
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biosynthesis of fungal meroterpenoids†
Yudai
Matsuda
and
Ikuro
Abe
*
Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, Japan. E-mail: abei@mol.f.u-tokyo.ac.jp
First published on 26th October 2015
Abstract
Covering: up to September 2015
Meroterpenoids are hybrid natural products that partially originate from the terpenoid pathway. The meroterpenoids derived from fungi display quite diverse structures, with a wide range of biological properties. This review summarizes the molecular bases for their biosyntheses, which were recently elucidated with modern techniques, and also discusses the plausible biosynthetic pathways of other related natural products lacking genetic information. (Complementary to the coverage of literature by Geris and Simpson in Nat. Prod. Rep., 2009, 26, 1063–1094.)
 Yudai Matsuda | Yudai Matsuda received his B.Sc. in 2010 and M.Sc. in 2012 from The University of Tokyo under the direction of Ikuro Abe. After a short period of experience in industry, he moved back to The University of Tokyo as a project researcher, and was then appointed as an assistant professor at the same university (2013–2015). Meanwhile, he obtained his Ph.D. in pharmaceutical sciences from The University of Tokyo in 2015. He is now a postdoc at Technical University of Denmark, working with Thomas O. Larsen. His research focuses on biosynthesis and biosynthetic engineering of fungal natural products. |
 Ikuro Abe | Ikuro Abe is Professor of Natural Products Chemistry at The University of Tokyo. He obtained his Ph.D. in 1989 from The University of Tokyo under the direction of Professor Yutaka Ebizuka, where he studied chemistry and biochemistry of natural products biosynthesis. After two years of postdoctoral research with Professor Guy Ourisson at the CNRS Institut de Chimie des Substances Naturelles, and mostly with Professor Michel Rohmer at the Ecole Nationale Supérieure de Chimie de Mulhouse (1989–1991), he went to the USA to work with Professor Glenn D. Prestwich at the State University of New York at Stony Brook (1991–1996) and then at The University of Utah (1996–1998) as a Research Assistant Professor. In 1998, he returned to Japan at University of Shizuoka, School of Pharmaceutical Sciences (1998–2009). In 2009, he then moved back to The University of Tokyo, Graduate School of Pharmaceutical Sciences as a Full Professor. His research interests mostly focus on exploring and engineering of natural products biosynthesis. He is a Fellow of The Royal Society of Chemistry. |
1 Introduction
Meroterpenoids are hybrid natural products partially derived from terpenoid pathways as the prefix “mero-” means “part, partial, and fragment”.1 On the basis of their definition, numerous compounds derived from animals, plants, bacteria, and fungi can be grouped into this class of natural products, which range from widely distributed molecules to species-specific secondary metabolites. Ubiquinone-10 (1, “coenzyme Q10”)2 and α-tocopherol (2, “vitamin E”)3 are examples of commonly occurring meroterpenoids, whereas the anti-cancer chemotherapy drug vinblastine (3),4 and a component of marijuana, tetrahydrocannabinol (4),5 are produced by specific plants. Merochlorin A (5)6 and teleocidin B-4 (6)7 are generated by the actinomycetes in the genus Streptomyces, and thus represent the bacterial meroterpenoids.
Among the meroterpenoid producers, fungi synthesize molecules with diverse chemical structures and a wide range of biological activities (Fig. 1).1 Mycophenolic acid (7), produced by several Penicillium species, is a strong inhibitor of inosine 5′-monophosphate dehydrogenase (IMPDH),8 and its derivative mycophenolate mofetil is clinically used as an immunosuppressant drug.9 Fumagillin (8), isolated from Aspergillus fumigatus,10 is used for the treatment of microsporidiosis and amebiasis,11 and has also been investigated as an anti-angiogenesis agent, since 8 inhibits methionine aminopeptidase-2 (MetAP2).12 Pyripyropene A (9),13 territrem B (10),14 and ascofuranone (11)15 are highly potent inhibitors of acyl-coenzyme A:cholesterol acyltransferase-2 (ACAT-2), acetylcholinesterase (AChE), and trypanosome alternative oxidase, respectively. Berkeleyacetal C (12) was originally isolated from Penicillium rubrum, based on its inhibitory activities against matrix metalloproteinase-3 (MMP-3) and caspase-1.16 Subsequently, 12 was found to exhibit anti-inflammatory activity by inhibiting nitric oxide (NO) production and inducible NO synthase (iNOS), in which the target molecule of 12 was elucidated to be interleukin-1 receptor-associated kinase-4 (IRAK4).17
 | ||
| Fig. 1 Representative fungal meroterpenoids. | ||
Although the above-described fungal meroterpenoids are all polyketide–terpenoid hybrid molecules, several other meroterpenoids are also derived from non-polyketide starters. For example, the kinesin Eg5 inhibitor terpendole E (13)18 contains an indole ring instead of a polyketide moiety as the non-terpenoid part, and thus is classified as an “indole-diterpene (-diterpenoid) alkaloid”. Compound 13 also serves as a biosynthetic intermediate of lolitrem B (14), which is produced by Neotyphodium lolii and is a potent tremorgenic neurotoxin.19
Besides their biological activities, structural diversity and complexity are important characteristics of the fungal meroterpenoids, as depicted in Fig. 1. These intriguing structures have attracted scientists for elucidation of their biosynthetic pathways, as well as the key reactions involved in their biosyntheses. Significantly, recent advances in genome-sequencing technologies and the development of tools for biosynthetic studies, as well as intensive researches at genetic and enzymatic levels, have allowed the discovery of many biosynthetic gene clusters for fungal meroterpenoids. This review is intended to continue the coverage of the literature previously reviewed in this journal by Geris and Simpson.1 It will mainly focus on the recent biosynthetic studies of fungal meroterpenoids for which the biosynthetic genes/enzymes have been elucidated, and also discuss the plausible biosynthetic mechanisms of some related meroterpenoids for which genetic information is lacking. Fungal meroterpenoids are quite often generated via the addition of a C5 isoprene unit, catalyzed by dimethylallyltryptophan synthases (DMATSs), but we will limit this review to the meroterpenoids originating from relatively long prenyl chains (more than C5) as the functions of DMATSs have been well summarized elsewhere.20,21
2 Pyripyropene A: a model case of fungal meroterpenoid biosynthesis
2.1 Biosynthesis of pyripyropene A
Due to their structural diversity, it is difficult to generalize the biosynthetic machinery of fungal meroterpenoids, but the biosynthesis of pyripyropene A (9), which has been recently fully elucidated (Fig. 2A), exemplifies one common strategy for the generation of this class of natural products. Basically, the biosynthesis can be divided into several phases: preparation of the non-terpenoid starter unit, prenylation of the non-terpenoid moiety, epoxidation of the olefin of the prenyl group, protonation-initiated cyclization of the prenyl portion, and various decorations of the cyclized intermediate. | ||
| Fig. 2 Biosynthesis of (A) pyripyropene A and (B) related compounds. | ||
Earlier studies of pyripyropene A biosynthesis, using isotope-labeled substrates, allowed an outline of the pathway to be proposed, albeit without genetic information.22 Subsequently, the biosynthetic gene clusters for 9 have been individually identified from two different fungal species, Aspergillus fumigatus Af293 (the pyr cluster) and Penicillium coprobium PF 1169 (the ppb cluster), and the biosynthesis was investigated, mainly by heterologous expression of the genes in Aspergillus oryzae.23–25 The two clusters indeed share the same gene organization, with high sequence identity with each corresponding gene in the other cluster. The first half of the biosynthesis of 9 was investigated using the pyr genes,23 whereas the latter half was elucidated using the ppb cluster.24,25 Considering the high similarity between the two clusters, it is reasonable that each corresponding enzyme plays the same role in the biosynthetic scheme.
In the pyripyropene A pathway, the non-terpenoid portion is synthesized by two enzymes, the coenzyme A (CoA)-ligase Pyr1/Ppb1 and the iterative type I polyketide synthase (PKS) Pyr2/Ppb2, from an unusual nicotinic acid (15) starter. In the first committed step, 15 is transformed into nicotinyl-CoA (16) by Pyr1/Ppb1, which is then used as the starter substrate for the PKS Pyr2/Ppb2, with ketosynthase, acyltransferase, and acyl carrier protein domains, but presumably no other functional domains, to yield 4-hydroxy-6-(3-pyridinyl)-2H-pyran-2-one (HPPO, 17) by condensation with two molecules of malonyl-CoA.
The α-pyrone 17 is subsequently farnesylated by the transmembrane prenyltransferase (PTase) Pyr6/Ppb7, using farnesyl pyrophosphate (FPP) as a prenyl donor, to provide 18. This reaction is followed by the epoxidation of the terminal olefin, catalyzed by the flavin-dependent monooxygenase (FMO) Pyr5/Ppb6. Interestingly, Pyr6/Ppb7 is homologous to UbiA, the 4-hydroxybenzoate polyprenyltransferase involved in ubiquinone biosynthesis,26 whereas Pyr5/Ppb6 shares sequence similarity with UbiF, UbiH, and UbiI, which are also oxidative enzymes required for ubiquinone biogenesis.27 This observation suggests that the gene cluster evolved from the ubiquinone pathway.
The resultant epoxide 19, synthesized by Pyr5/Ppb6, undergoes cyclization of its terpenoid moiety to yield deacetyl-pyripyropene E (20). The cyclization reaction, initiated by an acid-catalyzed epoxide ring-opening with participation by a neighboring π-bond, is catalyzed by Pyr4/Ppb5. These enzymes are membrane-bound proteins with seven-transmembrane helices, and therefore the enzyme can also serve as a transporter or has been evolved from transport proteins.28 Intriguingly, they share no sequence homology with any previously characterized terpene cyclases (TPCs). Thus, the detailed mechanism for the catalysis still remains unclear, but a mutational study has revealed that two conserved acidic amino acid residues, Glu63 and Asp218 (numbering in Pyr4), are essential for the cyclization, suggesting that these residues might serve as general acids that protonate the epoxide ring of 19.23 Importantly, a protein homologous to Pyr4 is also present in the actinomycete biosynthetic pathways, such as that for xiamycin A.29
The scaffold of the pyripyropenes is thus prepared, and is then subjected to the tailoring reactions. The acetyltransferase Pyr7/Ppb8 acetylates the hydroxyl group of deacetyl-pyripyropene E (20) to yield pyripyropene E (21), which is hydroxylated by the cytochrome P450 monooxygenase Pyr3/Ppb3 to produce 11-deacetyl-pyripyropene O (22). The other acetyltransferase Pyr8/Ppb9 introduces an acetyl group to this newly introduced hydroxyl group, thus producing pyripyropene O (23). Two distinct positions of 23 undergo hydroxylation reactions catalyzed by the other P450 Pyr9/Ppb4 for conversion into deacetyl-pyripyropene A (24), and the acetyltransferase Pyr8/Ppb9 again accepts 24 to complete the pathway for pyripyropene A (9).
As discussed later in this review, the fungal meroterpenoid pathways often employ biosynthetic enzymes similar to those for the production of 9 in their early biosynthetic stage. For example, the iterative type-I PKS, UbiA-like PTase, flavin-dependent epoxidase, and transmembrane TPC are often utilized as common components for meroterpenoids biosyntheses, thus facilitating the rapid discovery of putative biosynthetic gene clusters for meroterpenoids from the available genomic data.
2.2 Biosynthesis of territrems, arisugacins, and yaminterritrem B
The pyripyropene A pathway provides plausible schemes for the biosynthesis of molecules lacking genetic information, such as territrems A–C30 (25, 10, 26) and arisugacins A (27) and B (28) (Fig. 2B),31,32 which are all strong inhibitors of AChE, although they have similar carbon skeletons to that of 9. If 4-hydroxybenzoic acid (29) is used instead of nicotinic acid (15), and undergoes the same reactions utilized in the first five steps of the pyripyropene A pathway, then the cyclized compound 31 harboring the scaffolds of 10 and 25–28 would be obtained, via the pre-cyclization intermediate 30. Further modifications, including oxidations and methylations, should consequently generate 10 and 25–28. Meanwhile, yaminterritrem B (32) has a quite similar backbone structure to those of 10 and 25–28, but differs in that it contains the γ-pyrone moiety instead of the α-pyrone.33 Compound 32 also appears to be derived from 30, but the cyclization occurs in a different manner to provide 33, an isomer of 31, which undergoes several oxidations and ether linkage formation to generate 32 (Fig. 2B). Interestingly, the fungal strain producing 32 also synthesizes another meroterpenoid, yaminterritrem A (34), which harbors the α-pyrone motif.33The biosyntheses of 9, 10, 25–28, and 32 provide important insights into fungal meroterpenoid pathways that generate their structural diversity: the same intermediate can be cyclized into different compounds, and furthermore, the same or highly similar scaffolds can be tailored in versatile manners.
2.3 Biosynthesis of 15-deoxyoxalicines
Although HPPO (17) is farnesylated in the pyripyropene A biosynthesis, 17 can also undergo geranylgeranylation in other pathways. Very recently, the biosynthetic gene cluster for 15-deoxyoxalicine A (35) and B (36) was discovered from the genome of Penicillium canescens (the olc cluster), and their biosyntheses were investigated by targeted gene deletions.34 In the proposed pathway (Fig. 3), after the formation of 17 in the same way as that for pyripyropene A (9), 17 is geranylgeranylated by the UbiA-like PTase OlcH to yield 37. Interestingly, unlike the biosynthetic machinery for 9, the prenyl donor of the prenylation, geranylgeranyl pyrophosphate (GGPP), seems to be prepared by the GGPP synthase OlcC encoded by the olc cluster, whereas the pyr cluster possesses no corresponding gene. Subsequently, the FMO OlcE epoxidizes 37 to 38, which is cyclized into predecaturin E (39) by the transmembrane TPC OlcD. | ||
| Fig. 3 Biosynthesis of 15-deoxyoxalicines. | ||
The multicyclic intermediate 39 is then oxidized by the P450 OlcG to provide decaturin E (41) probably via40, which is the presumable product of OlcG. Two further rounds of oxidations by the P450 OlcJ and the short-chain dehydrogenase/reductase (SDR) OlcF transforms 41 into a predicted intermediate 43via42, which is followed by the hemiacetal formation to give decaturin C (44). Afterward, 44 undergoes hydroxylation to generate decaturin A (45) by the Fe(II)/α-ketoglutarate (α-KG)-dependent dioxygenase OlcK and the transmembrane protein OlcL. Finally, 45 is converted into the end product 36 with the aid of the P450 OlcB. Multicyclic 44, the deoxy analogue of 45, can also be accepted by OlcB, generating 35.
The mechanism for the spiro-lactone formation in the last step of the biosynthesis is proposed as follows.34 OlcB hydroxylates either of the methyl groups bound to C-28 to yield 46 or 47, which could induce the elimination of a water molecule and the oxidative rearrangement to form 35 or 36 (Fig. 3).
3 Meroterpenoids with an uncyclized terpenoid moiety
Although many biosynthetic pathways for fungal meroterpenoids involve the cyclization of their prenyl side chains, those with linear terpenoid moieties are also widely found in nature, including mycophenolic acid (7), yanuthone D (48),35 and aspernidine A (49).36 Thus, the gene clusters for these compounds do not encode the enzymes for the terpene cyclization. This section summarizes the molecular bases for the biosyntheses of 7, 48, and 49, which have recently been investigated.3.1 Biosynthesis of mycophenolic acid
The biosynthetic gene cluster of 7 was first identified in the genome of Penicillium brevicompactum IBT23078 (the mpa cluster),37 and subsequently was also discovered in P. brevicompactum NRRL 864 (the mpa′ cluster).38 The two clusters are almost identical, and each consists of seven open reading frames, with the same gene organization.The first three steps of the pathway were investigated using the mpa genes, by gene deletion and heterologous expression in Aspergillus nidulans (Fig. 4).37,39,40 The initial reaction is catalyzed by the type-I non-reducing PKS (NR-PKS) MpaC, which synthesizes 5-methylorsellinic acid (50, 5-MOA) from one molecule of acetyl-CoA, three molecules of malonyl-CoA, and one molecule of S-adenosylmethionine (SAM). In the next step, the unique chimeric protein of P450 monooxygenase and the hydrolase, MpaDE, transforms 50 into 5,7-dihydroxy-4-methylphthalide (52, DHMP), in which the P450 domain (MpaD) is responsible for the hydroxylation of the methyl group, whereas the hydrolase domain (MpaE) performs the lactonization to yield the phthalide 52. The bifunctionality of MpaDE was confirmed by the observation that the heterologous expression of MpaC and only the MpaD part of the chimeric enzyme resulted in the accumulation of 51. A similar fusion protein is also utilized to form a phthalide derivative in the biosynthesis of anditomin, as described later.41
 | ||
| Fig. 4 Biosynthesis of mycophenolic acid. | ||
The mid-stage of the mycophenolic acid pathway after DHMP (52) still remains to be elucidated, since there is no experimental evidence to date. The phthalide 52 would then be accepted by the PTase MpaA, which is homologous to Pyr6/Ppb7, and converted into its farnesylated form 53. The next step should be the oxidative olefin cleavage of the prenyl side chain to synthesize demethylmycophenolic acid (54), in a reaction that may be catalyzed by MpaH, which exhibits sequence homology to α/β-hydrolases.37 An isotope-feeding study suggested the mechanism for the oxidative cleavage, in which the olefin of 53 is transformed into the epoxide 55, the ketone 56, and the α-hydroxy ketone 57 in this order.42 Subsequent C–C bond cleavage would finally produce 54, but the actual intermediates for this conversion have yet to be identified.
Intriguingly, MpaH shares weak amino acid sequence identity (ca. 15% identity) with 1-H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase (HOD) and 1-H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase (QDO).43 HOD and QDO both have the α/β-hydrolase fold, but these enzymes serve as cofactor-independent dioxygenases that cleave the C–C double bond of their substrate quinoline derivatives, resulting in the formation of a carboxyl group. The reactions by HOD and QDO are similar to those in the mycophenolic acid pathway, in terms of the olefin cleavage followed by the carboxylate formation. Thus, MpaH could also perform its catalysis in a similar manner to that of HOD and QDO, but this should be confirmed by the future biochemical characterization of MpaH.
The last reaction in the biosynthetic scheme was established by biochemical experiments using the purified MpaG′ protein,38 in which the phenolic hydroxyl group of demethylmycophenolic acid (54) is methylated (Fig. 4). Investigations of the substrate tolerance of MpaG′ revealed that the enzyme is promiscuous, as it accepts several unnatural substrates.
3.2 Biosynthesis of yanuthone D
The farnesylated epoxy cyclohexenoids, yanuthones, are known as antifungal compounds targeting Candida albicans, and among them yanuthone D (48) is the most potent agent.44 The biosynthetic gene cluster for 48, consisting of ten open reading frames, was identified in the genome of Aspergillus niger, and was designated as the yan cluster.45 The yanuthone D pathway was studied by gene disruption, heterologous expression, and isotope-feeding experiments (Fig. 5).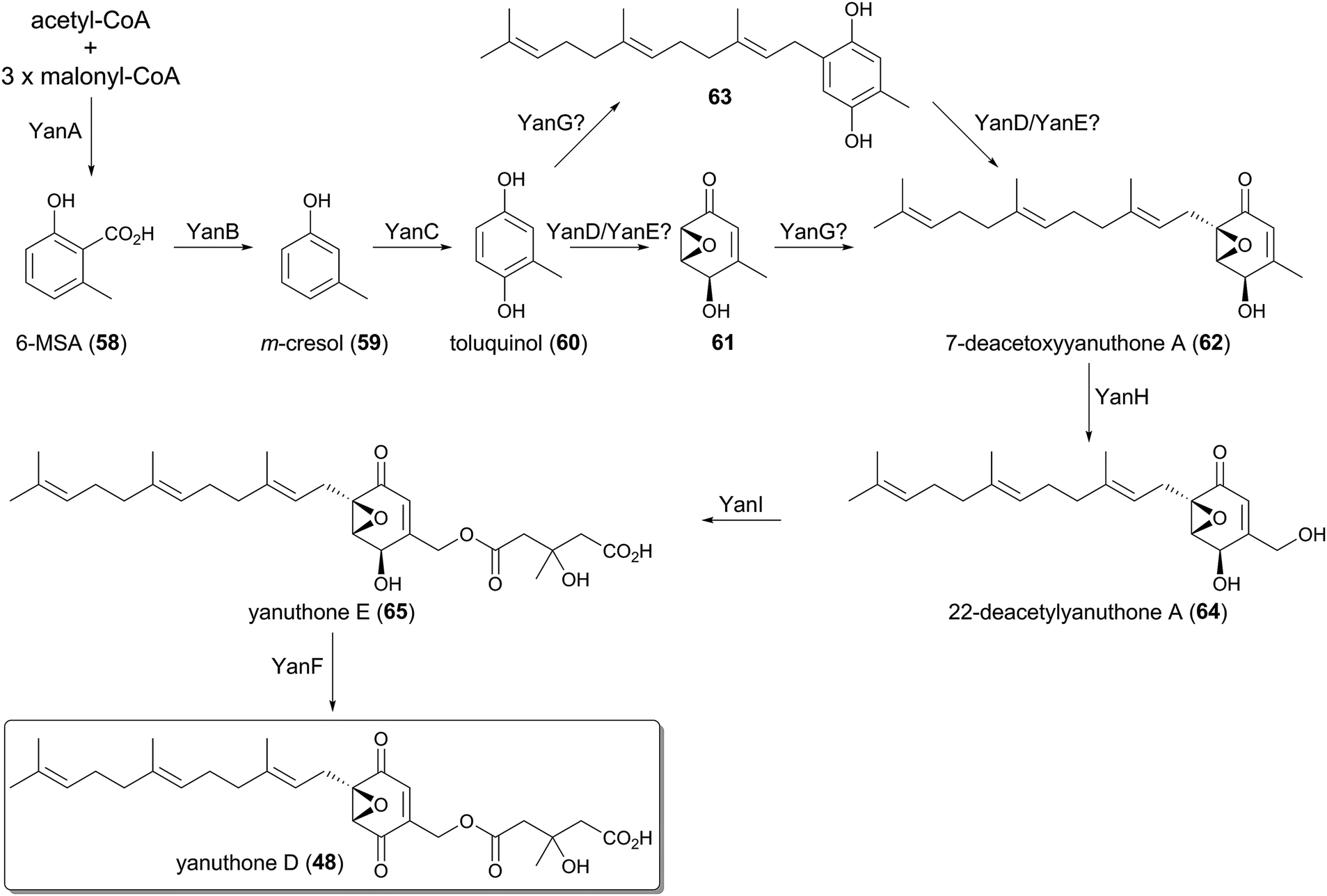 | ||
| Fig. 5 Biosynthesis of yanuthone D. | ||
The partially reducing PKS (PR-PKS) YanA initiates the pathway to form 6-methylsalicylic acid (58, 6-MSA). Compound 58 is subsequently transformed to m-cresol (59) by the decarboxylase YanB, and is further converted to toluquinol (60) by the P450 YanC. The involvement of 58 and 59 in the biosynthesis of yanuthone D (48) was validated, as the feeding of 58 or 59 restored the production of 48 upon the deletion of the PKS gene yanA. These results excluded the previous hypothesis that 48 is derived from shikimic acid.35
The next several steps still remain ambiguous since no biosynthetic intermediate has been obtained from the targeted gene deletions. It was proposed that toluquinol (60) undergoes epoxidation to generate 61, in a reaction that might be catalyzed by YanD, a putative dehydrogenase, and/or YanE with unknown function. Compound 61 is then farnesylated by the UbiA-like PTase YanG to yield 7-deacetoxyyanuthone A (62). The epoxidation is predicted to precede the farnesylation, since the prenylated toluquinol has never been isolated from the yanuthone D-producing strain or its mutants.45 However, considering that the C-prenylation reaction generally requires the π-electron of a prenyl acceptor to attack the allylic cation generated on the prenyl donor, 60 may also be accepted by YanG to yield 63 before the epoxidation occurs, as 61 loses a π-electron at the prenylation site due to the epoxide formation.
The late-stage biosynthesis of yanuthone D (48) is well established, since the precursors of 48 were successfully obtained by the gene disruptions. The P450 YanH hydroxylates 62 to 22-deacetylyanuthone A (64), and the introduced hydroxyl group is then mevalonylated by the membrane-bound O-acyltransferase YanI to generate yanuthone E (65). Lastly, the flavin-containing dehydrogenase YanF oxidizes the secondary alcohol of 65, to produce the ketone functionality and complete the biosynthesis.
3.3 Biosynthesis of aspernidine A
The prenylated isoindolinone alkaloid, aspernidine A (49), has been isolated as a bioactive compound with moderate antiproliferative activity.36 The pkf cluster, responsible for the biosynthesis of 49, was subsequently identified in the genome of A. nidulans.46 The cluster is relatively small with six genes altogether, and the functions of each gene were investigated by gene deletion experiments (Fig. 6). | ||
| Fig. 6 Biosynthesis of aspernidine A. | ||
In the biogenesis of aspernidine A (49), PkfA, an NR-PKS with a reductase (R) domain at its C-terminus, first synthesizes orsellinaldehyde (66). Compound 66 is subsequently converted into aspernidine D (67), in which the farnesylation should be performed by the UbiA-like PTase PkfE, whereas the other enzymes participating in the remaining transformations (hydroxylations and methylation) have yet to be discovered. The P450 PkfB introduces a hydroxyl group to yield aspernidine E (68), which is proposed to be further oxidized to the dialdehyde 69 by the dehydrogenase PkfF. The last several steps leading to 49 are still enigmatic since no enzyme for this scheme has been identified.
4 Meroterpenoids derived from 3,5-demethylorsellinic acid
Among the fungal meroterpenoids, those derived from a simple aromatic tetraketide 3,5-dimethylorsellinic acid (70, DMOA) display huge structural diversity. After the isolation of the first member of this group in 1976,47 >100 compounds have been described as DMOA-derived meroterpenoids. These molecules include andrastin A (71),48–50 austinol (72),51 terretonin (73),52 anditomin (74),53 novofumigatonin (75),54 tropolactone A (76),55 paraherquonin (77),56 and emervaridione (78)57 (Fig. 7). It is noteworthy that they often possess complex and intriguing skeletons, as exemplified by the spiro-lactone of 72, the intramolecular bridged-ring systems of 74 and 78, the orthoester of 75, and the tropone moiety of 76. | ||
| Fig. 7 Representative DMOA-derived meroterpenoids. | ||
The biosyntheses of these fascinating molecules have been investigated for decades, and recently the molecular bases for four DMOA-derived meroterpenoids (71–74) have been reported,41,58–65 enabling more in-depth predictions of the biosynthetic mechanisms for other related meroterpenoids. One of the major contributors to the structural diversity of DMOA-derived meroterpenoids is the presence of many cyclization patterns of the terpenoid moiety, as well as the divergence of the pathway before the cyclization. The other important factor is the existence of a variety of tailoring reactions that often are responsible for dynamic structural rearrangements. This section explains how a simple aromatic precursor DMOA (70) can be transformed into diverse and complex compounds.
4.1 Diversity in early-stage biosynthesis
The biosynthetic clusters for andrastin A (71), austinol (72), and terretonin (73) have been identified in the genomes of Penicillium chrysogenum (the adr cluster),63A. nidulans (the aus cluster),59 and Aspergillus terreus (the trt cluster),60 respectively, and their functions have been investigated by gene deletion and/or heterologous expression approaches. The biosyntheses of 71–73 adopt the same early-stage reactions before the cyclization. At the outset, the NR-PKSs AdrD/AusA/Trt4 produce DMOA (70) in a similar manner to that of MpaA in the mycophenolic acid pathway, but use one additional molecule of SAM for the catalysis. The polyketide 70 subsequently undergoes farnesylation by the UbiA-like PTase AdrG/AusN/Trt2 to 66, methyl-esterification by the methyltransferase AdrK/AusD/Trt5 to 80, and epoxidation by the FMO AdrH/AusM/Trt8 to 81 (Fig. 8).60,61 The epoxidation can precede the methylation, but in all of the cases, the methyl ester group is essential for the following cyclization step.61,63 In the next biosynthetic process, the TPCs AdrI/AusL/Trt1 accept 81 to perform the cyclization to provide the common tetracyclic carbocationic intermediate 82, which is then transformed into different products according to the position where the deprotonation occurs. Deprotonations from C-11 (Ha, AdrI), C-1′ (Hb, AusL), and C-9′ (Hc, Trt1) yield andrastin E (83), protoaustinoid A (84), and preterretonin A (85), respectively, thus demonstrating that the terpene cyclization diverges the pathway for 71–73 (Fig. 8 and 9).61,63 It is remarkable that the stereochemistries of the epoxidation and cyclization reactions are rigidly controlled by the membrane-bound FMOs and the novel family of TPCs, respectively. | ||
| Fig. 8 Early-stage biosynthesis of DMOA-derived meroterpenoids. | ||
 | ||
| Fig. 9 Comparison of the cyclization reactions in the DMOA-derived meroterpenoids biosynthesis. | ||
The biosynthesis of anditomin (74) involves a different strategy to prepare the substrate for the TPC (Fig. 8), which was revealed by the functional analysis of the and cluster discovered in Emericella variecolor.41 The opening process to produce DMOA (70) by the NR-PKS AndM is identical to that for 71–74, but in the second step, before prenylation occurs, 70 is converted to the phthalide 86 by AndK. AndK is a chimeric protein of P450 monooxygenase and hydrolase and is homologous to MpaDE in the biosynthesis of mycophenolic acid, and thus it catalyzes two-rounds of reactions to provide 86. The phthalide 86 is then transformed into the pentacyclic intermediate preandiloid A (89) via87 and 88, in which the UbiA-like PTase AndD, the FMO AndE, and the TPC AndB are responsible for the conversion (Fig. 8 and 9). Interestingly, the absolute configuration of the terpenoid moiety of 89 is 3S,5R,8S,9S,10S, whereas that for 83–85 it is 3R,5S,8S,9S,10R. These differences could be attributed to the distinct absolute configurations between the precyclization intermediates, 81 and 88; the absolute configuration of 81 can be deduced to be 6R and 10′R, and that of 88 would be 4R and 10′S. This observation indicates that the anditomin pathway utilizes an epoxidase that forms an epoxide ring with different stereochemistry from that of 71–73.
Furthermore, similar considerations for the stereochemistry can be applied to the biosynthesis of novofumigatonin (75) and tropolactone D (90) (Fig. 8 and 9).54,55 Compound 90 has a similar skeleton to that of preandiloid A (89) with 5R,8S,9S,10S configuration, but the stereochemistry of one methyl group at C-12 (C-3′ of 89) is opposite between the two compounds. This position is the site where the farnesyl group is attached, and therefore a PTase that generates 91, an enantiomer of 66, might be involved in the biosynthesis of 90. Compound 91 would subsequently be converted to the tetracyclic intermediate 93via92, in a similar manner to that observed in the anditomin pathway. Meanwhile, considering its structure, 75 could be synthesized from 96 as the product of a TPC, although no biosynthetic study or proposed biosynthetic pathway has been reported to date. The direct precursor of 96 should be 94, which would be cyclized to 96via a cationic intermediate 95. Compound 94 is an epimer of 81 with an (S)-epoxide, suggesting that the novofumigatonin biosynthesis pathway employs an epoxidase with different product selectivity from that of AdrH/AusM/Trt8.
Collectively, not only the cyclization patterns but also the presence of PTases and epoxidases with different stereo-preferences greatly contribute to the production of the structural diversity of this class of natural products. It should be noted that the prenylation reactions in the DMOA-derived meroterpenoid biosynthesis destroy the aromaticity of the substrate DMOA (70), generating a chiral center in the product. This is different from the other PTases described above, in which the aromaticity of the substrates remains after prenylation, thus contributing to the creation of structural diversity in the DMOA-derived metabolites.
4.2 Biosynthesis of andrastin A and related compounds
Andrastins, including andrastin A (71) and andrastins B–D (94–96) have been isolated as inhibitors of the protein farnesyltransferase,48–50,66 and they have relatively simple structures among the DMOA-derived meroterpenoids. Consistent with their simple skeletons, the first tetracyclic intermediate andrastin E (83) is converted to the end product 71 with the aid of only four enzymes (Fig. 10).63 First, 83 is oxidized into the ketone form 96 by the SDR AdrF, which is followed by reduction of the C-3 carbonyl to a β-hydroxyl group to provide andrastin F (97), a C-3 epimer of 83. The reaction is catalyzed by the ketoreductase AdrE, and the discovery of 97 as a product of AdrE ruled out the previous proposal that 97 is directly generated by the terpene cyclization.67 The β-alcohol of 97 is acetylated by the acetyltransferase AdrJ to yield 95, and the P450 AdrA finally oxidizes the C-23 methyl group in two successive reactions to produce 94 and 71. | ||
| Fig. 10 Biosynthesis of andrastin A and related compounds. | ||
Although the structure of andrastin A (71) is not very complex, 71 appears to be further derivatized into more complicated meroterpenoids in other fungi, which produce citreohybridone J (98), other citreohybridones,67–69 isocitreohybridones,67,69–71 and citreohybriddiones.70,72 Meanwhile, atlantinones A (99) and B (100) possess the same carbon skeleton as that of 71, but differ in that they have the 3R configuration.73,74 This suggests that 83 does not undergo C-3 epimerization in the atlantinones pathway, and the lactonization of 101 generated by oxidation of the C-23 methyl group of 83 would produce 99, which is hydroxylated for conversion into 100.
4.3 Biosynthesis of terretonin and related compounds
The trt cluster, responsible for the biosynthesis of terretonin (73) and its analogue terretonin C (102)75 lacking the methyl ester group, has been biochemically and molecular biologically analyzed by targeted gene deletion and heterologous expression in A. oryzae, leading to the complete determination of the pathway (Fig. 11).60–62,65 The cyclase Trt1-product, preterretonin A (85), is oxidized at the C-3 position to generate preterrenoid (103) by the SDR Trt9, which is highly homologous to AdrF involved in the andrastin A pathway. Compound 103 is further hydroxylated at C-16 by the FMO Trt3 to yield terrenoid (104), which undergoes multistep oxidations by the P450 Trt6, and subsequent D-ring expansion to produce terretonin H (106). The ring-expanded product 106 seems to be derived from 105 by a spontaneous reaction, but the detailed mechanism for this rearrangement has yet to be clarified. | ||
| Fig. 11 Biosynthesis of terretonin and related compounds. | ||
Interestingly, 104 can also be transformed into terretonin D (107), which is similar to 106 but contains an additional methyl ester group at the C-16 position, only in the presence of Trt14. An isotope-incorporation experiment with [methyl-13C]methionine excluded the possibility that Trt14 is a methyltransferase and indicated the involvement of the intramolecular transfer of the methoxy group.65 Trt14 shares almost no amino acid sequence identity with any biochemically characterized proteins, but appears to be structurally related to some isomerases as well as epoxide hydrolases.76–79 The detailed role of Trt14 also has yet to be elucidated, but Trt14 would somehow be engaged in the intramolecular rearrangement of the methoxy group to afford the terretonin scaffold. Both 106 and 107 are then oxidized in two sequential reactions by the Fe(II)/α-ketoglutarate (α-KG)-dependent dioxygenase Trt7, which is homologous to phytanoyl-CoA 2-hydroxylase (PhyH).80 In the Trt7-catalyzed reactions, 106 and 107 are first transformed into terretonin J (108) and A (109), and then further oxidized to terretonin C (102) and terretonin (73), respectively, to complete the biosynthesis.
Terretonin E (110) and F (111) have similar carbon skeletons to that of terretonin (73), but possess a distinct D-ring system,81,82 suggesting that they are biosynthesized in a similar but slightly different manner from that for 73. In the proposed biosyntheses of 110 and 111, 105 is utilized as the pathway intermediate as well, and is converted to 113via112 by the nucleophilic attack of water at the C-17 ketone of 105 and the subsequent lactonization (Fig. 11). In contrast, the newest member of this group of meroterpenoids is terretonin G (114), from a marine-derived Aspergillus sp.,83 and it is most likely derived from the hydrolysis of the D-ring lactone of 107 and the subsequent formation of a methyl ester (Fig. 11).
4.4 Biosyntheses of austinol/dehydroaustinol and related compounds
The biosyntheses of austinol (72) and its oxidized form dehydroaustinol (115) have attracted keen interest for biosynthetic studies because of their intriguing structures, including the unique spiro-lactone system.84–86 The biosyntheses of 72 and 115 are controlled by the aus cluster, which is separately located in two different regions on the chromosomes.59 Almost the entire austinol pathway was initially elucidated by targeted gene disruption experiments,58,59 and a subsequent heterologous expression study illuminated the missing link in the biosynthesis, to allow the proposal of the complete pathway for 72 (Fig. 12).64 | ||
| Fig. 12 Biosynthesis of austinol/dehydroaustinol and related compounds. | ||
The tetracyclic molecule protoaustinoid A (84) is oxidized into berkeleyone A (116) by the FMO AusB, which is highly homologous to Trt3. This reaction is followed by C-3 dehydrogenation catalyzed by the PhyH-like Fe(II)/α-KG-dependent dioxygenase AusE to generate preaustinoid A (117). In the next process, the FMO AusC inserts one oxygen atom between C-3 and C-4 to yield preaustinoid A1 (118), serving as a Baeyer–Villiger monooxygenase (BVMO). Subsequently, AusE again accepts 118 to perform two consecutive oxidations, in which 118 undergoes dehydrogenation to preaustinoid A2 (119) and oxidative rearrangement to preaustinoid A3 (120). Thus, AusE catalyzes multistep oxidation reactions before and after the Baeyer–Villiger reaction to generate the spiro-lactone ring system.
Meanwhile, in other meroterpenoid pathways, preaustinoid A1 (118), with a seven-membered lactone A-ring, is likely to be transformed into berkeleydione (121)87,88via berkeleyone B (122),89 probably in two successive reactions similar to those of the multifunctional dioxygenase AusE. Compound 121 seems to be a common biosynthetic intermediate of many fungal meroterpenoids, including paraherquonin (77),56 berkeleyacetals,16 miniolutelides,90,91 and dhilirolides.92,93 The possible mechanisms for the spiro-ring formation and the cycloheptadiene-formation of 121 are discussed later in this section.
The next several steps of the pathway are quite interesting, since they apparently involve three novel classes of proteins, AusJ, AusF, and AusH, which are all homologous to Trt14 in the terretonin biosynthesis and share almost no amino acid sequence homology to any characterized enzymes. AusJ catalyzes the isomerization of 120 to produce preaustinoid A4 (123) with an acetyl group, but the enzyme also seems to produce preaustinoid A5 (124), an isomer of 120 and 123, as a shunt product. Compound 124 is predicted to be further transformed into austinoneol A (125), but the reaction mechanism and the enzyme responsible for this conversion have never been proposed. Given the proposed mechanism for Trt14,65 it is possible that the reaction is also catalyzed by AusJ, in which AusJ performs the deacetylation by retro-Claisen cleavage. Intriguingly, the deletion mutant of ausF, encoding a protein homologous to AusJ (54% identity), accumulated 120 and 125 as the major metabolites, although it also produced austinol (72) and dehydroaustinol (115) in low yields. Thus, AusF was proposed to have a regulatory function in the biosynthesis,59 but it is also possible that AusJ and AusF work collaboratively to synthesize the pathway intermediate 123, and that AusJ alone is not sufficient to produce a good yield of 123.
Preaustinoid A4 (123) is then converted into isoaustinone (128), with the aid of the ketoreductase AusK and AusH, and, interestingly, 123 is transformed into the shunt metabolite (5′S)-isoaustinone (129), the C-5′ epimer of 128, in the absence of AusH. Due to the lack of intermediates between 123 and 128, the mechanism for the lactonization is still unclear, but it could be proposed as follows. AusK reduces the ketone functionality of the acetyl group to produce 126 with the 5′R configuration, which is spontaneously lactonized to yield 129. Contrary to this, in the presence of AusH, the apparently unstable intermediate 126 is accepted by AusH to undergo C-5′ epimerization to generate 127, and the following lactonization provides 128.
Finally, two P450 monooxygenases participate in the formation of austinol (72), in which AusI serves as the BVMO to yield austinolide (130) and AusG hydroxylates 130 to austinol (72). The very last step of the biosynthesis to provide dehydroaustinol (115) remains unresolved, since no enzyme for this conversion has been identified. At least, the enzyme(s) does not appear to be encoded in the aus cluster. In other fungi, the isoaustinone (128) and austinol/dehydroaustinol undergo further decorations, including hydroxylation and acetylation, to produce compounds such as austin (131), dehydroaustin (132),94 7-hydroxydehydroaustin (133),95 acetoxydehydroaustin (134),96 and 11-α-acetoxyisoaustinone (135).95
One of the most interesting reactions in the austinol pathway is the spiro-ring formation by AusE. Fe(II)/α-KG-dependent dioxygenases are known to initiate their reactions by abstracting a hydrogen atom from the substrate using the active site Fe(IV)-oxo species, which is generated by the oxidative decarboxylation of the cosubstrate α-KG.97 On the basis of the general mechanism of this class of dioxygenases, several scenarios for the spiro-lactone synthesis can be described (Fig. 13A). In the originally proposed mechanism,64 the C-5 hydrogen of the substrate preaustinoid A2 (119) is abstracted by the enzyme to produce the radical species 136, which is hydroxylated by the enzyme to generate the hydroxylated intermediate 137 bound to an Fe(II) species. Upon elimination of the C-5 hydroxyl group as a water molecule, the rearrangement and deprotonation from C-9 occur to afford the spiro-lactone preaustinoid A3 (120) (Path A). Subsequently, an alternative mechanism has been proposed, since the above-described mechanism requires a chemically unfavorable syn-rearrangement.98 The other scheme involves the cyclopropane intermediate 138, which is further rearranged to 139 with the spiro-lactone moiety. The enzymatic reaction is hereby completed by the rebound of the hydroxyl radical to 139 to yield 140, which undergoes spontaneous dehydration to produce 120 (Path B-1). However, it is still questionable whether the dehydration indeed occurs non-enzymatically, as 120 might be directly generated from 139 by the abstraction of the C-9 hydrogen atom (Path B-2).
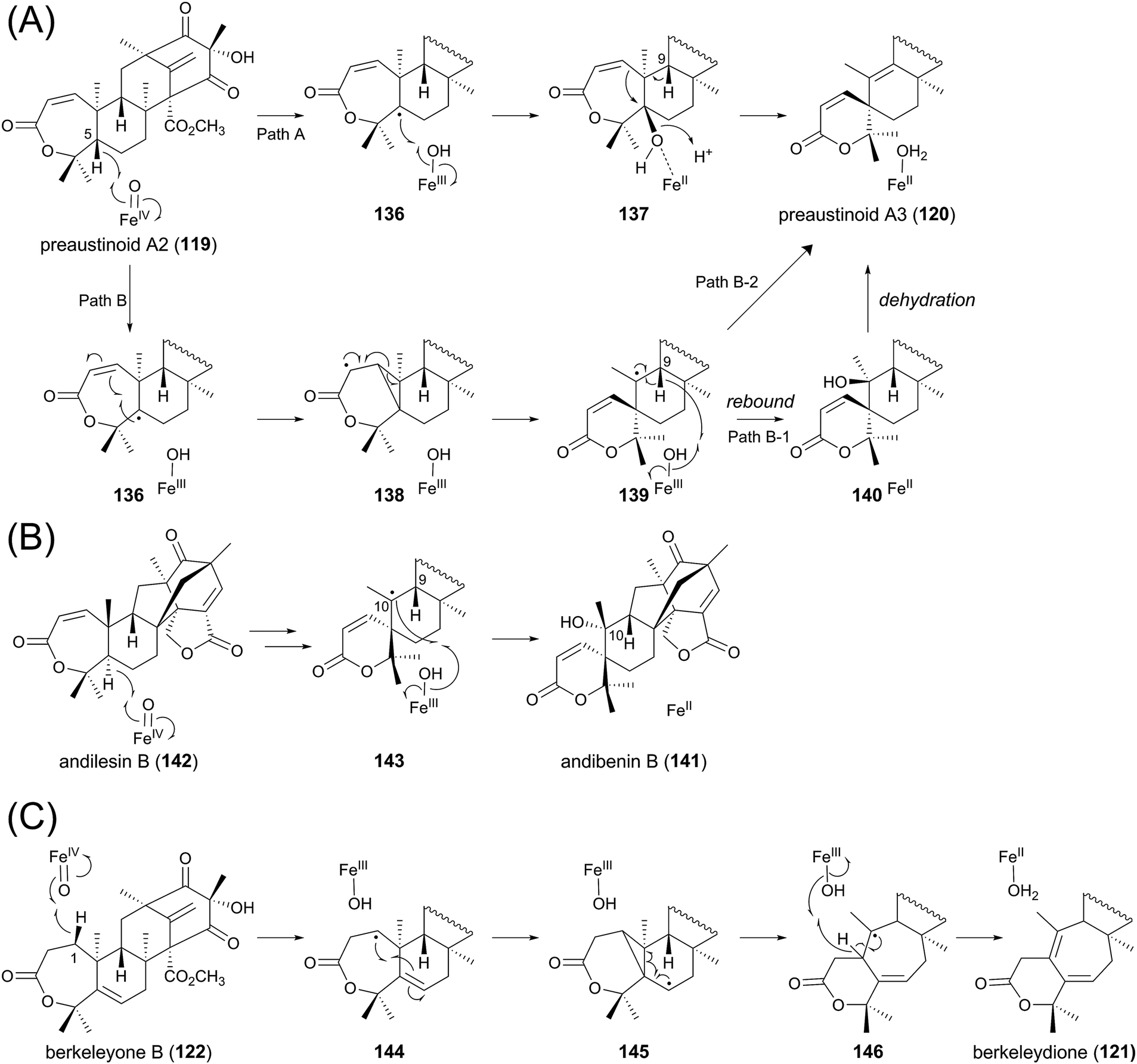 | ||
| Fig. 13 Proposed reaction mechanisms for formation of (A) preaustinoid A3, (B) andibenin B, and (C) berkeleydione. | ||
Although there is no experimental evidence to distinguish the three proposals described above, the biosynthetic study on andibenin B (141) provided the clue to solve this problem. Compound 141 contains a similar spiro-lactone moiety to that of 120, and is apparently derived from andilesin B (142) in a manner resembling that described for AusE. The previous study elucidated that the C-10 hydroxyl group of 141 can be labeled with 18O2.99 If the reaction employs the cationic pathway (similar to Path A), then this position should not be labeled with 18O2, as a water molecule would be used for the neutralization. This observation strongly suggests that the spiro-ring formation occurs by the radical-based pathway (similar to Path B), in which 143 exists as a reaction intermediate (Fig. 13B). One major difference regarding the carbaspirocycle-forming reactions in the austinol and andibenin pathways is that the two hydrogen atoms at C-5 and C-9 are both α-oriented in 119, whereas they have different orientations in 142. Thus, in the reaction for the synthesis of 141, the C-9 hydrogen is on the opposite side of the active center, and the enzyme cannot abstract the C-9 hydrogen. This could explain why only the C-10 α-hydroxylated product is obtained in the andibenin pathway. Importantly, there has been no report that 141 undergoes spontaneous dehydration, although its C-9 hydrogen and C-10 hydroxyl group are in more favorable positions for dehydration than those in 140. Collectively, Path B-2 in Fig. 13A would be the most plausible mechanism for the generation of preaustinoid A3 (120).
As mentioned above, berkeleydione (121) would also be produced by a similar mechanism from berkeleyone B (122) as the substrate (Fig. 13C). The reaction would be initiated by the abstraction of the C-1 hydrogen, yielding the substrate radical 144. The cyclopropane-forming reaction could then provide 145, which undergoes the carbon skeletal rearrangement to afford 146. The hydrogen abstraction and resultant introduction of the double bond would generate the product 121.
On the basis of the biosynthetic pathway and the enzymes for austinol (72) production, likely biosynthetic mechanisms of other related meroterpenoids can now be proposed. Here, the plausible biosyntheses of berkeleyacetal C (12) and dhilirolide A (147), which are both presumably derived from berkeleydione (121), are discussed (Fig. 14). In the berkeleyacetal C pathway, the exo-methylene of 121 would initially be oxidized into an aldehyde to generate 148. Retro-Claisen cleavage would then yield the ring-opened intermediate 149, and the recyclization of 149 would produce berkeleyacetal A (150),16 which possesses the same scaffold as 12. The existence of 148 is supported by the isolation of 22-epoxyberkeleydione (151) from several fungi.90,91 Indeed, since some P450s can oxidize an exo-olefin to both an epoxide and an aldehyde,100151 could be the shunt product generated at the same time as 148. Double bond isomerization and epoxidation would then transform 150 into berkeleyacetal B (152).16 The pathway from 152 to 12 involves C-7 oxidation and elimination of the methyl ester group, which would proceed via the demethylation of the methyl ester followed by the spontaneous decarboxylation of the β-keto acid 153. Meanwhile, the biosynthesis of 147 has been investigated by isotope-feeding studies, and a possible pathway has been proposed.93 However, the detailed mechanisms for the biosynthesis and the genuine intermediates have yet to be elucidated. In one rational explanation, 121 would first be converted to a known natural product dhilirolide D (154),92 in which the D- and E-rings should be synthesized in reactions similar to those catalyzed by AusJ and AusK in the austinol biosynthesis, but an AusH-like enzyme is not likely to be involved in the pathway. The next process would generate the C-9 cation, which induces the ring contraction and the acetyl group formation, to afford 155. Furthermore, ring-closure reactions could occur on 155 by two successive nucleophilic attacks, leading to the synthesis of dhilirolide J (156).93 Finally, two olefin epoxidations convert 156 to the end product dhilirolide A (147).
 | ||
| Fig. 14 Proposed biosynthesis of berkeleyacetal C and dhilirolide A. | ||
4.5 Biosyntheses of anditomin and related compounds
The biosyntheses of anditomin (74)53 and its biosynthetic precursors, andilesins A–C (157, 142, 158),101,102 have been intensively studied by labeling experiments because of their intriguing intramolecular bridged-ring system,99,103–106 which was proposed to be synthesized via a [4 + 2]-cycloaddition (Diels–Alder reaction) (Fig. 15). The proposed reaction also convinced synthetic chemists to perform biomimetic reactions.107 The complete biosynthetic pathway of 74 was recently established, mainly by the reconstitution of its biosynthesis in A. oryzae (Fig. 16), which confirmed that the Diels–Alder reaction is actually not involved in the biosynthesis.41 | ||
| Fig. 15 Previously proposed mechanism for the bridged-ring system formation in the anditomin/andilesins pathway. | ||
 | ||
| Fig. 16 Biosynthesis of anditomin and related compounds. | ||
In the anditomin pathway, the pentacyclic intermediate 89, generated by the TPC AndB, is oxidized at C-3 by the SDR AndC, which is homologous to AdrF and Trt9, to yield preandiloid B (159). AndA, the PhyH-like Fe(II)/α-KG-dependent dioxygenase, subsequently catalyzes two consecutive reactions to convert 159 to andiconin (161) via preandiloid C (160), by constructing the unique bridged framework characteristic of the andilesins. Intermediate 161 then undergoes Baeyer–Villiger oxidation to generate andilesin D (162) by the FMO AndJ, which is followed by ketoreduction by the SDR AndI to afford andilesin A (157). The acetyltransferase AndG then acetylates 157 to produce 163, which undergoes spontaneous acetate elimination to yield an enone andilesin B (142) due to the generation of a better leaving group. The enoylreduction of 142 by the SDR AndH and the following oxidative rearrangement by another α-KG-dependent dioxygenase AndF eventually produces andilesin C (158) and anditomin (74), completing the biosynthesis.
Andibenins A–C (164, 141, 165),102 analogues of andilesin with a spiro-lactone moiety, should be synthesized by a similar pathway to that of anditomin (74); andilesin A (157) could be initially oxidized to 164 by the mechanism shown in Fig. 13B, and subsequent dehydration and reduction would produce 141 and 165. Alternatively, 141 and 165 could be obtained via the direct oxidations of andilesin B (142) and andilesin C (158), respectively.
One of the most fascinating reactions in the anditomin pathway is the generation of the bicyclo[2.2.2]octane system by the multifunctional Fe(II)/α-KG-dependent dioxygenase AndA. In the proposed mechanism for this rearrangement (Fig. 17), the hydrogen atom of the C-12 methyl group of preandiloid C (160) is first abstracted to yield the substrate radical 166, and the subsequent C–O bond cleavage generates the resonance-stabilized radical species 167. The electrophilic radical of 167 at C-5′ would then form a C–C bond with the electron-rich olefin at C-12 to generate 168, which has the nucleophilic radical and the electron-deficient olefin. Thus, C–C bond formation occurs between C-8 and C-2′ to generate the product radical 169. The reaction by AndA is known to require ascorbate as an essential factor in vitro, and therefore the reducing agent should be used at the last step to reduce 169 to the product andiconin (161) as well as the ferric ion in the active site. Similarly, FtmF/FtmOx1 from A. fumigatus, which is the endoperoxide-generating α-KG-dependent dioxygenase, also reportedly requires a reducing agent for its reaction.108,109
 | ||
| Fig. 17 Proposed reaction mechanisms for AndA-catalyzed reaction and generation of the insuetolide scaffold. | ||
The above-discussed mechanism of AndA also provides insight into the biosynthesis of insuetolides A–C (170–172), which are analogous to andilesins but possess one additional inserted oxygen atom (Fig. 17).110 The scaffolds of 170–172 should be generated via a similar mechanism to that for AndA: oxygen rebound to 168 would occur, leading to the formation of 173 with a C-8 hydroxyl group. The resultant alcohol could spontaneously attack the electrophilic C-2′ to afford 174. Further modifications would synthesize 170–172.
4.6 Biosyntheses of fumigatonin and novofumigatonin
Despite the intriguing structures of fumigatonin (175)111 and novofumigatonin (75),54 isolated from A. fumigatus and Aspergillus novofumigatus, respectively, with the unusual orthoester moiety, neither biosynthetic studies nor a proposed pathway for these compounds have been reported. The expected intermediate 96 should undergo multistep decorations to afford 75 and 175. The modification of the A-ring would be performed in a similar manner to that seen in the austinol or anditomin pathways, while the oxidation of the C-13 methyl group resembles the reaction by AdrA in the andrastin A biogenesis. One possible scenario for the orthoester formation can be proposed as follows (Fig. 18). Oxidative cleavage of the C-1′–C-6′ olefin, ketoreduction at C-5′, and reduction of the double bond between C-3′ and C-4′ could provide the intermediates 176 and 177. In the novofumigatonin pathway, the C-5′ alcohol of 177 initially attacks the C-1′ carbonyl, and the resulting hemiorthoester anion then attacks the C-6′ ketone. Thus, the generated hemiacetal anion again attacks the C-7′ carbonyl to yield the molecular skeleton of novofumigatonin (75), along with the elimination of a methoxide. In contrast, the C-5′ hydroxyl group of the predicted intermediate of 175, 176, is acetylated and thus it is unable to initiate the reaction by the nucleophilic attack from the C-5′ alcohol. Alternatively, the C-13 hydroxyl group could be used as the nucleophile to initiate the similar successive nucleophilic attacks, providing the orthoester moiety of fumigatonin (175). | ||
| Fig. 18 Proposed reaction mechanisms for orthoester formation in the fumigatonin/novofumigatonin pathways. | ||
5 Indole-diterpenoids
Indole-diterpenoids are a large class of natural products with diverse structures; they are also hybrid molecules consisting of an indole-ring as a non-terpenoid group and a geranylgeranyl pyrophosphate (GGPP)-derived terpenoid moiety. The indole-diterpenoids are further classified into two major groups: paspaline (178)-derived compounds and non-paspaline-derived ones. For example, paxilline (179),112 janthitrem B (180),113 and sulpinine A (181)114 are generated from 178, whereas emindole SA (182),115 thiersinine A (183),116 radarin A (184),117 anominine (185),118 tubingensin B (186),119 and aflavinine (187)120 are synthesized via different pathways (Fig. 19). After the first discovery of the biosynthetic gene cluster for 179 in 2001,121 the gene clusters for aflatrem (188),122 terpendole K (189),123 lolitrem B (14),124,125 penitrem A (190)126,127 and, shearinine A (191)127 were reported and analyzed. These compounds are all synthesized via178, and no biosynthetic gene cluster for non-paspaline-derived the indole-diterpenoids has been identified. Although the gene clusters for indole-diterpenoids lack PKS genes, consistent with the fact that they lack a polyketide portion, the biosynthetic schemes are basically similar to those of the other meroterpenoids discussed above. In this section, the biosynthetic studies and pathways for paspaline-derived molecules are summarized (Fig. 20 and 21). | ||
| Fig. 19 Representative indole-diterpenoids. | ||
 | ||
| Fig. 20 Biosynthesis of paspaline. | ||
 | ||
| Fig. 21 Late-stage biosynthesis of paxilline-derived indole-diterpenoids. | ||
5.1 Biosynthesis of paxilline
The function of the biosynthetic gene cluster for paxilline (179) has been investigated by gene deletion, complementation, and heterologous reconstitution experiments, which established the complete biosynthetic route of 179.121,128–132 In the paxilline pathway, indole-2-glycerol phosphate (192) serves as the indole donor, which is geranylgeranylated by the PTase PaxC to yield 3-geranylgeranylindole (193). The prenylation reaction differs from the above-described meroterpenoid pathways in two aspects. First, the prenyl donor geranylgeranyl pyrophosphate (GGPP) is synthesized by the GGPP synthase PaxG encoded by the cluster. Second, PaxC is a soluble protein and shares almost no sequence homology with the transmembrane UbiA-like PTases that are often found in the meroterpenoid biosynthesis.The 3-geranylgeranylindole (193) intermediate then undergoes two rounds of epoxidation–cyclization processes to generate paspaline (178). The FMO PaxM initially forms an epoxide ring at C-10 and C-11 to produce 194, which is accepted by the TPC PaxB for cyclization into emindole SB (195). Epoxidation again occurs on 195 to provide 196, and PaxB-catalyzed cyclization produces 178. Originally, the bisepoxide 197 was thought to be the biosynthetic intermediate, and it has also been revealed that PaxB utilizes synthetically prepared 197 as a substrate to afford 178.131 However, the heterologous expression of paxC, paxG, and paxM in A. oryzae resulted in accumulation of 194, not 197, thus indicating that 197 is not used in the natural pathway. The biosynthesis up to 178 seems to be shared by the other biosyntheses for 14 and 188–191 (Fig. 20).
In the late-stage of the biosynthesis, two P450s are responsible for the generation of paxilline (179) from paspaline (178).130 PaxP catalyzes multistep oxidative reactions, including demethylation, to yield 13-desoxypaxilline (198), which is hydroxylated by PaxQ to afford 179 (Fig. 21).
Interestingly, the pax cluster encodes a protein homologous to DMATSs, PaxD, and the function of the PTase has been investigated by in vitro reactions using the purified enzyme.133,134 PaxD attaches a dimethylallyl group at either C-21 or C-22, and also produces diprenylated compounds at both of these positions.133 These compounds can indeed be detected in a paxilline-producing strain, but the productivity is quite low (∼0.5% of 179),133 and thus it might not be the intended metabolite in the pax pathway. An in vitro assay also elucidated that PaxD accepts 178 to perform dimethylallylation at either the C-21 or C-22 position to generate two mono-prenylated products.134
Furthermore, the involvement of paxA in paxilline biosynthesis is indicated, since the paxA-deletion mutant is unable to produce paxilline (179) and the deficiency is complemented by the reintroduction of paxA.132 PaxA is predicted to be a six-transmembrane protein and is also present in the other indole-diterpenoid biosynthetic gene clusters. However, reconstitution studies have revealed that paxA was not required for the production of 179,131 and therefore the role of paxA remains enigmatic.
5.2 Biosyntheses of aflatrem and shearinine A
Aflatrem (188) is synthesized via a very similar scheme to that for paxilline (179) since it utilizes 13-desoxypaxilline (198) as the pathway intermediate. The P450 AtmQ is highly homologous to PaxQ; however, it converts 198 into paspalinine (199) with an ether linkage.135,136 The DMATS-type PTase AtmD subsequently catalyzes reverse prenylation at either the C-20 or C-21 position to provide 188 and its isomer β-aflatrem (200), respectively (Fig. 21). In vitro experiments with purified AtmD illuminated that AtmD can also accept 178 and 179 as substrates to perform prenylation reactions.134 The entire pathway to produce 188 was reconstructed in A. oryzae, verifying the biosynthesis of 188.136Paspalinine (199) is also utilized in the biosynthesis of shearinine A (191), which is likely to undergo diprenylation by the DMATS-type PTase JanD to yield shearinine K (201).127 The subsequent oxidation ring closure may be performed by the FMO JanO and/or the P450 PJ-13, leading to the formation of 191 (Fig. 21).
5.3 Biosyntheses of terpendole K and lolitrem B
The biosyntheses of terpendole K (189) and lolitrem B (14) diverge from the paxilline or aflatrem pathways just after paspaline (178) is generated (Fig. 21).123,125,137 The P450 TerQ/LtmQ oxidizes 178 to generate the C-11 hydroxylated product terpendole E (13), which is accepted by the other P450 TerP/LtmP and converted to 13-desoxyterpendole I (202). TerQ/LtmQ again hydroxylates 202 to produce terpendole I (203). The last several steps in the terpendole K pathway are proposed to involve the PTase TerF and the P450 TerK, in which TerF attaches a dimethylallyl moiety to the C-27 alcohol, and TerK catalyzes the ether-forming ring closure reaction.123 Finally, an unidentified dehydrogenase would introduce the double bond between C-6 and C-7 to afford 189. Meanwhile, the lolitrem B biosynthesis utilizes the homologous proteins of TerF and TerK, LtmF, and LtmK, respectively, which are responsible for the same reactions to construct the eastern part of 14. Additionally, LtmE and LtmJ, another PTase and P450 set achieve the prenylations at C-20 and C-21 and the oxidative ring closure to afford the western portion of 14, and complete the biosynthesis.1375.4 Biosynthesis of penitrem A
Among the indole-diterpenoids, one of the most complex molecules, penitrem A (190), requires 17 dedicated genes for its biosynthesis.126 The biosynthetic gene cluster for 190 was initially identified in the genome of Penicillium simplicissimum (the ptm cluster),126 and was later discovered in Penicillium crustosum as well (the pen cluster).127 Since the complete pathway of 190 has been elucidated using the ptm genes, the functions of the Ptm enzymes are focused on below (Fig. 21).After the formation of paxilline (179) in the same manner as that by the Pax enzymes, 179 undergoes ketoreduction at C-10 by PtmH, C-20 dimethylallylation by PtmD, dehydration by PtmI and PtmV, and hydroxylation by PtmO to produce PC-M5 (204). Subsequently, the key transformation in the pathway, the construction of the bicyclo[3.2.0]heptane system, is catalyzed by the PTase PtmE to afford PC-M4 (205). Finally, four P450s, PtmK, PtmU, PtmL, and PtmJ, as well as the flavin-dependent halogenase PtmN synthesize the end product penitrem A (190), in which PtmK is engaged in the ring expansion, and PtmU generates the ether linkage to provide penitrem D (206).
6 Miscellaneous meroterpenoids
Although the above-described meroterpenoid biosyntheses generally involve prenylation at the early-stage of the biosynthesis and cyclization by transmembrane TPCs, meroterpenoids are sometimes generated via completely different machineries. This section focuses on the meroterpenoids that require the hybridization of terpenoid and non-terpenoid portions at a later stage and adopt different cyclization strategies for their biosynthesis.6.1 Biosynthesis of viridicatumtoxin
Viridicatumtoxin (207), isolated from Penicillium aethiopicum, represents tetracycline-like molecules derived from fungi, and reportedly possesses nephrotoxic or antitumor activities.138–140 The biosynthesis of 207 has been studied by gene deletion and in vitro analysis of selected enzymes (Fig. 22).141–144 The tetracyclic core of 207 is generated collaboratively by the NR-PKS VrtA, the Claisen cyclase VrtG, and the FMO VrtH, from one molecule of malonamoyl-CoA and nine molecules of malonyl-CoA, yielding 208.142 The P450 VrtE and the methyltransferase VrtF then convert 208 into 209, which is further geranylated by the DMATS-type PTase VrtC to generate 210.143 The very last step of the biosynthesis involves the cyclization of the geranyl moiety, and interestingly this process is catalyzed by the P450 VrtK and not by the canonical TPCs.144 Thus, the viridicatumtoxin pathway utilizes neither a transmembrane PTase nor a TPC to afford the cyclized terpenoid moiety. | ||
| Fig. 22 Biosynthesis of viridicatumtoxin. | ||
6.2 Biosynthesis of penigequinolones
Penigequinolones A (211) and B (212) are insecticidal compounds produced by various fungi, and possess a quinolone alkaloid core structure equipped with a cyclized C10 terpenoid moiety.145 The biosynthetic gene cluster for 211 and 212 (the pen cluster)146 was identified in Penicillium thymicola by searching for a similar gene cluster to the asq cluster responsible for the biosynthesis of aspoquinolones,147 which are analogues of 211 and 212. Indeed, the two pathways appear to share most of their biosynthetic machinery. The early stage of the biosynthesis was investigated with the asq cluster, whereas the mid-stage was elucidated by analyzing the pen cluster (Fig. 23). | ||
| Fig. 23 Biosynthesis of penigequinolones. | ||
Initially, the non-ribosomal peptide synthase (NRPS) AsqK/PenN condenses anthranilic acid (213) and O-methyl-L-tyrosine (214) to produce the benzodiazepinedione, 4′-methoxycyclopeptin (215). Subsequently, the PhyH-like dioxygenase AsqJ/PenM performs oxidative reactions on 215, and the following spontaneous structural rearrangement provides the quinolone core to generate 4′-methoxyviridicatin (216),147 which is further transformed into 217 by an uncharacterized pathway.
Considering the structures of 211 and 212, it was thought that the terpenoid motif of these compounds is derived from geranyl pyrophosphate (GPP), but interestingly, a biochemical experiment has revealed that PenI, which is the PTase that accepts 217 as a substrate, only produces the dimethylallylated molecule 218.146 In the next step, the FMO PenH catalyzes dehydrogenation to generate the conjugated diene 219, and the introduced exo-methylene hereby serves as a nucleophile required for the second prenylation by the DMATS-like PTase PenG. Thus, PenG catalyzes the prenyl elongation reaction to afford 220 with a C10 side chain, in which a water molecule is used to terminate the reaction.
In the following processes, the FMO PenE, which is homologous to PaxM in the paxilline biogenesis, catalyzes epoxidation at the terminal olefin to yield 221.146 The epoxide hydrolase PenJ then accelerates the 5-exo-tet cyclization to afford yaequinolone C (222), which is somehow converted into penigequinolones A (211) and B (212).
6.3 Biosynthesis of fumagillin
The biosynthesis of fumagillin (8) is significantly distinct from all of the other pathways described above, in that the terpenoid moiety is first cyclized and tailored before it is hybridized with the polyketide portion (Fig. 24). A similar synthetic strategy is also employed in other biosyntheses, such as the xenovulene A pathway.1,148,149 The fumagillin biosynthesis begins with the cyclization of FPP by the TPC Fma-TC to produce β-trans-bergamotene (223).150 The Fma-TC-catalyzed reaction is initiated by the elimination of a pyrophosphate group to generate the allylic cation, and therefore it can be classified as a class I TPC. However, Fma-TC is a transmembrane protein homologous to UbiA, and shares no sequence similarity with any known TPCs, although plants can produce several bergamotenes with soluble class I enzymes. At present, no other UbiA-like TPCs have been discovered, but they could also be present in the genomes of different fungal species. | ||
| Fig. 24 Biosynthesis of fumagillin. | ||
β-trans-Bergamotene (223) then undergoes multistep oxidations as well as structural rearrangement by the P450 Fma-P450 to yield 5-keto-demethoxyfumagillol (224), which is further converted to fumagillol (225) with the aid of the PhyH-like dioxygenase Fma-C6H, the methyltransferase Fma-MT, and the ketoreductase, Fma-KR.151 In contrast, the polyketide motif of fumagillin (8) is prepared by the highly reducing PKS Fma-PKS, which synthesizes an enzyme-bound dodecapentaenoyl group (226).150 The polyketide portion is then trans-esterified with 225 by the acyltransferase Fma-AT, providing prefumagillin (227). Finally, Fma-ABM, which is structurally related to the ABM superfamily of monooxygenase, catalyzes the oxidative cleavage of 227 to afford the end product 8.151
7 Conclusions and prospects
This review has summarized recent biosynthetic studies of fungal meroterpenoids at the genetic/enzymatic levels. Although the structures of fungal meroterpenoids are extremely diverse and often highly complex, their biosyntheses employ similar enzymes and mechanisms to synthesize the wide range of natural products. Thus, the fungal meroterpenoid pathways have revealed nature's sophisticated strategy to generate complexity by using simple and achiral starting units.Biosynthetic studies of unique molecules have indeed led to the discoveries of novel families of enzymes with intriguing activities, such as transmembrane TPCs (both class I and II) and isomerases including Trt14, AusJ, and AusH. However, it should be noted that fascinating reactions are often catalyzed or induced by ordinary enzymes on the basis of their sequences; indeed, oxidative enzymes are especially responsible for amazing transformations in many pathways, and are often multifunctional.98,152 For instance, PhyH-like Fe(II)/α-KG-dependent dioxygenases are widely utilized in fungal meroterpenoid biosyntheses, and their catalyzing/inducing reactions range from simple hydroxylation (as by Trt7 and Fma-C6H) to drastic structural rearrangement (as by AusE, AndA, and AsqJ).
Finally, due to the characterization of many biosynthetic gene clusters for fungal meroterpenoids and the rapid increase of available fungal genomes, we can now readily discover many putative gene clusters for meroterpenoid biosynthesis with uncharacterized functions. By means of well-established approaches in recent years including heterologous reconstitution techniques, metabolites derived from these gene clusters can be easily obtained, even though the clusters are silent. Furthermore, the genes in both characterized and uncharacterized gene clusters could be used as “genetic building blocks” to construct artificial metabolic pathways to afford unnatural meroterpenoids. Future studies on fungal meroterpenoids should accelerate not only the discovery of enzymes with unique activities but also the accumulation of knowledge to efficiently compile many biosynthetic genes to synthesize desired molecules.
8 Acknowledgements
The authors would like to express sincere appreciation to an excellent group of co-workers whose contributions are cited in the text, in particular to Dr Takayoshi Awakawa, Takahiro Mori, and Toshiyuki Wakimoto (The University of Tokyo). This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.9 References
- R. Geris and T. J. Simpson, Nat. Prod. Rep., 2009, 26, 1063–1094 RSC.
- R. Morton, Nature, 1958, 182, 1764–1767 CrossRef CAS PubMed.
- E. Fernholz, J. Am. Chem. Soc., 1938, 60, 700–705 CrossRef CAS.
- J. W. Moncrief and W. N. Lipscomb, J. Am. Chem. Soc., 1965, 87, 4963–4964 CrossRef CAS.
- Y. Gaoni and R. Mechoulam, J. Am. Chem. Soc., 1964, 86, 1646–1647 CrossRef CAS.
- L. Kaysser, P. Bernhardt, S.-J. Nam, S. Loesgen, J. G. Ruby, P. Skewes-Cox, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2012, 134, 11988–11991 CrossRef CAS PubMed.
- T. Awakawa, L. Zhang, T. Wakimoto, S. Hoshino, T. Mori, T. Ito, J. Ishikawa, M. E. Tanner and I. Abe, J. Am. Chem. Soc., 2014, 136, 9910–9913 CrossRef CAS PubMed.
- M. D. Sintchak, M. A. Fleming, O. Futer, S. A. Raybuck, S. P. Chambers, P. R. Caron, M. A. Murcko and K. P. Wilson, Cell, 1996, 85, 921–930 CrossRef.
- L. M. Shaw, H. W. Sollinger, P. Halloran, R. E. Morris, R. W. Yatscoff, J. Ransom, I. Tsina, P. Keown, D. W. Holt, R. Lieberman, A. Jaklitsch and J. Potter, Ther. Drug Monit., 1995, 17, 690–699 CrossRef CAS.
- M. C. McCowen, M. E. Callender and J. F. Lawlis, Science, 1951, 113, 202–203 CAS.
- J.-M. Molina, M. Tourneur, C. Sarfati, S. Chevret, A. de Gouvello, J.-G. Gobert, S. Balkan and F. Derouin, N. Engl. J. Med., 2002, 346, 1963–1969 CrossRef CAS PubMed.
- S. Liu, J. Widom, C. W. Kemp, C. M. Crews and J. Clardy, Science, 1998, 282, 1324–1327 CrossRef CAS.
- H. Tomoda, H. Nishida, Y. K. Kim, R. Obata, T. Sunazuka, S. Ōmura, J. Bordner, M. Guadliana, P. G. Dormer and A. B. Smith, J. Am. Chem. Soc., 1994, 116, 12097–12098 CrossRef CAS.
- J.-W. Chen, Y.-L. Luo, M.-J. Hwang, F.-C. Peng and K.-H. Ling, J. Biol. Chem., 1999, 274, 34916–34923 CrossRef CAS PubMed.
- N. Minagawa, Y. Yabu, K. Kita, K. Nagai, N. Ohta, K. Meguro, S. Sakajo and A. Yoshimoto, Mol. Biochem. Parasitol., 1996, 81, 127–136 CrossRef CAS.
- D. B. Stierle, A. A. Stierle and B. Patacini, J. Nat. Prod., 2007, 70, 1820–1823 CrossRef CAS PubMed.
- T. Etoh, Y. P. Kim, H. Tanaka and M. Hayashi, Eur. J. Pharmacol., 2013, 698, 435–443 CrossRef CAS PubMed.
- J. Nakazawa, J. Yajima, T. Usui, M. Ueki, A. Takatsuki, M. Imoto, Y. Y. Toyoshima and H. Osada, Chem. Biol., 2003, 10, 131–137 CrossRef CAS.
- R. T. Gallagher, A. D. Hawkes, P. S. Steyn and R. Vleggaar, J. Chem. Soc., Chem. Commun., 1984, 614–616 RSC.
- S.-M. Li, Nat. Prod. Rep., 2010, 27, 57–78 RSC.
- M. E. Tanner, Nat. Prod. Rep., 2015, 32, 88–101 RSC.
- H. Tomoda, N. Tabata, Y. Nakata, H. Nishida, T. Kaneko, R. Obata, T. Sunazuka and S. Ōmura, J. Org. Chem., 1996, 61, 882–886 CrossRef CAS.
- T. Itoh, K. Tokunaga, Y. Matsuda, I. Fujii, I. Abe, Y. Ebizuka and T. Kushiro, Nat. Chem., 2010, 2, 858–864 CrossRef CAS PubMed.
- J. Hu, H. Okawa, K. Yamamoto, K. Oyama, M. Mitomi and H. Anzai, J. Antibiot., 2011, 64, 221–227 CrossRef CAS PubMed.
- J. Hu, A. Furutani, K. Yamamoto, K. Oyama, M. Mitomi and H. Anzai, Biotechnol. Biotechnol. Equip., 2014, 28, 818–826 CrossRef CAS PubMed.
- W. Cheng and W. Li, Science, 2014, 343, 878–881 CrossRef CAS PubMed.
- M. H. Chehade, L. Loiseau, M. Lombard, L. Pecqueur, A. Ismail, M. Smadja, B. Golinelli-Pimpaneau, C. Mellot-Draznieks, O. Hamelin and L. Aussel, J. Biol. Chem., 2013, 288, 20085–20092 CrossRef CAS PubMed.
- J. M. Crawford and J. Clardy, Nat. Chem., 2010, 2, 805–807 CrossRef CAS PubMed.
- H. Li, Y. Sun, Q. Zhang, Y. Zhu, S.-M. Li, A. Li and C. Zhang, Org. Lett., 2015, 17, 306–309 CrossRef CAS PubMed.
- S.-S. Lee, F.-C. Peng, C.-M. Chiou and K. H. Ling, J. Nat. Prod., 1992, 55, 251–255 CrossRef.
- F. Kuno, K. Otoguro, K. Shiomi, Y. Iwai and S. Ōmura, J. Antibiot., 1996, 49, 742–747 CrossRef CAS.
- F. Kuno, K. Shiomi, K. Otoguro, T. Sunazuka and S. Ōmura, J. Antibiot., 1996, 49, 748–751 CrossRef CAS.
- C.-C. Liaw, Y.-L. Yang, C.-K. Lin, J.-C. Lee, W.-Y. Liao, C.-N. Shen, J.-H. Sheu and S.-H. Wu, Org. Lett., 2015, 17, 2330–2333 CrossRef CAS PubMed.
- J. Yaegashi, J. Romsdahl, Y.-M. Chiang and C. C. C. Wang, Chem. Sci., 2015, 6, 6537–6544 RSC.
- T. S. Bugni, D. Abbanat, V. S. Bernan, W. M. Maiese, M. Greenstein, R. M. van Wagoner and C. M. Ireland, J. Org. Chem., 2000, 65, 7195–7200 CrossRef CAS.
- K. Scherlach, J. Schuemann, H.-M. Dahse and C. Hertweck, J. Antibiot., 2010, 63, 375–377 CrossRef CAS PubMed.
- T. B. Regueira, K. R. Kildegaard, B. G. Hansen, U. H. Mortensen, C. Hertweck and J. Nielsen, Appl. Environ. Microbiol., 2011, 77, 3035–3043 CrossRef CAS PubMed.
- W. Zhang, S. Cao, L. Qiu, F. Qi, Z. Li, Y. Yang, S. Huang, F. Bai, C. Liu, X. Wan and S. Li, ChemBioChem, 2015, 16, 565–569 CrossRef CAS PubMed.
- B. G. Hansen, B. Salomonsen, M. T. Nielsen, J. B. Nielsen, N. B. Hansen, K. F. Nielsen, T. B. Regueira, J. Nielsen, K. R. Patil and U. H. Mortensen, Appl. Environ. Microbiol., 2011, 77, 3044–3051 CrossRef CAS PubMed.
- B. G. Hansen, E. Mnich, K. F. Nielsen, J. B. Nielsen, M. T. Nielsen, U. H. Mortensen, T. O. Larsen and K. R. Patil, Appl. Environ. Microbiol., 2012, 78, 4908–4913 CrossRef CAS PubMed.
- Y. Matsuda, T. Wakimoto, T. Mori, T. Awakawa and I. Abe, J. Am. Chem. Soc., 2014, 136, 15326–15336 CrossRef CAS PubMed.
- L. Colombo, C. Gennari, D. Potenza, C. Scolastico, F. Aragozzini and R. Gualandris, J. Chem. Soc., Perkin Trans. 1, 1982, 365–373 RSC.
- R. A. Steiner, H. J. Janssen, P. Roversi, A. J. Oakley and S. Fetzner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 657–662 CrossRef CAS PubMed.
- L. M. Petersen, D. K. Holm, P. B. Knudsen, K. F. Nielsen, C. H. Gotfredsen, U. H. Mortensen and T. O. Larsen, J. Antibiot., 2015, 68, 201–205 CrossRef CAS PubMed.
- D. K. Holm, L. M. Petersen, A. Klitgaard, P. B. Knudsen, Z. D. Jarczynska, K. F. Nielsen, C. H. Gotfredsen, T. O. Larsen and U. H. Mortensen, Chem. Biol., 2014, 21, 519–529 CrossRef CAS PubMed.
- J. Yaegashi, M. B. Praseuth, S.-W. Tyan, J. F. Sanchez, R. Entwistle, Y.-M. Chiang, B. R. Oakley and C. C. Wang, Org. Lett., 2013, 15, 2862–2865 CrossRef CAS PubMed.
- A. W. Dunn, R. A. Johnstone, B. Sklarz and T. J. King, J. Chem. Soc., Chem. Commun., 1976, 270a RSC.
- S. Ōmura, J. Inokoshi, R. Uchida, K. Shiomi, R. Masuma, T. Kawakubo, H. Tanaka, Y. Iwai, S. Kosemura and S. Yamamura, J. Antibiot., 1996, 49, 414–417 CrossRef.
- R. Uchida, K. Shiomi, J. Inokoshi, T. Sunazuka, H. Tanaka, Y. Iwai, H. Takayanagi and S. Ōmura, J. Antibiot., 1996, 49, 418–424 CrossRef CAS.
- K. Shiomi, R. Uchida, J. Inokoshi, H. Tanaka, Y. Iwai and S. Ōmura, Tetrahedron Lett., 1996, 37, 1265–1268 CrossRef CAS.
- T. J. Simpson, D. J. Stenzel, A. J. Bartlett, E. Obrien and J. S. E. Holker, J. Chem. Soc., Perkin Trans. 1, 1982, 2687–2692 RSC.
- J. P. Springer, J. W. Dorner, R. J. Cole and R. H. Cox, J. Org. Chem., 1979, 44, 4852–4854 CrossRef CAS.
- T. J. Simpson and M. D. Walkinshaw, J. Chem. Soc., Chem. Commun., 1981, 914–915 RSC.
- C. Rank, R. K. Phipps, P. Harris, P. Fristrup, T. O. Larsen and C. H. Gotfredsen, Org. Lett., 2008, 10, 401–404 CrossRef CAS PubMed.
- M. Cueto, J. B. MacMillan, P. R. Jensen and W. Fenical, Phytochemistry, 2006, 67, 1826–1831 CrossRef CAS PubMed.
- E. Okuyama, M. Yamazaki, K. Kobayashi and T. Sakurai, Tetrahedron Lett., 1983, 24, 3113–3114 CrossRef CAS.
- J. Liangsakul, S. Pornpakakul, E. Sangvichien, N. Muangsin and P. Sihanonth, Tetrahedron Lett., 2011, 52, 6427–6430 CrossRef CAS PubMed.
- M. L. Nielsen, J. B. Nielsen, C. Rank, M. L. Klejnstrup, D. K. Holm, K. H. Brogaard, B. G. Hansen, J. C. Frisvad, T. O. Larsen and U. H. Mortensen, FEMS Microbiol. Lett., 2011, 321, 157–166 CrossRef CAS PubMed.
- H.-C. Lo, R. Entwistle, C.-J. Guo, M. Ahuja, E. Szewczyk, J.-H. Hung, Y.-M. Chiang, B. Oakley and C. C. Wang, J. Am. Chem. Soc., 2012, 134, 4709–4720 CrossRef CAS PubMed.
- T. Itoh, K. Tokunaga, E. K. Radhakrishnan, I. Fujii, I. Abe, Y. Ebizuka and T. Kushiro, ChemBioChem, 2012, 13, 1132–1135 CrossRef CAS PubMed.
- Y. Matsuda, T. Awakawa, T. Itoh, T. Wakimoto, T. Kushiro, I. Fujii, Y. Ebizuka and I. Abe, ChemBioChem, 2012, 13, 1738–1741 CrossRef CAS PubMed.
- C.-J. Guo, B. P. Knox, Y.-M. Chiang, H.-C. Lo, J. F. Sanchez, K.-H. Lee, B. R. Oakley, K. S. Bruno and C. C. Wang, Org. Lett., 2012, 14, 5684–5687 CrossRef CAS PubMed.
- Y. Matsuda, T. Awakawa and I. Abe, Tetrahedron, 2013, 69, 8199–8204 CrossRef CAS PubMed.
- Y. Matsuda, T. Awakawa, T. Wakimoto and I. Abe, J. Am. Chem. Soc., 2013, 135, 10962–10965 CrossRef CAS PubMed.
- Y. Matsuda, T. Iwabuchi, T. Wakimoto, T. Awakawa and I. Abe, J. Am. Chem. Soc., 2015, 137, 3393–3401 CrossRef CAS PubMed.
- R. Uchida, K. Shiomi, J. Inokoshi, H. Tanaka, Y. Iwai and S. Ōmura, J. Antibiot., 1996, 49, 1278–1280 CrossRef CAS.
- S. Kosemura and S. Yamamura, Tetrahedron Lett., 1997, 38, 6221–6224 CrossRef CAS.
- S. Kosemura, K. Matsunaga, S. Yamamura, M. Kubota and S. Ohba, Tetrahedron Lett., 1991, 32, 3543–3546 CrossRef CAS.
- S. Kosemura, Tetrahedron Lett., 2002, 43, 1253–1256 CrossRef CAS.
- S. Kosemura, K. Matsunaga and S. Yamamura, Chem. Lett., 1991, 1811–1814 CrossRef CAS.
- S. Kosemura, Tetrahedron, 2003, 59, 5055–5072 CrossRef CAS.
- S. Kosemura, S. Matsuo and S. Yamamura, Phytochemistry, 1996, 43, 1231–1234 CrossRef CAS.
- X. Wang, J. G. Sena Filho, A. R. Hoover, J. B. King, T. K. Ellis, D. R. Powell and R. H. Cichewicz, J. Nat. Prod., 2010, 73, 942–948 CrossRef CAS PubMed.
- P. W. Dalsgaard, B. O. Petersen, J. Ø. Duus, C. Zidorn, J. C. Frisvad, C. Christophersen and T. O. Larsen, Metabolites, 2012, 2, 214–220 CrossRef CAS PubMed.
- G.-Y. Li, B.-G. Li, T. Yang, J.-H. Yin, H.-Y. Qi, G.-Y. Liu and G.-L. Zhang, J. Nat. Prod., 2005, 68, 1243–1246 CrossRef CAS PubMed.
- H.-S. Cho, N.-C. Ha, G. Choi, H.-J. Kim, D. Lee, K. S. Oh, K. S. Kim, W. Lee, K. Y. Choi and B.-H. Oh, J. Biol. Chem., 1999, 274, 32863–32868 CrossRef CAS PubMed.
- M. Arand, B. M. Hallberg, J. Zou, T. Bergfors, F. Oesch, M. J. van der Werf, J. A. de Bont, T. A. Jones and S. L. Mowbray, EMBO J., 2003, 22, 2583–2592 CrossRef CAS PubMed.
- A. Sultana, P. Kallio, A. Jansson, J. S. Wang, J. Niemi, P. Mäntsälä and G. Schneider, EMBO J., 2004, 23, 1911–1921 CrossRef CAS PubMed.
- A. Minami, T. Ose, K. Sato, A. Oikawa, K. Kuroki, K. Maenaka, H. Oguri and H. Oikawa, ACS Chem. Biol., 2014, 9, 562–569 CrossRef CAS PubMed.
- M. McDonough, K. Kavanagh, D. Butler, T. Searls, U. Oppermann and C. Schofield, J. Biol. Chem., 2005, 280, 41101–41110 CrossRef CAS PubMed.
- M. P. López-Gresa, N. Cabedo, M. C. González-Mas, M. L. Ciavatta, C. Avila and J. Primo, J. Nat. Prod., 2009, 72, 1348–1351 CrossRef PubMed.
- X.-H. Liu, F.-P. Miao, M.-F. Qiao, R. H. Cichewicz and N.-Y. Ji, RSC Adv., 2013, 3, 588–595 RSC.
- T. Fukuda, Y. Kurihara, A. Kanamoto and H. Tomoda, J. Antibiot., 2014, 67, 593–595 CrossRef CAS PubMed.
- T. J. Simpson, D. J. Stenzel, A. J. Bartlett, E. O'Brien and J. S. E. Holker, J. Chem. Soc., Perkin Trans. 1, 1982, 2687–2692 RSC.
- C. R. McIntyre, T. J. Simpson, D. J. Stenzel, A. J. Bartlett, E. O'Brien and J. S. E. Holker, J. Chem. Soc., Chem. Commun., 1982, 781–782 RSC.
- S. A. Ahmed, F. E. Scott, D. J. Stenzel, T. J. Simpson, R. N. Moore, L. A. Trimble, K. Arai and J. C. Vederas, J. Chem. Soc., Perkin Trans. 1, 1989, 807–816 RSC.
- D. B. Stierle, A. A. Stierle, J. D. Hobbs, J. Stokken and J. Clardy, Org. Lett., 2004, 6, 1049–1052 CrossRef CAS PubMed.
- A. Stierle, D. Stierle and D. Decato, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, o248 CAS.
- D. Stierle, A. Stierle, B. Patacini, K. McIntyre, T. Girtsman and E. Bolstad, J. Nat. Prod., 2011, 74, 2273–2277 CrossRef CAS PubMed.
- M. Iida, T. Ooi, K. Kito, S. Yoshida, K. Kanoh, Y. Shizuri and T. Kusumi, Org. Lett., 2008, 10, 845–848 CrossRef CAS PubMed.
- Y. Zhang, X.-M. Li, Z. Shang, C.-S. Li, N.-Y. Ji and B.-G. Wang, J. Nat. Prod., 2012, 75, 1888–1895 CrossRef CAS PubMed.
- E. D. de Silva, D. E. Williams, D. R. Jayanetti, R. M. Centko, B. O. Patrick, R. L. Wijesundera and R. J. Andersen, Org. Lett., 2011, 13, 1174–1177 CrossRef PubMed.
- R. M. Centko, D. E. Williams, B. O. Patrick, Y. Akhtar, M. A. Garcia Chavez, Y. A. Wang, M. B. Isman, E. D. de Silva and R. J. Andersen, J. Org. Chem., 2014, 79, 3327–3335 CrossRef CAS PubMed.
- T. J. Simpson, D. J. Stenzel, A. J. Bartlett, E. O'Brien and J. S. Holker, J. Chem. Soc., Perkin Trans. 1, 1982, 2687–2692 RSC.
- J. Arunpanichlert, V. Rukachaisirikul, S. Phongpaichit, O. Supaphon and J. Sakayaroj, Tetrahedron, 2015, 71, 882–888 CrossRef CAS PubMed.
- H. Hayashi, M. Mukaihara, S. Murao, M. Arai, A. Y. Lee and J. Clardy, Biosci., Biotechnol., Biochem., 1994, 58, 334–338 CrossRef CAS.
- E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef CAS PubMed.
- R. J. Cox, Nat. Prod. Rep., 2014, 31, 1405–1424 RSC.
- T. J. Simpson, S. A. Ahmed, C. R. McIntyre, F. E. Scott and I. H. Sadler, Tetrahedron, 1997, 53, 4013–4034 CrossRef CAS.
- D. Kumar, S. P. de Visser and S. Shaik, Chem.–Eur. J., 2005, 11, 2825–2835 CrossRef CAS PubMed.
- A. W. Dunn, R. A. W. Johnstone, B. Sklarz, L. Lessinger and T. J. King, J. Chem. Soc., Chem. Commun., 1978, 533–534 RSC.
- T. J. Simpson, J. Chem. Soc., Perkin Trans. 1, 1979, 2118–2121 RSC.
- J. S. E. Holker and T. J. Simpson, J. Chem. Soc., Chem. Commun., 1978, 626–627 RSC.
- A. J. Bartlett, J. S. Holker, E. O'Brien and T. J. Simpson, J. Chem. Soc., Chem. Commun., 1981, 1198–1200 RSC.
- T. J. Simpson, Tetrahedron Lett., 1981, 22, 3785–3788 CrossRef CAS.
- C. R. McIntyre, F. E. Scott, T. J. Simpson, L. A. Trimble and J. C. Vederas, J. Chem. Soc., Chem. Commun., 1986, 501–503 RSC.
- J. E. Spangler and E. J. Sorensen, Tetrahedron, 2009, 65, 6739–6745 CrossRef CAS PubMed.
- N. Steffan, A. Grundmann, S. Afiyatullov, H. Ruan and S.-M. Li, Org. Biomol. Chem., 2009, 7, 4082–4087 CAS.
- N. Kato, H. Suzuki, H. Takagi, M. Uramoto, S. Takahashi and H. Osada, ChemBioChem, 2011, 12, 711–714 CrossRef CAS PubMed.
- E. Cohen, L. Koch, K. M. Thu, Y. Rahamim, Y. Aluma, M. Ilan, O. Yarden and S. Carmeli, Bioorg. Med. Chem., 2011, 19, 6587–6593 CrossRef CAS PubMed.
- E. Okuyama, M. Yamazaki and Y. Katsube, Tetrahedron Lett., 1984, 25, 3233–3234 CrossRef CAS.
- J. P. Springer, J. Clardy, J. M. Wells, R. J. Cole and J. W. Kirksey, Tetrahedron Lett., 1975, 16, 2531–2534 CrossRef.
- J. Penn, R. Swift, L. J. Wigley, P. G. Mantle, J. N. Bilton and R. N. Sheppard, Phytochemistry, 1993, 32, 1431–1434 CrossRef CAS.
- J. A. Laakso, J. B. Gloer, D. T. Wicklow and P. F. Dowd, J. Org. Chem., 1992, 57, 2066–2071 CrossRef CAS.
- K. Nozawa, M. Yuyama, S. Nakajima, K.-i. Kawai and S.-i. Udagawa, J. Chem. Soc., Perkin Trans. 1, 1988, 2155–2160 RSC.
- C. Li, J. B. Gloer, D. T. Wicklow and P. F. Dowd, Org. Lett., 2002, 4, 3095–3098 CrossRef CAS PubMed.
- J. A. Laakso, J. B. Gloer, D. T. Wicklow and P. F. Dowd, J. Org. Chem., 1992, 57, 138–141 CrossRef CAS.
- J. B. Gloer, B. L. Rinderknecht, D. T. Wicklow and P. F. Dowd, J. Org. Chem., 1989, 54, 2530–2532 CrossRef CAS.
- M. R. TePaske, J. B. Gloer, D. T. Wicklow and P. F. Dowd, Tetrahedron Lett., 1989, 30, 5965–5968 CrossRef CAS.
- R. T. Gallagher, T. McCabe, K. Hirotsu, J. Clardy, J. Nicholson and B. J. Wilson, Tetrahedron Lett., 1980, 21, 243–246 CrossRef CAS.
- C. Young, L. McMillan, E. Telfer and B. Scott, Mol. Microbiol., 2001, 39, 754–764 CrossRef CAS.
- S. Zhang, B. Monahan, J. Tkacz and B. Scott, Appl. Environ. Microbiol., 2004, 70, 6875–6883 CrossRef CAS PubMed.
- T. Motoyama, T. Hayashi, H. Hirota, M. Ueki and H. Osada, Chem. Biol., 2012, 19, 1611–1619 CrossRef CAS PubMed.
- C. Young, M. Bryant, M. Christensen, B. Tapper, G. Bryan and B. Scott, Mol. Genet. Genomics, 2005, 274, 13–29 CrossRef CAS PubMed.
- C. A. Young, S. Felitti, K. Shields, G. Spangenberg, R. D. Johnson, G. T. Bryan, S. Saikia and B. Scott, Fungal Genet. Biol., 2006, 43, 679–693 CrossRef CAS PubMed.
- C. Liu, K. Tagami, A. Minami, T. Matsumoto, J. C. Frisvad, H. Suzuki, J. Ishikawa, K. Gomi and H. Oikawa, Angew. Chem., Int. Ed., 2015, 54, 5748–5752 CrossRef CAS PubMed.
- M. J. Nicholson, C. J. Eaton, C. Stärkel, B. A. Tapper, M. P. Cox and B. Scott, Toxins, 2015, 7, 2701–2722 CrossRef PubMed.
- L. McMillan, R. Carr, C. Young, J. Astin, R. Lowe, E. Parker, G. Jameson, S. Finch, C. Miles, O. McManus, W. A. Schmalhofer, M. L. Garcia, G. J. Kaczorowski, M. Goetz, J. S. Tkacz and B. Scott, Mol. Genet. Genomics, 2003, 270, 9–23 CrossRef CAS PubMed.
- S. Saikia, E. Parker, A. Koulman and B. Scott, FEBS Lett., 2006, 580, 1625–1630 CrossRef CAS PubMed.
- S. Saikia, E. Parker, A. Koulman and B. Scott, J. Biol. Chem., 2007, 282, 16829–16837 CrossRef CAS PubMed.
- K. Tagami, C. Liu, A. Minami, M. Noike, T. Isaka, S. Fueki, Y. Shichijo, H. Toshima, K. Gomi, T. Dairi and H. Oikawa, J. Am. Chem. Soc., 2013, 135, 1260–1263 CrossRef CAS PubMed.
- B. Scott, C. A. Young, S. Saikia, L. K. McMillan, B. J. Monahan, A. Koulman, J. Astin, C. J. Eaton, A. Bryant and R. E. Wrenn, Toxins, 2013, 5, 1422–1446 CrossRef CAS PubMed.
- C. Liu, M. Noike, A. Minami, H. Oikawa and T. Dairi, Appl. Microbiol. Biotechnol., 2014, 98, 199–206 CrossRef CAS PubMed.
- C. Liu, A. Minami, M. Noike, H. Toshima, H. Oikawa and T. Dairi, Appl. Environ. Microbiol., 2013, 79, 7298–7304 CrossRef CAS PubMed.
- M. J. Nicholson, A. Koulman, B. J. Monahan, B. L. Pritchard, G. A. Payne and B. Scott, Appl. Environ. Microbiol., 2009, 75, 7469–7481 CrossRef CAS PubMed.
- K. Tagami, A. Minami, R. Fujii, C. Liu, M. Tanaka, K. Gomi, T. Dairi and H. Oikawa, ChemBioChem, 2014, 15, 2076–2080 CrossRef CAS PubMed.
- S. Saikia, D. Takemoto, B. Tapper, G. Lane, K. Fraser and B. Scott, FEBS Lett., 2012, 586, 2563–2569 CrossRef CAS PubMed.
- R. Hutchison, P. Steyn and S. van Rensburg, Toxicol. Appl. Pharmacol., 1973, 24, 507–509 CrossRef CAS.
- C. Kabuto, J. Silverton, T. Akiyama, U. Sankawa, R. D. Hutchison, P. S. Steyn and R. Vleggaar, J. Chem. Soc., Chem. Commun., 1976, 728–729 RSC.
- M. Raju, G.-S. Wu, A. Gard and J. Rosazza, J. Nat. Prod., 1982, 45, 321–327 CrossRef CAS.
- Y.-H. Chooi, R. Cacho and Y. Tang, Chem. Biol., 2010, 17, 483–494 CrossRef CAS PubMed.
- Y. Li, Y.-H. Chooi, Y. Sheng, J. S. Valentine and Y. Tang, J. Am. Chem. Soc., 2011, 133, 15773–15785 CrossRef CAS PubMed.
- Y.-H. Chooi, P. Wang, J. Fang, Y. Li, K. Wu, P. Wang and Y. Tang, J. Am. Chem. Soc., 2012, 134, 9428–9437 CrossRef CAS PubMed.
- Y.-H. Chooi, Y. J. Hong, R. A. Cacho, D. J. Tantillo and Y. Tang, J. Am. Chem. Soc., 2013, 135, 16805–16808 CrossRef CAS PubMed.
- Y. Kimura, M. Kusano, H. Koshino, J. Uzawa, S. Fujioka and K. Tani, Tetrahedron Lett., 1996, 37, 4961–4964 CrossRef CAS.
- Y. Zou, Z. Zhan, D. Li, M. Tang, R. A. Cacho, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2015, 137, 4980–4983 CrossRef CAS PubMed.
- N. Ishikawa, H. Tanaka, F. Koyama, H. Noguchi, C. C. Wang, K. Hotta and K. Watanabe, Angew. Chem., Int. Ed., 2014, 53, 12880–12884 CrossRef CAS PubMed.
- A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, Chem. Commun., 2007, 4053–4055 RSC.
- M. E. Raggatt, T. J. Simpson and M. I. Chicarelli-Robinson, Chem. Commun., 1997, 2245–2247 RSC.
- H.-C. Lin, Y.-H. Chooi, S. Dhingra, W. Xu, A. M. Calvo and Y. Tang, J. Am. Chem. Soc., 2013, 135, 4616–4619 CrossRef CAS PubMed.
- H.-C. Lin, Y. Tsunematsu, S. Dhingra, W. Xu, M. Fukutomi, Y.-H. Chooi, D. E. Cane, A. M. Calvo, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2014, 136, 4426–4436 CrossRef CAS PubMed.
- R. V. Cochrane and J. C. Vederas, Acc. Chem. Res., 2014, 47, 3148–3161 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Professor Ushio Sankawa on the occasion of his 80th birthday. |
| This journal is © The Royal Society of Chemistry 2016 |