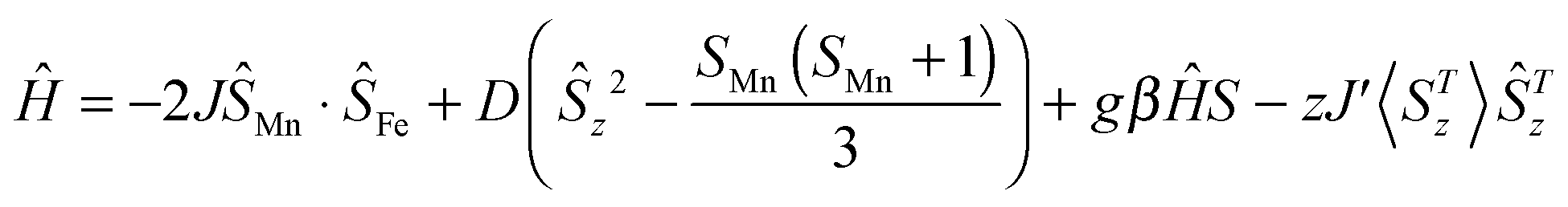DOI:
10.1039/C5NJ02132D
(Paper)
New J. Chem., 2016,
40, 365-371
Cyanide-bridged heterobimetallic magnetic complexes based on metalloporphyrinate and tricyanometalate building blocks†
Received
(in Victoria, Australia)
11th August 2015
, Accepted 26th October 2015
First published on 30th October 2015
Abstract
Three cyanide-bridged metalloporphyrinate complexes with the formula {[(Tp*)Fe(CN)3]2[Mn(TPP)]}[Mn(TPP)(MeOH)2] (1), {[(Tp)Cr(CN)3]2[Mn(TPP)]}[Mn(TPP)(MeOH)2] (2), and {[(Tp*)Fe(CN)3][Mn(TPyP)]}n (3) (Tp*− = hydrotris(3,5-dimethylpyrazol-1-yl)borate, Tp− = tris(pyrazolyl)hydroborate, TPP2− = tetra(phenyl)porphyrinate, and TPyP2− = tetra(4-pyridyl)porphyrinate) have been first synthesized based on tricyanometalate building blocks and metalloporphyrinate precursors. The crystal structures show that complexes 1 and 2 possess similar sandwich-like molecular structures composed of an anionic trimer and a cationic monomer. More interestingly, the introduction of manganese tetra(4-pyridyl)porphyrinate precursor results into the formation of an unprecedented neutral one-dimensional zigzag single chain complex 3. Variable temperature dc magnetic measurements reveal the ferromagnetic and antiferromagnetic interaction between MnIII and FeIII centers for complexes 1 and 3, respectively, and antiferromagnetic coupling between CrIII and MnIII centers for complex 2. While ac magnetic susceptibility measurements mainifest that only complex 3 shows the slow magnetic relaxation behavior of a single-molecule magnet.
Introduction
The interest in cyanide-bridged molecule-based magnetic materials with reduced dimensionality, such as single-molecule magnets (SMMs) and single-chain magnets (SCMs), has increased exponentially during the past two decades, owing to their fascinating molecular architectures and promising applications in magnetic storage, sensors, and nanoscale electronic devices.1–4
So far, many cyanide-bridged transition metal SMMs or SCMs have been reported.1,2,5,6 To obtain such low-dimensional cyanide-containing magnetic complexes, one of the synthetic strategies is to utilize modified cyanometalates as building blocks toward coordinatively unsaturated metal precursors containing various ancillary ligands, in which the magnetic coupling interaction between neighbouring paramagnetic metal centers can be easily controlled and anticipated.
It is worth noting that the selection of suitable ancillary functional ligands to construct the metal coordination precursors is the most effective method for rationally designing and synthesizing unusual cyanide-bridged molecular magnetic materials.5c,7 As a representative, the macrocyclic porphyrin and its derivatives, which have been intensively studied in the field of catalysis, magnetism, nonlinear optics and sensors, can provide a tunable platform to present multidentate coordination sites along with tailored functionality at the molecular periphery for assembly-directing various metal ions.7b,8,9 Furthermore, the introduction of such a highly conjugated and functionalized metalloporphyrinate precursor into the SMMs or SCMs will extend the research of the multifunctional molecular magnetic materials due to their intriguing structures and unique chemical/physical properties.
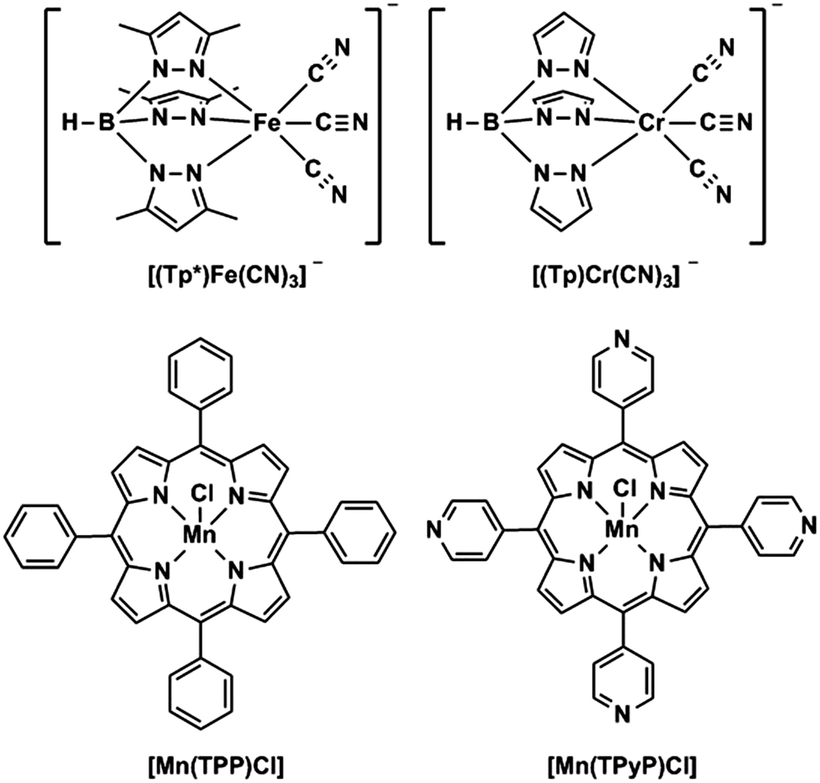
Herein, as a part of our continual efforts to develop new low-dimensional coordination compounds, three cyanide-bridged metalloporphyrinate complexes with the formula {[(Tp*)Fe(CN)3]2[Mn(TPP)]}[Mn(TPP)(MeOH)2] (1), {[(Tp)Cr(CN)3]2[Mn(TPP)]}[Mn(TPP)(MeOH)2] (2), and {[(Tp*)Fe(CN)3][Mn(TPyP)]}n (3) (Tp*− = hydrotris(3,5-dimethylpyrazol-1-yl)borate, Tp− = tris(pyrazolyl)hydroborate, TPP2− = tetra(phenyl)porphyrinate, and TPyP2− = tetra(4-pyridyl)porphyrinate) have been first synthesized by the reaction of various tricyanometalate building blocks and metalloporphyrinate precursors (Scheme 1). These complexes were structurally characterized and their spectroscopic and magnetic properties were also studied. To the best of our knowledge, cyanide-bridged transition metal complexes based on metalloporphyrinate are still scarce,10 and there are no reports about such cationic–anionic polynuclear or neutral one-dimensional (1D) cyanide-bridged metalloporphyrinate magnetic complexes mainly based on tricyanometalate building blocks. The meaningful structure–property relationship obtained will improve our understanding of the magnetic behaviors of novel cyanide-bridged molecular magnetic materials.
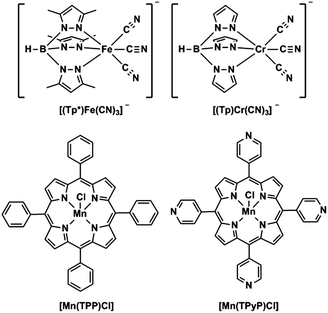 |
| | Scheme 1 Structures of tricyanometalate building blocks (top) and metalloporphyrinate compounds (bottom). | |
Experimental section
General methods
All reagents and solvents were purchased from commercial suppliers and used without further purification. (n-Bu4N)[(Tp*)Fe(CN)3],11 (n-Bu4N)[(Tp)Cr(CN)3],12 [Mn(TPP)Cl],13 and [Mn(TPyP)Cl]14 were synthesized according to literature procedures, respectively. Elemental analyses were carried out using an Elementar Vario MICRO analyzer. IR spectra were recorded on a Vector22 Bruker spectrophotometer with KBr pellets. UV-Vis spectra were obtained using a UV-3600 spectrophotometer. Magnetic susceptibility measurements for polycrystalline samples were performed using a Quantum Design MPMS-SQUID-VSM magnetometer. Direct current (dc) magnetic susceptibility data measurements were collected between 1.8 and 300 K for dc applied fields ranging from 0 to 70 kOe. Alternating-current (ac) magnetic susceptibility data measurements were carried out under an oscillating field of 2 Oe and ac frequencies ranging between 1 and 997 Hz. Diamagnetic corrections were applied for the sample holder and calculated using Pascal's constants.15
X-ray crystallography
All of the crystal structures were determined at 296(2) K on a Bruker APEX CCD diffractometer using monochromated Mo Kα radiation (λ = 0.71073 Å). Diffraction data analysis and reduction were performed within SMART and SAINT.16 Absorption corrections were applied by the SADABS program.17 The crystal structures were solved by direct methods and refined by full-matrix least-squares based on F2 using the SHELXTL program18 with anisotropic displacement parameters for all non-hydrogen atoms. Hydrogen atom positions were calculated geometrically and were riding on their respective atoms. The remaining solvent molecules were removed by the SQUEEZE routine in PLATON.19 More details for the data collections and structure refinements were given in Table 1. The selected bond lengths and bond angles were listed in Tables S1–S3 (ESI†). (CCDC 1417664 (1), 1417665 (2), and 1417666 (3)).
Table 1 Crystallographic data for complexes 1–3
| |
1
|
2
|
3
|
| Formula |
C126H108B2Fe2Mn2N26O2 |
C114H82.55B2Cr2Mn2N26O2.28 |
C58H46BFeMnN17 |
| FW |
2261.58 |
2088.50 |
1102.72 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2(1)/c |
P2(1)/c |
|
a/Å |
13.471(3) |
19.758(3) |
20.928(7) |
|
b/Å |
14.060(3) |
12.4162(17) |
14.437(4) |
|
c/Å |
18.498(4) |
22.828(3) |
20.777(6) |
| α/deg |
106.035(3) |
90 |
90 |
|
β/deg |
103.800(3) |
105.308(2) |
105.019(3) |
|
γ/deg |
92.507(3) |
90 |
90 |
|
V/Å3 |
3247.3(10) |
5401.5(13) |
6063(3) |
|
Z
|
1 |
2 |
4 |
|
ρ
calcd, g cm−3 |
1.156 |
1.284 |
1.208 |
|
T/K |
296(2) |
296(2) |
296(2) |
|
μ/mm−1 |
0.466 |
0.487 |
0.498 |
|
ν/deg |
1.19–25.50 |
1.85–25.51 |
1.73–25.51 |
|
F(000) |
1174 |
2149 |
2276 |
| Data/parameters |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 956/768 956/768 |
10006/662 |
11155/709 |
| GOF (F2) |
1.104 |
1.066 |
1.021 |
|
R
1, wR2 (I > 2σ(I)) |
0.0575, 0.1879 |
0.0623, 0.1737 |
0.1146, 0.2569 |
|
R
1, wR2 (all data) |
0.0727, 0.2004 |
0.0762, 0.1843 |
0.1345, 0.2674 |
Synthesis of {[(Tp*)Fe(CN)3]2[Mn(TPP)]}[Mn(TPP)(MeOH)2] (1).
The reaction mixture of [MnTPPCl] (17.58 mg, 0.025 mmol) and (n-Bu4N)[(Tp*)Fe(CN)3] (16.84 mg, 0.025 mmol) in 10 mL of methanol/acetone (1:1 v/v) was stirred at room temperature for 12 h. Brown block-shaped crystals of 1 were obtained after slow evaporation of the filtrate for several days. Yield: 65%. Anal. calcd for C126H108B2Fe2Mn2N26O2: C 66.92; H 4.81; N 16.10. Found: C 67.11; H 5.09; N 16.36. Selected IR data (KBr, cm−1): 3402(w), 2920(m), 2524(m), 2127(s), 1636(s), 1486(s), 1201(s), 1009(s), 799(m), 753(m), and 702(m). Selected UV-Vis data (CH2Cl2, λmax/nm): 341, 390, 481, 579, and 617.
Synthesis of {[(Tp)Cr(CN)3]2[Mn(TPP)]}[Mn(TPP)(MeOH)2] (2).
The brown block-shaped crystals of complex 2 were obtained by following the same procedure as that of complex 1 but using (n-Bu4N)[(Tp)Cr(CN)3] instead of (n-Bu4N)[(Tp*)Fe(CN)3]. Yield: 65%. Anal. calcd for C114H76B2Cr2Mn2N26O2: C 65.91; H 3.69; N 17.53. Found: C 66.19; H 3.85; N 17.84. Selected IR data (KBr, cm−1): 3411(w), 2917(m), 2492(m), 2132(s), 1628(s), 1495(m), 1308(s), 1206(m), 1029(m), 1010(s), 815(m), 757(m), and 707(m). Selected UV-Vis data (CH2Cl2, λmax/nm): 339, 392, 479, 580, and 617.
Synthesis of {[(Tp*)Fe(CN)3][Mn(TPyP)]}n (3).
The brown block-shaped crystals of complex 3 were obtained by following the same procedure for complex 1 but using [MnTPyPCl] instead of [MnTPPCl]. Yield: 45%. Anal. calcd for C58H46BFeMnN17: C 63.17; H 4.20; N 21.59. Found: C 63.50; H 4.39; N 21.85. Selected IR data (KBr, cm−1): 3397(w), 2932(m), 2531(m), 2128(s), 1640(s), 1599(m), 1425(m), 1311(s), 1225(m), 1116(s), 1011(m), 799(m), 732(m), and 687(m). Selected UV-Vis data (CH2Cl2, λmax/nm): 342, 373, 399, 476, 576, and 611.
Results and discussion
Synthesis and characterization
All the complexes were successfully synthesized based on tricyanometalate building blocks and various metalloporphyrinate precursors. By the equivalent molar reaction of five-coordinated [Mn(TPP)Cl] with [(Tp*)Fe(CN)3]− or [(Tp)Cr(CN)3]−, two sandwich-type trinuclear cyanide-bridged anionic complexes [FeIII–MnIII–FeIII]− (1), or [CrIII–MnIII–CrIII]− (2) were obtained, respectively, with a larger balance cation [Mn(TPP)(MeOH)2]+. However, the reaction of [(Tp*)Fe(CN)3]− with the manganese(III) tetra(4-pyridyl)porphyrinate precursor [Mn(TPyP)Cl] containing electron-donating pyridyl substituents results in the formation of a neutral 1D zigzag single chain complex 3. These compounds have been characterized by elemental analysis, IR, UV-Vis, and suitable single crystals for X-ray diffraction analysis were obtained by slow evaporation of the solution of these complexes in methanol and acetone. The electronic absorption spectra of metalloporphyrinate precursors and the targeted complexes in CH2Cl2 solution are almost identical to high intensity Soret bands at around 480 nm and weaker Q-bands above 575 nm (Fig. S1, ESI†), which can be assigned to the intra-ligand π–π* transitions of the porphyrin and its derivatives.13
Crystal structures of complexes 1 and 2
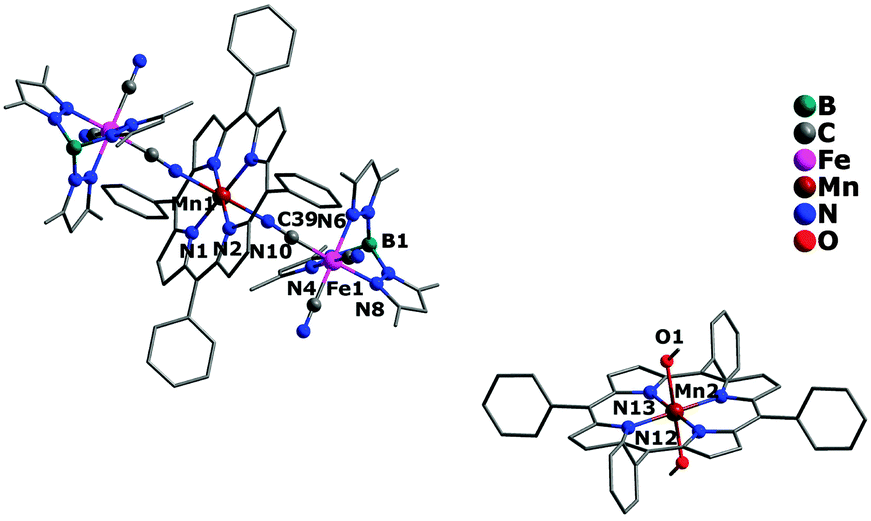
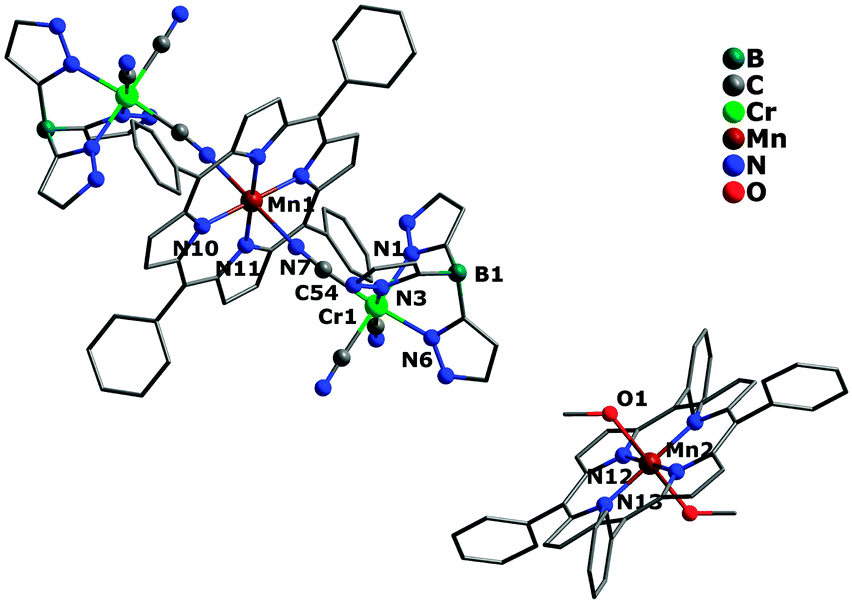
The crystal structures of complexes 1 and 2 are similar, crystallizing in the triclinic (1) and monoclinic (2) system with space group P and P2(1)/c, respectively. Both of them possess a similar sandwich-like molecular structure made up of an anionic trinuclear entity [(Tp*)Fe(CN)3]2[Mn(TPP)]− (1) or [(Tp)Cr(CN)3]2[Mn(TPP)]− (2) with the free [Mn(TPP)(MeOH)2]+ as a balance cation, respectively. In the anionic trinuclear unit, the tricyanometalate building block acts as a monodentate linker through one of its three cyanide groups towards the central MnIII ion from the Mn(TPP)+ fragment. The central Fe(1) or Cr(1) ion situates in a slightly distorted octahedral coordination environment, bonded by three N (pyrazolyl groups) atoms and three C (cyanide groups) atoms with an average Fe–N(pyrazolyl) bond length of 2.003 Å for 1, and a Cr–N(pyrazolyl) bond length of 2.052 Å for 2, while the Mn(1) ion is coordinated by six N atoms (four from the porphyrinate ligand located in a perfect equatorial plane and two axial position from the bridging cyanide groups) (Fig. 1 and 2). In addition, the distance of Mn(1)–N(pyrrolyl) is shorter than the Mn(1)–N(cyanide) bond length, and the corresponding Mn(1)–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond angle deviates significantly from linearity, indicating the obviously distorted octahedron surrounding the Mn(1) ion. In the countercationic segment [Mn(TPP)(MeOH)2]+, the central metal ion is also six-coordinated by four pyrrolyl nitrogen atoms of the porphyrinate ligand and two oxygen atoms from neutral methanol molecules. It should be noted that the highly distorted structure and the size of metalloporphyrinate precursor may hinder the formation of a one-dimensional (1D) chain structure. The intramolecular cyanide-bridged Fe(1)⋯Mn(1) distance for 1 is 5.263(5) Å, and the Cr(1)⋯Mn(1) distance for 2 is 5.432(6) Å, while the shortest intermolecular metal–metal separation among anionic trinuclear units is 13.471(4) (Mn⋯Mn), 8.153(3) (Fe⋯Fe), and 10.217(3) Å (Fe⋯Mn) for 1, and 12.416(3) (Mn⋯Mn), 8.320(4) (Cr⋯Cr), and 9.065(3) Å (Cr⋯Mn) for 2, respectively. Additionally, the intermolecular hydrogen-bond interactions can be found for complex 2 between the nitrogen atom of the cyanide group and the oxygen atom of the uncoordinated [Mn(TPP)(MeOH)2]+ (Fig. S2 and S3, ESI†).
C bond angle deviates significantly from linearity, indicating the obviously distorted octahedron surrounding the Mn(1) ion. In the countercationic segment [Mn(TPP)(MeOH)2]+, the central metal ion is also six-coordinated by four pyrrolyl nitrogen atoms of the porphyrinate ligand and two oxygen atoms from neutral methanol molecules. It should be noted that the highly distorted structure and the size of metalloporphyrinate precursor may hinder the formation of a one-dimensional (1D) chain structure. The intramolecular cyanide-bridged Fe(1)⋯Mn(1) distance for 1 is 5.263(5) Å, and the Cr(1)⋯Mn(1) distance for 2 is 5.432(6) Å, while the shortest intermolecular metal–metal separation among anionic trinuclear units is 13.471(4) (Mn⋯Mn), 8.153(3) (Fe⋯Fe), and 10.217(3) Å (Fe⋯Mn) for 1, and 12.416(3) (Mn⋯Mn), 8.320(4) (Cr⋯Cr), and 9.065(3) Å (Cr⋯Mn) for 2, respectively. Additionally, the intermolecular hydrogen-bond interactions can be found for complex 2 between the nitrogen atom of the cyanide group and the oxygen atom of the uncoordinated [Mn(TPP)(MeOH)2]+ (Fig. S2 and S3, ESI†).
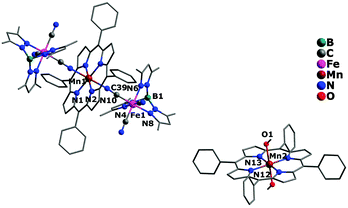 |
| | Fig. 1 Crystal structure of complex 1 with hydrogen atoms and solvent molecules omitted for clarity. | |
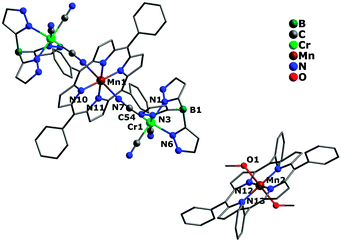 |
| | Fig. 2 Crystal structure of complex 2 with hydrogen atoms and solvent molecules omitted for clarity. | |
Crystal structure of complex 3
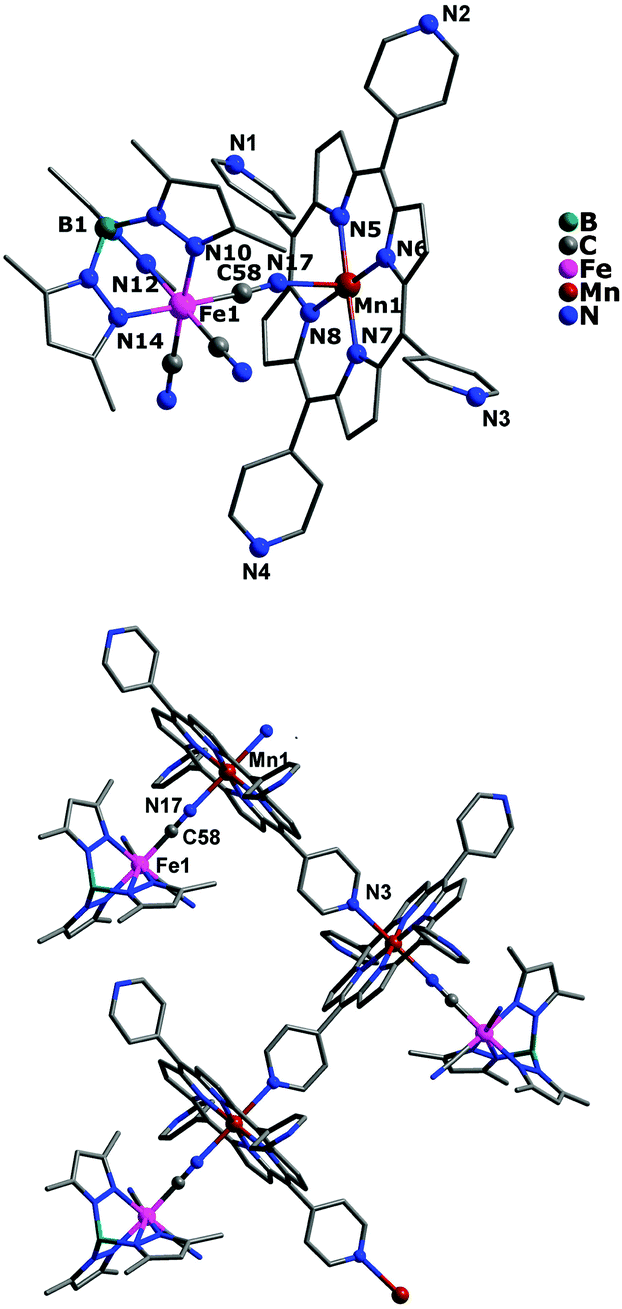
Unlike the above mentioned two cationic–anionic polynuclear complexes, complex 3 has been structurally characterized as a neutral 1D zigzag single chain crystallizing in the monoclinic system P2(1)/c space group. As shown in Fig. 3, each [(Tp*)Fe(CN)3]− entity acts as a monodentate ligand toward the [Mn(TPyP)]+ unit through one of its three cyanide groups, whereas the MnIII tetra(4-pyridyl)porphyrinate precursor is linked to each other by one pyridyl N atom coming from the tetra(4-pyridyl)porphyrinate ligand. The coordination geometries around both FeIII and MnIII ions are all octahedral, in which the FeIII ion is bonded by three pyrazolyl N atoms and three cyanide C atoms, while the MnIII ion is occupied by equatorial N4 donor atoms from the porphyrinate ligand, one apical N atom from the pyridyl group, and one cyanide N atom. The average Fe–N(pyrazolyl) and Mn–N bond lengths are 1.988 and 2.104 Å, respectively, and the shortest intrachain Fe⋯Mn, Mn⋯Mn and Fe⋯Mn distances are 5.238(4), 10.093(3), and 14.437(4) Å. However, in crystal packing (Fig. S4, ESI†), the shortest interchain metal–metal separations are 10.586(4) Å for Fe⋯Fe, 11.494(3) Å for Mn⋯Mn, and 11.399(3) Å for Fe⋯Mn, respectively.
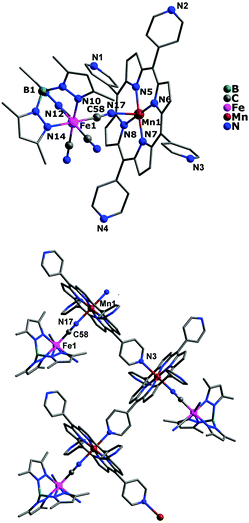 |
| | Fig. 3 Crystal structure of complex 3 for its asymmetric unit (top) and one-dimensional zigzag infinite chain (bottom) with hydrogen atoms and solvent molecules omitted for clarity, respectively. | |
Magnetic properties
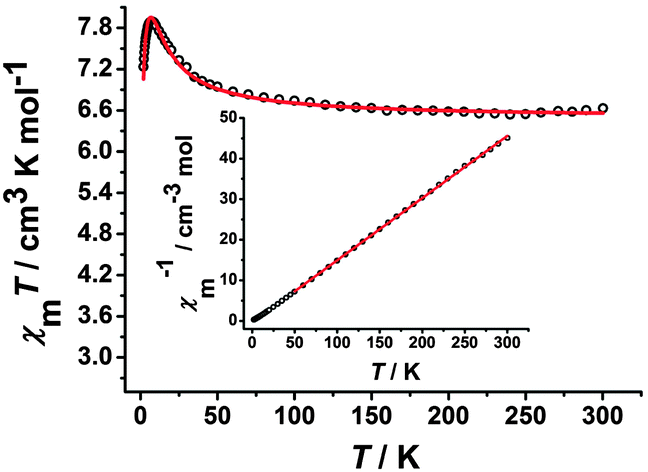
The variable-temperature dc magnetic susceptibilities for all of the complexes were measured under an applied dc field of 100 Oe in the temperature range of 1.8–300 K. For complex 1, the χMT value at room temperature is 6.65 cm3 K mol−1 (Fig. 4), which is close to the value of 6.75 cm3 K mol−1 expected for uncoupled two low-spin FeIII centers (S = 1/2) and two high-spin MnIII centers (S = 2) assuming g = 2.00. Upon cooling, the χMT value increases gradually and reaches the maximum value of 7.95 cm3 K mol−1 at 6.5 K, suggesting the presence of the ferromagnetic coupling between the FeIII and MnIII ions mediated by the cyanide group. While the χMT value starts to decrease rapidly to 7.23 cm3 K mol−1 at 1.8 K, probably ascribed to the intermolecular antiferromagnetic coupling and/or the zero-field splitting (ZFS) of the MnIII ion at such a low temperature region.6i,10d,20 On the basis of the trimeric model, the magnetic susceptibilities can be fitted accordingly by the following expression derived from the isotropic exchange spin Hamiltonian Ĥ = −2JŜMn1(ŜFe1 + ŜFe2), in which we assume an equivalent interaction between the central MnIII ion and the two FeIII ions, and neglect the magnetic interaction between the terminal FeIII ions.21 The free MnIII anion was included as a constant term (χMT = (Ng2β2/3k)SMn2(SMn2 + 1)). The best fit between 1.8 and 300 K gave g = 1.96, J = 2.95 cm−1 and zj′ = −0.025 cm−1 (R = 2.68 × 10−3). Furthermore, the magnetic susceptibility above 50 K obeys the Curie–Weiss law χM = C/(T − θ) with a Curie constant C = 6.54 cm3 K mol−1 and a Weiss constant θ = 2.46 K (the inset of Fig. 4). The C value corresponds to magnetically isolated FeIII and MnIII ions, while the positive θ also suggests dominant ferromagnetic coupling in complex 1. The field-dependent measurement of the magnetization up to 70 kOe was also carried out for 1 at 1.8 K. The magnetization increases rapidly at low field and then slowly reaches a maximum value of 7.71 Nβ (Fig. S5, ESI†), which further confirms that ferromagnetic coupling interactions exist between the FeIII and MnIII ions.10d
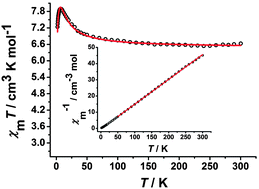 |
| | Fig. 4 Temperature-dependent χMT product for 1 at 100 Oe. The red solid line represents the best fits of the data. Inset: Plots of 1/χMversus T. The red solid line is the fitting result by Curie–Weiss law. | |
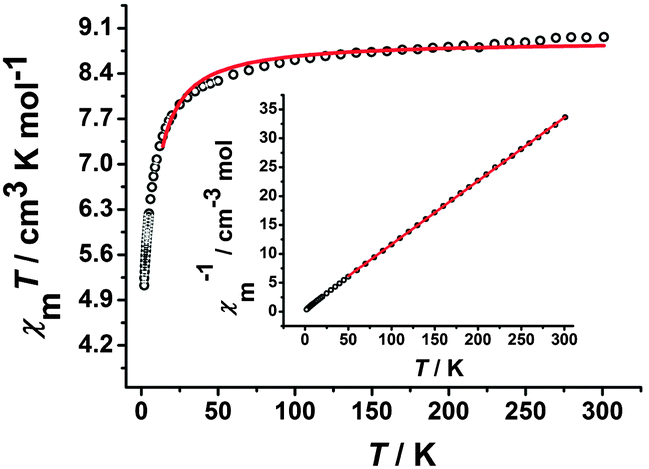
The temperature-dependent χMT product for complex 2 is shown in Fig. 5, the χMT value at 300 K is 8.97 cm3 K mol−1, which is basically consistent with the value of 9.75 cm3 K mol−1 for uncoupled two high-spin MnIII centers (S = 2) and two CrIII centers (S = 3/2) based on g = 2.00. Upon decreasing the temperature, the χMT value decreases gradually and then rapidly reaches the lowest value of 5.13 cm3 K mol−1 at 1.8 K. This behavior may be caused by the antiferromagnetic coupling between the CrIII and MnIII ions and/or the ZFS effect of the MnIII ion.10a On the basis of the trimeric model, the magnetic susceptibilities can be fitted accordingly by the following expression derived from the isotropic exchange spin Hamiltonian Ĥ = −2JŜMn1(ŜCr1 + ŜCr2), which includes only nearest-neighbour exchange and the interaction between the central MnIII ion and the two CrIII ions is equivalent.21 The free MnIII anion was included as a constant term (χMT = (Ng2β2/3k)SMn2(SMn2 + 1)). The best fit between 14 and 300 K gave g = 1.91 and J = −0.63 cm−1 (R = 7.32 × 10−3). The magnetic susceptibilities conform well to the Curie–Weiss law above 50 K, giving a Curie constant C = 9.11 cm3 K mol−1 and a Weiss constant θ = −5.95 K (inset of Fig. 5). The negative θ also confirms the antiferromagnetic coupling in complex 2. The field dependence of the magnetization of complex 2 measured at 1.8 K (Fig. S6, ESI†) with a value of 10.18 Nβ at 70 kOe is obviously lower than the value of uncoupled two MnIII (S = 2) and two CrIII (S = 3/2) based on g = 2.0, confirming the antiferromagnetic coupling interaction between CrIII and MnIII ions bridged by the cyanide group.10e
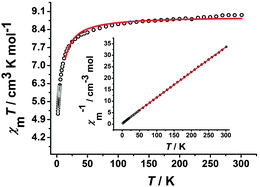 |
| | Fig. 5 Temperature-dependent χMT product for 2 at 100 Oe. The red solid line represents the best fits of the data. Inset: Plots of 1/χMversus T. The red solid line is the fitting result by Curie–Weiss law. | |
For neutral 1D single chain complex 3, the room-temperature χMT value is 3.35 cm3 K mol−1 (Fig. 6), close to the value of 3.38 cm3 K mol−1 expected for a low-spin FeIII center (S = 1/2) and a high-spin MnIII center (S = 2) assuming g = 2.00 and no exchange coupling. Between 300 and 60 K, the χMT product gradually decreases and reaches a minimum value of 3.20 at 60 K. While the χMT product increases to a maximum value of 3.24 cm3 K mol−1 at 16 K and then decreases further to 2.82 cm3 K mol−1 at 1.8 K, suggesting the presence of antiferromagnetic coupling between FeIII and MnIII centers with the cooperation of ZFS of the MnIII ion.6i Due to a long Mn⋯Mn distance of 10.093(3) Å within the chain, the intrachain magnetic interactions between MnIII and MnIII ions are very weak and can be negligible. Only the magnetic couplings between neighbouring FeIII and MnIII ions through cyanide bridges are considered. In order to evaluate the strength of intra-/inter-dimer magnetic coupling (J/zJ′) and the zfs parameter (D) of Mn(III), the best fit for the experimental magnetic data above 60 K is given based on the equation:6i
affording the corresponding parameters of
g = 2.00,
J = −1.01 cm
−1,
D = −1.34 cm
−1 and
zj′ = −0.01 cm
−1 (
R = 1.6 × 10
−4). It should be mentioned that based on the powder magnetic susceptibility data it is difficult to determine the sign of
DMn. The calculated
DMn value (−1.34 cm
−1) is normal for high-spin tetragonally elongated octahedral Mn(
III).
6j While the Curie constant
C = 3.40 cm
3 K mol
−1 and the Weiss constant
θ = −4.81 K can be obtained by fitting the magnetic susceptibilities above 50 K using the Curie–Weiss law (inset of
Fig. 6). The negative
θ indicates the presence of antiferromagnetic coupling in complex
3. The curve of the field-dependent magnetization for complex
3 at 1.8 K attains a maximum value of 3.53
Nβ at 70 kOe (Fig. S7, ESI
†), which is obviously lower than the value of the uncoupled Fe
IIIMn
III unit based on
g = 2.0, confirming again the existence of the antiferromagnetic coupling interaction between the neighbouring Fe
III and Mn
III ions and/or the ZFS effect of the Mn
III ion.
6i
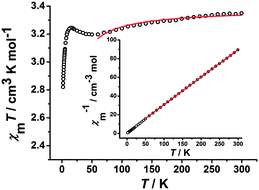 |
| | Fig. 6 Temperature-dependent χMT product for 3 at 100 Oe. The red solid line represents the best fits of the data. Inset: Plots of 1/χMversus T. The red solid line is the fitting result by Curie–Weiss law. | |
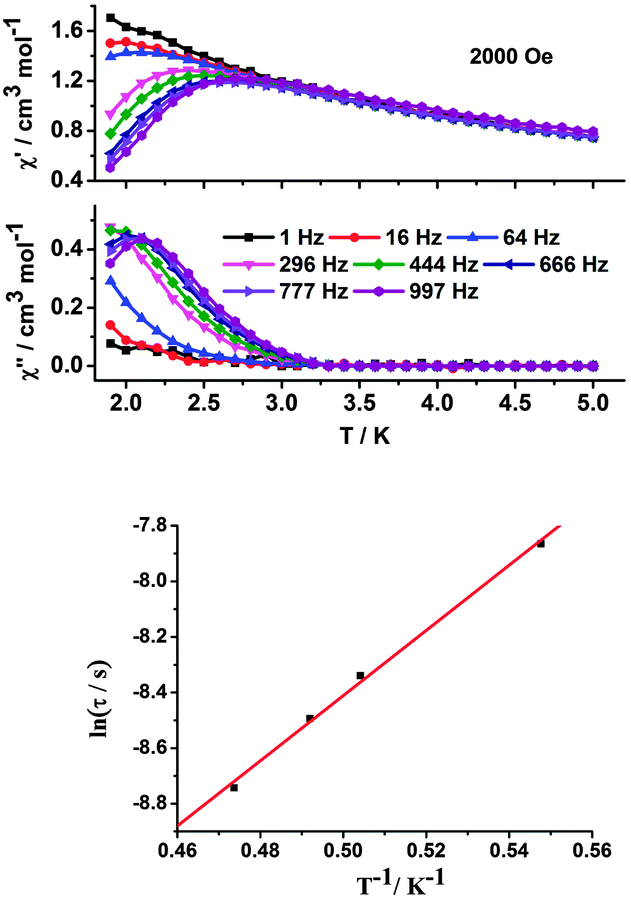
To investigate the magnetization dynamics, ac magnetic susceptibility measurements were performed on all the complexes at zero dc field. For complexes 1 and 2, neither peaks nor obvious temperature-dependent in-phase (χ′) and out-of-phase (χ′′) signals were observed at a frequency of 997 Hz and at temperatures down to 1.9 K (Fig. S8 and S9, ESI†). While the χ′′ component of the ac susceptibility for complex 3 increases with lowering temperature, suggesting the presence of slow relaxation effects. Unfortunately, the maxima on the χ′′ vs. T curves cannot be found down to 1.9 K, even at 997 Hz (Fig. S10, ESI†). Applying a static dc field of 2000 Oe, the clear peaks of the χ′′ signal can be easily found at frequencies higher than 444 Hz (Fig. 7, top), indicating the presence of thermally activated relaxation processes under such an external applied magnetic field. Fitting the data to the Arrhenius law [τ = τ0exp(Δ/kBT)] affords anisotropic energy barrier Δ/kB = 11.73 K and pre-exponential factor τ0 = 6.3 × 10−7 s (R = 0.998) (Fig. 7, bottom), which are consistent with the expected τ0 of 10−6–10−11 s for typical SMMs.22
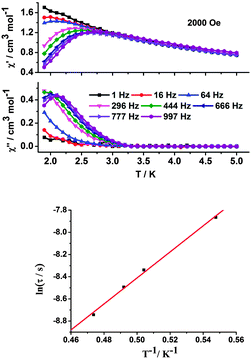 |
| | Fig. 7 Temperature-dependent in-phase (χ′) and out-of-phase (χ′′) ac susceptibilities for complex 3 under 2000 Oe dc field at 1–997 Hz. The solid lines are guides only (top) and lnτ versus T−1 plots for complex 3. Red line represents the best fit to the Arrhenius law (bottom). | |
Magneto-structural correlation
It has been experimentally shown that the magnetic coupling between low-spin FeIII and high-spin MnIII is ferromagnetic arising from the strict orthogonality of the magnetic orbitals (t2g in FeIIIvs. dz2 in MnIII) or antiferromagnetic arising from the orbital overlap between them.6i,23 The overall magnetic properties depend on which contribution is predominant. Some important structural parameters, such as the Mn–N(axial) lengths and Mn–NC angles, affect the magnetic exchange coupling. The analysis shows that the bending of the Mn–NC–Fe linkages and the rotation of the x and z axes for MnIII can enlarge the overlap and therefore weaken the ferromagnetic contribution. In our paper, the length of Mn–N(equatorial) is shorter than the Mn–N(axial), and the Mn–NC bond angle (168.8(3)° for 1 and 164.4(7)° for 3) deviates significantly from linearity, indicating the obviously distorted octahedron surrounding the central Mn ion, associated with the ferromagnetic and antiferromagnetic interaction between neighbouring MnIII and FeIII ions for complexes 1 and 3, respectively. By investigating some similar cyanide-bridged heterobimetallic FeIII–MnIII complexes reported previously,6i,10d,10f,23,24 it can also be found that the ferromagnetic interactions become weak while the antiferromagnetic interactions become strong with the bending of the Mn–NC angles. However, the antiferromagnetic coupling interaction can usually be investigated through the orbital overlap between t2g orbitals of CrIII and MnIII for the cyanide-bridged CrIII–MnIII complexes,25 similar to complex 3 as we reported.
Conclusions
Three cyanide-bridged heterobimetallic complexes have been synthesized based on tricyanometalate building blocks and various metalloporphyrinate precursors. Complexes 1 and 2 show similar molecular structures composed of an anionic trimer and a cationic monomer. However, the introduction of manganese(III) tetra(4-pyridyl)porphyrinate precursor results in the formation of an unprecedented neutral one-dimensional zigzag single chain complex 3. Variable temperature direct current magnetic measurements reveal the ferromagnetic and antiferromagnetic interaction between MnIII and FeIII centers for complexes 1 and 3, respectively, and antiferromagnetic coupling between CrIII and MnIII centers for complex 2. While dynamic magnetic properties confirm that only complex 3 exhibits the slow magnetic relaxation behavior of a single-molecule magnet. More studies focusing on designing and synthesizing novel cyanide-bridged magnetic complexes including such highly conjugated and functionalized metallomacrocyclic precursors with intriguing structures and unique chemical/physical properties are underway in our laboratory.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21401085 and 21401101), the Natural Science Foundation of Jiangsu Province (BK20140935), PAPD of Jiangsu Higher Education Institutions, and the Natural Science Foundation of Jiangsu Normal University (14XLR031).
Notes and references
-
(a) L. M. C. Beltran and J. R. Long, Acc. Chem. Res., 2005, 38, 325–334 CrossRef CAS PubMed;
(b) M. Ohba and H. Okawa, Coord. Chem. Rev., 2000, 198, 313–328 CrossRef CAS;
(c) M. Atanasov, P. Comba, S. Hausberg and B. Martin, Coord. Chem. Rev., 2009, 253, 2306–2314 CrossRef CAS;
(d) H.-L. Sun, Z.-M. Wang and S. Gao, Coord. Chem. Rev., 2010, 254, 1081–1100 CrossRef CAS;
(e) R. Lescouezec, L. M. Toma, J. Vaissermann, M. Verdaguer, F. S. Delgado, C. Ruiz-Pérez, F. Lloret and M. Julve, Coord. Chem. Rev., 2005, 249, 2691–2729 CrossRef CAS.
-
(a) J. H. Lim, J. H. Yoon, H. C. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2006, 45, 7424–7426 CrossRef CAS PubMed;
(b) C.-F. Wang, J.-L. Zuo, B. M. Bartlett, S. You, J. R. Long and X.-Z. You, J. Am. Chem. Soc., 2006, 128, 7162–7163 CrossRef CAS PubMed;
(c) S. Wang, J.-L. Zuo, H.-C. Zhou, H. J. Choi, Y. Ke, J. R. Long and X.-Z. You, Angew. Chem., Int. Ed., 2004, 43, 5940–5943 CrossRef CAS PubMed.
-
(a) S. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef PubMed;
(b) J. T. Culp, J.-H. Park, F. Frye, Y.-D. Huh, M. W. Meisel and D. R. Talham, Coord. Chem. Rev., 2005, 249, 2642–2648 CrossRef CAS.
-
(a) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS;
(b) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed;
(c) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed;
(d) D. A. Garanin and E. M. Chudnovsky, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 11102–11118 CrossRef CAS.
-
(a) S. Wang, X.-H. Ding, Y.-H. Li and W. Huang, Coord. Chem. Rev., 2012, 256, 439–464 CrossRef CAS;
(b) S. Wang, X.-H. Ding, J.-L. Zuo, X.-Z. You and W. Huang, Coord. Chem. Rev., 2011, 255, 1713–1732 CrossRef CAS;
(c) K. S. Pedersen, J. Bendix and R. Clérac, Chem. Commun., 2014, 50, 4396–4415 RSC;
(d) L. Cui, F. Zhu, C. F. Leong, J. Ru, F. Gao, D. M. D'Alessandro and J. Zuo, Sci. China: Chem., 2015, 58, 650–657 CrossRef CAS;
(e) E. Gungor, Y. Yahsi, H. Kara and A. Caneschi, CrystEngComm, 2015, 17, 3082–3088 RSC;
(f) Y.-Z. Zhang, P. Ferko, D. Siretanu, R. Ababei, N. P. Rath, M. J. Shaw, R. Clérac, C. Mathonière and S. M. Holmes, J. Am. Chem. Soc., 2014, 136, 16854–16864 CrossRef CAS PubMed;
(g) D.-Y. Wu and O. Sato, Z. Anorg. Allg. Chem., 2009, 635, 389–392 CrossRef CAS;
(h) T. Delgado, A. Tissot, C. Desnard, L. Guénée, P. Pattison and A. Hauser, Chem. – Eur. J., 2015, 21, 3664–3670 CrossRef CAS PubMed;
(i) X.-J. Xu, R.-R. Zhou, J. Wang, L. Li and J.-Q. Tao, Z. Anorg. Allg. Chem., 2015, 641, 490–494 CrossRef CAS.
-
(a) D. Pinkowicz, H. Southerland, X.-Y. Wang and K. R. Dunbar, J. Am. Chem. Soc., 2014, 136, 9922–9924 CrossRef CAS PubMed;
(b) Y.-Y. Chem, A.-Y. Hu, M.-Q. Shen, X.-Y. Tian and H. Zhou, Z. Anorg. Allg. Chem., 2014, 640, 2287–2291 CrossRef;
(c) P.-F. Zhuang, Y.-J. Zhang, H. Zheng, C.-Q. Jiao, L. Zhao, J.-L. Wang, C. He, C.-Y. Duan and T. Liu, Dalton Trans., 2015, 44, 3393–3398 RSC;
(d) J. J. Sokol, A. G. Hee and J. R. Long, J. Am. Chem. Soc., 2002, 124, 7656–7657 CrossRef CAS PubMed;
(e) M.-X. Yao, Q. Zheng, X.-M. Cai, Y.-Z. Li, Y. Song and J.-L. Zuo, Inorg. Chem., 2012, 51, 2140–2149 CrossRef CAS PubMed;
(f) M.-X. Yao, Z.-Y. Wei, Z.-G. Gu, Q. Zheng, Y. Xu and J.-L. Zuo, Inorg. Chem., 2011, 50, 8636–8644 CrossRef CAS PubMed;
(g) D. Zhang, S. Zhuo, P. Wang, X. Chen and J. Jiang, New J. Chem., 2014, 38, 5470–5479 RSC;
(h) H. Zhou, Y. Wang, F. Mou, X. Shen and Y. Liu, New J. Chem., 2014, 38, 5925–5934 RSC;
(i) Z.-H. Ni, J. Tao, W. Wernsdorfer, A.-L. Cui and H.-Z. Kou, Dalton Trans., 2009, 2788–2794 RSC;
(j) D. Visinescu, L. M. Toma, J. Cano, O. Fabelo, C. Ruiz-Pérez, A. Labrador, F. LIoret and M. Julve, Dalton Trans., 2010, 39, 5028–5038 RSC.
-
(a) E. L. Gavey and M. Pilkington, Coord. Chem. Rev., 2015, 296, 125–152 CrossRef CAS;
(b) G. Kumar and R. Gupta, Chem. Soc. Rev., 2013, 42, 9403–9453 RSC.
-
(a) C. M. B. Carvalho, T. J. Brocksom and K. T. de Oliveira, Chem. Soc. Rev., 2013, 42, 3302–3317 RSC;
(b) J. Otsuki, Coord. Chem. Rev., 2010, 254, 2311–2341 CrossRef CAS;
(c) K.-J. Lin, Angew. Chem., Int. Ed., 1999, 38, 2730–2732 CrossRef CAS;
(d) L. Pan, S. Kelly, X. Huang and J. Li, Chem. Commun., 2002, 2334–2335 RSC.
-
(a) W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC;
(b) H. L. Buckley and J. Arnold, Dalton Trans., 2015, 44, 30–36 RSC.
-
(a) D. Zhang, Z. Zhao, P. Wang and Z. Ni, CrystEngComm, 2013, 15, 2504–2511 RSC;
(b) S. Huh, K.-T. Youm, Y. J. Park, A. J. Lough, M. Ohba and M.-J. Jun, Bull. Korean Chem. Soc., 2005, 26, 1031–1032 CrossRef CAS;
(c) D. Zhang, L.-F. Zhang, Y. Chen, H. Wang, Z.-H. Ni, W. Wernsdorfer and J. Jiang, Chem. Commun., 2010, 46, 3550–3552 RSC;
(d) D. Zhang, H. Wang, L. Tian, H.-Z. Kou, J. Jiang and Z.-H. Ni, Cryst. Growth Des., 2009, 9, 3989–3996 CrossRef CAS;
(e) D. Zhang, H. Wang, Y. Chen, Z.-H. Ni, L. Tian and J. Jiang, Inorg. Chem. Commun., 2009, 12, 698–700 CrossRef CAS;
(f) G.-L. Li, J. Nie, H. Chen, Z.-H. Ni, Y. Zhao and L.-F. Zhang, Inorg. Chem. Commun., 2012, 19, 66–69 CrossRef CAS;
(g) X. Chen, S.-Q. Wu, A.-L. Cui and H.-Z. Kou, Chem. Commun., 2014, 50, 2120–2122 RSC.
- D. Li, S. Parkin, G. Wang, G. T. Yee and S. M. Holmes, Inorg. Chem., 2006, 45, 1951–1959 CrossRef CAS PubMed.
- T. D. Harris and J. R. Long, Chem. Commun., 2007, 1360–1362 RSC.
-
(a) E. Fagadar-Cosma, M. C. Mirica, I. Balcu, C. Bucovicean, C. Cretu, I. Armeanu and G. Fagadar-Cosma, Molecules, 2009, 14, 1370–1388 CrossRef CAS PubMed;
(b) L. Wang, Y. She, R. Zhong, H. Ji, Y. Zhang and X. Song, Org. Process Res. Dev., 2006, 20, 757–761 CrossRef.
- N. Datta-Gupta, J. C. Fanning and L. L. Dickens, J. Coord. Chem., 1976, 5, 201–207 CrossRef CAS.
-
E. A. Boudreaux and L. N. Mulay, Theory and Application of Molecular Paramagnetism, John Wiley & Sons, New York, 1976, pp. 491–494 Search PubMed.
-
Bruker; SMART, SAINT and XPREP: Area Detector Control and Data Integration and Reduction Software, Bruker Analytical Xray Instruments Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
-
G. M. Sheldrick, SADABS: Empirical Absorption and Correction Software, University of Göttingen, Göttingen, Germany, 2012 Search PubMed.
-
G. M. Sheldrick, SHELXS-2014 and SHELXL-2014, Program for X-ray Crystal Structure Determination, Göttingen University, Göttingen, Germany, 2014 Search PubMed.
- Platon Program: A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
-
(a) H. Miyasaka, H. Ieda, N. Matsumoto, K. Sugiura and M. Yamashita, Inorg. Chem., 2003, 42, 3509–3515 CrossRef CAS PubMed;
(b) H. J. Choi, J. J. Sokol and J. R. Long, Inorg. Chem., 2004, 43, 1606–1608 CrossRef CAS PubMed;
(c) H. Miyasaka, H. Okawa, A. Miyazaki and T. Enoki, Inorg. Chem., 1998, 37, 4878–4883 CrossRef CAS PubMed.
-
O. Kahn, Molecule Magnetism, VCH, Weinhem, Germany, 1993 Search PubMed.
-
(a) S. M. J. Aubin, Z. Sun, L. Pardi, J. Krzystek, K. Folting, L. C. Brunel, A. L. Rheingold, G. Christou and D. N. Hendrickson, Inorg. Chem., 1999, 38, 5329–5340 CrossRef CAS;
(b) K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS;
(c) N. Ishii, Y. Okamura, S. Chiba, T. Nogami and T. Ishida, J. Am. Chem. Soc., 2008, 130, 24–25 CrossRef CAS PubMed.
-
(a) H. Miyasaka, N. Matsumoto, H. Okawa, N. Re, E. Gallo and C. Floriani, J. Am. Chem. Soc., 1996, 118, 981–994 CrossRef CAS;
(b) M. Ferbinteanu, H. Miyasaka, W. Wernsdorfer, K. Nakata, K. Sugiura, M. Yamashita, C. Coulon and R. Clérac, J. Am. Chem. Soc., 2005, 127, 3090–3099 CrossRef CAS PubMed;
(c) Z.-H. Ni, L.-F. Zhang, V. Tangoulis, W. Wernsdorfer, A.-L. Cui, O. Sato and H.-Z. Kou, Inorg. Chem., 2007, 46, 6029–6037 CrossRef CAS PubMed.
-
(a) H.-R. Wen, C.-F. Wang, Y.-Z. Li, J.-L. Zuo, Y. Song and X.-Z. You, Inorg. Chem., 2006, 45, 7032–7034 CrossRef CAS PubMed;
(b) H. Miyasaka, H. Okawa, A. Miyazaki and T. Enoki, Inorg. Chem., 1998, 37, 4878–4883 CrossRef CAS PubMed;
(c) W.-W. Ni, Z.-H. Ni, A.-L. Cui, X. Liang and H.-Z. Kou, Inorg. Chem., 2007, 46, 22–33 CrossRef CAS PubMed.
-
(a) H. J. Choi, J. J. Sokol and J. R. Long, Inorg. Chem., 2004, 43, 1606–1608 CrossRef CAS PubMed;
(b) D. Zhang, H. Wang, Y. Chen, Z.-H. Ni, L. Tian and J. Jiang, Inorg. Chem., 2009, 48, 11215–11225 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The supplementary tables, figures, additional characterization data and X-ray crystallographic files. CCDC 1417664 (1), 1417665 (2), and 1417666 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj02132d |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.