
Poly(amic acid) salt-stabilized silver nanoparticles as efficient and recyclable quasi-homogeneous catalysts for the aqueous hydration of nitriles to amides†
Jun
Li
,
Guannan
Tang
,
Yuchen
Wang
,
Ya
Wang
,
Zixi
Li
and
Hengfeng
Li
*
School of Materials Science and Engineering, Central South University, Changsha, 410083 China. E-mail: lihf@csu.edu.cn; Fax: +86-731-88877692; Tel: +86-731-88877793
First published on 27th October 2015
Abstract
Water-soluble silver nanoparticles stabilized by poly(amic acid) salt (Ag–PAAS) were synthesized by a one-pot method and were characterized by using UV-vis absorption, transmission electron microscopy, powder X-ray diffraction, and ζ potential measurements. The Ag–PAAS catalyzed selective hydration of nitriles to amides in water allowed for not only highly efficient quasi-homogeneous catalytic reactions under mild conditions, but also an easy recovery and reuse of the catalyst attributed to the pH response of the PAAS.
1 Introduction
The significance of the hydration of nitriles to the corresponding amides under ecologically friendly conditions is undoubted in our current social environment.1 Nitrile hydration reactions are conventionally catalysed by concentrated acids or bases, which usually cause over-hydrolysis of amides to undesirable carboxylic acids and the formation of a massive amount of salts after the neutralization of the catalysts.1,2 Metal-mediated hydration of nitriles is a more promising alternative, especially owing to its better selectivity toward amides.2,3 A number of homogeneous transition-metal complexes have been reported for activating the hydration of nitriles.4–10 However, they usually suffered from various drawbacks, especially difficulties in the recovery and reuse of the catalyst, as well as the high cost of ligands and the special handling of sensitive complexes.4,5,7,9,10 In contrast, heterogeneous transition-metal nanocatalysts display many advantages such as high activity, and easy handling and recovery.2,11–14 To date, many heterogeneous catalytic systems have been found to be effective in the selective hydration of nitriles; however, they mainly comprised expensive noble metals (Ru, Pd, Au, etc.).12,15–18 From an economic point of view, developing efficient and inexpensive metal catalysts is still inciting chemists and materials scientists. Recently an increasing number of inexpensive and heterogeneous catalysts, such as MnO2, CeO2, Co3O4 and Ni nanoparticles, have been developed and have exhibited noticeable catalytic activity in the hydration of nitriles.18–22Owing to their relatively low cost and easy synthesis, Ag nanoparticles (Ag NPs) have been widely used as efficient catalysts for the selective oxidation of olefins and in the reduction of many organic dyes and nitroaromatic compounds.13,23–26 Since the first report on the hydration of nitriles to amides catalyzed by Ag NPs was published by Mitsudome et al.,2 Ag NPs have attracted more and more attention in the catalytic hydration of nitriles. Unfortunately, most of the reported Ag-based catalysts required high reaction temperature (>140 °C) and even an inert reaction atmosphere to function.2,11,13,14,27–29 Sherbow et al. found that quasi-homogeneous Ag NPs stabilized by the 1,3,5-triaza-7-phosphaadamantane were active for the hydration of nitriles at 90 °C, but the extremely low Ag loading and long reaction time (hundreds of hours) severely restrict their further development.27 Ag NP-grafted imine network materials showed high performance for the hydration of nitriles at 90–100 °C, though the materials preparation was somewhat tedious and time-consuming.30,31
Nowadays, a novel protocol based on “smart” nanocatalysts has been receiving significant attention, where quasi-homogeneous NPs are loaded on a stimuli-responsive polymer that endows the NPs with facile recovery and reusability by simple environmental changes, such as pH or temperature.32,33 The combination of the efficiency of a homogeneous catalyst and the durability of a heterogeneous catalyst might be one of the most exciting challenges in the search for more efficient catalytic systems. In this paper, we present a one-pot synthesis of Ag NPs stabilized by the pH-responsive poly(amic acid) salt (Ag–PAAS). The Ag–PAAS was found to be highly active for the catalytic hydration of various nitriles in an aqueous medium and was easily recycled after the reaction by its pH response. To the best of our knowledge, this is the first report on the use of recyclable and quasi-homogeneous Ag NPs for the catalytic hydration of nitriles in water.
2 Experimental
2.1 Materials and methods
Silver acetate (AgAc) was purchased from Aldrich with a purity of 99.99%. Nitrile compounds (from J&K Scientific) were used without further purification. Sodium borohydride (NaBH4), triethylamine (TEA), N,N-dimethylformamide (DMF), 4,4′-oxydianiline (ODA), 3,3′,4,4′-benzophenonetetracarboxylic dianhydride (BTDA) and any other reagents were obtained from Sinopharm Chemical Reagent Co., Ltd. All reagents were of analytical grade or better.UV-vis spectra were recorded on a Shimadzu UV-vis spectrophotometer (model UV-2450) at a scanning speed of 1 nm s−1. The concentration of the Ag–PAAS solution used for UV-vis absorption measurements was 0.3 mM. The morphology and size of the silver nanoparticles were characterized on a TITAN transmission electron microscope (TEM, model G2 60-300). The test samples were made by placing drops of Ag–PAAS solution on a carbon-coated copper grid and drying them at room temperature. Powder X-ray diffraction (XRD) patterns were collected using a Japan Rigaku X-Ray diffractometer (D/max 2500/PC). The XRD sample (denoted as Ag-PAA) was prepared by precipitating the Ag NPs in an acidic solution (pH ∼ 2), then washing the precipitate with acetone and acidic water several times, and finally drying it in a vacuum at room temperature. ζ potentials of the Ag NPs were measured on a Malvern Zetasizer nano S. The concentration of the Ag–PAAS solution used for ζ potential measurements was identical to that used in the UV-vis absorption measurements. Gas chromatography (GC-FID) was carried out on a Shimadzu gas chromatograph (GC-2010) equipped with a 30 m capillary column using N2 as the carrier. 1H NMR spectra were recorded on a Bruker nuclear magnetic resonance spectrometer (Avance III, 400 MHz). Inductively coupled plasma (ICP-OES) was recorded on a Thermo Fisher inductively coupled plasma optical emission spectrometer (iCAP 6300).
2.2 Synthesis of Ag–PAAS
First, water-soluble BTDA/ODA-based PAAS was synthesized by adding two molar equivalents of TEA to neutralize poly(amic acid) (PAA) obtained through polycondensation of ODA and BTDA in DMF (15 wt% solid, 1.65 dL g−1 inherent viscosity at 25 °C). Subsequently, an aqueous solution of AgAc (50 mM, 10 mL) was mixed with PAAS solution (10 mM based on PAA monomer, 25 mL) under 1000 rpm stirring in an ice-water bath. NaBH4 (50 mM, 15 mL) was then rapidly injected into the solution, affording a deep brown solution. The mixture was stirred for another 30 min to ensure the complete reduction of silver ions. The obtained Ag–PAAS solution was stored in a refrigerator at 4 °C for further use.2.3 General procedure for the catalytic reaction
In a typical experiment, the nitrile (1 mmol) and the Ag–PAAS solution (3 mL, 10 mM) were added to a Teflon-sealed tube and vigorously stirred at the desired temperature for a certain period of time under air. The course of the reaction was monitored by regularly taking samples of ∼30 μL, which, after extraction with ethyl acetate (3 × 2 mL), were analyzed by GC. In most cases, after the completion of the reaction, clean and extensive crystals of the corresponding amide were obtained by cooling the mixture at low temperature, which were separated by direct decantation and analyzed by 1H NMR. For those amides that did not precipitate, the product was extracted with ethyl acetate (3 × 2 mL) and dried over anhydrous MgSO4. The combined organic layers were then concentrated by rotary evaporation and the resulting residue was analyzed by 1H NMR. In some cases additional purification by column chromatography was required (silica gel with CH2Cl2 as the eluent).2.4 Recycling of the Ag–PAAS catalyst
In a typical experiment, pyrazinecarbonitrile (1 mmol) and the Ag–PAAS solution (3 mL, 10 mM) were introduced into a Teflon-sealed tube, and the reaction mixture was vigorously stirred at 90 °C for 1 h. After cooling at room temperature, drops of 1 M acetic acid were added until the solution reached pH ∼ 2, causing the Ag–PAAS catalyst to precipitate. The precipitate was then separated by filtration and washed with ethyl acetate prior to reuse. After that, the solid catalyst was mixed with fresh water (3 mL), and the pH of the mixture was adjusted to 10.0 with triethylamine, resulting in complete redispersion of the Ag–PAAS precipitate. Finally, the fresh nitrile was added and the tube was heated to 90 °C for the next hydration cycle.3 Results and discussion
Ag NPs were in situ formed in an ice-cold PAAS aqueous solution by the reduction of silver acetate with sodium borohydride (Scheme 1). First, Ag ions were loaded into poly(amic acid) (PAA) chains in aqueous solution by an ion-exchange reaction between the tertiary ammonium (TEA)+ and Ag ions to generate self-assembled PAA–Ag(I) complexes.34 Silver polycarboxylate thus formed was then converted into zero-valent Ag by fast reduction using sodium borohydride, which then nucleated and grew into Ag NPs. With the aid of powerful cation-complexing properties and a considerable steric hindrance effect of the PAA molecules,35,36 the ensuing Ag NPs are unexpectedly stable without any precipitation even after six months' storage under ambient conditions. Moreover, the preparation method as described above is simple and suitable for the scale-up production up to hundreds of milliliters at a time.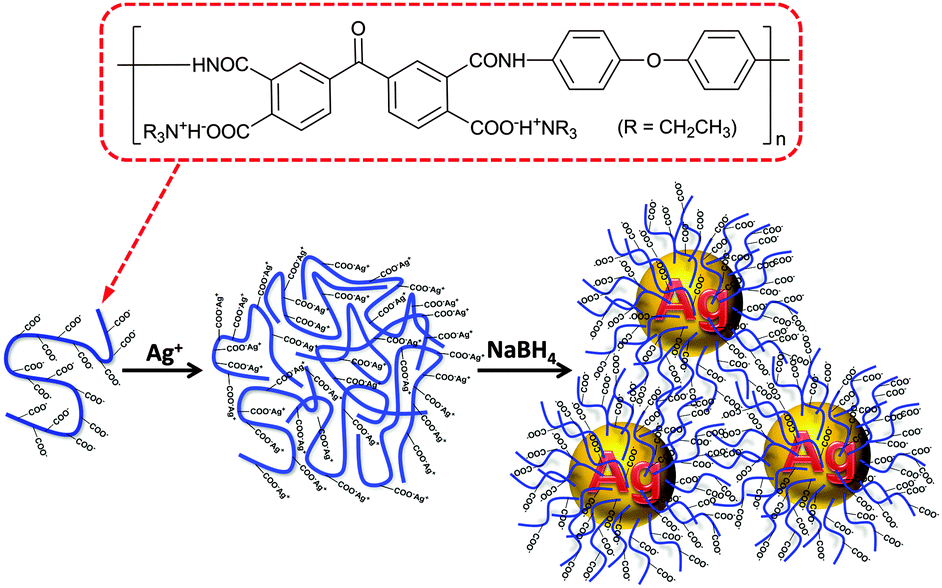 | ||
| Scheme 1 Schematic diagram for the synthesis of PAAS-stabilized Ag NPs. | ||
3.1 Characterization of the Ag–PAAS material
The TEM image (Fig. 1) shows that the spherical Ag NPs are highly dispersed in the PAAS solution with a diameter of 4.8 ± 1.5 nm. The high-resolution TEM image shows that the Ag NPs have well-defined crystalline planes with an interplanar d spacing of 0.24 nm, corresponding to the d111 (0.2359 nm) plane for face-centered cubic (fcc) silver.37 The broadening XRD pattern in Fig. 2 further evidences the existence of Ag nanocrystals, and the average crystallite size of the Ag NPs was calculated to be 5.6 nm using the Scherrer equation,37,38 which is in good agreement with the TEM results. It also implies here that the Ag NPs were well stabilized throughout the whole processing either by PAAS in the alkaline solution-phase or by PAA in the acid solid-phase, which is quite favorable for the following recyclable catalytic applications. | ||
| Fig. 1 (A) TEM image, (B) size distribution and (C) HRTEM image of PAAS-stabilized Ag NPs. | ||
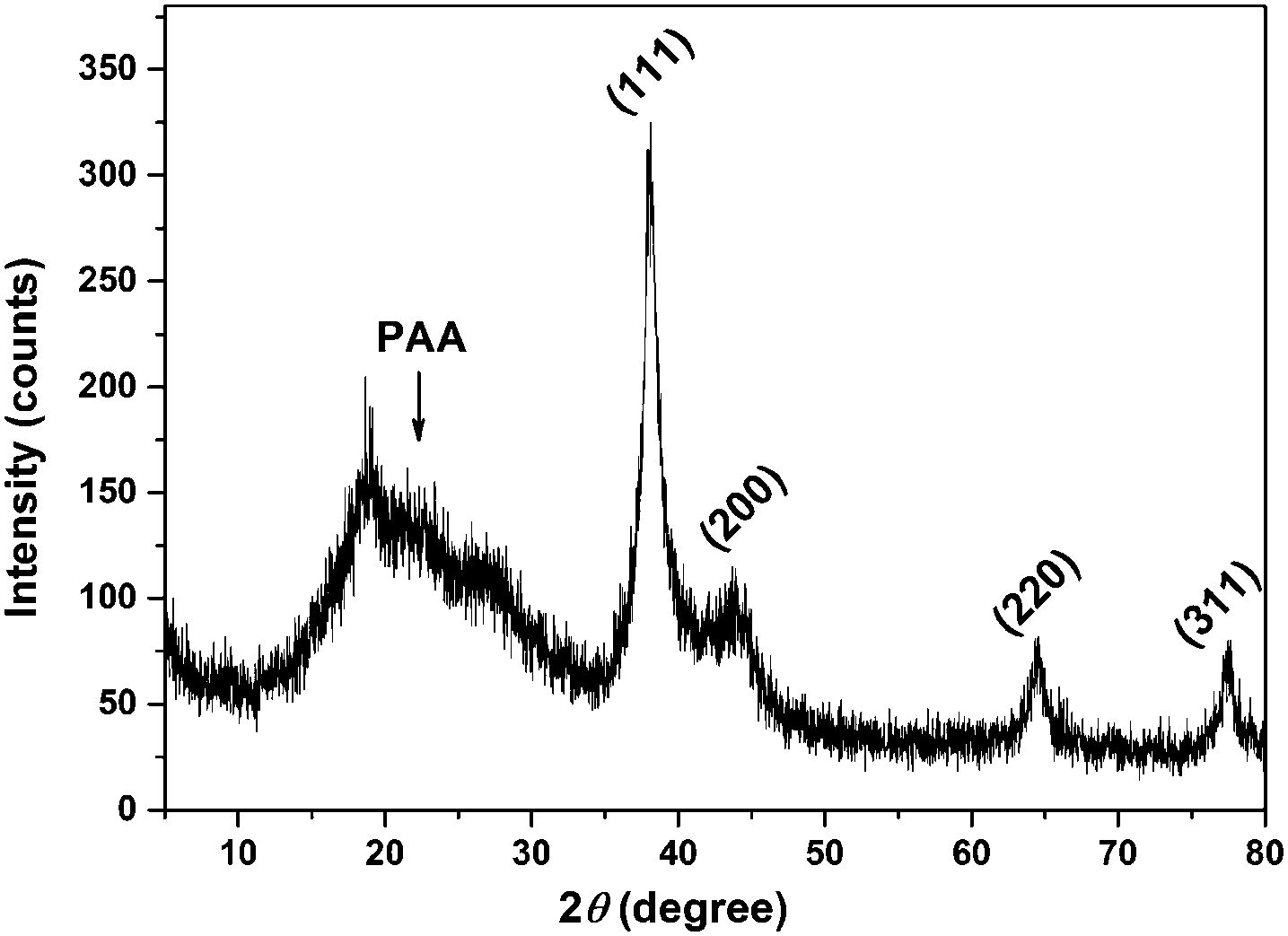 | ||
| Fig. 2 X-ray diffraction pattern of the Ag–PAA hybrid. | ||
The Ag–PAAS solution remained highly homogeneous in a wide pH range (pH > 3.5) and swiftly precipitated from the solution at pH < 3 as shown in Fig. 3(A) and (B). The corresponding surface plasmon bands of the PAAS-stabilized Ag NPs are presented in Fig. 3(D). When the pH of the solution was decreased, the UV-vis spectrum showed blue-shifts in the absorption peak, from 406 nm (pH 8.5) to 409 nm (pH 4.7), 410 nm (pH 3.5), and 412 nm (pH 2.9), probably as a result of the more compressive wrapping of the PAA molecule layers onto Ag NPs, derived from the incremental protonation of the carboxylic groups of PAA. Notably, the red brown precipitate was redispersed smoothly in water by increasing the pH of the solution to ∼10 (see Fig. 3(C)) and the same plasmon bands were recovered, implying that the size and shape of the Ag NPs remained unaltered during pH switching. The TEM image of the Ag NPs after redispersion (Fig. S1 in the ESI†) shows no obvious changes with respect to the dispersity and mean size of the NPs, further verifying the favorable effect of the pH-mediated response of Ag NPs by PAAS.
 | ||
| Fig. 3 Images of (A) fresh Ag–PAAS nanocomposite solution, (B) after addition of drops of 1 M acetic acid until the pH of solution decreased to ∼2 and (C) redispersion of precipitates after adjusting the pH of solution to ∼10 with triethylamine. (D) UV-vis spectra of the Ag–PAAS nanocomposite solution at different pH values. | ||
ζ potential measurements were used to study the stability of the Ag–PAAS dispersion as shown in Fig. 4. The Ag NPs were highly negatively charged at nearly neutral and high pH levels (pH 6–11) with a maximum ζ potential of up to 60 mV, which presumably originated from the coating of PAA chains with huge numbers of negatively charged and extended carboxylate groups, responsible for the extraordinary stability and homogeneity of the colloidal system owing to the electrostatic repulsion mechanism.32,39 A dramatic decrease in the charge density occurred as the pH decreased in the range 2.5–4.0, because of the increasing protonation of the PAA carboxylate moieties, resulting in the concomitant collapse of the polymer chains obscuring the solution gradually. At pH < 2.4, the ζ potential reached the minimum, indicating that the majority of PAA coatings on Ag NPs were protonated, leading to complete salting-out of the NPs from the solution. The lack of electrostatic charges on the NP surface (|ζ| < 20) generally brings about severe instability of NPs, but the pH-responsive dispersion of the Ag–PAAS here was highly reversible (see the UV-vis, TEM and XRD analyses mentioned above), and thus it is reasonable to assume that at lower pHs a denser wrapping of PAA on the Ag NPs should instead prevent the Ag NPs from agglomeration and the resulting Ag-PAA hybrid can be readily redissolved in relatively high pH media.
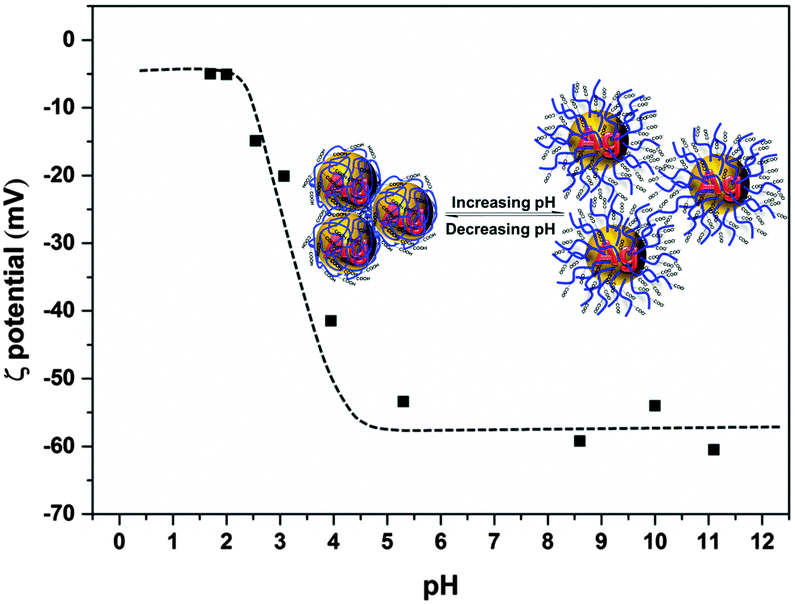 | ||
| Fig. 4 ζ potential measurements of Ag–PAAS solution at different pH values. | ||
3.2 Catalytic applications for the hydration of nitriles
Subsequently, the application of the Ag–PAAS was explored for the catalytic hydration of nitriles under aerobic conditions. First, the hydration of benzonitrile as a model substrate was performed to optimize the reaction conditions (Table 1). The reaction did not proceed at 90 °C even after 20 h in blank PAAS and NaBH4–PAAS solutions (entries 1 and 2), ensuring that the reaction was not catalyzed by PAAS and NaBH4. However, when Ag NPs were loaded onto PAAS and used as a catalyst for the hydration of benzonitrile, a dramatic conversion of nitrile to amide occurred within 9 h (entry 3). Various mixed solvents were used to optimize the mass transfer efficiency (entries 4–7). Unfortunately, protic solvents ethanol and iso-propanol afforded significantly lower conversions, and the addition of aprotic solvents such as N,N-dimethylformamide and pyridine further decreased the conversions. A prolonged period of time was required to obtain a good-to-excellent yield for the cases of lowering the catalyst dosage or reaction temperature, though the turnover frequency (TOF) increased moderately as the Ag loading was decreased (entries 8 and 9).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yielda (%) | TOFb (h−1) |
| Reaction conditions: benzonitrile (1 mmol), Ag catalyst (3 mol%), H2O (3 mL), at 90 °C, in air.a Yields were determined by GC and confirmed by 1H NMR. The selectivity of all the reactions is ∼100%.b Turnover frequencies ((mol benzamide/mol Ag)/time).c The original pH of the Ag–PAAS solution is 10.0.d At 80 °C.e Performed with 1 mol% Ag catalyst. | |||||
| 1 | PAAS | H2O | 20 | — | — |
| 2 | NaBH4–PAAS | H2O | 20 | Trace | — |
| 3 | Ag–PAAS | H2O | 9 | 90 | 3.33 |
| 4 | Ag–PAAS | EtOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) :2) |
22 | 21 | 0.32 |
| 5 | Ag–PAAS |
iPrOH/H2O (1:2) |
16 | 20 | 0.42 |
| 6 | Ag–PAAS | DMF/H2O (1:2) |
22 | 5 | 0.076 |
| 7 | Ag–PAAS | Pyridine/H2O (1:2) |
20 | 4 | 0.067 |
| 8d | Ag–PAAS | H2O | 12 | 83 | 2.31 |
| 9e | Ag–PAAS | H2O | 18 | 94 | 5.22 |
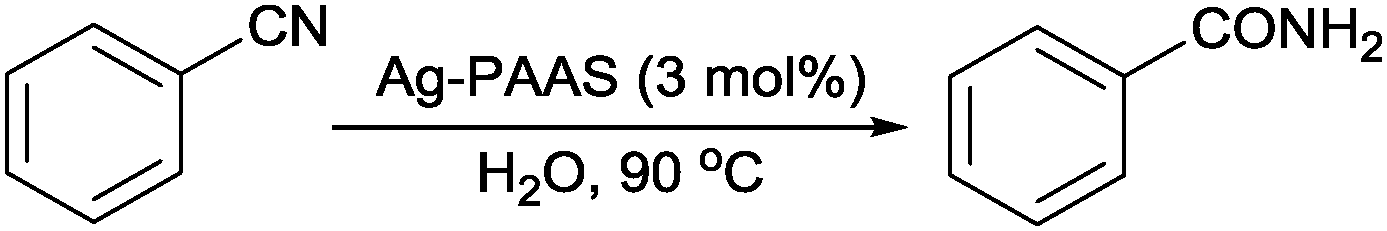
Under the optimized reaction conditions, the scope of the Ag–PAAS catalyst was then examined with regard to various types of nitriles (Table 2). The Ag–PAAS catalyst displayed high activity for the hydration of activated, inactivated, aliphatic, and heterocyclic nitriles in water. Benzonitrile derivatives bearing electron-withdrawing groups (entries 4–7) seemed more active than those bearing electron-donating groups (entries 2 and 3), except in the case of 4-chlorobenzonitrile and 4-bromobenzonitrile (the low solubility of halogen-substituted benzonitriles is frequently a problem in aqueous hydration reactions). Furthermore, less reactive aliphatic substrates were also efficiently hydrated to the corresponding amides in good to excellent yields (entries 8–10). Acetonitrile and acrylonitrile showed relatively low reactivity, due to the increased electron density of the nitrile carbon compared with that of benzonitriles. To our surprise, the hydration of cinnamonitrile smoothly afforded cinnamamide in 97% GC yield with an intact alkene bond in a short period of time, indicating that the replacement of a hydrogen atom of acrylonitrile by a phenyl group produced a dramatic increase in the electrophilicity of the nitrile carbon atom (entry 10). The metal-catalyzed hydration of heteroaromatic nitriles is usually difficult in homogeneous catalysis, because of the strong coordinating ability of the heteroatoms to the metal center.9,40–43 While in our experiments the reactions furnished a quantitative yield of the corresponding amides within only 1 h (entries 11–13). The hydration of 2-thiophenecarbonitrile afforded 97% GC yield even at 40 °C (entry 11). A large-scale experiment of the hydration of 2-cyanopyridine (10 mmol) with a smaller amount of Ag–PAAS catalyst (0.01 mmol, 0.1 mol%) showed a TOF of 1000 h−1 (entry 13). To the best of our knowledge, this value is unprecedented for the Ag-mediated hydration of nitriles at such a low temperature to date (see Table 3). Thus, the Ag–PAAS is catalytically active for the hydration of nitriles in air at relatively mild temperatures. In contrast, previously reported Ag NP catalysts required an inert atmosphere and high reaction temperatures (>140 °C) and only afforded low TOFs under mild conditions.
| Entry | Substrate | Time (h) | Product | Yielda (%) | TOFb (h−1) |
|---|---|---|---|---|---|
| Reaction conditions: substrate (1 mmol), Ag catalyst (3 mol%), H2O (3 mL), at 90 °C, in air.a Yields were determined by GC and confirmed by 1H NMR (isolated yields are given in parentheses). The selectivity is >99%.b Turnover frequencies ((mol product/mol Ag)/time).c Performed at 40 °C.d Nitrile (10 mmol), Ag catalyst (0.1 mol%), H2O (5 mL). | |||||
| 1 |

|
9 |
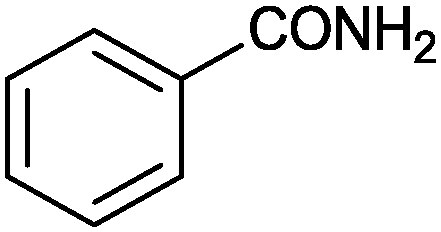
|
90 | 3.33 |
| 2 |

|
12 |
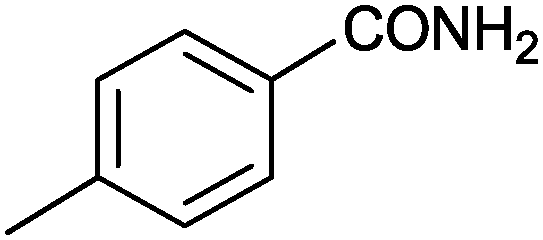
|
95 | 2.64 |
| 3 |

|
11 |
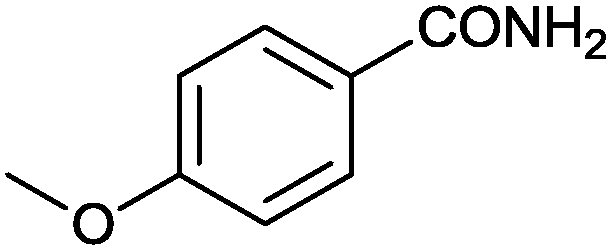
|
92 (88) | 2.79 |
| 4 |
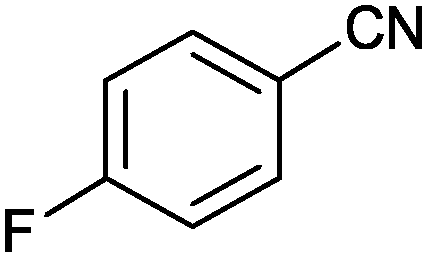
|
8 |
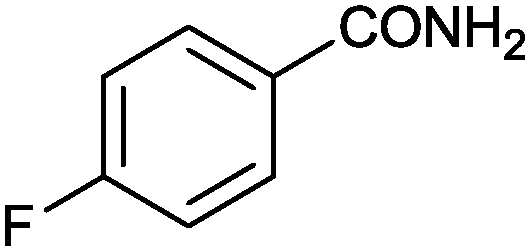
|
92 (87) | 3.96 |
| 5 |

|
10 |
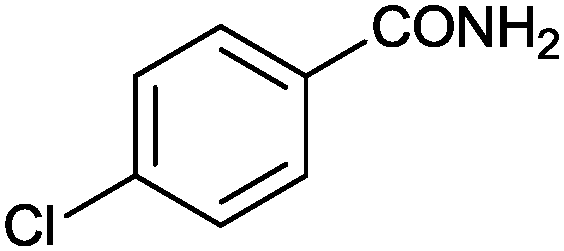
|
90 | 3 |
| 6 |
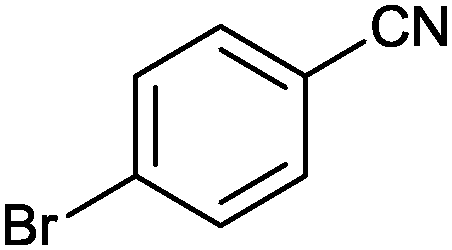
|
12 |
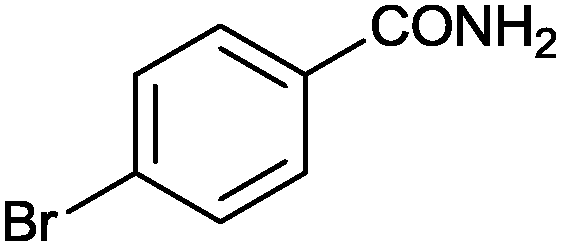
|
77 | 2.14 |
| 7 |
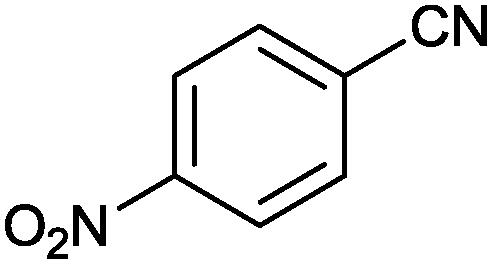
|
1 |
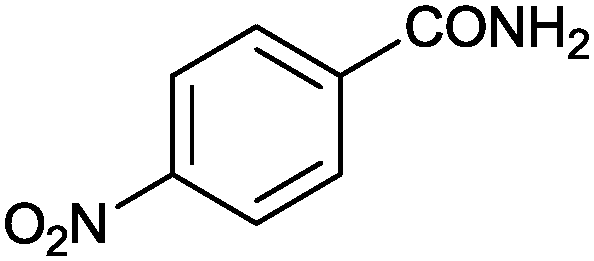
|
89 | 29.7 |
| 8 | CH3CN | 16 | CH3CONH2 | (73) | 1.52 |
| 9 |

|
15 |
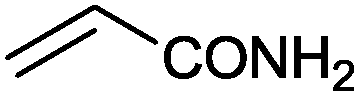
|
(88) | 1.96 |
| 10 |
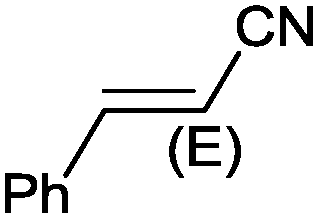
|
9 |
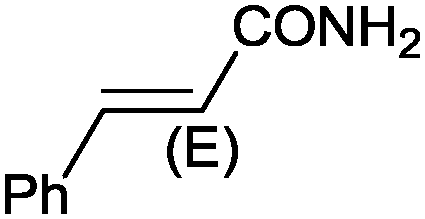
|
97 (90) | 3.59 |
| 11 |

|
1 |
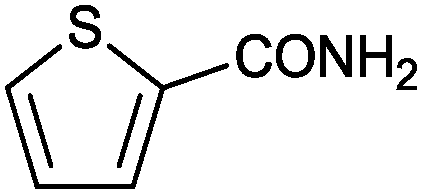
|
>99 | 33.3 |
| 10c | 97 | 3.23 | |||
| 12 |
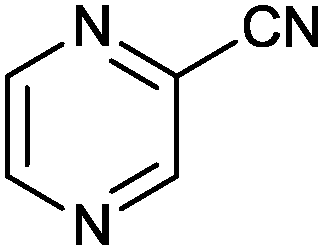
|
1 |
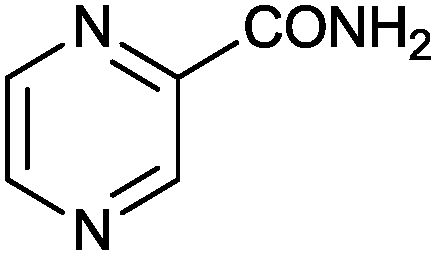
|
>99 (85) | 33.3 |
| 13 |
|
1 |
|
>99 | 33.3 |
| 1d | >99 | 1000 | |||
| Catalyst | Temp. (°C) | Time (h) | Catalyst loading (mol%) | Yield (%) | TOFa (h−1) | Ref. |
|---|---|---|---|---|---|---|
| a Turnover frequencies ((mol benzamide/mol Ag)/time) were calculated from the yield and time data in the papers by taking into account the total amount of metal used. b The content of Ag was estimated from the thermogravimetric analysis of the mPMF–Ag0 material displayed in the corresponding literature. | ||||||
| Ag–SiO2 | 140 | 0.4 | 1 | 37 | 93 | 28 |
| Ag–hydroxyapatite | 140 | 0.25 | 3 | 99 | 133 | 2 |
| mPMF–Ag0 | 90 | 7 | ∼5b | 96 | 2.7 | 31 |
| Au–NHC | 140 | 2 | 2 | 77 | 19.3 | 9 |
| Nafion–Ru | 175 | 12 | 4.2 | 95 | 1.9 | 42 |
| Ru–chitosan | 120 (microwave) | 1 | 1.5 | >99 | 67 | 15 |
| Ru complex | 100 | 7 | 5 | 24 | 0.68 | 41 |
| RuCl2(PTA)4 | 100 | 7 | 5 | 43 | 1.2 | 40 |
| Fe3O4@SiO2@SePh@Ru(OH)x | 120 | 7 | 4.3 | 87 | 2.9 | 43 |
| Pd/C-500Hox | 135 | 24 | 2 | 93 | 1.9 | 16 |
| RhCl(COD){P(NMe2)3} | 100 | 2 | 5 | 99 | 9.9 | 10 |
| Ag–PAAS | 90 | 1 | 0.1 | >99 | 1000 | This work |
Because the pH-induced precipitation–redispersion process of Ag–PAAS is highly reversible, the Ag–PAAS catalyst was easily separated by adjusting the pH of solution to ∼2 and then decanting the product phase. The dark precipitate of Ag–PAAS was collected and redissolved in 3 mL of water by increasing the pH to 10.0 with triethylamine. The activity of the recycled catalysts was examined for the hydration of pyrazinecarbonitrile. The high water solubility of its amide product generally makes it difficult to obtain a high isolated yield using a direct decantation method and to separate the catalyst from the reaction medium (similar to the case of 2-cyanopyridine). As shown in Fig. 5, the Ag–PAAS could be reused eight repeated times with loss of activity (<5%) being observed only after the sixth cycle (cumulative TON after the 8 cycles = 263). The TEM image of the catalyst after eight batches showed that the Ag NPs were still highly dispersed with slightly increased grain sizes (5–10 nm) (see Fig. S2 in the ESI†). ICP-OES analysis was used to determine the Ag content leaching into the product phase, resulting in an average residual concentration of 15.8 ng mL−1 (1.46 × 10−4 mM) per cycle, which is 0.00146% of the original concentration of the catalytic solution (10 mM). Such an extremely low loss of the catalyst during recycling combined with the high stability of NPs during catalysis due to the polymer stabilizer might be responsible for the excellent catalytic performance for the hydration of pyrazinecarbonitrile.
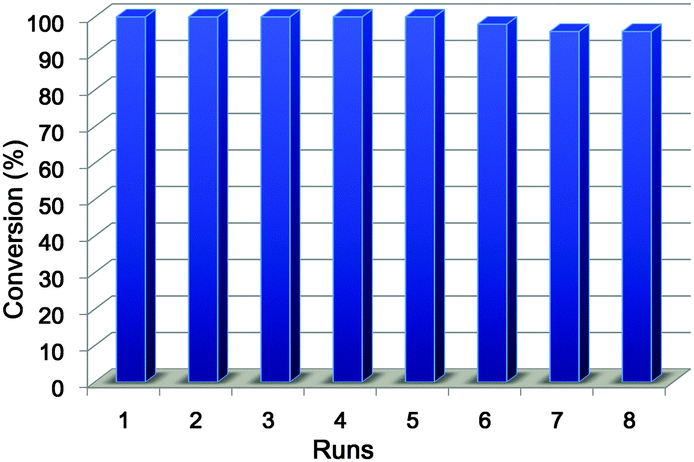 | ||
| Fig. 5 Reuse of the Ag–PAAS catalyst in the hydration of pyrazinecarbonitrile. Reaction conditions were identical to those indicated in Table 2 (1 h reaction time in each cycle). | ||
It is well accepted that the possible mechanism of the hydration of nitriles involves the coordination of water (or OH−) and aromatic nitrile on the “bifunctional” Ag surface.2,11,28 To elucidate the critical pathway of the Ag-catalyzed transformation in this study, the effect of the concentration of OH− on the catalytic activity is shown in Table 4. Decreasing the pH from the original 10.0 to 7.6 led to a 10-fold decrease in the reaction rate, indicating that a basic co-catalyst is essential for efficient catalysis. On the other hand, further increase in the pH up to 11.9 only slightly increased the TOF, indicating that an acceleration effect from the OH− reached saturation and some other unknown rate-limiting factors were present. Studies to elucidate the detailed mechanism of the catalytic action of the Ag–PAAS system are still in progress in our lab.
4 Conclusions
In summary, a novel quasi-homogeneous Ag NP-based catalyst was developed using PAAS as a stabilizer. The Ag–PAAS catalyst displayed high activity for the selective hydration of nitriles to amides in an aqueous medium under relatively mild conditions, especially for the hydration of heteroaromatic nitriles (a maximum turnover frequency of 1000 h−1 was obtained for the hydration of 2-cyanopyridine). Moreover, the pH response of PAAS afforded a reversible precipitation–redispersion switch of Ag NPs, allowing efficient recovery and recycling of the catalyst. The catalyst could be reused for eight runs without significant loss (<5%) in its initial catalytic activity, due to the extremely low Ag leaching during recovery handling and the high stability of Ag NPs during catalysis. The Ag–PAAS catalyst here combines the advantages of the high efficiency of a quasi-homogeneous catalyst and the easy recovery and reuse of a heterogeneous catalyst.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51573209), the Hunan Provincial Innovation Foundation for Graduates (CX2014B049) and the Free Exploration Project for Undergraduates (ZY2015613).Notes and references
- R. Garcia-Alvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46–66 RSC.
- T. Mitsudome, Y. Mikami, H. Mori, S. Arita, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 3258–3260 RSC.
- N. K. Thallaj, P.-Y. Orain, A. Thibon, M. Sandroni, R. Welter and D. Mandon, Inorg. Chem., 2014, 53, 7824–7836 CrossRef CAS PubMed.
- I. Ferrer, J. Rich, X. Fontrodona, M. Rodriguez and I. Romero, Dalton Trans., 2013, 42, 13461–13469 RSC.
- E. Tomas-Mendivil, F. J. Suarez, J. Diez and V. Cadierno, Chem. Commun., 2014, 50, 9661–9664 RSC.
- D. Kumar, C. A. Masitas, T. N. Nguyen and C. A. Grapperhaus, Chem. Commun., 2013, 49, 294–296 RSC.
- P. Crochet and V. Cadierno, Dalton Trans., 2014, 43, 12447–12462 RSC.
- M. L. Buil, V. Cadierno, M. A. Esteruelas, J. Gimeno, J. Herrero, S. Izquierdo and E. Oñate, Organometallics, 2012, 31, 6861–6867 CrossRef CAS.
- R. S. Ramón, N. Marion and S. P. Nolan, Chem. – Eur. J., 2009, 15, 8695–8697 CrossRef PubMed.
- E. Tomás-Mendivil, R. García-Álvarez, C. Vidal, P. Crochet and V. Cadierno, ACS Catal., 2014, 4, 1901–1910 CrossRef.
- E. L. Downs and D. R. Tyler, Coord. Chem. Rev., 2014, 280, 28–37 CrossRef CAS.
- S. Kumar and P. Das, New J. Chem., 2013, 37, 2987–2990 RSC.
- H. Woo, K. Lee, S. Park and K. Park, Molecules, 2014, 19, 699–712 CrossRef PubMed.
- A. Y. Kim, H. Bae, S. Park, S. Park and K. Park, Catal. Lett., 2011, 141, 685–690 CrossRef CAS.
- R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2014, 16, 2122–2127 RSC.
- K.-i. Shimizu, T. Kubo, A. Satsuma, T. Kamachi and K. Yoshizawa, ACS Catal., 2012, 2, 2467–2474 CrossRef CAS.
- Y.-M. Liu, L. He, M.-M. Wang, Y. Cao, H.-Y. He and K.-N. Fan, ChemSusChem, 2012, 5, 1392–1396 CrossRef CAS PubMed.
- T. Subramanian and K. Pitchumani, Catal. Commun., 2012, 29, 109–113 CrossRef CAS.
- C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2014, 16, 1060–1063 CrossRef CAS PubMed.
- M. Tamura, A. Satsuma and K.-i. Shimizu, Catal. Sci. Technol., 2013, 3, 1386–1393 CAS.
- Y. Gangarajula and B. Gopal, Chem. Lett., 2012, 41, 101–103 CrossRef CAS.
- K. Yamaguchi, Y. Wang, H. Kobayashi and N. Mizuno, Chem. Lett., 2012, 41, 574–576 CrossRef CAS.
- P. Christopher and S. Linic, ChemCatChem, 2010, 2, 78–83 CrossRef CAS.
- Z.-J. Jiang, C.-Y. Liu and L.-W. Sun, J. Phys. Chem. B, 2005, 109, 1730–1735 CrossRef CAS PubMed.
- R. Xu, D. Wang, J. Zhang and Y. Li, Chem. – Asian J., 2006, 1, 888–893 CrossRef CAS PubMed.
- J. Davarpanah and A. R. Kiasat, Catal. Commun., 2013, 41, 6–11 CrossRef CAS.
- T. J. Sherbow, E. L. Downs, R. I. Sayler, J. J. Razink, J. J. Juliette and D. R. Tyler, ACS Catal., 2014, 4, 3096–3104 CrossRef CAS.
- K.-i. Shimizu, N. Imaiida, K. Sawabe and A. Satsuma, Appl. Catal., A, 2012, 421–422, 114–120 CrossRef CAS.
- K. Kawai, H. Kawakami, T. Narushima and T. Yonezawa, J. Nanopart. Res., 2015, 17, 1–9, DOI:10.1007/s11051-014-2816-1.
- N. Salam, S. K. Kundu, R. A. Molla, P. Mondal, A. Bhaumik and S. M. Islam, RSC Adv., 2014, 4, 47593–47604 RSC.
- K. Ghosh, M. A. Iqubal, R. A. Molla, A. Mishra, Kamaluddin and S. M. Islam, Catal. Sci. Technol., 2015, 5, 1606–1622 CAS.
- Y. Yuan, N. Yan and P. J. Dyson, Inorg. Chem., 2011, 50, 11069–11074 CrossRef CAS PubMed.
- X. Wu, X. Chen, H. Guan, X. Wang and L. Chen, Catal. Commun., 2014, 51, 29–32 CrossRef CAS.
- S. Qi, X. Shen, Z. Lin, G. Tian, D. Wu and R. Jin, Nanoscale, 2013, 5, 12132–12135 RSC.
- J. Li, Y.-c. Fang, G.-w. He and H.-f. Li, J. Cent. South Univ., 2013, 20, 1475–1481 CrossRef CAS.
- N. Du, C. Wong, M. Feurstein, O. A. Sadik, C. Umbach and B. Sammakia, Langmuir, 2010, 26, 14194–14202 CrossRef CAS PubMed.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, John Wiley & Sons, Inc., 1983 Search PubMed.
- R. Jenkins and R. L. Snyder, Introduction to X-ray powder diffractometry, John Wiley & Sons, Inc., 1996 Search PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- W.-C. Lee and B. J. Frost, Green Chem., 2012, 14, 62–66 RSC.
- W.-C. Lee, J. M. Sears, R. A. Enow, K. Eads, D. A. Krogstad and B. J. Frost, Inorg. Chem., 2013, 52, 1737–1746 CrossRef CAS PubMed.
- G. K. S. Prakash, S. B. Munoz, A. Papp, K. Masood, I. Bychinskaya, T. Mathew and G. A. Olah, Asian J. Org. Chem., 2012, 1, 146–149 CrossRef CAS.
- H. Joshi, K. N. Sharma, A. K. Sharma, O. Prakash, A. Kumar and A. K. Singh, Dalton Trans., 2014, 43, 12365–12372 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TEM image, XPS data of the Ag NPs and the NMR data of the reaction products. See DOI: 10.1039/c5nj02497h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |