
V(IV), Fe(II), Ni(II) and Cu(II) complexes bearing 2,2,2-tris(pyrazol-1-yl)ethyl methanesulfonate: application as catalysts for the cyclooctane oxidation†
Telma F. S.
Silva
*ab,
Bruno G. M.
Rocha
a,
M. Fátima C.
Guedes da Silva
a,
Luísa M. D. R. S.
Martins
 *ac and
Armando J. L.
Pombeiro
*a
*ac and
Armando J. L.
Pombeiro
*a
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisboa, Portugal. E-mail: pombeiro@tecnico.ulisboa.pt
bÁrea Departamental de Física, Instituto Superior de Engenharia de Lisboa, Instituto Politécnico de Lisboa, R. Conselheiro Emídio Navarro, 1959-007, Lisboa, Portugal. E-mail: tsilva@sa.isel.pt
cÁrea Departamental de Engenharia Química, Instituto Superior de Engenharia de Lisboa, Instituto Politécnico de Lisboa, R. Conselheiro Emídio Navarro, 1959-007 Lisboa, Portugal. E-mail: lmartins@deq.isel.ipl.pt
First published on 17th November 2015
Abstract
Water-soluble compounds [VOCl2{CH3SO2OCH2C(pz)3}] (pz = pyrazol-1-yl) 1, [FeCl2{CH3SO2OCH2C(pz)3}] 2, [NiCl2{CH3SO2OCH2C(pz)3}] 3 and [Cu{CH3SO2OCH2C(pz)3}2](OTf)24 were obtained by reactions between the corresponding metal salts and 2,2,2-tris(pyrazol-1-yl)ethyl methanesulfonate, CH3SO2OCH2C(pz)3. They were isolated as air-stable solids and fully characterized by IR, FTIR, NMR (for 2), EPR (for 1), ESI-MS(+/−), elemental analysis and (for 4) single-crystal X-ray diffraction. In all, half- (1–3) or full-sandwich (4), compounds the C-scorpionate ligand shows the N,N,N-coordination mode. 3 and 4 appear to provide the first examples of a Ni(II) and a full-sandwich Cu(II) compound respectively, bearing that scorpionate ligand. Compound 3 is the first Ni(II) tris(pyrazol-1-yl)methane type complex to be applied as catalyst for the oxidation of alkanes. Compounds 1–4 exhibit catalytic activity for the peroxidative (with aq. H2O2) oxidation, in water/acetonitrile medium and under mild homogeneous conditions, of cyclooctane to the corresponding alcohol and ketone (yields up to ca. 27%). The effect of the presence of additives, such as nitric acid or pyridine, was studied.
Introduction
Hydrotris(pyrazol-1-yl)-borates or −methanes and their derivatives, usually known as B- or C-homoscorpionates, respectively, have become well-established as ligands in coordination chemistry, and their complexes have made important contributions in different areas, namely in catalysis.1 These compounds coordinate to transition metals and main group elements with the possibility of steric and electronic tuning by changing the 3- and/or 5-substituents of the pyrazolyl rings1c–f,2 and/or by functionalization of the central B or C atom. For example, replacement of the methinic proton by other functional groups can change the solubility characteristics of the ligands and their metal complexes, including the possibility of obtaining water-soluble complexes.3 In this work, our attention was focused on the methanesulfonate tris(pyrazol-1-yl)methane ligand with a high hydrophilic nature, CH3SO2OCH2C(pz)3 (pz = pyrazol-1-yl), aiming to extend the still rather limited number of known complexes with this scorpionate ligand. The first examples are [CrCl3{CH3SO2OCH2C(pz)3}],4 [CoCl2(H2O){CH3SO2OCH2C(pz)3}]3d and [CuCl2{CH3SO2OCH2C(pz)3}]2.2dHomogeneous catalytic oxidation reactions generally involve the use of soluble salts or transition metal complexes in combination with oxidants like O2, H2O2 or ROOH (R = alkyl). The industrial process for oxidation of cyclohexane uses dioxygen as O-donor in the presence, e.g., of cobalt salts, at temperatures above 150 °C with conversion of ca. 4% for a selectivity of 85%.5 Oxidation of saturated hydrocarbons using V(IV), Fe(II) and Cu(II) based C-scorpionate catalysts with hydrogen peroxide as oxidant was reported to give high conversion and selectivity.1g,h,2c,3c,6 Recently, Ni(II) complexes bearing bis(pyrazolyl)pyridine or tripodal 4N ligands were found7a,b to catalyse cyclohexane oxidation, although providing low yields of the oxygenated products and requiring stronger oxidants such as meta-chloroperbenzoic acid (m-CPBA).
Cyclooctane (and other cycloalkanes) oxidation to the corresponding cycloalcohol and cycloketone has been catalysed by polymer or silica-supported iron N,N-dimethyl-N,N-bis (pyridine-2-yl-methyl)-ethane-1,2-diamine (BPMEN) using dilute aqueous H2O2 as oxidant7c or copper coordination polymer capped polyoxometalate using tert-butyl hydroperoxide as an oxidant, under mild reaction conditions.7d In addition the iridium complexes of chelating triazoles and mesoionic carbenes act as catalysts for the oxidation of cyclooctane, under moderate conditions, to cyclooctanol and cyclooctanone.7e
Supported metalloporphyrins and metallophthalocyanines, e.g. anchored on functionalized aminoalkyl silica gel, polystyrene or montmorillonite K10, or encapsulated in zeolite NaX supercages as catalyst for cyclooctane oxidation.8 Mesoporous chromium-silicate materials showed high activity and selectivity for the oxidation of cyclooctane to cyclooctanone, using dilute aqueous H2O2 or tert-butyl hydroperoxide as an oxidant, under mild reaction conditions.9 Gold(III or I) complexes10 and Keggin-type polyoxotungstates11 catalyse the oxidation of cyclooctane (to the corresponding hydroperoxide, ketone and alcohol) by H2O2 in acetonitrile, at 60–80 °C. In addition, pyrazole Re complexes anchored in 3-aminopropyl functionalized silica gel12a or carbamated silica gel supported bis(maltolato)oxovanadium(IV or V) complexes12b act as catalysts for the oxidation of cyclooctane by molecular oxygen, under moderate conditions, to cyclooctanol and cyclooctanone.
In pursuit of our on-going work on the coordination chemistry of C-scorpionates and on the application of their complexes as catalysts for the partial oxidation of alkanes1g,h,6,13 we have now (i) studied the coordination behaviour of the scarcely used 2,2,2-tris(pyrazol-1-yl)ethyl methanesulfonate, towards V(III), Fe(II), Ni(II) and Cu(II) centres; (ii) prepared the first examples of half-sandwich Ni(II) and full-sandwich Cu(II) complexes bearing the above ligand; and (iii) investigated the catalytic ability of the derived V(IV), Fe(II), Ni(II) and Cu(II) complexes for the peroxidative (with aqueous H2O2) oxidation of cyclooctane under mild conditions.
Experimental
General materials and procedures
VCl3, FeCl2, NiCl2, Cu(OSO2CF3)2, pyrazole, sulphur trioxide–trimethylamine complex, pyridine, nitric acid, hydrogen peroxide (50% aqueous solution), cyclooctane, nitromethane, diphenylamine (Aldrich), acetonitrile, tetrahydrofuran, diethyl ether, chloroform, methanol (Lab-Scan) were used as received from the supplier. CH3SO2OCH2C(pz)3 was obtained according to published methods.2d,4Infrared spectra (4000–400 cm−1) were recorded on a BIO-RAD FTS3000MX in KBr pellets; wavenumbers are in cm−1; abbreviations: vs, very strong; s, strong; m, medium; w, weak. Far infrared spectra FIR (400–200 cm−1) were recorded on a Vertex 70 spectrophotometer in CsI pellets.
ESI+/ESI− mass spectra were obtained on a VARIAN 500-MS LC ion trap mass spectrometer (solvent: MeOH; flow: 20 μL min−1; needle spray voltage: ±5 kV, capillarity voltage: ±100 V; nebulizer gas (N2): 35 psi; drying gas (N2): 10 psi; drying gas temperature (N2): 350 °C). For the MS spectra description, M denotes the complex part of compounds 1–4.
1H and 13C{1H} NMR spectra were recorded on a Bruker Avance II+ 300 and 400 MHz (UltraShieldTM Magnet) at ambient temperature. δ values are in ppm relative to Me4Si (1H and 13C). EPR spectra were recorded on Bruker ESP 300E X-band spectrophotometer equipped with an ER 4111 VT variable-temperature unit.
UV-Visible spectroscopic measurements were carried out on a PerkinElmer Lambda 750 UV-Visible spectrophotometer.
The C, H, N and S elemental analyses were carried out by the Micro analytical service of the Instituto Superior Técnico.
The electrochemical experiments were performed on an EG&G PAR 273A potentiostat/galvanostat connected to personal computer through a GPIB interface. Cyclic voltammograms (CV) were obtained in 0.2 M [nBu4N][BF4]/NCMe, at a platinum disc working electrode (d = 1 mm). Controlled-potential electrolyses (CPE) were carried out in electrolyte solutions with the above-mentioned composition, in a three-electrode H-type cell. The compartments were separated by a sintered glass frit and equipped with platinum gauze working and counter electrodes. For both CV and CPE experiments, a Luggin capillary connected to a silver wire pseudo-reference electrode was used to control the working electrode potential. The CPE experiments were monitored regularly by cyclic voltammetry, thus assuring no significant potential drift occurred along the electrolyses. Ferrocene was used as an internal standard for the measurement of the oxidation potentials of the complexes; the redox potential values are quoted relative to the SCE by using as internal reference the ferrocene/ferricinium ([Fe(η5-C5H5)2]0/+) couple (Eox1/2 = 0.42 V vs. SCE in NCMe).14
Chromatographic measurements were carried out in a ‘HP 5890–Serie-II’; fused silica capillary column Hewlett-Packard; the stationary phase was polyethylene glycol: INNOWAX with parameters 25 m × 0.2 mm × 0.4 μm; carrier gas was helium with column pressure of 15 psi.
Synthesis and characterization of complexes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), ν(NN)], 1184 and 647 [s, ν(SO), ν(C–S)], 976 [s, ν(VO)]. Far-IR (CsI pellet, cm−1): 346 and 388 [m, ν(V–Cl)]. EPR (DMSO, r.t.): a = 101.3 G, g = 1.9879. ESI+: m/z 424 ([M–Cl]+), 358 ([M–Cl–pz]+). ε218 (H2O) = 1.57 × 10−4 L mol−1 cm−1. Anal. calc. for C12H14Cl2N6O4SV: (mw 460.2): C, 31.3; H, 3.1; N, 18.2; S, 7.0%. Found: C, 31.5; H, 3.5; N, 18.3; S, 7.3%.
C), ν(NN)], 1027 and 653 [s, ν(SO), ν(C–S)]. Far-IR (CsI pellet, cm−1): 308 and 228 [m, ν(Fe–Cl)]. 1H NMR (MeOD-d6): δ 7.66 [s, br, 3H, H3, CH3SO2OCH2C(pz)3], 7.35 [s, br, 3H, H5, CH3SO2OCH2C(pz)3], 6.40 [s, br, 3H, H4, CH3SO2OCH2C(pz)3], 5.63 [s, br, 2H, CH2, CH3SO2OCH2C(pz)3], 3.34 [s, br, 3H, Me, CH3SO2OCH2C(pz)3]. 13C{1H} NMR (MeOD-d6): δ 141.92 [s, C(3) and C(5) CH3SO2OCH2C(pz)3], 130.69 [s, C(4), CH3SO2OCH2C(pz)3], 106.92 [s, CH3SO2OCH2C(pz)3], 70.57 [s, CH3SO2OCH2C(pz)3], 37.12 [s, CH3SO2OCH2C(pz)3]. 1H NMR (CD3CN-d6): δ 7.71 [s, br, 3H, H3, CH3SO2OCH2C(pz)3], 7.38 [s, br, 3H, H5, CH3SO2OCH2C(pz)3], 6.41 [s, br, 3H, H4, CH3SO2OCH2C(pz)3], 5.69 [s, br, 2H, CH2, CH3SO2OCH2C(pz)3], 3.36 [s, br, 3H, Me, CH3SO2OCH2C(pz)3]. ESI+: m/z 414 ([M–Cl]+), 347 ([M–Cl–pz]+). ε200(H2O) = 8.57 × 10−4 L mol−1 cm−1. Anal. calc. for C12H14Cl2FeN6O3S (mw 449.1): C, 32.1; H, 3.1; N, 18.7; S, 7.1%. Found: C, 32.6; H, 3.6; N, 19.3; S, 7.3%.
C), ν(NN)], 1033 and 641 [s, ν(SO), ν(C–S)]. Far-IR (CsI pellet, cm−1): 298 and 265 [m, ν(Ni–Cl)]. ESI+: m/z 349 ([M–Cl–pz]+), 282 ([M–Cl–2pz]+). ε219 (H2O) = 1.60 × 10−4 L mol−1 cm−1. Anal. calc. for C12H14Cl2N6NiO3S (mw 451.9): C, 31.9; H, 3.1; N, 18.6; S, 7.1%. Found: C, 31.7; H, 3.3; N, 18.9; S, 6.8%.
C), ν(NN)], 1054 and 640 [s, ν(SO), ν(C–S)]. EPR (solid, 90 K): a = 74.3 G, g = 2.1278. ESI+: m/z 707 ([M–H+]+), 354 ([M]2+). ε219 (H2O) = 3.43 × 10−4 L mol−1 cm−1. Anal. calc. for C26H26CuF6N12S4O12 (mw 1004.4): C, 31.1; H, 2.6; N, 16.7; S, 12.8%. Found: C, 31.1; H, 2.6; N, 16.4; S, 12.6%.
C), ν(NN)], 1184 and 647 [s, ν(SO), ν(C–S)], 976 [s, ν(VO)]. Far-IR (CsI pellet, cm−1): 346 and 388 [m, ν(V–Cl)]. EPR (DMSO, r.t.): a = 101.3 G, g = 1.9879. ESI+: m/z 424 ([M–Cl]+), 358 ([M–Cl–pz]+). ε218 (H2O) = 1.57 × 10−4 L mol−1 cm−1. Anal. calc. for C12H14Cl2N6O4SV: (mw 460.2): C, 31.3; H, 3.1; N, 18.2; S, 7.0%. Found: C, 31.5; H, 3.5; N, 18.3; S, 7.3%.
C), ν(NN)], 1027 and 653 [s, ν(SO), ν(C–S)]. Far-IR (CsI pellet, cm−1): 308 and 228 [m, ν(Fe–Cl)]. 1H NMR (MeOD-d6): δ 7.66 [s, br, 3H, H3, CH3SO2OCH2C(pz)3], 7.35 [s, br, 3H, H5, CH3SO2OCH2C(pz)3], 6.40 [s, br, 3H, H4, CH3SO2OCH2C(pz)3], 5.63 [s, br, 2H, CH2, CH3SO2OCH2C(pz)3], 3.34 [s, br, 3H, Me, CH3SO2OCH2C(pz)3]. 13C{1H} NMR (MeOD-d6): δ 141.92 [s, C(3) and C(5) CH3SO2OCH2C(pz)3], 130.69 [s, C(4), CH3SO2OCH2C(pz)3], 106.92 [s, CH3SO2OCH2C(pz)3], 70.57 [s, CH3SO2OCH2C(pz)3], 37.12 [s, CH3SO2OCH2C(pz)3]. 1H NMR (CD3CN-d6): δ 7.71 [s, br, 3H, H3, CH3SO2OCH2C(pz)3], 7.38 [s, br, 3H, H5, CH3SO2OCH2C(pz)3], 6.41 [s, br, 3H, H4, CH3SO2OCH2C(pz)3], 5.69 [s, br, 2H, CH2, CH3SO2OCH2C(pz)3], 3.36 [s, br, 3H, Me, CH3SO2OCH2C(pz)3]. ESI+: m/z 414 ([M–Cl]+), 347 ([M–Cl–pz]+). ε200(H2O) = 8.57 × 10−4 L mol−1 cm−1. Anal. calc. for C12H14Cl2FeN6O3S (mw 449.1): C, 32.1; H, 3.1; N, 18.7; S, 7.1%. Found: C, 32.6; H, 3.6; N, 19.3; S, 7.3%.
C), ν(NN)], 1033 and 641 [s, ν(SO), ν(C–S)]. Far-IR (CsI pellet, cm−1): 298 and 265 [m, ν(Ni–Cl)]. ESI+: m/z 349 ([M–Cl–pz]+), 282 ([M–Cl–2pz]+). ε219 (H2O) = 1.60 × 10−4 L mol−1 cm−1. Anal. calc. for C12H14Cl2N6NiO3S (mw 451.9): C, 31.9; H, 3.1; N, 18.6; S, 7.1%. Found: C, 31.7; H, 3.3; N, 18.9; S, 6.8%.
C), ν(NN)], 1054 and 640 [s, ν(SO), ν(C–S)]. EPR (solid, 90 K): a = 74.3 G, g = 2.1278. ESI+: m/z 707 ([M–H+]+), 354 ([M]2+). ε219 (H2O) = 3.43 × 10−4 L mol−1 cm−1. Anal. calc. for C26H26CuF6N12S4O12 (mw 1004.4): C, 31.1; H, 2.6; N, 16.7; S, 12.8%. Found: C, 31.1; H, 2.6; N, 16.4; S, 12.6%.
Crystal structure determination
The X-ray diffraction data were collected using a Bruker AXS-KAPPA APEX II with graphite monochromated Mo-Kα radiation. Data were collected using omega scans of 0.5° per frame, and a full sphere of data was obtained. Cell parameters were retrieved using Bruker SMART software and refined using Bruker SAINT15a on all the observed reflections. Absorption corrections were applied using SADABS.15a Structure was solved by direct methods by using the SIR-97 package15b and refined with SHELXL-2014/7.15c Calculations were performed using the WinGX System-Version 1.80.03.15d The hydrogen atoms were inserted at geometrically calculated positions and included in the refinement using the riding-model approximation; Uiso(H) were defined as 1.2Ueq of the parent carbon atoms for phenyl and methylene residues and 1.5Ueq of the parent carbon atoms for the methyl groups. Least square refinements with anisotropic thermal motion parameters for all the non-hydrogen atoms and isotropic ones for the remaining atoms were employed. The structure contained disordered non coordinated trifluoromethanesulfonate anion which was refined with the use of PART instruction. The occupancy of its two fragments refined to a ratio of 44.2% and 55.8%. CCDC 1412828.Catalytic study
The catalytic oxidation reactions of complexes 1–4 were carried out in MeCN solvent (total volume is 10.0 mL) in thermostated Pyrex round bottom vessels and in open atmosphere, at 60 °C. The catalyst (1–4) was used and introduced into the reaction mixture in the form of a stock solution in acetonitrile prepared by dissolving it in acetonitrile. Cyclooctane and promoter (if any) were then added in this order and the reaction started with the addition of hydrogen peroxide in a portion. (CAUTION: the combination of air or molecular oxygen and H2O2 with organic compounds at elevated temperatures may be explosive!) Initial concentrations of cyclooctane, catalyst, H2O2 and promoter (if used) were 0.25, 10−3, 1 and 0.072 M, respectively. Solution samples were analysed by GC after addition of nitromethane (as a standard compound) using a method previously used by Shul'pin.16 This method allows the estimate of concentrations of formed alkyl hydroperoxide, alcohol and ketone. Attribution of peaks was made by comparison with chromatograms of authentic samples. For precise determination of the product concentrations only data obtained after the reduction of the reaction sample with PPh3 were typically used, taking into account that the original reaction mixture contained the three products: cycloalkyl hydroperoxide (as the primary product), ketone and alcohol. When used, the oxygen radical trap, Ph2NH, was applied in a stoichiometric amount relative to the oxidant.Results and discussion
Synthesis and characterization of complexes
Compounds [VOCl2{CH3SO2OCH2C(pz)3}] (pz = pyrazol-1-yl) 1, [FeCl2{CH3SO2OCH2C(pz)3}] 2, [NiCl2{CH3SO2OCH2C(pz)3}] 3 and [Cu{CH3SO2OCH2C(pz)3}2](OTf)24 were obtained from the reactions of VCl3, FeCl2, NiCl2 and Cu(OTf)2 (OTf = trifluoromethanesulfonate), respectively, with CH3SO2OCH2C(pz)3 (Scheme 1).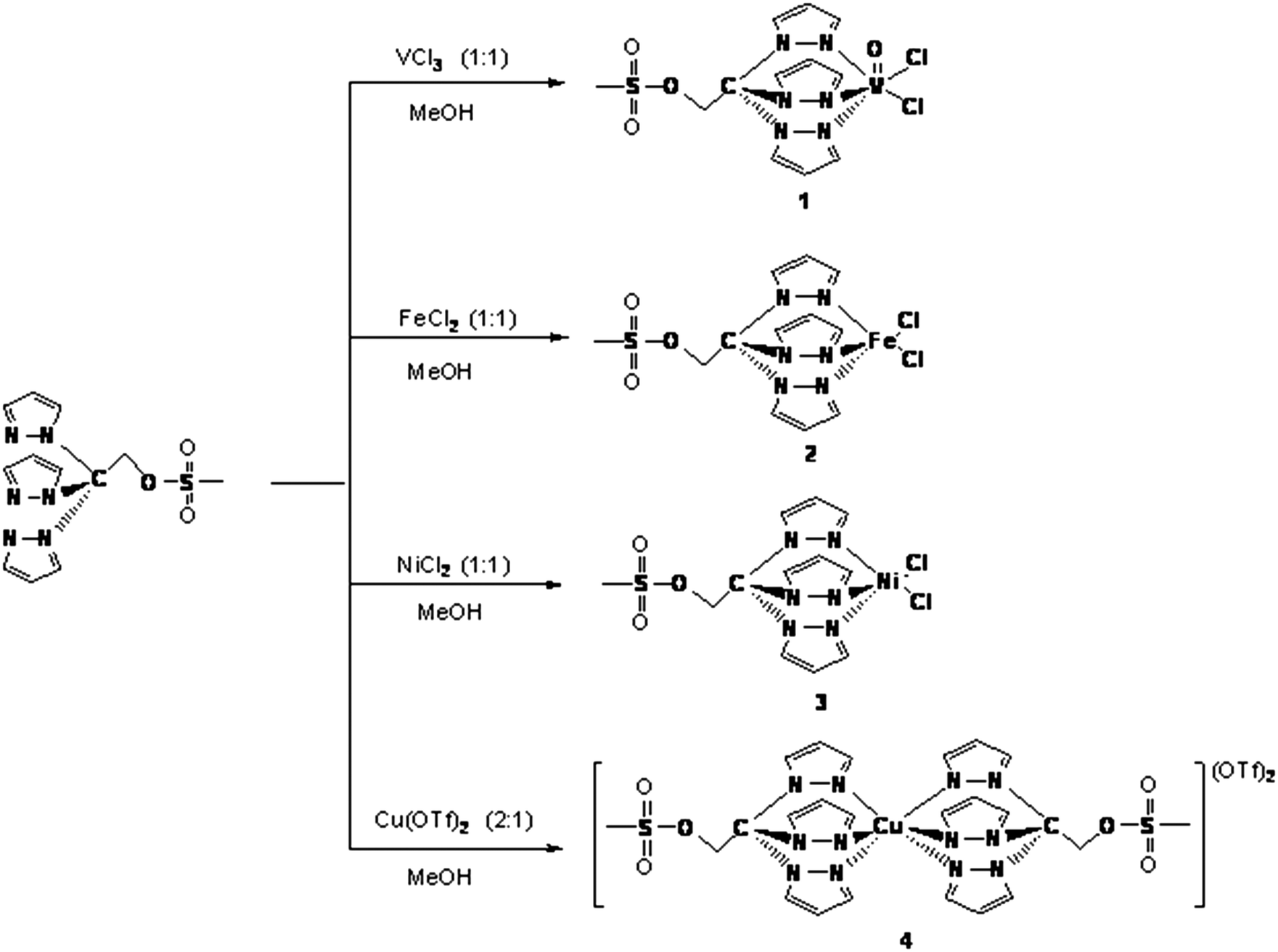 | ||
| Scheme 1 Synthesis and schematic representation of compounds 1–4. | ||
The structures of the compounds were authenticated taking into account the microanalytical results, IR and far-IR spectroscopic data, ESI-MS as well as EPR (for 1), NMR (for 2) and single crystal X-ray diffraction (for 4). In our compounds the scorpionate ligands act as N,N,N-tridentate chelators, disregarding a N,N-bidentate coordination mode previously found for [CuCl2{CH3SO2OCH2C(pz)3}]22d or a N,N,O-tridentate binding type as found for the related [Cu(TpmsPh)(CyNC)] (Cy = cyclohexyl) (TpmsPh = tris(3-phenylpyrazolyl)methane sulfonate).2f
The IR spectra of 1–4 exhibit ν(NC) at ca. 1522 cm−1 and ν(NN) in the range 1477–1413 cm−1 which are comparable to those obtained for other related scorpionate compounds.1d–f,2a,b,6b–f The ν(SO) or ν(C–S) vibrations of the methanesulfonate group are observed at the usual2d,17 ranges of 1184–1027 and 653–640 cm−1, respectively. The ν(VO) value at 976 cm−1 in 1 is comparable to those quoted for several oxovanadium(IV) complexes2c,6b,c and also for those bearing the tris(1-pyrazolyl)borate ligand, such as [VOCl{HB(3,5Me2pz)3}(Me2Hpz)].18
The characterization by FIR was done for all complexes 1–4.
The presence of the chloro ligands is confirmed by the occurrence, in their far-IR spectra, of two ν(M–Cl) bands at 388 and 346 (1), 308 and 228 (2), and 298 and 265 cm−1 (3) values comparable to those observed for other scorpionate chloro-copper complexes.2d
While the EPR spectrum of 1 shows the expected 8-line pattern of a paramagnetic V(IV) monomeric species (51V, I = 7/2) with the typical values19 of the g (1.9879) and a (101.3 G) parameters, that of 4 exhibits the usual pattern 63Cu (I = 3/2) also similar that of [CuCl2{HC(pz)3}].3c
The N5 coordination of Ni(II) is consistent with the 1H NMR that shows its typical paramagnetic nature (NiII, S = 1).2c
For complex 2, the equivalence of the pyrazolyl groups in solution is indicated by the unique broad resonances for each type of protons, i.e. for the 3, 4 or 5 position in the ring. Moreover, in the 13C{1H} NMR spectrum, the three carbon atoms from the pyrazolyl rings appear as singlets (141.92–130.69 ppm), and the carbon in apical position resonances at 106.92 ppm. The functional group CH3SO2OCH2– is detected at 70.57 and 37.12 ppm.
The ESI+ spectra of the compounds exhibit ion fragments at m/z corresponding to the loss of chloride [M–Cl]+ (in 1 and 2) and loss of a chloride and a pyrazolyl group [M–Cl–pz]+ (in 1–3). The molecular ion fragment could be observed only for the dicationic compound 4.
All the complexes are stable in air and display high solubility in water (solubility ca. 2–2.4 mg mL−1), an important feature towards their application (see below) as catalyst precursors in aqueous media.
X-ray molecular structure
Compound 4 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 1a), the asymmetric unit includes half of the molecule where the metal is sitting in an inversion centre, and one disordered trifluoromethanesulfonate counter-ion. The copper atom is coordinated by six N-pyrazolyl atoms in a slightly distorted octahedral geometry (quadratic elongation of 1.024 and angle variance of 48.62°2).20
(Fig. 1a), the asymmetric unit includes half of the molecule where the metal is sitting in an inversion centre, and one disordered trifluoromethanesulfonate counter-ion. The copper atom is coordinated by six N-pyrazolyl atoms in a slightly distorted octahedral geometry (quadratic elongation of 1.024 and angle variance of 48.62°2).20
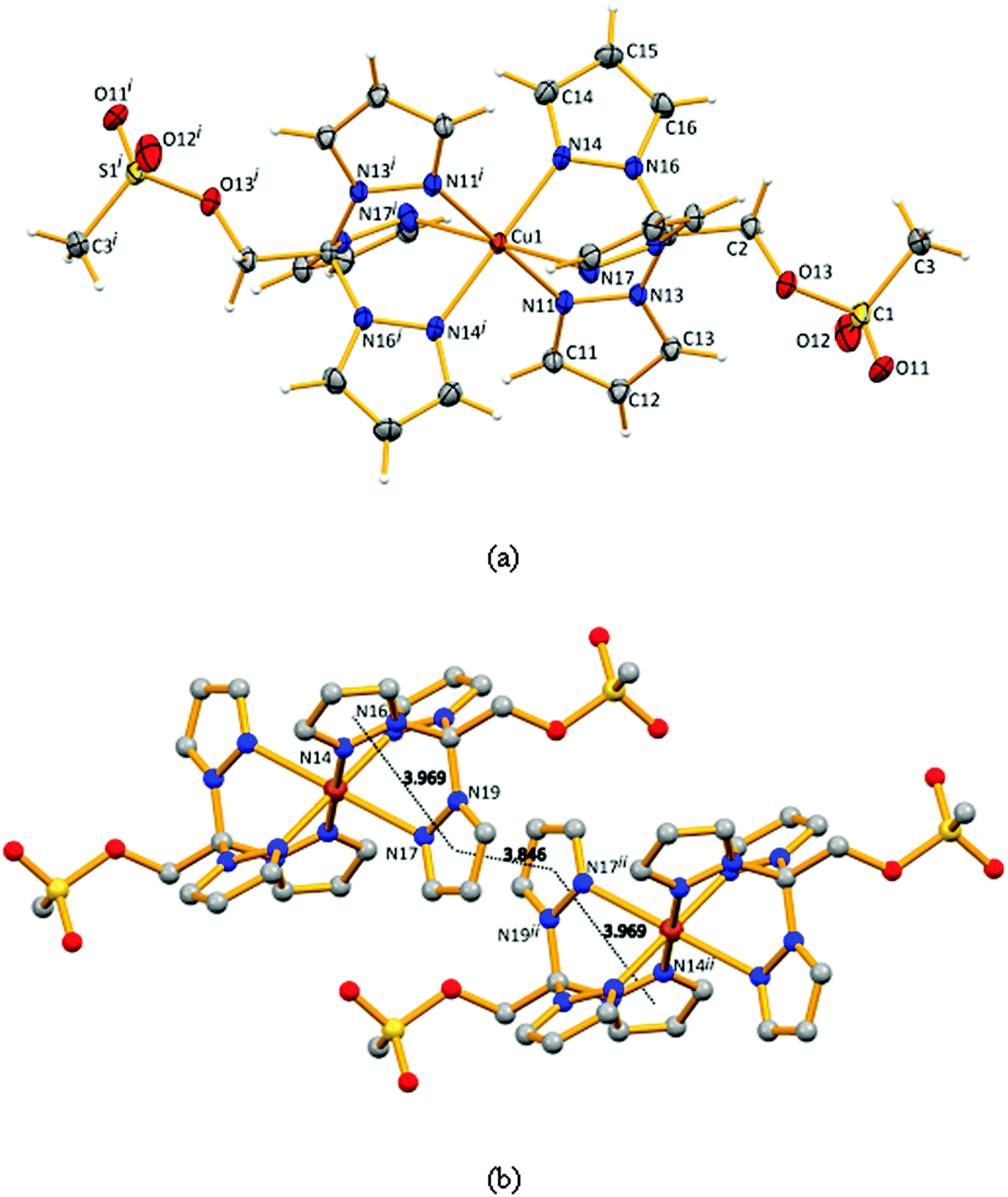 | ||
| Fig. 1 (a) Thermal ellipsoid diagrams of complex 4 with atomic numbering schemes. Ellipsoids are drawn at 30% probability and the sulfonate counter-ions are omitted. (b) Fragment of the crystal packing diagram (arbitrary view) of 4 showing the π⋯π contacts. Symmetry operation used to generate equivalent atoms: (i) −x, 1 − y, −z; (ii) 1 − x, 1 − y, −z. Selected bond distances (Å) and angles (°): N11–Cu1 1.999(3), N14–Cu1 2.021(3), N17–Cu1 2.347(3), O11–S1 1.436(4), O12–S1 1.411(4), O13–S1 1.600(3), C1–C2 1.540(5), C2–O13 1.446(5), C3–S1 1.741(5), N11–Cu1–N14 86.20(13), N11–Cu1–N17 86.20(13), N14–Cu1–N17 84.19(12), N11–Cu1–N14i 93.80(13), N11–Cu1–N17i 99.52(12), N14–Cu1–N17i 95.81(12). | ||
The bond distances in the basal plane of the compound involve the N11, N11i, N14 and the N14i atoms and assume values of 1.999(3) and 2.021(4) Å (Fig. 1, legend) which are shorter than the apical bond involving N17 (and N17i), 2.347(4) Å, as a consequence of Jahn–Teller effects. The minimum Cu⋯Cu distance equals the crystallographic a dimension (8.323 Å).
The conformations of the pyrazolyl rings differ in the molecule of 4. Indeed, the Cu–N–N–C torsion angles assume values of 142.5, 169.0 and 170.9°. In an ideal case, such angles would be 180° and the metal atom would reside in the planes defined by the pyrazolyl rings. Such a fact may result from the presence of the –CH2OSO2CH3 moiety which almost lies in the plane of one of the pyrazolyl rings, as confirmed by the value of the N16–C1–C2–O13 torsion angle of 168.3°, against 49.3° and −72.6° for N19–C1–C2–O13 and N13–C1–C2–O13, respectively (Table 1).
| 4 | |
|---|---|
| The crystal packing diagram in Fig. 1b shows the observed intermolecular π⋯π interactions which are also affected by the conformation of the rings. These interactions could stabilize the packing method and the coordination mode through the three pyrazolyl rings. | |
| Empirical formula | C26H28CuF6N12O12S4 |
| Formula weight | 1006.38 |
| Crystal system | Triclinic |
| Space group |
P |
| T (K) | 150(2) |
| a (Å) | 8.2321(7) |
| b (Å) | 9.8695(8) |
| c (Å) | 12.5776(10) |
| α (°) | 101.763(3) |
| β (°) | 103.543(2) |
| λ (°) | 100.003(3) |
| V (Å3) | 945.92(14) |
| Z | 1 |
| Density (cal.) (Mg m−3) | 1.767 |
| μ (mm−1) | 0.905 |
| F(000) | 511 |
| Rfl collected/unique/obs | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 880/5584/3812 880/5584/3812 |
| R int | 0.0320 |
| R 1, wR2 [I > 2σ(I)] | 0.0679, 0.1943 |
| R 1, wR2 (all data) | 0.1003, 0.2237 |
| GOF on F2 | 0.980 |
Electrochemical studies
In view of the potential application of the complexes 1–4 as catalyst precursors in oxidation processes (see below), their redox properties were studied by cyclic voltammetry (CV) (Table 2) and controlled potential electrolysis (CPE) in 0.2 M [nBu4N][BF4]/NCMe solutions at a Pt (disc or gauze, respectively) electrode.Complexes 1 and 2 exhibit single-electron irreversible cathodic waves at −0.78 and −0.38 V vs. SCE, respectively (Fig. 2 for 1) assigned to V(IV)/V(III) and to Fe(II)/Fe(I) reductions.
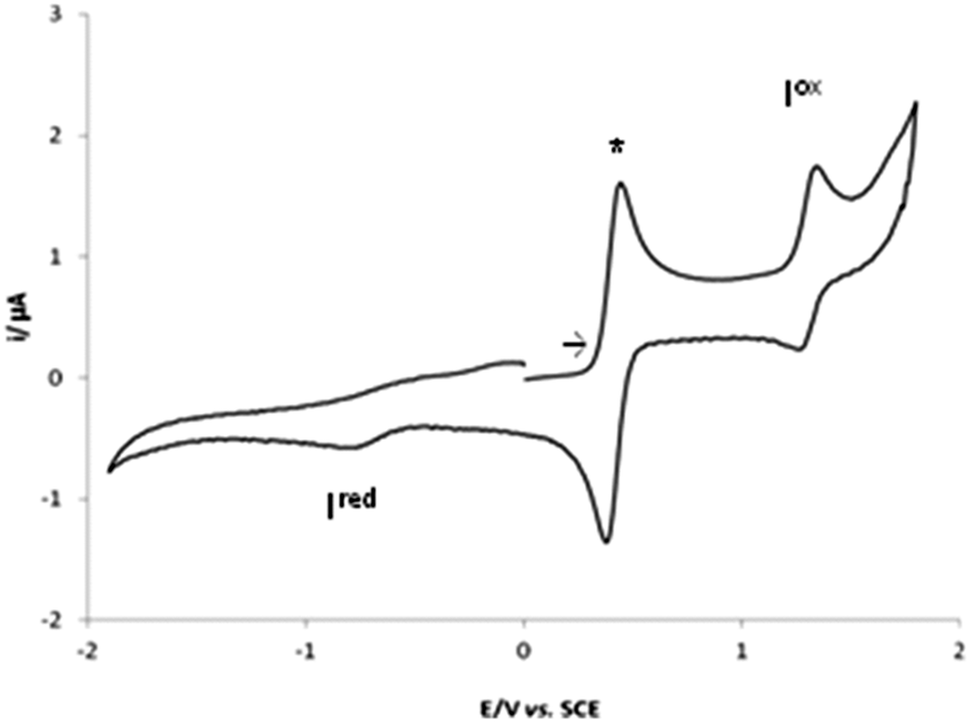 | ||
| Fig. 2 Cyclic voltammograms of complex 1 initiated by the anodic sweep, at a Pt electrode, in 0.2 M [nBu4N][BF4]/NCMe solution (v = 200 mV s−1), * [Fe(η5-C5H5)2]0/+. | ||
Complex 1 exhibits by CV a reversible monoelectronic (confirmed by CPE) anodic wave (Iox) at 1.35 V vs. SCE, assigned to the V(IV) to V(V) oxidation. Complex 2 presents a reversible oxidation wave (Iox) at 1.06 vs. SCE, conceivably due to the Fe(II)/Fe(III) oxidation.
For 3 and 4 no redox activity was found in the range from −2.00 to 2.00 V vs. SCE under the experimental conditions tested.
Cyclooctane oxidation
Compounds 1–4 act as catalyst precursors for the oxidation of cyclooctane with 50% aqueous H2O2, at 60 °C, in NCMe/H2O. The main oxidation products of cyclooctane is a mixture of cylooctanol, cyclooctanone, and cyclooctyl hydroperoxide (Scheme 2). The quantification can be performed by gas chromatography and, using a method often applied16 to ensure the precise quantification of the oxidation products present in the reaction solution, cyclooctyl hydroperoxide can be converted quantitatively to cyclohexanol by reduction with triphenylphosphine.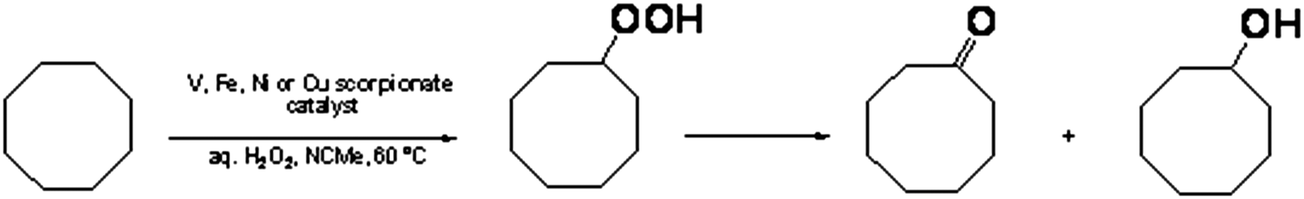 | ||
| Scheme 2 Cyclooctane oxidation to cyclooctyl hydroperoxide, cyclooctanone and cyclooctanol. | ||
The presence of cyclooctyl hydroperoxide was verified by additional GC injections of the samples before the treatment with triphenylphosphine.16 This method allowed us to estimate the real concentrations of alkyl hydroperoxide, alcohol and ketone.
It is known that alkyl hydroperoxides, such as cyclooctyl hydroperoxide, are decomposed in the chromatograph to provide corresponding alcohols and ketones as main products. Nevertheless, the alkyl hydroperoxides decomposition also affords various byproducts obtained in the C–C bond cleavage process, which are trace detectable when compared to alcohol and ketone products.
Fig. 3 displays the effect of reaction time (until 32 h) on the overall (cyclooctanol + cyclooctanone) products yields without additive. Whereas for the V (1) and Cu (4) complexes the maximum yield is observed for ca. 8 h reaction time (followed by a decrease conceivably due to overoxidation), for the Fe (2) and Ni (3) compounds there is a continuous increase of the yield products with time, although not so marked, as at the first hours.
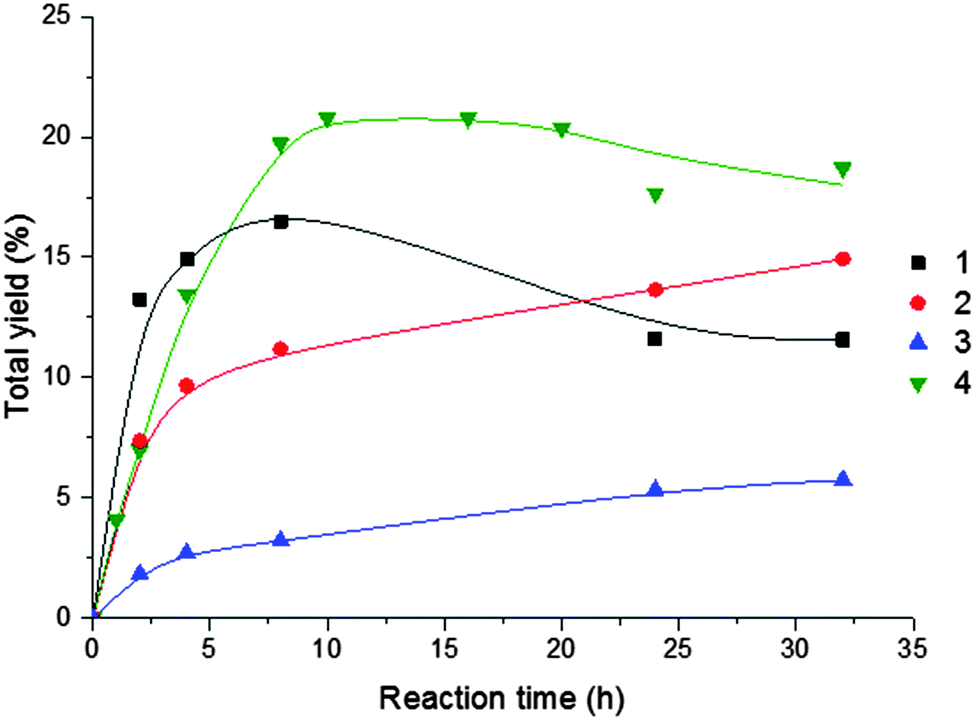 | ||
| Fig. 3 Effect of reaction time on the overall (cyclooctanol + cyclooctanone) products yield for oxidation of cyclooctane catalysed by 1–4. Reaction conditions are those of Table 3. | ||
The highest oxygenates yields in the first 20 h were provided by the Cu (4) (21%) and the V(1) (17%) compounds (Table 3, entries 5 and 4, respectively), but, after a prolonged time (30 h), the Fe (2) complex led to a 15% yield (Table 3, entry 9).
| Entry | Additive | — | Nitric acid | Pyridine | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | 1 | 2 | 3 | 4 | 1 | 2 | 4 | 1 | 4 | ||
| a Reaction conditions: cyclooctane (0.25 M), complex 1–4 (10−3 M), if used:{nitric acid or pyridine (0.072 M)} H2O2 (1.0 M) in acetonitrile at 60 °C; total volume of reaction mixture is 10 mL. b Amounts of cyclooctanone and cyclooctanol were determined after reduction of the aliquots with PPh3. c Cyclooctanone/cyclooctanol molar ratio. | |||||||||||
| 1 | Time (h) | 1 | 4 (2:98) |
5 (1:99) |
|||||||
| 2 | 2 | 13 (32:68) |
7 (6:94) |
2 (2:98) |
7 (2:98) |
19 (61:39) |
18 (59:41) |
7 (2:98) |
0.8 (0:100) |
16 (38:62) |
|
| 3 | 4 | 15 (34:66) |
10 (7:93) |
3 (2:98) |
13 (4:96) |
17 (73:27) |
19 (71:29) |
22 (2:98) |
2 (0:100) |
17 (60:40) |
|
| 4 | 8 | 17 (38:62) |
11 (9:91) |
3 (3:97) |
20 (8:92) |
15 (80:20) |
21 (73:27) |
24 (5:95) |
4 (0:100) |
16 (57:43) |
|
| 5 | 10 | 21 (30:70) |
25 (5:95) |
||||||||
| 6 | 16 | 21 (30:70) |
23 (6:94) |
||||||||
| 7 | 20 | 20 (31:69) |
24 (7:93) |
||||||||
| 8 | 24 | 12 (51:49) |
14 (11:89) |
5 (5:95) |
18 (34:66) |
16 (83:17) |
25 (79:21) |
27 (12:88) |
14 (1:99) |
14 (51:49) |
|
| 9 | 32 | 12 (56:44) |
15 (12:88) |
6 (6:94) |
19 (39:61) |
14 (84:16) |
20 (82:18) |
17 (14:86) |
12 (23:77) |
15 (53:47) |
|
Interestingly, the full-sandwich of Cu complex 4 presents a high catalytic activity in spite of its coordinative saturation, thus acting as a sufficiently labile catalyst precursor.
In the following studies, the most active compounds 4, 1 and 2 were used.
Effect of additives
The role of certain additives, in particular acids, on the catalytic oxidation of various cycloalkanes catalysed by Cu, Fe or V complexes is well known.1g,h,3c,6b–f,13,21 Strong accelerating effects of pyridine and nitric acid was also described for Os-catalyzed oxidation of alkanes and other compounds.22Hence, the influence of the presence of nitric acid (HNO3) and pyridine (py) on the cyclooctanol and cyclooctanone yields was investigated.
Addition of HNO3 had a promoting effect (Fig. 4a and Table 4) and enhanced total yields of 27% (4), 25% (2) and 19% (1) were obtained (Table 4). The favourable effect of acid presumably, as previously described for other systems,1g,h,3c,6b–f,13,21 can be associated to ligand protonation leading to coordinative insaturation of the metal, and eventually to assist proton-transfer in the formation of hydroperoxyl (HOO˙) and hydroxyl (HO˙) radicals1g,h,22 involved in the proposed mechanism (see below).
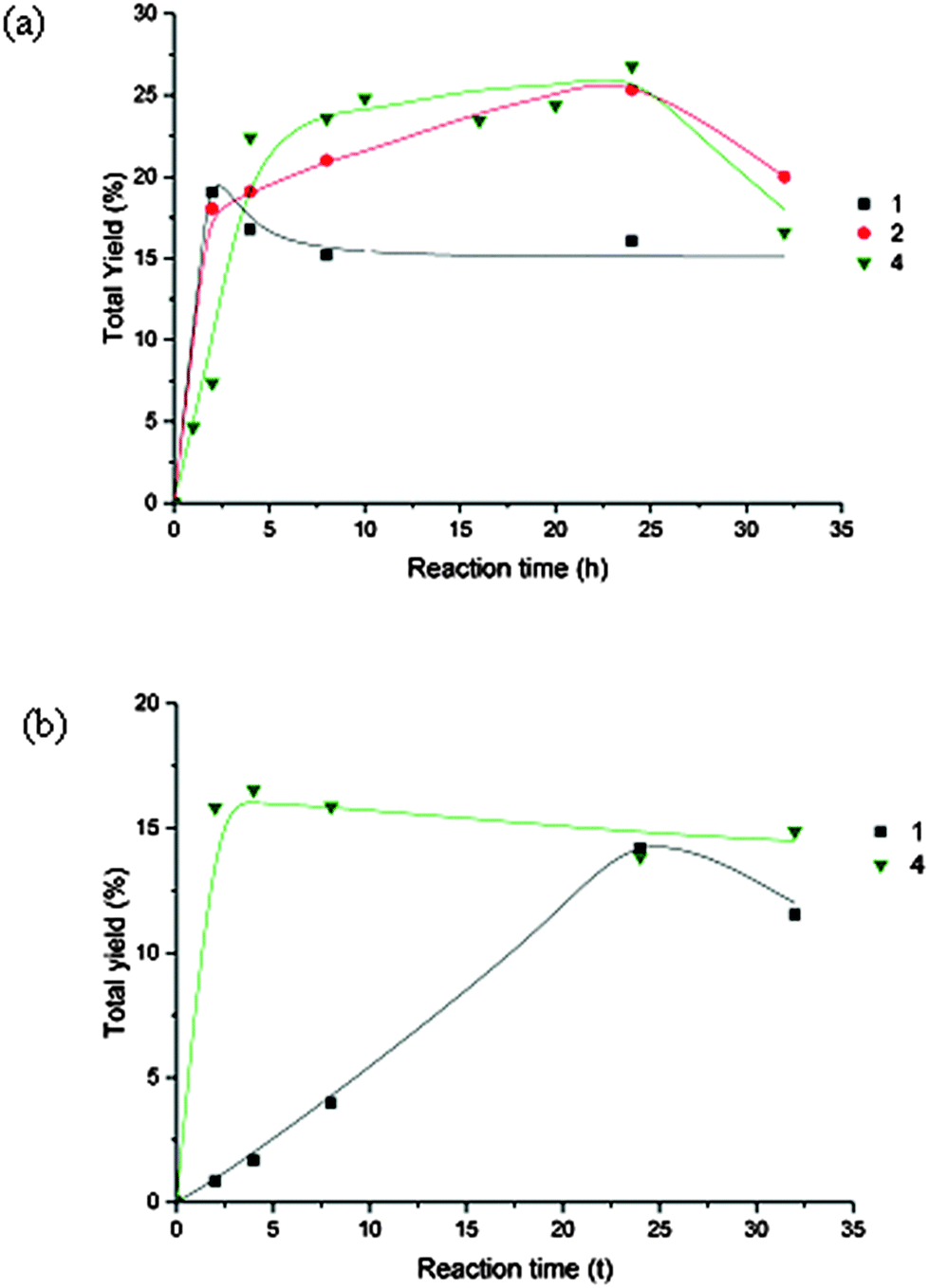 | ||
| Fig. 4 (a) Effect of HNO3 in the oxidation of cyclooctane catalysed by 1, 2 and 4 on the overall yield. Reaction conditions are those of Table 3. (b) Effect of pyridine in the oxidation of cyclooctane catalysed by 1 and 4 on the overall yield. Reaction conditions are those of Table 3. | ||
| Entry | Additive | Catalyst |
|---|---|---|
| 4 | ||
| a Reaction conditions: cyclooctane (0.25 M), complex 4 (10−3 M), Ph2NH (1 M), additive if used (0.072 M), H2O2 (1 M) in acetonitrile at 60 °C after 8 h; total volume of reaction mixture is 10 mL. b Amounts of cyclooctanone and cyclooctanol were determined after reduction of the aliquots with PPh3. c Cyclooctanone/cyclooctanol molar ratio. | ||
| 1 | — | 9.5 (15:85) |
| 2 | HNO3 | 3.3 (6:94) |
| 3 | Py | 10.6 (24:76) |
In contrast, pyridine has usually an inhibiting effect although in the case of 4 the reaction is accelerated in the first 2 h (Fig. 4b, Table 3).
The alcohol to ketone ratio (A/K) is known as an indicator for catalyst selectivity, reflecting the combined conversion of alkane either to directly the alcohol or to alkyl hydroperoxide, which subsequently is reduced to the alcohol as a result from the reduction with triphenylphosphine.16 Thus, using the same nomenclature recently introduced by Haslinger et al.,23 the A/K ratio is in fact a (alcohol + hydroperoxide)/ketone ratio (A + H)/K (Table S2, ESI†). Comparison of alcohol/ketone ratio before and after the reduction with PPh3, (Table S1, ESI†) allow us to identify cyclooctyl hydroperoxide. For complex 4, the (A + H)/K ratio along the reaction time was determined using conditions of Table 3. The ratio without reduction is drastically smaller compared to the values obtained after reduction with PPh3. We can grossly estimate that the presence in the reaction mixture of cyclooctyl hydroperoxide is, under these conditions, around ca. 90% for the first hours of reaction time and ca. 50% after 24 hours reaction time, which means that alkyl hydroperoxide is the major oxidation product. Nevertheless, once the decomposition of cyclooctyl hydroperoxide upon injection is neither quantitative, nor well defined, the reduction with triphenylphosphine is, as mentioned before, necessary prior to GC injection for a precise quantitative analysis.16
The presence of pyridine promotes the selectivity towards the ketone. For instance, in the case of 4 and for 2 h reaction time, pyridine leads to the formation of a cyclooctanone:cyclooctanol ratio of 38:62 (entry 2, Table 3), whereas in the presence of nitric acid (entry 2, Table 3) or without any additive (entry 2, Table 3), that ratio is 2:98.
In fact, if we take into consideration that, although that the alkyl hydro peroxides decomposition in GC to the corresponding alcohol and ketones is accepted as ca. 1:1 ratio, there are many factors (such as the injector type, temperature, substrates, etc.) that can substantially change this ratio, we can explain this ketone increase due to the presence of pyridine. In fact, is known the presence of such base induces the transformation of the hydroperoxide to ketone. A similar behaviour was reported for Os compounds.22–24
Effect of radical traps and mechanism
Cyclooctanol oxidation appears to involve a radical pathway with both C- and O-centred radicals, as observed for other C-scorpionate catalysed cycloalkane oxidations.1g,h In fact, the product yield decreases markedly when the reaction is carried out in the presence of either a C- or an O-centred radical trap (such as CBrCl3 or Ph2NH, respectively). Using 4, at 8 h reaction time, the effect of Ph2NH addition was more pronounced in the presence of HNO3 (3.3% yield, entry 2, Table 4), followed by the reaction in the absence of any promoter and by the reaction with the basic additive (entries 1–3, Table 4, to be compared with 20% yield in the absence of radical trap, entry 4, Table 3).The oxidation of cyclooctane (CyH) could proceed as follows (Scheme 3): metal-assisted decomposition of H2O2 could lead to the hydroxyl radical (HO˙) which, upon H-abstraction from cyclooctane (CyH), would form the cyclooctyl radical (Cy˙).22 The formation of the HO˙ radical should involve both Mn and Mn−1 redox states (the former leading to H2O2 oxidation to HOO˙ and the latter to the H2O2 reduction with liberation of HO˙), as well as proton-transfer steps among H2O2, hydroperoxo and peroxo metal-species, similarly to what has been indicated1g,h,25 for some V5+/4+, Re7+/6+, Cu2+/Cu1+ or Au1+/0 systems.
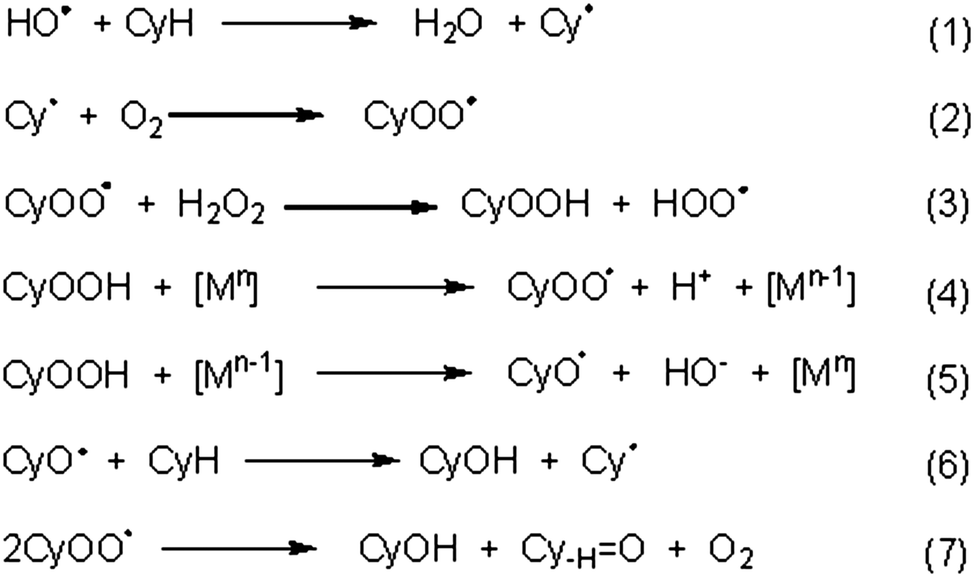 | ||
| Scheme 3 A possible mechanism path for oxidation of cyclooctane with H2O2 as oxidant. | ||
The cyclooctyl radical, on reaction with O2 or with a metal-peroxo intermediate species [M]-OOH, could form CyOO˙ (peroxyl radical) or CyOOH, respectively. CyOO˙ can also form CyOOH upon H-abstraction from H2O2,22,25 and CyOOH can decompose to the final products (cyclooctanol and cyclooctanone). Moreover, metal-promoted decomposition of CyOOH could lead to the peroxyl (CyOO˙) or the oxyl (CyO˙) radical and cyclooctanol could then be formed by H-abstraction from cyclooctane by CyO˙ or upon decomposition of CyOO˙ to both cyclooctanol and cyclooctanone.1g,h,22,25
Conclusions
We have found simple and convenient routes for the formation of water soluble dichloro-V (1), -Fe (2) and -Ni (3) complexes, and the full-sandwich Cu (4) compound, with the tri-hapto N,N,N-coordinated mode of 2,2,2-tris(pyrazol-1-yl)ethyl methanesulfonate, thus contributing towards the expansion of the still scarcely explored coordination chemistry of such a type of C-functionalized scorpionate.We have also showed the application of such a type of compounds as catalysts for the single-pot homogeneous cyclooctane oxidation, under mild conditions and with an environmentally friendly oxidant (H2O2). 4 provides the best activity which is promoted in acidic medium reaching remarkable yields (based on the alkane) up to ca. 27%.
Hence, the work also shows that a full-sandwich complex, such as 4, can present a high catalytic activity, if sufficiently labile to form an active species.
The reactions proceed via radical mechanisms with involvement of both C-centred and O-centred radicals.
Acknowledgements
This work has been partially supported by the Fundação para a Ciência e a Tecnologia (FCT), Portugal, and its PPCDT program (FEDER funded) through the research projects UID/QUI/00100/2013 and PTDC/QUI-QUI/119561/2010. BR is grateful to the Group V of Centro de Química Estrutural and the FCT, for the fellowships BL/CQE-2012-026, BL/CQE-2013-010 and the PhD fellowship of CATSUS PhD program (SFRH/BD/52370/2013). The authors gratefully acknowledge Dr Maria Cândida Vaz (IST) for the direction of the elemental analysis service, the Portuguese NMR Network (IST-UTL Centre) for providing access to the NMR facility, the Portuguese MS Network (IST Node) (Dr Conceição Oliveira) for the ESI measurements.References
- (a) S. Trofimenko, Scorpionates, The Coordination Chemistry of Polypyrazolylborate Ligands, Imperial College Press, London, 1999 Search PubMed; (b) C. Pettinari, Scorpionates II: Chelating Borate Ligands, Imperial College Press, London, 2008 Search PubMed; (c) C. Pettinari and C. Santini, in Comprehensive Coordination Chemistry II: From Biology to Nanotechnology, ed. J. A. McCleverty and T. J. Meyer, Elsevier Pergamon (APS), Amsterdam, 2003, vol. 1, p. 159 Search PubMed; (d) C. Pettinari and R. Pettinari, Coord. Chem. Rev., 2005, 249, 525–543 CAS; (e) H. R. Bigmore, C. S. Lawrence, P. Mountford and C. S. Tredget, Dalton Trans., 2005, 635–651 RSC; (f) A. Otero, J. Fernández-Baeza, A. Lara-Sánchez and L. F. Sánchez-Barba, Coord. Chem. Rev., 2013, 257, 1806–1868 CrossRef CAS; (g) L. M. D. R. S. Martins and A. J. L. Pombeiro, Coord. Chem. Rev., 2014, 265, 74–88 CrossRef CAS; (h) L. M. D. R. S. Martins, A. J. L. Pombeiro, Carbon-scorpionate Complexes in Oxidation Catalysis, in Advances in Organometallic Chemistry and Catalysis, The Silver/Gold Jubilee ICOMC Celebratory Book, ed. A. J. L. Pombeiro, J. Wiley & Sons, 2014, ch. 22, pp. 285–294 Search PubMed.
- (a) L. M. D. R. S. Martins, E. C. B. A. Alegria and A. J. L. Pombeiro, Synthesis and Biological Applications of Tris(pyrazol-1-yl)-methane and -borate Metal Complexes, in Ligands: Synthesis, Characterization and Role in Biotechnology, ed. P. Smolenski and P. Gawryszewska, Nova Science Publishers, Inc., New York, 2014, ch. 4, pp. 117–140 Search PubMed; (b) R. Wanke, P. Smoleński, M. F. C. Guedes da Silva, L. M. D. R. S. Martins and A. J. L. Pombeiro, Inorg. Chem., 2008, 47, 10158–10168 CrossRef CAS PubMed; (c) R. Wanke, M. F. C. Guedes da Silva, S. Lancianesi, T. F. S. Silva, C. Pettinari and A. J. L. Pombeiro, Inorg. Chem., 2010, 49, 7941–7952 CrossRef CAS PubMed; (d) T. F. S. Silva, G. S. Mishra, M. F. C. Guedes da Silva, R. Wanke, L. M. D. R. S. Martins and A. J. L. Pombeiro, Dalton Trans., 2009, 9207–9215 RSC; (e) B. G. M. Rocha, T. C. O. Mac Leod, M. F. C. Guedes da Silva, K. V. Luzyanin, L. M. D. R. S. Martins and A. J. L. Pombeiro, Dalton Trans., 2014, 43, 15192–15200 RSC; (f) B. G. M. Rocha, R. Wanke, M. F. C. Guedes da Silva, K. V. Luzyanin, L. M. D. R. S. Martins, P. Smolénski and A. J. L. Pombeiro, J. Organomet. Chem., 2012, 714, 47–52 CrossRef CAS; (g) E. C. B. Alegria, L. M. D. R. S. Martins, M. Haukka and A. J. L. Pombeiro, Dalton Trans., 2006, 4954–4961 RSC; (h) E. C. B. Alegria, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, J. Organomet. Chem., 2005, 690, 1947–1958 CrossRef CAS; (i) C. Dinoi, M. F. C. G. Silva, E. C. B. A. Alegria, P. Smoleński, L. M. D. R. S. Martins, R. Poli and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2010, 2415–2424 CrossRef CAS.
- (a) W. Klaui, M. Berghahn, G. Rheinwald and H. Lang, Angew. Chem., Int. Ed., 2000, 39, 2464–2466 CrossRef CAS; (b) W. Klaui, M. Berghahn, W. Frank, G. J. ReiB, T. Schonherr, G. Rheinwald and H. Land, Eur. J. Inorg. Chem., 2003, 2059–2070 CrossRef CAS; (c) T. F. S. Silva, L. M. D. R. S. Martins, E. C. B. A. Alegria and A. J. L. Pombeiro, Adv. Synth. Catal., 2008, 350, 706–716 CrossRef CAS; (d) T. F. S. Silva, L. M. D. R. S. Martins, M. F. C. Guedes da Silva, A. R. Fernandes, A. Silva, P. M. Borralho, S. Santos, C. M. P. Rodrigues and A. J. L. Pombeiro, Dalton Trans., 2012, 41, 12888–12897 RSC; (e) C. Pettinari, F. Marchetti, G. Lupidi, L. Quassinti, M. Bramucci, D. Petrelli, L. A. Vitali, M. F. C. Guedes da Silva, L. M. D. R. S. Martins, P. Smolenski and A. J. L. Pombeiro, Inorg. Chem., 2011, 50, 11173–11183 CrossRef CAS PubMed; (f) L. M. D. R. S. Martins, E. C. B. A. Alegria, P. Smoleński, M. L. Kuznetsov and A. J. L. Pombeiro, Inorg. Chem., 2013, 52, 4534–4546 CrossRef CAS PubMed.
- I. García-Orozco, R. Quijada, K. Vera and M. Valderrama, J. Mol. Catal. A: Chem., 2006, 260, 70–76 CrossRef.
- (a) Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 6th edn, 2002 Search PubMed; (b) W. Weissermel and H. J. Horpe, Ind. Org. Chem., VCH Press, Weinheim, 2nd edn, 1993 Search PubMed; (c) D. Mandelli, E. V. Spinacé and E. L. Pires, Appl. Catal., A, 2001, 211, 1–17 CrossRef.
- (a) G. S. Mishra, T. F. S. Silva, L. M. D. R. S. Martins and A. J. L. Pombeiro, Pure Appl. Chem., 2009, 81, 1217–1227 CrossRef CAS; (b) T. F. S. Silva, K. V. Luzyanin, M. V. Kirillova, M. F. C. Guedes da Silva, L. M. D. R. S. Martins and A. J. L. Pombeiro, Adv. Synth. Catal., 2010, 352, 171–187 CrossRef CAS; (c) T. F. S. Silva, T. C. O. Mac Leod, L. M. D. R. S. Martins, M. F. C. Guedes da Silva, M. A. Schiavon and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2013, 367, 52–60 CrossRef CAS; (d) T. F. S. Silva, M. F. C. Guedes da Silva, G. S. Mishra, L. M. D. R. S. Martins and A. J. L. Pombeiro, J. Organomet. Chem., 2011, 696, 1310–1318 CrossRef CAS; (e) L. M. D. R. S. Martins, A. Martins, E. C. B. A. Alegria, A. P. Carvalho and A. J. L. Pombeiro, Appl. Catal., A, 2013, 464, 43–50 CrossRef; (f) L. M. D. R. S. Martins, M. Peixoto de Almeida, S. A. C. Carabineiro, J. L. Figueiredo and A. J. L. Pombeiro, ChemCatChem, 2013, 5, 3847–3856 CrossRef CAS.
- (a) E. Tordina, M. List, U. Monkowius, S. Schindler and G. Knöra, Inorg. Chim. Acta, 2013, 402, 90–96 CrossRef PubMed; (b) N. Xing, L. T. Xu, X. Liu, Q. Wu, X. T. Ma and Y. H. Xing, ChemPlusChem, 2014, 79, 1198–1207 CrossRef CAS; (c) Y. Feng, E. G. Moschetta and C. W. Jones, Chem. – Asian J., 2014, 9, 3142–3152 CrossRef CAS PubMed; (d) F. Farzaneh and F. Moghzi, React. Kinet., Mech. Catal., 2015, 115, 175–185 CrossRef CAS; (e) S. Hohloch, S. Kaiser, F. L. Duecker, A. Bolje, R. Maity, J. Košmrljb and B. Sarkar, Dalton Trans., 2015, 44, 686–693 RSC.
- (a) J. Połtowicz, E. Tabor, K. Pamin and J. Haber, Inorg. Chem. Commun., 2005, 8, 1125–1127 CrossRef; (b) J. Haber, K. Pamin and J. Połtowicz, J. Mol. Catal. A: Chem., 2004, 224, 153–159 CrossRef CAS; (c) J. Połtowicz and J. Haber, J. Mol. Catal. A: Chem., 2004, 220, 43–51 CrossRef; (d) J. Haber, L. Matachowski, K. Pamin and J. Połtowicz, J. Mol. Catal. A: Chem., 2000, 162, 105–109 CrossRef CAS; (e) J. Połtowicz, K. Pamin, L. Matachowski, E. M. Serwicka, R. Mokaya, Y. Xia and Z. Olejniczak, Catal. Today, 2006, 114, 287–292 CrossRef.
- S. Samanta, N. K. Mal and A. Bhaumik, J. Mol. Catal. A: Chem., 2005, 236, 7–11 CrossRef CAS.
- G. B. Shul'pin, A. E. Shilov and G. Suss-Fink, Tetrahedron Lett., 2001, 42, 7253–7256 CrossRef.
- M. M. Q. Simões, I. C. M. S. Santos, M. S. S. Balula, J. A. F. Gamelas, A. M. V. Cavaleiro, M. Graça, P. M. S. Neves and J. A. S. Cavaleiro, Catal. Today, 2004, 91, 211–214 CrossRef.
- (a) G. S. Mishra, E. C. B. A. Alegria, L. M. D. R. S. Martins, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2008, 285, 92–100 CrossRef CAS; (b) G. S. Mishra, J. J. R. Frausto da Silva and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2007, 265, 56–59 CrossRef.
- (a) M. Peixoto de Almeida, L. M. D. R. S. Martins, S. A. C. Carabineiro, T. Lauterbach, F. Rominger, A. S. K. Hashmi, A. J. L. Pombeiro and J. L. Figueiredo, Catal. Sci. Technol., 2013, 3, 3056–3069 RSC; (b) T. F. S. Silva, L. M. D. R. S. Martins, M. F. C. Guedes da Silva, M. L. Kuznetsov, A. R. Fernandes, A. Silva, S. Santos, C. J. Pan, J. Lee, B. J. Hwang and A. J. L. Pombeiro, Chem. – Asian J., 2014, 9, 1132–1143 CrossRef CAS PubMed.
- (a) A. J. L. Pombeiro, M. F. C. Guedes da Silva and M. A. N. D. A. Lemos, Coord. Chem. Rev., 2001, 219, 53–80 CrossRef; (b) M. F. C. Guedes da Silva and A. J. L. Pombeiro, Electrochim. Acta, 2012, 82, 478–483 CrossRef CAS; (c) M. E. N. P. R. A. Silva, A. J. L. Pombeiro, J. J. R. Fraústo da Silva, R. Herrmann, M. Deus and R. E. Bozak, J. Organomet. Chem., 1994, 480, 81–90 CrossRef CAS; (d) M. E. N. P. R. A. Silva, A. J. L. Pombeiro, J. J. R. Fraústo da Silva, R. Herrmann, N. Deus, T. J. Castilho and M. F. C. Guedes da Silva, J. Organomet. Chem., 1991, 421, 75–90 CrossRef CAS.
- (a) Bruker, APEX2 & SAINT, AXS Inc., Madison, WI, 2004 Search PubMed; (b) A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (d) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- (a) G. B. Shul'pin, in Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, New York, 2nd edn, 2004, vol. 2, p. 215 Search PubMed; (b) G. B. Shul'pin and G. V. Nizova, React. Kinet. Catal. Lett., 1992, 48, 333–338 CrossRef; (c) G. B. Shul'pin, M. G. Matthes, V. B. Romakh, M. I. F. Barbosa, J. L. T. Aoyagi and D. Mandelli, Tetrahedron, 2008, 64, 2143–2152 CrossRef; (d) G. B. Shul'pin, Dalton Trans., 2013, 42, 12794–12818 RSC; (e) G. B. Shul'pin, Org. Biomol. Chem., 2010, 8, 4217–4228 RSC; (f) G. B. Shul'pin, Mini-Rev. Org. Chem., 2009, 6, 95–104 CrossRef.
- (a) W. Klaui, D. Schramm, W. Peters, G. Rheinwald and H. Land, Eur. J. Inorg. Chem., 2001, 1415–1424 CrossRef CAS; (b) C. A. Root, J. D. Hoeschele, C. R. Cornman, J. W. Kampf and V. L. Pecoraro, Inorg. Chem., 1993, 32, 3855–3861 CrossRef CAS; (c) M. R. Maurya, Coord. Chem. Rev., 2003, 237, 163–181 CrossRef CAS.
- (a) Z. Li, Y. Xing, Y. Cao, X. Zeng, M. Ge and S. Niu, Polyhedron, 2009, 28, 865–871 CrossRef CAS; (b) C. J. Carrano, M. Mohan, S. M. Holmes, R. Rosa, A. Butler, J. Charnock and C. D. Garner, Inorg. Chem., 1994, 33, 646–655 CrossRef CAS; (c) D. Collison, D. Eardley, F. Mabbs, A. Powell and S. Turner, Inorg. Chem., 1993, 32, 664–671 CrossRef CAS.
- M. R. Bond, L. M. Mokry, T. Otieno, J. Thompson and C. J. Carrano, Inorg. Chem., 1995, 34, 1894–1905 CrossRef CAS.
- K. Robinson, G. V. Gibbs and P. H. Ribbe, Science, 1971, 172, 567–570 CAS.
- (a) R. R. Fernandes, J. Lasri, M. M. F. C. Guedes da Silva, J. A. L. Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2011, 351, 100–111 CrossRef CAS; (b) S. Tanase, P. Marques-Gallego, W. R. Browne, R. Hage, E. Bouwman, B. L. Feringa and J. Reedijk, Dalton Trans., 2008, 2026–2033 RSC; (c) L. S. Shul'pina, M. V. Kirillova, A. J. L. Pombeiro and G. B. Shul'pin, Tetrahedron, 2009, 65, 2424–2429 CrossRef; (d) R. R. Fernandes, M. V. Kirillova, J. A. L. da Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Appl. Catal., A, 2009, 351, 100–107 CrossRef; (e) E. C. B. A. Alegria, M. V. Kirillova, L. M. D. R. S. Martins and A. J. L. Pombeiro, Appl. Catal., A, 2007, 317, 43–52 CrossRef CAS.
- (a) G. B. Shul'pin, A. R. Kudinov, L. S. Shul'pina and E. A. Petrovskaya, J. Organomet. Chem., 2006, 691, 837–845 CrossRef; (b) G. B. Shul'pin, Y. N. Kozlov, L. S. Shul'pina, A. R. Kudinov and D. Mandelli, Inorg. Chem., 2009, 48, 10480–10482 CrossRef PubMed; (c) G. B. Shul'pin, M. V. Kirillova, Y. N. Kozlov, L. S. Shul'pina, A. R. Kudinov and A. J. L. Pombeiro, J. Catal., 2011, 164–172 CrossRef; (d) G. B. Shul'pin, Y. N. Kozlov, L. S. Shul'pina, W. A. Carvalho and D. Mandelli, RSC Adv., 2013, 3, 15065–15074 RSC; (e) M. M. Vinogradov, L. S. Shul'pina, Y. N. Kozlov, A. R. Kudinov, N. S. Ikonnikov and G. B. Shul'pin, J. Organomet. Chem., 2015, 784, 52–61 CAS.
- S. Haslinger, A. Raba, M. Cokoja, A. Pothig and F. E. Kuhn, J. Catal., 2015, 331, 147–153 CrossRef CAS.
- G. B. Shul'pin, Y. N. Kozlov, L. S. Shul'pina and P. V. Petrovskiy, Appl. Organomet. Chem., 2010, 24, 464–472 Search PubMed.
- (a) G. B. Shul'pin, C. R. Chim., 2003, 6, 163–178 CrossRef; (b) G. B. Shul'pin, Y. N. Kozlov, G. V. Nizova, G. Süss-Fink, S. Stanislas, A. Kitaygorodskiy and V. S. Kulikova, J. Chem. Soc., Perkin Trans. 2, 2001, 1351–1371 RSC; (c) G. B. Shul'pin, G. Süss-Fink and J. R. L. Smith, Tetrahedron, 1999, 55, 5345–5358 CrossRef; (d) M. V. Kirillova, M. L. Kuznetsov, V. B. Romakh, L. S. Shul'pina, J. J. R. Fraústo da Silva, A. J. L. Pombeiro and G. B. Shul'pin, J. Catal., 2009, 267, 140–157 CrossRef CAS; (e) M. V. Kirillova, A. M. Kirillov, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2008, 3423–3427 CrossRef CAS; (f) M. V. Kirillova, A. M. Kirillov, P. M. Reis, J. A. L. Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, J. Catal., 2007, 248, 130–136 CrossRef CAS; (g) M. L. Kuznetsov and A. J. L. Pombeiro, Inorg. Chem., 2009, 48, 307–318 CrossRef CAS PubMed; (h) M. V. Kirillova, M. L. Kuznetsov, Y. N. Kozlov, L. S. Shul'pina, A. Kitaygorodskiy, A. J. L. Pombeiro and G. B. Shul'pin, ACS Catal., 2011, 1, 1511–1520 CrossRef CAS; (i) S. A. C. Carabineiro, L. M. D. R. S. Martins, M. Avalos-Borja, J. G. Buijnsters, A. J. L. Pombeiro and J. L. Figueiredo, Appl. Catal., A, 2013, 467, 279–290 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1412828. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj01865j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |