
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic approaches towards alkaloids bearing α-tertiary amines
Anastasia
Hager†
,
Nina
Vrielink†
,
Dominik
Hager
,
Julien
Lefranc
and
Dirk
Trauner
*
Fakultät für Chemie und Pharmazie, Ludwig-Maximilians-Universität München, and Munich Center for Integrated Protein Science, Butenandtstr. 5 – 13, 81377 München, Germany. E-mail: Dirk.Trauner@lmu.de
First published on 1st December 2015
Abstract
Covering: up to August 2015
Alkaloids account for some of the most beautiful and biologically active natural products. Although they are usually classified along biosynthetic criteria, they can also be categorized according to certain structural motifs. Amongst these, the α-tertiary amine (ATA), i.e. a tetrasubstituted carbon atom surrounded by three carbons and one nitrogen, is particularly interesting. A limited number of methods have been described to access this functional group and fewer still are commonly used in synthesis. Herein, we review some approaches to asymmetrically access ATAs and provide an overview of alkaloid total syntheses where those have been employed.
 Anastasia Hager | Anastasia Hager studied chemistry in Würzburg, conducting her Diploma thesis on natural product isolation and synthesis with Prof. G. Bringmann. After joining Prof. D. Trauner's research group for an internship at UC Berkeley, she moved to Munich for her PhD work that was centered on the development of biomimetic syntheses of complex natural products implementing cascade reactions. From 2013 until 2015 she was a postdoctoral fellow with Prof. D. Fiedler at Princeton University developing novel chemical tools to allow for investigations of inositol pyrophosphate signaling pathway. She now works as a laboratory head in process research at Bayer in Wuppertal. |
 Nina Vrielink | Nina Vrielink was born in 1987 in Nordhorn, Germany. She studied chemistry and biochemistry at LMU. During her Master's studies in chemistry in Munich, she joined the group of Prof. E. Tate at the Imperial College London for a research internship. In 2012, she joined the laboratory of Prof. D. Trauner for her Master's thesis and returned to conduct her Ph.D. studies starting in 2013. In 2014, she visited the laboratory of Prof. B. M. Stoltz for three months. She is currently pursuing natural product synthesis of α-tertiary-amine-containing alkaloids, supported by a scholarship from the Deutsche Telekom Foundation. |
 Dominik Hager | Dominik Hager studied chemistry in Würzburg, and in 2007, he joined the group of Prof. K. P. C. Vollhardt at UC Berkeley as a visiting researcher. Dominik performed his Diploma studies with Prof. G. Bringmann, working on the development of an axial-chirality transfer concept. For his graduate work he joined the laboratory of Prof. D. Trauner at the LMU. His research projects were centered on natural product synthesis. Between 2013 and 2015 he was a postdoctoral researcher with Prof. D. W. C. MacMillan at Princeton University developing new photochemical protocols. Currently, Dominik works as laboratory head for Bayer CropScience AG. |
 Julien Lefranc | Julien Lefranc was born in 1984 in Marseille, France. He studied chemistry at the Université de la Mediterranée, Marseille, and then moved to the Ecole Nationale Supérieure de Chimie de Montpellier in 2006. In 2008, he joined the group of Prof. J. Clayden at the University of Manchester where he obtained his PhD working on the syntheses of α-tertiary amines. In 2012, Julien joined the group of Prof. D. Trauner as a postdoctoral researcher where he worked on Natural Product synthesis. Since 2014, Julien has been working as a laboratory leader in Medicinal Chemistry for Bayer Healthcare in Berlin. |
 Dirk Trauner | Dirk Trauner was born and grew up in Linz, Austria. After studying biology and biochemistry at the University of Vienna, he joined Professor Johann Mulzer's group at the Free University of Berlin to pursue natural product synthesis. Subsequently, he became a postdoctoral fellow with Professor Samuel J. Danishefsky at the Memorial Sloan-Kettering Cancer Center in New York City. In 2000, Dirk joined the University of California, Berkeley, where he became an Associate Professor of chemistry (with tenure). In the summer of 2008, he moved to the University of Munich, where he currently resides as a Professor of Chemistry and Chemical Biology. His research interests range from organic synthesis and natural product chemistry to chemical neurobiology, optogenetics and photopharmacology. |
1 Introduction
Alkaloids have played an important role in the development of synthetic organic chemistry, pharmacology and medicine. Once considered to be metabolic waste products, they are now known to benefit their producers in various ways, e.g. as antimicrobials, antifeedants or as mediators of ecologically beneficial interactions.1 Though a limited number of amino acids are involved in their biosynthesis, alkaloids exhibit enormous structural variability, which is often increased through the incorporation of terpenoid and polyketide components and late-stage oxidative transformations.2 Reflecting their structural diversity and relatively weak basicity, alkaloids interact with a large variety of biological targets and have found many uses in human medicine.3,4 In addition, they have provided inspiration for countless synthetic drugs that borrow structural motifs from their natural counterparts.The α-tertiary amine (ATA) stands out among the structural features frequently found in alkaloids.5–8 For the purposes of this review and in keeping with the literature, we define an ATA as a nitrogen atom bound to a sp3-hybridized carbon that bears three additional carbon–carbon bonds. The nitrogen itself can be sp3-hybridized as part of a primary, secondary and tertiary amine. Broadening our definition, it can also be sp2- or sp-hybridized, e.g. in an amide or isonitrile. The tetrasubstituted carbon from which the C,N-bond branches out is often stereogenic, which makes ATAs particularly interesting from a synthetic point of view. Our definition puts emphasis on this particular C,N-bond and avoids the confusion that is often associated with the term ‘quaternary stereocenter’, which, strictly speaking, refers only to a carbon atom surrounded by four other carbons.
Fig. 1 shows some alkaloids and drugs with alkaloid-like properties that illustrate our definition and demonstrate that the nitrogen in ATAs (highlighted in red) can be substituted to various degrees. Memantine and huperzine A contain primary ATAs, whereas ketamine, MK-801 and histrionicotoxin 283A (HTX 283A) feature secondary ATAs, and lycopodine is representative of molecules containing a tertiary ATA. 2,2,6,6-Tetramethylpiperidine (TMP) and the alkaloid porantherine are examples for molecules featuring a twofold ATA. The dimeric alkaloid stephacidin B contains no fewer than four ATAs. Notably, the α-carbons are stereogenic in the majority of these compounds.
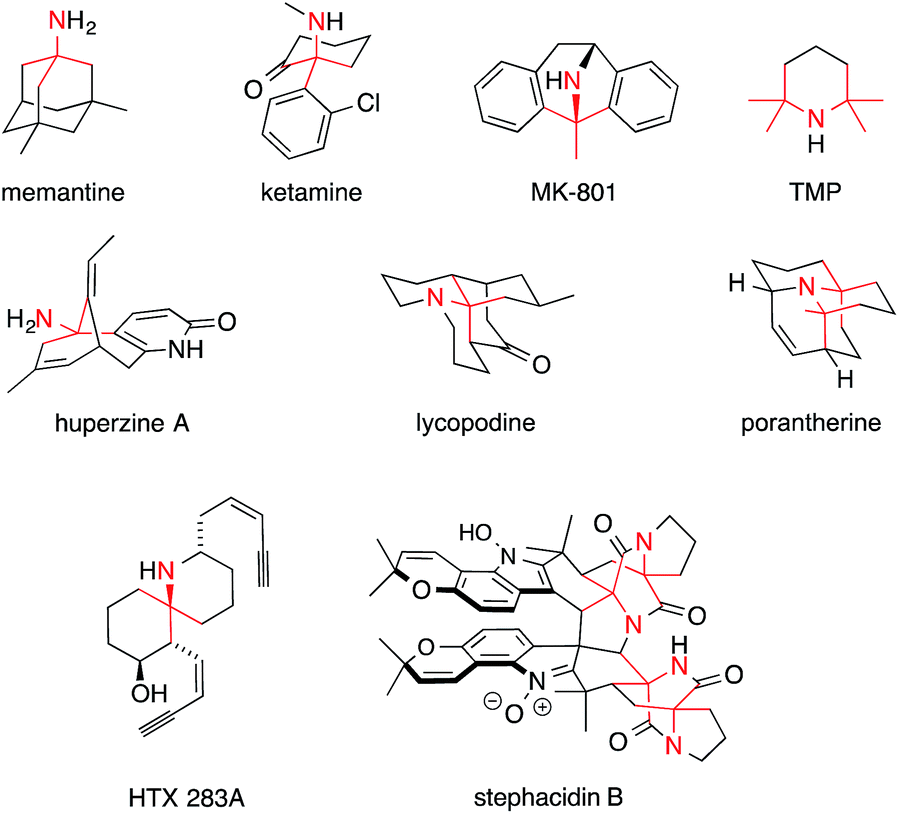 | ||
| Fig. 1 Alkaloids and synthetic molecules that contain the ATA motif. | ||
In this review, we wish to provide a brief survey of synthetic methods used to install the ATA motif and discuss their application in the total synthesis of alkaloids. The syntheses included here have been selected based on their historical significance, the intriguing structure of their target molecule, and the elegance and efficiency of the method used. The order of their presentation is somewhat arbitrary, mixing biosynthetic and taxonomic categories (such as Lycopodium alkaloids), with purely structural ones (such as quinolizidine alkaloids). Generally, we have aimed to proceed from simpler target molecules to more complex ones. While our review is by no means comprehensive, we hope to feature the most instructive examples for the establishment of ATAs and thus provide inspiration and valuable lessons for future work. We also hope that this review will benefit the design of synthetic pathways toward drugs and synthetic building blocks that contain α-tertiary amines.
2 Methods used for the installation of α-tertiary amines
Many approaches toward the installation of ATAs have been developed but only a relatively small subset of these has proven popular in alkaloid total synthesis. Here, we provide a brief survey of these methods, discussing them in general terms. We classify them according to the bond that is formed in the key step and the electronic nature of the nitrogen and carbon, respectively. However, it should be noted that not all of the syntheses discussed in this review fall into this simplified organizational scheme.2.1 A C,C-bond is formed in the step that generates the ATA
• The α-carbon is electrophilic. Some of the most commonly encountered methods involve the addition of carbon nucleophiles to activated imines and iminium ions (Scheme 1). They include Mannich reactions, Strecker reactions, aza-Prins reactions and the 1,2-addition of organometallic reagents to C,N-double bonds. N-Acyliminium ions are particularly powerful electrophiles in reactions of this type. A variant of the Heck olefination that involves enamines also falls into this category.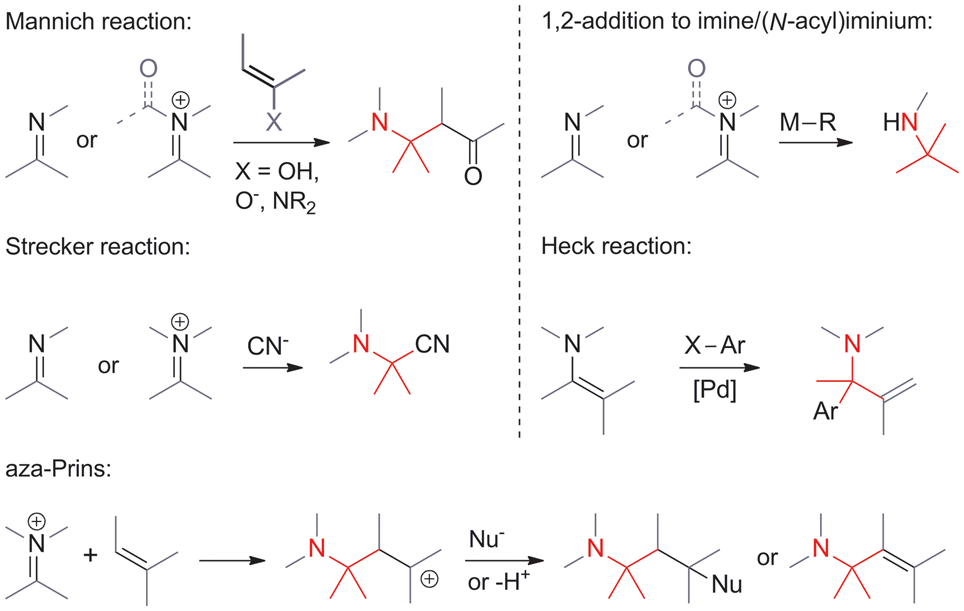 | ||
| Scheme 1 C,C-bond formation involving electrophilic α-carbons. | ||
• The α-carbon is nucleophilic. In an Umpolung of the above situation, the α-carbon can also serve as a nucleophile (Scheme 2). For instance, the alkylation of branched nitroalkanes or of deprotonated amino acid derivatives can be used to establish ATAs. Insertions of carbons into nucleophilic C,H-bonds next to a C,N-bond are a member of this general category as well.
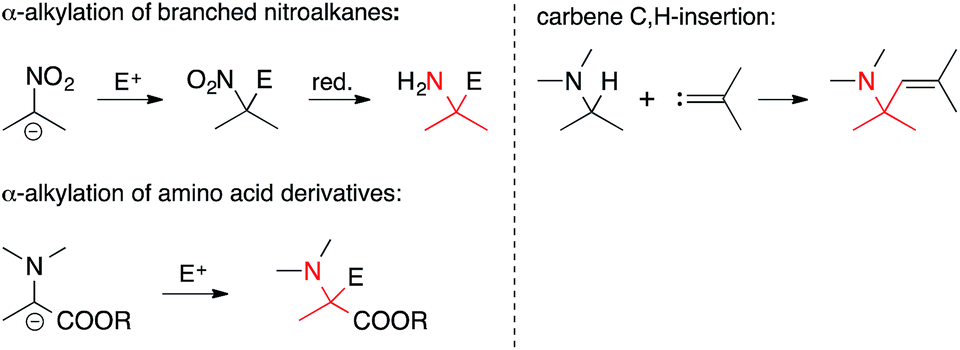 | ||
| Scheme 2 C,C-bond formation involving nucleophilic α-carbons. | ||
• Pericyclic reactions. Pericyclic reactions that form a C,C-bond in the key step have occasionally been employed to form ATAs (Scheme 3). They include Diels–Alder cycloadditions involving 1-aminodienes or 2-azadienes, as well as certain amino dienophiles, [2+2] cycloadditions, and 1,3-dipolar cycloadditions involving nitrones and azomethine imines.
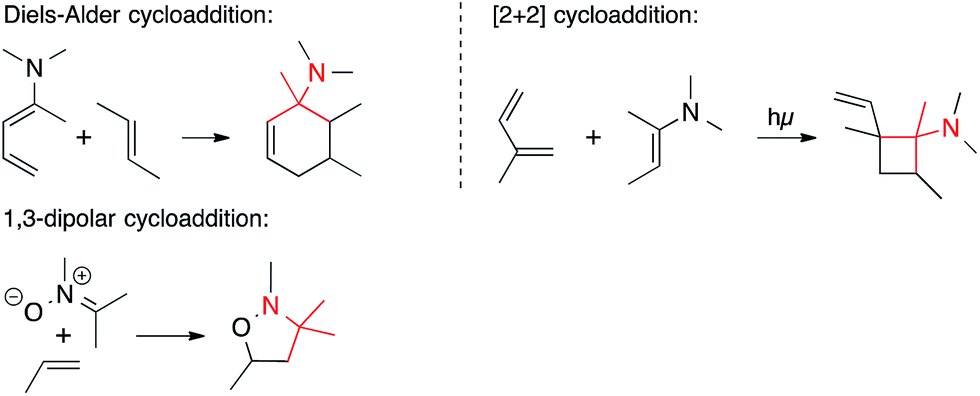 | ||
| Scheme 3 C,C-bond formation involving pericyclic reactions. | ||
• Radical reactions. Radical reactions establishing ATAs are relatively rare, but not unprecedented (Scheme 4). 5-endo-Trig and 6-endo-trig cyclizations as well as radical transfer allylations belong to this category.
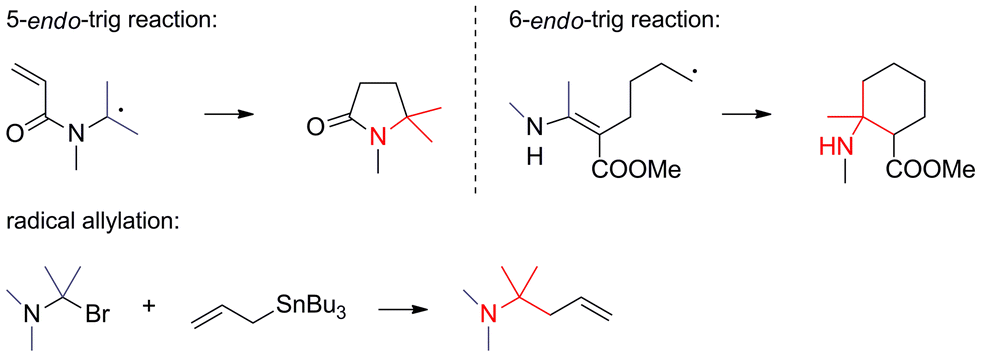 | ||
| Scheme 4 C,C-bond formation via radical reactions. | ||
2.2 A C,N-bond is formed in the step that generates the ATA
• The nitrogen is electrophilic. Rearrangements that involve electron-deficient nitrogen atoms are often encountered in the formation of ATAs (Scheme 5).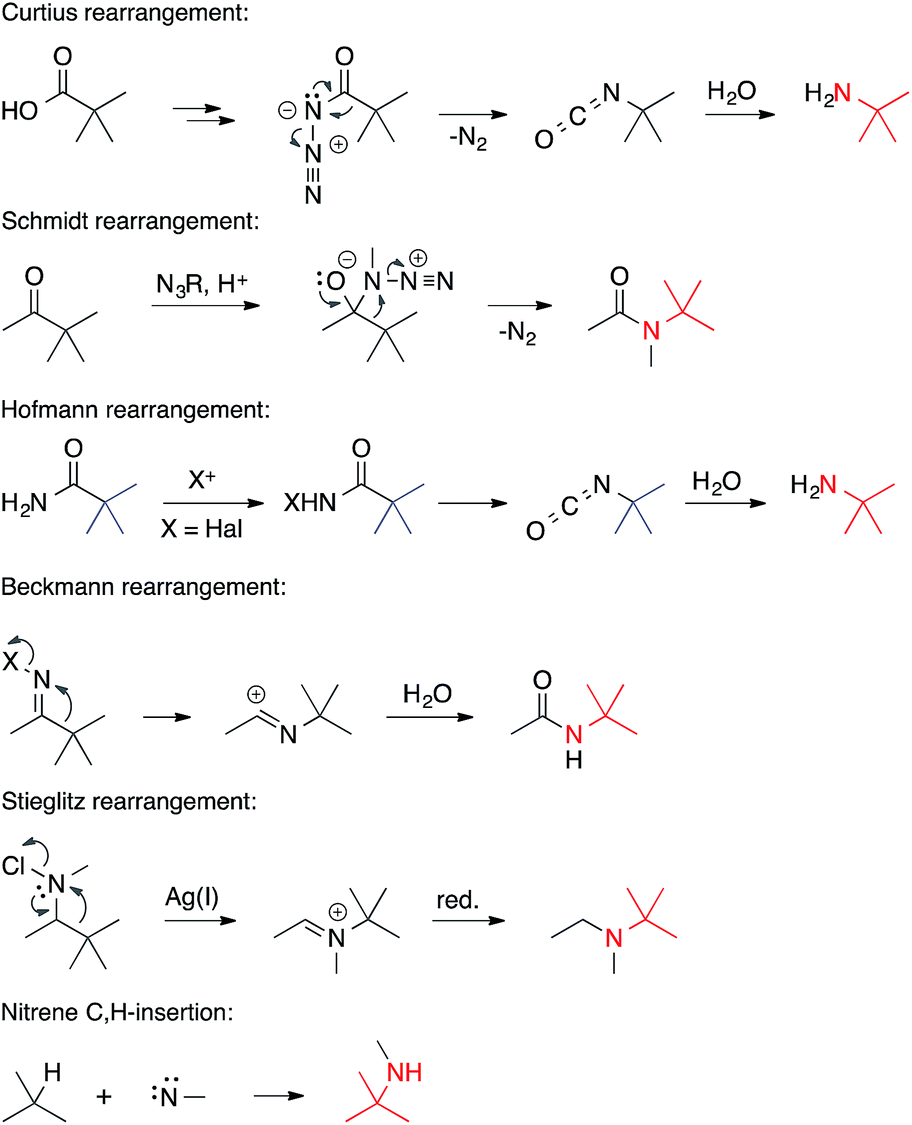 | ||
| Scheme 5 C,N-bond formation involving electrophilic nitrogens. | ||
They include the Curtius, Schmidt, Hofmann, Beckmann and Stieglitz rearrangements.5 Often, these reactions can be classified as [1,2] sigmatropic rearrangements. Related nucleophilic substitutions involving N-haloamines have been used as well. An electron-deficient nitrogen atom also plays a role in the insertion of nitrenes into C,H-bonds.
• The nitrogen is nucleophilic. The formation of ATAs through nucleophilic additions or substitutions involving nitrogen is fairly common (Scheme 6). The classical Michael addition falls into this category, as do SN2′ reactions and haloaminations. For obvious reasons, SN2 reactions are rare and mostly confined to intramolecular cases. Carbocations that react with a nucleophilic nitrogen occur in the aza-Prins reaction and the Ritter reaction. Oxidative dearomatizations have also been used in a few cases to establish ATAs.
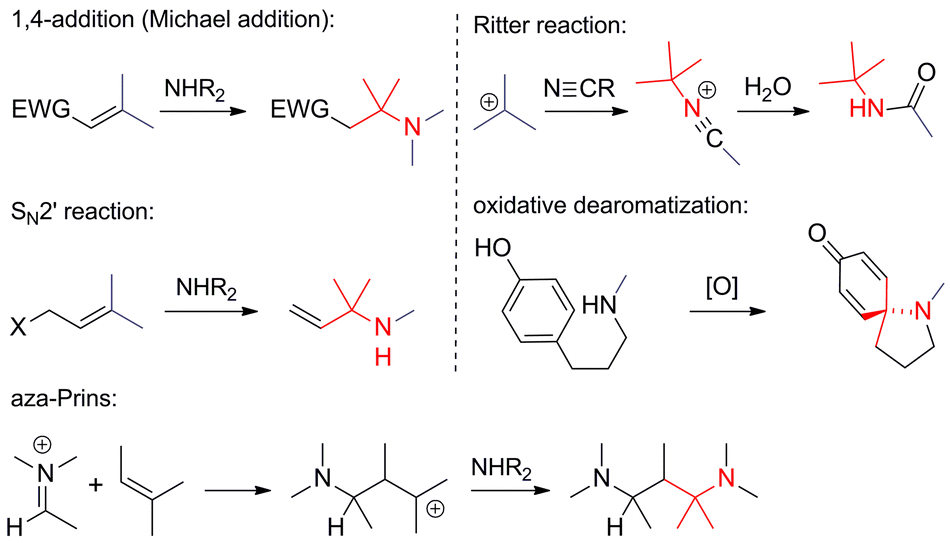 | ||
| Scheme 6 C,N-bond formation involving nucleophilic nitrogens. EWG = electron-withdrawing group. | ||
• Pericyclic reactions. Pericyclic reactions in which a C,N-bond is formed provide powerful means to establish ATAs (Scheme 7). Overman, Kazmaier–Claisen and [3,3] sigmatropic rearrangements of allylic isocyanides belong to this category. Divinyl cyclopropane rearrangements have also been used to establish ATAs.5
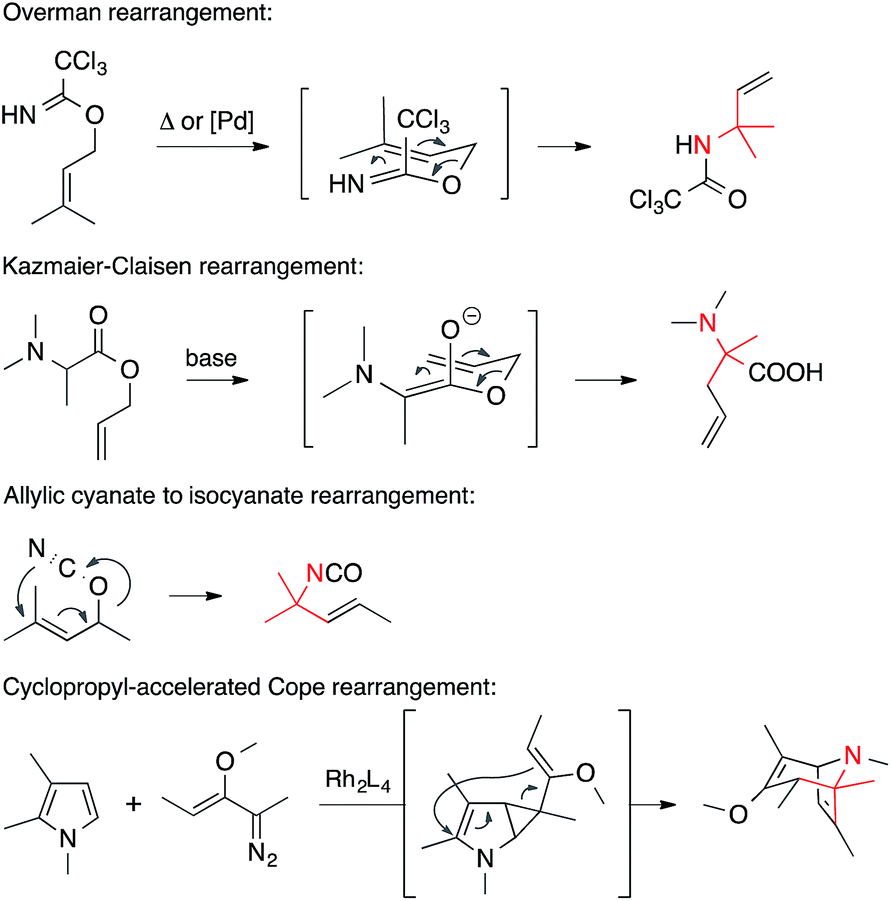 | ||
| Scheme 7 C,N-bond formation involving pericyclic reactions. | ||
Many more methods have emerged in recent years that can be used to create ATAs, such as reactions proceeding via C,H-activation9 and hydroaminations.10 Since they have not yet been employed in the total synthesis of alkaloids, they are not featured in this review. Other methods, such as the Mannich reaction, Curtius rearrangement and Michael reaction, have proven to be so popular in the total synthesis of alkaloids that we cannot include all instances where they have been employed in this review.
3 Homotropane alkaloids
One of the first applications of Mannich reactions in the construction of ATAs occurred during the synthesis of certain homotropane alkaloids. Three representatives, euphococcinine, N-methyl euphococcine and adaline, feature an ATA in the bridgehead position of a bicyclic framework (Fig. 2). These simple natural products are excreted by lady beetles (coccinellids) when threatened.11,12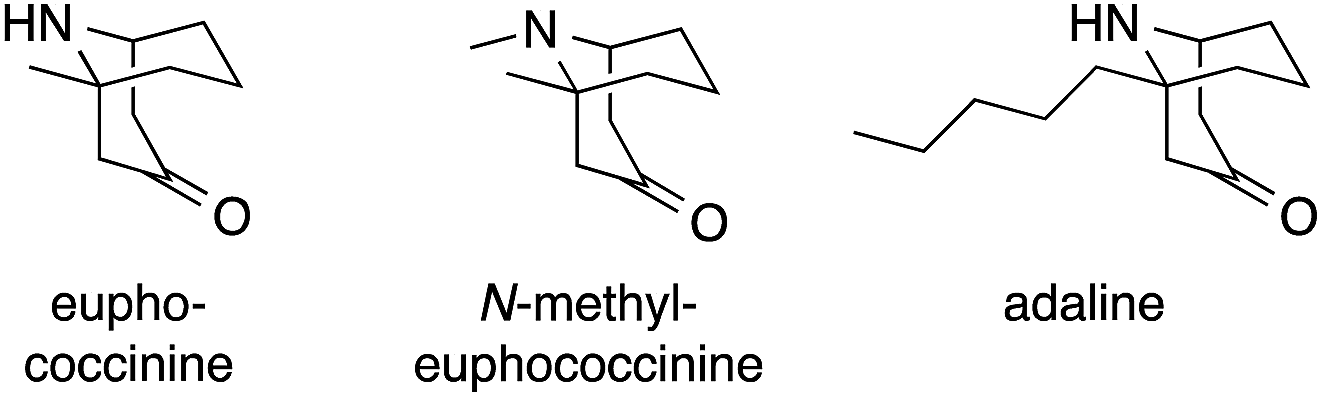 | ||
| Fig. 2 Representative ATA-containing homotropane alkaloids. | ||
In 1959, Alder synthesized N-methyl-euphococcinine using a protocol analogous to the famous tropinone syntheses of Robinson13,14 and Schöpf15 (Scheme 8a).16 Dehydropyrane 1 was converted into ketoaldehyde 2, which was then transformed into N-methyl-euphococcinine in a one-pot process (via iminium-intermediate 3).17–19 A similar strategy was later adopted to synthesize the structurally related alkaloid adaline.20
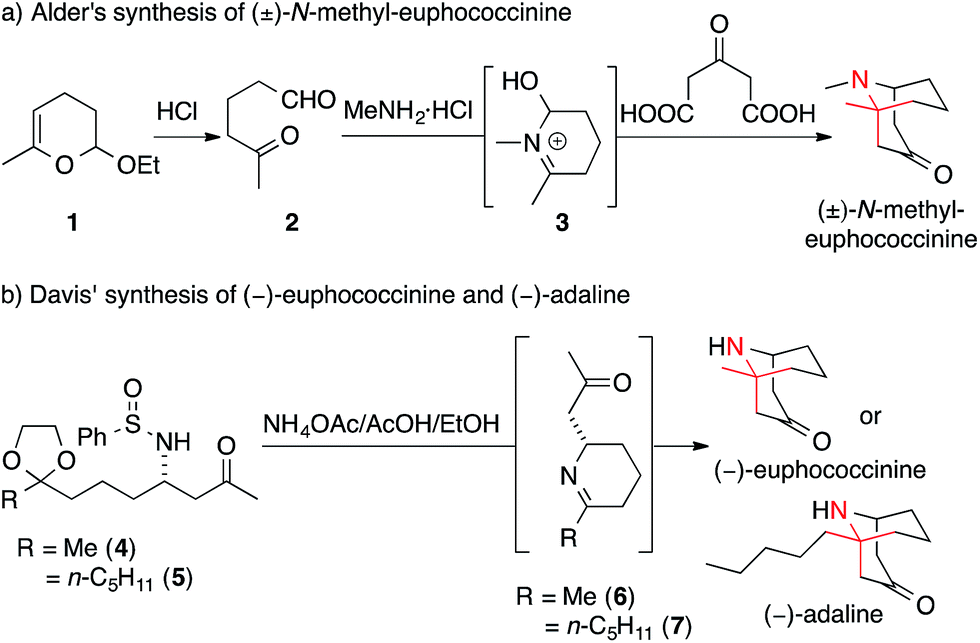 | ||
| Scheme 8 Homotropane alkaloid syntheses by Alder (1959) and Davis (2010). Ac = acetyl. | ||
Throughout the years, this biomimetic Mannich strategy was adopted in other syntheses of euphococcinine and adaline.21–23 Alternative approaches involved a 1,3-dipolar cycloaddition,24 addition to an N-acyliminium ion,25 Michael addition26,27 and allylic rearrangement of a cyanate to an isocyanate.28,29
In 2010, Davis published a biomimetic synthesis of (−)-euphococcinine and (−)-adaline in enantiopure form (Scheme 8b).30 The key steps of these syntheses involved the stereoselective formation of piperideine 6 and 7 from the enantiomerically pure N-sulfinyl aminoketones 4 and 5, respectively.31 An ensuing intramolecular Mannich reaction afforded the azabicyclononane natural products.
4 Histrionicotoxins
In 1971, Daly isolated six different alkaloids, termed histrionicotoxins (HTXs), from skin extracts of the Colombian poison arrow frog Dendrobates histrionicus (Fig. 3).32,33 They all contain a unique spirocyclic piperidine core and differ mostly in the length and the degree of saturation of the two side chains. Several histrionicotoxins were identified as inhibitors of nicotinic acetylcholine receptors,34–38 which, together with their attractive structures, prompted significant attention from the synthetic community.39 The low natural abundance of these alkaloids and the fact that the frogs do not secrete HTXs in captivity made an efficient synthetic approach all the more desirable.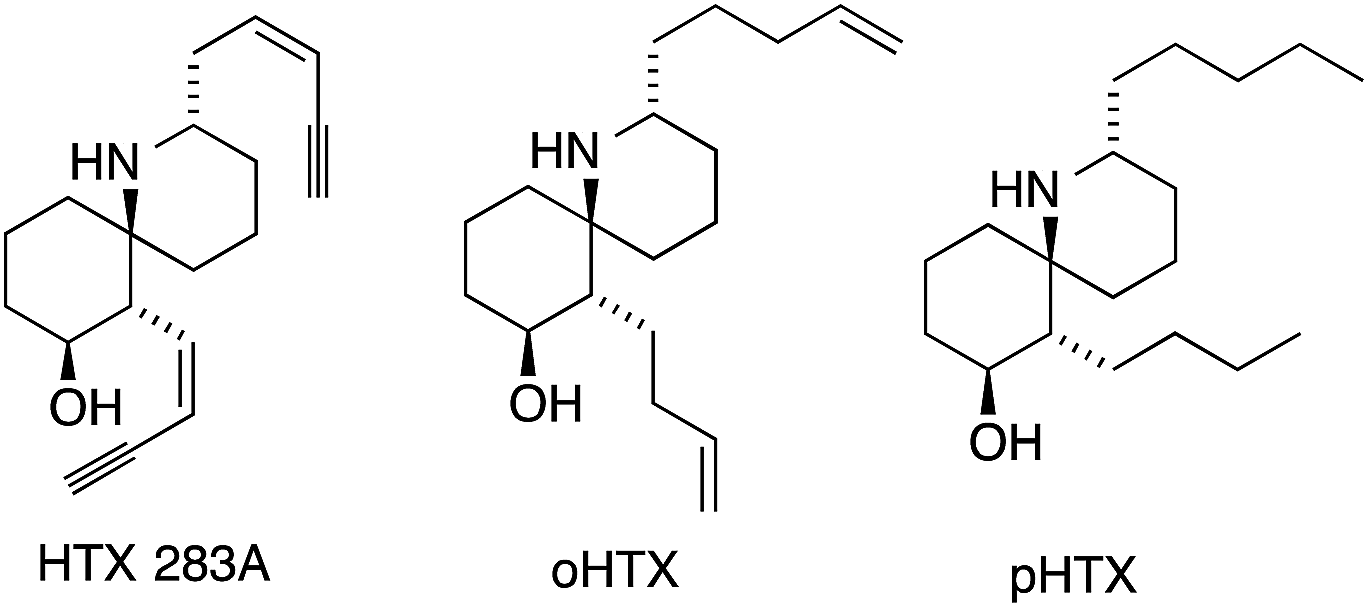 | ||
| Fig. 3 The major members of the histrionicotoxin family. | ||
The first total synthesis of histrionicotoxin alkaloids was reported by Kishi in 1975 (Scheme 9a).40–42 His synthesis of octahydrohistrionicotoxin (oHTX) utilized an intramolecular acid-catalyzed aza-Michael addition to set the ATA. Amide 8 was converted to a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of epimeric spiroketolactams 9 and 10. It was possible to transform 9 into the desired diastereoisomer 10 upon treatment with sodium methoxide.
:1 mixture of epimeric spiroketolactams 9 and 10. It was possible to transform 9 into the desired diastereoisomer 10 upon treatment with sodium methoxide.
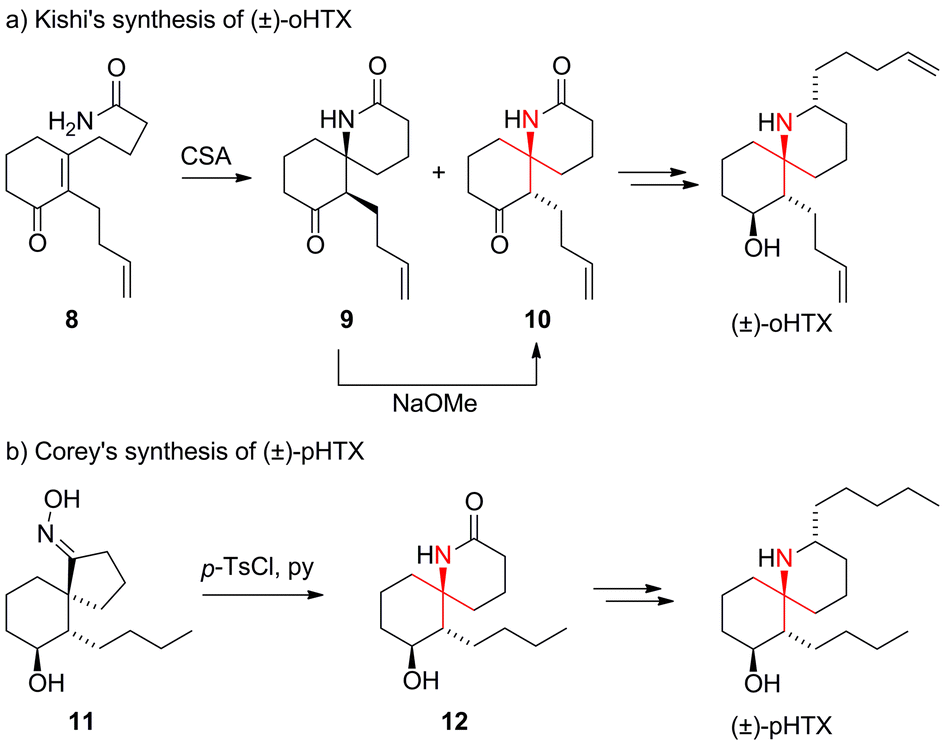 | ||
| Scheme 9 Early synthetic approaches to HTXs by Kishi (1975) and Corey (1975). CSA = camphorsulfonic acid, HTX = histrionicotoxin, p-TsCl = para-toluenesulfonyl chloride. | ||
In the same year, Corey reported the first racemic synthesis of perhydrohistrionicotoxin (pHTX), a synthetic HTX derivative (Scheme 9b).43 For the installation of the ATA, Corey used a Beckmann rearrangement that expanded spirocyclic oxime 11 to spirocyclic amide 12. Several other groups subsequently employed related ring expansion strategies.44–48
Stork reported a synthesis of HTX 283A using a Hofmann rearrangement to set the ATA (Scheme 10a).49 During this transformation, amide 13 was oxidized with bis(trifluoroacetoxy)iodobenzene to promote the alkyl migration, giving amine 14 after decarboxylation. More recently, Fukuyama reported an asymmetric synthesis of HTX 283A (Scheme 10b).50,51 The key carboxylic acid 15 underwent a stereospecific Curtius rearrangement to yield bicyclo[5.4.0]undecane 16, which could be converted into HTX 283A.
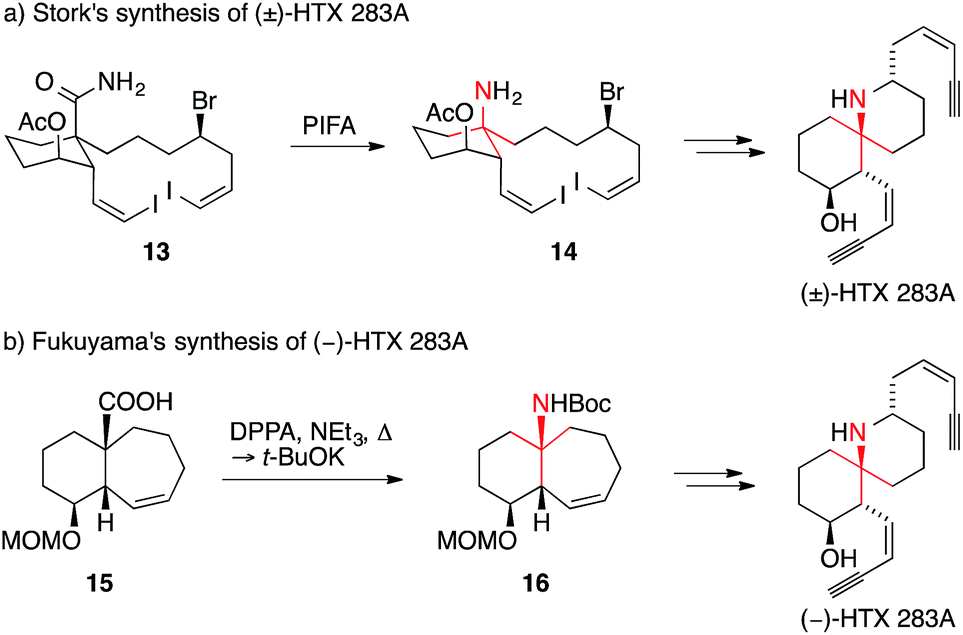 | ||
| Scheme 10 HTX 283A syntheses by Stork (1990) and Fukuyama (2011). HTX = histrionicotoxin, Ac = acetyl, Boc = tert-butyloxycarbonyl, t-Bu = tert-butyl, DPPA = diphenylphosphoryl azide, MOM = methoxymethyl, PIFA = (bis(trifluoroacetoxy)iodo)benzene. | ||
A particularly short and efficient synthesis of racemic HTX 283A was reported in 2006 by Stockman and Fuchs (Scheme 11).52 In their approach, the key intermediate 21 was formed from the symmetric ketodinitrile 17 using a cascade reaction. Ketone 17 was condensed with hydroxylamine yielding nitrone 18 after intramolecular Michael addition. Subsequent intramolecular [3+2] cycloaddition afforded isoxazolidine 19. Following its isolation, 19 was converted to its more stable regioisomer 21 through a retro-[3+2]/[3+2] cycloaddition process (via intermediate 20). This so-called ‘Holmes dinitrile’ (21) had been previously converted into HTX 283A.53,54
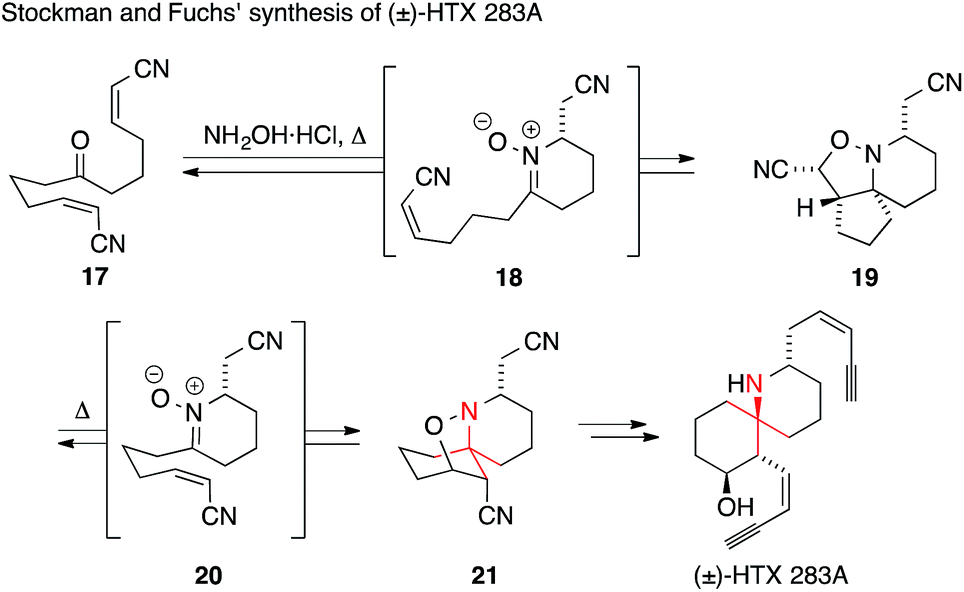 | ||
| Scheme 11 HTX 283A synthesis by Stockman and Fuchs (2006). HTX = histrionicotoxin. | ||
Additional strategies to set the ATA in the histrionicotoxins involved Michael reactions,41,55,56 Tsuji–Trost amination,57,58 iodoetherification,59–61 oxidative dearomatizations,62 Henry reactions,63 diastereoselective aziridation,64 1,2-carbon migration of hydroxyimines,65 [2,3] sigmatropic rearrangements,66 [3,3] sigmatropic rearrangements (Kazmaier–Claisen rearrangement)67 and [2+2] cycloadditions.68,69
5 Lycopodium alkaloids
The genus Lycopodium comprises more than 200 species of moss-like plants that have been used in traditional Chinese medicine for centuries.70 They have yielded a variety of ATA-containing alkaloids, some of which are featured in Fig. 4. Lycopodine, the first member of the family described in the literature,71 has proven to be a particularly attractive target for chemical synthesis. In 1968, only seven years after its structural elucidation, Stork72 and Ayer73,74 simultaneously published the first syntheses of this natural product. Ayer established the ATA using an intermolecular addition of a racemic Grignard reagent to tricyclic, Cv-symmetric iminium ion 22 to obtain 23 (Scheme 12a).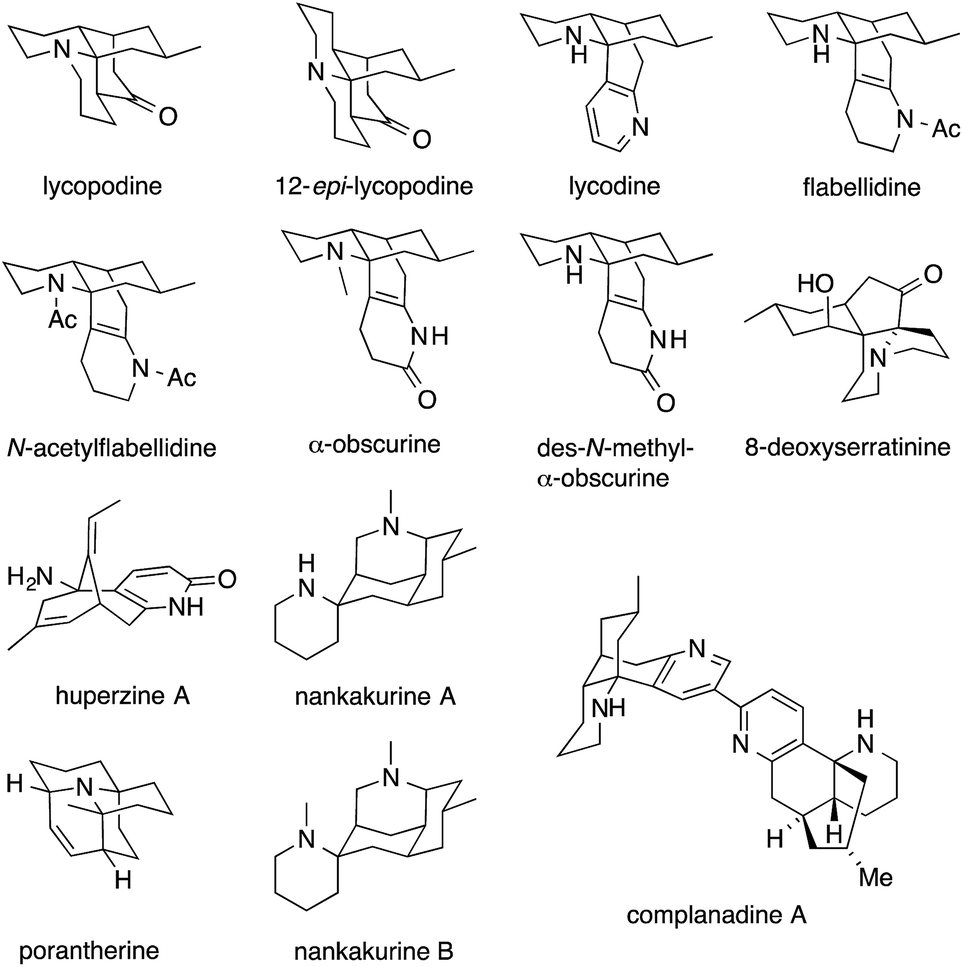 | ||
| Fig. 4 Representative Lycopodium alkaloids. | ||
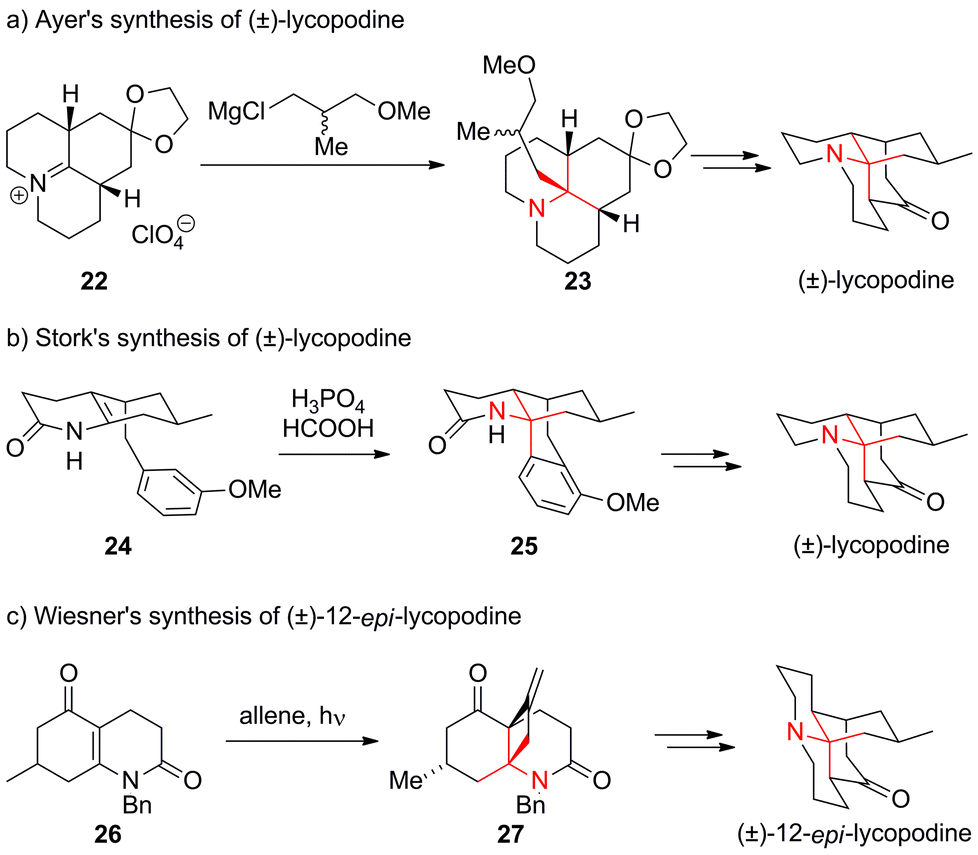 | ||
| Scheme 12 Representative lycopodine syntheses by Ayer (1968), Stork (1968) and Wiesner (1967). Bn = benzyl. | ||
Stork's synthesis involved a Pictet–Spengler type reaction, furnishing tetracyclic amide 25 (24 → 25, Scheme 12b). A similar cyclization approach was also utilized almost 30 years later in Padwa's asymmetric synthesis of lycopodine (not shown).75 Wiesner published a synthesis of 12-epi-lycopodine and annotinine using an innovative photochemical strategy (Scheme 12c).76–79 This work involved the conversion of vinylogous imide 26 into exo-methylene cyclobutane 27via a photochemical [2+2] cycloaddition of allene.
Heathcock established one of the most elegant and influential routes to lycopodine in 1982 (Scheme 13a).80–82 In a remarkable sequence, intermediate 28 underwent deprotection, condensation and intramolecular Mannich reaction to yield secondary amine 30, presumably via iminium ion 29. The installation of the α-tertiary amine and the formation of two out of four rings thus occurred in a single step, mimicking the proposed biosynthesis of this natural product. Subsequent optimization led to the shortest synthesis of lycopodine to date, consisting of only eight steps.82 Using a similar sequence, lycodine and lycodoline were prepared as well.82
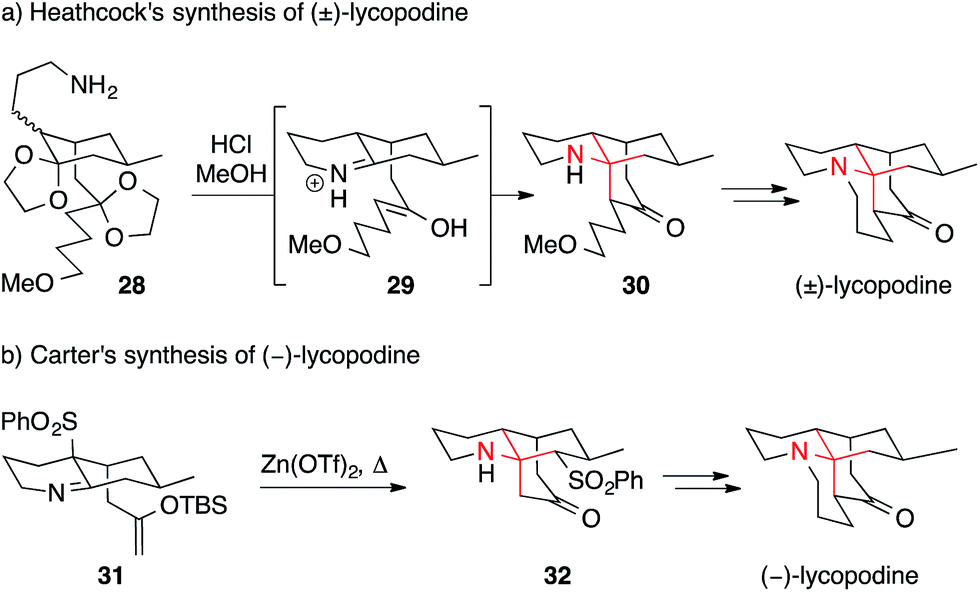 | ||
| Scheme 13 Lycopodine syntheses by Heathcock (1982) and Carter (2008). OTf = trifluoromethanesulfonate, TBS = tert-butyldimethylsilyl. | ||
Variations of Heathcock's strategy have been used in other synthetic approaches toward Lycopodium alkaloids, e.g. in syntheses of clavolonine by Evans (2005)84 and Fujioka (2011).83
One drawback of intramolecular Mannich reactions, however, is the need to simultaneously form an iminium ion and an enol. Thus, long reaction times of up to 18 days were needed.82 Recently, this problem was solved in an elegant way by Carter (Scheme 13b).85,86 Using an aza-Wittig approach, Carter was able to prepare and isolate the TBS–enol ether imine 31. Treatment of 31 with zinc triflate furnished the ATA and concomitantly resulted in the rearrangement of the sulfinyl residue yielding lycopodine precursor 32.
In 1985, Kraus published a route towards lycopodine that was based on the formation of a bridgehead olefin (Scheme 14a).87 Tertiary alkyl bromide 33 was treated with DBU and 3-amino-1-propanol to install amino ketone 34, which could be further transformed into the natural product in two additional steps using Heathcock's protocol. An equally unusual approach was reported by Grieco, who employed a Stieglitz rearrangement (Scheme 14b).88 To effect the reaction, N-chloroamine 35 was treated with silver tetrafluoroborate followed by cyanoborohydride. Many other syntheses of lycopodine have been accomplished utilizing different strategies, such as Michael additions, for the assembly of the ATA.89–91
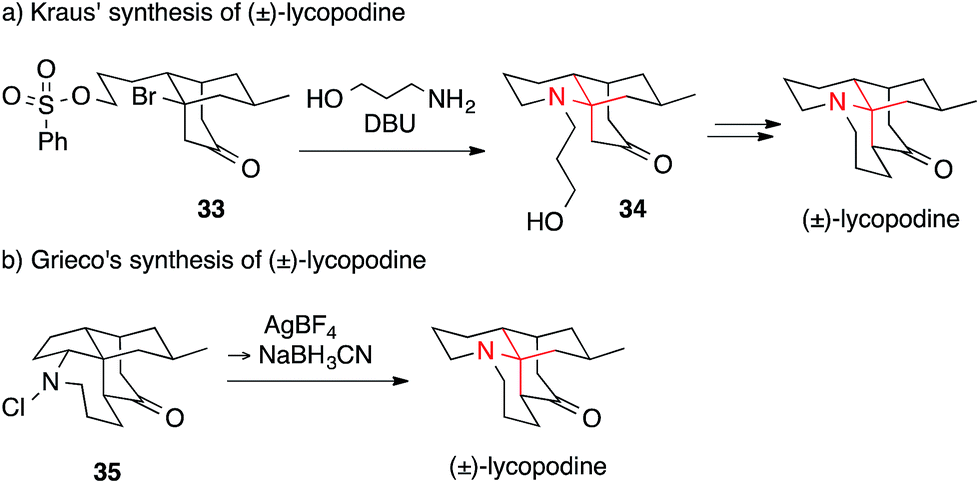 | ||
| Scheme 14 Lycopodine syntheses by Kraus (1985) and Grieco (1998). DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. | ||
Members of the lycodine class of natural products feature an ATA and a pyridine or pyridone moiety. The parent compound, lycodine,92 was first isolated from L. annotinum in 1958.93 Apart from the Heathcock synthesis mentioned above,82 several additional syntheses of lycodine have been achieved to date.94–97
Schumann used a classical Mannich strategy to access racemic lycodine, α-obscurine and N-acetylflabellidine (Scheme 15).98–100
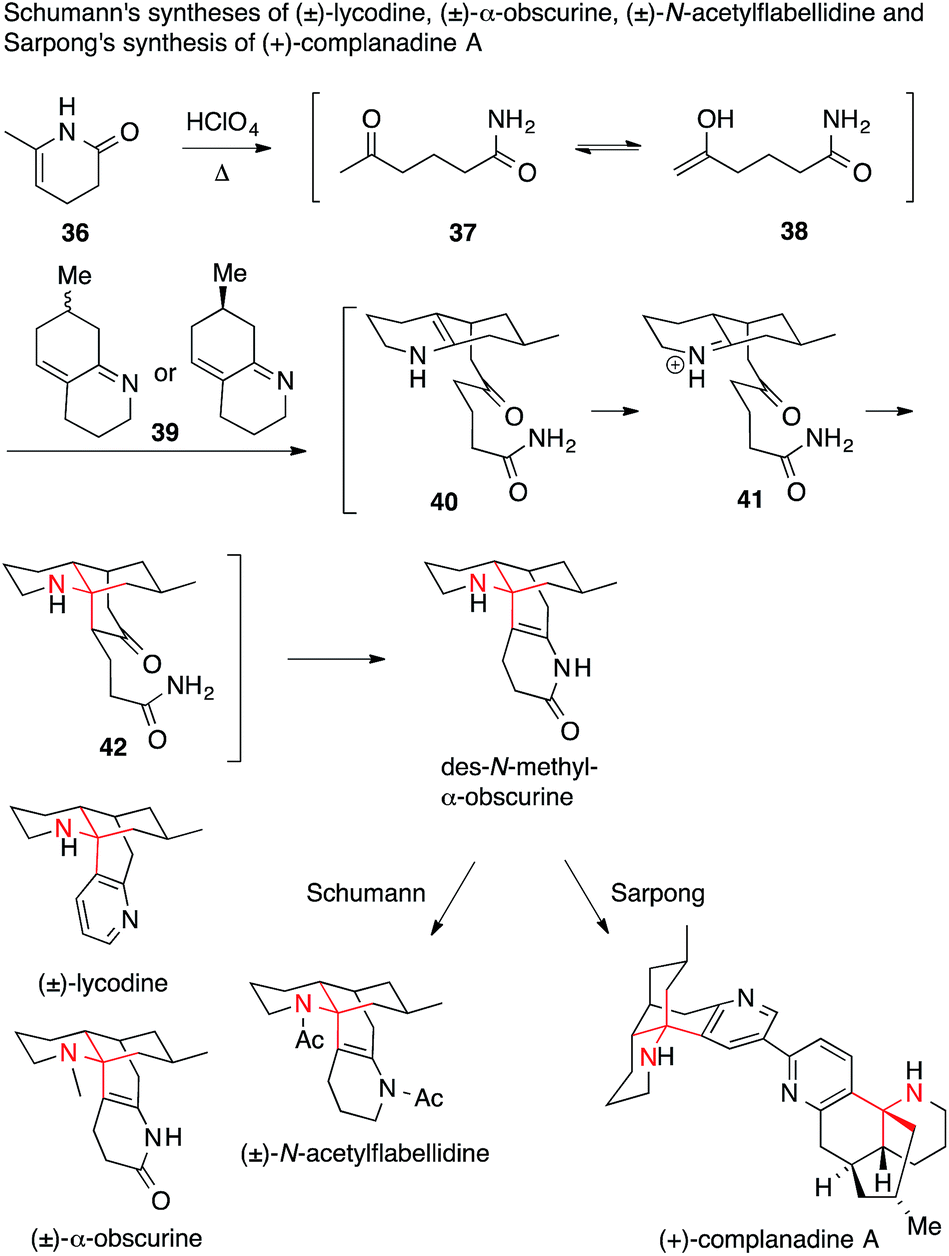 | ||
| Scheme 15 Synthesis of various Lycopodium alkaloids by Schumann (1982) and Sarpong (2010). Ac = acetyl. | ||
The mechanism of the key double Mannich reaction cascade was further explored almost 30 years later by Sarpong.94 He used the same cascade as an opening sequence in an asymmetric synthesis of enantiomerically pure (+)-complanadine A, a lycodine dimer, which was shown to enhance expression of nerve growth factor in human cells.101 It was found that cyclic enamide 36 opens to ketone 37 or enol 38 under acidic conditions, which adds to the unsaturated bicyclic imine 39. Protonation of the resulting enamine 40 triggers a second, intramolecular Mannich reaction to afford tricycle 42via the iminium ion 41. Finally, an intramolecular enamide formation furnished tetracyclic des-N-methyl-α-obscurine, containing the entire lycodine framework.
In an unusual approach, Tsukano and Hirama applied an intramolecular palladium-mediated Heck reaction between enecarbamate and pyridine triflate 43 to form the ATA, which yielded lycodine precursor 44 (Scheme 16a).95
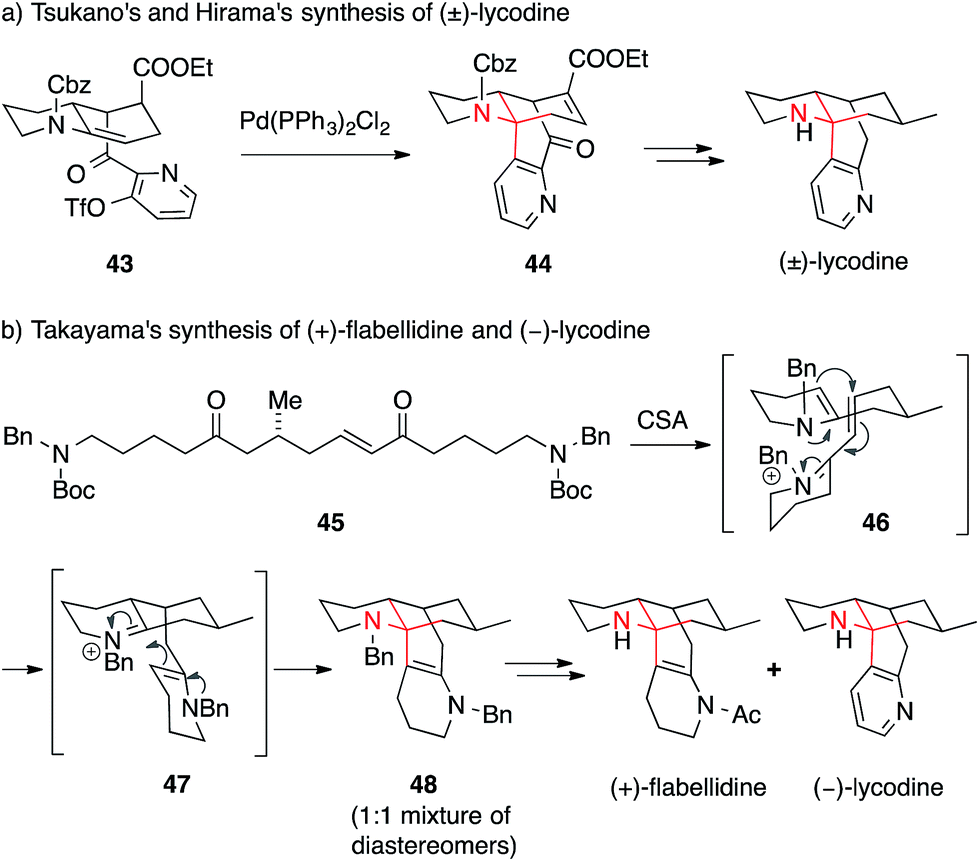 | ||
| Scheme 16 Syntheses of various Lycopodium alkaloids by Tsukano (2010) and Takayama (2014). Cbz = benzyloxycarbonyl, CSA = camphorsulfonic acid, Boc = tert-butyloxycarbonyl, Bn = benzyl, OTf = trifluoromethanesulfonate. | ||
Recently, another very short synthesis of (−)-lycodine as well as the closely related (+)-flabellidine was accomplished by Takayama (Scheme 16b).97 Starting from a linear precursor 45, he was able to assemble the whole tetracyclic skeleton 48 of both alkaloids in a cascade reaction involving a double condensation (45 → 46), a conjugate addition (46 → 47) followed by a Mannich reaction (47 → 48). In addition, Shair published an approach towards several members of the 7-membered-ring-containing Lycopodium alkaloids using a transannular Mannich reaction (not shown).102,103
One of the rare cases of a SN2 reaction in ATA formation can be found in Lei's recent synthesis of (−)-8-deoxyserratinine (Scheme 17).104 Tertiary alcohol 49 was converted into chloride 50, which was attacked intramolecularly by the free secondary amine (50 → 51). In 2014, Lei extended his strategy to a synthesis of the oxidised congener (−)-serratinine.105 Other approaches towards 8-deoxyserratinine and related alkaloids include a Schmidt rearrangement and an intramolecular epoxide opening.106–108
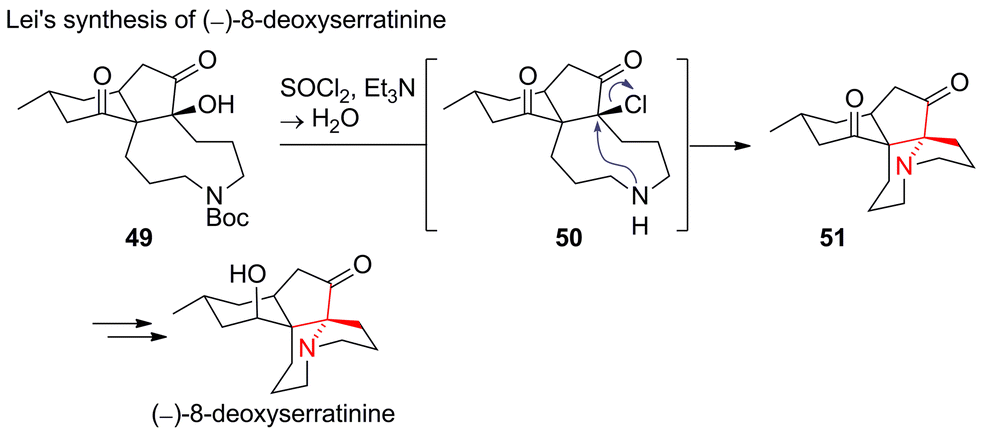 | ||
| Scheme 17 Lei's synthesis of (−)-8-deoxyserratinine (2014). Boc = tert-butyloxycarbonyl. | ||
In contrast to the multiple strategies used for the installation of ATAs in the Lycopodium alkaloids mentioned above, the methods used to access the medicinally important acetylcholine esterase inhibitor huperzine A are less diverse. Since the ATA in huperzine A is primary, it can be efficiently installed using a Curtius rearrangement. Indeed, synthetic efforts towards huperzine A were almost exclusively focused on carboxylic acid precursors, such as 52.109–116
The first synthesis of huperzine A was published by Kozikowski in 1989 (Scheme 18).109 First, he completed the core 52 wherein the primary amine is replaced by a methyl ester. After saponification, Curtius rearrangement (52 → 53) followed by double deprotection provided racemic huperzine A. In the following years, many huperzine A syntheses and several semi-syntheses were published.117,118 All of them featured a racemic or enantiomerically pure carboxylic acid derivative of precursor 53, keeping the Curtius rearrangement as the key step for the formation of the ATA.109–116 These efforts culminated in the recently published large-scale asymmetric synthesis of huperzine A.119
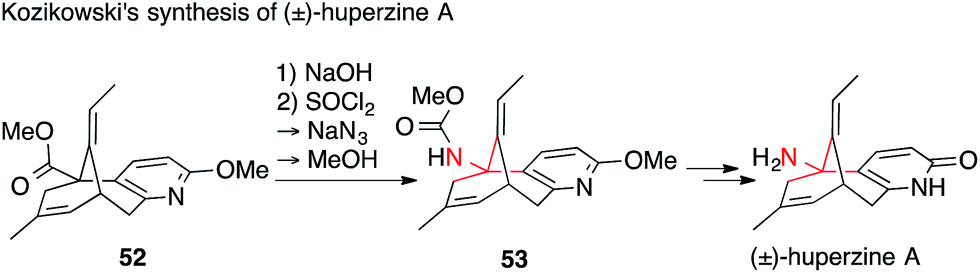 | ||
| Scheme 18 Huperzine A synthesis by Kozikowski (1989). | ||
A few groups, however, have been able to avoid Curtius rearrangements in the synthesis of huperzine A. Sun and Lin accessed the alkaloid using an intramolecular Heck reaction (54 → 55) (Scheme 19a),120 whereas the White group performed an elegant tandem intramolecular aza-Prins cyclization/cyclobutane fragmentation (56 → 53) to set the ATA in 53 (Scheme 19b).121
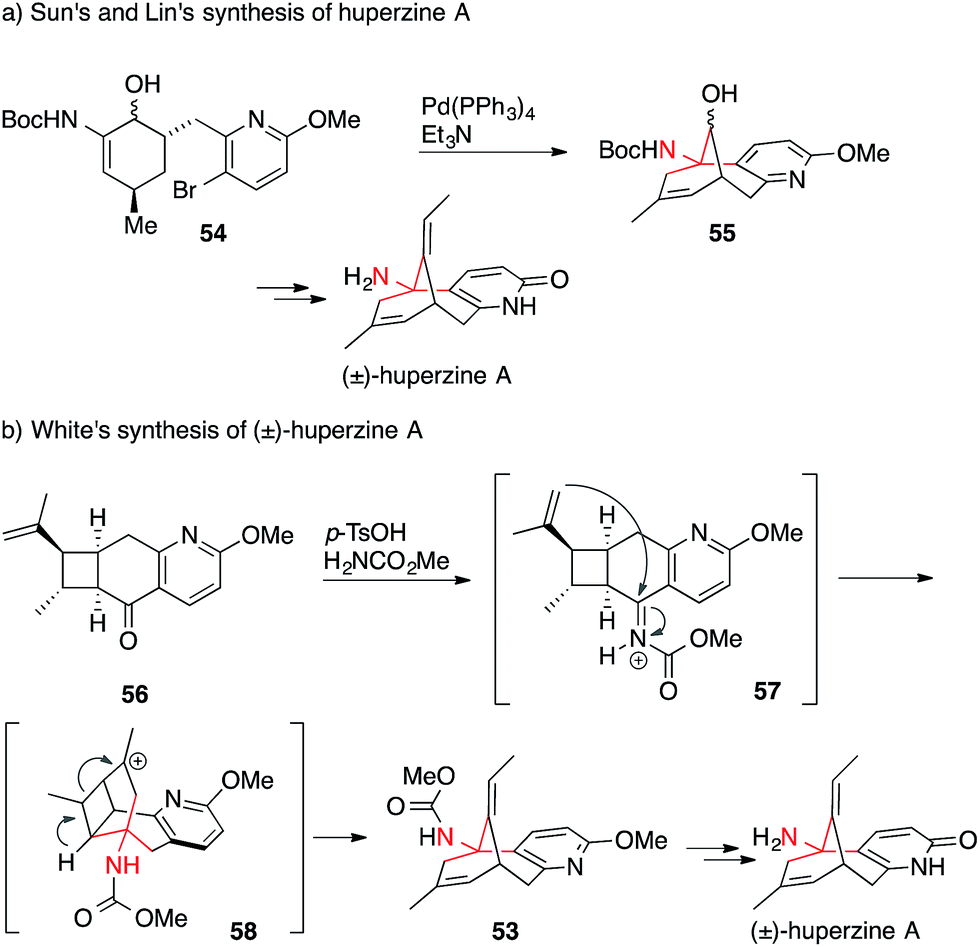 | ||
| Scheme 19 Huperzine A syntheses by Sun/Lin (2012) and White (2013). Boc = tert-butyloxycarbonyl, p-TsCl = para-toluenesulfonic acid. | ||
Two Lycopodium alkaloids recently isolated from Lycopodium hamiltonii, viz. the nankakurines A and B, have attracted broad interest in the synthetic community (Fig. 4).122,123 So far, two syntheses of these natural products have been reported. In 2008, Overman published an enantioselective synthesis of the misassigned original structure of nankakurine A (61) (Scheme 20a) followed by the syntheses of the reassigned structures of nankakurine A and B in 2010 (Scheme 20b).124,125
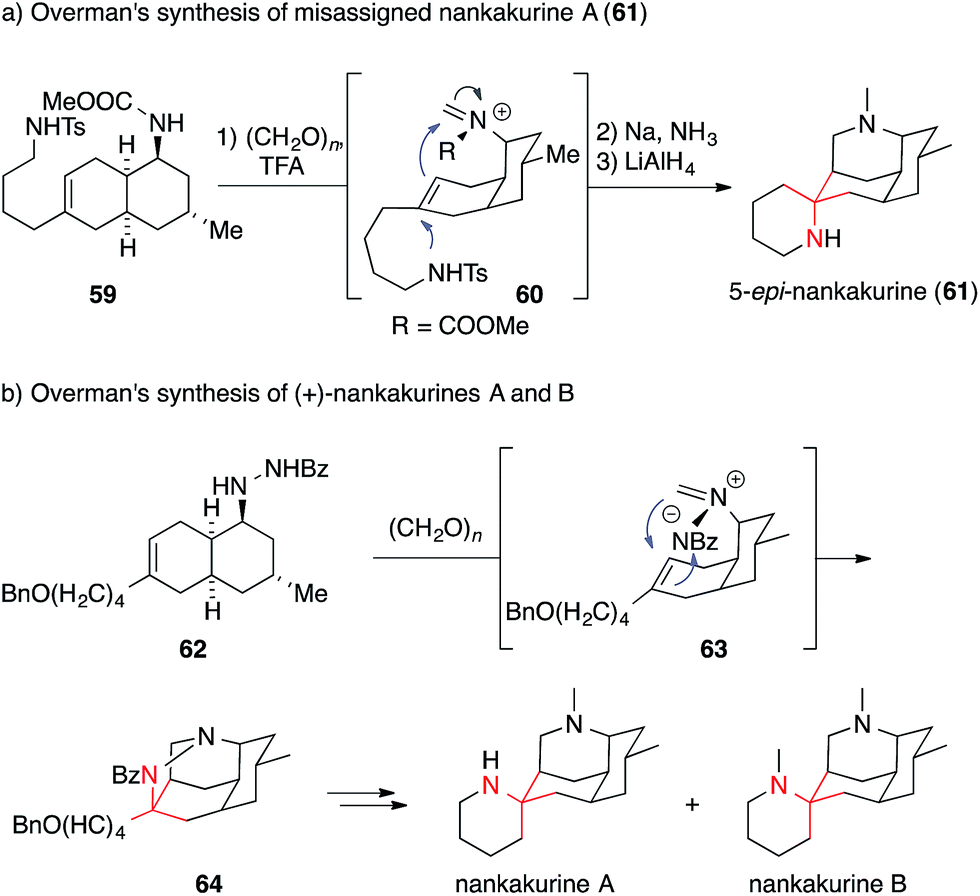 | ||
| Scheme 20 Overman's syntheses of misassigned nankakurine A (2008) and revised nankakurines A and B (2010). Bn = benzyl, Bz = benzoyl, TFA = trifluoroacetic acid, Ts = para-toluenesulfonyl. | ||
In the case of 5-epi-nankakurine (61), an aza-Prins reaction (59 → 60) was used, which allowed for the direct formation of both piperidine rings in 61 in one step starting from bicycle 59.124 This strategy, however could not be applied for the formation of actual nankakurine A. Its synthesis was accomplished utilizing an intramolecular 1,3-dipolar cycloaddition of an azomethine imine 63, formed in situ via condensation of 62 with formaldehyde. This reaction provided access to tetracyclic pyrazolidine 64, which, after SmI2 mediated N,N-bond cleavage, gave rise to nankakurines A and B.125 Two years later, Waters reported a racemic synthesis of nankakurines A and B using a Grignard addition to an iminium species derived from luciduline, which is easily accessible by total synthesis (not shown).126
Porantherine, the major alkaloid of the poisonous woody shrub Poranthera corymbosa, is structurally similar to the Lycopodium alkaloids, although not a member of the family (Fig. 4).127,128 Possessing two tertiary carbons attached to the same amine (twofold ATA), porantherine is a considerable synthetic challenge that has been met only twice thus far.129,130 Both syntheses are racemic and based on similar strategies for the assembly of the ATA motif, namely an addition to a ketimine followed by Mannich reaction. Corey published his synthesis of the natural product in 1974 (Scheme 21),129 only three years after its isolation. The first ATA was installed through addition of an organolithium compound to imine 65 to form 66, which then cyclized to the corresponding enamine 67 upon treatment with acid. The formation of the second ATA center through an intramolecular Mannich addition (via iminium ion 68) furnished ketone 69, which was eventually converted to the natural product.
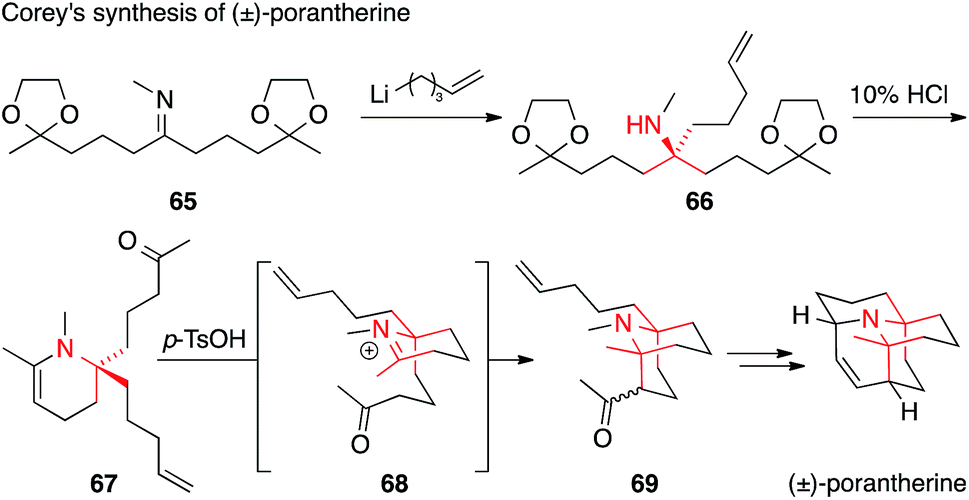 | ||
| Scheme 21 Porantherine synthesis by Corey (1974). p-TsOH = para-toluenesulfonic acid. | ||
A second synthesis of porantherine, published by Stevens in 1987, involved the addition of two alkyllithium compounds to an iminoether (not shown).130
6 Hasubanan alkaloids
The hasubanan alkaloids, isolated from various plant sources, are structurally related to the better-known morphine alkaloids but feature a pyrrolidine ring instead of a piperidine ring. They are comprised of over 40 family members, all of which share the same aza-propellane skeleton (Fig. 5).131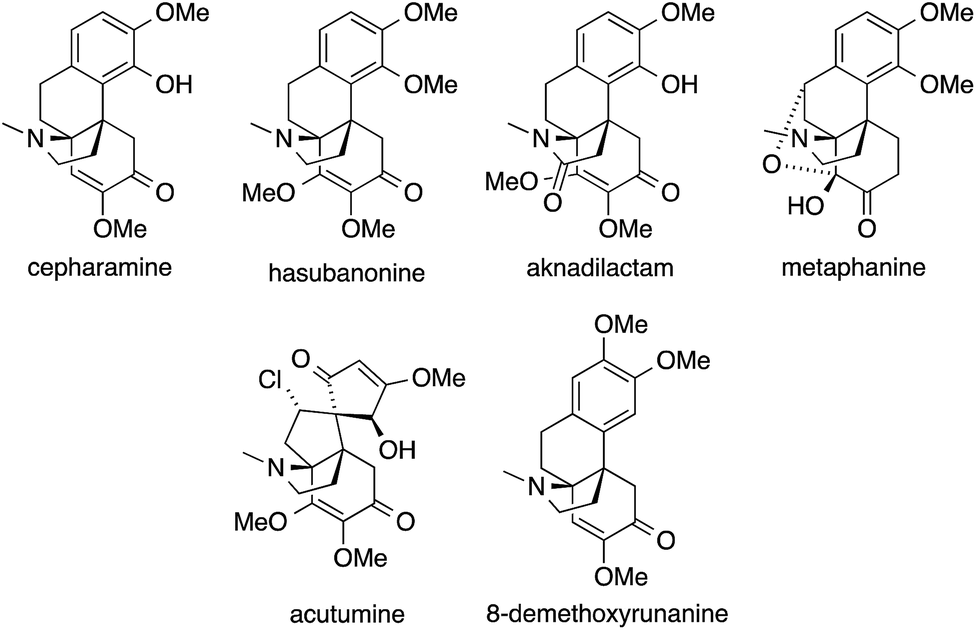 | ||
| Fig. 5 Representative hasubanan alkaloids. | ||
Although the physiological effects of the hasubanans are of less significance compared to the related morphine alkaloids, their beautiful structures stimulated numerous synthetic studies and several total syntheses have been reported to date.132–146
The first successful syntheses of (±)-cepharamine,132,135 (±)-hasubanonine,134,137 (±)-aknadilactam134 and (±)-metaphanine136,138 were published by Inubushi in the 1970s (Scheme 22a). Starting from tricyclic β-tetralone 70, the ATA was set via a cascade reaction involving an aza-Michael addition (71 → 74). Almost 30 years later, Schultz prepared (+)-cepharamine using a Hofmann-type rearrangement to introduce the ATA (not shown).139
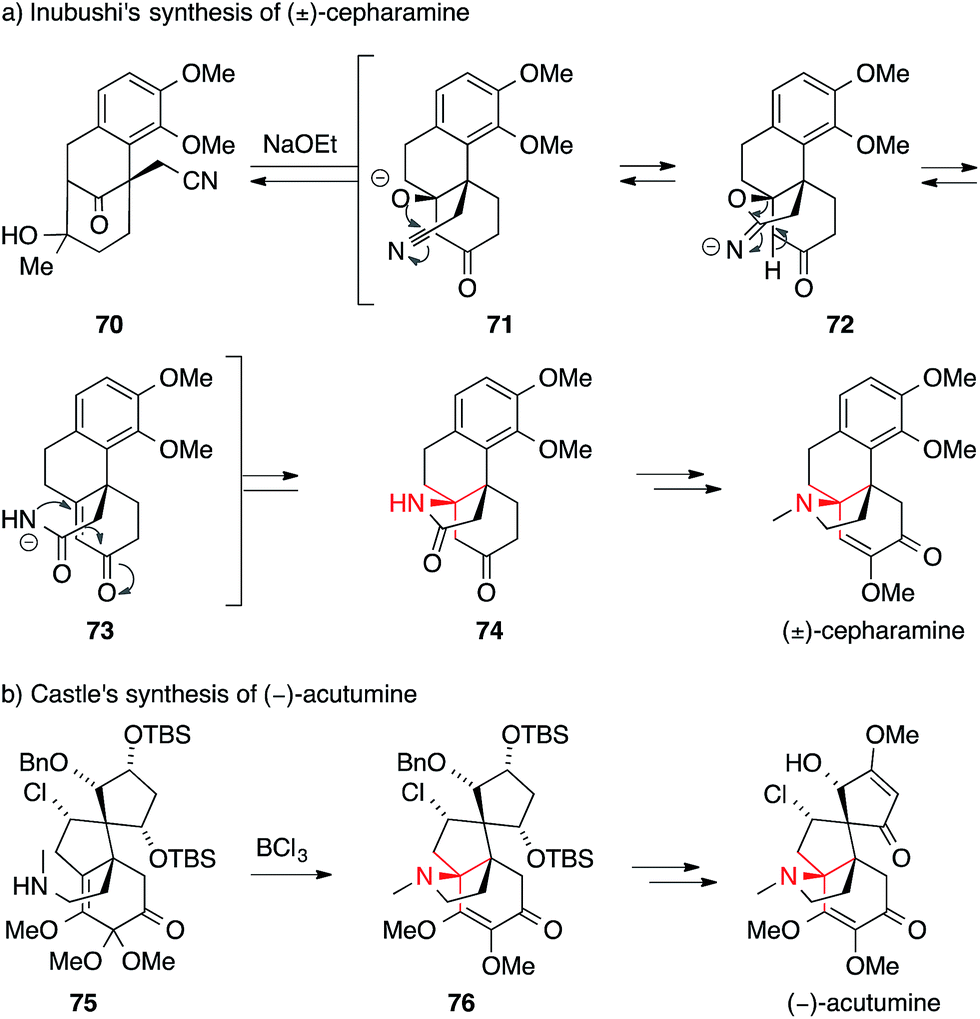 | ||
| Scheme 22 Syntheses of hasubanan alkaloids by Inubushi (1969) and Castle (2009). Bn = benzyl, TBS = tert-butyldimethylsilyl. | ||
Recent years have seen revived interest in hasubanans. An elegant method for the installation of the ATA was developed by Castle, who closed the pyrrolidine ring of isohasubanonine through a SN2′-reaction.140,147 Subsequently, this strategy was adapted to access (−)-acutumine (Scheme 22b).142 To this end, amine 75 was exposed to a Lewis acid to generate an allylic cation that was intercepted by the appended secondary amine. The resulting ATA-containing pyrrolidine 76 was then converted into (−)-acutumine.
In contrast to this approach, which sets the ATA at a relatively late stage in the synthesis, Reisman installed it at the beginning (Scheme 23a).144 Reaction of the chiral N-tert-butanesulfinimine 78 with phenethyl Grignard 77 provided sulfinamide 79 with a high degree of diastereoselectivity. Subsequently, 79 was converted into a series of hasubanan alkaloids such as (−)-8-demethoxyrunanine.
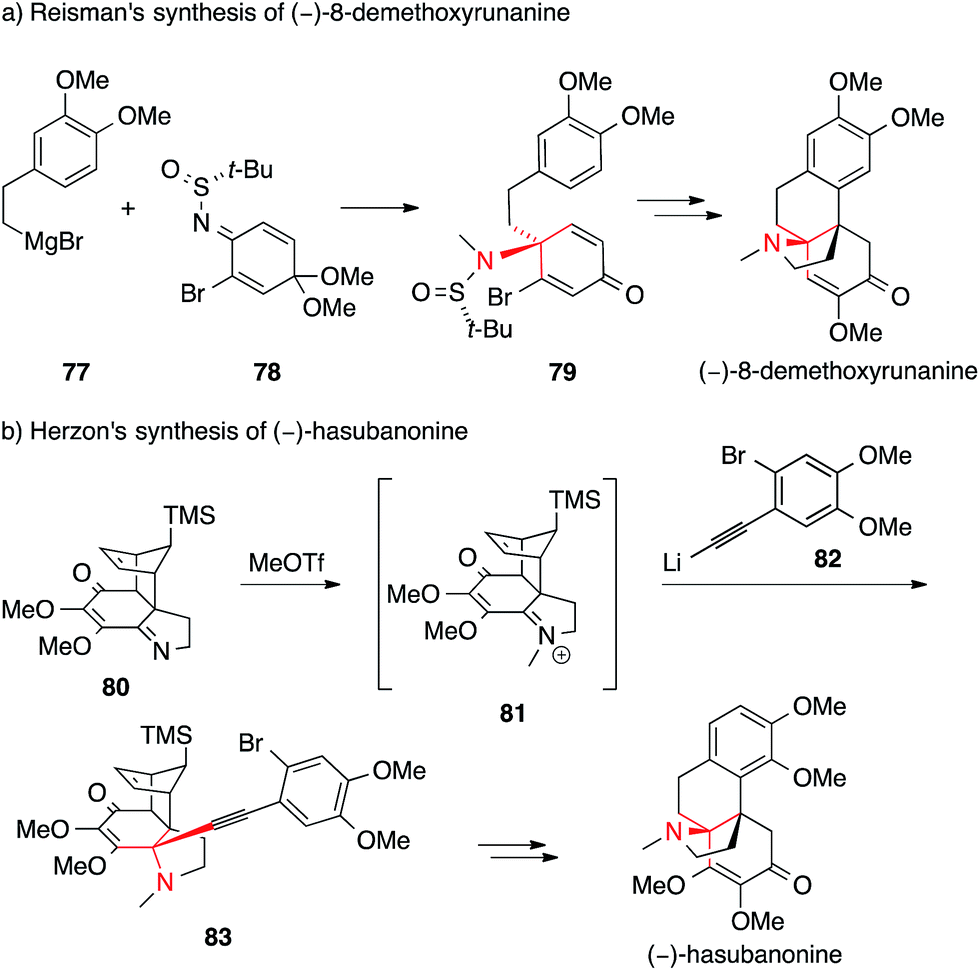 | ||
| Scheme 23 Syntheses of hasubanan alkaloids by Reisman (2011) and Herzon (2011). t-Bu = tert-butyl, TMS = trimethylsilyl, OTf = trifluoromethanesulfonate. | ||
The first enantioselective synthesis of hasubanonine was published by Herzon (Scheme 23b).143 Methylation of iminoquinone Diels–Alder adduct 80 (80 → 81), followed by addition of alkynyl lithium 82 gave amine 83, which was eventually transformed into optically pure (−)-hasubanonine. This strategy proved to be versatile, as many more hasubanan alkaloids, including (−)-runanine, (−)-delavayine, (+)-periglaucine B and (−)-acutumine, could be accessed by variation of the alkynyl species.143,145,146
7 Stemona alkaloids
Plants belonging to the family Stemonaceae, which are mostly found in Southeast Asia, have been used for centuries as insecticides and for the treatment of respiratory diseases.148–150 Phytochemical investigations led to the isolation of a variety of natural products known as Stemona alkaloids (Fig. 6).151,152 These polycyclic natural products possess highly complex structures weaving together pyrrolidines and butenolides, often through spiro fusions that contain ATAs.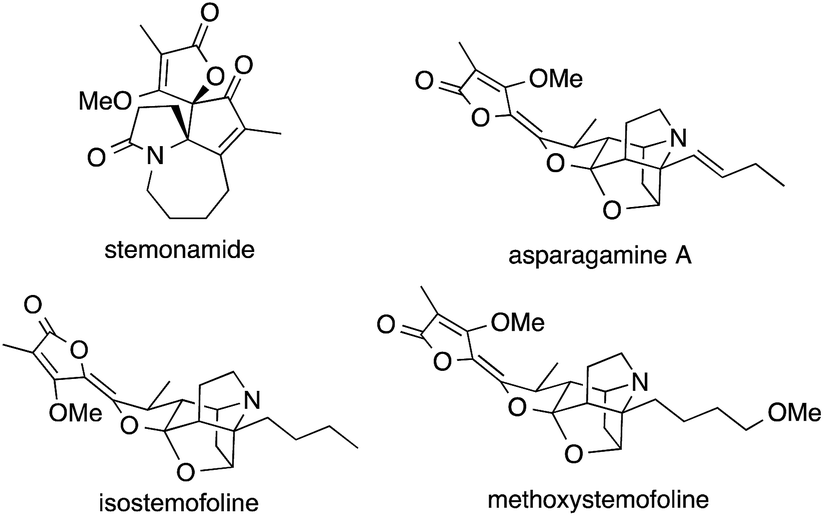 | ||
| Fig. 6 Representative Stemona alkaloids. | ||
The structural beauty of these molecules generated considerable interest in the synthetic community and stimulated the development of new synthetic methods for the installation of ATAs.151,152 The strategies employed range from classical additions to imines,153,154 to radical cyclization cascades,155,156 radical allylations,157 semipinacol-Schmidt cascades,158,159 Schmidt reactions,160 aza-Cope–Mannich reactions,161 cyclopropane-Cope rearrangements162 and catalytic 1,3-dipolar cycloadditions.163
The first synthesis of a Stemona alkaloid, viz. isostemofoline, was published by Kende in 1999 and employed a highly unusual and elegant approach.162 The ATA was formed via rhodium-catalyzed reaction of pyrrole 84 with vinyl diazoester 85. The resultant divinyl cyclopropane 86 underwent Cope rearrangement in situ to afford bicycle 87, which was then used as a key intermediate in the further assembly of the natural product (Scheme 24a).
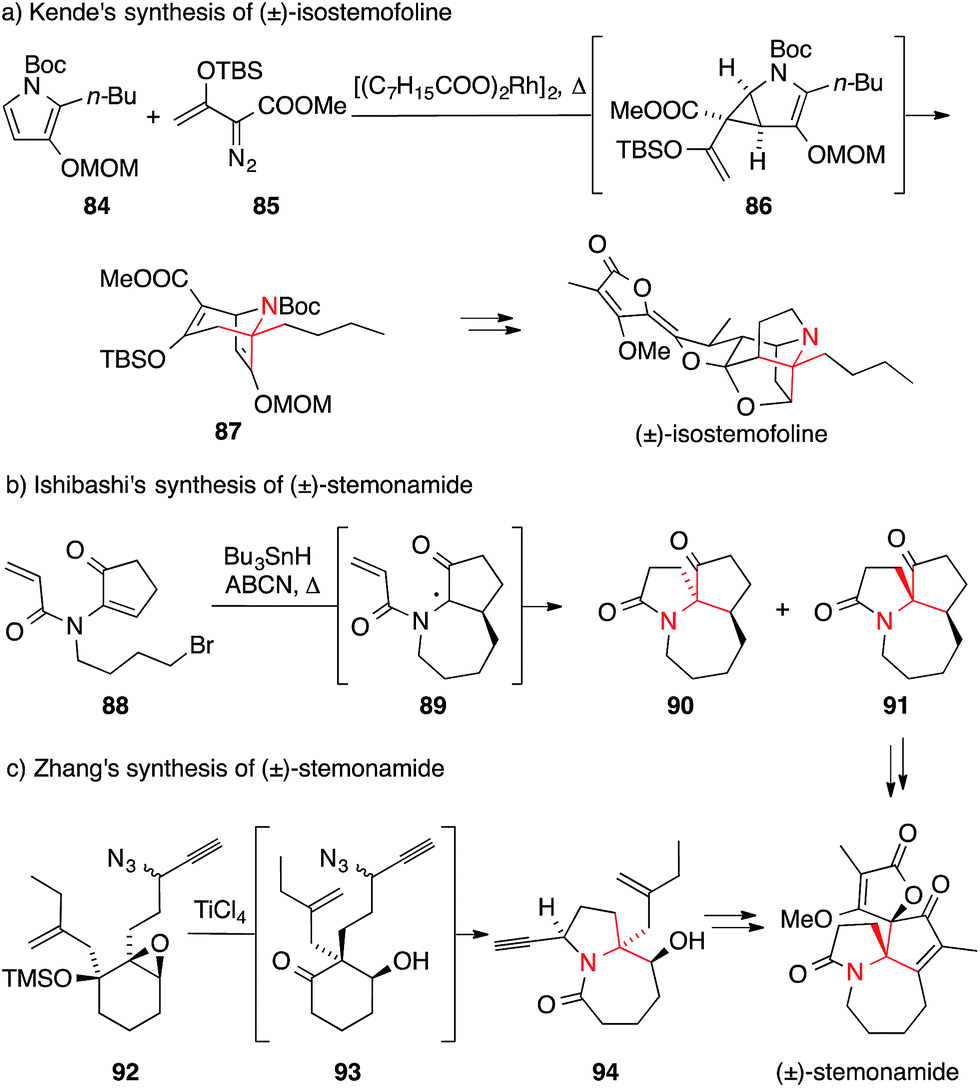 | ||
| Scheme 24 Syntheses of Stemona alkaloids by Kende (1999), Ishibashi (2008) and Zhang (2011). ABCN = 1,1′-azobis(cyclohexanecarbonitrile), Boc = tert-butyloxycarbonyl, MOM = methoxymethyl, TBS = tert-butyldimethylsilyl, TMS = trimethylsilyl. | ||
More recently, two synthetic approaches aimed at members of the stemonamine group were published. Ishibashi developed an entry to racemic stemonamide and isostemonamide as well as their reduced derivatives stemonamine and isostemonamine, based on a radical cascade as the key step for the formation of the ATA (Scheme 24b).155,156 Treatment of the achiral precursor 88 with tributyltin hydride and 1,1′-azobis(cyclohexanecarbonitrile) (ABCN) at elevated temperatures effected a 7-endo-trig cyclization that likely yielded radical 89 as the proposed intermediate, which in turn underwent an unusual 5-endo-trig cyclization providing access to a separable mixture of isomers 90 and 91. Further transformations of these tricyclic compounds furnished stemonamide and some of its congeners.
An alternative approach to stemonamide and related Stemona alkaloids was published by Zhang (Scheme 24c).159 Based on his systematic studies on the reactivity of α-hydroxy epoxides such as 92,164 he developed a powerful cascade that combines a semipinacol rearrangement with an Aubé–Schmidt reaction (92→94). The resulting amide 94 was obtained as a 5:1 mixture of diastereomers, reflecting the diastereomeric mixture of propargylic azides employed as substrates. Using this strategy and variations thereof, Zhang was able to synthesize stemonamide and three additional Stemona alkaloids, viz. maistemonine, stemonamine, and isomaistemonine.158–160
The only synthesis of asparagamine A, an unsaturated derivative of stemonamide, was achieved by Overman in 2003.161 He installed the ATA using his signature aza-Cope–Mannich cascade (Scheme 25a). The synthesis of a precursor molecule suitable for this transformation commenced with a Diels–Alder reaction, forming an ATA that later took part in the rearrangement. Treatment of 95 with excess paraformaldehyde at elevated temperatures generated iminium ion 96, which underwent the aza-Cope–Mannich sequence to afford the ATA of asparagamine A (98, via intermediate 97). More recently, a radical allylation (99 → 100) was utilized by Huang to set the ATA in (+)-methoxystemofoline at an early stage of the synthesis (Scheme 25b).157
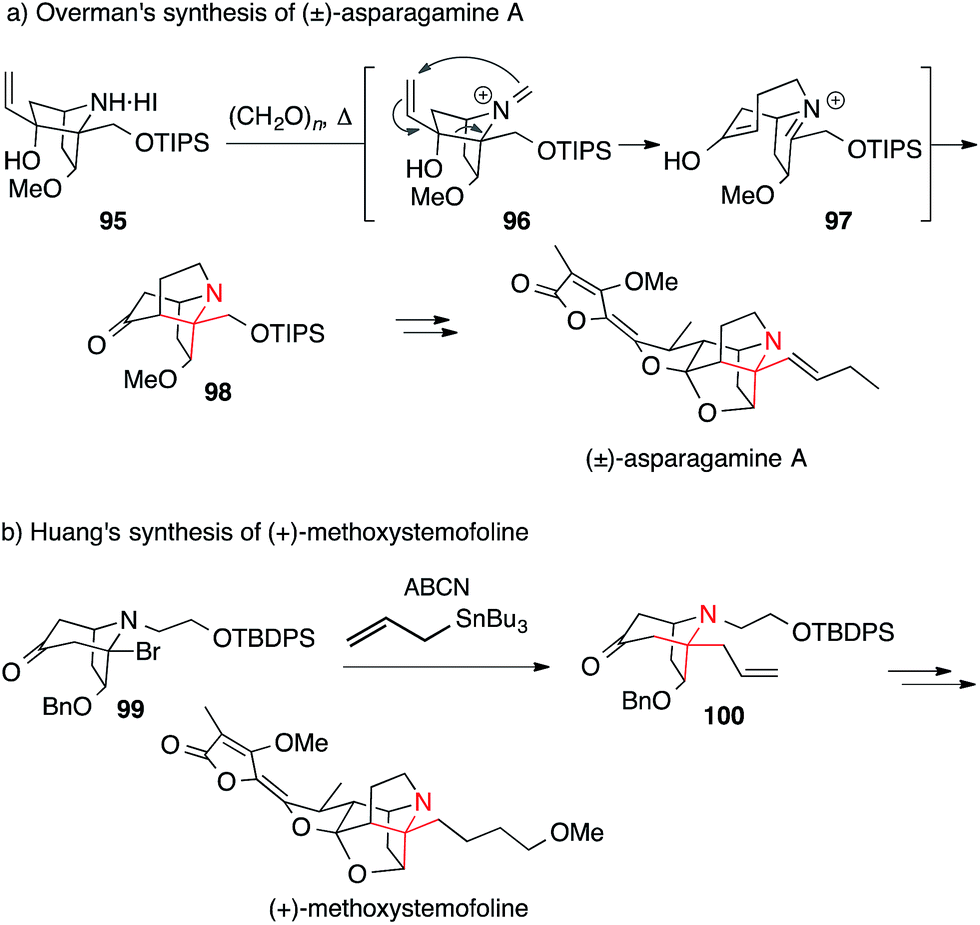 | ||
| Scheme 25 Syntheses of Stemona alkaloids by Overman (2003) and Huang (2015). ABCN = 1,1′-azobis(cyclohexanecarbonitrile), Bn = benzyl, TBDPS = tert-butyldiphenylsilyl, TIPS = triisopropylsilyl. | ||
8 Indole alkaloids
Indole alkaloids are a structurally and biosynthetically heterogeneous class of natural products characterized by an indole nucleus or derivative thereof. Several of them, albeit not the best known ones, contain ATAs (Fig. 7).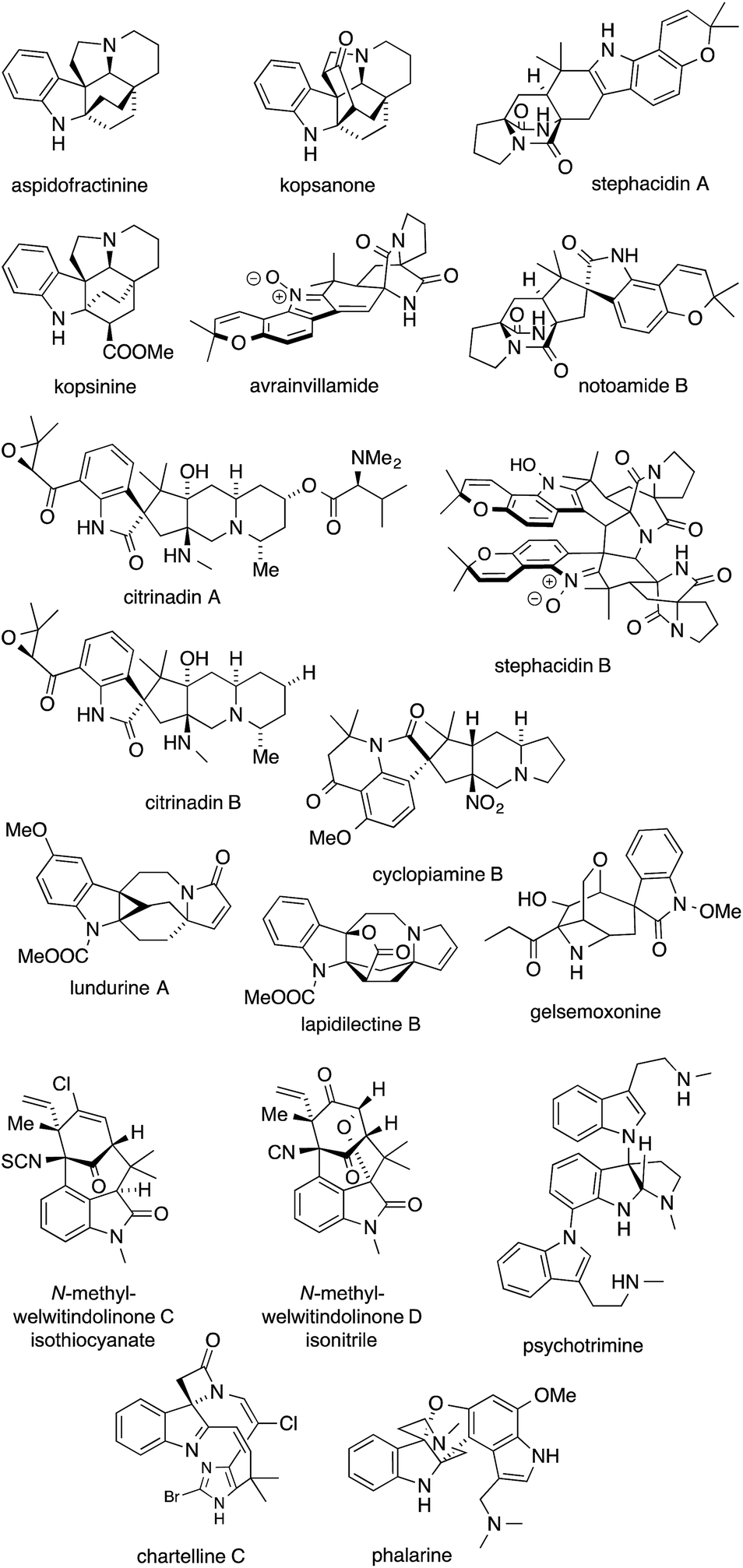 | ||
| Fig. 7 Indole alkaloids featuring ATAs. | ||
Kopsine, the first member of the so-called Kopsia alkaloids, was isolated as early as 1890,165 but it took several decades before its complex structure, and those of its congeners, could be elucidated.166–173 All members of this family possess an ATA incorporated in a bicyclo[2.2.2]octane system. Thus, the kopsanes seem predestined for Diels–Alder reactions, and few syntheses fail to employ a [4+2] cycloaddition strategy.174–176 The routes used can be divided into two main categories: (a) intermolecular Diels–Alder reactions177–180 and (b) intramolecular Diels–Alder reactions.181–185
The very first synthesis of (±)-aspidofractinine, completed in 1976, introduced an intermolecular Diels–Alder reaction to set the ATA using nitroethylene as a dienophile (not shown).177 Over time, phenyl vinyl sulfone emerged as a more practical dienophile178,179 and in 2009 the first enantioselective synthesis of (+)-aspidofractinine was reported by Spino using this reagent (Scheme 26a).180 In this case, imine 101 thermally isomerized to diene 102, which then underwent cycloaddition from the sterically more accessible convex side to afford sulfone 103.
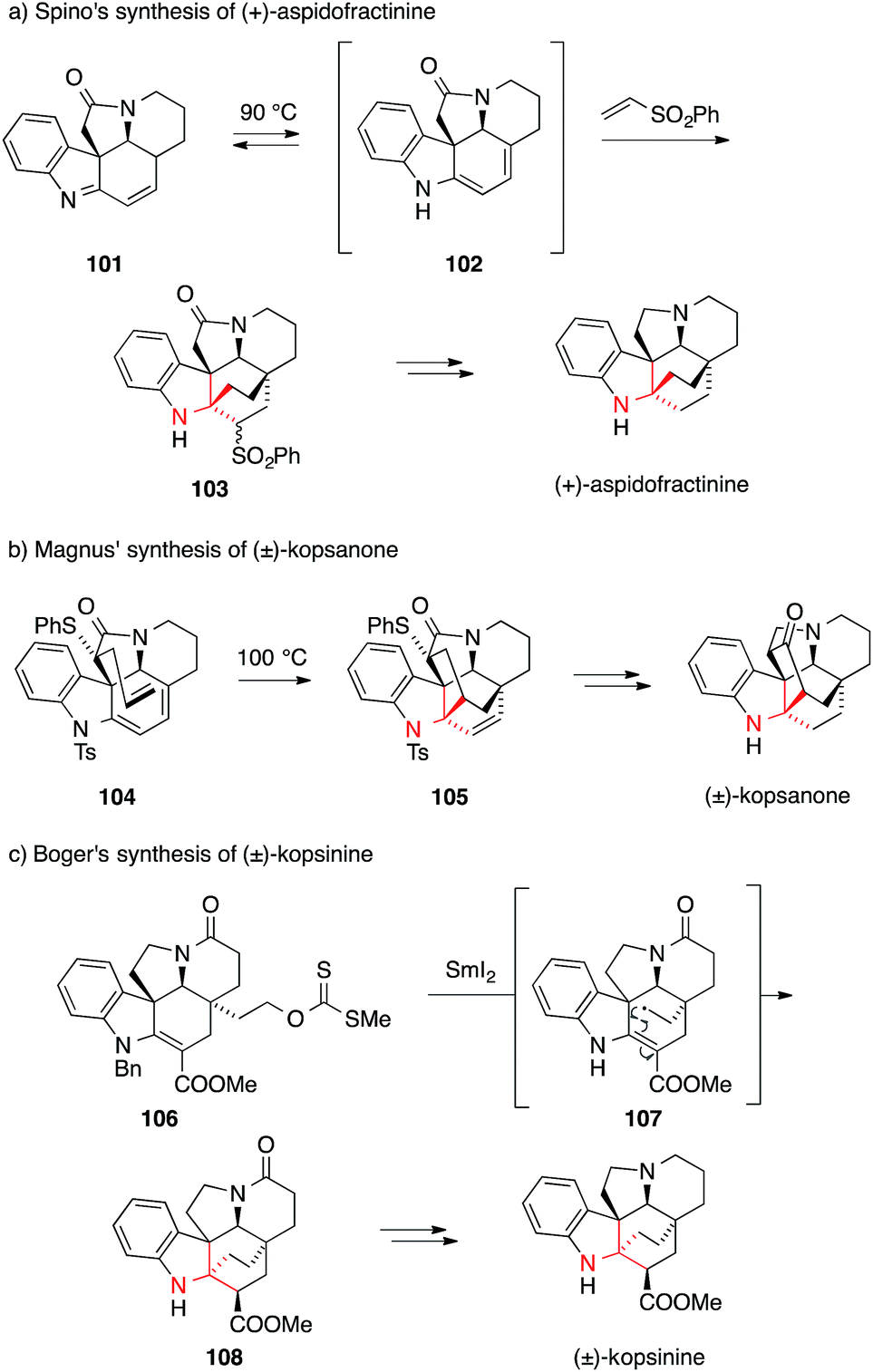 | ||
| Scheme 26 Kopsia alkaloid syntheses by Spino (2009), Magnus (1983) and Boger (2013). Bn = benzyl, Ts = p-toluenesulfonyl. | ||
The first successful intramolecular Diels–Alder approach to (±)-kopsanone and (±)-10,22-dioxokopsane was reported in 1983 by Magnus (Scheme 26b).181,182 They synthesized sulfide 104 as a suitable precursor, with the dienophile placed in the concave position. The cycloaddition reaction proceeded at 100 °C and provided intermediate 105, which was transformed into (±)-kopsanone in a few steps. Using a similar strategy, other indole alkaloids, (±)-kopsijasmine and (±)-kopsine, were prepared as racemates,184,185 as well as (−)-kopsinilam and (−)-kopsinine in enantiomerically pure form.183
In a recent example for an alternative approach by Boger, a powerful radical transannular cyclization was applied to install the ATA of kopsinine (Scheme 26c).176 Upon treatment of xanthate 106 with SmI2, ATA 108 was formed as a single diastereomer. Presumably, a primary radical intermediate 107 is formed, which undergoes a radical cyclization followed by reduction and diastereoselective protonation of the ester enolate.
Lapidilectine B and lundurine A are two structurally related Kopsia alkaloids that contain two ATAs. Although not originating from the same organism, they show a similar scaffold with a bridged 8-membered ring fused to an indoline on one side and a 5-membered ring on the other. Lapidilectine A was isolated by Awang from the leaves of the tree Kopsia lapidilecta in 1992.186,187 Lundurines A–D were isolated from the Malaysian tree Kopsia tenuis,188 and shown to be effective at bypassing multidrug resistance in vincristine-resistant KB cells.189
Qin accomplished the first enantioselective synthesis of (−)-lundurine A in 2014 (Scheme 27a).190 The first ATA was established via the addition of allylmagnesium bromide to an iminium ion generated by in situ alkylation of imine 109 to form tetracycle 110. In order to establish the two fully substituted stereocenters on the indoline of 112, Qin resorted to an unusual intramolecular Simmons–Smith cyclopropanation of diiodide 111. Two other racemic syntheses of lundurine A and B have been reported by Nishida.191–193 He employed a Curtius rearrangement and a 1,2-addition to an iminium ion for lundurine B193 and a Tsuji–Trost amination and an indoxyl bisalkylation for the synthesis of lundurine A (not shown).191,192
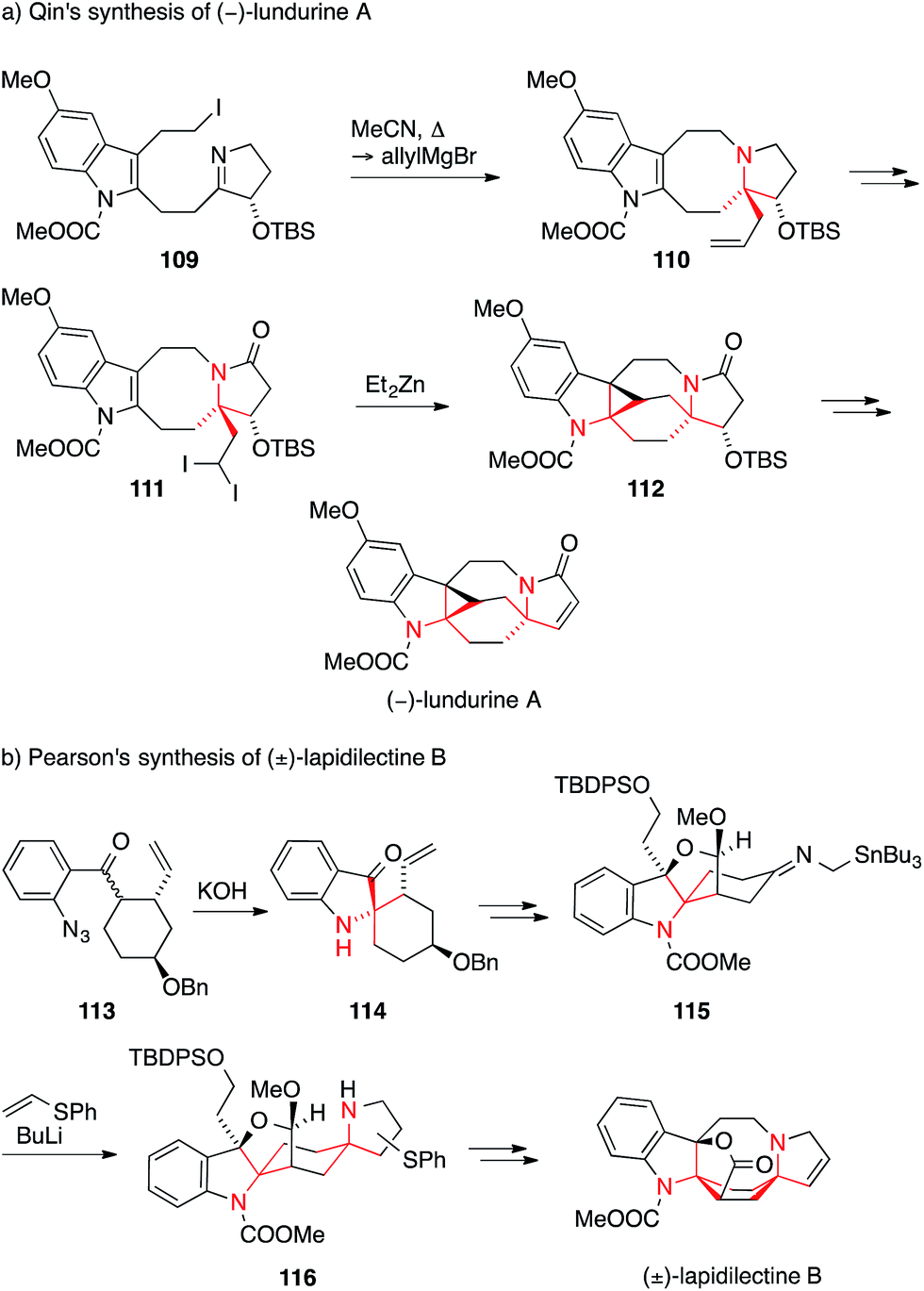 | ||
| Scheme 27 Syntheses of Kopsia alkaloids by Qin (2014) and Pearson (2001). Bn = benzyl, TBDPS = tert-butyldiphenylsilyl, TBS = tert-butyldimethylsilyl. | ||
In 2001, Pearson employed a Smalley cyclization of aryl ketone azide 113 to furnish the spiroindoxyl 114 (Scheme 27b).194,195 In the final steps of his lapidilectine B synthesis, he then used his trademark azaallyl anion [3+2] cycloaddition to establish the pyrrolidine ring (115 → 116) as an inconsequential mixture of regioisomers.
The cycloaddition approach has not been limited to the kopsane alkaloids. Other indole alkaloids, such as stephacidin A and the notoamides, which bear two ATAs, were prepared by a presumably biomimetic [4+2] cycloaddition.
Williams synthesized stephacidin A and notoamide B starting from imidate 117, which underwent base-mediated isomerization to 118 followed by intramolecular Diels–Alder reaction to afford diazabicyclo[2.2.2]octane 119 (Scheme 28).196 This remarkable reaction sets both ATAs in a single step. Later that year, stephacidin B was accessed via avrainvillamide using the same strategy.197
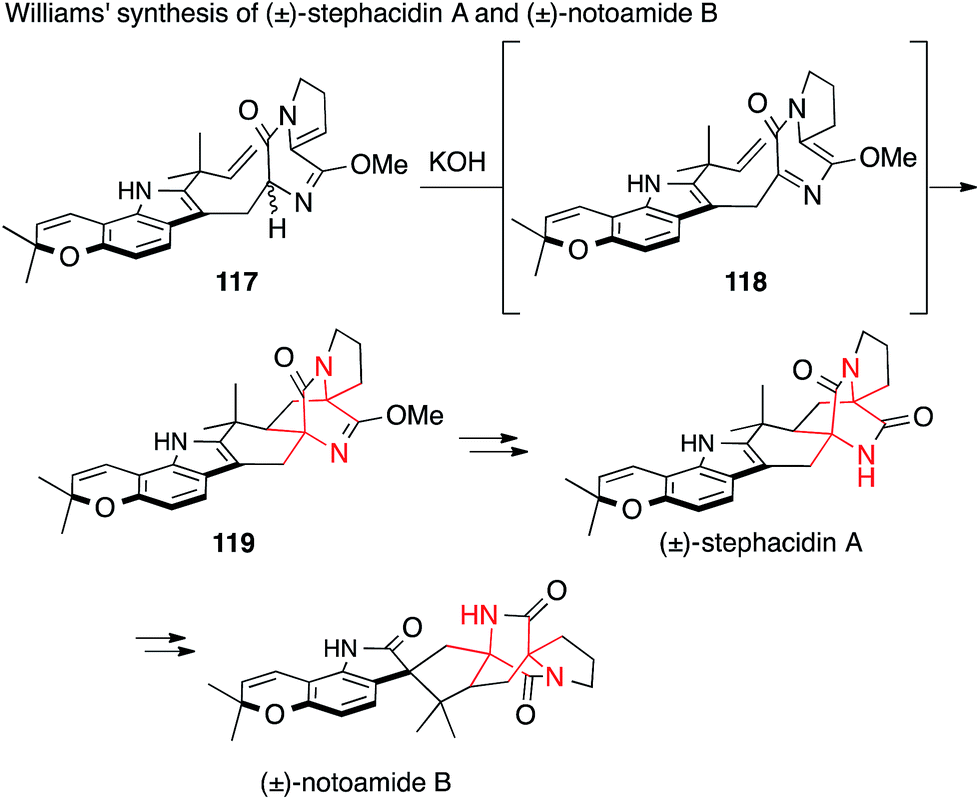 | ||
| Scheme 28 Synthesis of prenylated indole alkaloids by Williams (2007). | ||
In 2005, Baran used the α-alkylation of proline derivative 120 with complete chirality transfer, a method developed by Seebach,198 to set the first ATA of stephacidin A in 121 (Scheme 29).199 The second ATA present in 123 was installed by an intramolecular, stereocontrolled oxidative enolate coupling starting from diketopiperazine 122. Baran was then able to convert stephacidin A into avrainvillamide and stephacidin B following a biosynthetic proposal.200
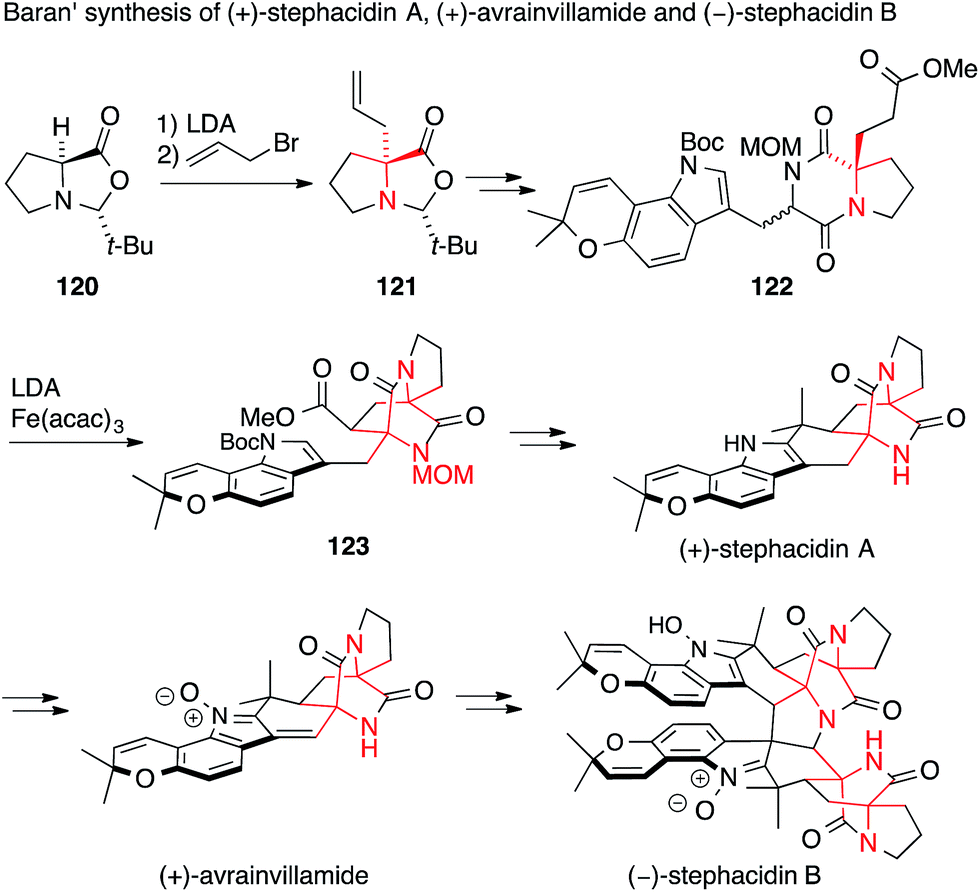 | ||
| Scheme 29 Synthesis of prenylated indole alkaloids by Baran (2005). acac = acetylacetonate, Boc = tert-butyloxycarbonyl, t-Bu = tert-butyl, LDA = lithiumdiisopropylamide. | ||
A second synthesis of avrainvillamide and stephacidin B was accomplished concurrently by Myers (Scheme 30).201 In this case, the first ATA was installed by a Strecker-type addition of TMS cyanide to enamine 124 to form the N-Boc amino nitrile 125. The second ATA was then set by a very unusual radical transfer cyclization. Abstraction of a hydrogen atom in 126, followed by loss of toluene, generates an aminoacyl radical which attacks the enamide double bond and ejects a phenylthiyl radical to form the diketopiperazine 127.
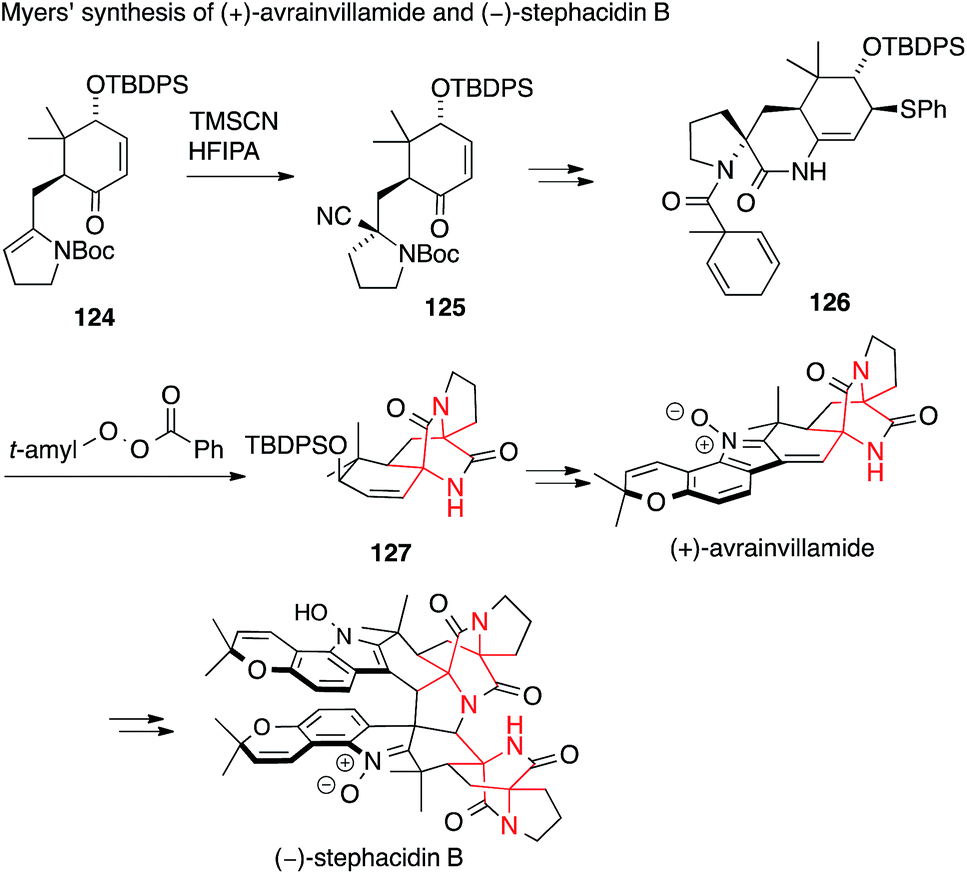 | ||
| Scheme 30 Synthesis of prenylated indole alkaloids by Myers (2005). t-amyl = tert-amyl (2-methylbutyl), Boc = tert-butyloxycarbonyl, HFIPA = 1,1,1,3,3,3-hexafluoroisopropyl alcohol, TBDPS = tert-butyldiphenylsilyl, TMSCN = trimethylsilyl cyanide. | ||
Several members of a related family of prenylated spirooxindole alkaloids, namely cyclopiamine, citrinadin A and citrinadin B, also feature an asymmetric ATA.202–204 In 2013, Martin and Wood reported the first syntheses of citrinadin A and B.205,206 In the case of citrinadin A, epoxide 128 was heated in the presence of methylamine to provide 1,2-amino alcohol 129 (Scheme 31a).205 Wood's approach employed an azide-mediated opening of epoxide 130 to establish the ATA in 131 (Scheme 31b).206 Both reactions are rare examples where an ATA has been set through a SN2 reaction.
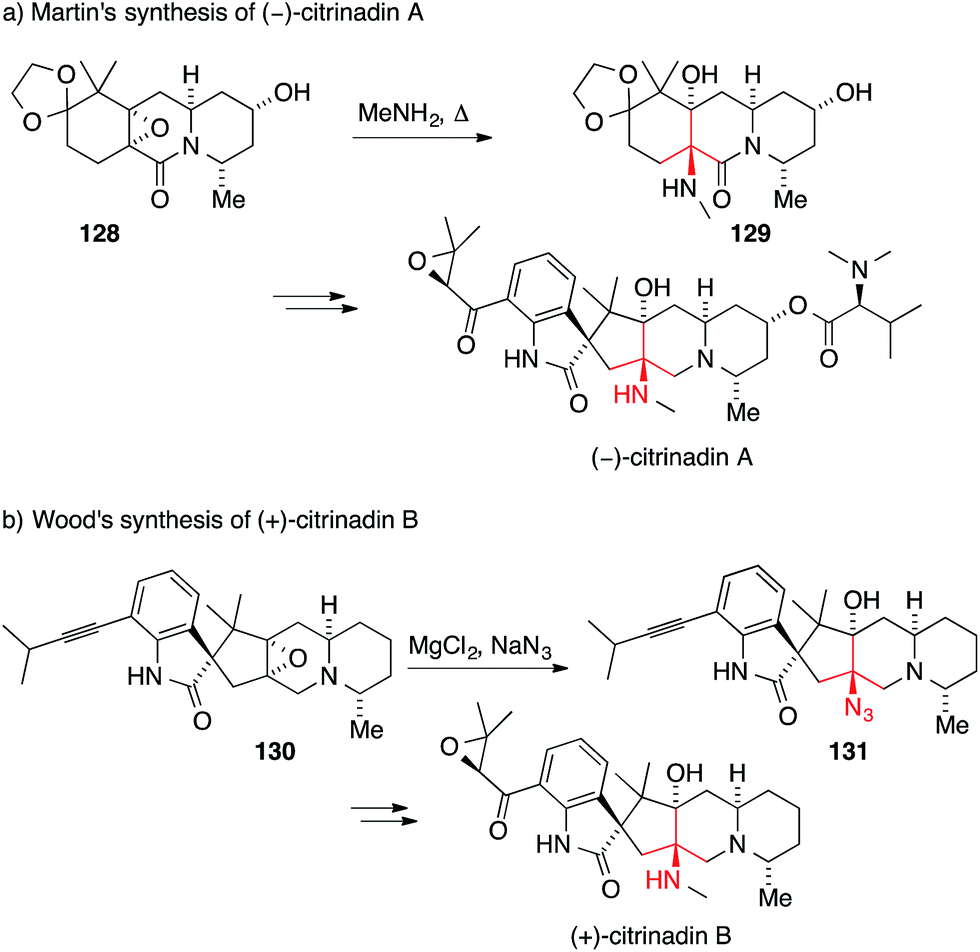 | ||
| Scheme 31 Syntheses of prenylated indole alkaloids by Martin (2013) and Wood (2013). | ||
More recently, Sarpong published his entry to the prenylated indole alkaloids cyclopiamine B and ent-citrinalin B (Scheme 32). The first ATA was set via a Hofmann rearrangement (132 → 133).207 The second, not asymmetric, ATA center was established by treating ent-citrinalin B with sodium hydride to effect the rearrangement of the chromanone to the tetrahydroquinolone moiety present in cyclopiamine via retro-Michael/Michael addition. Using a similar approach, he was then able to synthesize the structurally related alkaloids stephacidin A and notoamide B.208
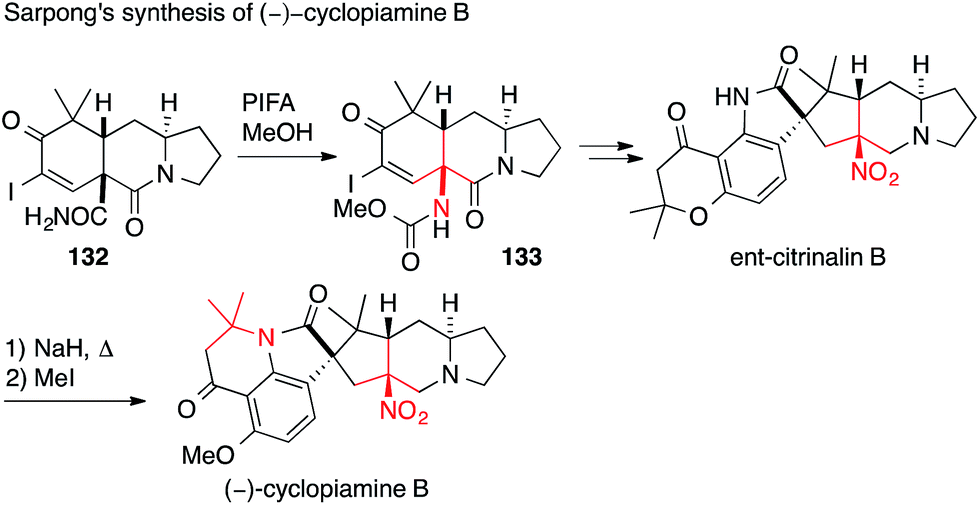 | ||
| Scheme 32 Synthesis of ent-citrinalin B and (−)-cyclopiamine B by Sarpong (2015). PIFA = (Bis(trifluoroacetoxy)iodo)benzene. | ||
Two alkaloids closely related to notoamide B, marcfortine B and C, were synthesized by Trost using a Michael addition and a radical cyclization to set the two ATAs (not shown).209,210
Gelsemoxonine is an indole alkaloid with an ATA that is part of a azetidine, a rare structural motif. It is also a member of the Gelsemium spirooxindole family, a large alkaloid family with highly compact, strained and complex structures, which have attracted considerable synthetic activity.211–213 In 2011, Fukuyama accomplished a total synthesis of gelsemoxonine that employed an intramolecular epoxide opening of 134 to install the ATA (Scheme 33a).214 Recently, Carreira published an elegant entry to gelsemoxonine, setting the ATA 136via a diastereoselective propynyllithium addition to isoxazoline 135 (Scheme 33b).215
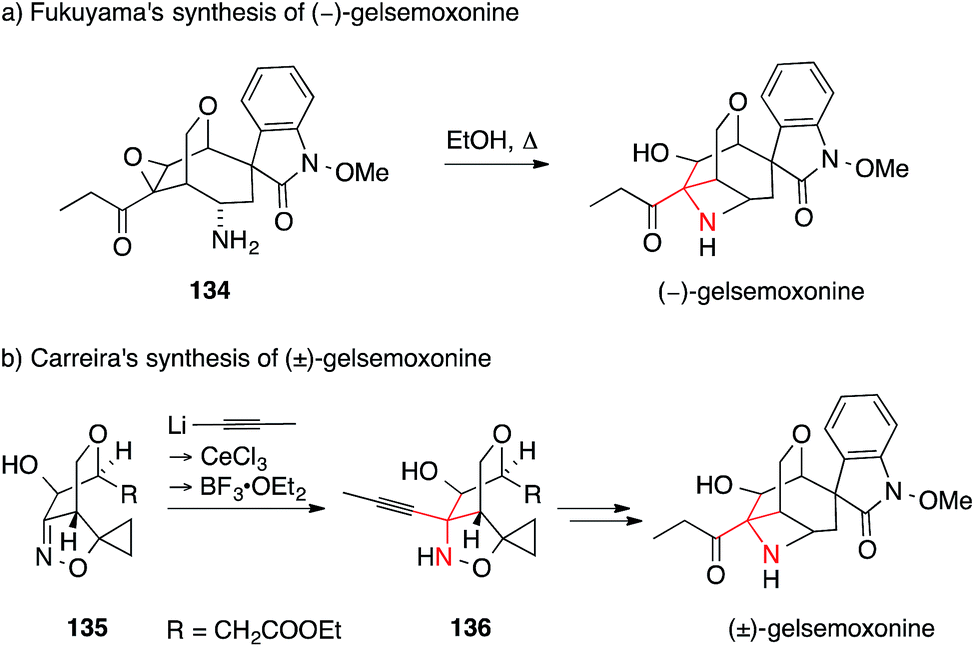 | ||
| Scheme 33 Syntheses of gelsemoxonine by Fukuyama (2011) and Carreira (2013). Boc = tert-butyloxycarbonyl. | ||
The welwitindolinones are another class of indole alkaloids with an ATA that is not part of the indole-derived moiety itself. The first welwitindolinone natural products (Fig. 7) were isolated by Moore in 1994 from the cyanobacteria Hapalosiphom welwitschii and Westiella intracta.216 All welwitindolinones known so far feature a [4.3.1] bicyclic framework, which, in some cases, contains a modified ATA that bears an isothiocyanate or isonitrile functional group.217,218
Being a considerable challenge for total synthesis, the welwitindolinones have become popular targets.219 The first total synthesis of N-methylwelwitindolinone D isonitrile was accomplished by Rawal in 2011220–222 using Kim's oxime rearrangement to install the isothiocyanide (137 → 138, Scheme 34a).223,224 Desulfuration of 138 then gave the naturally occuring isonitrile. Martin completed a synthesis that intercepts Rawal's synthesis in 2012.225
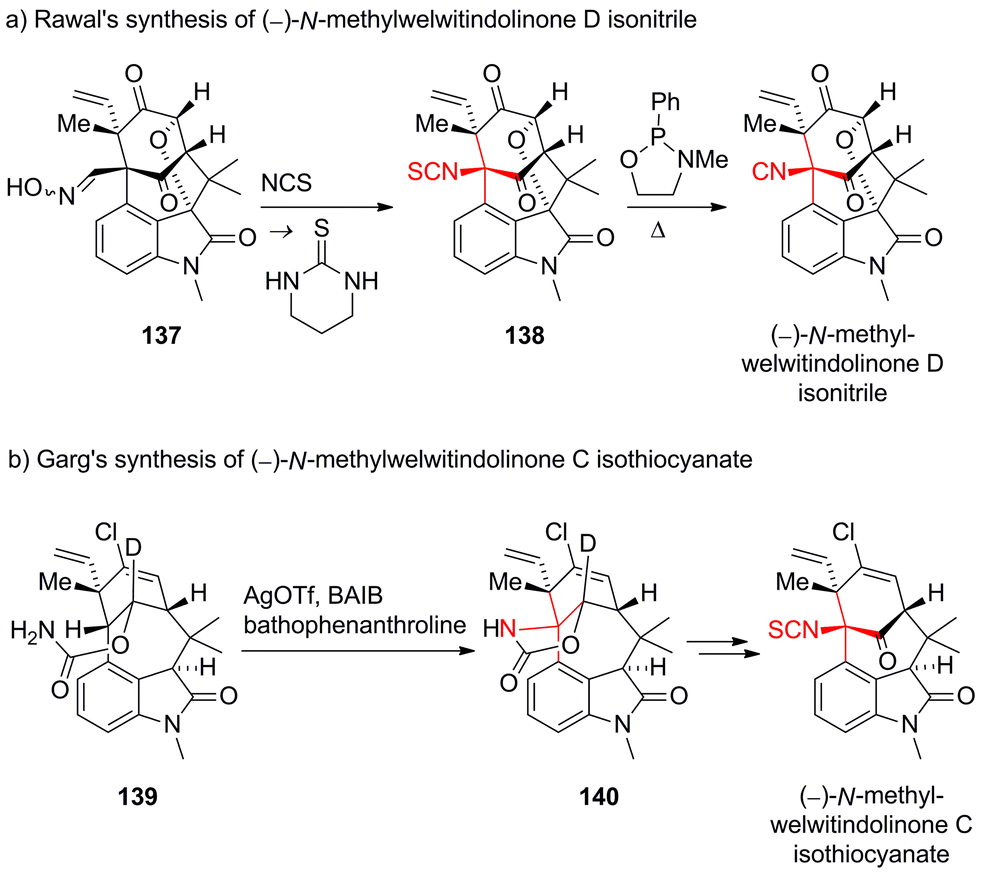 | ||
| Scheme 34 Syntheses of welwitindolinones by Rawal (2011) and Garg (2011). BAIB = (diacetoxyiodo)benzene, NCS = N-chlorosuccinimide, OTf = trifluoromethanesulfonate, Ph = phenyl. | ||
Garg's total synthesis of N-methylwelwitindolinone C isothiocyanate used an intramolecular Ag-mediated nitrene C,H-insertion of amide 139 as the critical step, which furnished carbamate 140 (Scheme 34b).226,227 To improve the regioselectivity and yield of this late stage transformation, the authors beautifully exploited the deuterium kinetic isotope effect.228
Both Rawal and Garg were able to subsequently synthesize several members of the welwitindolinone family by varying their initial strategies.222,228–230 In addition, Hatakeyama recently accomplished another synthesis of (−)-N-methylwelwitindolinone C isothiocyanate using an endgame similar to Rawal's.231
Two examples of reactions which have been specifically developed to set an ATA, both explored by the Baran laboratory, are shown in Scheme 35.
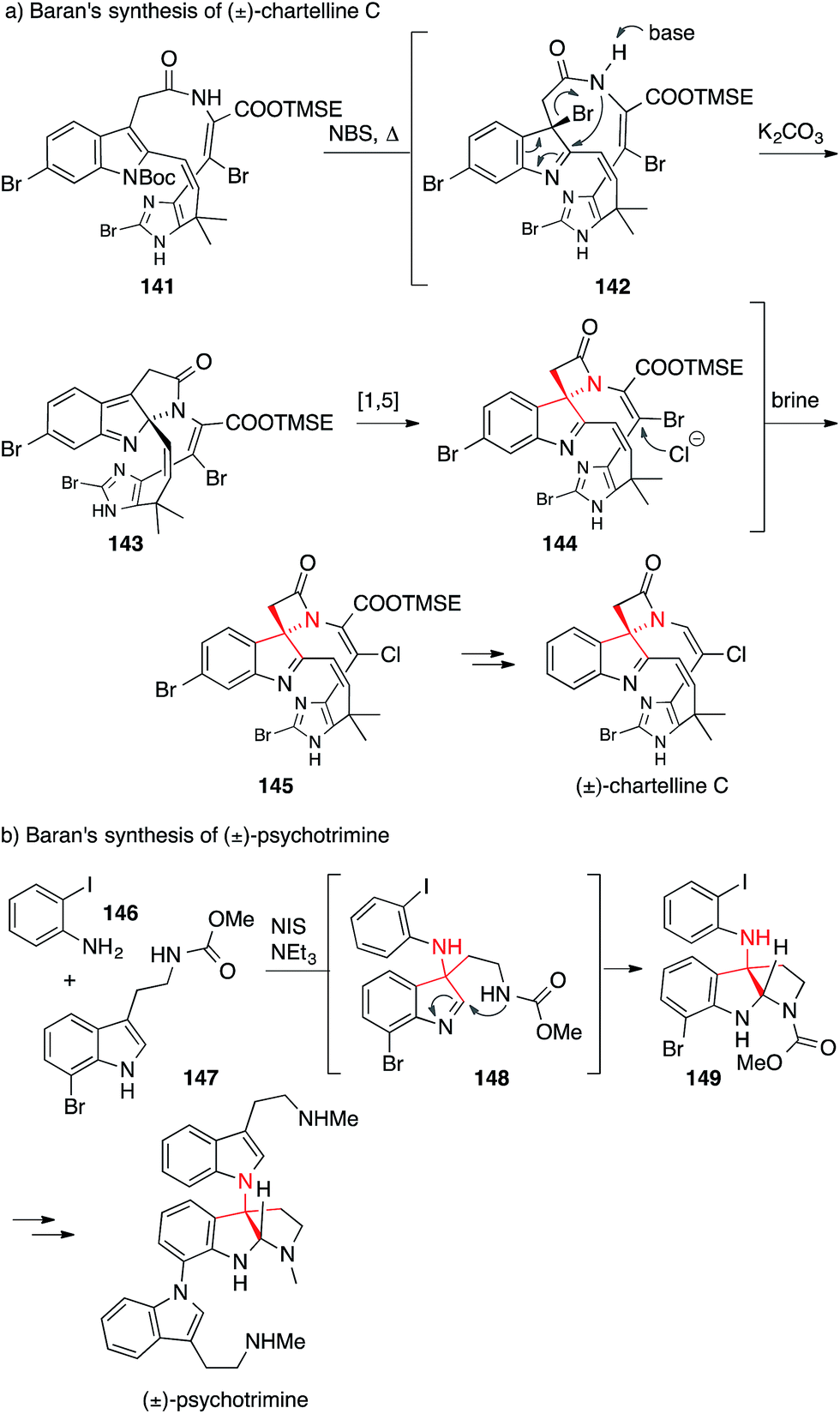 | ||
| Scheme 35 Baran's syntheses of chartelline C (2006) and psychotrimine (2008). Bn = benzyl, Boc = tert-butyloxycarbonyl, NBS = N-bromosuccinimide, NIS = N-iodosuccinimide, TMSE = 2-(trimethylsilyl)ethyl. | ||
In the synthesis of chartelline C, the ATA was set via a cascade reaction initiated by the bromination of indole 141 at 185 °C resulting in 142 (Scheme 35a). Amide attack furnished intermediate 143, which then rearranged in a 1,5-shift to give the ring contracted spiro-β-lactam 144.232,233 For the synthesis of psychotrimine, a coupling of indole 147 with 2-iodoaniline (146) was developed to yield 148, which then underwent further cyclization to give 149 (Scheme 35b).234 This method was also used for the syntheses of psychotetramine,235 kapakahine B and kapakahine F.236,237
Another interesting way to install an ATA in a structurally complex indole alkaloid was published by Danishefsky (Scheme 36).238 In his synthesis of the furanobisindole alkaloid phalarine, amino acid derivative 150 was treated with formaldehyde and acid to set the ATA in a diastereoselective fashion (150 → 152). It is not clear, however, whether this reaction proceeds via a 1,2-Wagner–Meerwein shift (possible intermediate 153) or a Pictet–Spengler reaction (possible intermediate 154) starting from iminium intermediate 151.
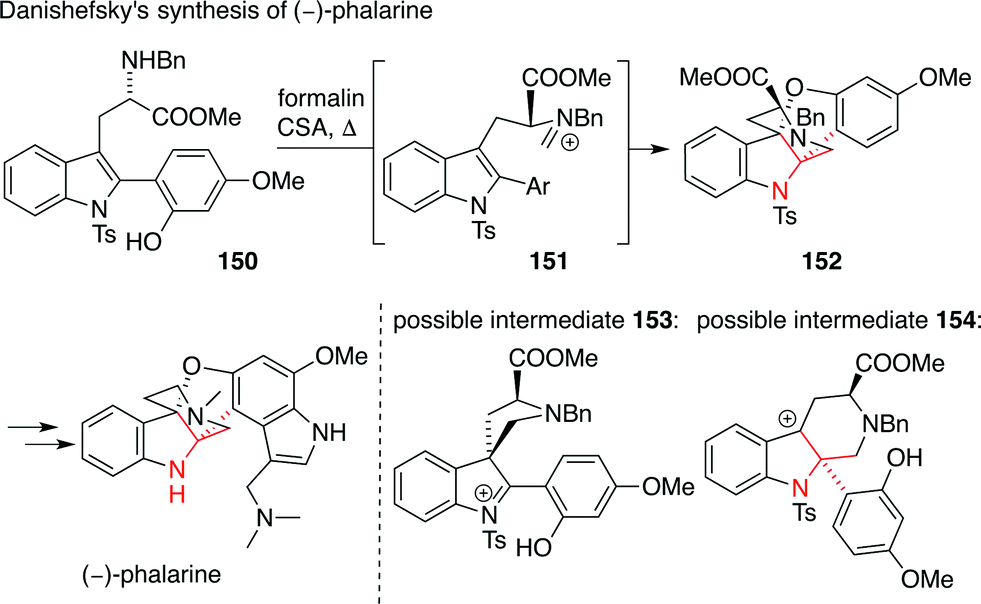 | ||
| Scheme 36 Danishefsky's synthesis of phalarine (2010). Bn = benzyl, CSA = camphorsulfonic acid. | ||
9 Cephalotaxines
Due to their interesting chemical structure and antileukemic activities, the cephalotaxines, isolated from the Japanese plum yew (Cephalotaxus harringtonii), have emerged as popular targets for natural product synthesis (Fig. 8).239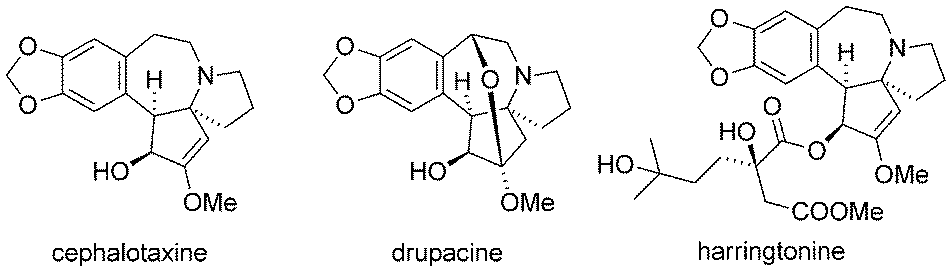 | ||
| Fig. 8 Representative cephalotaxines. | ||
The first synthesis of cephalotaxine itself was reported by Weinreb in 1972 (Scheme 37a).240 Conversion of enamine 155 into diketone 156 set the stage for a Lewis-acid catalyzed cyclization to yield tertiary amine 158 (via intermediate 157). Weinreb was able to synthesize cephalotaxine in six additional steps with an overall yield of 20%, setting a high bar for the following syntheses.
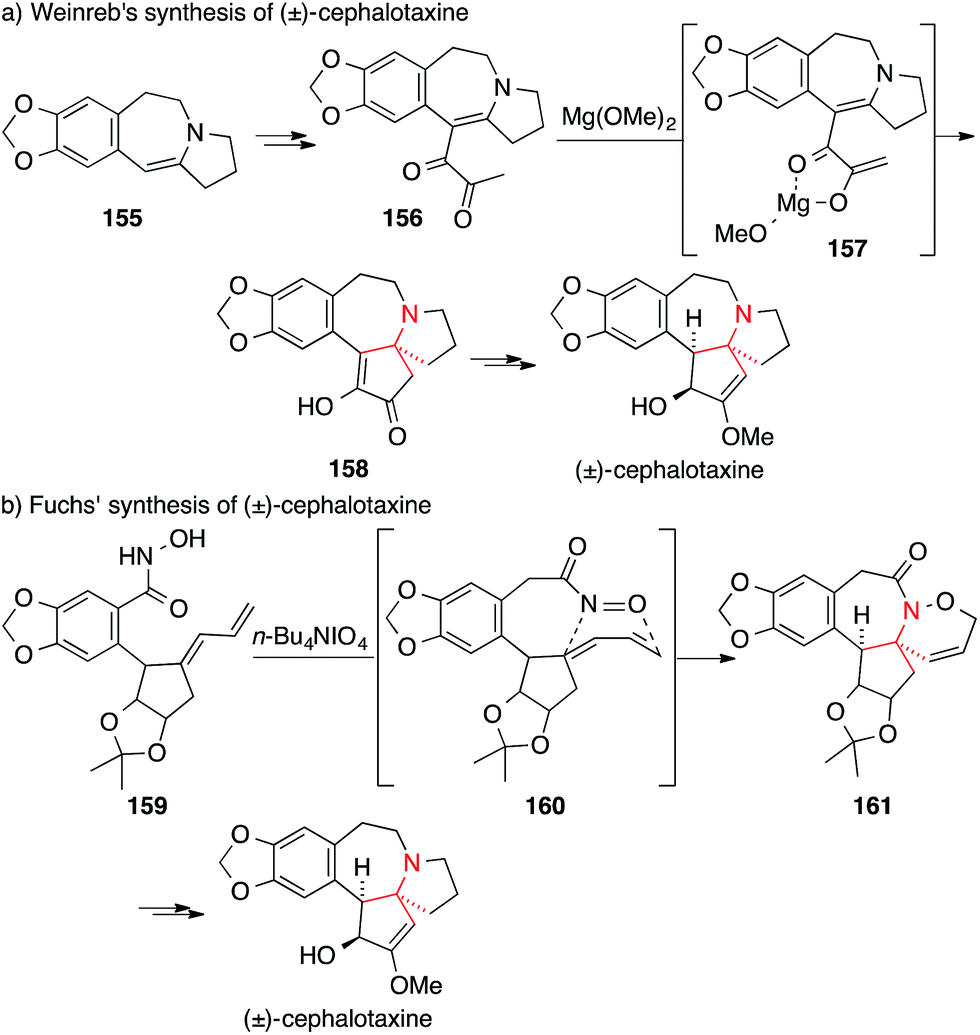 | ||
| Scheme 37 Syntheses of Cephalotaxine by Weinreb (1972) and Fuchs (1988). | ||
In 1988, Fuchs utilized an intramolecular [4+2] nitroso-Diels–Alder cycloaddition to assemble the benzazepine 161 from hydroxamic acid 159 (via intermediate 160, Scheme 37b).241
Tietze published a formal asymmetric synthesis of (−)-cephalotaxine in 1999 that is based on palladium catalysis (Scheme 38a).242 A Tsuji–Trost allylation on secondary amine 162 established the ATA and afforded 163 which was converted into cephalotaxine via a subsequent Heck reaction. A similar Tsuji–Trost allylation was used by Stoltz in 2007 in his synthesis of drupacine and cephalotaxine.243
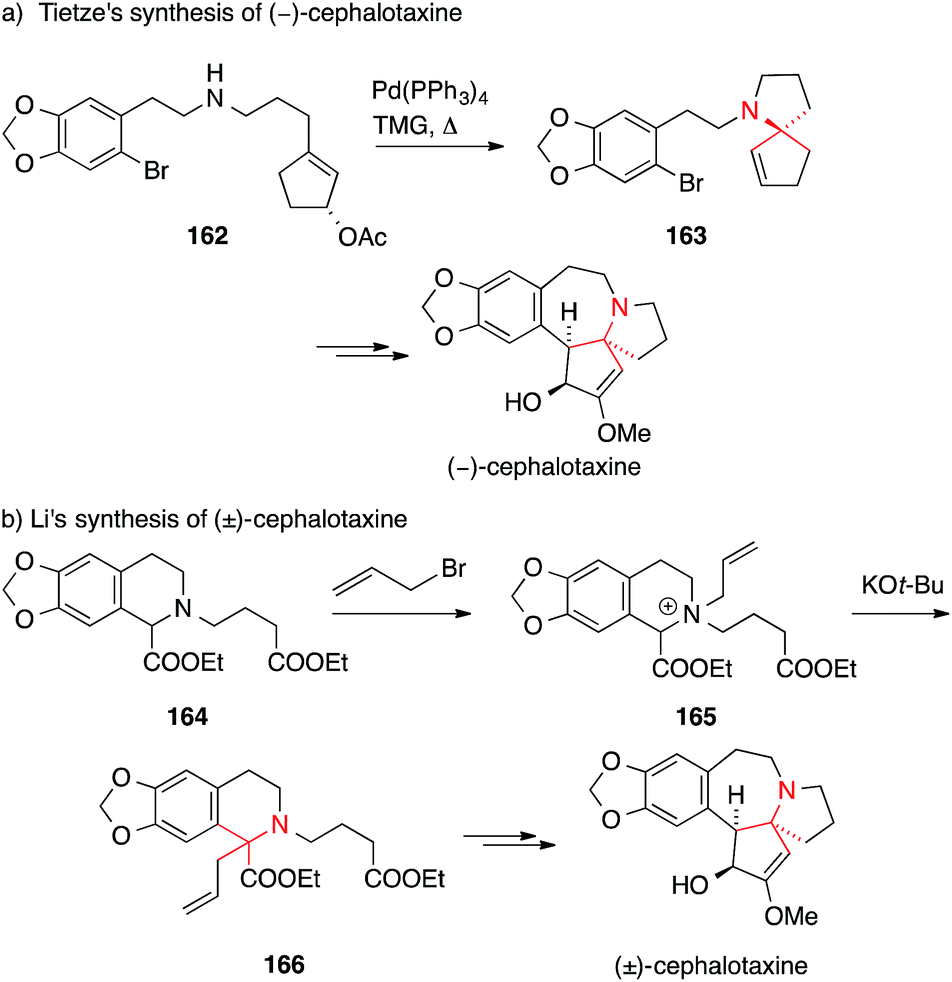 | ||
| Scheme 38 Syntheses of cephalotaxine by Tietze (1999) and Li (2003). Ac = acetyl, TMG = 1,1,3,3-tetramethylguanidine. | ||
A [2,3] sigmatropic rearrangement was utilized by Li to convert quaternary ammonium ion 165 to the α-tertiary amino ester 166 (164 → 166, Scheme 38b).244
A rather unusual approach for the asymmetric synthesis of (−)-cephalotaxine was pursued by Royer, who introduced the ATA on key intermediate 169via semipinacol rearrangement of chiral α-hydroxyiminium 168. The latter was generated by acid-catalyzed isomerization and protonation of pyrrolinone 167 (Scheme 39a).245
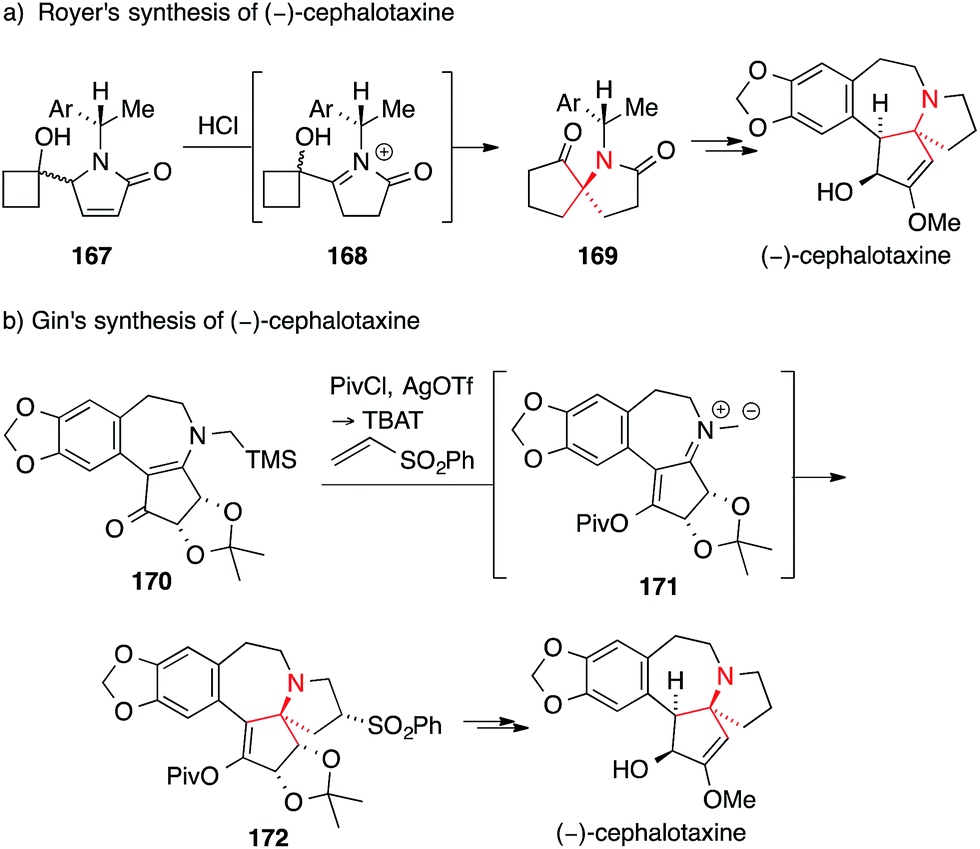 | ||
| Scheme 39 Syntheses of cephalotaxines by Royer (2004) and Gin (2006). Ar = aryl, OTf = trifluoromethanesulfonate, Piv = pivaloyl, TBAT = tetrabutylammonium difluorotriphenylsilicate. | ||
Another synthesis was developed by Gin, who transformed vinylogous amide 170 into azomethine ylide 171 which then underwent 1,3-dipolar cycloaddition with phenyl vinyl sulfone to yield 172 (Scheme 39b).246,247 The unexpected yet advantageous stereochemical outcome of this cycloaddition was confirmed by X-ray analysis.
In 2006, Mariano used a photochemical cyclization of a bicyclic pyridinium ion 173 to afford aziridine 174, which was then transformed into the natural product (Scheme 40a).248 Hayes' synthetic route towards (−)-cephalotaxine made use of an alkylidene carbene 1,5-C,H-insertion starting from ketone 175 to furnish spiro[4.4]azanonane 177, which corresponds to the skeleton of cephalotaxine (175 → 177, Scheme 40b).249
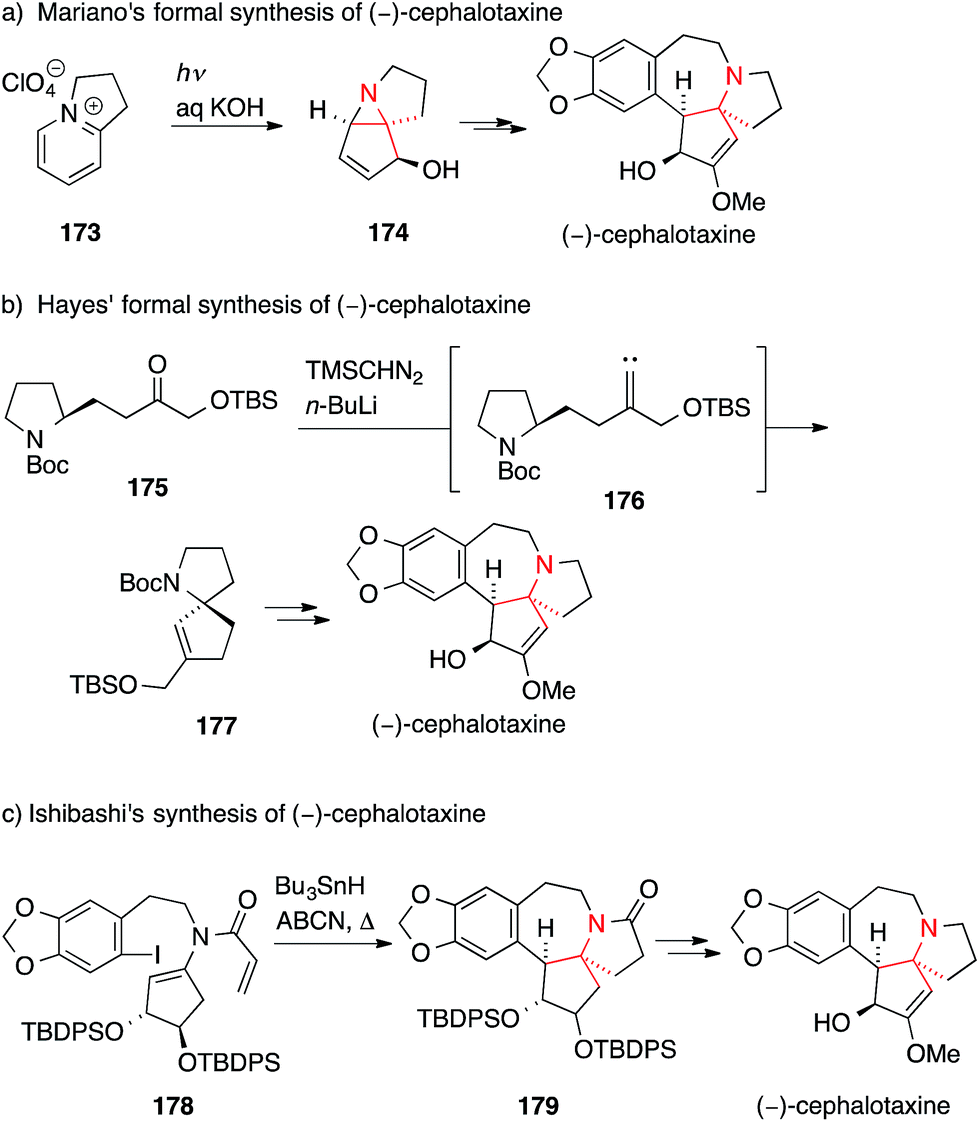 | ||
| Scheme 40 Syntheses of cephalotaxine by Mariano (2006), Hayes (2008) and Ishibashi (2008). ABCN = 1,1′-azobis(cyclohexanecarbonitrile), Boc = tert-butyloxycarbonyl, TBS = tert-butyldimethylsilyl, TBDPS = tert-butyldiphenylsilyl, TMS = trimethylsilyl. | ||
Ishibashi chose a radical cyclization approach to synthesize cephalotaxine (Scheme 40c).250 Treatment of aryl iodide 178 with tributyltin hydride and a radical initiator resulted in a 7-endo cyclization followed by a 5-endo cyclization that, after hydrogen transfer, yielded 179. Additional functional group manipulations allowed them to intercept an intermediate that had previously been used to synthesize (−)-cephalotaxine.251
A host of other methods have been used for the installation of the ATA in cephalotaxines.239 These include alkylation,252,253 Claisen rearrangement,254 Michael addition,255,256 Schmidt reaction,257 addition to an imine,258 transannular cyclization259 and oxidative rearrangement.251
10 Erythrina alkaloids
Erythrina alkaloids were discovered at the end of the 19th century, when extracts of Erythrina trees were found to possess curare-like neuromuscular activities.260 Due to their biological activities and interesting structures (Fig. 9), several total syntheses of these natural products have been carried out and many creative ways to install ATAs have been developed in this context.261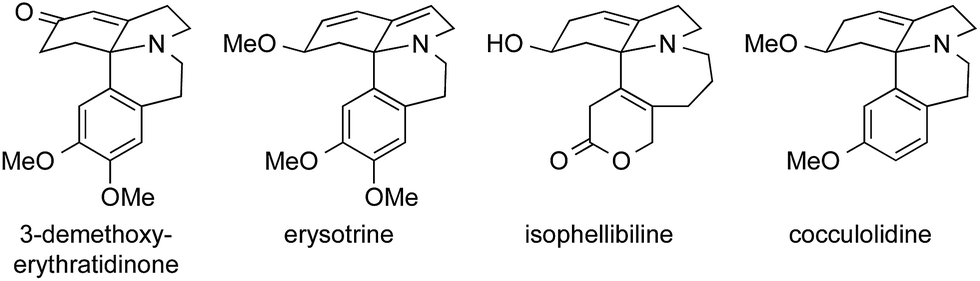 | ||
| Fig. 9 Representative Erythrina alkaloids. | ||
In 1990, the group of Ishibashi published the synthesis of (±)-3-demethoxyerythratidinone using an intramolecular Pummerer-like rearrangement of the enamine 180, setting the stage for a Pictet–Spengler-type reaction (181 → 182) to furnish the ATA (Scheme 41a).262 Thirteen years later, the same group published an oxidative radical cyclization starting from enamine 183 to obtain the skeleton of 3-demethoxyerythratidinone 182 (Scheme 41b).263
 | ||
| Scheme 41 Syntheses of Erythrina alkaloids by Ishibashi (1990 and 2003) and Tsuda (1992). Ac = acetyl, OTf = trifluoromethanesulfonate, TMS = trimethylsilyl, p-TsOH = para-toluenesulfonic acid. | ||
Tsuda's approach featured an intermolecular photochemical [2+2] cyclization to install the ATA, starting from bicycle 184 and diene 185 (Scheme 41c). In the following steps, a ring expansion of the four-membered ring in 186 furnished the six-membered ring by a formal 1,3-migration of a vinylcyclobutane, affording the scaffold of erysotrine.264
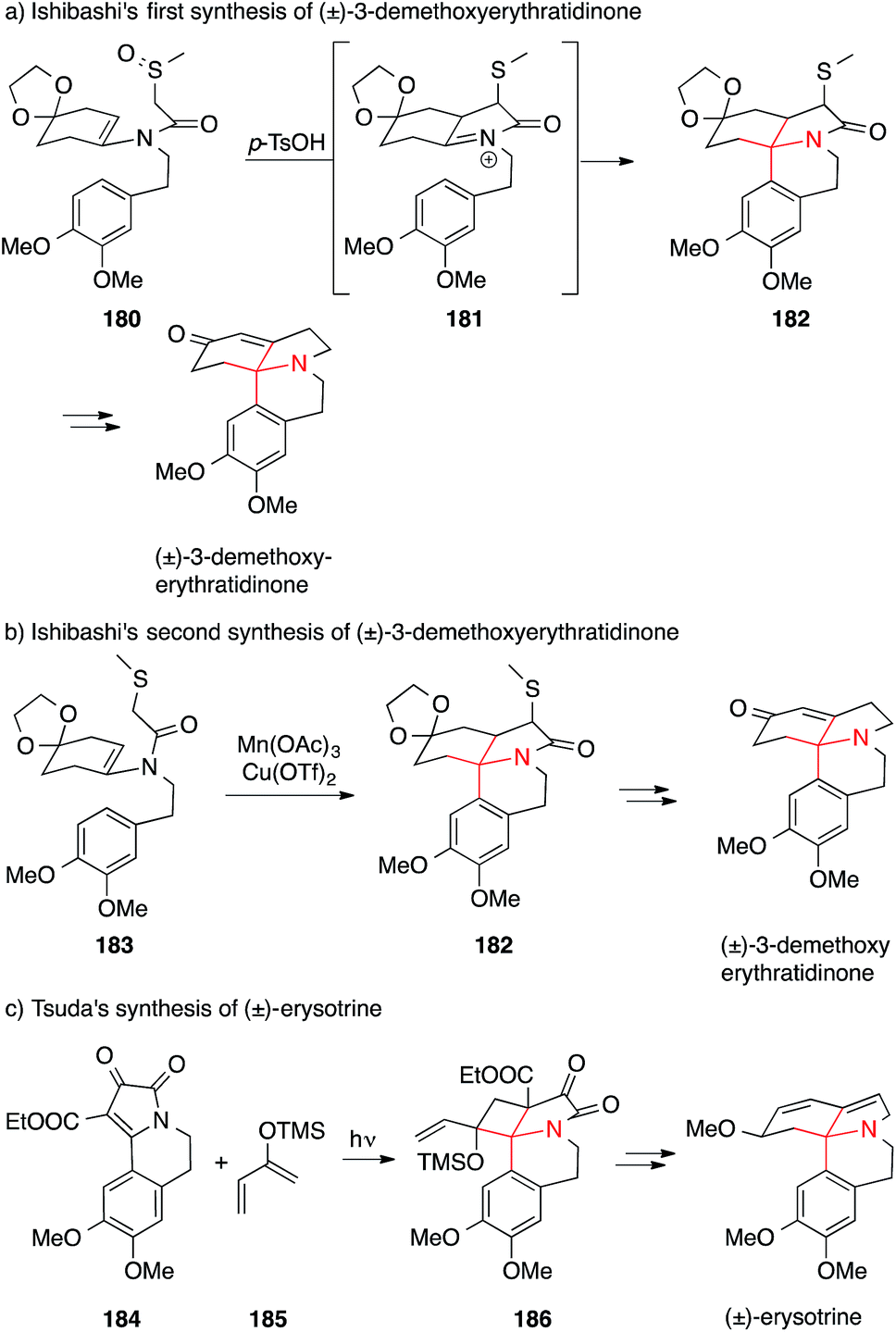
Funk accomplished the synthesis of isophellibiline via an approach that relies on pericyclic reactions (Scheme 42a).265 Heating of dioxine 187 resulted in retro-Diels–Alder reaction to afford dehydroalanine derivative 188, which then underwent intramolecular [4+2] cycloaddition to yield lactam 189. The latter was converted into isophellibinine in a few steps.
 | ||
| Scheme 42 Syntheses of Erythrina alkaloids by Funk (2012) and Sarpong (2013). | ||
Recently, Sarpong developed a new methodology to furnish ATAs and applied it to the synthesis of cocculolidine (Scheme 42b).266 Propargylic alcohol 190 underwent cycloisomerization upon heating to form benz[g]indolizinone 191 which was then transformed to cocculidine in two additional steps.
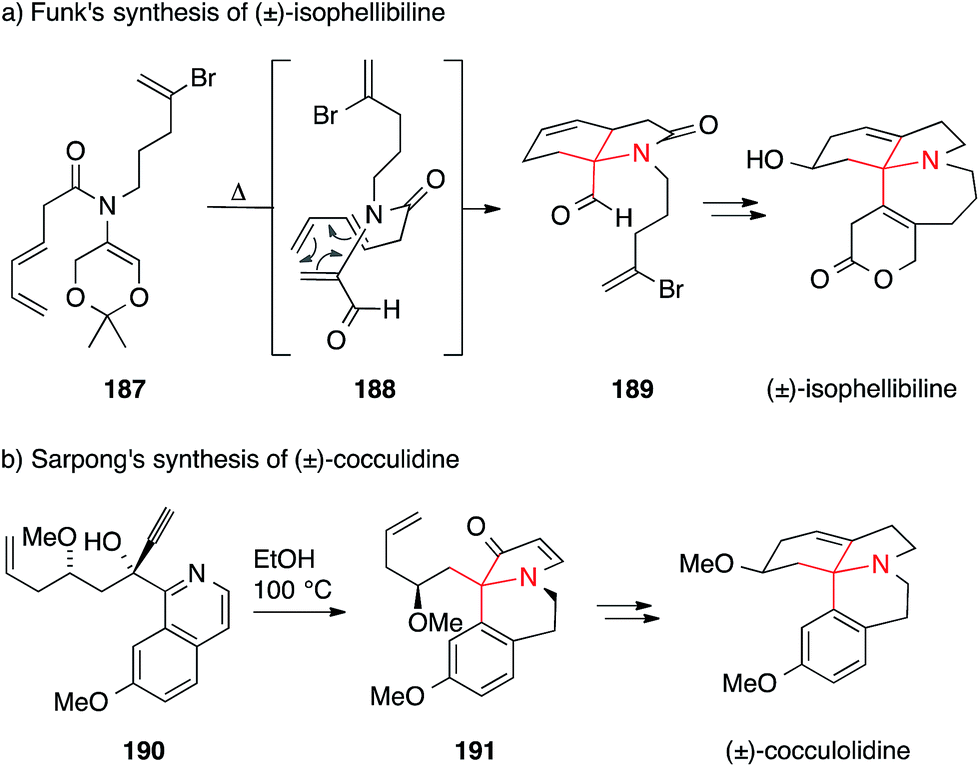
A short and elegant synthesis of 3-demethoxyerythratidinone was accomplished by Streuff in 2015 (Scheme 43a). He used a titanium(III)-catalyzed reductive Umpolung (192 → 194) to assemble the 1,1-disubstituted tetrahydroisoquinoline core of the Erythrina alkaloids.267 In the same year, Ciufolini set the same ATA via an oxidative dearomatizative cyclization of an oxazoline 195, yielding spiropiperidine 197 presumably via intermediate 196 (Scheme 43b).268,269
 | ||
| Scheme 43 Syntheses of Erythrina alkaloids by Streuff (2015) and Ciufolini (2015). HFIP = 1,1,1,3,3,3-hexafluoroisopropyl alcohol, PIFA = (bis(trifluoroacetoxy)iodo)benzene, TMS = trimethylsilyl. | ||
Other methods used to install the ATA in Erythrina alkaloids include 1,2-addition of organometallic reagents to sulfinimines or iminium ions,270–272 Schmidt reaction,273 Diels–Alder reactions,274 Michael addition,275N-acyliminium Pictet–Spengler reaction276–278 and Heck reaction.279
11 Indolizidine and quinolizidine alkaloids
A range of alkaloids that belong to the indolizidine and quinolizidine structural class feature an ATA in their carbon skeleton.280 They include natural products as diverse as the cylindricines,280,281 FR901483,282 himandrines, lepadiformines283 and halichlorine284 (Fig. 10).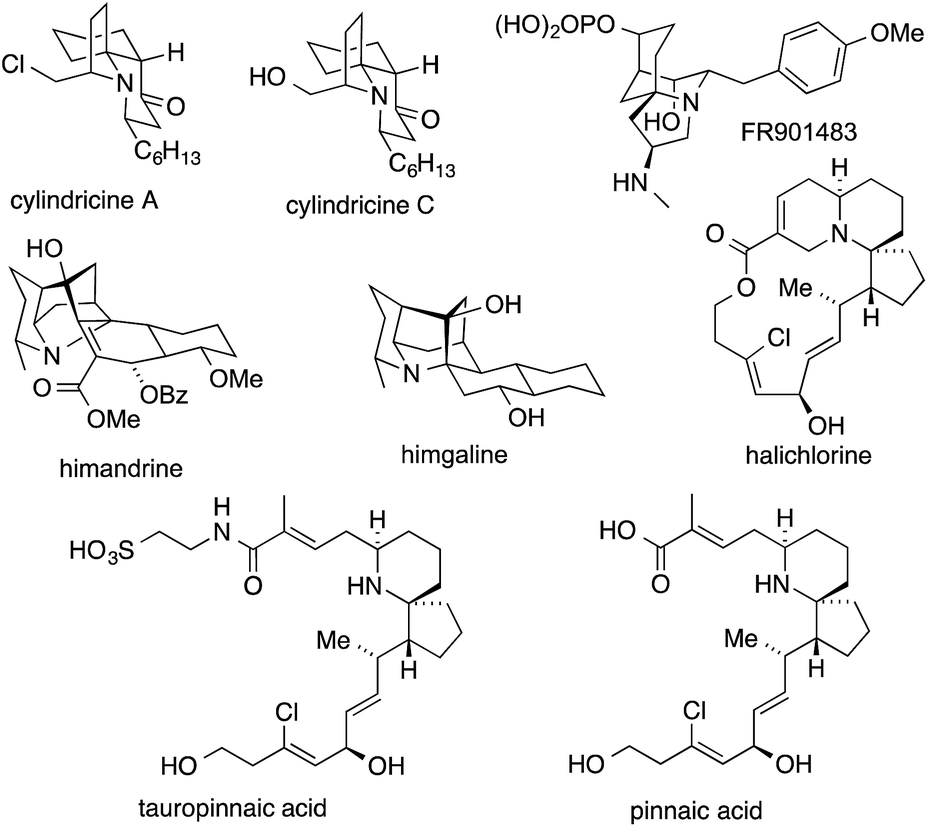 | ||
| Fig. 10 Selection of quinolizidine alkaloids, including halichlorine and the closely related alkaloids tauropinnaic acid and pinnaic acid. | ||
The first synthesis of cylindricine alkaloids (viz. cylindricine A, D and E) was accomplished by Snider utilizing a double Michael addition of ammonia to divinylketone 198 which gave the ATA 199, a direct precursor of cylindricine A (Scheme 44a).285 Variations of this approach have been used several times in the synthesis of cylindricines.286
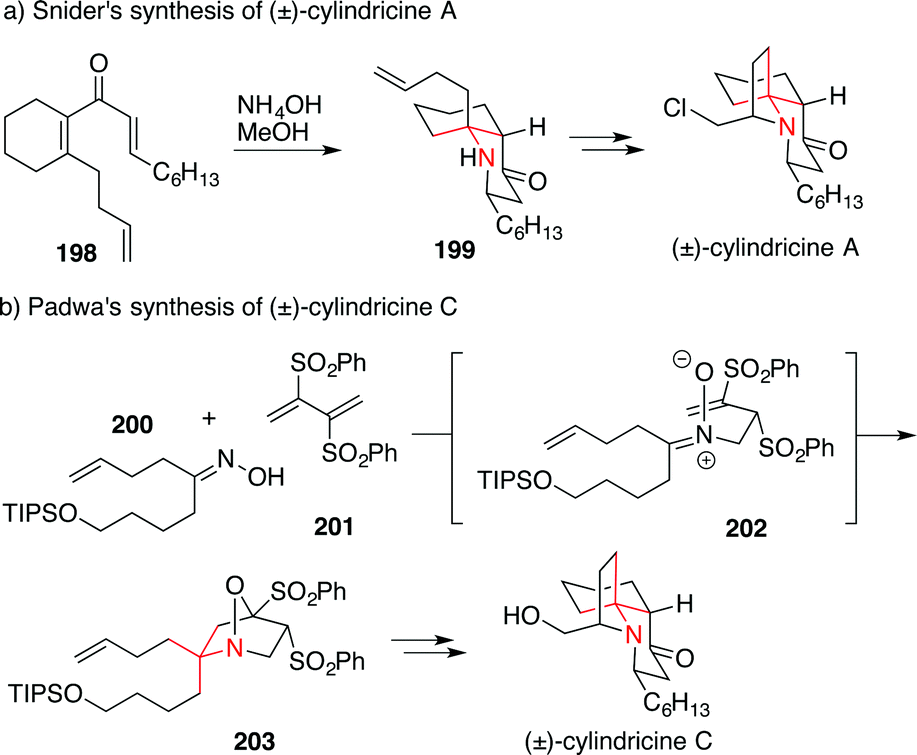 | ||
| Scheme 44 Synthesis of cylindricines by Snider (1997) and Padwa (2003). Boc = tert-butyloxycarbonyl, TBDPS = tert-butyldiphenylsilyl. | ||
In 2003, Padwa published a synthesis featuring a Michael addition/dipolar cycloaddition cascade between butadiene 201 and oxime 200 to form 203via intermediate 202 (Scheme 44b).287
The Hsung synthesis of enantiomerically pure cylindricine C relies on a nucleophilic attack of a diene on N-acyliminium ion 205 starting from ketone 204 (Scheme 45).288,289 This vinylogous aza-Prins approach was based on a synthesis published by Kibayashi in 2005.290
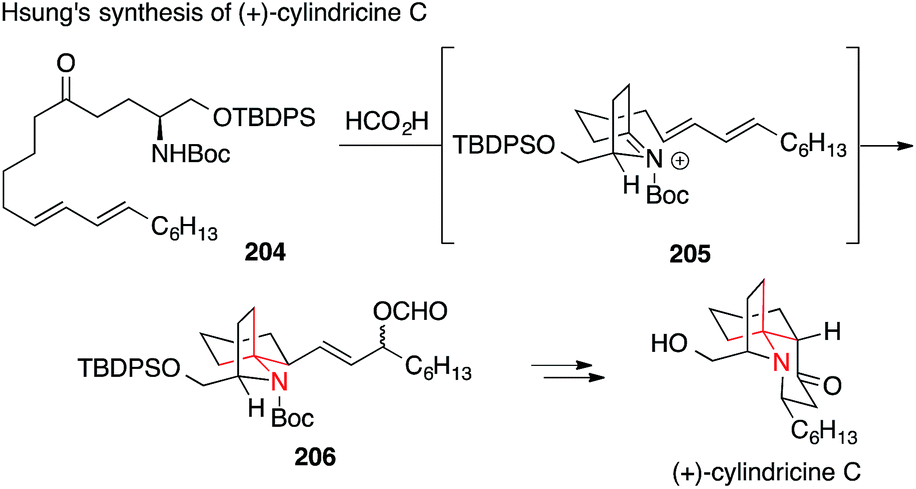 | ||
| Scheme 45 Synthesis of cylindricine C by Hsung (2004). Boc = tert-butyloxycarbonyl, TBDPS = tert-butyldiphenyl-silyl. | ||
Additional strategies to synthesize the ATA in cylindricine alkaloids involve mainly Michael additions,286,291,292 Grignard additions to an imine,293 a cycloaddition of an alkyne to a pyrrole derivative,294 and carboazidation.295
FR901483, an alkaloid isolated from the fermentation broth of a Cladobotryum species with an intricate tricyclic structure,282 proved to be an equally popular synthetic target. A biomimetic approach was employed by Sorensen in his enantioselective synthesis (Scheme 46a).296
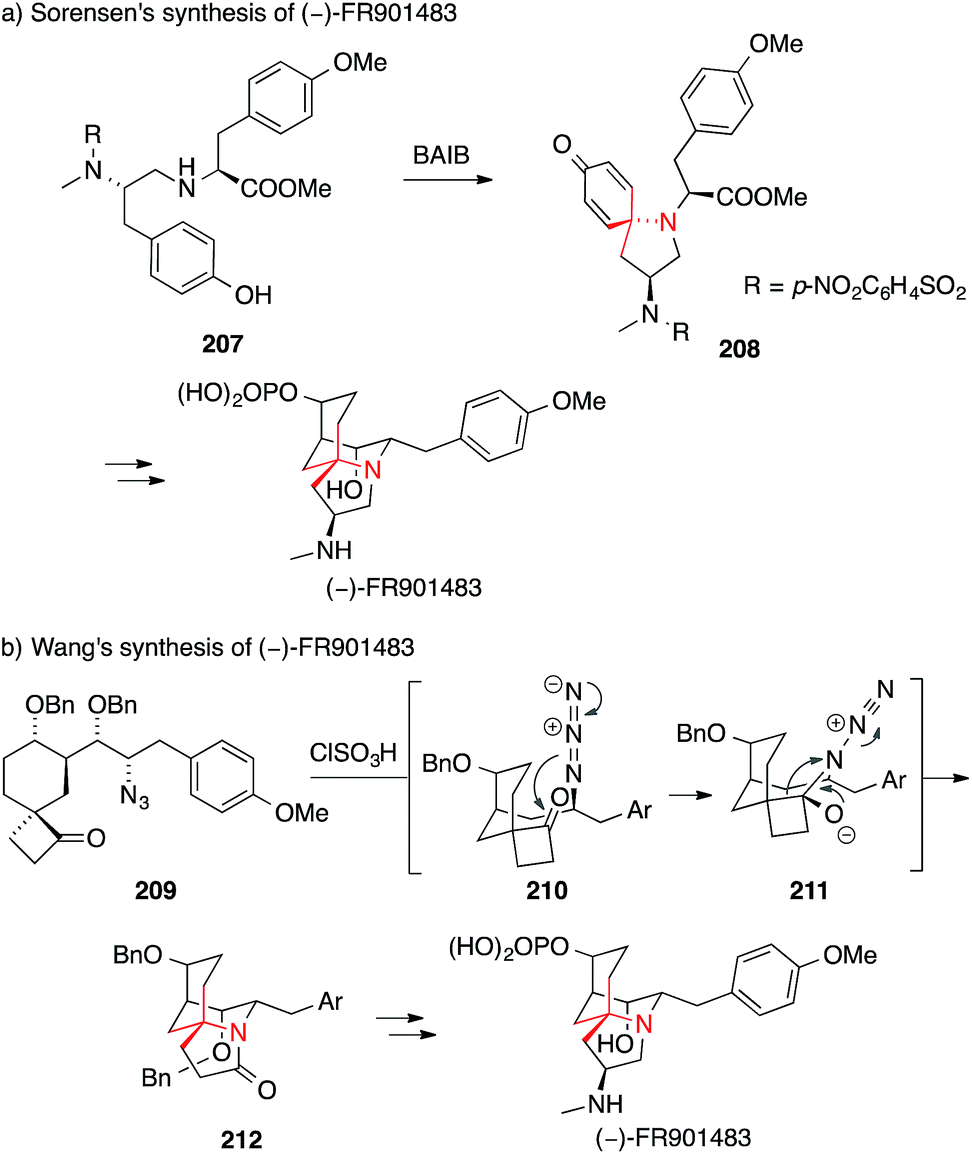 | ||
| Scheme 46 Syntheses of FR901483 by Sorensen (2000) and Wang (2012). Ar = aryl, BAIB = (diacetoxyiodo)benzene, Bn = benzyl, TBDPS = tert-butyldiphenylsilyl. | ||
The oxidative azaspiroannulation of amine 207 promoted by (diacetoxyiodo)benzene resulted in the formation of spiroamine 208, an intermediate on the way to the natural product. The same year, Ciufolini set the ATA via a closely related oxidative spiroannulation (not shown, for an example of the methodology see Scheme 43b).297
An alternative to this strategy was found by Wang.298 In this case, an Aubé–Schmidt reaction of azide 209 provided access to lactam 212, featuring the ATA of FR901483 (209 → 212, Scheme 46b). Additional synthetic strategies to set the ATA in FR901483 include a triple Michael addition,299 a one-pot bisalkylation,300,301 an aza-Cope rearrangement/Mannich cyclization302,303 and an oxidative dearomatization.297
The members of the galbulimima alkaloid family, such as himgaline and himandrine, also possess an ATA-containing quinolizidine core (Fig. 10). Exploring a biosynthetic hypothesis, Chackalamannil used an intramolecular Michael addition to convert GB 13 to himgaline via ketone 213 (Scheme 47a).304,305 In an interesting variation of this apporach, Movassaghi converted enone 214via its α-chloroester 215 to hexacyclic amine 216, which could then be transformed into himandrine. (Scheme 47b).304
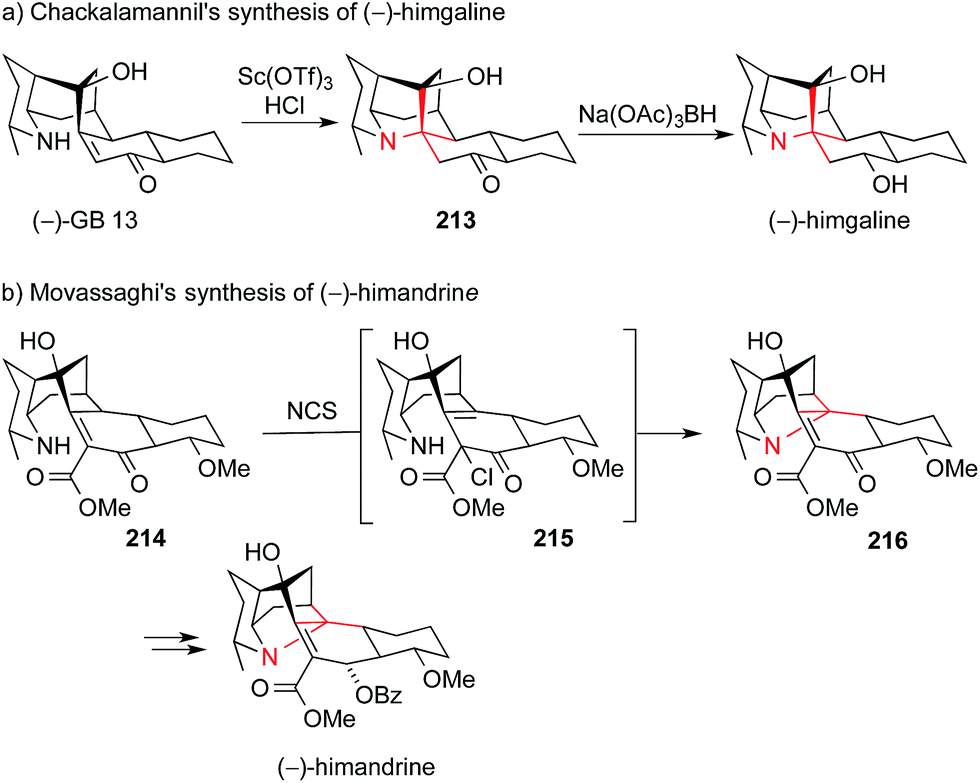 | ||
| Scheme 47 Synthesis of galbulimima alkaloids by Chackalamannil (2009) and Movassaghi (2009). Ac = acetyl, Bz = benzoyl, NCS = N-chlorosuccinimide, OTf = trifluoromethanesulfonate. | ||
In 1996, Uemura disclosed a small series of unusual marine alkaloids featuring ATAs. One of these compounds, halichlorine, was isolated from the marine sponge Halichondria okadai and was found to selectively inhibit the induction of vascular cell adhesion molecule-1 (VCAM-1).306 Pinnaic acid and tauropinnaic acid were recovered from bivalve Pinna muricata.307,308 All three molecules present a challenging 6-aza-spiro[4.5]decane core containing the ATA. The latter two lack a quinolizidine moiety, but are included in this chapter due to their close structural relationship.
Danishefsky and Trauner were the first to report the synthesis of (+)-halichlorine in 1999309 followed by a synthesis of pinnaic acid in 2001 (Scheme 48).310,311 They used Meyers' lactam 217 as a chiral precursor, which was combined with allyltrimethylsilane in a Sakurai reaction to install the ATA in 218. Intermediate 219 could be diversified to reach both halichlorine and pinnaic acid. These syntheses established the absolute configuration of halichlorine and confirmed the stereochemistry at C-14 and C-17 of pinnaic acid.
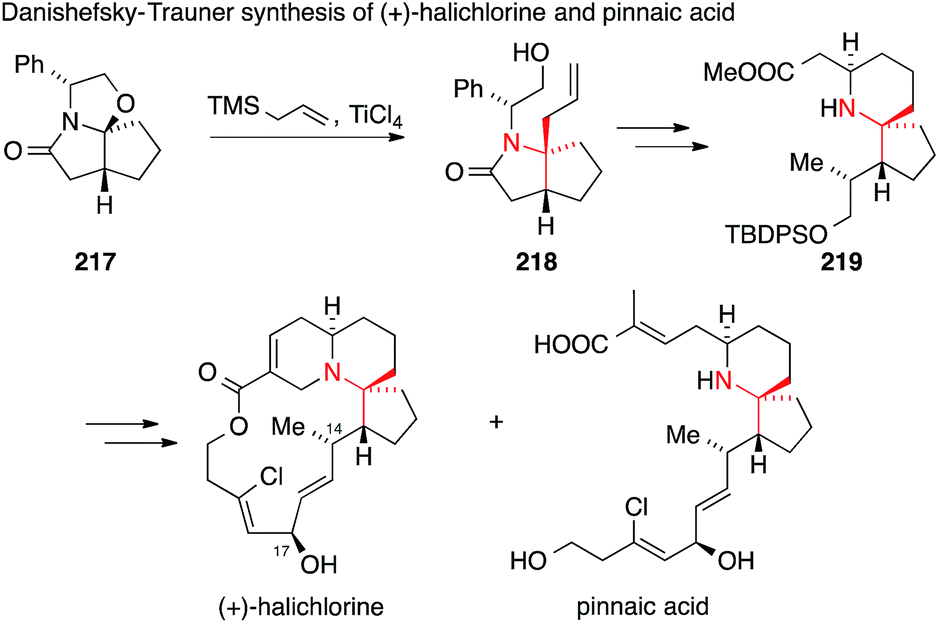 | ||
| Scheme 48 Synthesis of halichlorine and pinnaic acid by Danishefsky and Trauner (1999). TBDPS = tert-butyldiphenylsilyl, TMS = trimethylsilyl. | ||
A related apporach employing a different type of N-acyl iminium ion was used by Heathcock in 2004 for the synthesis of halichlorine, pinnaic acid and tauropinnaic acid (Scheme 49a).312 Treatment of carbamate acetal 220 with allyl trimethylsilane and titanium tetrachloride furnished ATA-bearing carbamate 221 with a high degree of stereoselectivity. This key intermediate could be transformed into all three natural products.
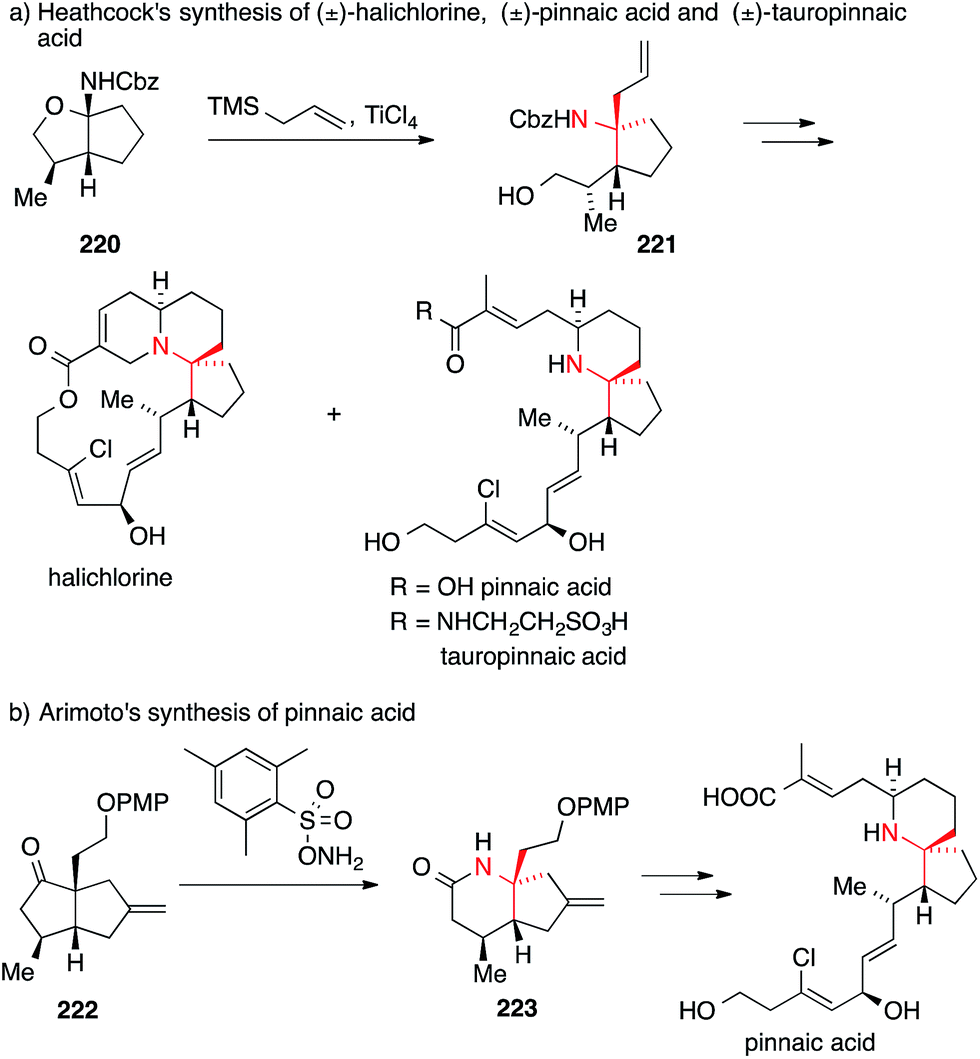 | ||
| Scheme 49 Syntheses of halichlorine, pinnaic acid and tauropinnaic acid by Heathcock (2004) and Arimoto (2007). Cbz = benzyloxycarbonyl, PMP = p-methoxyphenyl, TMS = trimethylsilyl. | ||
In 2007, Arimoto reported his version of an asymmetric synthesis of pinnaic acid using a Beckmann rearrangement to install the ATA (Scheme 49b).313 The enantiomerically pure bicyclic ketone 222 was treated with a bulky hydroxylamine reagent to afford the desired lactam 223, which was then converted into the natural product.
12 Lactacystine and salinosporamide
In 1991, Omura isolated the unusual natural product lactacystin from Streptomyces sp. OM-6519 and identified it as a proteasome inhibitor (Fig. 11).314,315 A structurally related β-lactone, salinosporamide A, which shows similar biological activity, was subsequently isolated from a marine bacterium, Salinispora tropica.316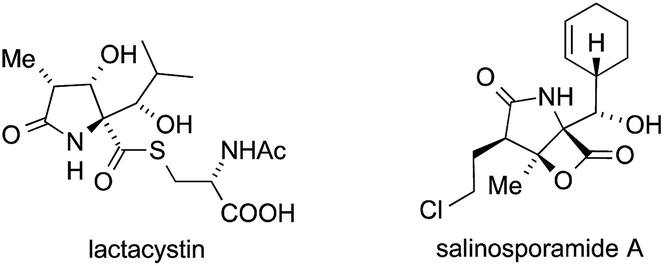 | ||
| Fig. 11 Lactacystin and salinosporamide A. | ||
Both compounds possess a densely functionalized γ-lactam core with three contiguous stereocenters, one of which is of the ATA type. Their significant biological activity has stimulated a large number of total syntheses,317 and a variety of methods for the installation of the ATA motif have been applied.318–345
In pioneering work, Corey reported five total syntheses of lactacystin between 1992 and 1998.318–320,326,327 The Corey group showed that the ATA can be installed using an aldol addition of α-amino acid derivative 224 (via intermediate 225, Scheme 50a). Other groups also contributed to this field in the 1990s.321–325,328 In most cases, the strategy applied for the installation of the ATA motif involved an alkylation or aldol reaction of an α-amino acid derivative.318–322,324,326–328 By contrast, Shibasaki introduced the ATA with a catalytic enantioselective Strecker reaction (Scheme 50b).317,334 In this work, phosphinoylimine 226 was converted to aminonitrile 227 using a gadolinium catalyst and the chiral ligand A.
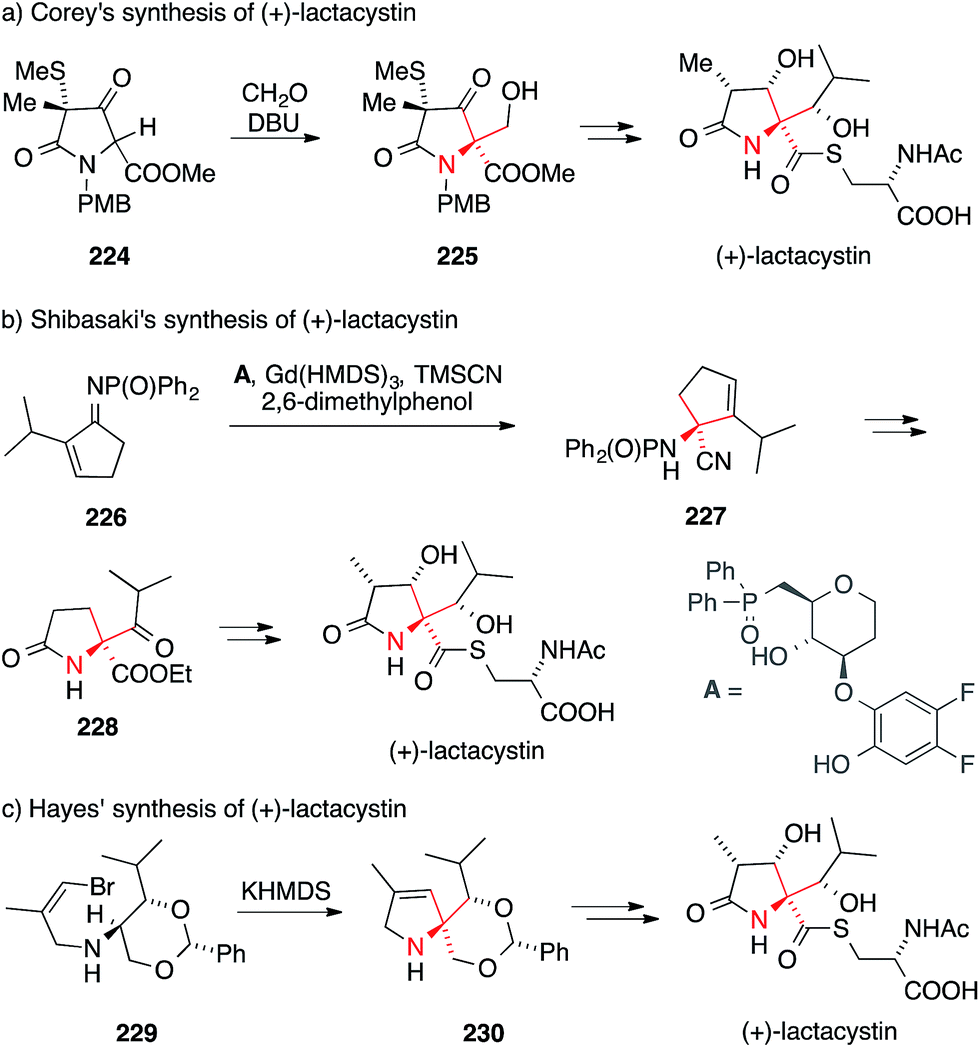 | ||
| Scheme 50 Syntheses of lactacystin by Corey (1992), Shibasaki (2006) and Hayes (2008). Ac = acetyl, DBU = 1,8-diazabicycloundec-7-ene, HMDS = bis(trimethylsilyl)amine, PMB = paramethoxybenzyl, TMS = trimethylsilyl. | ||
Another unusual approach was taken by Wardrop346 in his formal synthesis and Hayes338 in his total synthesis of lactacystin (Scheme 50c). Both groups explored an intramolecular carbene insertion into a C,H-bond to form the five-membered heterocyclic core. Hayes converted the enantiomerically pure vinyl bromide 229 to the corresponding vinylidene carbene, which underwent cyclization to afford 230 in high yield.
The first synthesis of salinosporamide was reported by Corey in 2004.330,331 In this case, the ATA was installed by alkylation of threonine-derived oxazoline 231 with chloromethyl benzyl ether (via intermediate 232, Scheme 51a).
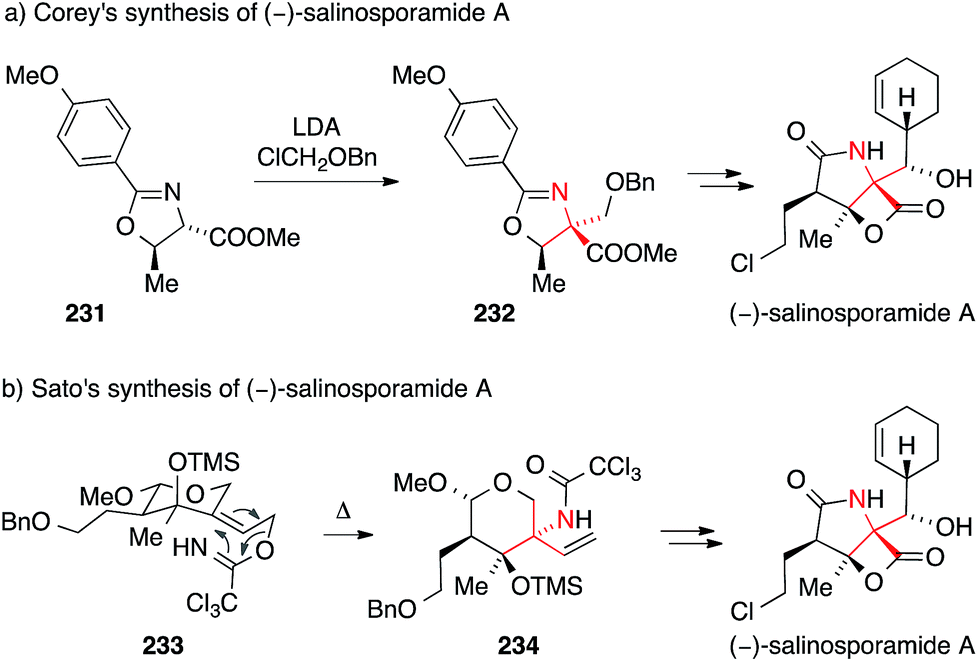 | ||
| Scheme 51 Syntheses of salinosporamide A by Corey (2004) and Sato (2011). Bn = benzyl, LDA = lithium diisopropylamide, TMS = trimethylsilyl. | ||
A more recent synthesis of salinosporamide A published by Sato and Chid uses a stereoselective Overman rearrangement to install the ATA (Scheme 51b).343 Heating of the highly functionalized trichloroacetimidate 233 provided trichloroacetamide 234 as a key intermediate.
Other syntheses of salinosporamide and lactacystine used different strategies for the formation of the ATA, such as intramolecular aldol reactions,336,337 intramolecular acylations,332 acid catalyzed cyclizations of malonates,335,339 indium-catalyzed Conia-ene reactions,341 C,H-alkynylation347 and enzymatic desymmetrizations of prochiral ATAs.340
13 Manzacidins
The manzacidins, a small family of bromopyrrole alkaloids, have attracted considerable attention from the synthetic community despite, or maybe because of their relatively simple structures. Manzacidins A–C (Fig. 12) were first isolated form the Okinawan sponge Hymeniacidon sp. by Kobayashi in 1991,348 followed by the isolation of manzacidin D from the ‘living fossil’ sponge Astrosclera willeyana349 and N-methylmanzacidin C from Axinella brevistyla.350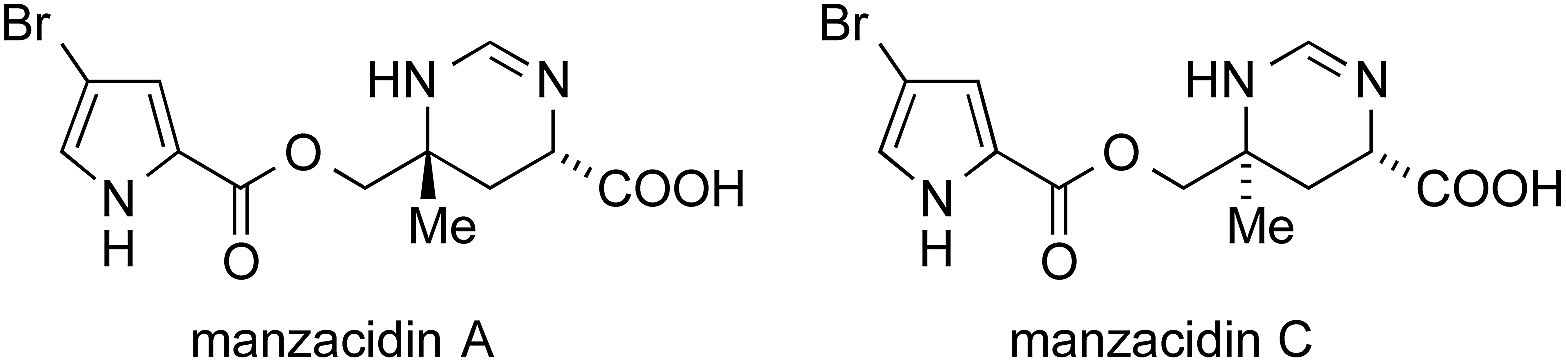 | ||
| Fig. 12 Manzacidin A and C. | ||
In 2000, Ohfune reported the synthesis of manzacidins A and C via a Strecker reaction and assigned the absolute configuration of these natural products (not shown).351 In 2002, DuBois synthesized manzazidin C using an elegant oxidative C,H-insertion that involved sulfamate 235 (via intermediate 236, Scheme 52a).352 One year later, he used a similar strategy to set the ATA in tetrodotoxin (see chapter 14, Scheme 54c).353
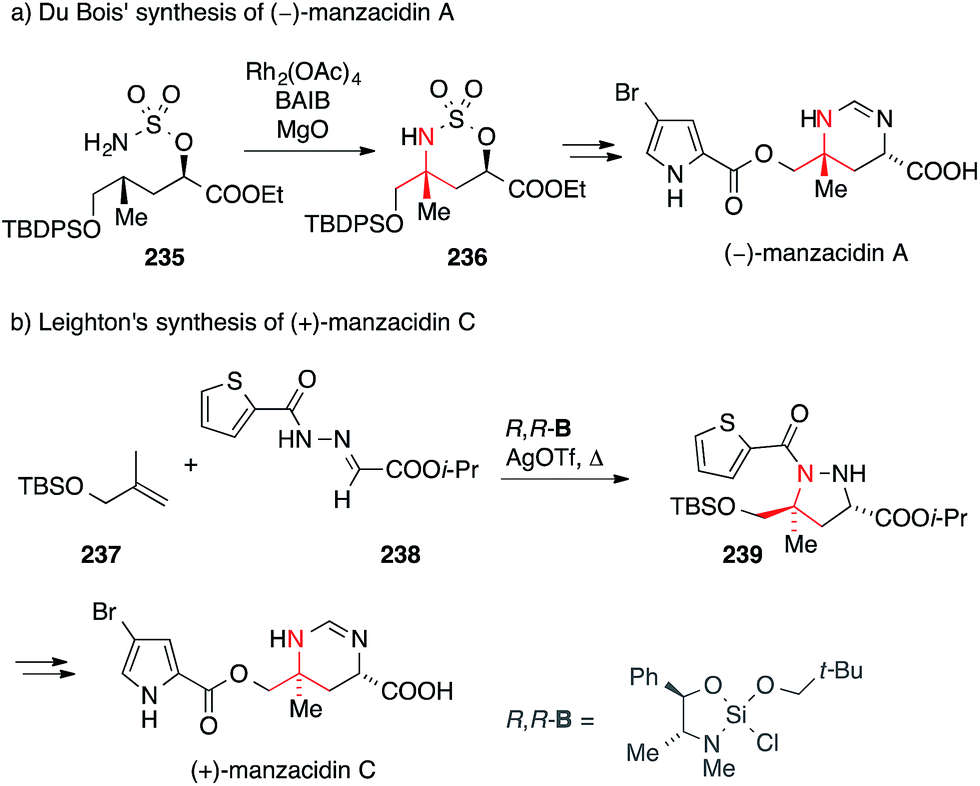 | ||
| Scheme 52 Syntheses of manzacidins by DuBois (2002) and Leighton (2008). Ac = acetyl, BAIB = (diacetoxyiodo)benzene, OTf = trifluoromethanesulfonate, i-Pr = iso-propyl, TBDPS = tert-butyldiphenylsilyl. | ||
Leighton accomplished the synthesis of manzacidin C employing their asymmetric silane-promoted [3+2] cycloaddition methodology (Scheme 52b).354 Exposure of alkene 237 and hydrazone 238 to chiral silane R,R-B gave pyrazolidine 239, thus setting both stereocenters of the target molecule, including the ATA, in a single step. Intermediate 239 was subsequently converted to manzacidin C via reductive N,N-bond cleavage.
A more recent formal synthesis of manzacidins A and C, published by Ichikawa, features a rare allyl cyanate/isocyanate rearrangement as the key step (Scheme 53).355 To this end, he synthesized carbamate 240, which was converted to allyl cyanate 241 by in situ dehydration. The subsequent rearrangement with chirality transfer gave isocyanate 242, which was then transformed into manzacidin A. The synthesis of manzacidin C was accomplished analogously from a diastereoisomer of carbamate 240.355
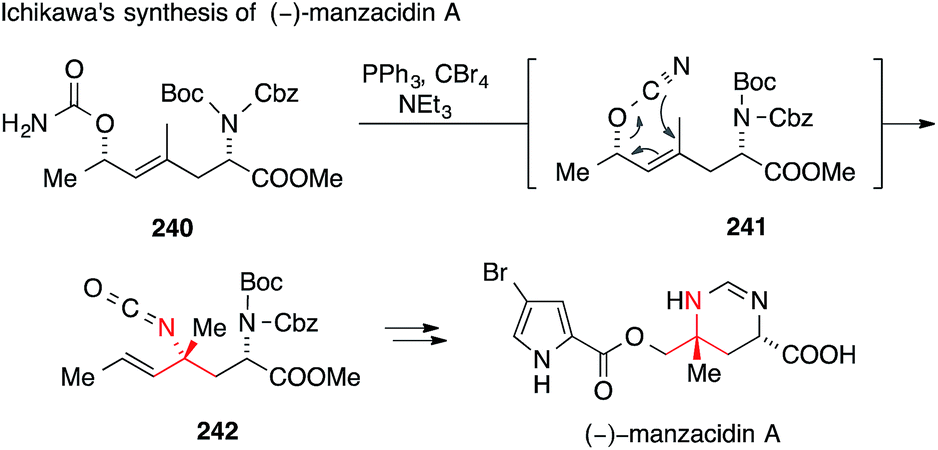 | ||
| Scheme 53 Synthesis of manzacidin A by Ichikawa (2012). Boc = tert-butyloxycarbonyl, Cbz = benzyloxycarbonyl. | ||
Several other synthetic approaches toward these molecules have been reported. These strategies for the installation of the ATA moiety involve diastereoselective nitrene insertion,352 1,3-dipolar cycloaddition,356,357 Hofmann rearrangement,358 diastereoselective iodocyclization,359,360 Grignard addition to an imine361 and a variety of other methods.362–366 Indeed, manzacidines remain targets of great interest for synthetic chemists. In 2015, Inoue published a synthesis of manzacidin A using a radical-based decarbonylative coupling (not shown).367 Recently the relative stereochemistry of manzacidin B, which possesses an additional stereocenter, was revised using total synthesis.362,363,365
14 Tetrodotoxin
Tetrodotoxin (TTX) was first isolated from the Fugu puffer fish in 1909.368,369 Its structure was independently reported by Hirata–Goto,370 Tsuda371 and Woodward372 in the 1960s. Their assignment was confirmed by X-ray crystallography, which also established the absolute configuration of the molecule.373,374 TTX features a highly functionalized heteroadamantane framework that contains an ortho-acid and is fused to a cyclic guanidinium moiety via an ATA motif. The molecule is an extremely powerful and selective blocker of voltage-gated sodium channels and is widely used as a research tool in neuroscience.375–379 Due to its intriguing structure and bioactivity, attempts to synthesize TTX have been made from an early stage and activity in this field has recently increased significantly.380The first total synthesis of TTX was accomplished by Kishi in 1972 (Scheme 54a).381–384 In his approach, the ATA was formed using a Beckmann rearrangement of oxime 243, which was synthesized using a regioselective Diels–Alder reaction. The resulting key intermediate 244 was converted into TTX using a series of stereoselective redox transformations, ring cleavage and the installation of the cyclic guanidine with newly developed methodology. Although the Kishi synthesis was not enantioselective, it still stands as one of the strategically most elegant approaches to a natural product featuring an ATA motif.
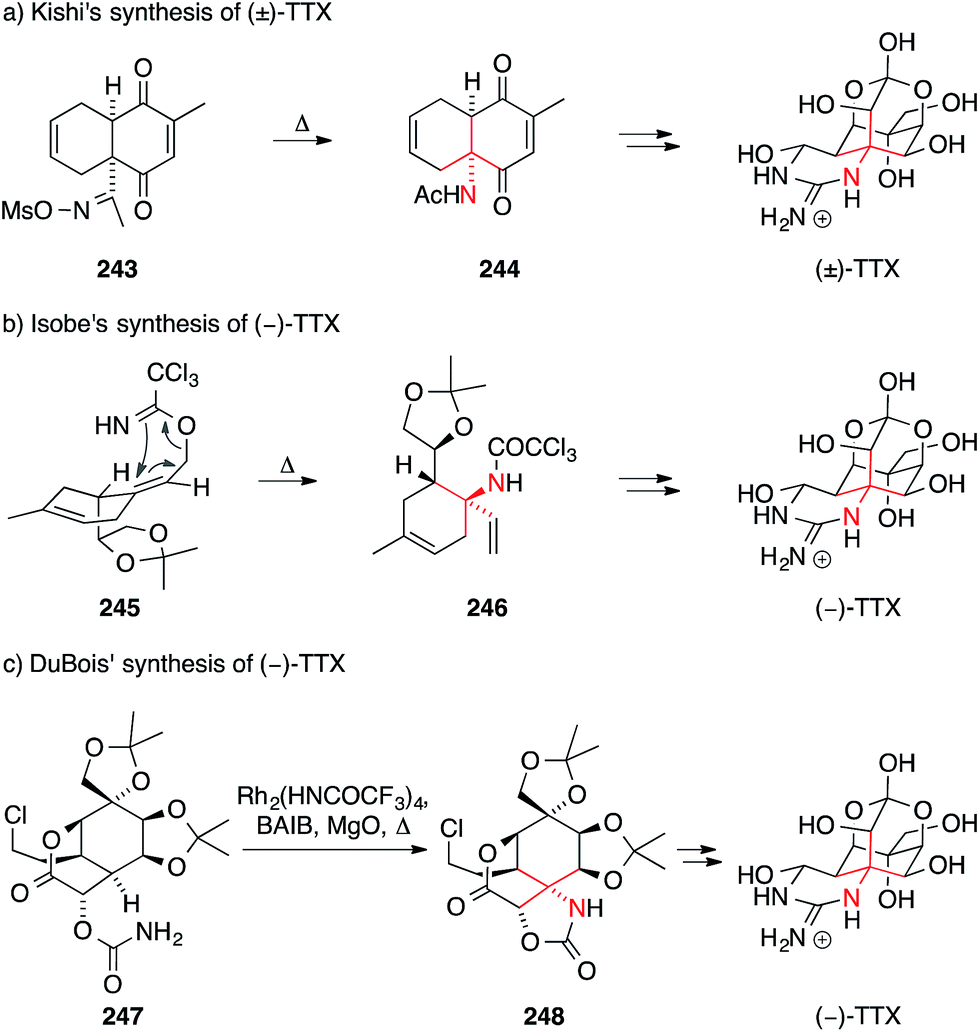 | ||
| Scheme 54 Syntheses of tetrodotoxin (TTX) by Kishi (1972), Isobe (2003) and DuBois (2003). Ac = acetyl, BAIB = (diacetoxyiodo)benzene, Ms = methanesulfonyl, Ph = phenyl,. | ||
After a 30 year lull, Isobe published the first enantioselective synthesis of TTX wherein the ATA motif was installed with a stereoselective Overman rearrangement (Scheme 54b).385–387 To this end, an allylic alcohol was converted to trichloroacetimidate 245, which underwent rearrangement to yield trichloroacetamide 246. Compound 246 bears all the carbon atoms of TTX and could be converted into the natural product in a series of steps.
Shortly thereafter, DuBois developed an enantioselective approach to TTX that involved his signature nitrene insertion chemistry (Scheme 54c).353 Exposure of the key intermediate, carbamate 247, to a hypervalent iodine reagent and magnesium oxide in the presence of a rhodium catalyst led to the formation of oxazolidinone 248, which bears the ATA motif. Insertion into other possible C,H-bonds was largely avoided through careful engineering of the substrate.
15 Miscellaneous alkaloids
ATA's occur in many other alkaloids that cannot easily be categorized along the biosynthetic and structural lines shown above. An example is gracilamine, which was isolated in 2005 by Ünver and Kaya from the Amaryllidacae species Galanthus gracilis.388 In 2012, the first synthesis of gracilamine was disclosed by Ma (Scheme 55a).389 It relies on a potentially biomimetic, stereoselective and intramolecular [3+2] cycloaddition, transforming 249 into the highly functionalized pyrrolidine 250.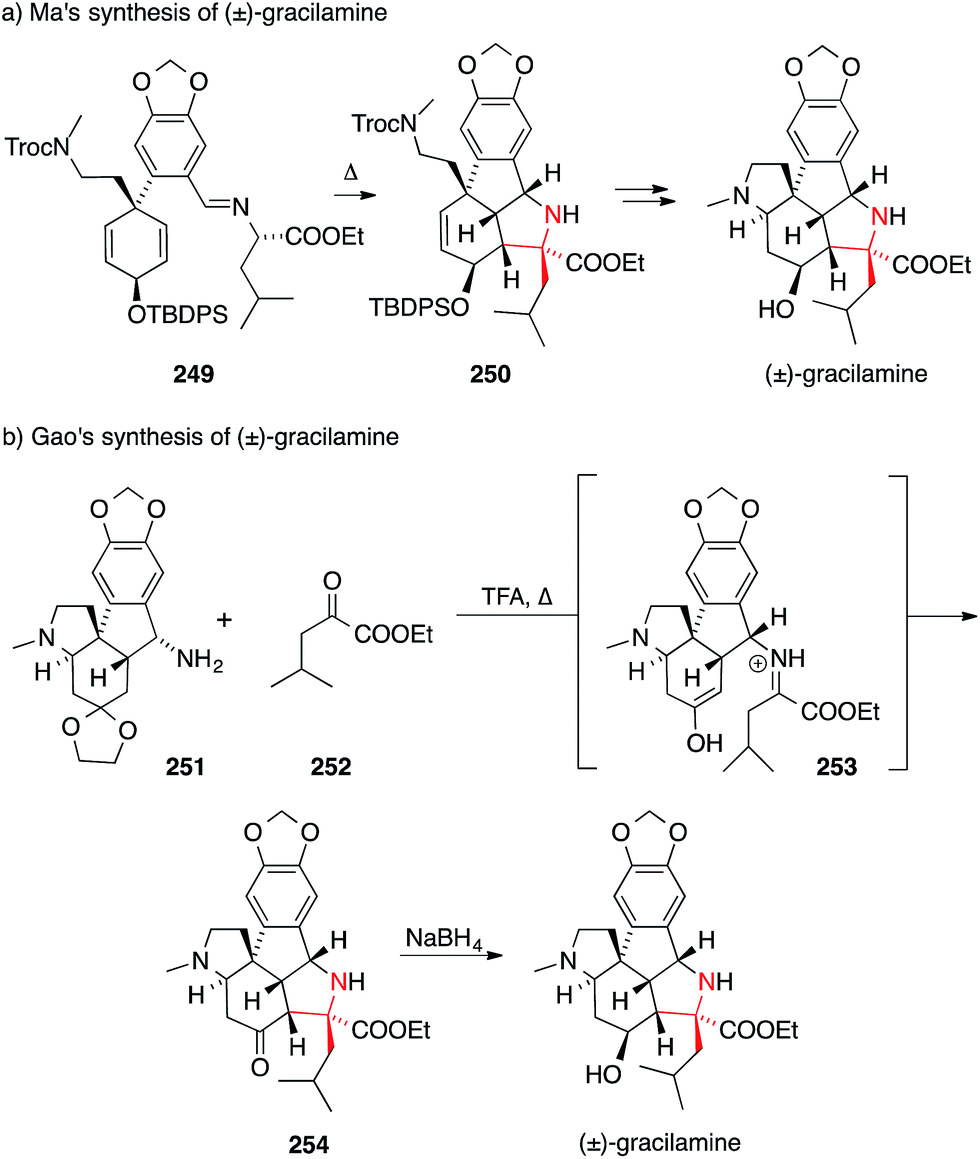 | ||
| Scheme 55 Syntheses of gracilamine by Ma (2012) and Gao (2014). TBDPS = tert-butyldiphenylsilyl, Troc = 2,2,2-trichlorethoxycarbonyl, TFA = trifluoroacetic acid. | ||
In a recent synthesis, Gao set the ATA via an intramolecular Mannich annulation (Scheme 55b).390 First, α-ketoester 252 was condensed with amine 251. The resulting iminium ion 253 then underwent a diastereoselective Mannich reaction to furnish the hexacyclic scaffold 254 of gracilamine.
The amathaspiramides A–F are a family of marine alkaloids isolated from the bryozoan Amathia wilsoni in 1999 (Fig. 13).391 They feature an unusual spirocyclic core consisting of a pyrrolidine fused to a pyrrolidinone moiety.391
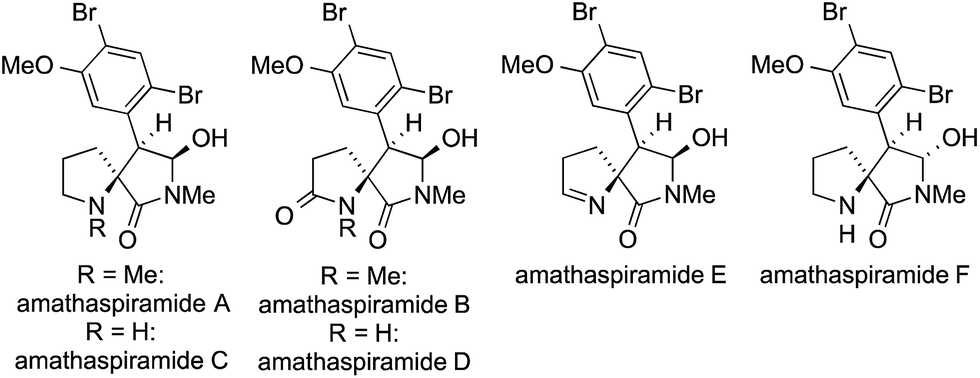 | ||
| Fig. 13 The amathaspiramides A–F. | ||
The first total synthesis of a member of this family, viz. amathaspiramide F, was disclosed by Trauner in 2002 (Scheme 56a).392 In this work, the proline-derived N,N-acetal 255 was converted to the corresponding silyl ketene acetal, which underwent a diastereoselective Michael addition to the nitro olefin 256, establishing the ATA of 257. Subsequently, Ohfune published his approach to amathaspiramide F which utilizes an enolate Claisen rearrangement for the same purpose (not shown).393
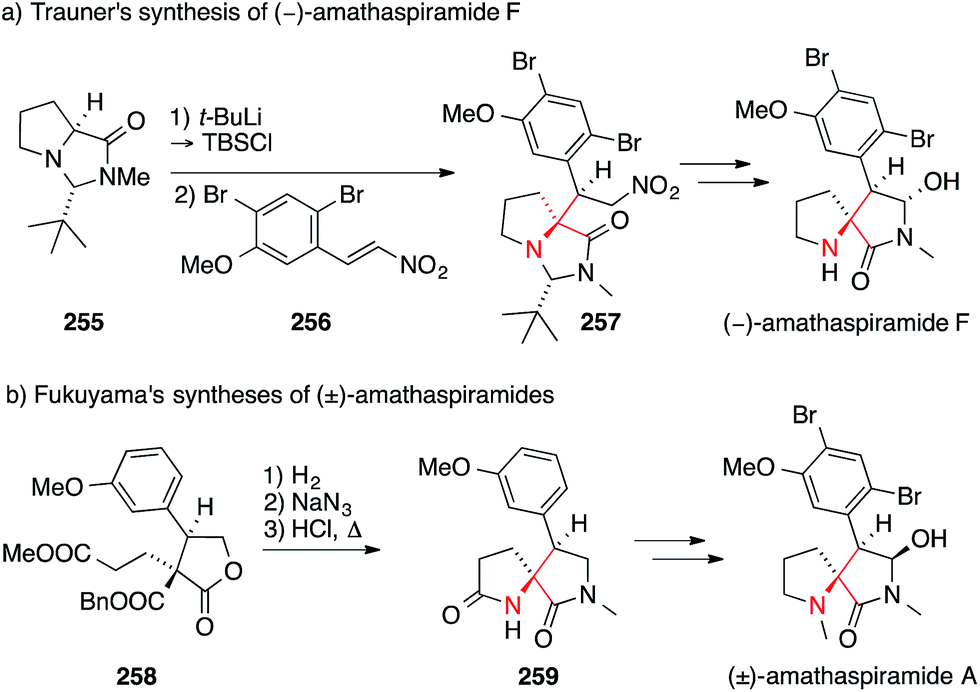 | ||
| Scheme 56 Syntheses of amathaspiramides by Trauner (2002) and Fukuyama (2012). Bn = benzyl, t-Bu = tert-butyl, TBS = tert-butyldimethylsilyl. | ||
In 2012, Fukuyama reported the asymmetric synthesis of the entire amathaspiramide family (Scheme 56b).394 In their work, the benzyl ester 258 bearing a quaternary stereocenter was first deprotected and the resulting acid converted to the corresponding amine via Curtius rearrangement. After hydrolysis of the resulting isocyanate, the intermediate amino ester underwent cyclization to afford the pyrrolidinone 259, which could be converted into all members of the family.
In Tambar's asymmetric synthesis of amanthaspiramide F, proline derivative 260 underwent a palladium-catalyzed allylic substitution of carbonate 261 to yield an intermediary quarternary ammonium ion 262, which, after deprotonation, engaged in a stereoselective [2,3] Stevens rearrangement via263 (Scheme 57a).395 The resulting ATA-containing pyrrolidine 264 was converted into amathaspiramide F.
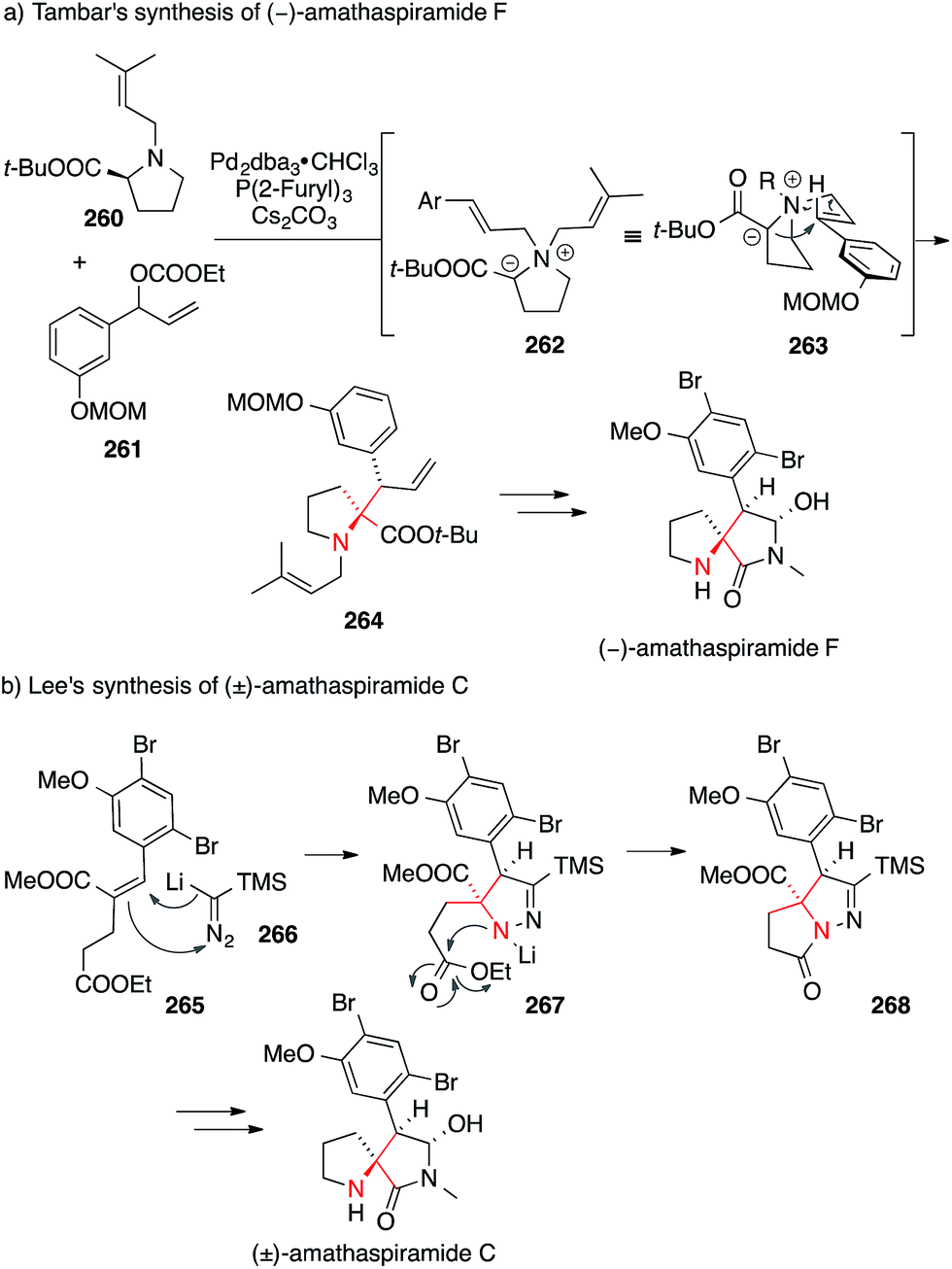 | ||
| Scheme 57 Syntheses of amathaspiramides by Tambar (2013) and Lee (2015). t-Bu = tert-butyl, dba = dibenzylideneacetone, MOM = methoxymethyl, TMS = trimethylsilyl. | ||
More recently, Lee used a formal [3+2] cycloaddition between lithium(trimethylsilyl)diazomethane 266 and α,β-unsaturated ester 265 to set the ATA in amanthaspiramide C (via intermediate 267, Scheme 57b).396 The N,N-bond in pyrazoline 268 was cleaved by treatment with p-TsOH and additional transformations led to the total synthesis of amathaspiramide C and the formal synthesis of all the other amathaspiramides.
16 Conclusions
Herein, we have provided a survey of syntheses that feature the installation of an α-tertiary amine (ATA) as a common thread. This structural motif is widespread amongst alkaloids and has physicochemical consequences, such as increased lipophilicity and chromatographic mobility that distinguishes its bearers from other basic amines. Since ATAs also occur in drug candidates and building blocks for functional materials, our review is intended to provide a useful reference for medicinal chemists and colleagues active in the material sciences. It may also provide a baseline for the development of additional and hopefully more efficient methods for the synthesis of target molecules containing α-tertiary amines.17 Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (SFB 749) for financial support. We are also grateful to the Deutsche Telekom Foundation (Ph.D. Scholarship to N. V.). We thank Felix Hartrampf, Benjamin Williams, Daniel W. Terwilliger, Dr Henry Toombs Ruane and David Konrad for helpful discussions and for proofreading the manuscript.18 Notes and references
- G. A. Cordell, The Alkaloids, Elsevier, London, 1998 Search PubMed.
- P. M. Dewick, Medicinal Natural Products; A Biosynthetic Approach, Wiley-VCH, Weinheim, third edn, 2009 Search PubMed.
- M. Wink, in Studies in Natural Products Chemistry, Elsevier, 2000, vol. 21, ch. Interference of Alkaloids with Neuroreceptors and Ion Channels, pp. 3–122 Search PubMed.
- G. A. Cordell, M. L. Quinn-Beattie and N. R. Farnsworth, Phytother. Res., 2001, 15, 183–205 CrossRef CAS PubMed.
- J. Clayden, M. Donnard, J. Lefranc and D. J. Tetlow, Chem. Commun., 2011, 47, 4624–4639 RSC.
- G. Dake, Tetrahedron, 2006, 62, 3467–3492 CrossRef CAS.
- S. A. A. El Bialy, H. Braun and L. F. Tietze, Synthesis, 2004, 2249–2262 CAS.
- S. H. Kang, S. Y. Kang, H.-S. Lee and A. J. Buglass, Chem. Rev., 2005, 105, 4537–4558 CrossRef CAS PubMed.
- A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600–604 CrossRef CAS PubMed.
- L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed.
- A. Glisan King and J. Meinwald, Chem. Rev., 1996, 96, 1105–1122 CrossRef PubMed.
- P. Laurent, B. Lebrun, J.-C. Braekman, D. Daloze and J. M. Pasteels, Tetrahedron, 2001, 57, 3403–3412 CrossRef CAS.
- R. Robinson, J. Chem. Soc., Trans., 1917, 111, 762–768 RSC.
- B. K. Blount and R. Robinson, J. Chem. Soc., 1933, 1511–1512 RSC.
- C. Schöpf and G. Lehmann, Liebigs Ann. Chem., 1935, 518, 1–37 CrossRef.
- K. Alder, H. Betzing, R. Kuth and H.-A. Dortmann, Liebigs Ann. Chem., 1959, 620, 73–87 CrossRef CAS.
- R. I. Longley and W. S. Emerson, J. Am. Chem. Soc., 1950, 72, 3079–3081 CrossRef CAS.
- K. Alder and H. A. Dortmann, Chem. Ber., 1953, 86, 1544–1554 CrossRef CAS.
- K. Alder, H. Wirtz and H. Koppelberg, Liebigs Ann. Chem., 1956, 601, 138–154 CrossRef CAS.
- B. Tursch, C. Chome, J. C. Braekman and D. Daloze, Bull. Soc. Chim. Belg., 1973, 82, 699–703 CrossRef CAS.
- D. H. Gnecco Medina, D. S. Grierson and H.-P. Husson, Tetrahedron Lett., 1983, 24, 2099–2102 CrossRef.
- C. Yue, J. Royer and H.-P. Husson, J. Org. Chem., 1992, 57, 4211–4214 CrossRef CAS.
- M. F. Mechelke and A. I. Meyers, Tetrahedron Lett., 2000, 41, 4339–4342 CrossRef CAS.
- E. C. Davison, A. B. Holmes and I. T. Forbes, Tetrahedron Lett., 1995, 36, 9047–9050 CrossRef CAS.
- T. Itoh, N. Yamazaki and C. Kibayashi, Org. Lett., 2002, 4, 2469–2472 CrossRef CAS PubMed.
- R. K. Hill and L. A. Renbaum, Tetrahedron, 1982, 38, 1959–1963 CrossRef CAS.
- T. C. Coombs, Y. Zhang, E. C. Garnier-Amblard and L. S. Liebeskind, J. Am. Chem. Soc., 2009, 131, 876–877 CrossRef CAS PubMed.
- S. Roy and C. Spino, Org. Lett., 2006, 8, 939–942 CrossRef CAS PubMed.
- M. Arbour, S. Roy, C. Godbout and C. Spino, J. Org. Chem., 2009, 74, 3806–3814 CrossRef CAS PubMed.
- F. A. Davis and R. Edupuganti, Org. Lett., 2010, 12, 848–851 CrossRef CAS PubMed.
- F. A. Davis, J. Org. Chem., 2006, 71, 8993–9003 CrossRef CAS PubMed.
- J. W. Daly, I. Karle, C. W. Myers, T. Tokuyama, J. A. Waters and B. Witkop, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1870–1875 CrossRef CAS.
- J. W. Daly, J. Nat. Prod., 1998, 61, 162–172 CrossRef CAS PubMed.
- E. X. Albuquerque, K. Kuba and J. Daly, J. Pharmacol. Exp. Ther., 1974, 189, 513–524 CAS.
- A. J. Lapa, E. X. Albuquerque, J. M. Sarvey, J. Daly and B. Witkop, Exp. Neurol., 1975, 47, 558–580 CrossRef CAS PubMed.
- A. T. Eldefrawi, M. E. Eldefrawi, E. X. Albuquerque, A. C. Oliveira, N. Mansour, M. Adler, J. W. Daly, G. B. Brown, W. Burgermeister and B. Witkop, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 2172–2176 CrossRef CAS.
- K. Takahashi, A. E. Jacobson, C. P. Mak, B. Witkop, A. Brossi, E. X. Albuquerque, J. E. Warnick, M. A. Maleque, A. Bavoso and J. V. Silverton, J. Med. Chem., 1982, 25, 919–925 CrossRef CAS PubMed.
- C. E. Spivak, M. A. Maleque, A. C. Oliveira, L. M. Masukawa, T. Tokuyama, J. W. Daly and E. X. Albuquerque, Mol. Pharmacol., 1982, 21, 351–361 CAS.
- A. Sinclair and R. A. Stockman, Nat. Prod. Rep., 2007, 24, 298–326 RSC.
- T. Fukuyama, L. V. Dunkerton, M. Aratani and Y. Kishi, J. Org. Chem., 1975, 40, 2011–2012 CrossRef CAS PubMed.
- M. Aratani, L. V. Dunkerton, T. Fukuyama, Y. Kishi, H. Kakoi, S. Sugiura and S. Inoue, J. Org. Chem., 1975, 40, 2009–2011 CrossRef CAS PubMed.
- S. C. Carey, M. Aratani and Y. Kishi, Tetrahedron Lett., 1985, 26, 5887–5890 CrossRef CAS.
- E. J. Corey, J. F. Arnett and G. N. Widiger, J. Am. Chem. Soc., 1975, 97, 430–431 CrossRef CAS PubMed.
- T. Ibuka, H. Minakata, Y. Mitsui, E. Tabushi, T. Taga and Y. Inubushi, Chem. Lett., 1981, 10, 1409–1412 CrossRef.
- T. Ibuka, Y. Mitsui, K. Hayashi, H. Minakata and Y. Inubushi, Tetrahedron Lett., 1981, 22, 4425–4428 CrossRef CAS.
- T. Ibuka, H. Minakata, Y. Mitsui, K. Hayashi, T. Taga and Y. Inubushi, Chem. Pharm. Bull., 1982, 30, 2840–2859 CrossRef CAS.
- N. Maezaki, H. Fukuyama, S. Yagi, T. Tanaka and C. Iwata, J. Chem. Soc., Chem. Commun., 1994, 1835–1836 RSC.
- D. Kim and C. Won Park, Chem. Commun., 1997, 2263–2264 RSC.
- G. Stork and K. Zhao, J. Am. Chem. Soc., 1990, 112, 5875–5876 CrossRef CAS.
- Y. Adachi, N. Kamei, S. Yokoshima and T. Fukuyama, Org. Lett., 2011, 13, 4446–4449 CrossRef CAS PubMed.
- Y. Adachi, N. Kamei, S. Yokoshima and T. Fukuyama, Org. Lett., 2014, 16, 1273 CrossRef CAS.
- M. S. Karatholuvhu, A. Sinclair, A. F. Newton, M.-L. Alcaraz, R. A. Stockman and P. L. Fuchs, J. Am. Chem. Soc., 2006, 128, 12656–12657 CrossRef CAS PubMed.
- G. M. Williams, S. D. Roughley, J. E. Davies, A. B. Holmes and J. P. Adams, J. Am. Chem. Soc., 1999, 121, 4900–4901 CrossRef CAS.
- E. C. Davison, M. E. Fox, A. B. Holmes, S. D. Roughley, C. J. Smith, G. M. Williams, J. E. Davies, P. R. Raithby, J. P. Adams, I. T. Forbes, N. J. Press and M. J. Thompson, J. Chem. Soc., Perkin Trans. 1, 2002, 1494–1514 RSC.
- M. Glanzmann, C. Karalai, B. Ostersehlt, U. Schön, C. Frese and E. Winterfeldt, Tetrahedron, 1982, 38, 2805–2810 CrossRef CAS.
- J. J. Venit, M. DiPierro and P. Magnus, J. Org. Chem., 1989, 54, 4298–4301 CrossRef CAS.
- S. A. Godleski, D. J. Heacock, J. D. Meinhart and S. van Wallendael, J. Org. Chem., 1983, 48, 2101–2103 CrossRef CAS.
- W. Carruthers and S. A. Cumming, J. Chem. Soc., Chem. Commun., 1983, 360–361 RSC.
- D. Tanner and P. Somfai, Tetrahedron Lett., 1985, 26, 3883–3886 CrossRef CAS.
- D. Tanner and P. Somfai, Tetrahedron, 1986, 42, 5657–5664 CrossRef CAS.
- D. Tanner, M. Sellen and J. E. Baeckvall, J. Org. Chem., 1989, 54, 3374–3378 CrossRef CAS.
- D. J. Wardrop, W. Zhang and C. L. Landrie, Tetrahedron Lett., 2004, 45, 4229–4231 CrossRef CAS.
- R. W. Fitch and F. A. Luzzio, Ultrason. Sonochem., 1997, 4, 99–107 CrossRef CAS PubMed.
- O. Y. Provoost, S. J. Hedley, A. J. Hazelwood and J. P. A. Harrity, Tetrahedron Lett., 2006, 47, 331–333 CrossRef CAS.
- P. Compain, J. Goré and J.-M. Vatèle, Tetrahedron, 1996, 52, 6647–6664 CrossRef CAS.
- D. Tanner, L. Hagberg and A. Poulsen, Tetrahedron, 1999, 55, 1427–1440 CrossRef CAS.
- S. Kim, H. Ko, T. Lee and D. Kim, J. Org. Chem., 2005, 70, 5756–5759 CrossRef CAS PubMed.
- D. L. Comins, Y.-M. Zhang and X. Zheng, Chem. Commun., 1998, 2509–2510 RSC.
- J. D. Winkler and P. M. Hershberger, J. Am. Chem. Soc., 1989, 111, 4852–4856 CrossRef CAS.
- X. Q. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752–772 RSC.
- K. Bödeker, Liebigs Ann. Chem., 1881, 208, 363–367 CrossRef.
- G. Stork, R. A. Kretchmer and R. H. Schlessinger, J. Am. Chem. Soc., 1968, 90, 1647–1648 CrossRef CAS PubMed.
- W. A. Ayer, W. R. Bowman, G. A. Cooke and A. C. Soper, Tetrahedron Lett., 1966, 7, 2021–2026 CrossRef.
- W. A. Ayer, W. R. Bowman, T. C. Joseph and P. Smith, J. Am. Chem. Soc., 1968, 90, 1648–1650 CrossRef CAS PubMed.
- A. Padwa, M. A. Brodney, J. P. Marino and S. M. Sheehan, J. Org. Chem., 1997, 62, 78–87 CrossRef CAS PubMed.
- K. Wiesner and L. Poon, Tetrahedron Lett., 1967, 8, 4937–4940 CrossRef.
- H. Dugas, M. E. Hazenberg, Z. Valenta and K. Wiesner, Tetrahedron Lett., 1967, 8, 4931–4936 CrossRef.
- K. Wiesner, V. Musil and K. J. Wesner, Tetrahedron Lett., 1968, 9, 5643–5646 CrossRef.
- K. Wiesner, L. Poon, I. Jirkovský and M. Fishman, Can. J. Chem., 1969, 47, 433–444 CrossRef CAS.
- C. H. Heathcock, E. Kleinman and E. S. Binkley, J. Am. Chem. Soc., 1978, 100, 8036–8037 CrossRef CAS.
- E. Kleinman and C. H. Heathcock, Tetrahedron Lett., 1979, 20, 4125–4128 CrossRef.
- C. H. Heathcock, E. F. Kleinman and E. S. Binkley, J. Am. Chem. Soc., 1982, 104, 1054–1068 CrossRef CAS.
- K. Nakahara, K. Hirano, R. Maehata, Y. Kita and H. Fujioka, Org. Lett., 2011, 13, 2015–2017 CrossRef CAS PubMed.
- D. A. Evans and J. R. Scheerer, Angew. Chem., Int. Ed., 2005, 44, 6038–6042 CrossRef CAS PubMed.
- H. Yang, R. G. Carter and L. N. Zakharov, J. Am. Chem. Soc., 2008, 130, 9238–9239 CrossRef CAS PubMed.
- H. Yang and R. G. Carter, J. Org. Chem., 2010, 75, 4929–4938 CrossRef CAS PubMed.
- G. A. Kraus and Y. S. Hon, J. Am. Chem. Soc., 1985, 107, 4341–4342 CrossRef CAS.
- P. A. Grieco and Y. Dai, J. Am. Chem. Soc., 1998, 120, 5128–5129 CrossRef CAS.
- E. Wenkert, B. Chauncy, K. G. Dave, A. R. Jeffcoat, F. M. Schell and H. P. Schenk, J. Am. Chem. Soc., 1973, 95, 8427–8436 CrossRef CAS.
- E. Wenkert and C. A. Broka, J. Chem. Soc., Chem. Commun., 1984, 714–715 RSC.
- G. A. Kraus and Y. S. Hon, Heterocycles, 1987, 25, 377–386 CrossRef CAS.
- W. A. Ayer and L. S. Trifonov, in The Alkaloids, ed. G. A. Cordell and A. Brossi, Academic, New York, 1994, vol. 45, pp. 233–266 Search PubMed.
- F. A. L. Anet and C. R. Eves, Can. J. Chem., 1958, 36, 902–909 CrossRef CAS.
- D. F. Fischer and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926–5927 CrossRef CAS PubMed.
- C. Tsukano, L. Zhao, Y. Takemoto and M. Hirama, Eur. J. Org. Chem., 2010, 4198–4200 CrossRef CAS.
- C. Yuan, C.-T. Chang and D. Siegel, J. Org. Chem., 2013, 78, 5647–5668 CrossRef CAS PubMed.
- M. Azuma, T. Yoshikawa, N. Kogure, M. Kitajima and H. Takayama, J. Am. Chem. Soc., 2014, 136, 11618–11621 CrossRef CAS PubMed.
- D. Schumann, H.-J. Müller and A. Naumann, Liebigs Ann. Chem., 1982, 2057–2061 CrossRef CAS.
- D. Schumann, H. J. Muller and A. Naumann, Liebigs Ann. Chem., 1982, 1700–1705 CrossRef CAS.
- D. Schumann and A. Naumann, Liebigs Ann. Chem., 1983, 220–225 CrossRef CAS.
- H. Morita, K. I. Ishiuchi, A. Haganuma, T. Hoshino, Y. Obara, N. Nakahata and J. I. Kobayashi, Tetrahedron, 2005, 61, 1955–1960 CrossRef CAS.
- B. B. Liau and M. D. Shair, J. Am. Chem. Soc., 2010, 132, 9594–9595 CrossRef CAS PubMed.
- A. S. Lee, B. B. Liau and M. D. Shair, J. Am. Chem. Soc., 2014, 136, 13442–13452 CrossRef CAS PubMed.
- H. Li, X. Wang and X. Lei, Angew. Chem., Int. Ed., 2012, 51, 491–495 CrossRef CAS PubMed.
- J. Zhang, J. Wu, B. Hong, W. Ai, X. Wang, H. Li and X. Lei, Nat. Commun., 2014, 5, 4614 CAS.
- Y.-R. Yang, Z.-W. Lai, L. Shen, J.-Z. Huang, X.-D. Wu, J.-L. Yin and K. Wei, Org. Lett., 2010, 12, 3430–3433 CrossRef CAS PubMed.
- Y.-R. Yang, L. Shen, J.-Z. Huang, T. Xu and K. Wei, J. Org. Chem., 2011, 76, 3684–3690 CrossRef CAS PubMed.
- S.-H. Hou, Y.-Q. Tu, L. Liu, F.-M. Zhang, S.-H. Wang and X.-M. Zhang, Angew. Chem., Int. Ed., 2013, 52, 11373–11376 CrossRef CAS PubMed.
- Y. Xia and A. P. Kozikowski, J. Am. Chem. Soc., 1989, 111, 4116–4117 CrossRef CAS.
- L. Qian and R. Ji, Tetrahedron Lett., 1989, 30, 2089–2090 CrossRef CAS.
- A. P. Kozikowski, Y. Xia, E. R. Reddy, W. Tuckmantel, I. Hanin and X. C. Tang, J. Org. Chem., 1991, 56, 4636–4645 CrossRef CAS.
- A. P. Kozikowski, G. Campiani, P. Aagaard and M. McKinney, J. Chem. Soc., Chem. Commun., 1993, 860–862 RSC.
- I. Y. C. Lee, M. H. Jung, H. W. Lee and J. Y. Yang, Tetrahedron Lett., 2002, 43, 2407–2409 CrossRef CAS.
- T. Koshiba, S. Yokoshima and T. Fukuyama, Org. Lett., 2009, 11, 5354–5356 CrossRef CAS PubMed.
- M. K. M. Tun, D.-J. Wüstmann and S. B. Herzon, Chem. Sci., 2011, 2, 2251–2253 RSC.
- S. M. A. Sohel and T. Opatz, ARKIVOC, 2014, i, 92–108 Search PubMed.
- Y. Hirasawa, J. Kobayashi and H. Morita, Heterocycles, 2009, 77, 679–729 CrossRef CAS.
- G. T. Ha, R. K. Wong and Y. Zhang, Chem. Biodiversity, 2011, 8, 1189–1204 CAS.
- S. R. Tudhope, J. A. Bellamy, A. Ball, D. Rajasekar, M. Azadi-Ardakani, H. S. Meera, J. M. Gnanadeepam, R. Saiganesh, F. Gibson, L. He, C. H. Behrens, G. Underiner, J. Marfurt and N. Favre, Org. Process Res. Dev., 2012, 16, 635–642 CrossRef CAS.
- R. Ding, B.-F. Sun and G.-Q. Lin, Org. Lett., 2012, 14, 4446–4449 CrossRef CAS PubMed.
- J. D. White, Y. Li, J. Kim and M. Terinek, Org. Lett., 2013, 15, 882–885 CrossRef CAS PubMed.
- Y. Hirasawa, H. Morita and J. Kobayashi, Org. Lett., 2004, 6, 3389–3391 CrossRef CAS PubMed.
- Y. Hirasawa, J. Kobayashi, Y. Obara, N. Nakahata, N. Kawahara, Y. Goda and H. Morita, Heterocycles, 2006, 68, 2357–2364 CrossRef CAS.
- B. L. Nilsson, L. E. Overman, J. R. de Alaniz and J. M. Rohde, J. Am. Chem. Soc., 2008, 130, 11297–11299 CrossRef CAS PubMed.
- R. A. Altman, B. L. Nilsson, L. E. Overman, J. R. de Alaniz, J. M. Rohde and V. Taupin, J. Org. Chem., 2010, 75, 7519–7534 CrossRef CAS PubMed.
- X. Cheng and S. P. Waters, Org. Lett., 2010, 12, 205–207 CrossRef CAS PubMed.
- W. A. Denne, S. R. Johns, J. A. Lamberton and A. M. Mathieson, Tetrahedron Lett., 1971, 12, 3107–3108 CrossRef.
- W. A. Denne, S. R. Johns, J. A. Lamberton and A. M. Mathieson, Tetrahedron Lett., 1973, 14, 794 CrossRef.
- E. J. Corey and R. D. Balanson, J. Am. Chem. Soc., 1974, 96, 6516–6517 CrossRef CAS.
- D. M. Ryckman and R. V. Stevens, J. Am. Chem. Soc., 1987, 109, 4940–4948 CrossRef CAS.
- M. Matsui, in The Alkaloids: Chemistry and Pharmacology, ed. B. Arnold, Elsevier Academic Press, 1988, vol. 33, pp. 307–347 Search PubMed.
- Y. Inubushi, T. Ibuka and M. Kitano, Tetrahedron Lett., 1969, 10, 1611–1614 CrossRef.
- D. A. Evans, C. A. Bryan and G. M. Wahl, J. Org. Chem., 1970, 35, 4122–4127 CrossRef CAS.
- T. Ibuka, K. Tanaka and Y. Inubushi, Tetrahedron Lett., 1970, 11, 4811–4814 CrossRef.
- Y. Inubushi, M. Kitano and T. Ibuka, Chem. Pharm. Bull., 1971, 19, 1820–1841 CrossRef CAS.
- T. Ibuka, K. Tanaka and Y. Inubushi, Tetrahedron Lett., 1972, 13, 1393–1396 CrossRef.
- T. Ibuka, K. Tanaka and Y. Inubushi, Chem. Pharm. Bull., 1974, 22, 782–798 CrossRef CAS.
- T. Ibuka, K. Tanaka and Y. Inubushi, Chem. Pharm. Bull., 1974, 22, 907–921 CrossRef CAS.
- A. G. Schultz and A. Wang, J. Am. Chem. Soc., 1998, 120, 8259–8260 CrossRef CAS.
- S. B. Jones, L. He and S. L. Castle, Org. Lett., 2006, 8, 3757–3760 CrossRef CAS PubMed.
- F. Li, S. S. Tartakoff and S. L. Castle, J. Am. Chem. Soc., 2009, 131, 6674–6675 CrossRef CAS PubMed.
- F. Li, S. S. Tartakoff and S. L. Castle, J. Org. Chem., 2009, 74, 9082–9093 CrossRef CAS PubMed.
- S. B. Herzon, N. A. Calandra and S. M. King, Angew. Chem., Int. Ed., 2011, 50, 8863–8866 CrossRef CAS PubMed.
- K. V. Chuang, R. Navarro and S. E. Reisman, Angew. Chem., Int. Ed., 2011, 50, 9447–9451 CrossRef CAS PubMed.
- S. M. King, N. A. Calandra and S. B. Herzon, Angew. Chem., Int. Ed., 2013, 52, 3642–3645 CrossRef CAS PubMed.
- N. A. Calandra, S. M. King and S. B. Herzon, J. Org. Chem., 2013, 78, 10031–10057 CrossRef CAS PubMed.
- D. K. Nielsen, L. L. Nielsen, S. B. Jones, L. Toll, M. C. Asplund and S. L. Castle, J. Org. Chem., 2009, 74, 1187–1199 CrossRef CAS PubMed.
- G.-W. Qin and R.-S. Xu, Med. Res. Rev., 1998, 18, 375–382 CrossRef CAS PubMed.
- R.-S. Xu, in Studies in Natural Products Chemistry, ed. R. Atta ur, Elsevier, 2000, vol. vol. 21, Part B, pp. 729–772 Search PubMed.
- H. S. Chung, P. M. Hon, G. Lin, P. P. H. But and H. Dong, Planta Med., 2003, 69, 914–920 CrossRef CAS PubMed.
- R. Aloise Pilli and M. da Conceição Ferreira de Oliveira, Nat. Prod. Rep., 2000, 17, 117–127 RSC.
- R. A. Pilli, G. B. Rosso and M. da Conceição Ferreira de Oliveira, Nat. Prod. Rep., 2010, 27, 1908–1937 RSC.
- A. S. Kende, J. I. Martin Hernando and J. B. J. Milbank, Org. Lett., 2001, 3, 2505–2508 CrossRef CAS PubMed.
- A. S. Kende, J. I. Martin Hernando and J. B. J. Milbank, Tetrahedron, 2002, 58, 61–74 CrossRef CAS.
- T. Taniguchi and H. Ishibashi, Tetrahedron, 2008, 64, 8773–8779 CrossRef CAS.
- T. Taniguchi, G. Tanabe, O. Muraoka and H. Ishibashi, Org. Lett., 2008, 10, 197–199 CrossRef CAS PubMed.
- P.-Q. Huang, S.-Y. Huang, L.-H. Gao, Z.-Y. Mao, Z. Chang and A.-E. Wang, Chem. Commun., 2015, 51, 4576–4578 RSC.
- Y.-M. Zhao, P. Gu, Y.-Q. Tu, C.-A. Fan and Q. Zhang, Org. Lett., 2008, 10, 1763–1766 CrossRef CAS PubMed.
- Z.-H. Chen, Z.-M. Chen, Y.-Q. Zhang, Y.-Q. Tu and F.-M. Zhang, J. Org. Chem., 2011, 76, 10173–10186 CrossRef CAS PubMed.
- Z.-H. Chen, Y.-Q. Zhang, Z.-M. Chen, Y.-Q. Tu and F.-M. Zhang, Chem. Commun., 2011, 47, 1836–1838 RSC.
- M. Brüggemann, A. I. McDonald, L. E. Overman, M. D. Rosen, L. Schwink and J. P. Scott, J. Am. Chem. Soc., 2003, 125, 15284–15285 CrossRef PubMed.
- A. S. Kende, T. L. Smalley and H. Huang, J. Am. Chem. Soc., 1999, 121, 7431–7432 CrossRef CAS.
- C. Fang, C. S. Shanahan, D. H. Paull and S. F. Martin, Angew. Chem., Int. Ed., 2012, 51, 10596–10599 CrossRef CAS PubMed.
- P. Gu, Y.-M. Zhao, Y. Q. Tu, Y. Ma and F. Zhang, Org. Lett., 2006, 8, 5271–5273 CrossRef CAS PubMed.
- M. Greshoff, Ber. Dtsch. Chem. Ges., 1890, 23, 3537–3550 CrossRef.
- A. R. Battersby and H. Gregory, J. Chem. Soc., 1963, 22–32 RSC.
- C. Djerassi, H. Schmid, W. G. Kump, A. R. Battersby, J. M. Wilson, R. J. Owellen, D. J. Lecount and H. Budzikiewicz, Helv. Chim. Acta, 1963, 46, 742–751 CAS.
- B. W. Bycroft, D. Schumann, M. B. Patel and H. Schmid, Helv. Chim. Acta, 1964, 47, 1147–1152 CrossRef CAS.
- D. Schumann, B. W. Bycroft and H. Schmid, Experientia, 1964, 20, 202–203 CrossRef CAS PubMed.
- H. Achenbach and K. Biemann, J. Am. Chem. Soc., 1965, 87, 4944–4950 CrossRef CAS.
- J. M. F. Filho, B. Gilbert, M. Kitagawa, L. A. P. Leme and L. J. Durham, J. Chem. Soc. C, 1966, 1260–1266 RSC.
- B. M. Craven, B. Gilbert and L. A. P. Leme, Chem. Commun., 1968, 955–956 RSC.
- B. Craven, Acta Crystallogr., Sect. B: Struct. Sci., 1969, 25, 2131–2139 CrossRef CAS PubMed.
- H. Kinoshita, T. Ohnuma, T. Oishi and Y. Ban, Chem. Lett., 1986, 15, 927–930 CrossRef.
- D. Cartier, M. Ouahrani and J. Lévy, Tetrahedron Lett., 1989, 30, 1951–1954 CrossRef CAS.
- J. Xie, A. L. Wolfe and D. L. Boger, Org. Lett., 2013, 15, 868–870 CrossRef CAS PubMed.
- Y. Ban, Y. Honma and T. Oishi, Tetrahedron Lett., 1976, 17, 1111–1114 CrossRef.
- M. E. Kuehne and P. J. Seaton, J. Org. Chem., 1985, 50, 4790–4796 CrossRef CAS.
- E. Wenkert and S. Liu, J. Org. Chem., 1994, 59, 7677–7682 CrossRef CAS.
- D. Gagnon and C. Spino, J. Org. Chem., 2009, 74, 6035–6041 CrossRef CAS PubMed.
- T. Gallagher and P. Magnus, J. Am. Chem. Soc., 1983, 105, 2086–2087 CrossRef CAS.
- P. Magnus, T. Gallagher, P. Brown and J. C. Huffman, J. Am. Chem. Soc., 1984, 106, 2105–2114 CrossRef CAS.
- P. Magnus and P. Brown, J. Chem. Soc., Chem. Commun., 1985, 184–186 RSC.
- P. Magnus, I. R. Matthews, J. Schultz, R. Waditschatka and J. C. Huffman, J. Org. Chem., 1988, 53, 5772–5776 CrossRef CAS.
- P. Magnus, T. Katoh, I. R. Matthews and J. C. Huffman, J. Am. Chem. Soc., 1989, 111, 6707–6711 CrossRef CAS.
- K. Awang, T. Sévenet, A. Hamid, A. Hadi, B. David and M. Païs, Tetrahedron Lett., 1992, 33, 2493–2496 CrossRef CAS.
- K. Awang, T. Sévenet, M. Païs and A. H. A. Hadi, J. Nat. Prod., 1993, 56, 1134–1139 CrossRef CAS.
- T.-S. Kam, K. Yoganathan and C.-H. Chuah, Tetrahedron Lett., 1995, 36, 759–762 CrossRef CAS.
- T.-S. Kam, K.-H. Lim, K. Yoganathan, M. Hayashi and K. Komiyama, Tetrahedron, 2004, 60, 10739–10745 CrossRef CAS.
- S. Jin, J. Gong and Y. Qin, Angew. Chem., Int. Ed., 2015, 54, 2228–2231 CrossRef CAS PubMed.
- S. Arai, M. Nakajima and A. Nishida, Angew. Chem., Int. Ed., 2014, 53, 5569–5572 CrossRef CAS PubMed.
- S. Arai, M. Nakajima and A. Nishida, Angew. Chem., Int. Ed., 2014, 53, 14295 CrossRef CAS.
- M. Hoshi, O. Kaneko, M. Nakajima, S. Arai and A. Nishida, Org. Lett., 2014, 16, 768–771 CrossRef CAS PubMed.
- W. H. Pearson, I. Y. Lee, Y. Mi and P. Stoy, J. Org. Chem., 2004, 69, 9109–9122 CrossRef CAS PubMed.
- W. H. Pearson, Y. Mi, I. Y. Lee and P. Stoy, J. Am. Chem. Soc., 2001, 123, 6724–6725 CrossRef CAS PubMed.
- T. J. Greshock, A. W. Grubbs, S. Tsukamoto and R. M. Williams, Angew. Chem., Int. Ed., 2007, 46, 2262–2265 CrossRef CAS PubMed.
- G. D. Artman III, A. W. Grubbs and R. M. Williams, J. Am. Chem. Soc., 2007, 129, 6336–6342 CrossRef PubMed.
- D. Seebach, M. Boes, R. Naef and W. B. Schweizer, J. Am. Chem. Soc., 1983, 105, 5390–5398 CrossRef CAS.
- P. S. Baran, C. A. Guerrero, N. B. Ambhaikar and B. D. Hafensteiner, Angew. Chem., Int. Ed., 2005, 44, 606–609 CrossRef CAS PubMed.
- P. S. Baran, C. A. Guerrero, B. D. Hafensteiner and N. B. Ambhaikar, Angew. Chem., Int. Ed., 2005, 44, 3892–3895 CrossRef CAS PubMed.
- S. B. Herzon and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 5342–5344 CrossRef CAS PubMed.
- R. F. Bond, J. C. A. Boeyens, C. W. Holzapfel and P. S. Steyn, J. Chem. Soc., Perkin Trans. 1, 1979, 1751–1761 RSC.
- M. Tsuda, Y. Kasai, K. Komatsu, T. Sone, M. Tanaka, Y. Mikami and J. I. Kobayashi, Org. Lett., 2004, 6, 3087–3089 CrossRef CAS PubMed.
- T. Mugishima, M. Tsuda, Y. Kasai, H. Ishiyama, E. Fukushi, J. Kawabata, M. Watanabe, K. Akao and J. Kobayashi, J. Org. Chem., 2005, 70, 9430–9435 CrossRef CAS PubMed.
- Z. Bian, C. C. Marvin and S. F. Martin, J. Am. Chem. Soc., 2013, 135, 10886–10889 CrossRef CAS PubMed.
- K. Kong, J. A. Enquist, M. E. McCallum, G. M. Smith, T. Matsumaru, E. Menhaji-Klotz and J. L. Wood, J. Am. Chem. Soc., 2013, 135, 10890–10893 CrossRef CAS PubMed.
- E. V. Mercado-Marin, P. Garcia-Reynaga, S. Romminger, E. F. Pimenta, D. K. Romney, M. W. Lodewyk, D. E. Williams, R. J. Andersen, S. J. Miller, D. J. Tantillo, R. G. S. Berlinck and R. Sarpong, Nature, 2014, 509, 318–324 CrossRef CAS PubMed.
- E. V. Mercado-Marin and R. Sarpong, Chem. Sci., 2015, 6, 5048–5052 RSC.
- B. M. Trost, D. A. Bringley, T. Zhang and N. Cramer, J. Am. Chem. Soc., 2013, 135, 16720–16735 CrossRef CAS PubMed.
- B. M. Trost, N. Cramer and H. Bernsmann, J. Am. Chem. Soc., 2007, 129, 3086–3087 CrossRef CAS PubMed.
- W. G. Beyersbergen van Henegouwen, R. M. Fieseler, F. P. J. T. Rutjes and H. Hiemstra, Angew. Chem., Int. Ed., 1999, 38, 2214–2217 CrossRef.
- H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36–51 CrossRef CAS.
- X. Zhou, T. Xiao, Y. Iwama and Y. Qin, Angew. Chem., Int. Ed., 2012, 51, 4909–4912 CrossRef CAS PubMed.
- J. Shimokawa, T. Harada, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2011, 133, 17634–17637 CrossRef CAS PubMed.
- S. Diethelm and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 8500–8503 CrossRef CAS PubMed.
- K. Stratmann, R. E. Moore, R. Bonjouklian, J. B. Deeter, G. M. L. Patterson, S. Shaffer, C. D. Smith and T. A. Smitka, J. Am. Chem. Soc., 1994, 116, 9935–9942 CrossRef CAS.
- J. I. Jimenez, U. Huber, R. E. Moore and G. M. L. Patterson, J. Nat. Prod., 1999, 62, 569–572 CrossRef CAS PubMed.
- J. C. Menéndez, in Bioactive Heterocycles V, ed. M. Khan, Springer, Berlin Heidelberg, 2007, vol. 11, ch. 63, pp. 63–101 Search PubMed.
- J. L. Wood, Nat. Chem., 2012, 4, 341–343 CrossRef CAS PubMed.
- J. A. MacKay, R. L. Bishop and V. H. Rawal, Org. Lett., 2005, 7, 3421–3424 CrossRef CAS PubMed.
- V. Bhat, K. M. Allan and V. H. Rawal, J. Am. Chem. Soc., 2011, 133, 5798–5801 CrossRef CAS PubMed.
- K. M. Allan, K. Kobayashi and V. H. Rawal, J. Am. Chem. Soc., 2012, 134, 1392–1395 CrossRef CAS PubMed.
- K. Jae Nyoung and E. K. Ryu, Tetrahedron Lett., 1993, 34, 8283–8284 CrossRef.
- J. N. Kim, K. S. Jung, H. J. Lee and J. S. Son, Tetrahedron Lett., 1997, 38, 1597–1598 CrossRef CAS.
- T.-H. Fu, W. T. McElroy, M. Shamszad and S. F. Martin, Org. Lett., 2012, 14, 3834–3837 CrossRef CAS PubMed.
- X. Tian, A. D. Huters, C. J. Douglas and N. K. Garg, Org. Lett., 2009, 11, 2349–2351 CrossRef CAS PubMed.
- A. D. Huters, K. W. Quasdorf, E. D. Styduhar and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 15797–15799 CrossRef CAS PubMed.
- K. W. Quasdorf, A. D. Huters, M. W. Lodewyk, D. J. Tantillo and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 1396–1399 CrossRef CAS PubMed.
- N. A. Weires, E. D. Styduhar, E. L. Baker and N. K. Garg, J. Am. Chem. Soc., 2014, 136, 14710–14713 CrossRef CAS PubMed.
- E. D. Styduhar, A. D. Huters, N. A. Weires and N. K. Garg, Angew. Chem., Int. Ed., 2013, 52, 12422–12425 CrossRef CAS PubMed.
- K. Komine, Y. Nomura, J. Ishihara and S. Hatakeyama, Org. Lett., 2015, 17, 3918–3921 CrossRef CAS PubMed.
- P. S. Baran, R. A. Shenvi and C. A. Mitsos, Angew. Chem., Int. Ed., 2005, 44, 3714–3717 CrossRef CAS PubMed.
- P. S. Baran and R. A. Shenvi, J. Am. Chem. Soc., 2006, 128, 14028–14029 CrossRef CAS PubMed.
- T. Newhouse and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 10886–10887 CrossRef CAS PubMed.
- K. Foo, T. Newhouse, I. Mori, H. Takayama and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 2716–2719 CrossRef CAS PubMed.
- T. Newhouse, C. A. Lewis and P. S. Baran, J. Am. Chem. Soc., 2009, 131, 6360–6361 CrossRef CAS PubMed.
- T. Newhouse, C. A. Lewis, K. J. Eastman and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 7119–7137 CrossRef CAS PubMed.
- C. Li, C. Chan, A. C. Heimann and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 1448–1450 CrossRef CAS PubMed.
- H. Abdelkafi and B. Nay, Nat. Prod. Rep., 2012, 29, 845–869 RSC.
- S. M. Weinreb and J. Auerbach, J. Am. Chem. Soc., 1975, 97, 2503–2506 CrossRef CAS.
- T. P. Burkholder and P. L. Fuchs, J. Am. Chem. Soc., 1988, 110, 2341–2342 CrossRef CAS.
- L. F. Tietze and H. Schirok, J. Am. Chem. Soc., 1999, 121, 10264–10269 CrossRef CAS.
- Q. Liu, E. M. Ferreira and B. M. Stoltz, J. Org. Chem., 2007, 72, 7352–7358 CrossRef CAS PubMed.
- W.-D. Z. Li and Y.-Q. Wang, Org. Lett., 2003, 5, 2931–2934 CrossRef CAS PubMed.
- L. Planas, J. Pérard-Viret and J. Royer, J. Org. Chem., 2004, 69, 3087–3092 CrossRef CAS PubMed.
- J. D. Eckelbarger, J. T. Wilmot and D. Y. Gin, J. Am. Chem. Soc., 2006, 128, 10370–10371 CrossRef CAS PubMed.
- J. D. Eckelbarger, J. T. Wilmot, M. T. Epperson, C. S. Thakur, D. Shum, C. Antczak, L. Tarassishin, H. Djaballah and D. Y. Gin, Chem.–Eur. J., 2008, 14, 4293–4306 CrossRef CAS PubMed.
- Z. Zhao and P. S. Mariano, Tetrahedron, 2006, 62, 7266–7273 CrossRef CAS.
- A. Hameed, A. J. Blake and C. J. Hayes, J. Org. Chem., 2008, 73, 8045–8048 CrossRef CAS PubMed.
- T. Taniguchi and H. Ishibashi, Org. Lett., 2008, 10, 4129–4131 CrossRef CAS PubMed.
- M. E. Kuehne, W. G. Bornmann, W. H. Parsons, T. D. Spitzer, J. F. Blount and J. Zubieta, J. Org. Chem., 1988, 53, 3439–3450 CrossRef CAS.
- M. Ikeda, S. A. A. El Bialy, K. Hirose, M. Kotake, T. Sato, S. M. M. Bayomi, I. A. Shehata, A. M. Abdelal, L. M. Gad and T. Yakura, Chem. Pharm. Bull., 1999, 47, 983–987 CrossRef CAS PubMed.
- S. Suga, M. Watanabe and J. Yoshida, J. Am. Chem. Soc., 2002, 124, 14824–14825 CrossRef CAS PubMed.
- S. Yasuda, T. Yamada and M. Hanaoka, Tetrahedron Lett., 1986, 27, 2023–2026 CrossRef CAS.
- H. Ishibashi, M. Okano, H. Tamaki, K. Maruyama, T. Yakura and M. Ikeda, J. Chem. Soc., Chem. Commun., 1990, 1436–1437 RSC.
- L. F. Tietze, H. Braun, P. L. Steck, S. A. A. El Bialy, N. Tölle and A. Düfert, Tetrahedron, 2007, 63, 6437–6445 CrossRef CAS.
- Y.-M. Zhao, P. Gu, H.-J. Zhang, Q.-W. Zhang, C.-A. Fan, Y.-Q. Tu and F.-M. Zhang, J. Org. Chem., 2009, 74, 3211–3213 CrossRef CAS PubMed.
- Y. Koseki, H. Sato, Y. Watanabe and T. Nagasaka, Org. Lett., 2002, 4, 885–888 CrossRef CAS PubMed.
- X. Lin, R. W. Kavash and P. S. Mariano, J. Am. Chem. Soc., 1994, 116, 9791–9792 CrossRef CAS.
- K. Folkers and R. T. Major, J. Am. Chem. Soc., 1937, 59, 1580–1581 CrossRef CAS.
- E. Reimann, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Falk and G. W. Kirby, Springer, Vienna, 2007, vol. 88, ch. 1, pp. 1–62 Search PubMed.
- H. Ishibashi, T. Sato, M. Takahashi, M. Hayashi, K. Ishikawa and M. Ikeda, Chem. Pharm. Bull., 1990, 38, 907–911 CrossRef CAS.
- S. Chikaoka, A. Toyao, M. Ogasawara, O. Tamura and H. Ishibashi, J. Org. Chem., 2003, 68, 312–318 CrossRef CAS PubMed.
- T. Sano, J. Toda, T. Ohshima and Y. Tsuda, Chem. Pharm. Bull., 1992, 40, 873–878 CrossRef CAS.
- R. L. Funk and J. Belmar, Tetrahedron Lett., 2012, 53, 176–178 CrossRef CAS PubMed.
- S. T. Heller, T. Kiho, A. R. H. Narayan and R. Sarpong, Angew. Chem., Int. Ed., 2013, 52, 11129–11133 CrossRef CAS PubMed.
- H.-T. Luu, S. Wiesler, G. Frey and J. Streuff, Org. Lett., 2015, 17, 2478–2481 CrossRef CAS PubMed.
- N. A. Braun, M. Ousmer, J. D. Bray, D. Bouchu, K. Peters, E.-M. Peters and M. A. Ciufolini, J. Org. Chem., 2000, 65, 4397–4408 CrossRef CAS PubMed.
- M. Paladino, J. Zaifman and M. A. Ciufolini, Org. Lett., 2015, 17, 3422–3425 CrossRef CAS PubMed.
- T. Kawasaki, N. Onoda, H. Watanabe and T. Kitahara, Tetrahedron Lett., 2001, 42, 8003–8006 CrossRef CAS.
- J. M. Joo, R. A. David, Y. Yuan and C. Lee, Org. Lett., 2010, 12, 5704–5707 CrossRef CAS PubMed.
- K. V. Chuang, R. Navarro and S. E. Reisman, Chem. Sci., 2011, 2, 1086–1089 RSC.
- M. A. le Dreau, D. Desmaele, F. Dumas and J. d'Angelo, J. Org. Chem., 1993, 58, 2933–2935 CrossRef CAS.
- Y. Yoshida, K. Mohri, K. Isobe, T. Itoh and K. Yamamoto, J. Org. Chem., 2009, 74, 6010–6015 CrossRef CAS PubMed.
- K. Shimizu, M. Takimoto, Y. Sato and M. Mori, J. Organomet. Chem., 2006, 691, 5466–5475 CrossRef CAS.
- H. Fukumoto, T. Esumi, J. Ishihara and S. Hatakeyama, Tetrahedron Lett., 2003, 44, 8047–8049 CrossRef CAS.
- Q. Wang and A. Padwa, Org. Lett., 2006, 8, 601–604 CrossRef CAS PubMed.
- F. Zhang, N. S. Simpkins and A. J. Blake, Org. Biomol. Chem., 2009, 7, 1963–1979 CAS.
- J. H. Rigby, C. Deur and M. J. Heeg, Tetrahedron Lett., 1999, 40, 6887–6890 CrossRef CAS.
- J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS PubMed.
- S. M. Weinreb, Chem. Rev., 2006, 106, 2531–2549 CrossRef CAS PubMed.
- K. Sakamato, E. Tsujii, F. Abe, T. Nakanishi, M. Yamashite, N. Shigematsu, S. Izumi and M. Okuhara, J. Antibiot., 1996, 49, 37–44 CrossRef.
- J. F. Biard, S. Guyot, C. Roussakis, J. F. Verbist, J. Vercauteren, J. F. Weber and K. Boukef, Tetrahedron Lett., 1994, 35, 2691–2694 CrossRef CAS.
- D. L. J. Clive, M. L. Yu, J. Wang, V. S. C. Yeh and S. Z. Kang, Chem. Rev., 2005, 105, 4483–4514 CrossRef CAS PubMed.
- B. B. Snider and T. Liu, J. Org. Chem., 1997, 62, 5630–5633 CrossRef CAS.
- B. M. Trost and M. T. Rudd, Org. Lett., 2003, 5, 4599–4602 CrossRef CAS PubMed.
- A. C. Flick, M. J. A. Caballero and A. Padwa, Org. Lett., 2008, 10, 1871–1874 CrossRef CAS PubMed.
- J. Liu, R. P. Hsung and S. D. Peters, Org. Lett., 2004, 6, 3989–3992 CrossRef CAS PubMed.
- J. Liu, J. J. Swidorski, S. D. Peters and R. P. Hsung, J. Org. Chem., 2005, 70, 3898–3902 CrossRef CAS PubMed.
- H. Abe, S. Aoyagi and C. Kibayashi, J. Am. Chem. Soc., 2005, 127, 1473–1480 CrossRef CAS PubMed.
- J. F. Liu and C. H. Heathcock, J. Org. Chem., 1999, 64, 8263–8266 CrossRef CAS PubMed.
- G. A. Molander and M. Rönn, J. Org. Chem., 1999, 64, 5183–5187 CrossRef CAS.
- T. Arai, H. Abe, S. Aoyagi and C. Kibayashi, Tetrahedron Lett., 2004, 45, 5921–5924 CrossRef CAS.
- G. Pandey and V. Janakiram, Chem.–Eur. J., 2015, 21, 13120–13126 CrossRef CAS PubMed.
- G. Lapointe, K. Schenk and P. Renaud, Org. Lett., 2011, 13, 4774–4777 CrossRef CAS PubMed.
- G. Scheffler, H. Seike and E. J. Sorensen, Angew. Chem., Int. Ed., 2000, 39, 4593–4596 CrossRef CAS.
- M. Ousmer, N. A. Braun and M. A. Ciufolini, Org. Lett., 2001, 3, 765–767 CrossRef CAS PubMed.
- A.-J. Ma, Y.-Q. Tu, J.-B. Peng, Q.-Y. Dou, S.-H. Hou, F.-M. Zhang and S.-H. Wang, Org. Lett., 2012, 14, 3604–3607 CrossRef CAS PubMed.
- S. Ieda, T. Kan and T. Fukuyama, Tetrahedron Lett., 2010, 51, 4027–4029 CrossRef CAS.
- H.-H. Huo, H.-K. Zhang, X.-E. Xia and P.-Q. Huang, Org. Lett., 2012, 14, 4834–4837 CrossRef CAS PubMed.
- H.-H. Huo, X.-E. Xia, H.-K. Zhang and P.-Q. Huang, J. Org. Chem., 2013, 78, 455–465 CrossRef CAS PubMed.
- K. M. Brummond and J. L. Lu, Org. Lett., 2001, 3, 1347–1349 CrossRef CAS PubMed.
- K. M. Brummond and S.-P. Hong, J. Org. Chem., 2005, 70, 907–916 CrossRef CAS PubMed.
- M. Movassaghi, M. Tjandra and J. Qi, J. Am. Chem. Soc., 2009, 131, 9648–9650 CrossRef CAS PubMed.
- D. A. Evans, D. J. Adams and E. E. Kwan, J. Am. Chem. Soc., 2012, 134, 8162–8170 CrossRef CAS PubMed.
- M. Kuramoto, C. Tong, K. Yamada, T. Chiba, Y. Hayashi and D. Uemura, Tetrahedron Lett., 1996, 37, 3867–3870 CrossRef CAS.
- T. Chou, M. Kuramoto, Y. Otani, M. Shikano, K. Yazawa and D. Uemura, Tetrahedron Lett., 1996, 37, 3871–3874 CrossRef CAS.
- J. Kobayashi and M. Ishibashi, Chem. Rev., 1993, 93, 1753–1769 CrossRef CAS.
- D. Trauner, J. B. Schwarz and S. J. Danishefsky, Angew. Chem., Int. Ed., 1999, 38, 3542–3545 CrossRef CAS.
- M. W. Carson, G. Kim and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4453–4456 CrossRef CAS.
- M. W. Carson, G. Kim, M. F. Hentemann, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4450–4452 CrossRef CAS.
- H. S. Christie and C. H. Heathcock, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12079–12084 CrossRef CAS PubMed.
- S. Xu, H. Arimoto and D. Uemura, Angew. Chem., Int. Ed., 2007, 46, 5746–5749 CrossRef CAS PubMed.
- S. Omura, K. Matsuzaki, T. Fujimoto, K. Kosuge, T. Furuya, S. Fujita and A. Nakagawa, J. Antibiot., 1991, 44, 117–118 CrossRef CAS PubMed.
- S. Omura, T. Fujimoto, K. Otoguro, K. Matsuzaki, R. Moriguchi, H. Tanaka and Y. Sasaki, J. Antibiot., 1991, 44, 113–116 CrossRef CAS PubMed.
- R. H. Feling, G. O. Buchanan, T. J. Mincer, C. A. Kauffman, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2003, 42, 355–357 CrossRef CAS PubMed.
- M. Shibasaki, M. Kanai and N. Fukuda, Chem.–Asian J., 2007, 2, 20–38 CrossRef CAS PubMed.
- E. J. Corey and G. A. Reichard, J. Am. Chem. Soc., 1992, 114, 10677–10678 CrossRef CAS.
- E. J. Corey and G. A. Reichard, Tetrahedron Lett., 1993, 34, 6973–6976 CrossRef CAS.
- E. J. Corey and S. Choi, Tetrahedron Lett., 1993, 34, 6969–6972 CrossRef CAS.
- T. Sunazuka, T. Nagamitsu, K. Matsuzaki, H. Tanaka, S. Omura and A. B. Smith, J. Am. Chem. Soc., 1993, 115, 5302 CrossRef CAS.
- H. Uno, J. E. Baldwin and A. T. Russell, J. Am. Chem. Soc., 1994, 116, 2139–2140 CrossRef CAS.
- N. Chida, J. Takeoka, N. Tsutsumi and S. Ogawa, J. Chem. Soc., Chem. Commun., 1995, 793–794 RSC.
- T. Nagamitsu, T. Sunazuka, H. Tanaka, S. Ōmura, P. A. Sprengeler and A. B. Smith, J. Am. Chem. Soc., 1996, 118, 3584–3590 CrossRef CAS.
- N. Chida, J. Takeoka, K. Ando, N. Tsutsumi and S. Ogawa, Tetrahedron, 1997, 53, 16287–16298 CrossRef CAS.
- E. J. Corey, W. Li and T. Nagamitsu, Angew. Chem., Int. Ed., 1998, 37, 1676–1679 CrossRef CAS.
- E. J. Corey, W. Li and G. A. Reichard, J. Am. Chem. Soc., 1998, 120, 2330–2336 CrossRef CAS.
- J. S. Panek and C. E. Masse, Angew. Chem., Int. Ed., 1999, 38, 1093–1095 CrossRef CAS.
- H. Ooi, N. Ishibashi, Y. Iwabuchi, J. Ishihara and S. Hatakeyama, J. Org. Chem., 2004, 69, 7765–7768 CrossRef CAS PubMed.
- L. R. Reddy, P. Saravanan and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 6230–6231 CrossRef CAS PubMed.
- L. R. Reddy, J.-F. Fournier, B. V. S. Reddy and E. J. Corey, Org. Lett., 2005, 7, 2699–2701 CrossRef CAS PubMed.
- A. Endo and S. J. Danishefsky, J. Am. Chem. Soc., 2005, 127, 8298–8299 CrossRef CAS PubMed.
- E. P. Balskus and E. N. Jacobsen, J. Am. Chem. Soc., 2006, 128, 6810–6812 CrossRef CAS PubMed.
- N. Fukuda, K. Sasaki, T. V. R. S. Sastry, M. Kanai and M. Shibasaki, J. Org. Chem., 2006, 71, 1220–1225 CrossRef CAS PubMed.
- N. P. Mulholland, G. Pattenden and I. A. S. Walters, Org. Biomol. Chem., 2006, 4, 2845–2846 CAS.
- G. Ma, H. Nguyen and D. Romo, Org. Lett., 2007, 9, 2143–2146 CrossRef CAS PubMed.
- T. Ling, V. R. Macherla, R. R. Manam, K. A. McArthur and B. C. M. Potts, Org. Lett., 2007, 9, 2289–2292 CrossRef CAS PubMed.
- C. J. Hayes, A. E. Sherlock, M. P. Green, C. Wilson, A. J. Blake, M. D. Selby and J. C. Prodger, J. Org. Chem., 2008, 73, 2041–2051 CrossRef CAS PubMed.
- N. P. Mulholland, G. Pattenden and I. A. S. Walters, Org. Biomol. Chem., 2008, 6, 2782–2789 CAS.
- T. Fukuda, K. Sugiyama, S. Arima, Y. Harigaya, T. Nagamitsu and S. Ōmura, Org. Lett., 2008, 10, 4239–4242 CrossRef CAS PubMed.
- K. Takahashi, M. Midori, K. Kawano, J. Ishihara and S. Hatakeyama, Angew. Chem., Int. Ed., 2008, 47, 6244–6246 CrossRef CAS PubMed.
- Y. Sato, H. Fukuda, M. Tomizawa, T. Masaki, M. Shibuya, N. Kanoh and Y. Iwabuchi, Heterocycles, 2010, 81, 2239–2246 CrossRef CAS.
- Y. Kaiya, J.-I. Hasegawa, T. Momose, T. Sato and N. Chida, Chem.–Asian J., 2011, 6, 209–219 CrossRef CAS PubMed.
- N. Satoh, S. Yokoshima and T. Fukuyama, Org. Lett., 2011, 13, 3028–3031 CrossRef CAS PubMed.
- H. Nguyen, G. Ma, T. Gladysheva, T. Fremgen and D. Romo, J. Org. Chem., 2011, 76, 2–12 CrossRef CAS PubMed.
- D. J. Wardrop and E. G. Bowen, Chem. Commun., 2005, 5106–5108 RSC.
- S. Yoshioka, M. Nagatomo and M. Inoue, Org. Lett., 2015, 17, 90–93 CrossRef CAS PubMed.
- J. Kobayashi, F. Kanda, M. Ishibashi and H. Shigemori, J. Org. Chem., 1991, 56, 4574–4576 CrossRef CAS.
- T. Jahn, G. M. König, A. D. Wright, G. Wörheide and J. Reitner, Tetrahedron Lett., 1997, 38, 3883–3884 CrossRef CAS.
- S. Tsukamoto, K. Tane, T. Ohta, S. Matsunaga, N. Fusetani and R. W. M. van Soest, J. Nat. Prod., 2001, 64, 1576–1578 CrossRef CAS.
- K. Namba, T. Shinada, T. Teramoto and Y. Ohfune, J. Am. Chem. Soc., 2000, 122, 10708–10709 CrossRef CAS.
- P. M. Wehn and J. Du Bois, J. Am. Chem. Soc., 2002, 124, 12950–12951 CrossRef CAS PubMed.
- A. Hinman and J. Du Bois, J. Am. Chem. Soc., 2003, 125, 11510–11511 CrossRef CAS PubMed.
- K. Tran, P. J. Lombardi and J. L. Leighton, Org. Lett., 2008, 10, 3165–3167 CrossRef CAS PubMed.
- Y. Ichikawa, K. Okumura, Y. Matsuda, T. Hasegawa, M. Nakamura, A. Fujimoto, T. Masuda, K. Nakano and H. Kotsuki, Org. Biomol. Chem., 2012, 10, 614–622 CAS.
- T. Kano, T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2006, 128, 2174–2175 CrossRef CAS PubMed.
- M. P. Sibi, L. M. Stanley and T. Soeta, Org. Lett., 2007, 9, 1553–1556 CrossRef CAS PubMed.
- Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2006, 128, 3928–3930 CrossRef CAS PubMed.
- J. C. S. Woo and D. B. MacKay, Tetrahedron Lett., 2003, 44, 2881–2883 CrossRef CAS.
- C. Drouin, J. C. S. Woo, D. B. MacKay and R. M. A. Lavigne, Tetrahedron Lett., 2004, 45, 7197–7199 CrossRef CAS.
- J. C. Lanter, H. F. Chen, X. Q. Zhang and Z. H. Sui, Org. Lett., 2005, 7, 5905–5907 CrossRef CAS PubMed.
- T. Shinada, E. Ikebe, K. Oe, K. Namba, M. Kawasaki and Y. Ohfune, Org. Lett., 2007, 9, 1765–1767 CrossRef CAS PubMed.
- T. Shinada, E. Ikebe, K. Oe, K. Namba, M. Kawasaki and Y. Ohfune, Org. Lett., 2010, 12, 2170 CrossRef CAS.
- K. Oe, T. Shinada and Y. Ohfune, Tetrahedron Lett., 2008, 49, 7426–7429 CrossRef CAS.
- K. Sankar, H. Rahman, P. P. Das, E. Bhimireddy, B. Sridhar and D. K. Mohapatra, Org. Lett., 2012, 14, 1082–1085 CrossRef CAS PubMed.
- T. Yoshimura, T. Kinoshita, H. Yoshioka and T. Kawabata, Org. Lett., 2013, 15, 864–867 CrossRef CAS PubMed.
- M. Nagatomo, H. Nishiyama, H. Fujino and M. Inoue, Angew. Chem., Int. Ed., 2015, 54, 1537–1541 CrossRef CAS PubMed.
- Y. Tahara, J. Pharm. Soc. Jpn., 1909, 29, 587–625 Search PubMed.
- Y. Tahara, Biochem. Z., 1911, 30, 255–275 Search PubMed.
- T. Goto, Y. Kishi, S. Takahash and Y. Hirata, Tetrahedron, 1965, 21, 2059–2088 CrossRef CAS PubMed.
- K. Tsuda, O. Amakasu, S. Ikuma, M. Kawamura, C. Tamura, R. Tachikawa and K. Sakai, Chem. Pharm. Bull., 1964, 12, 1357–1374 CrossRef CAS PubMed.
- R. B. Woodward and J. Z. Gougoutas, J. Am. Chem. Soc., 1964, 86, 5030 CrossRef CAS.
- A. Furusaki, Y. Tomiie and I. Nitta, Bull. Chem. Soc. Jpn., 1970, 43, 3325–3331 CrossRef CAS.
- A. Furusaki, Y. Tomiie and I. Nitta, Bull. Chem. Soc. Jpn., 1970, 43, 3332–3341 CrossRef CAS.
- F. A. Fuhrman, Ann. N. Y. Acad. Sci., 1986, 479, 1–14 CrossRef CAS PubMed.
- C. Y. Kao, Ann. N. Y. Acad. Sci., 1986, 479, 52–67 CrossRef CAS PubMed.
- W. Ulbricht, H. H. Wagner and J. Schmidtmayer, Ann. N. Y. Acad. Sci., 1986, 479, 68–83 CrossRef CAS PubMed.
- R. E. Weiss and R. Horn, Ann. N. Y. Acad. Sci., 1986, 479, 152–161 CrossRef CAS PubMed.
- B. K. Krueger, J. F. Worley and R. J. French, Ann. N. Y. Acad. Sci., 1986, 479, 257–268 CrossRef CAS PubMed.
- J. Chau and M. A. Ciufolini, Mar. Drugs, 2011, 9, 2046–2074 CrossRef CAS PubMed.
- Y. Kishi, F. Nakatsubo, M. Aratani, T. Goto, S. Inoue and H. Kakoi, Tetrahedron Lett., 1970, 11, 5129–5132 CrossRef.
- Y. Kishi, F. Nakatsubo, M. Aratani, T. Goto, S. Inoue, H. Kakoi and S. Sugiura, Tetrahedron Lett., 1970, 11, 5127–5128 CrossRef.
- Y. Kishi, M. Aratani, T. Fukuyama, F. Nakatsub, T. Goto, S. Inoue, H. Tanino, S. Sugiura and H. Kakoi, J. Am. Chem. Soc., 1972, 94, 9217–9219 CrossRef CAS PubMed.
- Y. Kishi, T. Fukuyama, M. Aratani, F. Nakatsub, T. Goto, S. Inoue, H. Tanino, S. Sugiura and H. Kakoi, J. Am. Chem. Soc., 1972, 94, 9219–9221 CrossRef CAS PubMed.
- T. Nishikawa, M. Asai, N. Ohyabu, N. Yamamoto, Y. Fukuda and M. Isobe, Tetrahedron, 2001, 57, 3875–3883 CrossRef CAS.
- N. Ohyabu, T. Nishikawa and M. Isobe, J. Am. Chem. Soc., 2003, 125, 8798–8805 CrossRef CAS PubMed.
- T. Nishikawa, D. Urabe and M. Isobe, Angew. Chem., Int. Ed., 2004, 43, 4782–4785 CrossRef CAS PubMed.
- N. Ünver and G. İ. Kaya, Turk. J. Chem., 2005, 29, 547–553 Search PubMed.
- S. Tian, W. Zi and D. Ma, Angew. Chem., Int. Ed., 2012, 51, 10141–10144 CrossRef CAS PubMed.
- Y. Shi, B. Yang, S. Cai and S. Gao, Angew. Chem., Int. Ed., 2014, 53, 9539–9543 CrossRef CAS PubMed.
- B. D. Morris and M. R. Prinsep, J. Nat. Prod., 1999, 62, 688–693 CrossRef CAS PubMed.
- C. C. Hughes and D. Trauner, Angew. Chem., Int. Ed., 2002, 41, 4556–4559 CrossRef CAS.
- K. Sakaguchi, M. Ayabe, Y. Watanabe, T. Okada, K. Kawamura, T. Shinada and Y. Ohfune, Tetrahedron, 2009, 65, 10355–10364 CrossRef CAS.
- K. Chiyoda, J. Shimokawa and T. Fukuyama, Angew. Chem., Int. Ed., 2012, 51, 2505–2508 CrossRef CAS PubMed.
- A. Soheili and U. K. Tambar, Org. Lett., 2013, 15, 5138–5141 CrossRef CAS PubMed.
- M. O'Connor, C. Sun and D. Lee, Angew. Chem., Int. Ed., 2015, 127, 10101–10104 CrossRef.
Footnote |
| † The authors contributed equally to the review. |
| This journal is © The Royal Society of Chemistry 2016 |