
Organic nanotubes and belt shaped molecules based on norbornadiene tethers
Pablo A.
Denis
*a and
Michael
Yanney
b
aComputational Nanotechnology, DETEMA, Facultad de Química, UDELAR, CC 1157, 11800 Montevideo, Uruguay. E-mail: pablod@fq.edu.uy; Fax: +589229241906; Tel: +59899714280
bDepartment of Chemistry, Mississippi State University, Mississippi MS 39762, USA
First published on 23rd October 2015
Abstract
We have employed density functional theory to study and characterize a new family of belt shaped molecules which use the norbornadiene tether and aromatic molecules as linkers. Our results indicated that the inclusion of the norbornadiene unit eliminates the strain commonly associated with the synthesis of belt-shaped molecules. Polymerization of the latter proved to be an effective method towards the bottom-up synthesis of organic nanotubes with uniform properties. The band gap of the infinite nanotubes proposed can be engineered by changing the molecule which links the norbornadiene tethers. In effect, the gap of the pyrene/norbornadiene and coronene/norbornadiene based nanotubes were 2.9 eV and 0.9 eV, respectively, as indicated by HSE calculations. Among the six belts assayed, two can be used to separate the magic fullerenes C180 and C240, given that large interaction energies were found upon complexation.
1. Introduction
The bottom-up approach suggested in the book Engines of Creation by Drexler1 is the more rational path for the production of new nanomaterials with identical physicochemical properties. The challenges associated with the bottom-up synthesis are difficult to surmount. However, the relentless efforts made by chemists have provided several successful examples which may pave the way towards the bottom-up synthesis of nanomaterials. One of the earlier examples is the rational synthesis of C60 reported by Scott et al.2 As regards the bottom-up production of one dimensional carbon based materials, we can highlight the studies by Jasti,3 Ishii4 and Xia5 which showed that highly strained molecules such as cycloparaphenylenes can be prepared. These molecules constitute the basic building blocks of carbon nanotubes and thus, their production in large quantities may facilitate the availability of carbon nanotubes with specific diameters and chiralities. Using a different approach, Scott et al.6 created a C50H10 capped (5,5) carbon nanotube. In the same vein, another recent example is the synthesis of (3,1) chiral edge graphene nanoribbons.7 These investigators obtained the latter nanoribbons by the thermally induced polymerization of 10,10′-dibromo-9,90-bianthryl monomers over Cu{111}. The one dimensional carbon nanomaterials reported to date have not been limited to the structures which resemble a rolled graphene sheet. For example, very recently porphyrins have been assembled into wires8 and nanotubes.9 Also, carbon based nanotubes may be obtained using recently synthesized belt persistent tubular molecules: (P)-(12,8)-[4]-cyclo-2,8-crysenylene ([4]CC)10 and (12,8)-[4]cyclo-2,8-anthanthrenylene ([4]CA),11 prepared by Isobe et al.10 and Matsuno et al.,11 respectively. It was shown that these two molecules bind C60 with similar affinity, even though the components of the free energy change were markedly different. Finally, it is important to comment on the recent work by Liu et al.12 in which three C-shaped strips that can embrace a pillar[5]arene macrocycle were synthesized. The new stripped host was a far better receptor for electron deficient guests than the free macrocycle.Theoretical calculations have been of tremendous help in designing fullerene receptors13–21 and new carbon nanomaterials for electronics. One of the more recent examples is the prediction of the outstanding electronic properties of biphenylene based nanoribbons and sheets by Hudspeth et al.,22 which was corroborated four years later, when Schlutter et al.23 devised a protocol to produce biphenylene based nanoribbons. These compounds display promising electronic and hydrogen storage properties.21–28 The molecules that can be used to build nanomaterials is almost unlimited as recently showed by Gutzler and Perepichka.29 For example we can highlight: (a) the remarkable studies dealing with tetra[8]circulene in which Baryshnikov et al.30,31 used tetraoxa[8]circulene to design one and two dimensional materials. These systems can complex very strongly with alkaline, alkaline-earth elements,32 and d-block metals.33 (b) The carbon multishell cages formed by C20, C28 or C60, and the infinite high genera multi-tori, both recently proposed by Diudea et al.34,35
The construction of belt shaped molecules can be made in the same way as in cycloparaphenylenes, where the same hexagonal ring is repeated until a ring is formed. Although the strain that is introduced can be quite significant, the synthesis of these molecules has been achieved.3–5 A different approach is that proposed by Klarner,36,37 more than 20 years ago, when his group prepared sterically rigid macrocycles using pressure induced repetitive Diels–Alder reactions. Bis-dienophiles and bis-dienes reacted to give stable ring36 or clip37 shaped molecules. Following the idea of constructing new belt shaped molecules which can host fullerenes and enable the production of infinite one dimensional materials for electronics, we reasoned that tubular structures can be formed if norbornadiene is used as a tether to link aromatic compounds such as benzene, pyrenene, coronene or 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene (exTTF). Norbornadiene has recently been shown by Yanney et al.38 to be the key block in constructing new fullerene receptors with corannulene pincers which displayed outstanding affinity towards C60. By means of first principle calculations we have characterized six belt shaped molecules and using two of them we have built two infinite one dimensional materials with band gaps that enable their use in molecular electronics. In addition to the characterization of these materials, we describe possible synthetic routes towards their production. Finally, we show that some of them are capable of binding higher fullerenes like C180 and C240 with high affinity.
2. Methods
In the first part of the work we investigated the belt shaped molecules presented in Fig. 1 by means of M06-2X39,40 calculations performed using Gaussian 09.41 The basis set selected was the 6-31G*42 and the ultrafine grid was used for all calculations. For the smallest molecules studied we assayed whether they can host C60. The M06-2X/6-31G* results were not corrected by the basis set superposition error because we have shown that these results are almost identical to those obtained at the M06-2X/6-311G* + BSSE level of theory,13,15,18,19 a fact that was also observed by Risthauss and Grimme43 very recently. For all structures we calculated vibrational frequencies and confirmed that they were real. When possible, free energy changes were estimated under normal conditions, i.e. 298 K and 1 atm.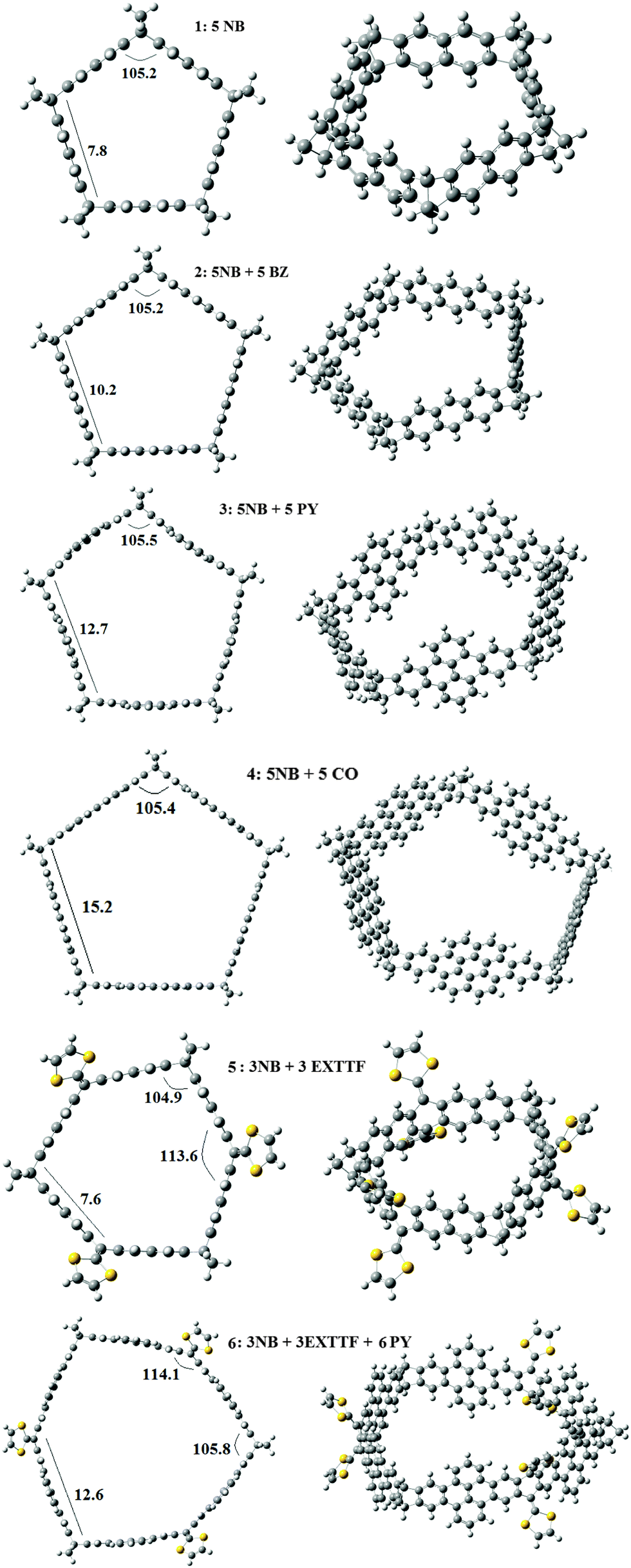 | ||
| Fig. 1 Optimized structures determined for the six belt shaped molecules investigated, at the M06-2X/6-31G* level. (NB = nobornadiene, BZ = benzene, PY = pyrene, CO = coronene, EXTTF = 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene). (Distances in Angstrom and angles in degrees.) | ||
The second part of the work dealt with the infinite tubular structure that can be formed with molecules which have the pyrene and coronene linkers. These structures were studied with the M06-L39,40 functional, because the inclusion of exact exchanges complicates the execution of M06-2X periodic calculations. In all cases, the structures were optimized with 100 k-points and electronic properties were obtained with a higher density, namely 1000 k-points.
For the sake of completeness, we estimated the band gaps of the latter infinite tubular structures with the aid of the HSEH1PBE44,45 method, which was specifically designed to correct the underestimation of band gaps which plagues most density functional based calculations. In addition to the Gaussian based calculations, we also performed periodic VDW-DF calculations as implemented in SIESTA.46,47 The double-zeta basis set with polarization functions was selected for all the spin polarized VDW-DF48 calculations. We fixed the orbital confining cut-off to 0.01 Ry and the split norm used was 0.15. The interaction between ionic cores and valence electrons was described by the Troullier–Martins norm conserving pseudopotentials.49 The lattice parameters were optimized for all structures. Monkhorst–Pack sampling of 1000 × 1 × 1 was performed for the infinite nanotubes. Band structures were calculated using 100 points along the Γ–X paths for the nanotubes.
3. Results and discussion
3.1 Finite belt shaped molecules
Given that the norbornadiene (NB) tether has recently been employed to prepare new fullerene receptors,38 and considering that this molecule has the suitable amount of flexibility, neither too large or too small, it can facilitate the formation of rigid structures. The ring shaped molecules presenting this tether may have triangular, rectangular, pentagonal, hexagonal, etc. cross sections, depending on the number of NB molecules used. Bearing in mind that the C(sp2)C(sp3)C(sp2) bond angle in NB is 107.3°, at the M06-2X/6-31G* level of theory, and considering that the integral bond angles of a square, pentagon and hexagon are 90.0, 108 and 120°, in first place we studied the formation of molecules with pentagonal cross sections. The six molecules studied are presented in Fig. 1. There are four molecules with five NB molecules that are linked by two benzenes in 1, three benzenes in 2, pyrene in 3 and coronene in 4. In the case of 5 and 6 we constructed polygons with six sides. This was achieved by combining NB and exTTF molecules, which show different stacking possibilities.50 In belt 5, we used three NB and three exTTF molecules, while 6 was obtained from 5 by introducing a pyrene molecule joining exTTF and NB on each side.In all cases the angle at each NB vertex is only 3° far from the internal angle of a pentagon, showing that the strain associated with the formation of the belts is small. The pentagonal shaped molecules 1–4 offer an interesting variety of sizes. The side of the pentagon is 7.8, 10.2, 12.7 and 15.2 Å, for 1, 2, 3 and 4, respectively. Thus, different guests may be introduced in each belt, as we will show below. The molecules which incorporate the exTTF unit have sides of 7.6 and 12.6 Å, for 5 and 6, respectively. Although the side of 5 is comparable in length to 1, the presence of one more side offers enhanced possibilities to host guests such as C60, as we will show later.
The HOMO and LUMO orbitals of the six molecules studied are presented in Fig. 2 and 3. In the cases of 1, 3 and 4, the LUMO is doubly degenerated, so any excitation will cause Jahn–Teller distortion. The HOMO of 1 and 3 are delocalized along the belt whereas in the case of 4, the HOMO is mostly located on the coronene links. On the other hand, 2, 5 and 6 present a doubly degenerated HOMO. When exTTF (5 and 6) is present, the HOMO is completely located on the exTTF linkers. The LUMO of 5 involves the entire belt, in contrast with 6, which does not have any contribution of the norbornadiene linkers in the LUMO. In the particular case of 5 we evaluated the UV spectra at the CAMB3LYP/6-31G* level of theory. This molecule has two excitations with large oscillator strengths located at 333 and 340 nm, which involve excitations from the doubly degenerated HOMOs to the LUMO.
 | ||
| Fig. 2 HOMO and LUMO corresponding to the belt shaped molecules 1, 2 and 3, at the M06-2X/6-31G* level. | ||
 | ||
| Fig. 3 HOMO and LUMO corresponding to the belt shaped molecules 4, 5 and 6, at the M06-2X/6-31G* level. | ||
3.2 Infinite one dimensional structures
Some of the molecules presented above can be used as building blocks to construct one dimensional materials. For example, 3 can be repeated in order to obtain a nanotube. Such a structure is presented in Fig. 4. The polymerization may be achieved in a similar way to that presented by Chen et al.,51 for the synthesis of 13-armchair nanoribbons, by the condensation for brominated anthracene derivatives. For the 1D material constructed with 3, the unit cell size is 8.6 Å and the periodic structure is obtained from 3 by removing 10 H atoms from the five pyrene linkers. The new tubular structures display interesting electronic properties as indicated by the band structure and density of states gathered in Fig. 4. At the M06-L/6-31G* level, the band gap is 2.6 eV and it climbs to 2.9 eV when the HSEH1PBE/6-31G* method is employed. In both cases the gap is direct. | ||
| Fig. 4 Optimized unit cell determined for the infinite nanotube formed by joining the ring 5NB + Pyrene molecules (distances in Å). | ||
Important changes occur when the pyrene linker is replaced by coronene. The largest periodic structure designed was obtained by the elimination of 20 hydrogen atoms from the coronene linkers. The structure and electronic properties are presented in Fig. 5. The unit cell size is similar to that obtained when the pyrene linker is used, namely 8.58 Å. However, the band gap experiences tremendous diminution. In effect the estimated band gap was 0.6 eV at the M06-L/6-31G* level and 0.9 eV when the more accurate HSEH1PBE method was selected. Therefore, the band gap of the pentagonal shaped nanotubes derived from NB can be tuned by using different linkers and thus it will be possible to prepare carbon based nanomaterials with uniform properties without the necessity of the complicated synthetic procedures which involve the production of nanotubes with specific chiralities. Finally, we would like to note that although we have not calculated the phonon DOS of the two 1D materials assayed, it is reasonable to expect real vibrational frequencies given that the finite systems to start the polymerization are stable. In order to proof this hypothesis, we studied a hydrogen terminated nanotube constructed with 2 unit cells of 3. At the PM6 level, all vibrational frequencies were real, thus supporting our statement about the stability of the polymerized structures.
 | ||
| Fig. 5 Optimized unit cell determined for the infinite nanotube formed by joining the ring 5NB + 5 Coronene (distances in Å). | ||
3.3 Inclusion complexes with fullerenes
The formation of inclusion complexes with fullerenes is a promising avenue for the preparation of donor–acceptor complexes that can be used in photochemistry. In first place, we tested whether C60 can fit in any of the six belt structures prepared. In Fig. 6, we show the structure of C60@1 determined at the M06-2X/6-31G* level of theory. The fullerene molecule is clearly too large to fit inside 1 and thus it interacts with C60 mainly through CH–π interactions employing its H atoms. The weak interaction is evidenced by a free energy change of −2.7 kcal mol−1. Using ring 5, whose side length is similar to that of 1, but has one more side that dramatically changes the picture. At the M06-2X/6-31G* level, the free energy change is −23.8 kcal mol−1 when C60@5 is formed and the deformation energy experienced by 5 upon complexation is only 0.8 kcal mol−1. This value indicates a significant interaction between the host and the guest. Yet, it is important to recall that in a recent work Isobe et al.10 showed that most functionals significantly overestimate the interaction between belt shaped molecules based on crysenylene and C60. | ||
| Fig. 6 Optimized structure determined for the supramolecular complexes C60@1 and C60@5, at the M06-2X/6-31G* level (free energies, enthalpies and entropies in kcal mol−1). | ||
In general, most of the studies on the host/guest chemistry of fullerenes focused on C60 and C70. Recent investigations have been trying to change that picture. On the experimental side, Yb@C82 isomers were trapped with Ni(II) octaphenylporphyrins52 and the complexation of Sc3N@C80 with Zn-Bisporphyrins.53 As regards theoretical investigations, we have shown that C90 is the best fullerene that can fit inside the C60H28 buckycatcher while C180 and C240 are too large for the guests available.
For this reason, we studied whether the belt shaped molecules proposed in this work can host the latter two large fullerenes. The structures studied are shown in Fig. 7. At the M06-2X/6-31G* level of theory we found that the formation of C180@3 is accompanied by an energy change of −77.7 kcal mol−1. Although this value is quite large, when C240 is hosted by 4 and C240@4 is formed, the energy change is −80.0 kcal mol−1. Finally, we tested if 6, the largest belt is a good host for C240 but the interaction energy was only −63.1 kcal mol−1, so 4 seems to be the best home for C240, instead of 6. We note that due to the large number of atoms in the systems we could not evaluate vibrational frequencies, but an important reduction of the energy change is expected when entropic terms are considered. Finally, it is important to mention that we tried to encapsulate C240 inside 3, but this fullerene was too large to give an interaction energy that can compete with that corresponding to C180@3. Therefore, if mixtures of C180 and C240 are present it is likely that 3 and 4 can be used to selectively interact with them, respectively.
 | ||
| Fig. 7 Optimized structure determined for the supramolecular complexes C180@3, C240@4, and C240@6 at the M06-2X/6-31G* level (interaction energies in kcal mol−1, vibrational frequencies were not calculated due to the size of the system). | ||
3.4 Proposed synthesis of 3
Nanostructure 3 can be achieved by starting with 4,9-dibromopyrene 7 which can be synthesized following Blatter and Dieter's method.54 Treating compound 7 with sodium amide in the presence of potassium tert-butoxide followed by a reaction with furan will lead to compound 8. Addition of 3,6-bis-2-pyridyl-1,2,4,5-tetrazine to 8 generates a key starting material (9, pyrenodifuran) for the belt formation. The subsequent steps which are similar to the published procedure by Yanney et al.38 are a sequence of repetitive Diels–Alder reactions between norbornadiene 10 and pyrenodifuran 9. The anticipated final steps in the belt formation will also be a Diels–Alder reaction between the diene end (A) and the dienophilic end (B) of chain 12 which would be followed by dehydration using p-toluene sulfonic acid to give 3. All the processes are described in Scheme 1. | ||
| Scheme 1 Proposed synthesis of 3. | ||
4. Conclusions
By means of first principles calculations we have characterized a new family of belt shaped molecules. The following are considered to be the major findings of the work:1. The nobornadiene tether is an efficient molecule that can be used to produce a large number of ring shaped molecules which by polymerization may lead to the rational synthesis of organic nanotubes with uniform properties.
2. The band gap of the infinite nanotubes proposed can be engineered by changing the molecule which links the norbornadiene tethers. The band gap of the pyrene based nanotube was lowered from 2.9 eV to 0.9 eV when coronene was used as a linker.
3. The molecules proposed showed a weak interaction with C60. Among the six molecules proposed, two belts may be used to separate the magic fullerenes C180 and C240, given that large interaction energy was found upon complexation.
Acknowledgements
The author thanks PEDECIBA Quimica, CSIC and ANII Uruguayan institutions for financial support.References
- E. K. Drexler, Engines of Creation, Anchor Books, 1986 Search PubMed.
- L. T. Scott, M. M. Boorum, B. J. McMahon, S. Hagen, J. Mack, J. Blank, H. Wegner and A. de Meijere, Science, 2002, 295, 1500–1503 CrossRef CAS PubMed.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed.
- Y. Ishii, Y. Nakanishi, H. Omachi, S. Matsuura, K. Matsui, H. Shinohara, Y. Segawa and K. Itami, Chem. Sci., 2012, 3, 2340 RSC.
- J. Xia, M. R. Golder, M. E. Foster, B. M. Wong and R. Jasti, J. Am. Chem. Soc., 2012, 134, 19709–19715 CrossRef CAS PubMed.
- L. T. Scott, E. A. Jackson, Q. Zhang, B. D. Steinberg, M. Bancu and B. Li, J. Am. Chem. Soc., 2012, 134, 107–110 CrossRef CAS PubMed.
- P. Han, K. Akagi, F. F. Canova, H. Mutoh, S. Shiraki, K. Iwaya, P. S. Weiss, N. Asao and T. Hitosugi, ACS Nano, 2014, 9, 9181–9187 CrossRef PubMed.
- P. Neuhaus, A. Cnossen, J. Q. Gong, L. M. Herz and H. L. Anderson, Angew. Chem., Int. Ed., 2015, 54, 7344–7348 CrossRef CAS PubMed.
- J. E. Lovett, M. Hoffmann, A. Cnossen, A. T. J. Shutter, H. J. Hogben, J. E. Warren, S. I. Pascu, C. W. M. Kay, C. R. Timmel and H. L. Anderson, J. Am. Chem. Soc., 2009, 131, 13852–13859 CrossRef CAS PubMed.
- H. Isobe, K. Nakamura, S. Hitosugi, S. Sato, H. Tokoyama, H. Yamakado, K. Ohnobd and H. Kono, Chem. Sci., 2015, 6, 2746 RSC.
- T. Matsuno, S. Sato, R. Iizuka and H. Isobe, Chem. Sci., 2015, 6, 909 RSC.
- X. Liu, Z. J. Weinert, M. Sharafi, C. Liao, J. Li and S. T. Schneebeli, Angew. Chem., Int. Ed., 2015, 54 DOI:10.1002/anie.201506793.
- P. A. Denis, RSC Adv., 2013, 3, 25296 RSC.
- D. Josa, J. Rodríguez-Otero, E. M. Cabaleiro-Lago, L. A. Santos and T. C. Ramalho, J. Phys. Chem. A, 2014, 118, 9521–9528 CrossRef CAS PubMed.
- P. A. Denis, New J. Chem., 2014, 38, 5608 RSC.
- D. Josa, L. A. dos Santos, I. Gonzalez-Veloso, J. Rodríguez-Otero, E. M. Cabaleiro-Lago and T. C. Ramalho, RSC Adv., 2014, 4, 29826–29833 RSC.
- G. Casella and G. Saielli, New J. Chem., 2011, 35, 1453–1459 RSC.
- P. A. Denis, Chem. Phys. Lett., 2011, 516, 82 CrossRef CAS.
- P. A. Denis, J. Phys. Org. Chem., 2014, 27, 918–925 CrossRef CAS.
- M. Yanney and A. Sygula, Tetrahedron Lett., 2013, 54, 2604 CrossRef CAS.
- L. N. Dawe, T. A. AlHujran, H.-A. Tran, J. I. Mercer, E. A. Jackson, L. T. Scott and P. E. Georghiou, Chem. Commun., 2012, 48, 5563–5565 RSC.
- M. A. Hudspeth, B. E. Whitman, V. Barone and J. E. Peralta, ACS Nano, 2010, 4, 4565–4570 CrossRef CAS PubMed.
- F. Schlutter, T. Nishiuchi, V. Enkelman and K. Mullen, Angew. Chem., Int. Ed., 2014, 53, 1538–1542 CrossRef PubMed.
- P. A. Denis, J. Phys. Chem. C, 2014, 118, 24976–24982 CAS.
- P. A. Denis and F. Iribarne, Comput. Theor. Chem., 2015, 1062, 30–35 CrossRef CAS.
- N. Karaush, G. V. Baryshnikov and B. F. Minaev, Chem. Phys. Lett., 2014, 612, 229–233 CrossRef CAS.
- G. Brunetto, P. A. S. Autreto, L. D. Machado, B. I. Santos, R. P. B. dos Santos and D. S. Galvao, J. Phys. Chem. C, 2012, 116, 12810–12813 CAS.
- E. Perim, R. Paupitz, P. A. S. Autreto and D. S. Galvao, J. Phys. Chem. C, 2014, 118, 23670 CAS.
- R. Gutzler and D. F. Perepichka, J. Am. Chem. Soc., 2013, 135, 16585 CrossRef CAS PubMed.
- G. V. Baryshnikov, B. F. Minaev, N. N. Karaush and V. A. Minaeva, RSC Adv., 2014, 4, 25843–25851 RSC.
- G. V. Baryshnikov, B. F. Minaev, N. N. Karaush and V. A. Minaeva, Phys. Chem. Chem. Phys., 2014, 16, 6555–6559 RSC.
- N. N. Karaush, G. V. Baryshnikov and B. F. Minaev, RSC Adv., 2015, 5, 24299–24305 RSC.
- N. N. Karaush, G. V. Baryshnikov, V. A. Minaeva and B. F. Minaev, New J. Chem., 2015, 39, 7815–7821 RSC.
- M. V. Diudea, A. Bende and C. L. Nagy, Phys. Chem. Chem. Phys., 2014, 16, 5260–5269 RSC.
- M. V. Diudea and B. Szefler, Phys. Chem. Chem. Phys., 2012, 14, 8111–8115 RSC.
- J. Benkhoff, R. Boese, F.-G. Klarner and A. E. Wigger, Tetrahedron Lett., 1994, 35, 73–76 CrossRef CAS.
- F.-G. Klarner, J. Panitzky, D. Blaser and R. Boese, Tetrahedron, 2001, 57, 3673–3687 CrossRef CAS.
- M. Yanney, F. R. Fronczek and A. Sygula, Angew. Chem., Int. Ed., 2015, 54, 11153–11156 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- W. Hehre, L. Radom, P. v. R Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- T. Risthaus and S. Grimme, J. Chem. Theory Comput., 2013, 9, 1580–1591 CrossRef CAS.
- V. Barone and G. E. Scuseria, J. Chem. Phys., 2004, 121, 10376–10379 CrossRef CAS PubMed.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 120, 7274–7280 CrossRef CAS PubMed.
- J. M. Soler, E. Artacho, J. D. Gale, A. Garcia, J. Junquera, P. Ordejon and D. Sanchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- P. Ordejon, E. Artacho and J. M. Soler, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, R10441 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schroder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef CAS.
- P. A. Denis and F. Iribarne, Struct. Chem., 2015, 26, 171–176 CrossRef CAS.
- Y.-C. Chen, D. G. de Oteyza, Z. Pedramrazi, C. Chen, F. R. Fischer and M. F. Crommie, ACS Nano, 2013, 7, 6123–6128 CrossRef CAS PubMed.
- M. Suzuki, Z. Slanina, N. Mizorogi, X. Lu, S. Nagase, M. M. Olmstead, A. L. Balch and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 18772–18778 CrossRef CAS PubMed.
- L. P. Hernandez-Eguia, E. C. Escudero-Adan, J. R. Pinzon, L. Echegoyen and P. Ballester, J. Org. Chem., 2011, 76, 3258 CrossRef CAS PubMed.
- K. Blatter and A. Dieter, Synthesis, 1989, 356–359 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |