
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyurethane foams doped with stable silver nanoparticles as bactericidal and catalytic materials for the effective treatment of water
Berta
Domènech
a,
Kharla
Ziegler
b,
Núria
Vigués
c,
Wojciech
Olszewski
de,
Carlo
Marini
d,
Jordi
Mas
c,
Maria
Muñoz
b,
Dmitri N.
Muraviev
b and
Jorge
Macanás
 *f
*f
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Spain
bDepartment of Chemistry, Universitat Autònoma de Barcelona, Campus UAB, 08193 Bellaterra, Spain
cDepartment of Genetics and Microbiology, Universitat Autònoma de Barcelona, UAB, 08193 Bellaterra, Spain
dALBA Synchrotron Light Source, Ctra. BP1413 Km. 3,3, 08290 Cerdanyola del Vallès, Spain
eFaculty of Physics, University of Bialystok, 1L K. Ciolkowskiego Str., 15-245 Białystok, Poland
fDepartment of Chemical Engineering, Universitat Politècnica de Catalunya, C/Colom 1, 08222, Terrassa, Spain. E-mail: jorge.macanas@upc.edu; Tel: +34 937398239
First published on 19th February 2016
Abstract
The development of reusable dual-purpose nanocomposite foams for catalytic and bactericidal water treatment is reported. Small non-aggregated silver nanoparticles were made using Intermatrix Synthesis inside a polyurethane foam, which was chosen as a suitable polymeric matrix due to its high chemical and mechanical stability and industrial applicability. The antibacterial activity of the obtained nanocomposites was evaluated against suspensions of Gram-negative bacteria (E. coli), showing ideal bactericidal features for being applied to water disinfection. The catalytic activity of nanocomposites was also evaluated through a model reaction carried out under flow conditions. The possibility of reusing the catalytic material was evaluated in 3 consecutive cycles and, for all of them, no significant loss of efficiency was found. Moreover, the leakage of the active species to the media was evaluated under accelerated ageing conditions (3 h in an ultrasonic bath) and a negligible amount of silver was found outside the matrix. The chemical stability of the as-prepared nanoparticles was also evaluated by XANES and any modification in the chemical structure of silver nanoparticles was detected, even after storing the samples for two years under dry conditions.
1. Introduction
About 780 million people in the world have no access to clean and potable water1 and ca. 3.4 million people die each year from water, sanitation, and hygiene-related causes.2 These figures dictate the need for the development of innovative technologies and materials for the production of safe potable water.Thanks to the development of nanomaterials, new approaches are being established in wastewater treatment.3 A paradigmatic example of the use of nanomaterials in sanitation related issues is the development of antibacterial systems that profit from the particular properties of silver nanoparticles (AgNPs or more generally nanosilver) which are supposed to release Ag+,4 even if the exact antibacterial mechanisms are still not completely understood nowadays.5,6 In fact, nanosilver is widely used today because of its antibacterial activity in wound dressings to protect patients with severe burns against infections and in consumer products such as food packaging (not yet allowed in EU), sports textiles or deodorants, where nanosilver reduces undesired odours.7–9
Nonetheless, the main drawback which still limits the wide application of nanomaterials, and thus of nanosilver, is their insufficient stability related to the manipulation of objects on the nanoscale and their high tendency to self-aggregate.10 In this regard, the use of polymer–metal nanocomposites has been demonstrated to be an attractive approach for obtaining and using nanomaterials.10,11 The superior stability of such nanocomposites in terms of low or even negligible release of nanoparticles allows the possibility of reusing the active species without losing efficiency. All the same, it is crucial to adequately choose the polymeric matrix to successfully synthesize such nanocomposites considering some limitations and requirements, since the polymeric matrix should (i) be chemically compatible with the active species; (ii) bear flexible chains able to accommodate the final nanoparticle; (iii) allow the interaction with the nanoparticle or the nanoparticle precursor; and (iv) be insoluble in water but with an appropriate swelling ratio.12
Under such considerations, the use of polyurethane foams (PUFs) could be a good approach since these materials exhibit high stability against chemical degradation, high mechanical durability, good swelling behaviour, ease of separation from a solution, and they are also one of the most cost-effective available polymers.13,14 To date, very few studies have been focused on the development of nanocomposites based on polyurethane matrices.15–23 These publications have demonstrated that the incorporation of nanoobjects (i.e. Metal Nanoparticles, MNPs, or carbon nanotubes) could significantly improve the mechanical or thermal properties of polyurethane materials and their further applicability.18,19 In some of the previous studies, the incorporation of AgNPs into PUFs was achieved either by in situ or ex situ approaches.11,17,20,23,24
Among the existing techniques for the fabrication of nanocomposites,11,25 there is the Intermatrix Synthesis (IMS) approach,12,24 based on the immobilization of nanoparticle precursors (metal ions or complexes) in a polymeric matrix followed by a chemical or electrochemical reduction yielding the formation of the final nanoparticles. IMS allows coping with some of the main drawbacks of nanocomposite preparation such as self-aggregation and Ostwald ripening.26 It is noteworthy to remark that IMS has been originally applied to ion exchange polymeric matrices (fibres, thin films, membranes and resin beads),24 because the ion exchange sites are able to immobilize ionic precursors of the nanoparticles. However, very recently, the successful application of this methodology to polymeric matrices without ionogenic functional groups was also achieved,29 although, further investigation was still necessary in order to get a comprehensive knowledge of the properties and stability of these new nanocomposites.
Thus, this work is focused on the IMS synthesis of AgNPs in polyurethane foams (PUFs), a non-ionic polymeric matrix and, in order to optimize the synthesis, some pre-treatments of the pristine PUF were tested and evaluated with regard to the final amount of nanoparticles and their performance. Besides, even though the nanocomposites described in the related literature20–22 were only considered to be applied for disinfection purposes, other purposes (such as degradation of hazardous compounds or particle filtration) become also crucial when considering a material to be used in water treatment. Accordingly, our goal was to assess a dual-purpose material as a water treatment filter: as an antimicrobial material and as a catalyst for the degradation of hazardous compounds in water. The antibacterial activity was considered against suspensions of the Gram-negative Escherichia coli whereas, in order to evaluate the additional catalytic functionality, a model reaction was carried out under different flow conditions: the reduction of p-nitrophenol (4-NP) to p-aminophenol (4-AP) in the presence of NaBH4 and a metallic catalyst.28,29
Considering that the environmental and health safety risks (sometimes referred to as nanosafety9) due to leaked metal nanoparticles are still unclear,30–33 special attention was paid to the stability of the nanocomposites obtained. In this sense, the leakage of the active species to the media was evaluated during the experiments and under artificial aging conditions and the possibility of reusing catalytic nanocomposites was also assessed in consecutive cycles. Furthermore, by X-ray Absorption Near Edge Spectroscopy (XANES), the chemical modification of AgNPs was researched after applying extreme aging conditions to the nanocomposites and even after 2 years of storage of the samples.
2. Materials and methods
2.1. Materials
Commercial open-cell polyurethane foams (PUFs) with an average density of about 18.5 g dm−3 were obtained from Comercial del Caucho (Spain).For bacterial tests, Escherichia coli (CGSC 5073 K12), Luria-Bertani medium (LB) and phosphate buffered saline (PBS) from Life Technologies were used. HNO3 was purchased from Fisher Chemical, whereas the other reagents (AgNO3, NaBH4, 4-nitrophenol, solvents, acetone, NaOH) were all from Panreac. Deionized water was used for all the experiments.
2.2. Pre-treatment of PUFs
Foams were washed with acetone and then with distilled water to eliminate the possible commercial post-treatments applied to the material. The resulting material was designated as “pristine PUF” and samples were cut out either in a cubic form (1 cm3) or in a disk shape (3 cm diameter, 1 cm width, ca. 7 cm3). Different pre-treatments were applied to these samples by their immersion in 25 mL of a reagent solution (i.e. HNO3 1M, HNO3 3M, NaOH 1 M, and NaOH 3 M) for 2 h, under agitation.27 Afterwards, samples were washed several times with distilled water.2.3. Synthesis of AgNPs
The synthesis of AgNPs in the polymeric matrices using the IMS methodology12,27 consisted of two steps: (1) loading of the material with the metal ions (AgNO3 0.4 M solution) and (2) reduction of metal ions (by using NaBH4 0.5 M solution) to build zero-valent MNPs.2.4. Characterization of nanocomposites
In order to determine the exact metal content in the prepared nanocomposites, two replicates of samples of known weight were digested with concentrated HNO3. The resulting solutions were appropriately diluted and analysed using Inductively Coupled Plasma Mass Spectrometry (ICP-MS, Agilent 7500).High Resolution Transmission Electron Microscopy (HR-TEM) was performed using a Jeol JEM-2011 HR-TEM, and images were used to establish the real distribution of the AgNPs in the matrix, as well as to evaluate the size and shape of the obtained MNPs. Before HR-TEM observation, nanocomposite samples were embedded in an epoxy resin and ultra-thin slices were obtained using an ultramicrotome (Leica EM UC6 with a 35° diamond knife, Diatome).
By using an image processing software (Adobe Professional) it was possible to measure the size of ca. 300 AgNPs per sample and to represent the corresponding size distribution histograms. Histograms were fitted to a three parameter Gaussian curve to find out the AgNP average diameter.
2.5. Evaluation of nanocomposite stability
In order to evaluate the aging of nanocomposites and the eventual leakage of AgNPs, some samples were treated either by long-term immersion in warm water or by ultrasounds, as specified in Table 1. Two replicates of cubic samples of 1 cm3 were deposited in a vessel with 10 mL of distilled water and placed for different times in the corresponding bath: an ultrasonic cleaner (Branson 1200) or an incubator at 37 °C (Aquatron Infors AG, shaker incubator). As ultrasounds can heat the solution, temperature variation in the ultrasonic bath was also monitored during the experiment.| Aging treatment | T (°C) | Solution composition | Duration (h) |
|---|---|---|---|
| Ultrasonic bath | 19–43 | Deionised water | 3 |
| Thermostatic stirred bath | 37 | Deionised water | 24 |
After each experiment, silver contents in both the solution (nAg,solution) and matrix (nAg,matrix) were analysed by ICP-MS so as to determine the silver release percentage (R%), see eqn (1). Matrix samples were digested as aforementioned. Typically, two replicates per time and sample were analysed.
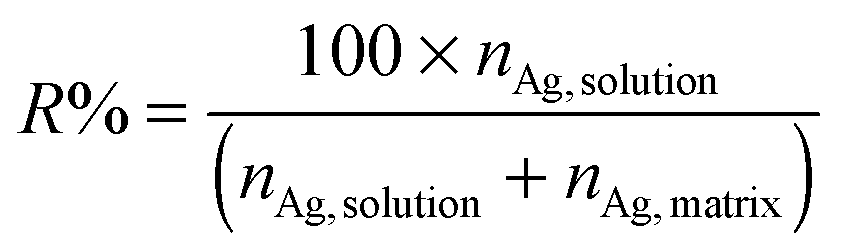 | (1) |
Besides, in order to assess the chemical stability of the AgNPs obtained in terms of chemical speciation,34,35 XANES was performed to freshly prepared samples (PUF–AgNPsnew), samples stored under dry conditions for two years (PUF–AgNPsold), and samples treated with ultrasounds (PUF–AgNPsUS), as specified in Table 2. Additionally, PUFs simply loaded with silver ions (PUF–Ag+) were also evaluated.
| Sample | Treatment | Storage |
|---|---|---|
| PUF–Ag+ | None | ∼1 week in 25 mL AgNO3 0.4 M |
| PUF–AgNPsnew | None | <24 h (before XAS measurements) under dry conditions |
| PUF–AgNPsUS | Ultrasonic bath for 30 min | <24 h (before XAS measurements) in deionised water |
| PUF–AgNPsold | None | 2 years under dry conditions |
XANES spectra were acquired at the Ag K-edge (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 514 eV) at CLAESS, the X-ray absorption and emission spectroscopy beamline at ALBA synchrotron facility (Barcelona, Spain). To do so, four identical samples were compacted in an eppendorf tube so as to reach an amount of sample that provided the minimal absorption signal and the tube was aligned with the beam. The wiggler source was monochromatized using a double crystal Si(311) monochromator cooled by liquid nitrogen. Higher harmonic contribution was rejected using a collimating and a focusing mirror by selecting on both the Rhodium coating using a rejection angle of 2.0 mrad and 0.72 mrad, respectively. This setting corresponds to a beam size of 200 × 200 μm2 at the sample position. The spectra were collected in transmission mode by means of the ionization chambers, filled with appropriate gases. Data normalization and linear combination have been done using the Athena package.36 The linear combination range was −50 to +150 eV around the absorption edge. The goodness of fit has been evaluated by the R-factor.
514 eV) at CLAESS, the X-ray absorption and emission spectroscopy beamline at ALBA synchrotron facility (Barcelona, Spain). To do so, four identical samples were compacted in an eppendorf tube so as to reach an amount of sample that provided the minimal absorption signal and the tube was aligned with the beam. The wiggler source was monochromatized using a double crystal Si(311) monochromator cooled by liquid nitrogen. Higher harmonic contribution was rejected using a collimating and a focusing mirror by selecting on both the Rhodium coating using a rejection angle of 2.0 mrad and 0.72 mrad, respectively. This setting corresponds to a beam size of 200 × 200 μm2 at the sample position. The spectra were collected in transmission mode by means of the ionization chambers, filled with appropriate gases. Data normalization and linear combination have been done using the Athena package.36 The linear combination range was −50 to +150 eV around the absorption edge. The goodness of fit has been evaluated by the R-factor.
2.6. Antimicrobial properties
The procedure to evaluate the bactericidal activity of the Ag-nanocomposites is described elsewhere.37E. coli CGSC 5073 was grown overnight in LB medium at 37 °C. An aliquot of 20 mL of culture was centrifuged for 10 minutes at 10000 × g (Eppendorf 5804R centrifuge). The supernatant liquid was removed and the pellet was suspended again in 10 mL of phosphate buffer saline (PBS) at pH 7.4. The bacterial suspension was then serially diluted down to 107 colony-forming units (CFU) per mL. The initial concentration was measured by plating on LB agar.
To determine the bactericidal properties of the as-prepared nanocomposites and bare matrices, cubic samples were immersed in 20 mL of bacterial suspension and maintained at 37 °C with gentle agitation (160 rpm). Periodically, 100 μL of each suspension were collected at different times and counted using plating and incubation on LB agar at 37 °C.
Besides, in order to evaluate the possible effect of the AgNPs or Ag+ ions released to the medium during the experiments, cubic samples were immersed in 20 mL of PBS and maintained for 24 h at 37 °C with gentle agitation. After this time, samples were removed, and an Escherichia coli pellet was suspended again in the solution. Aliquots of 100 μL were collected at different times and viable microorganisms were counted after cultivation on LB medium. The bactericidal activity of the material was in relation to the positive control (raw materials without NPs). Two replicates per sample were analysed and manipulations were performed under sterile conditions.
2.7. Catalytic activity
The reduction of nitroaromatic compounds in water was evaluated by monitoring a model reaction: the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) by NaBH4.28 Kinetics were followed using a UV-Vis spectrometer (Pharmacia LKB Novaspec II spectrometer, λ = 390 nm). Experiments were carried out as described: a disk-shaped nanocomposite sample was placed in a filter holder connected to a peristaltic pump that forced the test solution (4-NP 0.5 mM and NaBH4 500 mM) to pass through the nanocomposite at different flow rates: 0.4, 1.7, 4.1 and 6.8 mL min−1. Samples were used for up to 3 cycles with fresh solutions to evaluate their possible reuse.The reaction was assumed to follow pseudo-first order kinetics due to the high excess of reducing agent (1000:1) and the normalized absorbance (Abst/Abs0) was plotted versus time, and Abst and Abs0 being the solution absorbance at a defined time (t) and at the beginning of the experiment, respectively.38
3. Results and discussion
3.1. Nanocomposite characterization
After applying the IMS procedure to the PUF samples they turned dark, directly attesting to the successful loading of the AgNPs in the polymeric matrix. A quantitative value of the metal loading was determined using ICP-MS and the results are shown in Table 3.| mg Ag per g matrix | ||
|---|---|---|
| Cubes | Disks | |
| Pristine PUF | 16.7 ± 1.4 | 16 ± 3 |
| HNO3 1 M | 12.7 ± 0.2 | 13.4 ± 1.5 |
| HNO3 3 M | 11.0 ± 0.1 | 10.9 ± 0.3 |
| NaOH 1 M | 19.6 ± 1.0 | 20.0 ± 1.3 |
| NaOH 3 M | 21 ± 2 | 21 ± 5 |
As it can be seen, the total metal amount of the nanocomposites was found in the range of 11 to 21 mg Ag per g matrix. Very slight differences were found depending on the shape of the sample, proving the high reproducibility of the IMS technique, and in both cases a clear influence of the pre-treatment on the metal loading capacity was found. When compared to pristine PUFs, an acid pre-treatment caused a lower metal incorporation (up to 30% of reduction) while PUFs that were immersed in alkaline solutions were able to incorporate a higher metal amount (about 21 mg g−1, 25% of increase with respect to pristine PUFs).
From the results it comes that pre-treatments provide a way for tuning the total metal content in nanocomposites. Nevertheless, this fact is not related to any increase of the number of ion exchange sites since it was previously demonstrated that the use of such pre-treatments did not affect the chemical composition of PUFs as new functional groups were not detected by Infrared Spectroscopy (the obtained spectra for treated samples were identical to those of pristine PUFs).38 Taking this into account, the observed loading differences can be attributed to the possible coordination of Ag+ ions with lone electron pairs of nitrogen atoms of the polymeric structure: when an acid pre-treatment is applied, the nitrogen atom is mostly protonated and the formation of coordination bonds between the nitrogen and the Ag+ is hindered. Therefore, less Ag+ ions remain immobilized in the matrix and less AgNPs can be formed. Conversely, by a basic pre-treatment the number of binding positions is maximized. The same idea of coordination bonds was reported by Jain et al.20 but they incorporated ex situ fabricated AgNPs into PUFs and their reasoning considered the formation of coordination bonds between the nitrogen lone pairs and the pre-formed AgNPs. Consequently, it seems clear that the chemical structure of PUFs plays an important role in both the formation and stabilization of the AgNPs.
It is worth mentioning that by using the IMS approach, the total metal content was up to ten-fold higher than the previously reported values corresponding to similar systems where ex situ formed AgNPs were incorporated into PUFs.18,20,22 Moreover, compared with other synthetic methods,24,25 the MNPs obtained using IMS did not require any stabilizer other than the polymeric matrix itself and it was possible to easily tune the AgNP loading in the nanocomposites by applying simple chemical treatments to the matrix.
Besides, TEM images of the cross-section of samples were taken in order to evaluate the size, morphology and AgNP location, which are crucial parameters to understand the properties of the final nanocomposites. Fig. 1 shows that AgNPs were well dispersed in the matrix, without any clear agglomeration. Moreover, from the size distribution histograms, a narrow size distribution of the nanoparticles was observed, although the average diameter was not the same for all the samples (ranging from 6 to 10 nm).
 | ||
| Fig. 1 TEM images of the cross-section of samples and the corresponding average diameter of AgNPs. | ||
Nanocomposites showed far larger AgNPs on the surface while smaller ones were observed inside the matrix, as it is shown on the right column of Fig. 1 for the nanocomposites made of pristine PUFs. The size of inner particles was in the range of 6 to 10 nm. Still, acid or basic pre-treatments seemed not to dramatically affect the size and distribution of the AgNPs since any well-defined correlation between the treatments applied and the measured average diameter could be observed.
3.2. Stability of the nanocomposites
Since one of the most serious concerns associated with the production and final uses of MNPs in real-life applications deals with the possibility of their uncontrollable release to the medium under treatment,31 in this work it was considered essential to evaluate the stability of the obtained nanocomposites.To determine the eventual loss of AgNPs to the medium, cubic samples underwent different aging treatments (see Table 1) and the amount of metal found in solution (nAg,solution) was compared with the remaining amount in the sample (nAg,matrix). This study was limited to those treatments that involved the highest concentrations of reactants (e.g. HNO3 3 M and NaOH 3 M) and the results were compared with the pristine PUF nanocomposites.
As it can be observed in Fig. 2, after 3 h under ageing conditions provided by sonication, the total Ag release calculated by eqn (1) did not exceed 0.2% for any of the nanocomposites. On the other hand, after immersing the samples for 24 h at 37 °C, the amount of lost Ag was higher but it never exceeded 1% of the total amount of metal in the nanocomposites. It is significant that the basic pre-treatment (NaOH 3 M) allowed a better preservation of the nanoparticles inside the polymeric matrix for both ageing treatments (ultrasonic or thermostatic bath with agitation). Again, this higher stability is in agreement with the Ag+ coordination due to a nitrogen atom lone electron pair, which can retain ionic species as well as AgNPs as suggested previously.20 Accordingly, the basic pre-treatment would be judged as preferable for a final application since it allows a higher loading of AgNPs and provides a stronger stabilization, even if all the release values are very low.
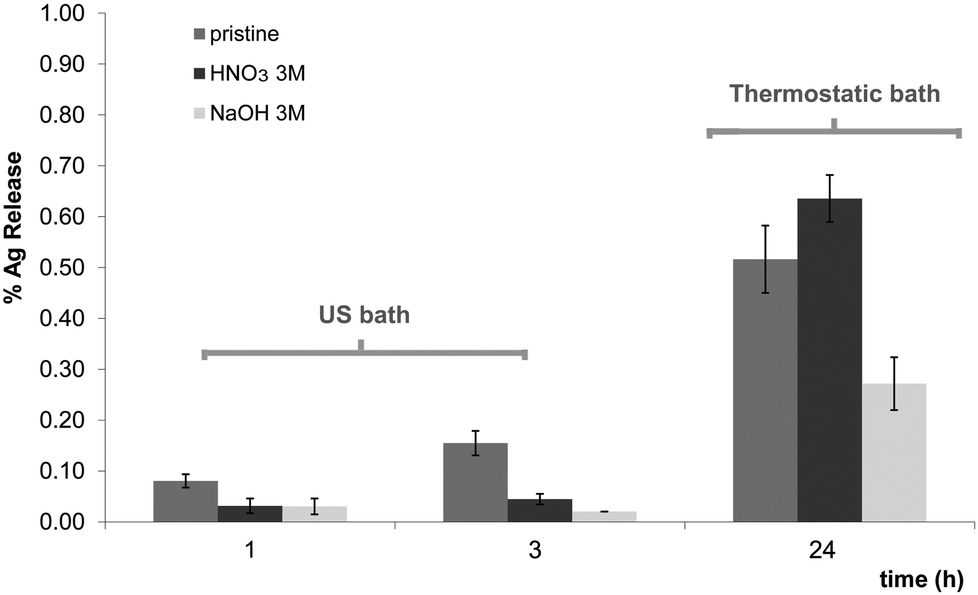 | ||
| Fig. 2 % Ag release in the media. Left: after 1 and 3 h in the ultrasonic bath, and right: after 24 h in a thermostatic bath at 37 °C, 160 rpm. The results are presented with the corresponding standard deviation. | ||
Besides, the influence of the ultrasound ageing treatment on AgNP size distribution was also considered and Table 4 reports the AgNP average diameters measured from TEM images of different samples before and after different ultrasonic bath treatment.
| Sample | 0 h | 3 h |
|---|---|---|
| Pristine | 9.4 ± 0.2 | 4.70 ± 0.03 |
| HNO3 1 M | 6.8 ± 0.1 | 6.4 ± 0.3 |
| HNO3 3 M | 10.4 ± 0.2 | 8.4 ± 0.2 |
| NaOH 1 M | 6.3 ± 0.2 | 6.4 ± 0.1 |
| NaOH 3 M | 9.4 ± 0.1 | 4.9 ± 0.4 |
The experimental results evidenced that after 3 h of ultrasonic treatment, the AgNP average diameter slightly diminished from 6–10 nm to 4.5–6.5 nm except for one of the samples whose tendency was not perfectly consistent (HNO3 3 M), and which underwent only a moderate reduction of the average size (from 10.4 ± 0.2 to 8.4 ± 0.2). Note that samples that originally contained AgNPs of about 6 nm did not dramatically change their size whereas bigger particles (>6 nm) became clearly smaller.
The size change can be attributed to: (i) the loss of the larger unstable AgNPs to the medium; (ii) to their partial and superficial oxidation to Ag+ that migrate to solution or (iii) to the splitting of big AgNPs into smaller ones. In our opinion, the first hypothesis is less likely to happen taking into account the extremely low (<0.2%) reduction of Ag content in the samples after ultrasound treatment. Thus, either nanoparticles break into smaller ones or they are partially oxidized, what would be reasonable since they reduce their size. The released Ag+ could be retained by the matrix functional groups as mentioned before.
Anyhow, the use of ultrasounds can be understood as an interesting post-treatment for the synthesized nanocomposites, useful to remove the unstable AgNPs or to reduce the average diameter of AgNPs, which would result in a greater catalytic surface and, accordingly, in a substantially improved reactivity. Moreover, such post-treatment will somehow guarantee that further AgNP loss to the reacting media will be minimized.
In order to ensure the chemical speciation of silver inside PUFs, synchrotron experiments were carried out comparing the spectra of samples specified in Table 2 with the spectra of pure standards, concretely, metallic silver foil (Ag) and solid inorganic salts such as Ag2O and AgNO3. The obtained results of the XANES data treatment and curve deconvolution (see Fig. 3a) are reported in Fig. 3b for the four nanocomposite samples detailed in the Experimental section. Regarding the goodness of fit, all the R-factor values were lower than 0.001, indicating very good fits for all the samples. Several conclusions can be drawn from these results. From Fig. 3b it becomes clear that some of the silver loaded into the matrix before any reduction (PUF–Ag+ sample) is readily oxidized to Ag2O, up to 34%. Since samples have been loaded with AgNO3, the appearance of this oxidized species could be in fact the result of the contact to atmospheric oxygen. Regarding the rest of the samples, which underwent a chemical reduction step, the first thing to be noticed is that the main species is, for all of them, Ag0. When comparing the distribution of silver species for the aged samples (PUF–AgNPsUS and PUF–AgNPsold), the first thing that becomes clear is that AgNPs in the zero oxidation state also prevail, even after the US treatment of the samples and 2 years of storage, testifying for the great stability of the final nanocomposite obtained. Comparing the Ag0 contribution in PUF–AgNPsUS (82%) and PUF–AgNPsold (76%) samples with newly prepared PUF–AgNPsnew (76%) it is possible to state that no significant differences can be observed. The small increase of Ag0 seen in PUF–AgNPsUS (82%) samples can be ascribed considering the corresponding fitting error (see error bars of each measure in Fig. 3b).
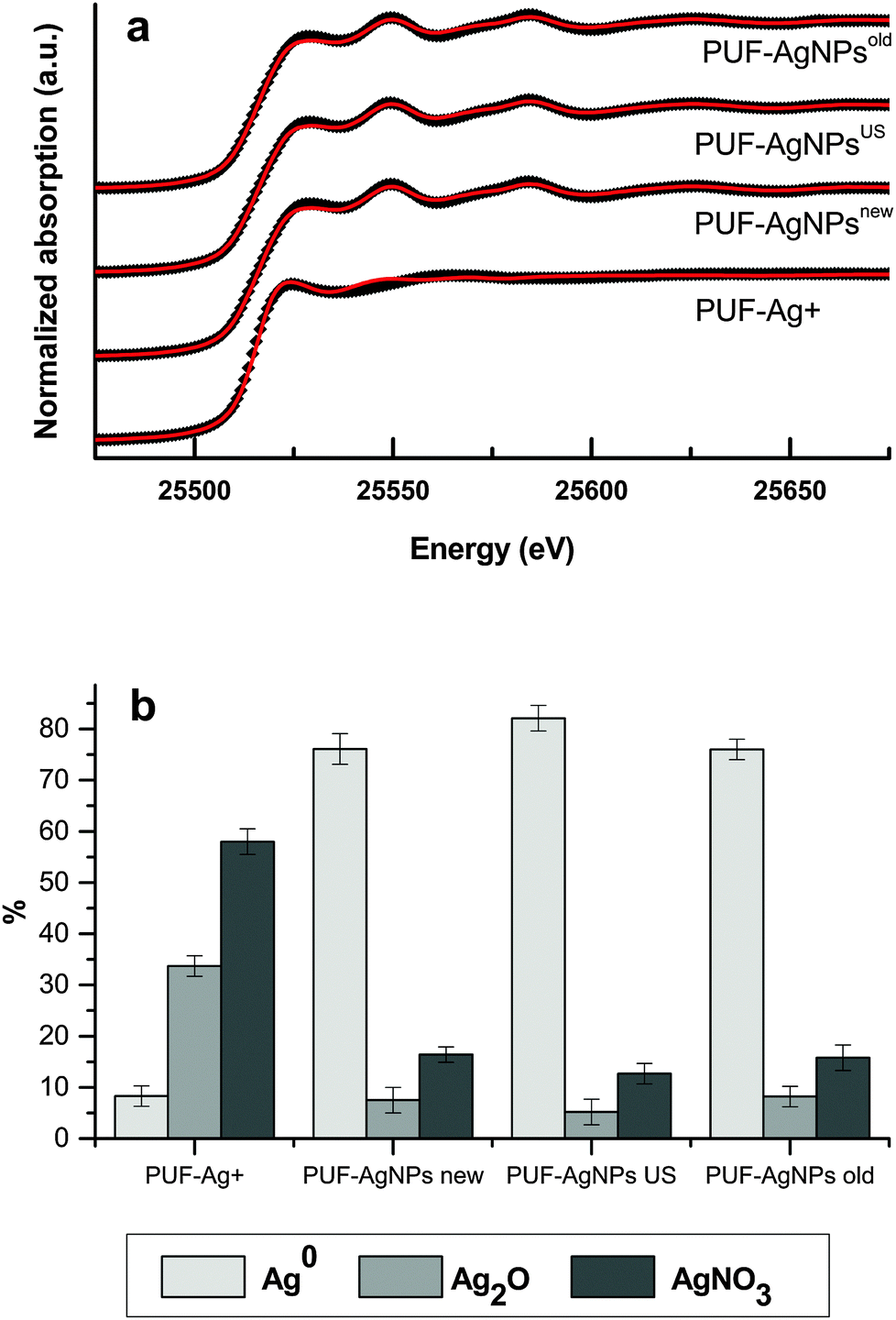 | ||
| Fig. 3 (a) Normalized Ag K-edge XANES spectra of PUF samples (black dots) and corresponding fit (red solid line). (b) Silver speciation in different samples. | ||
Conversely, it is worthy mentioning that, as it happened to unreduced samples, Ag2O is also found in those samples that undergo a chemical reduction although its percentage is much lower (<10%) than for PUF–Ag+ (∼34%). Also, quite unexpectedly, reduced samples contain a non-negligible amount of unreduced Ag+ in the form of AgNO3 (<17%). These facts altogether cause that the amount percentage of Ag0 is not higher than 82% in the reduced samples, far from what would be expected a priori (close to 100%). Having in mind that the reduction of Ag+ is carried out with an excess of NaBH4 and, therefore the reduction of ions should be almost complete, there are several facts that must be considered to understand these results. Firstly, the most likely explanation to the presence of Ag2O species is that Ag0 particles are oxidized in several degrees by contact with water or oxygen from air, after preparation. Secondly, the presence of remaining AgNO3 in reduced samples can be explained due to two factors that may be acting altogether. First, a barrier effect of the matrix could impede that inner Ag+ ions could be totally reduced: note that while Ag+ and NO3− ions can freely diffuse during the loading step, BH4− ions might not be able to reach inner Ag+ because of the formation of zero-valent metal particles. Secondly, taking into account that the deconvolution spectra had been obtained by the limited use of three references (Ag0, AgNO3 and Ag2O), the signal assigned to AgNO3 in reduced samples could be due to other non-reduced forms of silver such as ionic interstitial species inside nanoparticles or insoluble borates (being borate ions the by-products of BH4− decomposition) which have not been considered in the adjustment of the deconvolution spectra.
In reduced samples, the ratio between oxidized species (AgNO3/Ag2O) is maintained close to 1.9, while for PUF–Ag+ it is slightly higher, and equal to 2.2. Since the release of silver species from the matrix was found to be very low (<1%) according to ICP-MS measurements, it is presumable that oxidized species are kept by the matrix in a similar way that Ag+ ions are retained in the loading step. This is a major issue since it demonstrates that even if AgNPs are not perfectly stable their migration to the environment is minimal.
3.3. Antimicrobial properties
To date, there have been few but encouraging attempts to prepare AgNP-containing antimicrobial materials based on PUFs.20–22 Following a procedure described elsewhere37–39 cubic samples (1 cm3) of AgNP-containing nanocomposites made of pristine PUFs were exposed to the cultivation medium. Samples without AgNPs were also evaluated to ascertain any possible effect of the polymeric matrix, such as adsorption of the microorganisms on the matrix. Besides, the antibacterial properties of released silver were also estimated by using a water solution where nanocomposites were immersed for 24 h.The cell viability percentage is plotted versus time in Fig. 4 where it is possible to see that the cell population is not affected by the presence of pristine PUFs without AgNPs. Even if small variations in the number of cells can be observed, they can be attributed to the experimental error of CFU counting or to cell adsorption onto the polymeric matrix. As expected, the percentage of cell viability strongly decreased with time for test tubes where the solution was in contact either with PUF–AgNPs (or Ag2O) or with released AgNP/Ag+. In fact, nanocomposite samples (PUF–AgNPs) were able to kill 100% of bacteria in less than 6.5 h.
 | ||
| Fig. 4 Cell viability versus time. Initial dose: 107 CFU mL−1 in PBS. | ||
From the obtained results it was possible to calculate the mortality rate, expressed as the number of CFU deactivated per second in a single milliliter of the treated solution. For the PUF–AgNPs the calculated value was ca. 1000 CFU mL−1 s−1. Although in previous publications focused on the antibacterial application of PUF–AgNP nanocomposites20–22 this rate is not explicit, it can be calculated from the reported data and it is possible to state that the nanocomposites here-developed showed a mortality rate 3–6 fold higher. For instance, from the data reported by Phong et al., the calculated mortality rate that attained the highest value was about 300 CFU mL−1 s−1.22 This intensification of the bactericidal activity could be first accredited to the higher load of AgNPs found in the nanocomposites obtained using the IMS procedure. However, when comparing the results here presented with other nanocomposites containing Ag prepared using the same synthetic methodology, but in different polymeric matrices (fibers,27,37 membranes,39 and resin beads40) and with higher AgNP loads, the mortality rate of the PUF–AgNPs appeared to be up to 2 orders of magnitude higher. This exceptional result states not only for the synthetic methodology, but also for the suitable usefulness of the polymeric matrix employed.
Moreover, from the results obtained for the Ag released from the sample (after 24 h in 20 mL of a PBS solution under gentle agitation at 37 °C) a second conclusion can be drawn: the little amount of metal released to the media (either AgNPs or Ag2O or Ag+) was enough to kill all the microorganisms in a period as short as 2 h. This means that once the solution is not in contact with the nanocomposite (either because it is missing, deactivated, or exhausted), a high remaining mortality rate is still active provided that the contact between the nanocomposites and the fluid is long enough. Hence, these nanocomposites can be understood as Ag reservoirs with a remaining antibacterial effect and, consequently, they can provide a residual disinfection effect prolonging the possible use of the nanocomposite filter.
3.4. Catalytic activity
Taking into account the proved efficiency of nanosilver for bactericidal processes and looking for an entire evaluation of the nanocomposites, the developed materials were also evaluated as catalytic filters, envisaging an advanced dual-purpose water treatment. Though the use of AgNPs for catalytic applications is not the most frequent application of such nanomaterials, preliminary experimental results concerning the catalytic activity of PUF–AgNP nanocomposites were performed in batch configuration and demonstrated their ability to catalyze the reduction of 4-NP to 4-AP in basic media.27 In that case, no dramatic differences were found for acid or basic pre-treatments even if the latter showed a shorter induction period. However, it was still necessary to evaluate their catalytic performance under flow conditions as well as their possible reutilization.Since the nanocomposite used for these catalytic experiments had to be placed in a circular filter holder, disk-shaped foams were used in this case (see Table 2). The catalytic activity of all samples (nanocomposites made of pristine, NaOH 1 M, NaOH 3 M, HNO3 1 M and HNO3 3 M) was first evaluated at a constant flow rate (1.7 mL min−1) and the corresponding results are shown in Fig. 5a. Plotted data of normalized absorbance at 390 nm (maximum of absorbance of 4-NP) indicated that the catalytic performance was very similar for all the nanocomposites disregarding the endured pre-treatment. This situation is quite controversial since, a priori, one might expect that those nanocomposites with higher silver content (NaOH 1 M or NaOH 3 M) would have performed better. However, as no significant differences were found it is possible to assume that all the samples exceed the minimal amount of catalyst that ensured the catalytic reaction at that flow rate, being this value 11 mg g−1 or lower.
 | ||
| Fig. 5 Catalytic evaluation of the nanocomposites (a) with different pre-treatments and (b) under different flow conditions for the nanocomposite made of pristine PUFs. | ||
The higher porosity of the PUFs along with the AgNP location are beneficial aspects for the catalytic reaction if we compare them with other systems such as Nafion nanocomposites containing AgNPs reported previously.39 Nafion nanocomposites contained a higher amount of catalytic metal (ca. 52 mg g−1) but the dense structure of the perfluorinated matrix combined with the local distribution of AgNPs (note that a lower amount of catalyst was on the surface) played a negative role in the final catalytic application.
From a practical point of view, one can reason that those nanocomposites prepared with PUFs that underwent an acid pre-treatment could be preferable as the amount of precious metal is the lowest (ca. 11 mg g−1) but the catalytic efficiency is the same. Anyhow, in terms of simplicity, nanocomposites prepared using pristine PUFs are even more advantageous as they can be prepared without difficulty and without any additional step. This is why the impact of different flow conditions (0.4, 1.7, 4.1 and 6.8 mL min−1) was evaluated by using exclusively those nanocomposites made of pristine PUFs.
The results presented in Fig. 5b show the variation of the normalised absorbance for such experiments and a remarkable effect can be seen: the higher the flow, the fastest the reaction, albeit the trend was not linear. This effect can be explained because, under higher flow conditions, a higher amount of reactants flow through the nanocomposite for the same period of time and, therefore, the conversion of reactants to products is faster.
To verify the above-mentioned good results regarding AgNP stability, samples were evaluated in 3 different consecutive catalytic cycles of 4-NP reduction. The results for nanocomposites made of pristine PUFs working under different flow conditions are presented in Fig. 6.
 | ||
| Fig. 6 Comparison between catalytic cycles under different flow conditions of the nanocomposite made of pristine PUFs. ■ run#1, ● run#2, and ⋄ run#3. | ||
The effect of flow conditions in each cycle remains the same as before and it is noteworthy that almost no variation in the reaction rates between cycles was observed. The single experiment that was not perfectly consistent was the first run performed at 1.7 mL min−1, which can be attributed to experimental errors.
4. Conclusions
The preparation of silver nanoparticles inside polyurethane foams using Intermatrix Synthesis allowed obtaining small non-aggregated nanoparticles (diameters between 6–10 nm) all over the matrix. The total metal loading as well as the nanoparticle size were tuned by applying simple acid or basic pre-treatments to the polymeric matrix, without affecting the number of ion exchange sites.Nanocomposites were demonstrated to be very stable since the released nanosilver never exceeded 1%, being the more stable samples those that underwent a basic treatment, probably due to the establishment of coordination bonds between the lone electron pairs of the nitrogen atom of the urethane group and either Ag+ or silver nanoparticles.
Synchrotron experiments revealed that the distribution of silver species for aged samples (after ultrasound treatment and after 2 years of storage) was similar to newly prepared samples, testifying for the great stability of the final nanocomposites obtained. Nevertheless, some oxidized species, identified either as AgNO3 or Ag2O, were found in all the samples at different proportions.
Regarding the antibacterial activity, Ag-containing nanocomposites were able to wipe out all of the microorganisms present in solution in quite short times, even shorter than other previous publications using similar nanocomposites. Moreover, the bactericidal effect of small amounts of released silver provides an additional disinfection effect.
As it was meant, the developed nanocomposites also exhibited durable catalytic activity when evaluating the reduction of a nitroaromatic compound in water. It was found that the catalytic efficiency improves when increasing the flow and this trend was confirmed even after 3 consecutive catalytic cycles.
Accordingly, it was possible to achieve the targeted stable dual-function reusable polymer–metal nanocomposites by using a low-cost readily available polymer that can be used for highly effective and low-cost new water treatments.
Acknowledgements
This work was supported by CTQ2014-54553-C3-2-R and CTQ2014-61809-EXP projects from the Ministry of Economy and Competitiveness of Spain (MINECO). Special thanks are due to Servei de Microscòpia from Universitat Autònoma de Barcelona. XANES experiments were performed at the BL22 CLAESS beamline at ALBA Synchrotron with the collaboration of ALBA staff.References
- UNICEF and World Health Organization, Progress on Drinking-Water and Sanitation: 2012 Update, 2012.
- World Health Organization Safer Water, Better Health: Costs, Benefits, and Sustainability of Interventions to Protect and Promote Health, 2008.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- E. T. Hwang, J. H. Lee, Y. J. Chae, Y. S. Kim, B. C. Kim, B.-I. Sang and M. B. Gu, Small, 2008, 4, 746–750 CrossRef CAS PubMed.
- A. Panáček, L. Kvítek, R. Prucek, M. Kolář, R. Večeřová, N. Pizúrová, V. K. Sharma, T. J. Nevěčná and R. Zbořil, J. Phys. Chem. B, 2006, 110, 16248–16253 CrossRef PubMed.
- O. Choi, K. K. Deng, N.-J. Kim, Jr, L. Ross, R. Y. Surampalli and Z. Hu, Water Res., 2008, 42, 3066–3074 CrossRef CAS PubMed.
- S. W. P. Wijnhoven, W. J. G. M. Peijnenburg, C. A. Herbert, W. I. Hagens, A. G. Oomen, E. H. W. Heugens, B. Roszek, J. Bisschops, I. Gosens, D. Van De Meent, S. Dekkers, W. H. De Jong, M. Van Zijverden, A. J. A. M. Sips and R. E. Geertsma, Nanotoxicology, 2009, 3, 109–138 CrossRef CAS.
- P. Hartemann, P. Hoet, A. Proykova, T. Fernandes, A. Baun, W. De Jong, J. Filser, A. Hensten, C. Kneuer, J.-Y. Maillard, H. Norppa, M. Scheringer and S. Wijnhoven, Mater. Today, 2015, 18, 122–123 CrossRef.
- SCENIHR (Scientific Committee on Emerging and Newly Identified Health Risks), Nanosilver: safety, health and environmental effects and role in antimicrobial resistance, 11 June 2014.
- B. A. Rozenberg and R. Tenne, Prog. Polym. Sci., 2008, 33, 40–112 CrossRef CAS.
- Metal–Polymer Nanocomposites, ed. L. Nicolais and G. Carotenuto, John Wiley and Sons Ltd, 2004 Search PubMed.
- D. N. Muraviev, J. Macanás, M. Farre, M. Muñoz and S. Alegret, Sens. Actuators, B, 2006, 118, 408–417 CrossRef CAS.
- Discovering Polyurethanes, ed. K. K. Uhlig, Hanser Publishers, Munich, 1999 Search PubMed.
- H. Ulrich, Urethane Polymers, in Kirk-Othmer Encyclopedia of Chemical Technology, ed. Kirk-Othmer, Wiley, Michigan, 5th edn, 2004 Search PubMed.
- L. Madaleno, R. Pyrz, A. Crosky, L. R. Jensen, J. C. M. Rauhe, V. Dolomanova, A. M. M. V. de Barros Timmons, J. J. Cruz Pinto and J. Norman, Composites, Part A, 2013, 44, 1–7 CrossRef CAS.
- J. G. Han, Y. Q. Xiang and Y. Zhu, J. Inorg. Organomet. Polym. Mater., 2014, 24, 283–290 CrossRef CAS.
- V. Apyari, P. Volkov and S. Dmitrienko, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2012, 3, 1–7 Search PubMed.
- C.-W. Chou, S.-H. Hsu, H. Chang, S.-M. Tseng and H.-R. Lin, Polym. Degrad. Stab., 2006, 91, 1017–1024 CrossRef CAS.
- H. Deka, N. Karak, R. D. Kalita and A. K. Buragohain, Polym. Degrad. Stab., 2010, 95, 1509–1517 CrossRef CAS.
- P. Jain and T. Pradeep, Biotechnol. Bioeng., 2005, 90, 59–63 CrossRef CAS PubMed.
- G. Mulongo, J. Mbabazi, P. Nnamuyomba and S. Hak-Chol, Nanosci. Nanotechnol., 2012, 1, 40–42 CrossRef.
- N. T. P. Phong, N. V. K. Thanh and P. H. Phuong, J. Phys.: Conf. Ser., 2009, 187, 1–8 Search PubMed.
- W. Mackay, US Pat., US 8852639 B2, 2014.
- B. Domènech, J. Bastos-Arrieta, A. Alonso, M. Muñoz, D. N. Muraviev and J. Macanás, Ion Exchange Technologies, ed. A. Kilislioglu, Intech, 2012, pp. 35–72 Search PubMed.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS PubMed.
- S. B. Simonsen, I. Chorkendorff, S. Dahl, M. Skoglundh, J. Sehested and S. Helveg, J. Am. Chem. Soc., 2010, 132, 7968–7975 CrossRef CAS PubMed.
- B. Domènech, K. Ziegler, J. Macanás, F. Carrillo, M. Muñoz and D. N. Muraviev, Nanoscale Res. Lett., 2013, 8, 1–6 CrossRef PubMed.
- J. Macanás, L. Ouyang, M. L. Bruening, M. Muñoz, J.-C. Remigy and J.-F. Lahitte, Catal. Today, 2010, 156, 181–186 CrossRef.
- N. Goswami, M. L. Rahman, M. E. Huque and M. Qaisuddin, J. Chem. Technol. Biotechnol., 1984, 34, 195–202 CrossRef CAS.
- P. J. A. Borm and D. Berube, Nano Today, 2008, 3, 56–59 CrossRef CAS.
- Q. Li, S. Mahendra, D. Y. Lyon, L. Brunet, M. V. Liga, D. Li and P. J. J. Alvarez, Water Res., 2008, 42, 4591–4602 CrossRef CAS PubMed.
- S. F. Hansen and A. Baun, Nat. Nanotechnol., 2012, 7, 409–411 CrossRef CAS PubMed.
- P. V. AshaRani, G. Low Kah Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2008, 3, 279–290 CrossRef PubMed.
- E. Lombi, E. Donner, K. G. Scheckel, R. Sekine, C. Lorenz, N. Von Goetz and F. Nowack, Chemosphere, 2014, 111, 352–358 CrossRef CAS PubMed.
- L. Wang, T. Zhang, P. Li, W. Huang, J. Tang, P. Wang, J. Liu, Q. Yuan, R. Bai, B. Li, K. Zhang, Y. Zhao and C. Chen, ACS Nano, 2015, 9, 6532–6547 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Alonso, N. Vigues, X. Muñoz-Berbel, J. Macanás, M. Muñoz, J. Mas and D. N. Muraviev, Chem. Commun., 2011, 47, 10464–10466 RSC.
- B. Domènech, M. Muñoz, D. N. Muraviev and J. Macanás, Nanoscale Res. Lett., 2011, 6, 1–5 CrossRef PubMed.
- B. Domènech, N. Vigués, J. Mas, M. Muñoz, D. N. Muraviev and J. Macanás, Solvent Extr. Ion Exch., 2013, 32, 301–315 CrossRef.
- A. Alonso, X. Muñoz-Berbel, N. Vigués, R. Rodríguez-Rodríguez, J. Macanás, M. Muñoz, J. Mas and D. N. Muraviev, Adv. Funct. Mater., 2013, 23, 2450–2458 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |