
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC,N-Chelated organotin(IV) azides: synthesis, structure and use within click chemistry†
Petr
Švec
*a,
Karel
Bartoš
b,
Zdeňka
Růžičková
a,
Petra
Cuřínová
c,
Libor
Dušek
d,
Jan
Turek
e,
Frank
De Proft
e and
Aleš
Růžička
a
aDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Cz-532 10, Pardubice, Czech Republic. E-mail: petr.svec2@upce.cz
bHigh School of Chemistry Pardubice, Poděbradská 94, CZ-530 09, Pardubice, Czech Republic
cDepartment of Analytical and Material Chemistry, Institute of Chemical Process Fundamentals, Czech Academy of Sciences, v.v.i, Rozvojová 135/1, Cz-165 02, Prague, Czech Republic
dInstitute of Environmental and Chemical Engineering, Faculty of Chemical Technology, University of Pardubice, Studentská 573, CZ-532 10, Pardubice, Czech Republic
eEenheid Algemene Chemie (ALGC), Member of the QCMM VUB-UGent Alliance Research Group, Vrije Universiteit Brussel, Pleinlaan 2, 1050 Brussels, Belgium
First published on 2nd March 2016
Abstract
A set of tri- and diorganotin(IV) azides bearing 2-(N,N-dimethylaminomethyl)phenyl as a C,N-chelating ligand (LCN) has been prepared and structurally characterized. Triorganotin(IV) azides of the type LCNR2SnN3 (R = n-Bu (1) and Ph (2)) and (LCN)2(n-Bu)SnN3 are monomeric both in solution and in the solid state. The central tin atom in these species is five-coordinated with distorted trigonal bipyramidal geometry. Diorganotin(IV) azides of the type LCNRSn(N3)2 (R = n-Bu and Ph) are monomeric with trigonal bipyramidal geometry around the tin atom as well. Finally, (LCN)2Sn(N3)2 contains a six-coordinated tin atom with heavily distorted octahedral geometry due to the presence of two LCN units. The potential use of selected organotin(IV) azides 1 and 2 as useful building blocks within click chemistry was investigated. The reactions of 1 and 2 with various nitriles resulted in the formation of corresponding triorganotin(IV) tetrazolides (i.e. κ-N1: LCN(n-Bu)2Sn(5-MeCN4), LCNPh2Sn(5-MeCN4), LCN(n-Bu)2Sn(5-Me2NCH2CN4), LCNPh2Sn(5-Me2NCH2CN4); and κ-N2: LCN(n-Bu)2Sn(5-t-BuCN4), LCNPh2Sn(5-t-BuCN4), LCN(n-Bu)2Sn(5-PhCN4), LCNPh2Sn(5-PhCN4)). Similarly, the reaction of 1 and 2 with cyclooctyne provided corresponding C,N-chelated di-n-butyl/diphenyltin(IV) κ-N1 4,5,6,7,8,9-hexahydrocycloocta[d][1,2,3]triazol-1-ides. All azido complexes and products of the [3+2] cycloaddition reactions were characterized by the combination of elemental analysis, mass spectrometry, IR spectroscopy, multinuclear NMR spectroscopy and, in the case of crystalline materials, XRD analysis. In addition, DFT calculations were carried out within the click chemistry reactions in order to corroborate the preferred formation of the respective tetrazolide regioisomer.
1. Introduction
The first organotin(IV) azides were prepared conveniently in the 1960s (i) by the reaction of organotin(IV) chlorides with NaN31 or (ii) by the reaction of organotin(IV) amines with HN3.2 In the solid state, these species have mainly polymeric structures via bridging azide groups. The majority of polymeric organotin(IV) azides (Fig. 1A) reveal a distorted trigonal bipyramidal geometry around the central tin atom (e.g. Me3SnN3,3 Ph3SnN3,4 Bn3SnN35 or (Cy3Sn)2N3(OH)6). Organotin(IV) azides, usually bearing bulky (organic) substituents, behave as monomeric species with tetrahedrally coordinated tin centers as demonstrated, for example, for (t-Bu)3SnN3 and (t-Bu)2Sn(N3)27 or (Neo)3SnN3.8 The presumed polymerization of (organo)tin(IV) azides may also be prevented by the formation of adducts with ligands containing donor atoms (such as O and N) as shown for Ph3SnN3·L (L = HMPA, Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, pyridine, etc.), Sn(N3)4·2py or Sn(N3)4·bipy4 or by the incorporation of chelating ligand(s) within the molecule. The reactivity of some stannylenes (tin(II) complexes) towards Me3SiN3 has been studied from an experimental9 as well as theoretical point of view.9c,10 We have recently described the solution behaviour and solid state structures of two monomeric tin(IV) azides bearing either chelating or bulky bis(trimethylsilyl)amido ligands. These species were obtained by the oxidation of the corresponding stannylene with Me3SiN3.9c
O, pyridine, etc.), Sn(N3)4·2py or Sn(N3)4·bipy4 or by the incorporation of chelating ligand(s) within the molecule. The reactivity of some stannylenes (tin(II) complexes) towards Me3SiN3 has been studied from an experimental9 as well as theoretical point of view.9c,10 We have recently described the solution behaviour and solid state structures of two monomeric tin(IV) azides bearing either chelating or bulky bis(trimethylsilyl)amido ligands. These species were obtained by the oxidation of the corresponding stannylene with Me3SiN3.9c
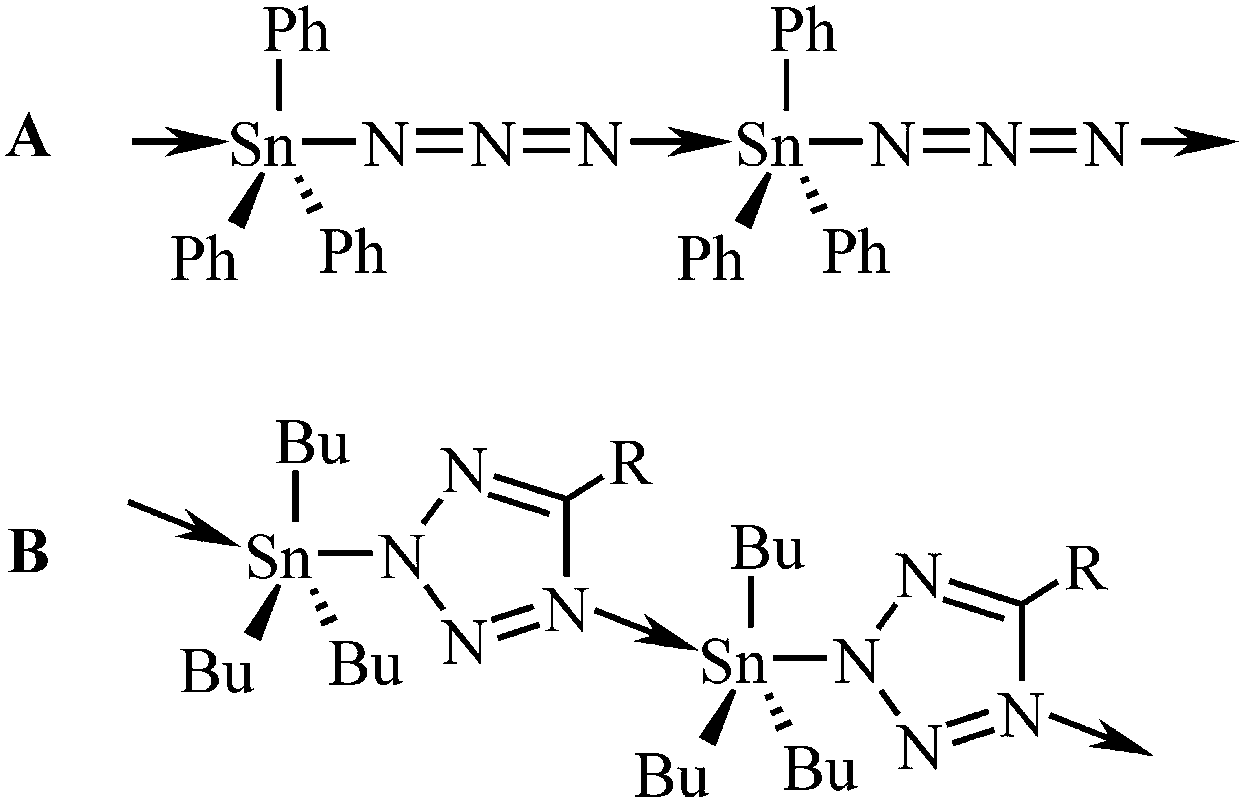 | ||
| Fig. 1 Structural drawings of polymeric triorganotin(IV) azide (A) and tetrazolide (B; Bu = n-Bu, R = (CH2)nSnR3). | ||
In contrast, the family of tin(II) compounds bearing the azide moiety is known to a lesser extent. These species involve, for example, [(Mes)2DAP]SnN3 (DAP = diazapentadienyl),11 [(n-Pr)2ATI]SnN3 (ATI = aminotroponiminate)12 or [HB(3,5-(CF3)2Pz)3]AgSn(N3)[(n-Pr)2ATI] ([HB(3,5-(CF3)2Pz)3] = hydrotris(3,5-bis(trifluoromethyl)pyrazolyl)borate)13 described by Dias and co-workers.
The term “click chemistry” was coined by Sharpless in 200114 and this area of chemistry is undergoing an exponential growth because of its benefits.15 In fact, the well-known 1,3-dipolar azide–alkyne Huisgen cycloaddition can be considered as the first described “click reaction”.16 The aspects of click chemistry reactions have been investigated by theoretical methods as well.17 To the best of our knowledge, the possible use of organotin(IV) azides in the [3+2] cycloaddition reaction with a nitrile was first described by Sisido et al. in 1971. The first N-trialkyltin(IV)-5-substituted tetrazoles were thus prepared.18 This chemistry has been further expanded mainly by Molloy19 who described the formation of oligomers,19c linear polymers19b (Fig. 1B) or layered supramolecular arrays19c constituted of the triorganotin(IV) tetrazolide units.
The incorporation of a C,N-chelating ligand within a class of (organo)tin(IV) compounds gives rise to an intramolecular interaction between tin and nitrogen (i.e. the N→Sn coordination). As a consequence of this effect, these species usually tend to form monomeric molecular structures with mainly five- or six-coordinated tin centers.20,21 Synthesis, structural studies as well as possible applications of a plethora of C,N-chelated organotin(IV) halides and pseudohalides containing the 2-(N,N-dimethylaminomethyl)phenyl ligand have been reported up to now.22–28 In this paper, we report on the expansion of the family of known monomeric tri- and diorganotin(IV) azides and their ability to undergo the [3+2] cycloaddition with nitriles or alkynes.
2. Results and discussion
2.1 Synthesis and general remarks
Six novel C,N-chelated organotin(IV) azides [i.e. LCN(n-Bu)2SnN3 (1), LCNPh2SnN3 (2), (LCN)2(n-Bu)SnN3 (3), LCN(n-Bu)Sn(N3)2 (4), LCNPhSn(N3)2 (5) and (LCN)2Sn(N3)2 (6)] were prepared by the reaction of the corresponding C,N-chelated organotin(IV) halides with an excess of sodium azide in the water/diethyl ether (or water/dichloromethane, respectively) mixture (Scheme 1). Compounds 1–6 do not hydrolyze in moist air and are exceptionally stable species. Thus, for example, the hexane solution of 1 can be irradiated with UV light (253.7 nm) overnight in the air without decomposition. Furthermore, 1 is thermally robust, showing no signs of dinitrogen liberation upon heating up to 300 °C for one hour. On the other hand, these organotin(IV) azides react with hydrochloric acid to give corresponding organotin(IV) chlorides and hydrazoic acid as a volatile by-product. In general, the organotin(IV) pseudohalides bearing n-Bu substituent(s) could be isolated in higher yields (up to 85% in the case of 1), while the presence of the phenyl substituents or the second LCN moiety led to a significant decrease of the isolated yield (e.g. only 51% in the case of 6). A possible explanation is that the species bearing the n-Bu substituent(s) are easier to extract from the heterogeneous reaction mixture. Unfortunately, the attempted synthesis of the proposed C,N-chelated tin(IV) tris(azide) LCNSn(N3)3 resulted in the formation of a mixture of several inseparable tin-containing compounds.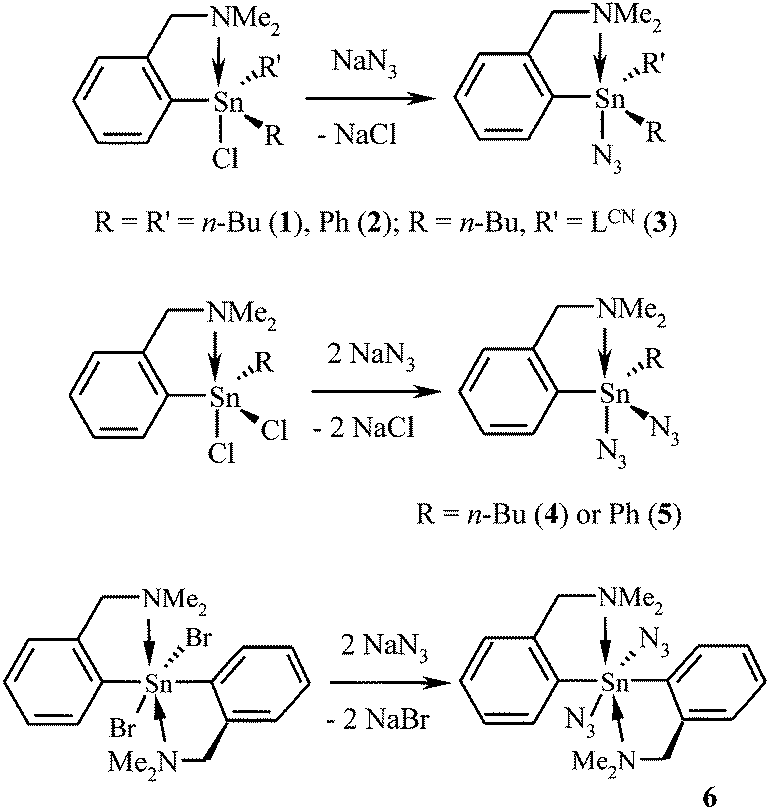 | ||
| Scheme 1 Preparation of C,N-chelated organotin(IV) azides 1–6. Reactions are carried out in a water/diethyl ether (valid for 1, 3 and 4) or a water/dichloromethane (valid for 2, 5 and 6) mixture of solvents. | ||
The reactivity of prepared organotin(IV) azides 1 and 2 towards species containing either the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N or the CC bond (Schemes 2 and 3) has been studied within the scope of click chemistry. It is a well-known fact that the course of a click chemistry reaction can be promoted by addition of a Cu(I) catalyst29 or by heating up the reaction mixture.30 According to our findings, surprisingly, none of the studied click reactions could be promoted by the Cu(I) catalyst (i.e. CuI, a mixture of CuSO4 and sodium ascorbate, and [NHC]CuBr31 were tested for this purpose). Therefore, all click chemistry experiments had to be performed at elevated temperatures, similar to those described in the literature.19 Depending on the boiling point of the reagent employed, either an excess or a strictly stoichiometric amount of organic nitrile or cyclooctyne was used. The corresponding products (i.e. organotin(IV) tetrazolides 7–14 and organotin(IV) triazolides 15–16) were prepared by the [3+2] cycloaddition reactions in essentially quantitative yields and sufficient purity. On the other hand, prolonged reaction times were required (starting from two days in the case of 15 and 16 up to twenty days in the case of 13) when compared with literature reports.19 Such extremely long reaction times were required, for example, for the preparation of 1,4-disubstituted Δ2-tetrazolin-5-ones.32 All reactions, except for the synthesis of 13 and 14, could be carried out in the air. When trying to crystallize species 14 from a chloroform solution, acidolysis (due to the action of the hydrochloric acid present in the solvent) occurred providing several single crystals of the corresponding 5-N,N-dimethylaminomethyl-1H-tetrazole 14a which were characterized by XRD analysis (see Fig. S4 and S5 in the ESI†). LCNPh2SnCl was further identified as a side product by multinuclear NMR spectroscopy in the solution.
N or the CC bond (Schemes 2 and 3) has been studied within the scope of click chemistry. It is a well-known fact that the course of a click chemistry reaction can be promoted by addition of a Cu(I) catalyst29 or by heating up the reaction mixture.30 According to our findings, surprisingly, none of the studied click reactions could be promoted by the Cu(I) catalyst (i.e. CuI, a mixture of CuSO4 and sodium ascorbate, and [NHC]CuBr31 were tested for this purpose). Therefore, all click chemistry experiments had to be performed at elevated temperatures, similar to those described in the literature.19 Depending on the boiling point of the reagent employed, either an excess or a strictly stoichiometric amount of organic nitrile or cyclooctyne was used. The corresponding products (i.e. organotin(IV) tetrazolides 7–14 and organotin(IV) triazolides 15–16) were prepared by the [3+2] cycloaddition reactions in essentially quantitative yields and sufficient purity. On the other hand, prolonged reaction times were required (starting from two days in the case of 15 and 16 up to twenty days in the case of 13) when compared with literature reports.19 Such extremely long reaction times were required, for example, for the preparation of 1,4-disubstituted Δ2-tetrazolin-5-ones.32 All reactions, except for the synthesis of 13 and 14, could be carried out in the air. When trying to crystallize species 14 from a chloroform solution, acidolysis (due to the action of the hydrochloric acid present in the solvent) occurred providing several single crystals of the corresponding 5-N,N-dimethylaminomethyl-1H-tetrazole 14a which were characterized by XRD analysis (see Fig. S4 and S5 in the ESI†). LCNPh2SnCl was further identified as a side product by multinuclear NMR spectroscopy in the solution.
 | ||
| Scheme 2 Preparation of C,N-chelated organotin(IV) tetrazolides 7–14 (for R = n-Bu: ′R = Me (7), t-Bu (9), Ph (11), Me2NCH2 (13); for R = Ph: ′R = Me (8), t-Bu (10), Ph (12), Me2NCH2 (14)). A = C,N-chelated organotin(IV) 5-′R-tetrazol-1-ide regioisomer; B = C,N-chelated organotin(IV) 5-′R-tetrazol-2-ide regioisomer. | ||
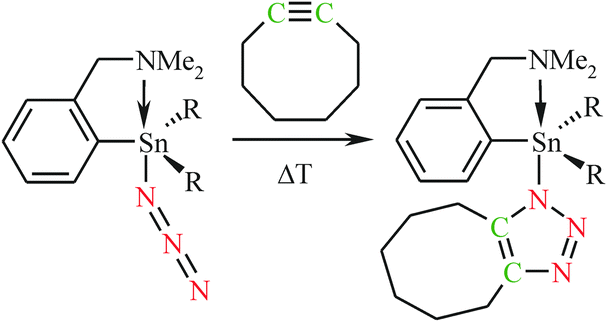 | ||
| Scheme 3 Preparation of C,N-chelated organotin(IV) triazolides 15 (R = n-Bu) and 16 (R = Ph). | ||
As discussed above, cyclooctyne proved to be more reactive in the studied click reactions than organic nitriles. On the other hand, when linear acetylenes (i.e. PhCCPh, MeCCMe and PhCCH, respectively) were used instead of cyclooctyne, the formation of the target organotin(IV) triazolides, even when the reaction mixtures were heated to reflux for one week, was not observed. These reactivity issues were also investigated from a theoretical point of view (vide infra). In principle, the considered click chemistry reactions may result in the formation of a mixture of tetrazolide regioisomers as demonstrated in Scheme 2. Fortunately, exclusive formation of organotin(IV) 5-R-tetrazol-1-ides 7, 8, 13 and 14 was observed when a small (Me) or flexible (Me2NCH2) substituent was bound to the nitrile moiety. As one would expect, organotin(IV) 5-R-tetrazol-2-ide regioisomers 9–12 were formed when nitriles bearing more bulky organic groups (t-Bu and Ph) were employed. These findings were the basis of additional DFT calculations (vide infra).
2.2 Spectroscopic studies
All 1H NMR spectra of 1–6 were recorded in CDCl3 (see Scheme S1, ESI† for the NMR numbering). Furthermore, in some cases (1 and 2), the NMR measurements were performed again in benzene-d6 in order to improve the differentiation of resonances in the aromatic region of the spectra. Both multiplicity and integral intensities of signals are in perfect accordance with presumed structures. The resonances of aromatic protons (H(6)) are accompanied by the characteristic tin satellites with reasonable 117/119Sn–1H coupling constants (see Table S1 and detailed NMR characterization in the ESI†). In principle, diorganotin(IV) bis(azides) exhibit larger coupling constants (3J(119Sn, 1H) values that vary from 84 Hz for 4 to 102 Hz for 6, respectively) than the triorganotin(IV) azides (ranging from 64 Hz for 1 to 73 Hz in case of 2) which is in good agreement with previously published results.22–28,33 The 1H NMR spectrum of 6 bearing two LCN substituents reveals an AX spin pattern at 3.77 and 3.49 ppm (Δδ = 142 Hz, 2J(1H, 1H) = 14 Hz) due to the CH2N fragments which is a typical phenomenon for this family of compounds.24,28The 1H NMR spectra of compounds 7–16 measured in CDCl3 are quite similar to the spectra of the corresponding 1 and 2 exhibiting additional resonances due to the R substituent originating from the organic nitrile or cyclooctyne.
The presence of the desired species was detected in the 13C NMR spectra of 1–16 recorded in CDCl3. All relevant carbon resonances of LCN and n-Bu or Ph substituents are accompanied by tin satellites (see Table S1 and detailed NMR characterization in the ESI†). As discussed above for the 1H NMR spectra, significantly larger 1J(119/117Sn, 13C) coupling constants are observed for the diorganotin(IV) azides 4–6 than for the triorganotin(IV) ones (1–3) which is in accordance with previously published data.22–28,33 Species 7–16 further exhibit resonances of the corresponding alkyl/aryl substituents (Me, Ph, t-Bu and Me2NCH2, respectively) or the C8 rings at reasonable regions of the carbon NMR spectra.
The formation of the respective tetrazolide ring of 7–14 is confirmed by the presence of a resonance found at higher frequencies (ranging from 158.1 ppm for 8 to 172.4 ppm in the case of 10) when compared to the signal of the starting organic nitrile (CN fragment at ca. 110–125 ppm).34 Similarly, a broad signal of the triazolide rings at 144.8 and 144.1 ppm is observed in the case of 15 and 16, respectively, along with the disappearance of the corresponding signal of the starting cyclooctyne (ca. 95 ppm).
The 119Sn NMR spectra of 1–6 measured in CDCl3 exhibit only one resonance significantly shifted to lower frequencies with respect to the starting C,N-chelated organotin(IV) halides (Table S1, ESI†). The same trend was described recently for other structurally related organotin(IV) pseudohalides bearing the C,N-chelating ligand.28 From another point of view and based on previous results,22–28,33 we assume that all organotin(IV) azides 1–6 are monomeric in solution. Owing to the presence of the C,N-chelating ligand(s), the central tin atom in compounds 1–5 is five-coordinate with a distorted trigonal bipyramidal geometry. In spite of the presence of two C,N-chelating ligands in the case of 3, we assume that only one of them is involved in the intramolecular N→Sn coordination and the presumed equilibrium between coordinated and non-coordinated bonding modes is too fast to be seen on the relatively slow NMR time scale. On the other hand, the central tin atom in monomeric 6 is clearly six-coordinate due to the presence of two C,N-chelating ligands that are both involved in the intramolecular N→Sn interaction. This hypothesis is based on the recorded 119Sn chemical shift value (−312.6 ppm). The resonance is shifted highly upfield when compared to the corresponding (LCN)2SnCl2 (δ(119Sn) = −252.8 ppm) or (LCN)2SnBr2 (δ(119Sn) = −271.2 ppm) where the tin atom is also six-coordinated.24 Generally, the coordination number of the tin atom does not increase upon the addition of a potentially coordinating solvent (THF-d8) since the 119Sn NMR chemical shift values differ only by a few ppm when compared to values recorded in CDCl3 (see the ESI,† Experimental part for 1 and 2).
The 119Sn chemical shift values of monomeric 7–16 are shifted more or less to lower frequencies with respect to the corresponding C,N-chelated organotin(IV) azides 1 and 2 (Table S1, ESI†). Based on these values we assume that the central tin atom remains five-coordinated in all cases.
Low temperature NMR measurements of 8 were carried out in order to investigate its potential isomerisation (Scheme 2). Two signals were observed in the 119Sn NMR spectrum (−217.7 and −223.4 ppm) at −40 °C (as the lowest temperature due to solubility reasons) in the mutual ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. Unfortunately, this phenomenon is not visible in the 1H and 13C NMR spectra. This could be connected to the fact that two isomers probably coexist at low temperature, while the stable one is observable as a sole species at room temperature with the 1H and 13C NMR spectra being insensitive to that process.
:10. Unfortunately, this phenomenon is not visible in the 1H and 13C NMR spectra. This could be connected to the fact that two isomers probably coexist at low temperature, while the stable one is observable as a sole species at room temperature with the 1H and 13C NMR spectra being insensitive to that process.
The infrared spectra of novel organotin(IV) azides 1–6 reveal very strong bands in the range of 2050 cm−1 (2) to 2066 cm−1 (4) which are characteristic for the ν(NNN) stretching vibration. These bands diminish upon the formation of the tetrazolide or triazolide ring. All assignments were in good agreement with the literature.35
Similarly to the MS spectral patterns of starting chlorides or related carboxylates,36 where the most intense isotopic clusters correspond to the formation of [M−X]+ (X = halide or carboxy group) particles as a result of the disconnection of the most labile bond, the measured HR ESI-MS spectra of 1–6 usually provide the [M−N3]+ peaks (100% intensity, see the ESI† page S19) with characteristic isotopic tin cluster in the positive ion mode. The [M+Na]+ ions are also observed but with a significantly lower intensity along with other minor particles. The MS spectra of 7, 8, 10 and 11 did not provide relevant structural data about the formation or stability of relevant regioisomers of organotin(IV) tetrazolides.
2.3 Solid state structural studies
The molecular structures of 2, 4, 6, 8, 10, 11 and 14a have been established using single-crystal XRD techniques. Suitable single crystals were obtained from the corresponding chloroform or dichloromethane solution by slow evaporation of the solvent in the air.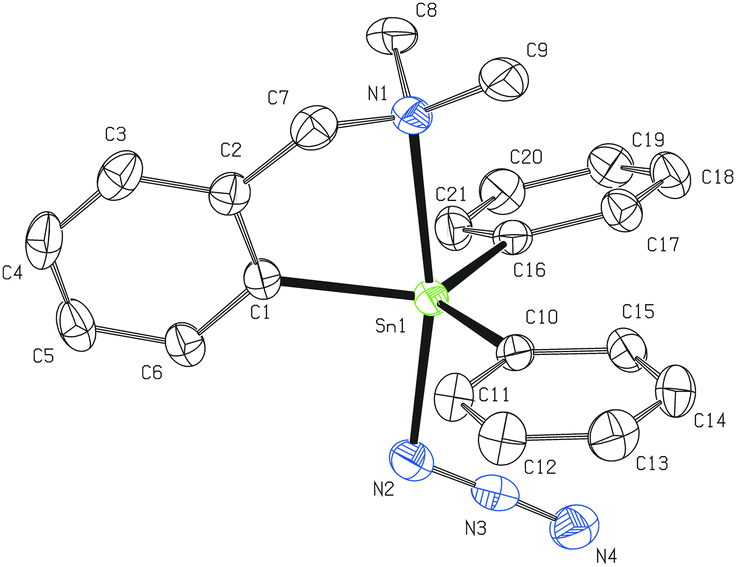 | ||
| Fig. 2 Molecular structure of 2 (ORTEP presentation, 50% probability level). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Sn1–N1 2.4590(19), Sn1–N2 2.2298(19), N2–N3 1.194(3), N3–N4 1.150(3), Sn1–C1 2.1140(19), Sn1–C10 2.1222(19), Sn1–C16 2.1269(19); N1–Sn1–N2 168.03(6), Sn1–N2–N3 111.22(14), N2–N3–N4 178.0(2), C1–Sn1–C10 115.44(7), C1–Sn1–C16 121.46(7), C10–Sn1–C16 122.09(7). | ||
Generally, the bonding in the M–N3 species may be described by two main canonical forms: M–NNN and M–N–NN. In addition, an ionic resonance form M+[N3]− may also contribute to a certain degree to the overall bonding pattern.11,12,38 The structural data of 2 suggest that based on the covalent radii39 of nitrogen atoms there is no dominant canonical form of the Sn-azide moiety (N2–N3 = 1.194(3) Å, N3–N4 = 1.150(3) Å). This bonding pattern is also consistent with the results obtained from the theoretical calculations.12
Two non-equivalent (axial and equatorial) azide moieties can be found in the molecular structure of 4 in which the tin atom is again five-coordinated with heavily distorted trigonal bipyramidal geometry (Fig. 3). Owing to the presence of the two mentioned azide units, the intramolecular N→Sn coordination is even stronger (Sn1–N1 = 2.410(2) Å) than in the case of 2. The N1–Sn1–N2 interatomic angle of 169.83(8)° differs only subtly from the ideal flat angle. The Sn1–N2 bond (2.183(2) Å) is noticeably longer than the Sn1–N5 bond (2.114(2) Å) due to the trans-effect of the donor pendant arm of the C,N-chelating ligand. Again, there is no dominant canonical form of the Sn-azide moiety (N2–N3 = 1.198(3) Å and N3–N4 = 1.147(3) Å; N5–N6 = 1.205(4) Å and N6–N7 = 1.145(4) Å, respectively). Both azide substituents are almost linear (175.6(3)° and 176.3(3)°) and again bent at the nitrogen N2 and N5 (Sn1–N2–N3 = 119.87(19)° and Sn1–N5–N6 = 120.7(2)°, respectively). A detailed investigation of the crystal packing in 4 revealed the presence of intermolecular N→Sn (3.278 Å, ∑rvdW (Sn, N) ≈ 3.9 Å)40 and N⋯H–C (3.285 Å, 122.74°) short contacts resulting from the interaction of the equatorial azide moiety from one molecule with the tin atom and the n-Bu substituent of the second molecule giving rise to an infinite chain (Fig. S1, ESI†).
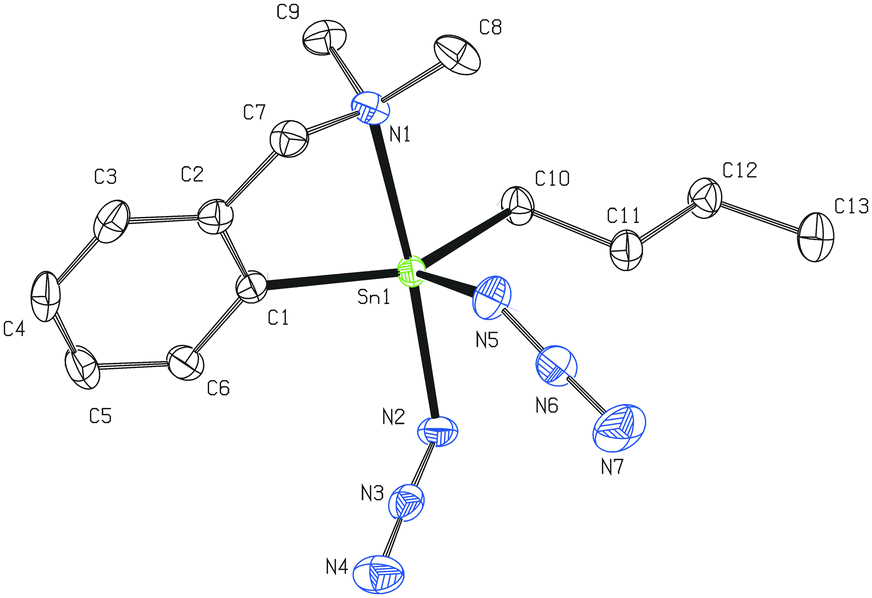 | ||
| Fig. 3 Molecular structure of 4 (ORTEP presentation, 50% probability level). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Sn1–N1 2.410(2), Sn1–N2 2.183(2), Sn1–N5 2.114(2), N2–N3 1.198(3), N3–N4 1.147(3), N5–N6 1.205(4), N6–N7 1.145(4), Sn1–C1 2.116(3), Sn1–C10 2.126(3); N1–Sn1–N2 169.83(8), Sn1–N2–N3 119.87(19), Sn1–N5–N6 120.7(2), N2–N3–N4 176.3(3), N5–N6–N7 175.6(3), C1–Sn1–C10 144.16(10), C1–Sn1–N5 104.74(9), C10–Sn1–N5 108.65(9). | ||
The tin atom in monomeric 6 is octahedrally coordinated as a result of the C,N-chelate bonding of two LCN groups and two cis-bonded azide units (Fig. 4). The C1 and C10 atoms are mutually in trans positions, while the intramolecularly coordinated nitrogen atoms are bonded in a cis fashion. The main deviation from the ideal octahedral geometry is the C1–Sn1–C10 (157.22(11)° vs. ideal 180°) angle. The interatomic distances Sn1–N1 (2.487(3) Å) and Sn1–N2 (2.606(3) Å) mutually differ but are still within the range expected for intramolecular N→Sn interactions. In 6 each nitrogen of the ligand pendant arm is disposed trans to one azide moiety (N1–Sn1–N3 = 164.64(11)° and N2–Sn1–N6 = 169.32(12)°). Both azide units exhibit nearly ideal linear arrangement (N3–N4–N5 = 176.2(4)° and N6–N7–N8 = 175.6(4)°, respectively) and, similarly to the situation described for all previous compounds, the Sn–N3 fragments are bent at nitrogen (Sn1–N3–N4 = 119.0(2) and Sn1–N6–N7 = 120.7(3)°, respectively). No dominant canonical form can be described for the Sn-azide moiety in 6 (N3–N4 = 1.205(5) Å and N4–N5 = 1.140(6) Å; N6–N7 = 1.197(4) Å and N7–N8 = 1.149(5) Å, respectively), too.
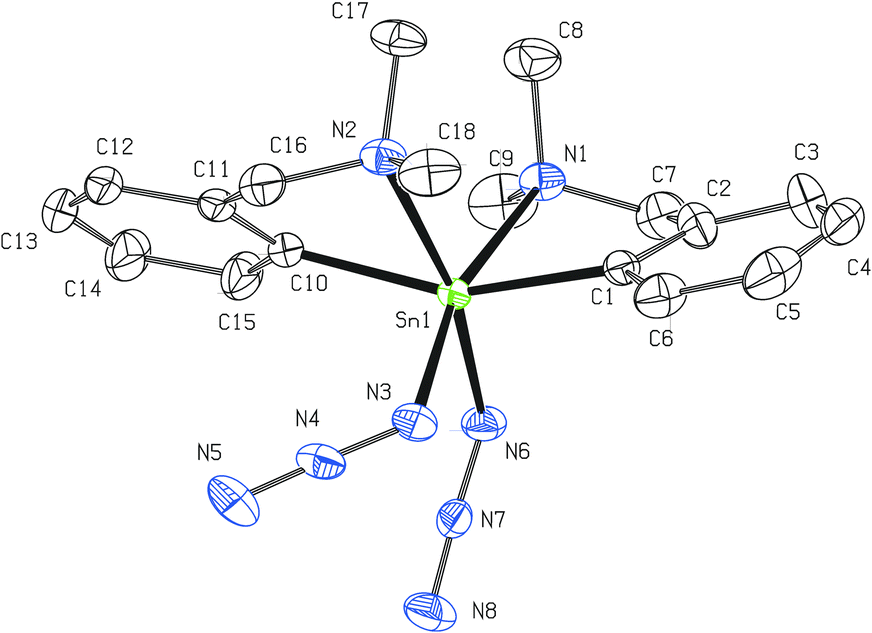 | ||
| Fig. 4 Molecular structure of 6 (ORTEP presentation, 40% probability level). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Sn1–N1 2.487(3), Sn1–N2 2.606(3), Sn1–N3 2.178(3), Sn1–N6 2.165(3), N3–N4 1.205(5), N4–N5 1.140(6), N6–N7 1.197(4), N7–N8 1.149(5), Sn1–C1 2.112(3), Sn1–C10 2.129(4); N1–Sn1–N3 164.64(11), N2–Sn1–N6 169.32(12), Sn1–N3–N4 119.0(2), Sn1–N6–N7 120.7(3), C1–Sn1–C10 157.22(11), N3–N4–N5 176.2(4), N6–N7–N8 175.6(4). | ||
From another point of view, the Sn–N3 interatomic distance in 2 (2.2298(19) Å) is somewhat longer when compared to the values reported for the monomeric Ph3SnN3·HMPA (2.217(6) Å) and polymeric Ph3SnN3 (2.210(3) Å).4a This phenomenon could be simply explained by the strong trans-effect of the pendant arm of the LCN ligand.21 On the other hand, diorganotin(IV) bis(azides) 4 and 6 reveal much shorter tin-azide interatomic distances (up to 2.114(2) Å for 4 and 2.165(3) Å in the case of 6). Such Sn–N3 interatomic distances are comparable with those reported for both monomeric (e.g. (t-Bu)2Sn(N3)2; 2.156(3) and 2.141(2) Å)7 and some polymeric organotin(IV) azides (e.g. Bn3SnN3; 2.16(1) Å).5
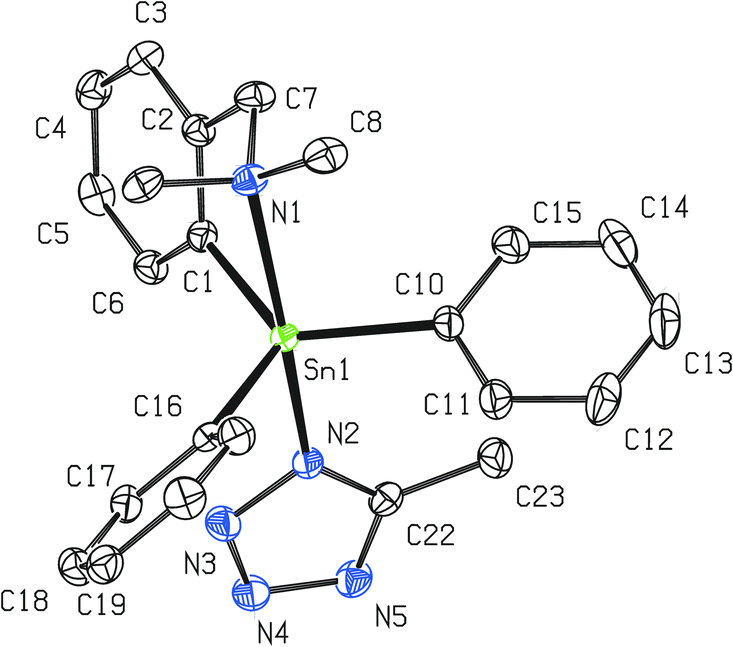 | ||
| Fig. 5 Molecular structure of 8 (ORTEP presentation, 40% probability level). Solvating CHCl3 and water molecules as well as all hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]; only selected parameters of one representative molecule are shown, the rest of them is presented in Fig. S2 (ESI†): Sn1–N1 2.466(5), Sn1–N2 2.263(5), N2–N3 1.361(6), N3–N4 1.302(7), N4–N5 1.358(6), N2–C22 1.344(7), N5–C22 1.315(8), Sn1–C1 2.126(5), Sn1–C10 2.126(5), Sn1–C16 2.132(5); N1–Sn1–N2 169.33(13), Sn1–N2–N3 116.7(3), N2–N3–N4 108.5(4), N3–N4–N5 109.6(4), N4–N5–C22 105.8(4), N2–C22–N5 110.8(4), C22–N2–N3 105.4(4), C1–Sn1–C10 122.89(18), C1–Sn1–C16 123.09(18), C10–Sn1–C16 112.57(19). | ||
The vicinity of the central tin atom with distorted trigonal bipyramidal geometry in 8 is preserved which is in accordance with the 119Sn NMR studies. The average intramolecular N→Sn interaction of ca. 2.46 Å is very strong and comparable with that discussed for 2 (2.459(19) Å). The average axial N–Sn–N interatomic angle of ca. 169.3° is even closer to the ideal 180° than in 2 (168.03(6)). The CN4 tetrazolide ring is planar with a maximum torsion angle of 1° among all atoms. All interatomic distances in the CN4 tetrazolide ring exhibit reasonable values (see the caption of Fig. 5) that were described for such heterocyclic systems.39,41 On the other hand, it is evident that at least partial π-electron delocalization must be considered due to the decrease of differences between formal N–N, NN, C–N and CN bond lengths39,41 or when compared to the interatomic distances reported for other organotin(IV) tetrazolides.19b,c
Contrary to 8, the bulky t-butyl substituent requires the formation of C,N-chelated dialkyl/aryltin(IV) 5-t-butyltetrazol-2-ide regioisomers 9 and 10. The vicinity of the central tin atom in 10 adopts the same geometry as in the case of 8 with a slightly weaker intramolecular N→Sn coordination (2.522(2) Å; Fig. 6). Surprisingly, all nitrogen–nitrogen and carbon–nitrogen interatomic distances in the planar tetrazolide ring are mutually even closer (ranging from 1.321 to 1.345 Å) which suggests a higher degree of π-electron delocalization. Essentially the same trend was found for the molecular structure of 11 (Fig. 7), which bears two n-Bu substituents on the central five-coordinated tin atom. All other interatomic distances and angles lie within the usual range for this class of compounds. In the case of 11, the Ph substituent is nearly co-planar with the N4C ring (maximum torsion angle of 4.8°).
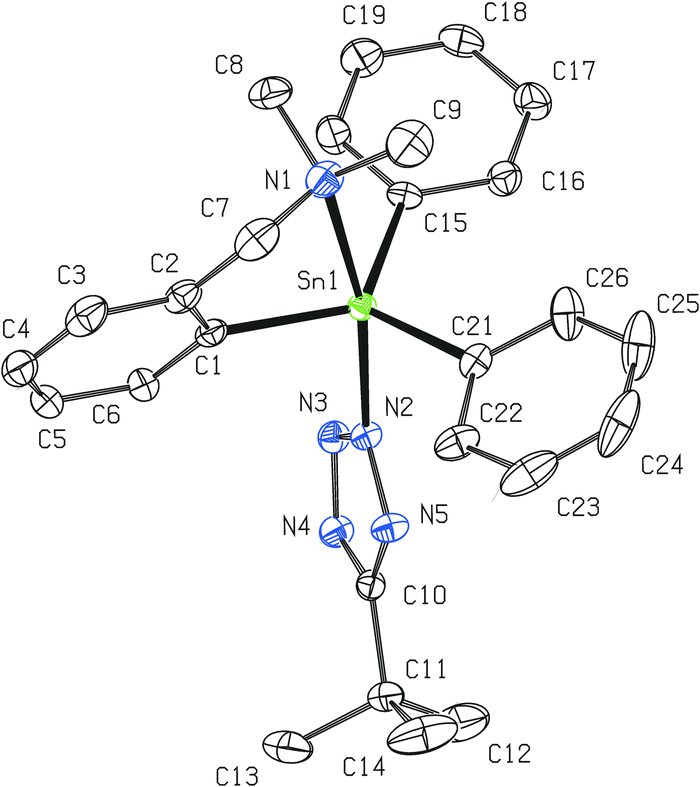 | ||
| Fig. 6 Molecular structure of 10 (ORTEP presentation, 40% probability level). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Sn1–N1 2.522(2), Sn1–N2 2.2218(19), N2–N3 1.321(3), N3–N4 1.331(3), N2–N5 1.343(3), N4–C10 1.345(3), N5–C10 1.329(3), Sn1–C1 2.123(2), Sn1–C15 2.126(2), Sn1–C21 2.119(2); N1–Sn1–N2 167.50(7), Sn1–N2–N3 126.52(15), N2–N3–N4 107.59(18), N3–N2–N5 111.43(18), N3–N4–C10 105.82(18), N2–N5–C10 103.41(18), C1–Sn1–C15 123.10(8), C1–Sn1–C21 117.80(9), C15–Sn1–C21 116.66(9). | ||
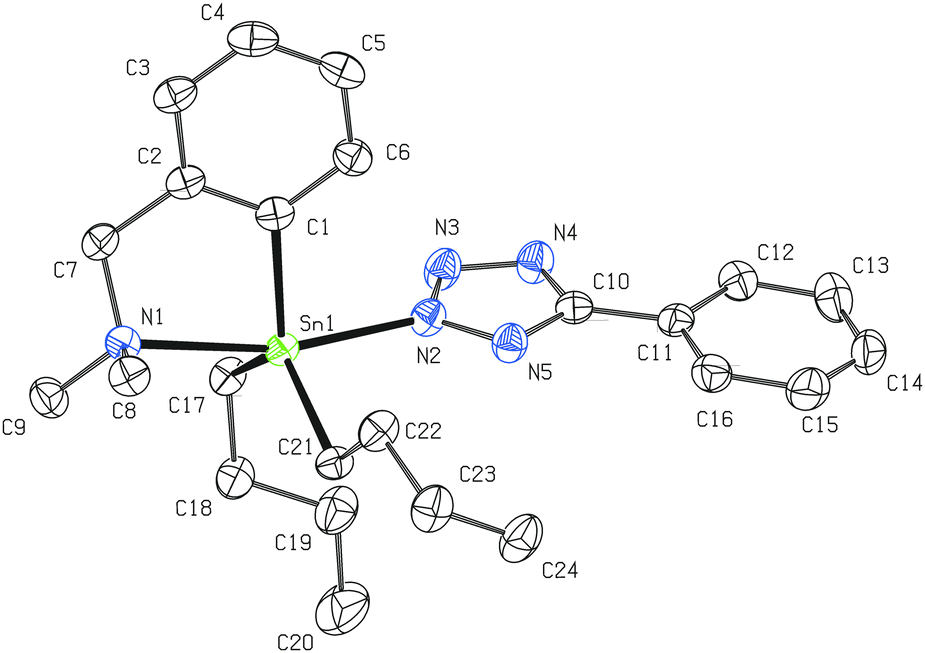 | ||
| Fig. 7 Molecular structure of 11 (ORTEP presentation, 50% probability level). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Sn1–N1 2.480(2), Sn1–N2 2.280(2), N2–N3 1.319(3), N3–N4 1.332(3), N2–N5 1.344(3), N4–C10 1.346(3), N5–C10 1.332(3), Sn1–C1 2.138(2), Sn1–C17 2.144(2), Sn1–C21 2.141(2); N1–Sn1–N2 166.37(7), Sn1–N2–N3 123.29(15), N2–N3–N4 108.21(19), N3–N2–N5 111.22(19), N3–N4–C10 105.20(19), N2–N5–C10 103.21(18), C1–Sn1–C17 126.61(9), C1–Sn1–C21 117.30(8), C17–Sn1–C21 119.85(9). | ||
Finally, several single crystals of free 5-N,N-dimethylaminomethyl-1H-tetrazole 14a (Fig. S4 and S5, ESI†) were obtained upon unintentional acidolysis of 14. It was found that 14a crystallizes as a centrosymmetric dimer due to the H-bonding between the N1 and N5 atoms. In this particular case the mutual differences between interatomic distances are significantly larger than discussed for 10 and 11, respectively.
2.4 Theoretical investigations
The formation of different regioisomers with respect to the substitution pattern of the nitrile reagent in the [3+2] cycloaddition reaction has prompted a theoretical study on selected compounds (8, 10, 11, 14, 15 and 16). The optimized geometries of all complexes (for more details see Fig. S6 and S7, ESI†) are in good agreement with the XRD data with a slightly underestimated intramolecular N→Sn interaction (ca. 7%, for comparison of the theoretical and experimental data see Table S5, ESI†).The DFT calculations confirmed that for the complexes formed in the reaction of organotin(IV) azides with nitriles bearing either a small or a flexible substituent (8 and 14), the organotin(IV) 5-R-tetrazol-1-ide regioisomers are 0.4 (8) and 2.9 (14) kcal mol−1 lower in energy than the organotin(IV) 5-R-tetrazol-2-ide regioisomers. On the other hand, the organotin(IV) 5-R-tetrazol-2-ide regioisomers are predicted to be more stable when synthesized from nitriles with bulkier substituents (t-Bu and Ph for 10 and 11, respectively) which again correlates very well with the experimental findings. The relatively small energy differences between the studied regioisomers suggest that the exclusive formation of only one regioisomer in all studied reactions is affected by the kinetic effects similarly as described in the previous studies.42 Alternatively, the regiospecificity of the studied reactions can be explained on the basis of the solid-state effects resulting in the crystallization of only one regioisomer from a solution where both isomers are present in a very rapid equilibrium.
Since the desired organotin(IV) triazolides were only formed in the reaction with cyclooctyne, the reactions of organotin(IV) azides 1 and 2 with cyclooctyne and substituted acetylenes were examined (the energy and Gibbs free energy differences for all the studied cycloaddition reactions are collected in Table S4, ESI†). A mutual comparison of the ΔG values of the cycloaddition reactions reveals that the reactions of the organotin(IV) azides with the cyclic and acyclic alkynes are significantly more exergonic than the reactions with nitriles. Nevertheless, the previous studies showed that a high activation barrier kinetically hinders the formation of the respective triazolide complexes.17b Therefore, the higher reaction rate of cyclooctyne can be explained by the bond angle deformation to ca. 160°, which is distorted toward the transition state of the cycloaddition reaction, resulting in a dramatic rate acceleration.43
3. Conclusion
We have expanded the family of known C,N-chelated organotin(IV) pseudohalides by the introduction of the azide substituent(s). The possible use of these compounds within the click chemistry (i.e. preparation of various organotin(IV) triazolides and tetrazolides) has been successfully authenticated. In addition, the C,N-chelated triorganotin(IV) azides 1 and 2 may be utilized as mild azidation reagents in the synthetic chemistry. Furthermore, C,N-chelated organotin(IV) azides may be used for bioorthogonal labeling of biologically active species. Finally, tri- and tetrazolides can serve as a source of heterocycles by simple protonolysis or via conversion with E–Cl bond(s) analogously to our fluorination agents described earlier.44All mentioned species are monomeric both in solution and in the solid state. Theoretical calculations proved the regiospecificity of the formation of the respective tetrazolides as well as a higher activation barrier of the reactions of azides with acetylenes.
4. Experimental section
4.1 General remarks
All reactions were carried out in the air unless stated otherwise. All reagents and solvents were obtained from commercial sources (Sigma-Aldrich). Caution! Sodium azide is extremely toxic. Handle with care! LCN(n-Bu)2SnCl,22 LCNPh2SnCl,23,25b (LCN)2(n-Bu)SnCl,22 LCN(n-Bu)SnCl2,22 LCNPhSnCl2,24 (LCN)2SnBr224 and cyclooctyne45 were prepared according to published procedures.4.2 NMR spectroscopy
The NMR spectra were recorded from solutions in C6D6, THF-d8 and CDCl3 on a Bruker Ascend™ 500 spectrometer (equipped with a Z-gradient 5 mm Prodigy cryoprobe) at frequencies 1H (500.13 MHz), 13C{1H} (125.76 MHz) and 119Sn{1H} (186.50 MHz) at 295 K. The solutions were obtained by dissolving approximately 40–60 mg of each compound in 0.6 mL of deuterated solvent. The values of 1H chemical shifts were calibrated to residual signals of CDCl3 (δ(1H) = 7.27 ppm), C6D6 (δ(1H) = 7.16 ppm) or THF-d8 (δ(1H) = 3.58 ppm). The values of 13C chemical shifts were calibrated to signals of CDCl3 (δ(13C) = 77.2 ppm), C6D6 (δ(13C) = 128.39 ppm) or THF-d8 (δ(13C) = 67.57 ppm). The 119Sn chemical shift values are referenced to external neat tetramethyl stannane (δ(119Sn) = 0.0 ppm). Positive chemical shift values denote shifts to the higher frequencies relative to the standards.4.3 Crystallography
The X-ray data (Tables S2 and S3, ESI†) for colourless crystals of 2, 4, 6, 8, 10, 11 and 14a were obtained at 150 K using an Oxford Cryostream low-temperature device on a Nonius KappaCCD diffractometer with MoKα radiation (λ = 0.71073 Å), a graphite monochromator in ϕ and χ scan modes. Data reductions were performed using DENZO-SMN.46 The absorption was corrected by integration methods.47 Structures were solved by direct methods (Sir92)48 and refined by full matrix least-square based on F2 (SHELXL-97).49 Hydrogen atoms were mostly localized on a difference Fourier map, however to ensure uniformity of the treatment of the crystal, all hydrogen atoms were recalculated into idealized positions (riding model) and assigned temperature factors Hiso(H) = 1.2Ueq (pivot atom) or of 1.5Ueq for the methyl moiety with C–H = 0.96, 0.97, and 0.93 Å for methyl, methylene and hydrogen atoms in aromatic rings, respectively, and 0.82 Å for N–H groups. The dynamic disorder of the co-crystallized solvent (CHCl3) in the structure of 8 was treated by standard methods.494.4 ESI MS analysis
The MS spectra were measured on a Bruker MicrOTOF-QIII apparatus using the ESI source in the positive mode. The measurement parameters were adjusted as follows: the capillary voltage was 4200 V and the end plate offset was −500 V. The collision cell RF was 350 Vpp. The nebulizer gas (N2) pressure was 1.6 bar and the flow of drying gas (N2, heated to 180 °C) was 8 l min−1. Scans of MS spectra were carried out in the mass range of m/z 80–1550. For the HRMS measurements, the calibration on sodium formate clusters was used. The samples were delivered by direct infusion using a syringe pump (Kd-Scientific, KDS-100-CE, 0.5 mL Hamilton syringe, flow rate 180 μL min−1) coupled to the MicrOTOF-QIII mass spectrometer. For the MS/MS measurements, the precursor ions were isolated by the collision cell energy of 2 eV in the window of 2u and the product ions were obtained with collision cell energy increasing from 15 to 40 eV, depending on the compound studied.4.5 IR spectroscopy
The IR spectra of neat samples were recorded on a Bruker Vector 22 FT-IR spectrometer using the ATR method at ambient temperature.4.6 DFT calculations
All calculations were carried out using Density Functional Theory (DFT) as implemented in the Gaussian09 quantum chemistry program.50 Geometry optimizations were carried out at the M06/cc-pVDZ(cc-pVDZ-pp for Sn) level of theory and the energies of the optimized structures were re-evaluated by additional single point calculations on each of all optimized geometries using the triple-ζ-quality cc-pVTZ(-pp) basis set.51 Analytical vibrational frequencies within the harmonic approximation were computed using the cc-pVDZ basis set to confirm appropriate convergence to well-defined minima or saddle points on the potential energy surface. The Gibbs free energies Gsolv(cc-pVTZ) used to calculate the energy differences reported in this article (Table S4, ESI†) have been computed with the following protocol:| Gsolv(cc-pVTZ) = G(cc-pVTZ) + SC | (1) |
| G(cc-pVTZ) = E(cc-pVTZ) + TC | (2) |
| TC = G(cc-pVDZ) − E(cc-pVDZ) | (3) |
| SC = Esolv(cc-pVDZ) − E(cc-pVDZ) | (4) |
4.7 Synthesis
The general procedure for the synthesis of 1–16 is described. For details concerning both synthesis and spectroscopic characterization of all compounds see the ESI.†Method A. Starting C,N-chelated organotin(IV) azide 1 or 2 was heated to reflux for the required period of time with an excess of organic nitrile in toluene giving pure 7–14 after evaporation of all volatiles in vacuo. Essentially quantitative yields of the desired products were obtained.
Method B. Starting C,N-chelated organotin(IV) azide 1 or 2 and a stoichiometric amount of organic nitrile or cyclooctyne were sealed under vacuum in the NMR tube in benzene-d6. Afterwards, the NMR tube was heated to 100 °C for a required period of time to achieve a complete conversion towards the desired products (11–16) of the [3+2] cycloaddition. Quantitative yields of the respective products were obtained.
Acknowledgements
The authors would like to thank the Czech Science Foundation for the financial support of this work (project P207/12/0223). J. T. would like to acknowledge the financial support of FWO, under the scheme FWO Pegasus Marie Curie fellowship (Project No. 12T6615N). F. D. P. wishes to acknowledge the Free University of Brussels (VUB) and the Research Foundation Flanders (FWO) for continuous support to his group.References
- (a) J. G. A. Luijten, M. J. Janssen and G. J. M. van der Kerk, Recl. Trav. Chim. Pays-Bas, 1962, 81, 202 CrossRef CAS; (b) J. S. Thayer and R. West, Inorg. Chem., 1964, 3, 406 CrossRef CAS.
- J. Lorberth, H. Krapf and H. Noth, Chem. Ber., 1967, 100, 3511 CrossRef CAS.
- R. Allmann, R. Hohlfeld, A. Waskowska and J. Lorberth, J. Organomet. Chem., 1980, 192, 353 CrossRef CAS.
- (a) I. Wharf, R. Wojtowski, C. Bowes, A.-M. Lebius and M. Onyszchuk, Can. J. Chem., 1998, 76, 1827 CrossRef CAS; (b) R. Campbell, M. F. Davis, M. Fazakerley and P. Portius, Chem. – Eur. J., 2015, 21, 1 CrossRef.
- A. C. Burke, M. F. Mahon and K. C. Molloy, Appl. Organomet. Chem., 2003, 17, 735 CrossRef CAS.
- R. A. Howie, J. L. Wardell and S. M. S. V. Wardell, Appl. Organomet. Chem., 2005, 19, 356 CrossRef CAS.
- D. Hänssgen, M. Jansen, C. Leben and T. Oster, J. Organomet. Chem., 1995, 494, 223 CrossRef.
- J. A. S. Bomfim, C. A. L. Filgueiras, R. A. Howie, J. N. Low, J. M. S. Skakle, J. L. Wardell and S. M. S. V. Wardell, Polyhedron, 2002, 21, 1667 CrossRef CAS.
- (a) A. M. Khmaruk and A. M. Pinchuk, Zh. Org. Khim., 1983, 19, 883 CAS; (b) P. B. Hitchcock, M. F. Lappert, A. V. Protchenko and P. G. H. Uiterweerd, Dalton Trans., 2009, 353 RSC; (c) P. Švec, Z. Padělková, M. Alonso, F. De Proft and A. Růžička, Can. J. Chem., 2014, 92, 434 CrossRef.
- M. S. Nechaev, THEOCHEM, 2008, 862, 49 CrossRef CAS.
- A. E. Ayers, T. M. Klapötke and H. V. R Dias, Inorg. Chem., 2001, 40, 1000 CrossRef CAS.
- A. E. Ayers, D. S. Marynick and H. V. R. Dias, Inorg. Chem., 2000, 39, 4147 CrossRef CAS PubMed.
- H. V. R. Dias and A. E. Ayers, Polyhedron, 2002, 21, 611 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- For example see: (a) M. G. Finn and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1231 RSC; (b) R. A. Evans, Aust. J. Chem., 2007, 60, 384 CrossRef CAS; (c) P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338 RSC; (d) Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262 RSC; (e) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS PubMed; (f) Z. P. Demko and K. B. Sharpless, Org. Lett., 2001, 3, 4091 CrossRef CAS PubMed.
- (a) R. Huisgen, Proc. Chem. Soc., London, 1961, 357 Search PubMed; (b) R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565 CrossRef.
- (a) C. G. Gordon, J. L. Mackey, J. C. Jewett, E. M. Sletten, K. N. Houk and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 9199 CrossRef CAS PubMed; (b) F. Schoenebeck, D. H. Ess, G. O. Jones and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 8121 CrossRef CAS PubMed.
- K. Sisido, K. Nabika and T. Isida, J. Organomet. Chem., 1971, 33, 337 CrossRef CAS.
- (a) M. F. Mahon, K. C. Molloy and P. C. Waterfield, J. Organomet. Chem., 1989, 361, C5 CrossRef CAS; (b) S. J. Blunden, M. F. Mahon, K. C. Molloy and P. C. Waterfield, J. Chem. Soc., Dalton Trans., 1994, 2135 RSC; (c) M. Hill, M. F. Mahon and K. C. Molloy, J. Chem. Soc., Dalton Trans., 1996, 1857 RSC; (d) M. Hill, M. F. Mahon, J. McGinley and K. C. Molloy, J. Chem. Soc., Dalton Trans., 1996, 835 RSC; (e) A. Goodger, M. Hill, M. F. Mahon, J. McGinley and K. C. Molloy, J. Chem. Soc., Dalton Trans., 1996, 847 RSC.
- (a) G. van Koten, C. A. Schaap and J. G. Noltes, J. Organomet. Chem., 1975, 99, 157 CrossRef CAS; (b) G. van Koten, J. G. Noltes and A. L. Spek, J. Organomet. Chem., 1976, 118, 183 CrossRef CAS; (c) G. van Koten, J. T. B. H. Jastrzebski, J. G. Noltes, A. L. Spek and J. C. Schoone, J. Organomet. Chem., 1978, 148, 233 CrossRef CAS.
- J. T. B. H. Jastrzebski and G. van Koten, Adv. Organomet. Chem., 1993, 35, 241 CrossRef CAS.
- A. Růžička, V. Pejchal, J. Holeček, A. Lyčka and K. Jacob, Collect. Czech. Chem. Commun., 1998, 63, 977 CrossRef.
- A. Růžička, R. Jambor, J. Brus, I. Císařová and J. Holeček, Inorg. Chim. Acta, 2001, 323, 163 CrossRef.
- P. Novák, Z. Padělková, L. Kolářová, I. Císařová, A. Růžička and J. Holeček, Appl. Organomet. Chem., 2005, 19, 1101 CrossRef.
- (a) R. A. Varga, A. Rotar, M. Schurmann, K. Jurkschat and C. Silvestru, Eur. J. Inorg. Chem., 2006, 1475 CrossRef CAS; (b) R. A. Varga, M. Schurmann and C. Silvestru, J. Organomet. Chem., 2001, 623, 161 CrossRef CAS; (c) A. Rotar, R. A. Varga, K. Jurkschat and C. Silvestru, J. Organomet. Chem., 2009, 694, 1385 CrossRef CAS; (d) R. A. Varga, K. Jurkschat and C. Silvestru, Eur. J. Inorg. Chem., 2008, 708 CrossRef CAS.
- (a) P. Novák, J. Brus, I. Císařová, A. Růžička and J. Holeček, J. Fluorine Chem., 2005, 126, 1531 CrossRef; (b) P. Novák, Z. Padělková, I. Císařová, L. Kolářová, A. Růžička and J. Holeček, Appl. Organomet. Chem., 2006, 20, 226 CrossRef.
- C. Coza, A. Stegarescu, R. Şuteu and A. Silvestru, J. Organomet. Chem., 2015, 777, 71 CrossRef CAS.
- P. Švec, Z. Růžičková, P. Vlasák, J. Turek, F. De Proft and A. Růžička, J. Organomet. Chem., 2016, 801, 14 CrossRef.
- (a) L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Sci. Adv., 2015, 1, e1500304 Search PubMed; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- (a) R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, 1984, pp. 1–176 Search PubMed; (b) K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS PubMed.
- J. Turek, I. Panov, P. Švec, Z. Růžičková and A. Růžička, Dalton Trans., 2014, 43, 15465 RSC.
- J.-M. Vandensavel, G. Smets and G. L'abbé, J. Org. Chem., 1973, 38, 675 CrossRef CAS.
- (a) P. Švec, Z. Padělková, P. Štěpnička, A. Růžička and J. Holeček, J. Organomet. Chem., 2011, 696, 1809 CrossRef; (b) P. Švec, E. Černošková, Z. Padělková, A. Růžička and J. Holeček, J. Organomet. Chem., 2010, 695, 2475 CrossRef; (c) P. Švec, Z. Padělková, A. Růžička, T. Weidlich, L. Dušek and L. Plasseraud, J. Organomet. Chem., 2011, 696, 676 CrossRef; (d) P. Švec, E. Černošková, Z. Padělková, A. Růžička and J. Holeček, J. Organomet. Chem., 2010, 695, 2475 CrossRef; (e) P. Švec, R. Olejník, Z. Padělková, A. Růžička and L. Plasseraud, J. Organomet. Chem., 2012, 708, 82 CrossRef.
- (a) C. J. Pouchert and J. Behnke, The Aldrich Library of 13C and 1H FT NMR Spectra, Aldrich Chemical Company, Inc., USA, 1993, vol. 1 Search PubMed; (b) H. J. Reich, online NMR database accessible free of charge at http://www.chem.wisc.edu/areas/organic/index-chem.htm.
- (a) G. Socrates, Infrared Characteristic Group Frequencies, John Wiley & Sons, Chichester, UK, 1980 Search PubMed; (b) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, New York, USA, 3rd edn, 1978 Search PubMed.
- (a) M. Holčapek, L. Kolářová, A. Růžička, R. Jambor and P. Jandera, Anal. Chem., 2006, 78, 4210 CrossRef PubMed; (b) L. Kolářová, M. Holčapek, R. Jambor, L. Dostál, M. Nádvorník and A. Růžička, J. Mass Spectrom., 2004, 39, 621 CrossRef PubMed.
- H. A. Bent, J. Chem. Phys., 1959, 33, 1258 CrossRef.
- V. N. Khrustalev, I. A. Portnyagin, N. N. Zemlyansky, I. V. Borisova, Y. A. Ustynyuk and M. Y. Antipin, J. Organomet. Chem., 2005, 690, 1056 CrossRef CAS.
- (a) P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186 CrossRef PubMed; (b) P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770 CrossRef PubMed.
- (a) A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS; (b) B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210 CrossRef CAS PubMed.
- (a) R. Turner, A. D. Jarrett, P. Goebel and B. J. Mallon, J. Am. Chem. Soc., 1972, 95, 790 CrossRef; (b) K. Shea and J. S. Kim, J. Am. Chem. Soc., 1992, 114, 4846 CrossRef CAS.
- (a) P. Švec and A. Růžička, Main Group Met. Chem., 2011, 34, 7 CrossRef; (b) P. Švec, A. Eisner, L. Kolářová, T. Weidlich, V. Pejchal and A. Růžička, Tetrahedron Lett., 2008, 49, 6320 CrossRef.
- L. Brandsma and H. D. Verkruijsse, Synthesis, 1978, 290 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CAS.
- P. Coppens, in Crystallographic Computing, ed. F. R. Ahmed, S. R. Hall and C. P. Huber, Copenhagen, Munksgaard, Denmark, 1970, pp. 255–270 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (b) T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007 CrossRef; (c) K. A. Peterson, J. Chem. Phys., 2003, 119, 11099 CrossRef CAS.
- (a) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed; (b) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All synthetic details, selected NMR and crystallographic parameters and additional figures of molecular structures as well as the computational details with optimized structures. CCDC 1432629–1432635 numbers for compounds 2, 4, 6, 8, 10, 11 and 14a. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj03187g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |