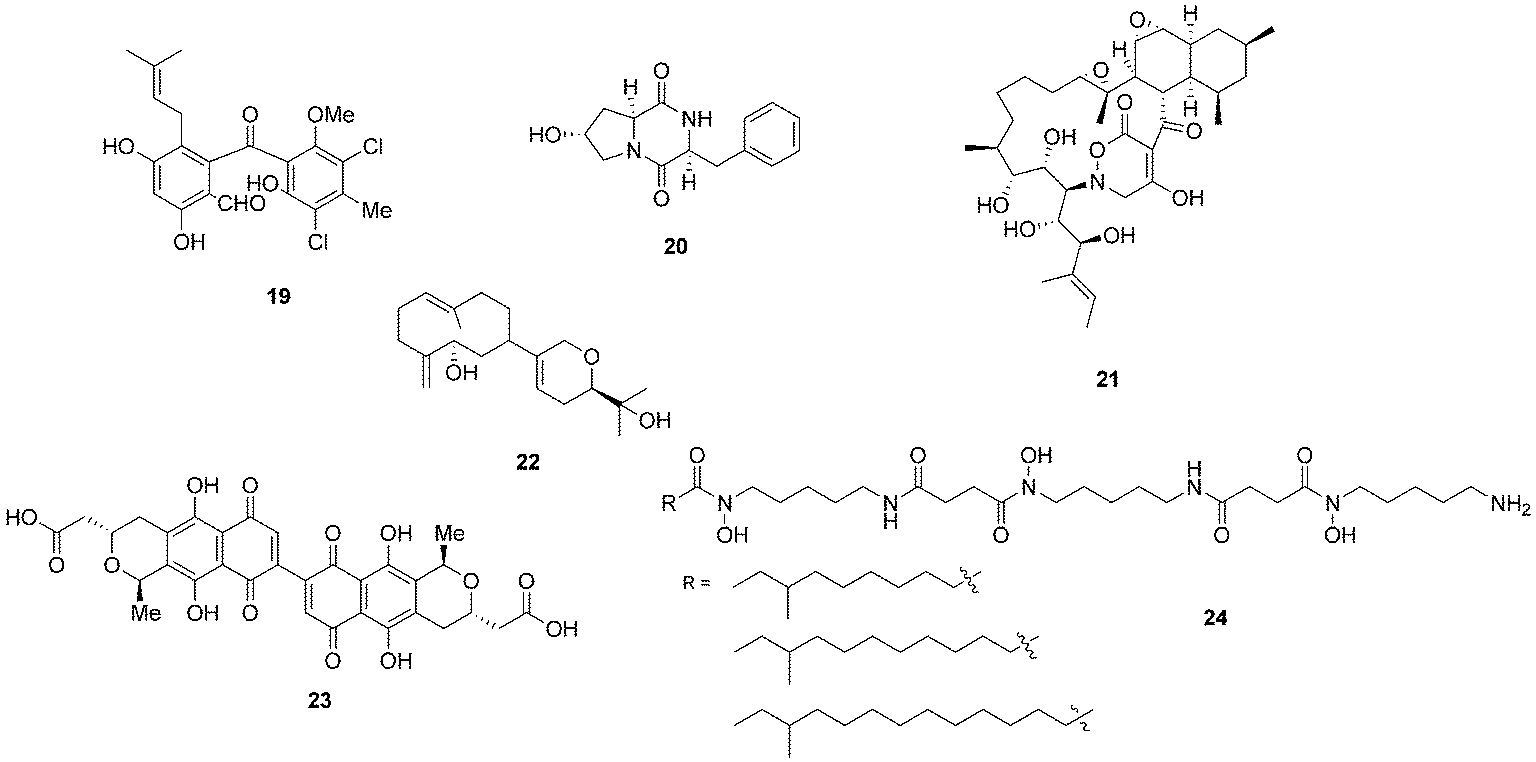 | ||
| Fig. 7 Pestalone (19),43 diketopiperazine (20),44 alchivemycin A (21),45 lobocompactol (22),46 actinorhodin (23)47 and acyl-desferrioxamines (24).48 | ||
Co-culturing provides a useful approach not only for eliciting the production of novel natural products so that they may be assigned to orphan gene clusters, but it can also be used for the induction of silent natural products the production titres of which may be too low to detect. The production of the diterpene lobocompactol (22), produced by Streptomyces cinnabarinus PK209, was investigated in co-culture with Alteromonas sp. KNS-16, a lobocompactol-resistant strain,46 and it was found that the production titres of this compound were increased 10-fold. The authors demonstrated that the amount of challenger inoculum is an important factor to consider when performing co-culture experiments. Although all levels of challenger inoculum resulted in production, it was recognised that an optimum inoculum strength existed for the production of lobocompactol (22) with an inoculum of 105 cells of challenger resulting in peak production levels (25% greater than when a 106 inoculum was used, and approximately 300% greater than 104 inoculum was used).46
The much studied model actinomycete S. coelicolor has also been subjected to co-culturing studies, and it has been demonstrated that Myxococcus xanthus induces the overproduction of actinorhodin (23) by 20-fold.47 In elegant research on S. coelicolor by Dorrestein and coworkers, employing nanospray desorption electrospray ionisation (NanoDESI) and matrix-assisted desorption ionisation time of flight (MALDI-TOF) imaging spectroscopy, the researchers were able to capture a global overview of the chemical cross talk of this model organism as it interacted with five other actinomycetes.48 The five different interactions revealed 227 compounds associated with S. coelicolor, with only 50% of these being known S. coelicolor metabolites. The newly associated S. coelicolor compounds included a number of novel acyl-desferrioxamines (24) which had previously not been observed. Strikingly it was demonstrated that there was huge variation in the portfolio of compounds produced by S. coelicolor as it interacted with the different actinomycetes, demonstrating that such responses are extremely specific. The revelation of the diversity of the chemical landscape, the incredible dynamics of these systems and the rich series of new compounds indicates that interspecies cross-talk and co-culturing will be a very valuable avenue in the search for new natural products.
3.4 HDAC inhibitors
Histone deacetylases (HDACs) are a large and ubiquitous family of proteins that play an essential role in modulating gene expression through deacetylation of histone proteins.49 Found in all domains of life,50 HDACs act to make regulatory genetic elements more or less accessible through the deacetylation of histone proteins which pack the DNA. Through modifying accessibility to the gene, gene transcription is controlled.49 Consequently, HDAC inhibitors can have a stimulatory or inhibitory effect on the expression of the corresponding genes.51 There have been many successful examples of the use of HDAC inhibitors to awake cryptic biosynthetic clusters in fungi.52,53 Cichewicz reviews this area of fungal epigenome manipulation, in which he is a key player.54 Following this pioneering work in fungi, researchers have recently started to apply the use of HDAC inhibitors to bacteria.49 Looking solely at the effect of HDAC inhibitors on the production of actinorhodin (ACT, 23) and the RED pigment (acknowledged elsewhere to comprise of both undecylprodigiosin (25) and streptorubin B (26)49b), Moore et al. screened a range of HDAC inhibitors to identify a number of compounds which elicit production of either one or both of these RED or ACT pigments.49 Elicitors included sodium butyrate (27), valproic acid (28), apicidin (29) and quercetin (30) (Table 1). The use of sodium butyrate as a chemical elicitor was further validated when applied to Streptomyces KY5 cultures, leading to a significant increase of antifungal activity (Fig. 8 and 9).49| HDAC inhibitor | Effective concentration | Stock solution | Effects |
|---|---|---|---|
| Sodium butyrate (27) | 25 mM | 100 mM in water | Strong induction of ACT (23), slight induction of the RED pigment |
| Valproic acid (28) | 1 mM | 10 mM in DMSO | Strong induction of ACT (23) |
| Apicidin (29) | 1 mM | 1 mg mL−1 in 50% DMSO | Strong induction of ACT (23) |
| Quercetin (30) | 10 mM | 100 mM in 5% DMSO | Slight induction of ACT (23) |
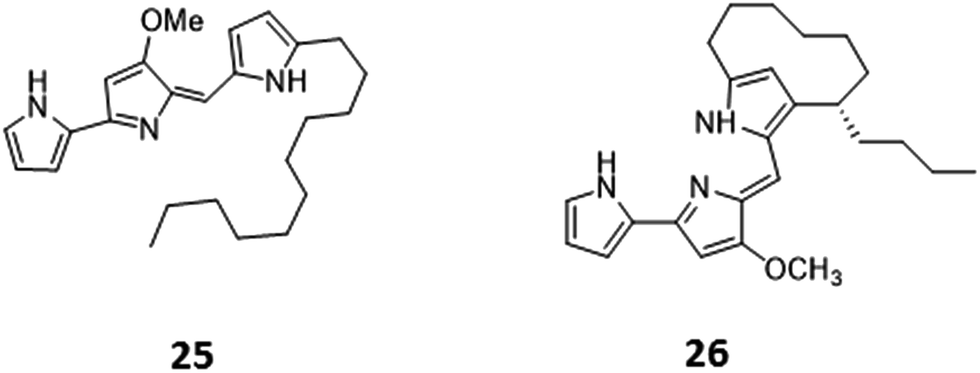 | ||
| Fig. 8 Undecylprodigiosin (25) and streptorubin B (26). | ||
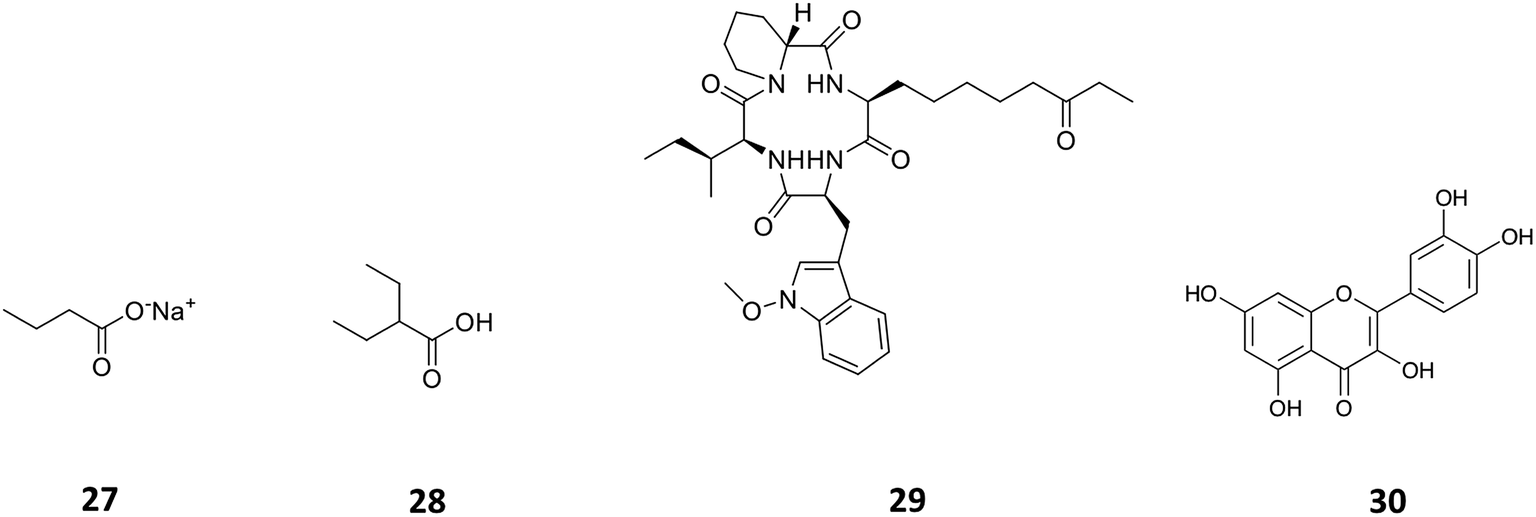 | ||
| Fig. 9 The HDAC inhibitors, sodium butyrate (27), valproic acid (28), apicidin (29), quercetin (30). | ||
From the few published examples in the use of HDAC inhibitors in bacterial systems, they been shown to improve or reduce the level of production of known natural products and their potential to be used for revealing cryptic natural products remains to be demonstrated. Although the exact mechanism of biosynthetic pathways induction or repression is not yet fully understood, the use of HDAC inhibitors provides a cheap and easy method to screen both bacteria and fungi for the production of secondary metabolites.
3.5 Chemical elicitors
Sub-inhibitory concentrations (SIC) of antibiotics have been shown to regulate gene expression at a transcriptional level, affecting 5–10% of all transcripts.55 This effect is observed with concentrations as low as 1% of MIC values.4,56,57 Transcriptomic studies using a combination of microarrays and reporter assays, have demonstrated that sub-inhibitory concentrations of antibiotics can activate quorum sensing regulators including those belonging to the LuxR family.55 Many phenotypic changes have been observed in the presence of sub-inhibitory concentrations of antibiotics, including the induction of biofilm formation and increased bacterial motility.58 SICs of antibiotics are being used as a tool to activate the biosynthesis of cryptic natural products. In a marine Streptomyces spp., a novel series of phenazines named streptophenazines (31, Fig. 10), differing in their alkyl chain, were produced in response to stimulation with SICs of either tetracycline (32) or bacitracin (33).59 Interestingly, it was found that whilst tetracycline had a positive effect on 5 of the 8 phenazines, elevating titres between 2.6 and 10.7-fold, it had a negative impact on streptophenazine biosynthesis (31), leading to a decrease in production to 80% of the original titre. Pulse feeding with bacitracin (33) at 0.1 μg mL−1 increased titres of streptophenazine (31) 2.2-fold.59 It is noteworthy that the same chemical modulator can have both a stimulatory and inhibitory effect on members of the same family of natural products, with the profile of streptophenazines produced dependent upon which antibiotic was being used to elicit their production.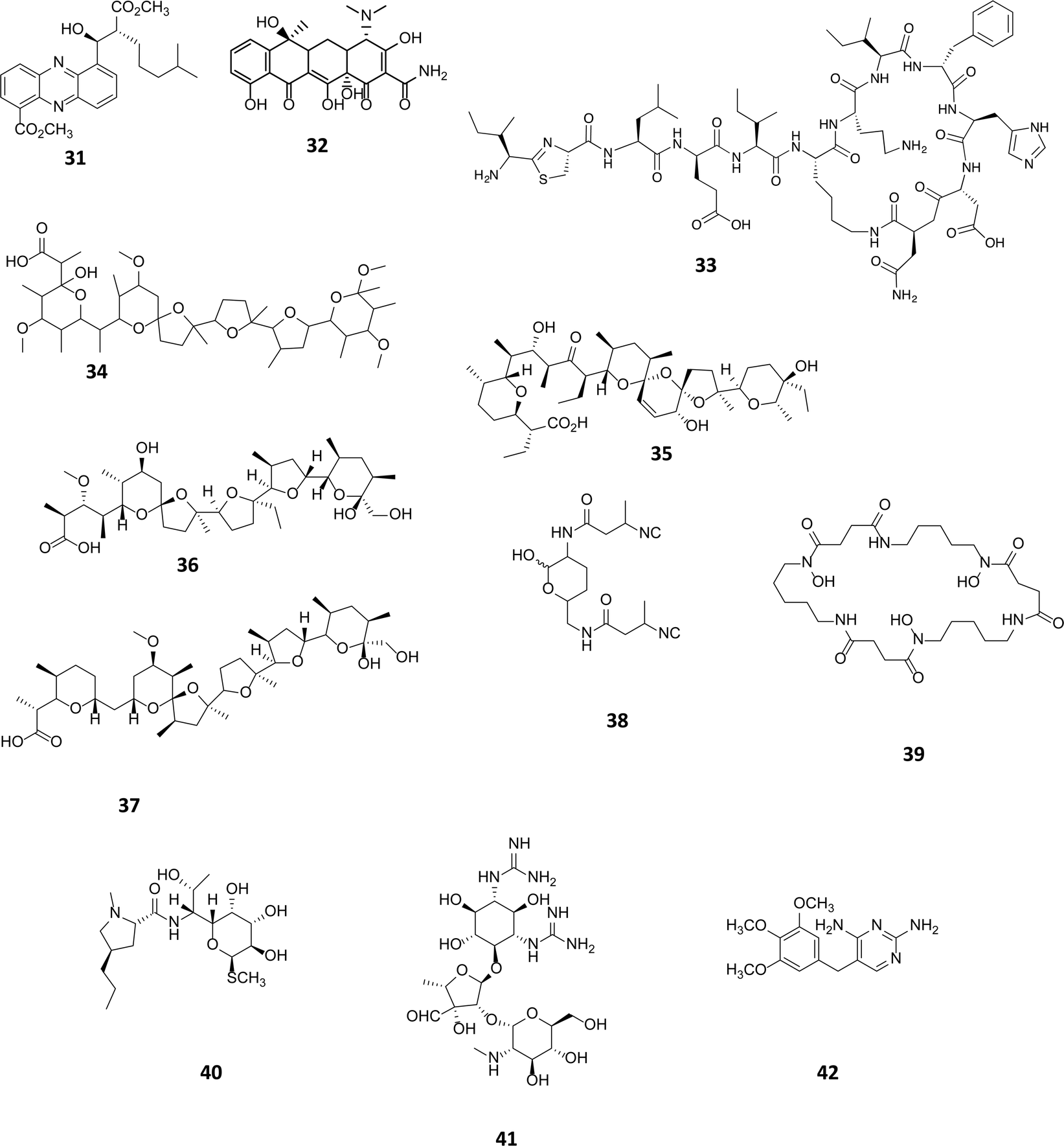 | ||
| Fig. 10 Streptophenazines (31),59 SF2768 (38),61 streptomycin (41).63 Sub-inhibitory concentration (SIC) stimulants tetracycline (32), bacitracin (33), promomycin (34), salinomycin (35), monensin (36), nigercin (37), desferrioxamine E (39), lincomycin (40) and trimethoprim (42). | ||
A range of polyether antibiotics have also been observed to induce antibiotic production in Streptomyces spp. Promomycin (34), a polyether natural product produced by Streptomyces scabrisporus strain 153, was found to induce antibiotic production in a number of environmental isolates when administered at SICs.60 Other polyethers, salinomycin (35), monensin (36) and nigercin (37) had similar effects, inducing antibiotic production in isolated strains when administered at SICs.60 Recently, one of the antibiotics whose production was induced by the administration of SICs of monensin (36) to the producing strain, Streptomyces griseorubiginosus strain 574, was identified as the isonitrile antibiotic SF2768 (38).61 Although not an antibiotic, the commercially available siderophore desferrioxamine E (39) produced by a number of Streptomyces spp., has been shown to induce and elevate the biosynthesis of some metabolites. The addition of 3 μg to a filter disc, creating a diffusion gradient adjacent to an environmental isolate, induced the production of an unknown yellow pigment and antibiotic.62 Treating cultures with lincomycin (40), which alters the transcriptional machinery, was seen to impact secondary metabolite production elevating streptomycin (41) production levels in Streptomyces griseus from 40–100 μg mL−1. Lincomycin binds to the ribosome of Gram-positive bacteria, and the authors postulate that although regulation of metabolite production in actinomycetes is highly complex, modification of the translational apparatus is likely to be key to inducing antibiotic production,63 further examples of such effects on secondary metabolite production are discussed in Section 4.1. In a medium throughput approach to looking for small molecule activators, using a lacZ fusion reporter assay, Seyedsayamdost explored the effect of sub-inhibitory concentrations of a library of 640 small molecules, many of which were clinically utilised antibiotics. Using the cryptic malleilactone (mal) cluster in Burkholderia thailandensis, a genetically tractable strain, as test bed malL essential to the biosynthesis of the metabolite was fused to lacZ and used as a reporter. In these studies it was demonstrated that low concentrations of the antibiotic trimethoprim (42) resulted in the induction of at least 5 biosynthetic gene clusters including mal. The mechanism of upregulation could be due to the elevated transcription of malR, an orphan luxR transcriptional regulator that could bind to small molecule elicitors or perhaps, as a series of synthetic as well as natural antibiotics elicited upregulation, a stress-induced mechanism could be operational.64
4 Genetic modification approaches
Genetic modification of bacterial strains represents an alternative approach to accessing novel biosynthetic pathways or improving the titres of known natural products. Approaches range from the technology light and non-specific induction of spontaneous mutations within the organism through the use of mutagenic compounds or radiation to informed and directed metabolic engineering.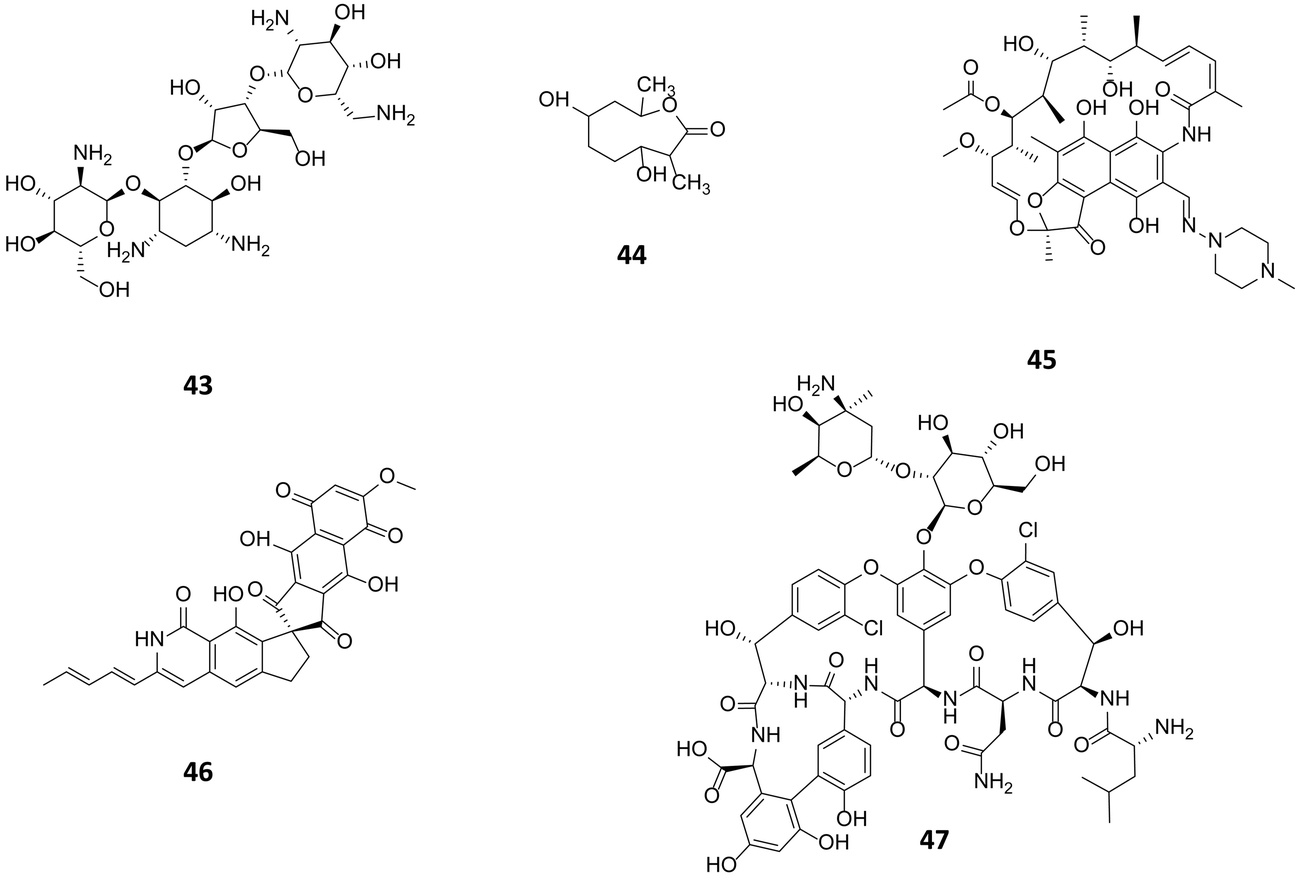 | ||
| Fig. 11 Paromomycin (43) used at high concentration resulted in a ribosomal mutation giving rise to substantial elevation of actinorhodin production in S. coelicolor. Following a similar approach actinolactomycin (44),68 and norvancomycin (47)69 are also produced in higher titre; and rifampicin (45) to unlock the production of fredericamycin (46) through the ribosomal mutation. | ||
4.1 Ribosome engineering and alteration in the transcriptional machinery
The concept of ribosome engineering in order to elevate or unlock metabolite production was introduced for the first time when Shima and coworkers observed that a S. lividans spontaneous mutant, resistant to streptomycin, was able to produce the antibiotic actinorhodin (23). This was significant as actinorhodin was not produced by the wild type strain. The observed mutation, a substitution of a lysine residue with a glutamate residue at position 88 in the ribosomal protein S12 responsible for streptomycin resistance, was found to alter translational accuracy (Fig. 12A).65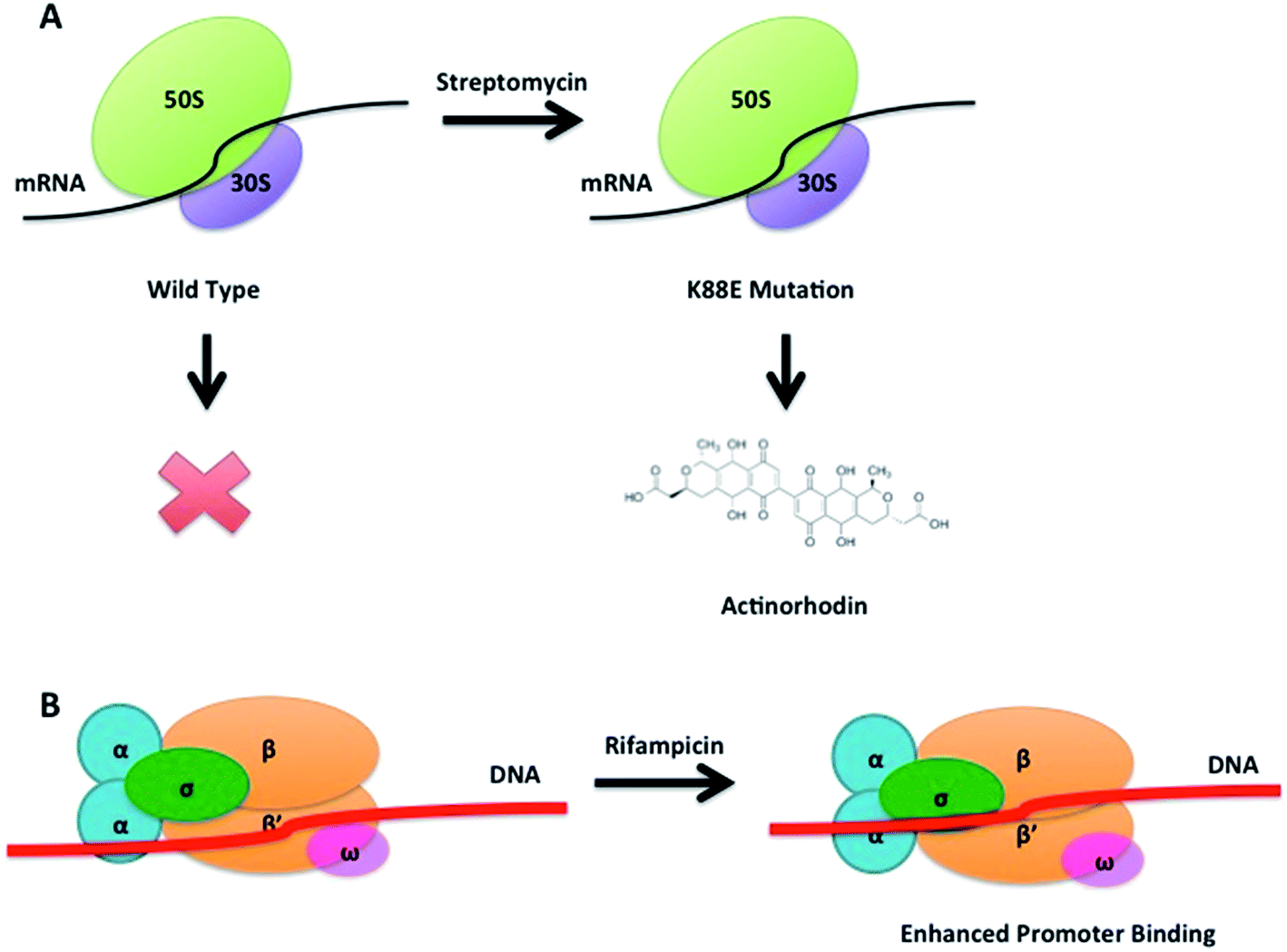 | ||
| Fig. 12 Antibiotic induced spontaneous K88E mutation in the 12S protein of the small ribosomal subunit leading to actinorhodin production in S. lividans (A). Rifampicin induced spontaneous mutations in the transcriptional machinery enhance the strength of promoter binding by the RNA polymerase (B). | ||
Multiple point mutations, affecting both transcriptional and translational machineries, may arise when bacteria are cultured on media containing different antibiotics. High concentrations of antibiotics must be used (depending on the bacterium tested, up to 100-fold of MIC values) in order to generate spontaneous mutants that overproduce a compound. The frequency of mutation in a microbial population is usually 10−6 to 10−8, therefore 109 or more cells must be plated in order to isolate mutant colonies after several days of incubation.66 These findings have been rationalised in order to select mutants with improved production of known compounds or to activate silent gene clusters encoding novel secondary metabolites. Utilising such an approach, a S. coelicolor strain known to be resistant to rifampicin, streptomycin and gentamicin, was grown in media containing up to eight different antibiotics: mutants were selected by screening for resistance. A 280-fold increase (1.63 g L−1) of actinorhodin (23) production was observed in a clone able to grow in the presence of all the tested antibiotics. Sequence analysis of this mutant showed the insertion of a glycine residue at position 92 of the ribosomal protein S12 conferring resistance to paromomycin (43, Fig. 11); however, mutations responsible for resistance to the other antibiotics were not identified.67
A similar approach was adopted in which an actinomycetes strain was cultured in media containing high concentrations of streptomycin. This not only resulted in the enhanced production of actinolactomycin (44), an anticancer agent, but additional peaks indicating the production of putative novel metabolites were observed by HPLC.68 Ochi has demonstrated, through generating rifampin resistant mutants of a series of actinomycetes, an up to 70-fold increase in the transcription level of biosynthetic gene clusters could be seen, with many of the mutants producing metabolites that could not be detected in the parent, wild-type strains. Rifampin resistance mutants were observed to develop at a frequency of 10−7 to 10−8; sequencing of these mutants showed that all mutations occurred in the rpoB gene encoding the RNAP β-subunit. The team were able to demonstrate that this strategy could be successfully applied to seven out of eight actinomycetes test bed strains.70 Similarly, by culturing the marine bacterium, Streptomyces somaliensis SCS10 ZHH with 300 μg mL−1 rifampicin (45), a mutation in the β subunit of RNA polymerase occurred unlocking the production of fredericamycin (46), a compound with potent toxicity. Using this approach followed by a response surface methodology (RSM) media optimisation, the group were able to go from cluster silence to achieving titres of 680 mg L−1.71 This strategy represents an approach to ribosome modification that does not require molecular biology, or the ability to transform the strain. Combinatorial mutations in ribosomal proteins and RNA polymerase can also increase secondary metabolite production through, for example, enhancing promoter binding (Fig. 12B): for example Amycolatopsis orientalis with mutations in both transcriptional and translational machinery showed a 46% increase of in the production of norvancomycin (47).69 These and many other examples illustrate the power of serendipitous and informed ribosome and RNA polymerase engineering in increasing titres of known bioactive compounds and for activating cryptic gene clusters for the isolation of novel compounds.72
4.2 Manipulation of pathway-specific and global regulators
Bacterial secondary metabolism gene clusters are generally transcribed from an inducible promoter under the control of regulators, activators and repressors; these modulate the expression of the gene cluster in response to environmental changes. Pathway-specific regulators act on the biosynthetic genes of their respective clusters whilst global regulators, are located outside the biosynthetic gene cluster and have a pleiotropic effect acting simultaneously on different metabolic pathways involved in various cellular functions. The overexpression of activators and silencing of repressors represent straightforward ways to enhance or unlock the expression of gene clusters of interest (Fig. 13). | ||
| Fig. 13 Simplified scheme of activator and repressor mediated regulation. | ||
Gene transcription is silenced by repressors bound to operator sites preventing the RNA polymerase from starting the transcription of the gene. By sequestering repressors or down-regulating their production, transcription can be activated. The directed transcription of gene clusters can also be achieved by up regulating their respective activators, which are able to recruit the RNA polymerase to their promoter site.
Synthetic modular regulatory elements with dynamic range of strength were successfully applied to activate and overproduce the cryptic lycopene in a predictable manner in Streptomyces avermitilis.72b Using the strategy of replacing the natural promoter of a series of gene clusters of interest with a strong inducible or constitutive promoter, by employing a cluster expression plasmid pCEP vector containing the first 300–600 bp of the biosynthetic cluster, enabling the introduction of the new promoter via homologous integration, the Bode group have reported a general tenfold increase in production of bacterial compounds including a series GameXPeptides, indigoidine.73 They have used this strategy to determine the previously unknown products of biosynthetic gene clusters. Through combining this approach with total synthesis and isotopic labelling strategies they have revealed and confirmed the product of the largest continuous bacterial NRPS discovered so far, consisting of 15 consecutive modules encoded by the 4.91 kol gene, and aptly named “Kolossin”.74 This formylated peptide is metabolically costly for the producing bacterium Photorhabdus luminescens to generate, and the natural mechanisms for activation of this pathway and the role of this compound remain to be determined.74 Salas employs a similar approach, to that utilised by Bode, of inserting strong constitutive promoters; in this way he explores five of the twenty seven gene clusters bioinformatically predicted in the genome of the common heterologous expression host, Streptomyces albus.75 Brady and co-workers also adopt the approach of refactoring series of promoters within the natural producing organism, developing replacement cassettes for the multiplexed exchange of constitutively active promoters in place of all native promoters within silent gene clusters. This approach, employing bidirectional promoter cassettes, benefits from using quick homologous recombination of the promoter region within a yeast auxotroph followed by its reengineering in to the natural producing strain. In such a manner the group used a series of single cassette insertions to replace each of the three natural bidirectional promoter regions involved in indolotryptoline biosynthesis with synthetic promoter cassettes, whilst at the same time repairing a base deletion in one of the genes that had rendered it non functional; the product of the previously dead cluster has been named lazarimide.76
In streptomycetes, overexpression of the pathway-specific regulators SARP (antibiotic regulatory protein family) has been shown to trigger the production of secondary metabolites. The pathway specific regulator ccaR, a member of the SARP family, is responsible for the regulation of clavulanic acid (48, Fig. 14) in Streptomyces clavuligerus.77 Overexpression of ccaR in a high copy number vector or integration in the chromosome of the strain, resulted in 2.25 and 9.4 fold increase of clavulanic production respectively.78 A further example, is the use of the consecutive overexpression of the three regulatory genes sgcR1, sgcR2 and sgcR3 for the production of enediyne C-1027 (49). These genes have been identified to belong to the AraC family of transcriptional regulators.79 Curiously, whilst their overexpression on an individual and consecutive basis resulted in increased C-1027 production, overexpression of these regulators in parallel had no noticeable effect on production, highlighting the complex interplay between regulators.
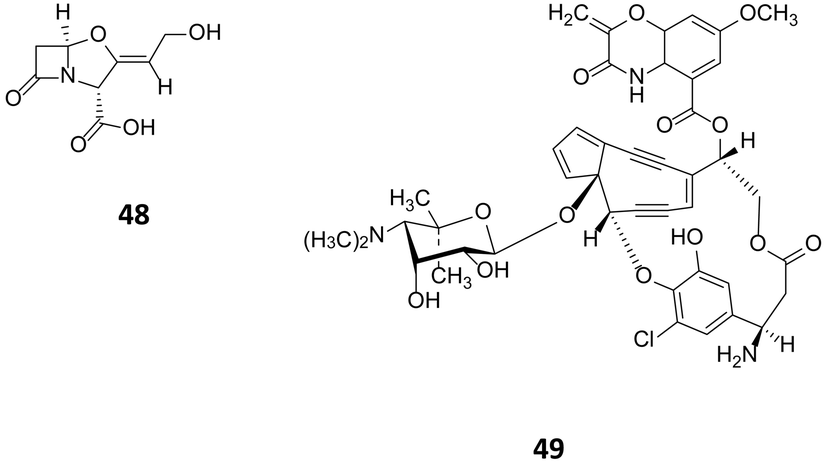 | ||
| Fig. 14 Clavulanic acid (48)77 accessed through over expression of the regulator cca and enediyne C-1027 (49)79 produced following the consecutive over expression of sgcR1, sgcR2 and sgcR3. | ||
Silencing transcriptional repressors provides another useful means for increasing production. A rather innovative and unusual method that has been successfully demonstrated to increase antibiotic production is the use of decoy oligonucleotides.80 McArthur and Bibb developed a series of double stranded oligonucleotides containing the regulatory element used by the repressor involved in the regulation of actinorhodin (23) biosynthesis. These oligonucleotides were engineered so that they formed stable structures able to compete for binding of the repressor protein with the regulatory element in the genomic DNA (Fig. 15). This technology has applicability as either a molecular switch to tune antibiotic production or as a tool with which to investigate antibiotic regulators.
In addition to pathway specific regulators, global regulators play an important role in the initiation of production of secondary metabolites since, when activated, they produce a pleiotropic effect ‘switching on’ or ‘off’ numerous metabolic pathways.81 Mutations in the dasR global regulator, belonging to the GntR family, was shown to enhance the production of antibiotics up to 4-fold in S. coelicolor;82 more interestingly, complete deletion of the dasR unlocked a cryptic gene cluster encoding a type I polyketide synthase not observed in the S. coelicolor wild-type strain.82 Despite the great potential for manipulating global regulators for the discovery of novel bioactive compounds, this approach is challenging as downstream events induced may affect vital cellular functions.
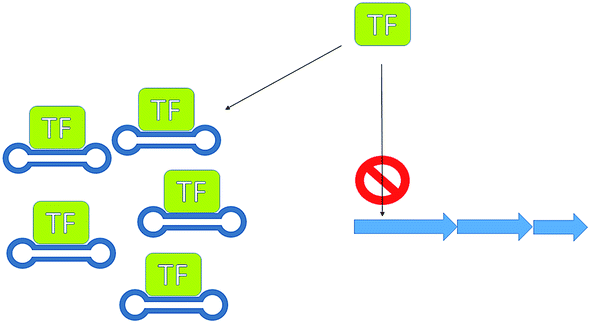 | ||
| Fig. 15 Transcription factor decoys. Using dumbbell oligo-nucleotides, to mimic genetic regulatory elements, thereby sequestering transcription factors, preventing them from binding to their regulatory element. | ||
4.3 Quorum sensing as a tool for the regulation of secondary metabolism
In the natural environment, bacteria live in multispecies communities and communication using signalling molecules is utilised in order to modulate activities of individual cells within the population. Such interaction, referred to as quorum sensing (QS), is population-density dependent since signal molecules need to reach a threshold level in order to trigger a concerted response.83 QS signal molecules are also known as “autoinducers” since they activate their own synthesis. Members of the LuxI/-type protein families are commonly distributed among Gram-negative bacteria. It has been well documented that P. aeruginosa utilises multiple lux-like QS systems expressed in a hierarchical manner and their activation triggers virulence factor and secondary metabolites production, such as pyrocyanin (50) and rhamnolipid (51, Fig. 16).84,85 Liu and coworkers demonstrated that the production of the antibiotic pyrrolnitrin (52) is dependent on the lux-like QS based signalling in Serratia plymuthica. They observed that insertional mutation in the splI gene led to undetectable quantities of antibiotic, but, after complementation with an active copy of the gene, pyrrolnitrin (52) production at the wild type level (1.14 mg mL−1) was restored.86 In a similar way, Erwinia carotovora produces the carbapenem antibiotics (53).87 The triggered production of other compounds including fosfomycin (54),88 geldanamycin (55),89 pimaricin (56),90 pikromycin (57),91 rapamycin (6)92 and stambomycin (58)93 (encoded by one of the largest known polyketide clusters, spanning ∼ 150 kbp) and many more, through the manipulation of lux-like systems is a popular approach showing that the production of natural products in bacterial strains can often be readily manipulated by engineering the promoters under the control of QS signal molecules. Although in most of the cases QS signalling has a positive effect for the production of secondary metabolites, in B. thailandensis the genetic manipulation of the luxR homologue thaA and the corresponding transcription factor binding region resulted in the activation of an NRPS/PKS hybrid gene cluster and production of its corresponding product, thailandamide A (59) whilst in the wild type strain, thailandamide A (59) biosynthesis was not observed except in low titres during early growth phase.94 This indicates that for thailandamide A biosynthesis, the complex ThaA/Acylated Homoserine Lactone (AHL) is acting as a negative regulator. | ||
| Fig. 16 Pyrocyanin (50),84 rhamnolipid (51),85 pyrrolnitrin (52),86 carbapenem antibiotics (53). The production of compounds including fosfomycin (54),88 geldanamycin (55),89 pimaricin (56),90 pikromycin (57), stambomycin (58) and thailandamide A (59)94 has been successfully triggered through QS manipulation. γ-butyrolactone (GBL). | ||
In a recent study to unlock a cryptic biosynthetic cluster encoding novel naphthalenic octaketide ansamycins, Shen and co-workers firstly investigated the introduction of the strong constitutive promoter ermE* (in a strategy similar to that reported by Bode in 4.2), this resulted in partial activation of the pathway with an increase in the transcriptional levels of only two of the genes. Analysis of the biosynthetic cluster indicated that a putative positive regulator of the LuxR family of transcription factors was encoded, nam1. Upon constitutive over-expressing of nam1 by placing it under the control of the ermE* promoter, a 12–30% increase in the levels of transcription of all of the genes within the biosynthetic pathway could be seen, and the encoded natural products produced in quantities sufficient for characterisation.95
In a similar fashion to AHL-mediated quorum sensing in Gram-negative bacteria, γ-butyrolactone (GBL) sensing in the Gram-positive streptomycetes is well documented, with these acylated lactones being structurally similar to AHLs.96Streptomyces spp. are therefore considered unusual amongst Gram-positive bacteria as most Gram-positive bacteria utilise peptide signalling based on two component systems.97 Just as with AHL's in Gram-negative bacteria, GBLs have been demonstrated to play an important role in regulating secondary metabolite production.
Amongst the 14 cryptic biosynthetic clusters identified in S. coelicolor, is a putative polyketide biosynthetic cluster named cryptic polyketide (cpk). Within this gene cluster resides a GBL receptor ScbR2, which acts as a transcriptional repressor.98 Gottelt et al. created a scbR2 knock out which resulted in a mutant capable of producing two novel metabolites: yellow cryptic polyketide (yCPK) and antibiotic cryptic polyketide (abCPK).98 The yellow metabolite, named coelimycin (60), was subsequently structurally characterised by Challis and co-workers.99
Except for the well-studied GBL, new signalling molecules have been reported and used to activate cryptic gene clusters. Corre and coworkers discovered a family of 5 new 2-alkyl-4-hydroxymethylfuran-3-carboxylic acids (AHFCAs), exemplified as methylenomycin furans (MMF), which are the products of the mmfR–mmfLHP–mmyR genes, could induce the production of methylenomycin (61) in S. coelicolor.100 Genes homologous to mmfLHP were found in Streptomyces venezuelae ATCC 10712 by BLAST searches. Inactivation of the mmyR homologue gbnR induced production of novel metabolites, named gaburedins (62).101 Avenolide, (4S,10R)-10-hydroxy-10-methyl-9-oxo-dodec-2-en-1,4-olide, triggered avermectin (63) production at nanomolar concentrations in Streptomyces avermitilis.102 Similarly, two butenolide signalling molecules SRB1 and SRB2 induced lankacidin (64) and lankamycin (65) production in Streptomyces rochei (Fig. 17).103
 | ||
| Fig. 17 Coelimycin P1 (60), methylenomycin A (61), gaburedin (62), avermectins (63), lankacidins (64) and lankamycin (65). | ||
5 Heterologous expression
The heterologous expression of genes encoding the biosynthesis of bioactive compounds in an “unnatural host” can prove challenging: hurdles of transcription and translation in the foreign host must first be overcome, enzymes must be correctly folded and may require post-transcriptional modifications crucial for their function within the biosynthetic pathway. However, increasingly, the heterologous expression of biosynthetic pathways and generation of the natural product is becoming an established approach to confirm that a biosynthetic gene cluster is responsible for the biogenesis of a particular metabolite of interest.104,105 Series of useful, engineered heterologous expression hosts are available,106–108 and this approach is also becoming a mainstream method through which to unlock silent or cryptic gene clusters that have been identified by genome mining and this has been summarized in a recent reviews of heterologous expression of bacterial natural products.109 Heterologous expression also represents a powerful tool for the discovery of novel compounds through cloning of DNA from metagenomic libraries.110,111 Metagenomic investigations by Piel on uncultured/unculturable sponge associated organisms provide evidence, for the first time, that a single member of the Theonella swinhoei microbiome community is the source of almost all of the polyketide and peptide metabolites isolated from the sponge host.112 Heterologous expression of genes from this member, named E. factor TSY1 may enable rapid access to this rich chemistry.A straightforward approach for heterologous expression is to clone clusters of interest under inducible promoters (this is a complementary approach to the introduction of strong constitutive promoters into the wild type producing organism). However, this requires detailed information of the target gene cluster, codon and substrate usage and is perhaps the most labour-intense approach described in this review. PKS and NRPs megasynthases are usually encoded by gene clusters that can reach over 100 kb in length. For example, the gene cluster encoding the antifungal nystatin is 125 kb in length.113 Cloning such large clusters is not without challenge, often even with a modest 30 kb cluster, a degree of luck is required for the entire cluster to be found included within a single cosmid. Frequently the cluster can be split between several cosmids which then require ‘stitching’ together. Many successful examples exist as previously reviewed.109 For example Bian et al. demonstrated the activation of a cryptic biosynthetic gene cluster by heterologous expression, successfully expressing an 18 kb cryptic luminmycin biosynthetic pathway from Photorhabdus luminescens in E. coli.114 From a 12 L culture of the heterologous expression system, 3.0 mg luminmycin A (66), 0.5 mg luminmycin B (67) and 0.4 mg luminmycin C (68) were isolated (Fig. 18); luminmycin A (66) had not been detected in the original extract from P. luminescens. Further engineering is often required in the heterologous host in order to unlock the silent or cryptic biosynthesis pathways; for example the replacement of promoters native to the pathway with stronger ones. Promoter selection is determined by the choice of expression host actI/actIII and ermE in Streptomyces spp. and Pm in Pseudomonas spp.115 Body provides an excellent and detailed review regarding the harnessing of transcription for unlocking cryptic gene clusters and places particular emphasis on promoter engineering.115 Utilising such an approach Luo et al. reconstructed an entire cryptic gene cluster from Streptomyces griseus in S. lividans by inserting optimized promoters upstream of the genes.116 The authors demonstrated the transcription of the gene cluster in the heterologous host resulting in the production of two polycyclic tetramate macrolactam compounds, not seen in the native host, alteramide A (69) and a novel tetramate (70). Apart from promoter optimization in heterologous expression systems, in many cases, regulatory genes need to be engineered (overexpression of upregulators ones, and deletion of down regulators). One such example is the heterologous expression study by Yamanaka et al. By genome reading the authors found a 67 kb NRPS biosynthetic gene cluster, predicted to produce a novel lipopeptide, in the marine actinomycete Saccharomonospora sp. CNQ-490. No such lipopeptide was identified in the native strain.117 After heterologous expression of the engineered gene cluster with deletion of a LuxR-type negative transcriptional regulator in S. coelicolor, 4 L of fermentation broth was reported to have yielded ∼60 mg of a series of at least 8 closely related lipopeptides; from this mixture 4 mg taromycin A (71), a novel lipopeptide was purified. The rapid and direct in vitro cloning of the 41 kbp gene cluster encoding conglobactin, and its subsequent heterologous expression, exemplifies a particularly expeditious and promising approach.117b
 | ||
| Fig. 18 Luminmycin A–C (66–68) isolated through heterologous expression studies in E. coli, 0.5 mg luminmycin B (67) and 0.4 mg luminmycin C (68) were isolated; luminmycin A (66) had not been detected in the original extract from the natural host P. luminescens.114 Alteramide A (69) and a novel tetramate (70) accessed through heterologous expression and promoter engineering in S. lividans.116 Heterologous expression of a gene cluster from marine actinomycete Saccharomonospora sp. CNQ-490 into S. coelicolor accompanied by deletion of a LuxR-type negative transcriptional regulator in S. coelicolor, results in taromycin A (71) production.117 | ||
6 Summary
Today, in the light of data from the many recently sequenced microbes and metagenomic libraries, we are even more aware of the wealth of untapped biosynthetic potential that exists. These data imply that, in terms of bioactive metabolite isolation and identification, so far only the very tip of the iceberg has been scraped and numerous valuable natural products remain to be discovered. These are exciting times, with new approaches to unlocking the treasures of previously uncultivable bacteria being explored and the discovery of teixobactin, a new antibiotic to which there was no detectable resistance.118Here we provide, in one place, an overview and guide of all of the main approaches available for unlocking and harnessing this biosynthetic potential of bacteria and revealing or increasing production of these previously unexplored bacterial natural products. Approaches overviewed range from easily accessible, low-tech and low-cost methods such as media manipulation and co-culturing, which can be strikingly effective, to the more focused and demanding molecular biological strategies of engineering organisms to produce a specific product. Unsurprisingly there is no single approach that can be used reliably to unlock every cryptic pathway.
Advances in genome annotation software are making it increasingly easy for natural products encoded by bacterial genomes to be predicted, providing a valuable tool for the identification of novel natural products. Yet in silico prediction only works faultlessly for highly canonical natural products. Currently information relating to biosynthetic clusters is distributed through the literature, and there is a call for the systematic deposition of such data to enable greater ease of retrieval and improved mining of sequence data.119 There is still much to be learnt about fundamentals of biosynthesis and for biosynthetically unprecedented assemblies in silico predictions are not possible. Heterologous expression provides a useful, but sometimes challenging approach to enable the production of a single series of compounds from a cryptic cluster of interest thereby enabling the compound's identification; such an approach represents the only way for accessing the chemical pallet of unculturable microbes. Metabolic switching in actinomycetes and the regulation of secondary metabolite production is highly complex and varied120–122 and such expression systems often require careful tailoring in order to enable production of a specific compound of interest; this can often be a bottle-neck to the discovery process.
Recent studies show that bacterial cross talk and the specific activation of certain biosynthetic pathways is far more complex than we had previously appreciated, and there is much research to be carried out before we can rationally utilise this powerful language. Rewards in the discovery of novel bioactive natural products are likely to be high. Recent reviews in this area have been published.123,124 Over the past 5 years due to genome sequencing and advances in molecular biological and analytical tools there has been a striking re-invigoration of natural product research. The relatively recent area of unlocking cryptics is advancing at an impressive rate enabling access to previously unidentified compounds.
7 References
- A. Simon, K. Traynor, K. Santos, G. Blaser, U. Bode and P. Molan, J. Evidence-Based Complementary Altern. Med., 2009, 6, 165–173 CrossRef PubMed.
- A. Fleming, Br. J. Exp. Pathol., 1929, 10, 226–236 CAS.
- E. Chain, H. W. Florey, A. D. Gardner, N. G. Heatley, M. A. Jennings, J. Orr-Ewing and A. G. Sanders, Lancet, 1940, ii, 226–228 CrossRef CAS.
- D. C. Hodgkin, Adv. Sci., 1949, 6, 85–89 CAS.
- J. W. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed.
- A. A. Brakhage and V. Schroeckh, Fungal Genet. Biol., 2011, 48, 15–22 CrossRef CAS PubMed.
- P. Wiemann and N. P. Keller, J. Ind. Microbiol. Biotechnol., 2014, 41, 301–313 CrossRef CAS PubMed.
- T. Netzker, J. Fischer, J. Weber, D. J. Mattern, C. C. Konig, V. Valiante, V. Schroeckh and A. A. Brakhage, Front. Microbiol., 2015, 6, 299 Search PubMed.
- F. Y. Lim and N. P. Keller, Nat. Prod. Rep., 2014, 31, 1277–1286 RSC.
- A. A. Brakhage, Nat. Rev. Microbiol., 2013, 11, 21–32 CrossRef CAS PubMed.
- D. C. Anyaogu and U. H. Mortensen, Front. Microbiol., 2015, 6, 77 Search PubMed.
- F. Zhu, C. Qin, L. Tao, X. Liu, Z. Shi, X. Ma, J. Jia, Y. Tan, C. Cui, J. Lin, C. Tan, Y. Jiang and Y. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12943–12948 CrossRef CAS PubMed.
- S. D. Bentley, K. F. Chater, A. M. Cerdeno-Tarraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hildalgo, T. Hornsby, S. Howarth, C. H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O'Neil, E. Rabbinowitsch, M. A. Raiandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill and D. A. Hopwood, Nature, 2002, 417, 141–147 CrossRef PubMed.
- M. Nett, H. Ikeda and B. S. Moore, Nat. Prod. Rep., 2009, 26, 1362–1384 RSC.
- H. Gross, Appl. Microbiol. Biotechnol., 2007, 75, 267–277 CrossRef CAS PubMed.
- K. Scherlach and C. Hertweck, Org. Biomol. Chem., 2009, 7, 1753–1760 CAS.
- F. J. Ribeiro, D. Przybylski, S. Yin, T. Sharpe, S. Gnerre, A. Abouelleil, A. M. Berlin, A. Montmayeur, T. P. Shea, B. J. Walker, S. K. Young, C. Russ, C. Nusbaum, I. MacCallum and D. B. Jaffe, Genome Res., 2012, 22, 2270–2277 CrossRef CAS PubMed.
- M. G. Watve, R. Tickoo, M. M. Jog and B. D. Bhole, Arch. Microbiol., 2001, 176, 386–390 CrossRef CAS PubMed.
- G. L. Challis, J. Ind. Microbiol. Biotechnol., 2014, 41, 219–232 CrossRef CAS PubMed.
- C. N. Boddy, J. Ind. Microbiol. Biotechnol., 2014, 41, 443–450 CrossRef CAS PubMed.
- A. C. Letzel, S. J. Pidot and C. Hertweck, Nat. Prod. Rep., 2013, 30, 392–428 RSC.
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS PubMed.
- A. Starcevic, J. Zucko, J. Simunkovic, P. F. Long, J. Cullum and D. Hranueli, Nucleic Acids Res., 2008, 36, 6882–6892 CrossRef CAS PubMed.
- S. Anand, M. V. Prasad, G. Yadav, N. Kumar, J. Shehara, M. Z. Ansari and D. Mohanty, Nucleic Acids Res., 2010, 38, W487–W496 CrossRef CAS PubMed.
- M. H. Li, P. M. Ung, J. Zajkowski, S. Garneau-Tsodikova and D. H. Sherman, BMC Bioinf., 2009, 10, 185 CrossRef PubMed.
- P. Caffrey, ChemBioChem, 2003, 4, 654–657 CrossRef CAS PubMed.
- R. Reid, M. Piagentini, E. Rodriguez, G. Ashley, N. Viswanathan, J. Carney, D. V. Santi, C. R. Hutchinson and R. McDaniel, Biochemistry, 2003, 42, 72–79 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS PubMed.
- S. Pidot, K. Ishida, M. Cyrulies and C. Hertweck, Angew. Chem., Int. Ed. Engl., 2014, 53, 7856–7859 CrossRef CAS PubMed.
- D. D. Nguyen, C. H. Wu, W. J. Moree, A. Lamsa, M. H. Medema, X. Zhao, R. G. Gavilan, M. Aparicio, L. Atencio, C. Jackson, J. Ballesteros, J. Sanchez, J. D. Watrous, W. Phelan, C. van de Wiel, R. D. Kersten, S. Mehnaz, R. de Mot, E. A. Shank, P. Charusanti, H. Nagarajan, B. M. Duggan, B. S. Moore, N. Bandeira, B. O. Paisson, K. Pogliano, M. Gutierrez and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2611–E2620 CrossRef CAS PubMed.
- H. B. Bode, B. Bethe, R. Hofs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS PubMed.
- H. Zahner, Angew. Chem., Int. Ed. Engl., 1977, 16, 687–694 CrossRef CAS PubMed.
- T. A. Knappe, U. Linne, S. Zirah, S. Rebuffat, X. Xie and M. A. Marahiel, J. Am. Chem. Soc., 2008, 130, 11446–11454 CrossRef CAS PubMed.
- M. E. Rateb, W. E. Houssen, M. Arnold, M. H. Abdelrahman, H. Deng, W. T. Harrison, C. K. Okoro, J. A. Asenjo, B. A. Andrews, G. Ferguson, A. T. Bull, M. Goodfellow, R. Ebel and M. Jaspars, J. Nat. Prod., 2011, 74, 1491–1499 CrossRef CAS PubMed.
- M. E. Rateb, W. E. Houssen, W. T. Harrison, H. Deng, C. K. Okoro, J. A. Asenjo, B. A. Andrews, A. T. Bull, M. Goodfellow, R. Ebel and M. Jaspars, J. Nat. Prod., 2011, 74, 1965–1971 CrossRef CAS PubMed.
- S. Lautru, R. J. Deeth, L. M. Bailey and G. L. Challis, Nat. Chem. Biol., 2005, 1, 265–269 CrossRef CAS PubMed.
- G. L. Challis and J. Ravel, FEMS Microbiol. Lett., 2000, 187, 111–114 CrossRef CAS PubMed.
- J. B. McAlpine, B. O. Bachmann, M. Piraee, S. Tremblay, A.-M. Alarco, E. Zazopoulos and C. M. Farnet, J. Nat. Prod., 2005, 68, 493–496 CrossRef CAS PubMed.
- H. Gross, V. O. Stockwell, M. D. Henckels, B. Nowak-Thompson, J. E. Loper and W. H. Gerwick, Chem. Biol., 2007, 14, 53–63 CrossRef CAS PubMed.
- A. Mearns-Spragg, M. Bregu, K. G. Boyd and J. G. Burgess, Lett. Appl. Microbiol., 1998, 27, 142–146 CAS.
- J. G. Burgess, E. M. Jordan, M. Bregu, A. Mearns-Spragg and K. G. Boyd, J. Biotechnol., 1999, 70, 27–32 CrossRef CAS PubMed.
- M. Cueto, P. R. Jensen, C. Kauffman, W. Fenical, E. Lobkovsky and J. Clardy, J. Nat. Prod., 2001, 64, 1444–1446 CrossRef CAS.
- F. Fdhila, V. Vázquez, J. L. Sánchez and R. Riguera, J. Nat. Prod., 2003, 66, 1299–1301 CrossRef CAS PubMed.
- H. Onaka, Y. Mori, Y. Igarashi and T. Furumai, Appl. Environ. Microbiol., 2011, 77, 400–406 CrossRef CAS PubMed.
- J. Y. Cho and M. S. Kim, Biosci., Biotechnol., Biochem., 2012, 76, 1849–1854 CrossRef CAS PubMed.
- J. Pérez, J. Muñoz-Dorado, A. F. Braña, L. J. Shimkets, L. Sevillano and R. I. Santamarí, Microb. Biotechnol., 2011, 4, 175–183 CrossRef PubMed.
- M. F. Traxler, J. D. Watrous, T. Alexandrov, P. C. Dorrestein and R. Kolter, mBio, 2013, 4, e00459–13 CrossRef PubMed.
- (a) J. M. Moore, E. Bradshaw, R. F. Seipke, M. I. Hutchings and M. McArthur, Methods Enzymol., 2012, 517, 367–385 CAS; (b) S.-J. Mo, P. K. Sydor, C. Corre, M. M. Alhamadsheh, a. E. Stanley, S. w. Haynes, L. Song, K. A. Reynolds and G. L. Challis, Chem. Biol., 2008, 15, 137–148 CrossRef CAS PubMed.
- D. D. Leipe and D. Landsman, Nucleic Acids Res., 1997, 25, 3693–3697 CrossRef CAS PubMed.
- R. Maegueron, P. Trojer and D. Reinberg, Curr. Opin. Genet. Dev., 2005, 15, 163–176 CrossRef PubMed.
- R. B. Williams, J. C. Henrikson, A. R. Hoover, A. E. Lee and R. H. Cichewicz, Org. Biomol. Chem., 2008, 6, 1895–1897 CAS.
- K. M. Fisch, A. F. Gillaspy, M. Gipson, J. C. Henrikson, A. R. Hoover, L. Jackson, F. Z. Najar, H. Wägele and R. H. Cichewicz, J. Ind. Microbiol. Biotechnol., 2009, 36, 1199–1213 CrossRef CAS PubMed.
- R. H. Cichewicz, Nat. Prod. Rep., 2010, 27, 11–22 RSC.
- E. B. Goh, G. Yim, W. Tsui, J. McClure, M. G. Surette and J. Davies, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 17025–17030 CrossRef CAS PubMed.
- G. Yim, H. H. Wang and J. Davies, Philos. Trans. R. Soc., B, 2007, 362, 1195–1200 CrossRef CAS PubMed.
- J. Davies, G. B. Spiegelman and G. Yim, Curr. Opin. Microbiol., 2006, 9, 445–453 CrossRef CAS PubMed.
- W. C. Ratcliff and R. F. Denison, Science, 2011, 332, 547–548 CrossRef CAS PubMed.
- (a) M. I. Mitova, G. Lang, J. Wiese and J. F. Imhoff, J. Nat. Prod., 2008, 71, 824–827 CrossRef CAS PubMed; (b) Revised structure of streptophenazine A confirmed through total synthesis: Z. Yang, X. Jin, M. Guaciaro, B. F. Molino, U. Mocek, R. Reategui, J. Rhea and T. Morley, Org. Lett., 2011, 13, 5436–5439 CrossRef CAS PubMed.
- S. Amano, T. Morota, Y. Kano, H. Narita, T. Hashidzume, S. Yamamoto, K. Mizutani, S. Sakuda, K. Furihata, H. Takano-Shiratori, H. Takano, T. Beppu and K. Ueda, J. Antibiot., 2010, 63, 486–491 CrossRef CAS PubMed.
- S. Amano, T. Sakurai, K. Endo, H. Takano, T. Beppu, K. Furihata, S. Sakuda and K. Ueda, J. Antibiot., 2011, 64, 703 CrossRef CAS PubMed.
- K. Yamanaka, H. Oikawa, H. Ogawa, K. Hosono, F. Shinmachi, H. Takano, S. Sakuda, T. Beppu and K. Ueda, Microbiology, 2005, 151, 2899–2905 CrossRef CAS PubMed.
- Y. Imai, S. Sato, Y. Tanaka, K. Ochi and T. Hosaka, Appl. Environ. Microbiol., 2015, 81, 3869–3879 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7266–7271 CrossRef CAS PubMed.
- J. Shima, A. Hesketh, S. Okamoto, S. Kawamoto and K. Ochi, J. Bacteriol., 1996, 178, 7276–7284 CAS.
- K. Ochi, S. Okamoto, Y. Tozawa, T. Inaoka, T. Hosaka, J. Xu and K. Kurosawa, Adv. Appl. Microbiol., 2004, 56, 155–184 CAS.
- G. Wang, T. Hosaka and K. Ochi, Appl. Environ. Microbiol., 2008, 74, 2834–2840 CrossRef CAS PubMed.
- X. Han, C. Cui, Z. Yao and M. Yang, Period. Ocean Univ. China, 2010, 40, 47–52 CAS.
- Y. Wang, Y. Liu, Y. Zhu, S. Liu, C. Cai and P. Xu, Chin. J. Antibiot., 2006, 31, 243–246 CAS.
- Y. Tanaka, K. Kasahara, Y. Hirose, K. Murakami, R. Kugimiya and K. Ochi, J. Bacteriol., 2013, 195, 2959–2970 CrossRef CAS PubMed.
- Y. Zhang, H. Huang, S. Xu, B. Wang, J. Ju, H. Tan and W. Li, Microb. Cell Fact., 2015, 14, 64 CrossRef PubMed.
- (a) K. Ochi and T. Hosaka, Appl. Microbiol. Biotechnol., 2013, 97, 87–98 CrossRef CAS PubMed; (b) C. Bai, Y. Zhang, X. Zhao, Y. Hu, S. Xiang, J. Miao, C. Lou and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2015 DOI:10.1073/pnas.1511027112.
- E. Bode, A. O. Brachmann, C. Kegler, R. Simsek, C. Dauth, Q. Zhou, M. Kaiser, P. Klemmt and H. B. Bode, ChemBioChem, 2015, 16, 1115–1119 CrossRef CAS PubMed.
- H. B. Bode, A. O. Brachmann, K. B. Jadhav, L. Seyfarth, C. Dauth, S. W. Fuchs, M. Kaiser, N. R. Waterfield, H. Sack, S. H. Heinemann and H. D. Arndt, Angew. Chem., Int. Ed. Engl., 2015, 54, 10352–10355 CrossRef CAS PubMed.
- C. Olano, I. Garcia, A. Gonzalez, M. Rodriguez, D. Rozas, J. Rubio, M. Sanchez-Hidalgo, A. F. Brana, C. Mendez and J. A. Salas, Microb. Biotechnol., 2014, 7, 242–256 CrossRef CAS PubMed.
- D. Montiel, H. S. Kang, F. Y. Chang, Z. Charlop-Powers and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8953–8958 CrossRef CAS PubMed.
- I. Santamarta, A. Rodríguez-García, R. Pérez-Redondo, J. F. Martín and P. Liras, J. Bacteriol., 2002, 184, 3106–3113 CrossRef CAS PubMed.
- T. V. Hung, S. Malla, B. C. Park, K. Liou, H. C. Lee and J. K. Sohng, J. Microbiol. Biotechnol., 2007, 17, 1538–1545 CAS.
- L. Wang, Y. Hu, Y. Zhang, S. Wang, Z. Cui, Y. Bao, W. Jiang and B. Hong, BMC Microbiol., 2009, 9, 14 CrossRef PubMed.
- M. McArthur and M. J. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1020–1025 CrossRef CAS PubMed.
- S. Gottesman, Annu. Rev. Genet., 1984, 18, 415–441 CrossRef CAS PubMed.
- S. Rigali, F. Titgemeyer, S. Barends, S. Mulder, A. W. Thomae, D. A. Hopwood and G. P. van Wezel, EMBO Rep., 2008, 9, 670–675 CrossRef CAS PubMed.
- M. B. Miller and B. L. Bassler, Annu. Rev. Microbiol., 2001, 55, 165–199 CrossRef CAS PubMed.
- A. Latfi, M. K. Winson, M. Foglino, B. W. Bycroft, G. S. Stewart, A. Lazdunski and P. Williams, Proc. Natl. Acad. Sci. U. S. A., 1995, 17, 333–343 Search PubMed.
- M. K. Winson, M. Camara, A. Latifi, M. Foglino, S. R. Chhabra, M. Daykin, M. Bally, V. Chapon, G. P. Salmond, B. W. Bycroft, A. Lazdunski, G. S. Stewart and P. Williams, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9427–9431 CrossRef CAS.
- X. Liu, M. Bimerew, Y. Ma, H. Müller, M. Ovadis, L. Eberl, G. Berg and L. Chernin, FEMS Microbiol. Lett., 2007, 270, 299–305 CrossRef CAS PubMed.
- S. J. McGowan, A. M. Barnard, G. Bosgelmez, M. Sebaihia, N. J. Simpson, N. R. Thomson, D. E. Todd, M. Welch, N. A. Whitehead and G. P. Salmond, Mol. Microbiol., 2005, 55, 526–545 CrossRef CAS PubMed.
- R. D. Woodyer, Z. Shao, P. M. Thomas, N. L. Kelleher, J. A. Blodgett, W. W. Metcalf, W. A. van der Donk and H. Zhao, Chem. Biol., 2006, 13, 1171–1182 CrossRef CAS PubMed.
- W. He, J. Lei, Y. Liu and Y. Wang, Arch. Microbiol., 2008, 189, 501–510 CrossRef CAS PubMed.
- N. Antón, J. Santos-Aberturas, M. V. Mendes, S. M. Guerra, J. F. Martín and J. F. Aparicio, Microbiology, 2007, 153(9), 3174–3183 CrossRef PubMed.
- D. J. Wilson, Y. Xue, K. A. Reynolds and D. H. Sherman, J. Bacteriol., 2001, 183, 3468–3475 CrossRef CAS PubMed.
- E. Kuscer, N. Coates, I. Challis, M. Gregory, B. Wilkinson, R. Sheridan and H. Petkovic, J. Bacteriol., 2007, 189, 4756–4763 CrossRef CAS PubMed.
- L. Laureti, L. Song, S. Huang, C. Corre, P. Leblond, G. L. Challis and B. Aigle, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6258–6263 CrossRef CAS PubMed.
- K. Ishida, T. Lincke, S. Behnken and C. Hertweck, J. Am. Chem. Soc., 2010, 132, 13966–13968 CrossRef CAS PubMed.
- S. Li, Y. Li, C. Lu, J. Zhang, J. Zhu, H. Wang and Y. Shen, Org. Lett., 2015, 17, 3706–3709 CrossRef CAS PubMed.
- C. Fuqua and E. P. Greenberg, Nat. Rev. Mol. Cell Biol., 2002, 3, 685–695 CrossRef CAS PubMed.
- E. Takano, R. Chakraburtty, T. Nihira, Y. Yamada and M. J. Bibb, Mol. Microbiol., 2001, 41, 1015–1028 CrossRef CAS PubMed.
- M. Gottelt, S. Kol, J. P. Gomez-Escribano, M. Bibb and E. Takano, Microbiology, 2010, 156(8), 2343–2353 CrossRef CAS PubMed.
- J. P. Gomez-Escribano, L. Song, D. J. Fox, V. Yeo, M. J. Bibb and G. L. Challis, Chem. Sci., 2012, 3, 2716–2720 RSC.
- C. Corre, L. Song, S. O'Rourke, K. F. Chater and G. L. Challis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17510–17515 CrossRef CAS PubMed.
- J. D. Sidda, L. Song, V. Poon, M. Al-Bassam, O. Lazos, M. J. Buttner, G. L. Challis and C. Corre, Chem. Sci., 2014, 5, 86–89 RSC.
- S. Kitani, K. T. Miyamoto, S. Takamatsu, E. Herawati, H. Iguchi, K. Nishitomi, M. Uchida, T. Nagamitsu, S. Omura, H. Ikeda and T. Nihira, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16410–16415 CrossRef CAS PubMed.
- K. Arakawa, N. Tsuda, A. Taniguchi and H. Kinashi, ChemBioChem, 2012, 13, 1447–1457 CrossRef CAS PubMed.
- E. J. Rackham, S. Gruschow, A. E. Ragab, S. Dickens and R. J. M. Goss, ChemBioChem, 2010, 11, 1700–1709 CrossRef CAS PubMed.
- W. Zhang, B. Ostash and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16828–16833 CrossRef CAS PubMed.
- J. P. Gomez-Escribano and M. J. Bibb, Microb. Biotechnol., 2011, 4, 207–215 CrossRef CAS PubMed.
- M. Komatsu, T. Uchiyama, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2646–2651 CrossRef CAS PubMed.
- P. Lindberg, S. Park and A. Melis, Metab. Eng., 2010, 12, 70–79 CrossRef CAS PubMed.
- R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2010, 37, 759–772 CrossRef CAS PubMed.
- Z. Feng, J. H. Kim and S. F. Brady, J. Am. Chem. Soc., 2010, 132, 11902–11903 CrossRef CAS PubMed.
- D. Kallifidas, H. S. Kang and S. F. Brady, J. Am. Chem. Soc., 2012, 134, 19552–19555 CrossRef CAS PubMed.
- M. C. Wilson, T. Mori, C. Rückert, A. R. Uria, M. J. Helf, K. Takada, C. Gernert, U. A. E. Steffens, N. Heycke, S. Schmitt, C. Rinke, E. J. N. Helfrich, A. O. Brachmann, C. Gurgui, T. Wakimoto, M. Kracht, M. Crüsemann, U. Hentschel, I. Abe, S. Matsunaga, J. Kalinowski, H. Takeyama and J. Piel, Nature, 2014, 506, 58–62 CrossRef CAS PubMed.
- T. Brautaset, O. N. Sekurova, H. Sletta, T. E. Ellingsen, A. R. StrLm, S. Valla and S. B. Zotchev, Chem. Biol., 2000, 7, 395–403 CrossRef CAS PubMed.
- X. Bian, A. Plaza, Y. Zhang and R. Muller, J. Nat. Prod., 2012, 75, 1652–1655 CrossRef CAS PubMed.
- D. C. Stevens, T. P. Hari and C. N. Boddy, Nat. Prod. Rep., 2013, 30, 1391–1411 RSC.
- Y. Luo, H. Huang, J. Liang, M. Wang, L. Lu, Z. Shao, R. E. Cobb and H. Zhao, Nat. Commun., 2013, 4, 2894 Search PubMed.
- (a) K. Yamanaka, K. A. Reynolds, R. D. Kersten, K. S. Ryan, D. J. Gonzalez, V. Nizet, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1957–1962 CrossRef CAS PubMed; (b) Y. Zhou, A. C. Murphy, M. Samborskyy, P. Prediger, L. C. Dias and P. F. Leadlay, Chem. Biol., 2015, 22, 745–754 CrossRef CAS PubMed.
- L. L. Ling, T. Schneidler, A. J. Peoples, A. L. Spoering, I. Engles, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millet, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- M. H. Medema, R. Kottmann, P. Yilmaz, M. Cummings, J. B. Biggins, K. Blin, I. de Bruijn, Y. H. Chooi, J. Claesen, R. C. Coates, P. Cruz-Moreales, S. Duddela, S. Dusterhus, D. J. Edwards, D. P. Fewer, N. Garg, C. Geiger, J. P. Gomez-Escribano, A. Greule, M. Hadjithomas, A. S. Haines, E. J. Helfrich, M. L. Hillwig, K. Ishida, A. C. Jones, C. S. Jones, K. Jungmann, C. Kegler, H. U. Kim, P. Kotter, D. Krug, J. Masschelein, A. V. Melnik, S. M. Mantovani, E. A. Monroe, M. Moore, N. Moss, H. W. Nutzmann, G. Pan, A. Pati, D. Petras, F. J. Reen, F. Fosconi, Z. Rui, Z. Tian, N. J. Tobias, Y. Tsunematsu, P. Wiemann, E. Wyckoff, X. Yan, G. Yim, F. Yu, Y. Xie, B. Aigle, A. K. Apel, C. J. Balibar, E. P. Balskus, F. Barona-Gomez, A. Bechthold, H. B. Bode, R. Borriss, S. F. Brady, A. A. Brakhage, P. Caffrey, Y. Q. Cheng, J. Clardy, R. J. Cox, R. de Mot, S. Donadio, M. S. Donia, W. A. van der Donk, P. C. Dorrestein, S. Doyle, A. J. Driessen, M. Ehling-Schulz, K. D. Entian, M. A. Fischbach, L. Gerwick, W. H. Gerwick, H. Gross, B. Gust, C. Hertweck, M. Hofte, S. E. Jensen, J. Ju, L. Katz, L. Kaysser, J. L. Klassen, N. P. Keller, J. Kormanec, O. P. Kuipers, T. Kuzuyama, N. C. Kyrpides, H. J. Kwon, S. Lautru, R. Lavigne, C. Y. Lee, B. Linquan, X. Liu, W. Liu, A. Luzhetskyy, T. Mahmud, Y. Mast, C. Mendez, M. Metsa-Ketela, J. Mickelfield, D. A. Mitchell, B. S. Moore, L. M. Moreira, R. Muller, B. A. Neilan, M. Nett, J. Nielsen, F. O'Gara, H. Oikawa, A. Osbourn, M. S. Osburne, B. Ostash, S. M. Payne, J. L. Pernodet, M. Petricek, J. Piel, O. Ploux, J. M. Raaijmakers, J. A. Salas, E. K. Schmitt, B. Scott, R. F. Seipke, B. Shen, D. H. Sherman, K. Sivonen, M. J. Smanski, M. Sosio, E. Stegmann, R. D. Sussmuth, K. Tahlan, C. M. Thomas, Y. Tang, A. W. Truman, M. Viaud, J. D. Walton, C. T. Walsh, T. Weber, G. P. van Wezel, B. Wilkinson, J. M. Willey, W. Wohlleben, G. D. Wright, N. Ziemert, C. Zhang, S. B. Zotchev, R. Breitling, E. Takano and F. O. Glockner, Nat. Chem. Biol., 2015, 11, 625–631 CrossRef CAS PubMed.
- K. Nieselt, F. Battke, A. Herbig, P. Bruheim, A. Wentzel, O. M. Jakobsen, H. Sletta, M. T. Alam, M. E. Merlo, J. Moore, W. A. Omara, E. R. Morrissey, M. A. Juarez-Hermosillo, A. Rodriguez-Garcia, M. Nentwich, L. Thomas, M. Igbal, R. Legaie, W. H. Gaze, G. L. Challis, R. C. Jansen, L. Dijkhuizen, D. A. Rand, D. L. Wild, M. Bonin, J. Reuther, W. Wohlleben, M. C. Smith, N. J. Burroughs, J. F. Martin, D. A. Hodgson, E. Takano, R. Breitling, T. E. Ellingsen and E. M. Wellington, BMC Genomics, 2010, 11, 10 CrossRef PubMed.
- L. Lo Grasso, S. Maffioli, M. Sosio, M. Bibb, A. M. Puglia and R. Alduina, J. Bacteriol., 2015, 197, 2536–2544 CrossRef CAS PubMed.
- E. J. Sherwood and M. J. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2500–E2509 CrossRef CAS PubMed.
- P. J. Rutledge and G. L. Challis, Nat. Rev. Microbiol., 2015, 13, 509–523 CrossRef CAS PubMed.
- U. R. Abdelmohsen, T. Grkovic, S. Balasubramanian, M. S. Kamel, R. J. Quinn and U. Hentschel, Biotechnol. Adv., 2015, 33, 798–811 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |