
CO2 capture in the presence of water vapour in MIL-53(Al)†‡
Mayra
Sánchez-Serratos
a,
Peter A.
Bayliss
b,
Ricardo A.
Peralta
a,
Eduardo
González-Zamora
*c,
Enrique
Lima
a and
Ilich A.
Ibarra
*a
aInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del. Coyoacán, 04510, México D. F., Mexico. E-mail: argel@unam.mx
bSchool of Chemistry, University of Nottingham, University Park, NG7 2RD, UK
cDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, San Rafael Atlixco 186, Col. Vicentina, Iztapalapa, C. P. 09340, México D. F., Mexico
First published on 6th November 2015
Abstract
MIL-53(Al) shows a CO2 capture of 3.5 wt% by kinetic uptake experiments, under anhydrous conditions at 30 °C. When this material is exposed to water vapour (20% RH and 30 °C), there is a considerable 1.5-fold increase in the CO2 capture up to 5.2 wt%.
Global warming and the resulting climate change is one of the biggest threats that our society has to solve. The cumulative carbon dioxide (CO2) emissions in the atmosphere are continuously rising due to anthropogenic activities and these, inadvertently, generate the undesirable greenhouse gas effect.1 The accelerating global energy demands and consumption of carbon-based fuels are the main causes of the increasing CO2 levels.2 To address these problems many countries have been motivated to invest in capturing and permanently sequestering CO2, requiring the development of new methods for efficient CO2 capture.3
The absorption of CO2 by aqueous solutions of amines, which take advantage of the Lewis acidity of CO2, have been widely studied. However they also have many major limitations as an absorbent for industrial CO2 capture due to thermal instability, and corrosion on vessels and pipelines.4 Therefore, the use of porous solids as an alternative medium for the adsorption of CO2 is a timely research area. The search for materials with a high adsorption capacity, structural stability, high tolerance to humidity, fast sorption kinetics and mild regeneration properties, remains a major challenge for practical applications.
Porous coordination polymers (PCPs) or metal–organic frameworks (MOFs) are amongst the most promising candidates for gas separation; due to the ability to selectively adsorb small molecules. This selectivity can be tuned as a function of the topology and chemical composition of the micropores.5,6 Although PCPs can exhibit high CO2 capacity and selectivity in the absence of water, many gas separation processes involve the exposure to water vapour. Water molecules can compete with gas molecules for the active sites (within PCPs) or disrupt the bonding between the organic ligand and metal, resulting in the collapse of the structure.7 Therefore capturing CO2 from real flue gas (high humidity and high temperature) is indeed a great challenge. Recently, a considerable number of PCPs have been reported with relatively good stability to water, for example: UiO-66,8 NOTT-401,9 MIL-100,10 MIL-101,11 MIL-5312 and InOF-1.13 A water stable MOF (Cu(bcppm)H2O, H2bcppm = bis(4-(4-carboxyphenyl)-1H-pyrazolyl)methane) that also showed exceptionally selective separations for CO2 over N2 was reported by Doonan et al.14
In addition to causing structural instability, direct contact between water and PCPs can seriously reduce their gas storage capacity; with exposure to water often unfavourable to gas separations.15 The effect of water on CO2 capture in PCPs has only recently been investigated.16–19 Llewellyn and co-workers20 investigated the CO2 adsorption in some PCPs under different relative humidities. Yaghi et al.21 showed that the presence of hydroxyl functional groups increase the affinity of the framework for water.
MIL-53 frameworks, first reported by Serre et al.,22 are a very interesting and well-studied series of PCPs. In the present work we have chosen an Al(III) based, water-stable12 material entitled MIL-53(Al), to study the CO2 capture in the presence of water vapour. This material is built up of infinite trans chains of corner-sharing (via OH groups) AlO4(OH)2 octahedra interconnected by BDC2− ligands (H2BDC = 1,4-benzenedicarboxylic acid).
We recently reported the synthesis of MIL-53(Al) via a continuous flow process, using solely water as the reaction medium and requiring a residence time of only 5–6 min.23 Through this approach we obtained large amounts of MIL-53(Al) which was calcined (extraction of terephthalic acid from within the pores) by heating in the oven at 330 °C for 3 days. Thermogravimetric analysis (calcined MIL-53(Al)) (see Fig. S1, ESI‡) and bulk powder X-ray diffraction patterns (see Fig. S2, ESI‡) of the calcined MIL-53(Al) confirmed that the material retains its structural integrity upon terephthalic acid removal. N2 adsorption isotherms for activated MIL-53(Al) at 77 K were used to calculate the BET surface area (0.01 < P/P0 < 0.04) of 1096 m2 g−1.
Dynamic and isothermal CO2 experiments were carried out on MIL-53(Al) (see Experimental). Fig. 1 (left) shows the kinetic uptake experiments at 30 °C and 50 °C. These two capture temperatures were chosen because they are of great interest in post-combustion CO2 capture processes.3b The weight gain, which represents the amount of CO2 captured, was higher at 30 °C than at 50 °C. At 30 °C the maximum uptake of 3.5 wt% was reached after 10 min and remained constant until the end of the experiment (120 min). At 50 °C the uptake was measured to be 2.1 wt% which was also reached after around 10 min (Fig. 1, left). As the temperature is increased (from 30 to 50 °C), the weight of CO2 adsorbed decreased.
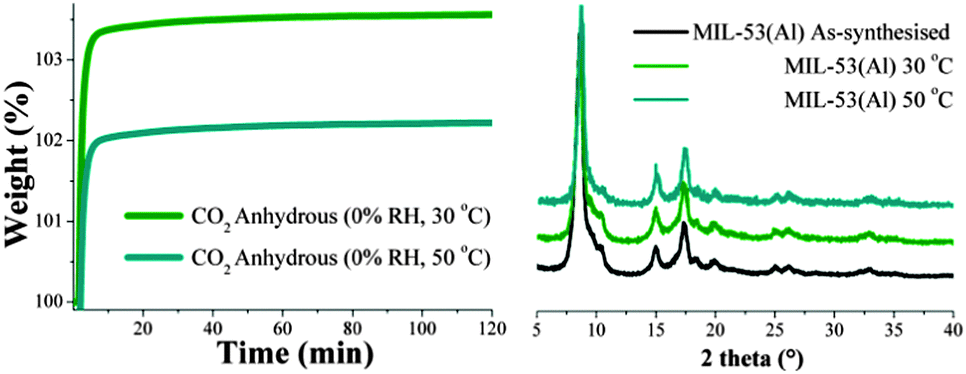 | ||
| Fig. 1 (left) Kinetic uptake experiments performed at different temperatures (30 and 50 °C) with a CO2 flow of 60 mL min−1; (right) PXRD patterns of MIL-53(Al) samples before and after the kinetic CO2 isotherms. | ||
In order to confirm the decrease in CO2 capture with temperature was not due to sample degradation, PXRD measurements were carried out on both samples after CO2 capture experiments. Fig. 1 (right) shows that the crystallinity of the samples was retained after each CO2 capture experiment.
For comparison, dynamic and isothermal CO2 experiments were run on an inorganic mesoporous molecular sieve entitled MCM-41. MCM-41 is a very well-known porous material that has been used in a range of fields including; ion exchange, catalysis, sensing, drug delivery and gas adsorption.24 The chemical environment and pore dimensions of MCM-41 (a mesoporous inorganic material), are very different to those of MIL-53(Al) (a microporous inorganic–organic material). Therefore, a comparison of the CO2 uptake properties of both materials can provide a better understanding of the main characteristics required for an ideal CO2 capturing material.
A sample of MCM-4125 was placed in a thermobalance (see Experimental) and at 30 °C exhibited a maximum CO2 uptake of 1.0 wt% (see Fig. S3, ESI‡). This uptake was reached after 40 min and remained constant until 120 min (end of the experiment). At 50 °C the CO2 uptake was 0.6 wt% and was also reached after around 40 min (see Fig. S3, ESI‡). PXRD experiments demonstrated the retention of the sample crystallinity after each CO2 capture experiments (see Fig. S4, ESI‡).
Motivated by the very interesting results previously reported by Llewellyn et al.,20 (5-fold increase in CO2 uptake for MIL-100(Fe) in the presence of water); kinetic isotherm experiments were carried out at different temperatures and relative humidities. A sample of MIL-53(Al) was placed into a humidity-controlled thermobalance and after activation of the material, the equipment was allowed to stabilise at 20% RH and 30 °C. A constant CO2 flow (60 mL min−1) was started and the change in weight of the sample was measured for 120 minutes. To measure the water uptake in the absence of CO2, this experimental procedure was repeated on a different sample of MIL-53(Al), without the constant CO2 flow. Fig. 2 (left) shows the kinetic uptake experiments at 30 °C for CO2 + H2O (20% RH) and only H2O (20% RH). In both isotherms, the material shows an increase in weight with time, due the contributions of CO2 + H2O or only H2O, respectively.
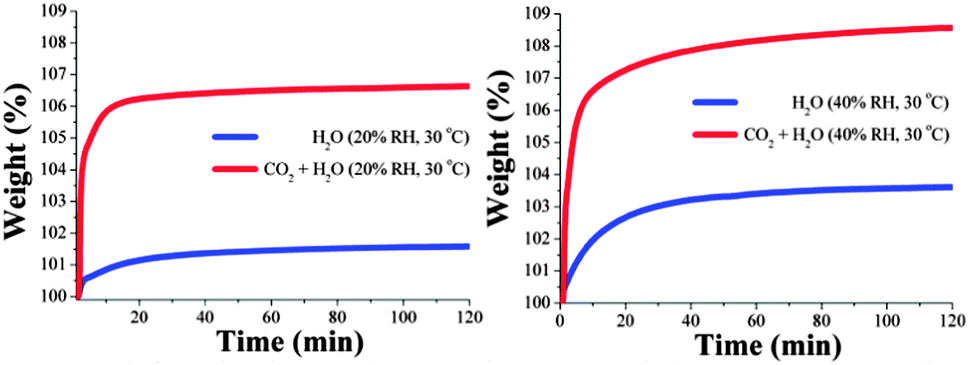 | ||
| Fig. 2 (left) Kinetic uptake experiments carried out at 30 °C using MIL-53(Al) and 20% RH with CO2 + H2O (red line) and only H2O (blue line); (right) kinetic uptake experiments carried out at 30 °C and 40% RH with CO2 + H2O (red line) and only H2O (blue line). | ||
In order to find the maximum CO2 capture at 20% RH and 30 °C, we need to remove the contribution of water from the total weight increase. The CO2 capture at 20% RH is simply the difference between the two isotherms (CO2 + H2O and H2O). In Fig. 2 (left) the gradual weight increase (for CO2 + H2O and H2O) starts at 0 min and stabilises at ∼20 min interestingly, under anhydrous conditions the CO2 uptake reached stability faster, at approximately 10 min (see Fig. 1, left). This equilibrium discrepancy is due to the nature of the water vapour adsorption process that in general takes considerably more time to reach equilibrium than the gas adsorption process in microporous materials.26 From 10 min to 120 min (end of the experiment), the maximum amounts of CO2 + H2O and only H2O captured were 106.7 wt% and 101.5 wt%, respectively and by taking the difference of these two values the CO2 capture in the material was ∼5.2 wt%. Therefore, there was a 1.5 fold increase in the CO2 capture from 3.5 wt% under anhydrous conditions to 5.2 wt% with 20% RH. This enhancement in CO2 uptake in the presence of water can be explained by CO2 confinements effects induced by bulky molecules (H2O).27 Indeed, Walton and co-workers28 proposed that functional groups (such as OH) act as directing agents for water in the pores, which allows for more efficient packing.
In order to obtain additional evidence that the increase in uptake is an increase in the CO2 capture and not an increase in the amount of water adsorbed, we carried out an additional kinetic isotherm experiment. Thus, an activated MIL-53(Al) sample (vide supra) was placed into a humidity-controlled thermobalance (at 30 °C) and the equipment was stabilised at 20% RH. After the equilibrium was reached, we open a constant CO2 flow (60 mL min−1), Fig. 3. In Fig. 3 the gradual weight increase (only H2O) starts at 0 min and stabilises at around 20 min. From 20 min to 95 min the H2O uptake was constant (plateau) and at 95 min the CO2 flow was opened and a sharp weight gain, which reached stability at approximately 110 min, was observed (see Fig. 3). From 110 min to 180 min (end of the experiment), the maximum amount of CO2 captured corresponds to ∼5.2 wt%.
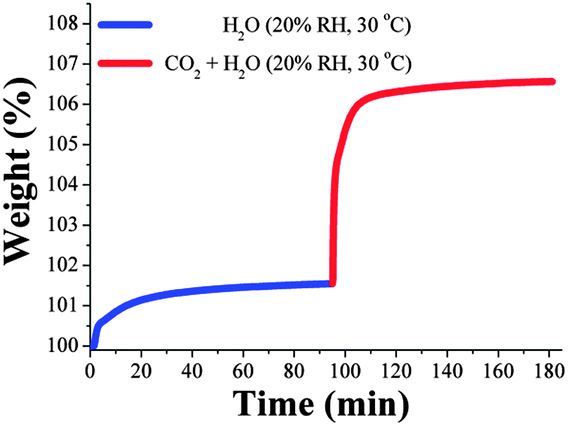 | ||
| Fig. 3 Kinetic uptake experiment carried out at 30 °C and 20% RH with H2O (blue line) and CO2 (red line). | ||
Later, kinetic uptake experiments were performed on an activated sample of MIL-53(Al) at 30 °C and 40% RH. Fig. 2 (right) shows a gradual weight increase (for CO2 + H2O and H2O) which starts at 0 min and stabilises at around 40 min. Thus, when the relative humidity (RH) was increased from 20 to 40% the stabilisation time was also considerably increased. At 40% RH the maximum quantities of CO2 + H2O and H2O captured were 108.5 wt% and 103.7 wt%, respectively and by taking the difference of these two values the CO2 capture was approximately 4.8 wt%.
This value represents an increase in CO2 capture relative to the anhydrous value of 3.5 wt% but a decrease from the value of 5.2 wt% under 20% RH. We rationalised that at higher water loadings (e.g. 40% RH) the directing effect of the OH groups is reduced due to an increase in the water disorder (within the pore) caused by thermal agitation and therefore, diminishing the CO2 capture.
To explore the effect of temperature on the capture of CO2 under humid conditions, kinetic uptake experiments were performed on activated samples of MIL-53(Al) at 20% RH and 50 °C. This humidity was chosen as a higher CO2 capture and shorter stabilisation time were obtained at 20% RH than 40% RH for the same material at 30 °C. Fig. 4 shows the gradual weight increase for CO2 + H2O and H2O under these conditions. The total CO2 capture value was 3.1 wt% with a stabilisation time of ∼20 min. Thus, the CO2 capture at 50 °C was approximately 1.5 fold increased: from 2.1 wt% under anhydrous conditions to 3.1 wt% with 20% RH.
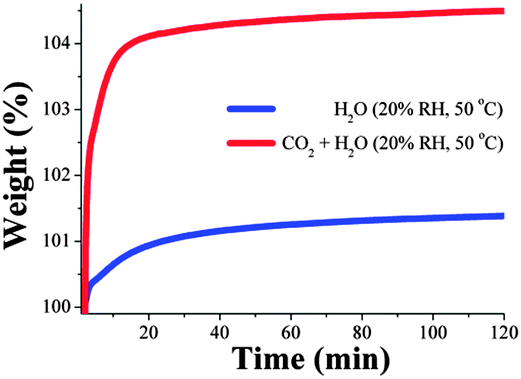 | ||
| Fig. 4 Kinetic uptake experiments carried out at 50 °C and 20% RH with CO2 + H2O (red line) and only H2O (blue line). | ||
To confirm that there was no sample degradation, PXRD measurements and N2 adsorption isotherms (BET surface area) were carried out on all the samples after CO2 capture experiments (see Fig. S5, ESI‡), which demonstrated that the crystallinity and the surface area of the samples were retained. These results are very promising for the application of PCPs in a more realistic CO2 capture scenario: humidity conditions and relatively high temperature (50 °C).
Kinetic isotherm experiments were also carried out on activated samples of MCM-41 at 30 °C with 20% RH and 40% RH (see Fig. S6 and S7, ESI‡). The total CO2 capture values were 1.1 and 1.2 wt% respectively and in both cases the stabilisation times were around 50 min. These values represent a small CO2 capture improvement, (from anhydrous conditions to 20 and 40% RH), from 1.0 to 1.1 wt% at 20% RH and 1.2 wt% at 40% RH. PXRD measurements confirmed the retention of the crystallinity after each CO2 experiment (see Fig. S8, ESI‡).
For MIL-53(Al) the capture of CO2 in the presence of water is considerably higher than that of MCM-41 and the stabilisation time is also shorter. Since MIL-53(Al) is a microporous material, the confinement effects27 can play a greater role than in the mesoporous material MCM-41 as we previously observed in Sc(III)-based water stable microporous materials.29
In addition, we decided to run a CO2 uptake experiment (60 mL min−1) at 20% RH and 30 °C on a non-porous sample to provide a direct CO2 capture comparison to MIL-53(Al), a microporous material. PCM-1430 was chosen and a sample was activated at 150 °C for 1 h, under a flow of N2 gas, (since PCM-14 is a non-porous material, when activated between 25–150 °C). Under anhydrous conditions, from 0 to 120 min the maximum CO2 uptake was 0.3 wt% (see Fig. S9, ESI‡). From 0 min to 120 min the maximum CO2 uptake (under 20% RH) was 0.7 wt% (see Fig. S10, ESI‡). This result corroborated that there is no increase in CO2 sequestration in a non-porous material when the relative humidity is 20% at 30 °C.
Finally, we run CO2 sorption studies under static mode on MIL-53 (see Fig. S11, ESI‡). The CO2 capture was 9.6 wt% which is considerably higher than under dynamic conditions (3.5 wt%). However, the main objective of the present work is to demonstrate, in a more realistic scenario, how MIL-53(Al) performs when it is exposed to a constant CO2 flow gas (60 mL min−1) and under humidity conditions. Additionally, we carried out a water uptake, static, experiment at 30 °C (see Fig. S12, ESI‡) which showed a good correlation with the dynamic experiment (at 20 RH% P/P0, the water uptake was 1.5 wt%).
In summary, the Al(III) coordination polymer MIL-53(Al) shows, by kinetic isotherm experiments, a total CO2 uptake of 3.5 wt% at 30 °C, which was rapidly reached after only approximately 10 min. CO2 uptakes were measured using MIL-53(Al) under different relative humidity conditions (20 and 40% RH) and temperatures (30 and 50 °C), displaying a maximum CO2 capture of approximately 5.2 wt% (20% RH and 30 °C). Significantly, this CO2 capture under humid conditions represents a 1.5-fold increase in comparison to anhydrous conditions. Both MCM-41 and PCM-14 exhibit lower CO2 capture in the presence of water, suggesting that the microporosity provided by MIL-53(Al) is fundamental for this capture process. The CO2 confinement effects induced by H2O27 can occur within the micropores of MIL-53(Al) which combined with the directing effect of the hydroxo functional groups allow CO2 to be accommodated more efficiently.28 MIL-53(Al) is an ideal candidate for use in more realistic post-combustion CO2 capture scenarios as it can be produced in a very short time, via continuous flow reactions,23 and can capture CO2 at relatively high temperatures (50 °C) in the presence of water.
Acknowledgements
The authors thank Dr A. Tejeda-Cruz (X-ray; IIM-UNAM). I. A. I thanks CONACyT (212318) and PAPIIT UNAM (IN100415), Mexico for financial support. E. G.-Z. thanks CONACyT (156801) and CONACyT (236879), Mexico for financial support. Thanks to U. Winnberg (ITAM and ITESM) for scientific discussions. We gratefully acknowledge the receipt of a University of Nottingham 2012 EPSRC Doctoral Prize to P. A. B. We thank Prof. M. Schröder for his encouragement and Prof. M. Poliakoff and R. A. Howie for prompting us to carry out the experiment shown in Fig. 3. We also thank RAH for advice on preparing the manuscript.Notes and references
- J. T. Litynski, S. M. Klara, H. G. McIlvried and R. D. Srivastava, Environ. Int., 2006, 32, 128 CrossRef CAS PubMed.
- M. Z. Jacobson, Energy Environ. Sci., 2009, 2, 148 CAS.
- (a) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, Z. T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed; (b) D. M. Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef PubMed.
- (a) G. T. Rochelle, Science, 2009, 325, 1652 CrossRef CAS PubMed; (b) F. Karadas, M. Atilhan and S. Aparicio, Energy Fuels, 2010, 24, 5817 CrossRef CAS.
- (a) S. Yang, G. S. B. Martin, G. J. J. Titman, A. J. Blake, D. R. Allan, N. R. Champness and M. Schröder, Inorg. Chem., 2011, 50, 9374 CrossRef CAS PubMed; (b) A. J. Nuñez, L. N. Shear, N. Dahal, I. A. Ibarra, J. W. Yoon, Y. K. Hwang, J.-S. Chang and S. M. Humphrey, Chem. Commun., 2011, 47, 11855 RSC.
- (a) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O’Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed; (b) A. M. Bohnsack, I. A. Ibarra, P. W. Hatfield, J. W. Yoon, Y. K. Hwang, J.-S. Chang and S. M. Humphrey, Chem. Commun., 2011, 47, 4899 RSC; (c) P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80 CrossRef CAS PubMed.
- (a) J. A. Greathouse and M. D. Allendorf, J. Am. Chem. Soc., 2006, 128, 1067 Search PubMed; (b) S. S. Han, S.-H. Choi and A. C. T. van Duin, Chem. Commun., 2010, 46, 5713 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- H. A. Lara-García, M. R. Gonzalez, J. H. González-Estefan, P. Sánchez-Camacho, E. Lima and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 442 RSC.
- K. A. Cychosz and A. J. Matzger, Langmuir, 2010, 26, 17198 CrossRef CAS PubMed.
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537 CrossRef CAS.
- J. Liu, F. Zhang, X. Zou, G. Yu, N. Zhao, S. Fan and G. Zhu, Chem. Commun., 2013, 49, 7430 RSC.
- J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696 RSC.
- W. M. Bloch, R. Babaro, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441 CrossRef CAS PubMed.
- (a) S. S. Nagarkar, A. K. Chaudhari and S. K. Ghosh, Inorg. Chem., 2012, 51, 572 CrossRef CAS PubMed; (b) H. J. Choi, M. Dincă, A. Daily and J. R. Long, Energy Environ. Sci., 2010, 3, 117 RSC.
- (a) J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2011, 27, 11451 CrossRef CAS PubMed; (b) A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368 CrossRef CAS PubMed; (c) H. Jasuja, Y.-G. Huang and K. S. Walton, Langmuir, 2012, 28, 16874 CrossRef CAS PubMed; (d) H. Jasuja, J. Zang, D. S. Sholl and K. S. Walton, J. Phys. Chem. C, 2012, 116, 23526 CrossRef CAS; (e) J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y.-G. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642 RSC.
- J. Liu, Y. Wang, A. I. Benin, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2010, 26, 14301 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 1 Search PubMed.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575 CrossRef CAS PubMed.
- E. Soubeyrand-Lenoir, C. Vagner, J. W. Yoon, P. Bazin, F. Ragon, Y. K. Hwang, C. Serre, J.-S. Chang and P. L. Llewellyn, J. Am. Chem. Soc., 2012, 134, 10174 CrossRef CAS PubMed.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369 CrossRef CAS PubMed.
- (a) G. Férey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guégan, Chem. Commun., 2003, 2976 RSC; (b) T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373 CrossRef CAS PubMed.
- P. A. Bayliss, I. A. Ibarra, E. Pérez, S. Yang, C. C. Tang, M. Poliakoff and M. Schröder, Green Chem., 2014, 16, 3796 RSC.
- X. S. Zhao, G. Q. Lu and G. J. Millar, Ind. Eng. Chem. Res., 1996, 35, 2075 CrossRef CAS.
- Q. Cai, W.-Y. Lin, F.-S. Xiao, W.-Q. Pang, X.-H. Chen and B. S. Zou, Micropor. Mesopor. Mater., 1999, 32, 1 CrossRef CAS.
- I. P. O’koye, M. Benham and K. M. Thomas, Langmuir, 1997, 13, 4054 CrossRef.
- (a) N. L. Ho, F. Porcheron and R. J.-M. Pellenq, Langmuir, 2010, 26, 13287 CrossRef CAS PubMed; (b) L. N. Ho, J. Perez-Pellitero, F. Porcheron and R. J.-M. Pellenq, Langmuir, 2011, 27, 8187 CrossRef CAS PubMed; (c) L. N. Ho, S. Clauzier, Y. Schuurman, D. Farrusseng and B. Coasne, J. Phys. Chem. Lett., 2013, 4, 2274 CrossRef CAS.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606 CrossRef CAS PubMed.
- (a) M. R. Gonzalez, J. H. González-Estefan, H. A. Lara-García, P. Sánchez-Camacho, E. I. Basaldella, H. Pfeiffer and I. A. Ibarra, New J. Chem., 2015, 39, 2400 RSC; (b) H. A. Lara-García, M. R. Gonzalez, J. H. González-Estefan, P. Sánchez-Camacho, E. Lima and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 442 RSC.
- I. A. Ibarra, K. E. Tan, V. M. Lynch and S. M. Humphrey, Dalton Trans., 2012, 41, 3920 RSC.
Footnotes |
| † Dedicated to Professor Pedro Bosch on the occasion of his retirement after 38 years of outstanding career. |
| ‡ Electronic supplementary information (ESI) available: TGA data, PXRDP data and kinetic uptake experiments. See DOI: 10.1039/c5nj02312b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |