
Efficient solid phase extraction of codeine from human urine samples using a novel magnetic molecularly imprinted nanoadsorbent and its spectrofluorometric determination
Tayyebeh
Madrakian
*,
Fariba
Fazl
,
Mazaher
Ahmadi
and
Abbas
Afkhami
Faculty of Chemistry, Bu-Ali Sina University, Hamedan, Iran. E-mail: madrakian@basu.ac.ir; madrakian@gmail.com; Fax: +98-811-8257407; Tel: +98-811-8257407
First published on 12th October 2015
Abstract
From a medical or clinical point of view, to assess toxicity, adverse effects, interactions and therapeutic efficiency, monitoring drug levels in body fluids, such as urine and plasma, has become increasingly necessary. This study reports on a novel method for determination of trace concentration levels of codeine phosphate in human urine samples. In this regard, a solid phase extraction step, based on a novel magnetic molecularly imprinted polymer nanoadsorbent, has been developed to be used prior to spectrofluorometric analysis of the drug. After optimization of various factors, such as pH, contact time, adsorbent dosage and desorbing solvent, the results showed that using the proposed method sensitive and accurate determination of the drug in the concentration range of 2.0–500.0 ng mL−1 with the detection limit of 0.67 ng mL−1 is achievable. The urine sample analysis results show that a powerful method for codeine phosphate determination has been proposed.
Introduction
Codeine (methyl morphine) is one of the most important constituents of opium and can be prepared from morphine by methylation. Its phosphate form (COP) is usually used for the treatment of low or moderate pain. Compared with morphine, the antitussive effect of codeine is approximately one-third that of morphine, and its analgesic action is only one-twentieth that of morphine.1 It is stronger than general antipyretic analgesics, and its function duration is similar to morphine. These properties lead to a wide prescription of codeine in cough syrups and tablets. It has moderate analgesic and weak cough suppressant effects. The analgesic effects are observed because only trace amounts of morphine are formed after the administration of codeine.2 However, long-term use of codeine is addictive and excessive intake can even cause death.3,4 Therefore, it is very necessary to analyze its content in urine or biological fluids.Various approaches, such as gas chromatography tandem mass spectrometry (GC-MS),5–7 high performance liquid chromatography (HPLC) with ultraviolet detection8,9 and tandem mass spectrometric detection (HPLC-MS),10 chemiluminescence,11,12 capillary electrophoresis,13,14 thin-layer chromatography (TLC),15 fluorescence (FL),16,17 electrochemistry1,18 and chemometrics methods,19 have been reported for COP determination. Most of these methods need a pretreatment step (such as liquid–liquid extraction or solid phase extraction) to enhance some analytical parameters such as detection limit (sensitivity) and selectivity.
Today, solid-phase extraction (SPE) is widely used for the extraction and preconcentration of analytes in various environmental, food and biological samples. It is the most popular clean-up technique due to factors, such as convenience, cost, time saving and simplicity, and it is the most accepted sample pretreatment method today.20–25 At present, there are several types of sorbents for SPE, including normal-phase, reversed-phase, ionic, and other special sorbents. However, due to their unsatisfactory selectivity, these traditional sorbents usually cannot efficiently separate analytes in complex biological or environmental samples.26
A relatively new development in the area of SPE is the use of molecularly imprinted polymers (MIPs) for sample clean-up and development of selective and sensitive analytical methods.26–28 MIPs are synthetic polymers possessing specific cavities designed for a target molecule and are synthesized by the polymerization of different components. With the development of molecularly imprinted adsorbents, some disadvantages of MIPs have emerged and greatly restricted its application. It was exciting for scientists to find that giving MIP magnetic properties was an effective approach to overcome the disadvantages of MIPs.26,29,30 When used as a SPE sorbent, magnetic MIP beads can be dispersed into solution directly and separated via an external magnetic field, avoiding the operation of making packed columns with the traditional SPE adsorbents. Accordingly, an interest has been raised in the preparation and application of magnetic MIPs in recent years.26,31–36 These studies greatly extended the preparation method of magnetic MIPs and made them more efficient as SPE adsorbents. However, the structure of the magnetic supporter (core), rather than a single Fe3O4 particle as reported in the previous studies, should be better designed to improve the magnetic performance and enhance the stability of MIPs.
In the present study, a highly selective magnetic COP-imprinted polymer nanoadsorbent has been synthesized using magnetite nanospheres (MNSs) as the magnetic core, a synthesized aminoimide monomer (AI) as the functional monomer, ethylene glycol dimethacrylate (EGDMA) as the cross-linker, ammonium persulfate as the initiator, and COP as the template. The prepared nanoadsorbent has been fully characterized and its affinity for COP was experimentally evaluated. After optimization of various affecting factors (such as pH, adsorbent dosage and contact time), the adsorbent was successfully used for extraction of COP from infected human urine samples.
Experimental
Apparatus
Infrared spectra of the polymeric adsorbent were collected using a FT-IR spectrometer (Perkin-Elmer model Spectrum GX) with the spectral range of 4000–400 cm−1. A transmission electronic microscope (TEM, Philips-CM10) obtained the nanographs of the adsorbent with measurements operating at 100 kV. The crystal structure of the synthesized materials was determined by an X-ray diffractometer (XRD, 38066 Riva, d/G via M. Misone, 11/D (TN) Italy) at ambient temperature. A Metrohm model 713 pH-meter was used for the pH adjustments. A 40 kHz universal ultrasonic cleaner water bath (RoHS, Korea) was used. A Knauer 1050 HPLC pump and a Knauer 2850 PDA detector with Macherey nagel column C18 (150 mm × 2.5 mm, particle size 4 μm) was used. For the instrumental control, data collection and processing, chromate software was employed. A Perkin Elmer (LS50B) luminescence spectrometer was used for the determination of COP at λem = 348 nm (λex = 214 nm).Chemicals
Alborz Bulk Pharmaceutical Company (Kaveh Industrial zone, Saveh, Iran) provided COP. The standard solution of 200 mg L−1 COP was prepared with doubly distilled water (DDW) and stored at 4 °C in the dark, and working standard solutions of different COP concentrations were prepared daily by diluting the stock solution with DDW water. Ethylene glycol (EG) and polyethylene glycol (PEG, fw ∼8000 g mol−1) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The AI functional monomer was synthesized according to our recently reported procedure. All the other chemicals were purchased from Merck Company (Merck, Darmstadt, Germany). DDW water was used throughout this study. Britton–Robinson universal pH buffer was used for pH adjustment of the working solutions. The buffer was a mixture of 0.04 M H3BO3, 0.04 M H3PO4 and 0.04 M CH3COOH that was titrated to the desired pH with NaOH and/or HCl solutions.Synthesis of the aminoimide functional monomer (Scheme 1a)
The aminoimide monomer used for preparation of the MIP layer was synthesized according to our recently reported procedure with some minor modifications (Scheme 1a).34 Briefly, the aminoimide monomer was synthesized by slow addition of maleic anhydride (1 g) to the solution of ethylenediamine (1 mL) in DDW (20 mL). The solution was heated to 120 °C for 1 h, until all the water was removed and ethylenediamine reacted with maleic anhydride through ring opening. Then, the unsaturated aminoimide monomer was prepared by heating the reaction product to 140 °C for 2 h.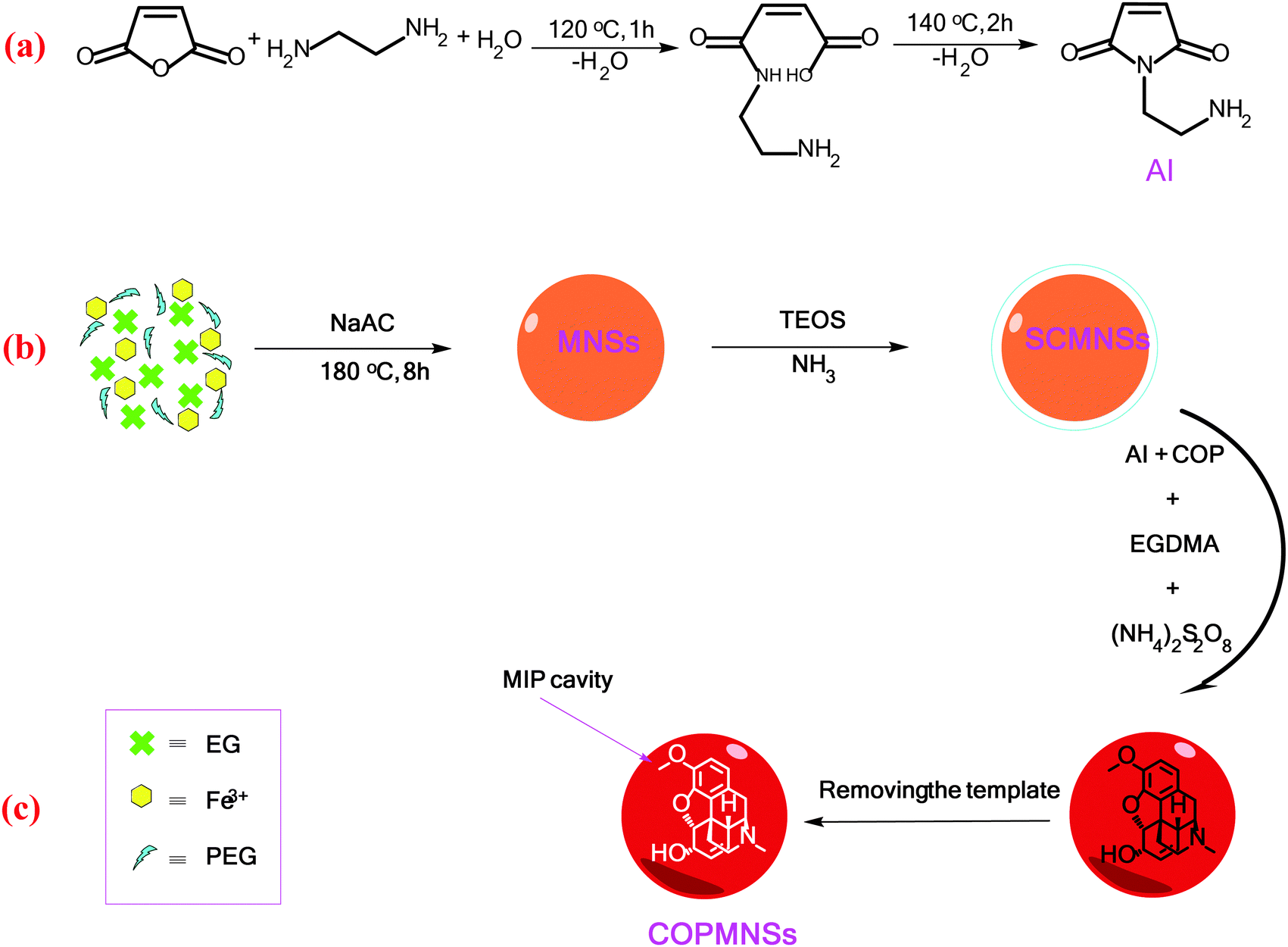 | ||
| Scheme 1 Reactions involved in the synthesis of (a) the aminoimide monomer, (b) MNSs and (c) COPMNSs adsorbents. | ||
Synthesis of the COP imprinted magnetic nanoadsorbent:
MNSs were coated with a silica layer to prevent iron oxide cores from leaching into the mother system under any acidic circumstances. Briefly, 0.10 g of MNSs nanospheres were dispersed in a mixture of ethanol (50 mL), DDW water (10 mL), and concentrated ammonia aqueous solution (3 mL), followed by the addition of tetraethylorthosilicate (TEOS, 2.0 mL). After stirring at room temperature for 1.5 h, the silica coated MNSs nanospheres (SCMNSs) were separated and washed with ethanol and DDW water.38
SPE procedure for COP and its spectrofluorometric determination
To a 25.0 mL sample solution containing COP and 10.0 mL Britton–Robinson buffer solution of pH 6.5, 0.09 g of COPMNSs was added. The solution was shaken at room temperature for 35.0 min. Subsequently, the COP loaded COPMNSs were separated from the mixture within 60 s with a permanent hand-held magnet. The residual amount of the drug in the solution was determined spectrofluorometrically at λem = 348 nm (λex = 214 nm). The adsorption percentage, i.e., the drug removal efficiency (% Re), was calculated using the following equation: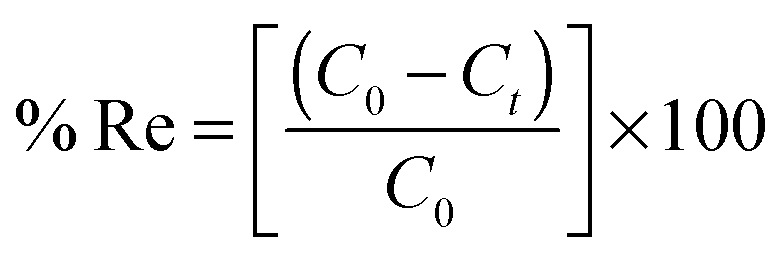 | (1) |
Preconcentration studies for the determination of trace amounts of COP were performed by adding 110.0 mL of the solution containing 2.0–500.0 ng mL−1 COP and 60 mL of Britton–Robinson buffer of pH 6.5 to 0.09 g of COPMNSs; the solution was stirred for 35.0 min. The concentration of COP decreased with time due to adsorption by the adsorbent. The drug loaded nanospheres were separated by magnetic decantation, and desorption was performed with 2.0 mL of methanol. The concentration of COP in the resulting solution was measured spectrofluorometrically at λem = 348 nm (λex = 214 nm).
Adsorption isotherm studies
An adsorption isotherm describes the fraction of sorbate molecules that are partitioned between liquid and solid phase at equilibrium. Adsorption of COP by COPMNSs and NIPMNSs nanospheres was modeled using Langmuir and Freundlich adsorption isotherms.Freundlich isotherm
The Freundlich isotherm is applicable to both monolayer (chemisorption) and multilayer adsorption (physisorption) and is based on the assumption that the adsorbate adsorbs onto the heterogeneous surface of an adsorbent.39 The linear form of the Freundlich equation is expressed as: | (2) |
Langmuir isotherm
The Langmuir isotherm assumes monolayer adsorption on a uniform surface with a finite number of adsorption sites.40 Once a site is filled, no further sorption can occur at that site. Furthermore, the surface will eventually reach a saturation point where the maximum adsorption of the surface will be achieved. The linear form of the Langmuir equation may be written as: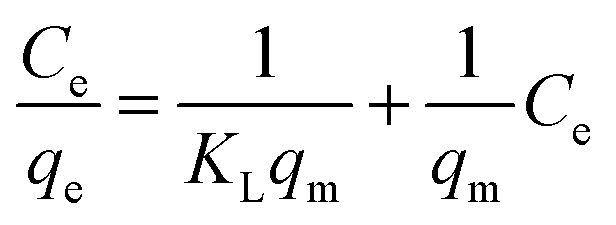 | (3) |
Urine samples pretreatment
Post-dose urine samples were collected from healthy donors who received one 60 mg dose of codeine phosphate orally. All urine samples were stored at −20 °C. Then, 110 mL of each sample was adjusted to pH 6.5 using 0.01–0.1 mol L−1 HCl and/or NaOH and directly subjected to the SPE procedure. All experiments were performed in compliance with the relevant laws and institutional guidelines, and Behbood Institute (Hamedan, Iran) has approved the experiments. Furthermore, the urine samples were spiked with COP at ng mL−1 concentration levels.Results and discussion
To synthesize COPMNSs, MNSs were first synthesized by a solvothermal reduction method. Then, the synthesized nanospheres were coated with a layer of SiO2 using TEOS silanization agent. Finally, the surface of the synthesized SCMNSs was imprinted with COP using the aminoimide monomer as the functional monomer, EGDMA as the cross-linker, COP as the template and ammonium persulfate as the polymerization reaction initiator.Characterization of the synthesized adsorbents
The FT-IR spectra of the products were obtained to verify the formation of the expected products. The related spectra are shown in Fig. 1. The characteristic absorption band of Fe–O in magnetite (around 598 cm−1) was observed in Fig. 1a. A strong peak at 1087 cm−1 in Fig. 1b is attributed to Si–O in SiO2. The new absorption peaks at almost 1452 and 1725 cm−1 in Fig. 1c are assigned to C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands, respectively, in the polymer-coated final product (COPMNSs). Moreover, new absorption peaks at 2956 and 3000 cm−1 are related to the stretching modes of the aliphatic C–H group.26 Based on the abovementioned results, it can be concluded that the fabrication procedure has been successfully performed.
O bands, respectively, in the polymer-coated final product (COPMNSs). Moreover, new absorption peaks at 2956 and 3000 cm−1 are related to the stretching modes of the aliphatic C–H group.26 Based on the abovementioned results, it can be concluded that the fabrication procedure has been successfully performed.
 | ||
| Fig. 1 FT-IR spectra of (a) MNSs, (b) SCMNSs and (c) COPMNSs. | ||
The XRD pattern of the COPMNSs in Fig. 2 shows diffraction peaks that are indexed to (311), (400), (511) and (440) reflection characteristics of the cubic spinel phase of magnetite (JCPDS powder diffraction data file no. 79-0418), revealing that the resultant nanospheres are mostly composed of magnetite. The average crystallite size of the COPMNSs nanospheres was estimated to be 16 nm from the XRD data according to the Scherer equation.23
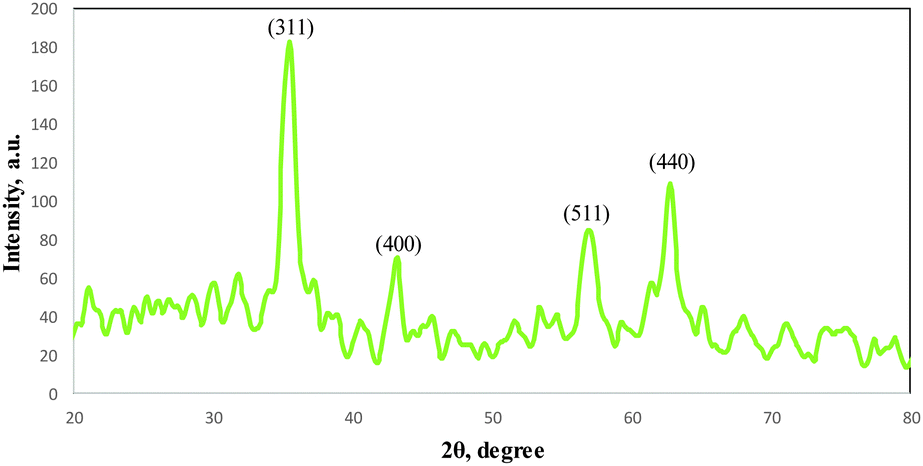 | ||
| Fig. 2 XRD pattern for the COPMNSs nanospheres. | ||
The TEM image of the COPMNSs in Fig. 3b, in comparison with SCMNSs image (Fig. 3a), indicates that the MNSs nanospheres are enwrapped rather homogeneously in an SiO2 shell, and further wrapped by the MIP layer. The average size of the synthesized nanospheres was estimated to be about 95 nm.
 | ||
| Fig. 3 TEM images of (a) SCMNSs and (b) COPMNSs nanospheres. | ||
Point of zero charge (pHPZC) of the COPMNSs adsorbent
The pHPZC of the COPMNSs was determined in degassed 0.01 mol L−1 NaNO3 solution at room temperature. Aliquots of 20.0 mL 0.01 mol L−1 NaNO3 were mixed with 0.03 g of the nanoparticles in several beakers. The initial pH of the solutions was adjusted at 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0 and 10.0 using 0.01–0.1 mol L−1 of HNO3 and/or NaOH solutions as appropriate. The initial pHs of the solutions were obtained, and the beakers were covered with parafilm and shaken for 24 h. The final pH values were obtained and the differences between the initial and final pH (ΔpH) of the solutions were plotted against their initial pH values. The pHPZC corresponds to the pH where ΔpH = 0.26 The pHPZC for the COPMNSs obtained was 6.4 and was determined using the abovementioned procedure. The results are shown at Fig. 4. | ||
| Fig. 4 Point of zero charge of the COPMNSs adsorbent. | ||
Effect of various factors affecting the COP removal efficiency
Various factors that could potentially affect the analyte removal efficiency (i.e. pH, contact time and nanospheres dosage) were optimized and here are the detailed results: | ||
| Fig. 5 Amount of COP uptake per gram of adsorbent at different pH values (Conditions: 0.04 g of COPMNSs, 25 mL of 2.0 mg L−1 of COP, agitation time of 45 min, N = 3). | ||
Effect of various factors affecting the COP preconcentration efficiency
The aim of this step is to provide the highest preconcentration factor and concentration of the adsorbed analyte into the minimum possible volume of desorbing solvent. In this regard, various factors that can potentially affect the COP's desorbing efficiency (i.e. type and volume of desorbing solvent, desorbing time and initial sample volume) have been optimized. The detailed results are given below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.01 mol L−1 HCl (1:1 v/v) and mixture of methanol:0.01 mol L−1 NaOH (1:1 v/v)) were performed. After adsorption of COP, the adsorbent was magnetically separated and washed with DDW. Then, 2.0 mL of the eluent was added to the COP loaded COPMNSs. Samples were collected after 5.0, 10.0, 15.0, 20.0, 25.0, 30.0 and 45.0 min contact times to evaluate the COP recovery. The results (Fig. 6) showed that methanol is the most effective as a back-extracting solvent and can be used for the quantitative recovery of the analyte. Desorption rate was found to be rapid as almost 98% desorption completed at almost 20.0 min.
:0.01 mol L−1 HCl (1:1 v/v) and mixture of methanol:0.01 mol L−1 NaOH (1:1 v/v)) were performed. After adsorption of COP, the adsorbent was magnetically separated and washed with DDW. Then, 2.0 mL of the eluent was added to the COP loaded COPMNSs. Samples were collected after 5.0, 10.0, 15.0, 20.0, 25.0, 30.0 and 45.0 min contact times to evaluate the COP recovery. The results (Fig. 6) showed that methanol is the most effective as a back-extracting solvent and can be used for the quantitative recovery of the analyte. Desorption rate was found to be rapid as almost 98% desorption completed at almost 20.0 min.
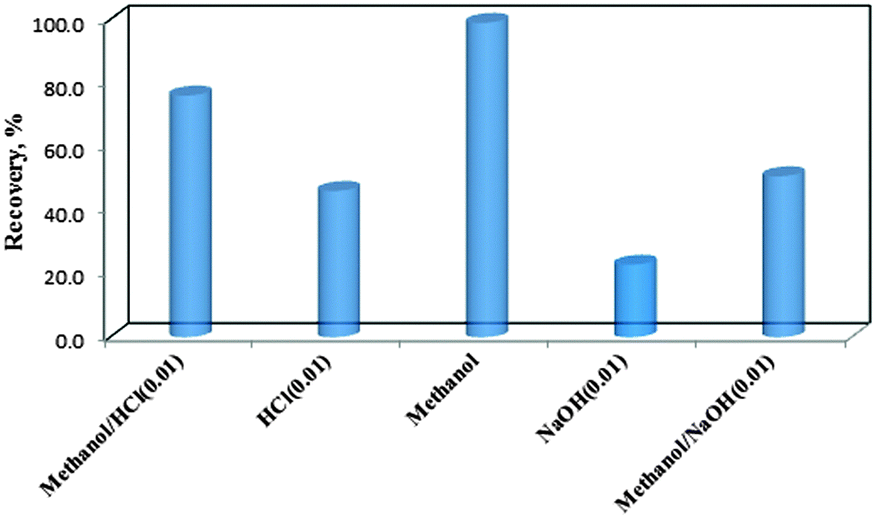 | ||
| Fig. 6 Effect of various eluents on COP recovery. | ||
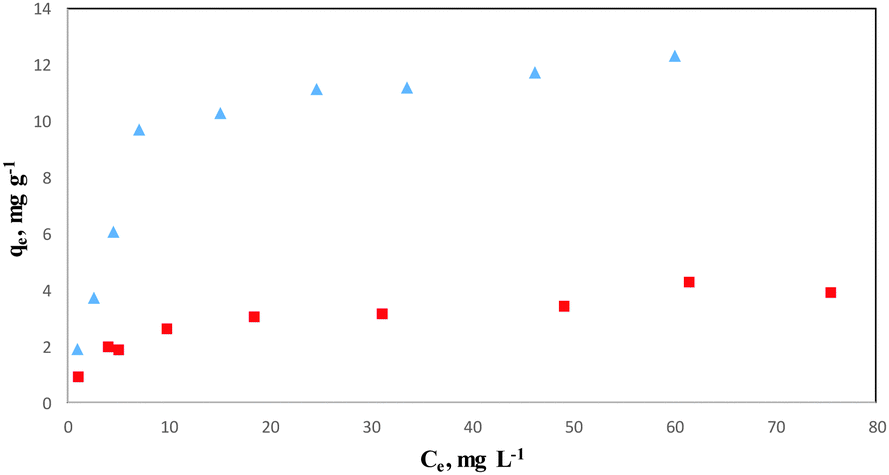 | ||
Fig. 7 Isothermal adsorption curves of COP on ( ) COPMNSs and ( ) COPMNSs and ( ) NIPMNSs adsorbents. ) NIPMNSs adsorbents. | ||
| Isotherm models | Langmuir | Freundlich | ||||
|---|---|---|---|---|---|---|
| K L (L mg−1) | q max (mg g−1) | R 2 | K f (mg1−1/n L1/n g−1) | 1/n (L g−1) | R 2 | |
| COPMNSs | 2.55 | 13.44 | 0.997 | 1.01 | 0.42 | 0.859 |
| NIPMNSs | 0.75 | 4.06 | 0.992 | 0.08 | 0.32 | 0.952 |
The higher correlation coefficient obtained for the Langmuir model (>0.99) indicates that the experimental data are better fitted into this model, and the adsorption of COP onto COPMNSs adsorbents is more compatible with Langmuir assumptions, i.e., adsorption occurs at specific homogeneous sites within the adsorbent. According to the results (Table 1), the maximum amount of COP that can be adsorbed by COPMNSs and NIPMNSs was found to be 13.44 and 4.06 mg g−1, respectively, at pH 6.5. The relatively high adsorption capacity of COPMNSs in comparison with NIPMNSs, shows that the adsorption of COP molecules occurs at a large number of specific homogeneous sites within the adsorbent (specific cavities of the MIP layer), besides the non-specific interactions, which are approximately identical for both the COPMNSs and the NIPMNSs adsorbents.
Analytical applications
A calibration graph was constructed from spectrofluorometric measurements of the desorbed COP after performing its adsorption/separation under the optimum conditions as described above. The calibration graph was linear in the range of 2.0–500.0 ng mL−1 for a sample volume of 110.0 mL. The calibration equation is IF = 0.714C + 0.791 with a determination coefficient of 0.995 (n = 10), where IF is the fluorescence intensity of the eluate at λem = 348 nm (λex = 214 nm) and C is the concentration of the analyte in ng mL−1.The limit of detection, defined as LOD = 3Sb/m, where LOD, Sb and m are the limit of detection, standard deviation of the blank and the slope of the calibration graph, respectively, was found to be 0.67 ng mL−1 of COP. As the analyte in 110.0 mL of the sample solution was concentrated into 2.0 mL, a maximum preconcentration factor of 55.0 was achieved in this method. The relative standard deviations (RSD) for 100.0 and 10.0 ng mL−1 of the analyte were 0.91% and 1.73% (n = 5), respectively.
The analytical applicability of the proposed method was evaluated by determining the analyte in human urine samples. The results are given in Table 2. The results of the spiked samples show good recoveries of the proposed method and suggest that the method is a good accurate candidate for COP determination in the investigated samples. Furthermore, the results were compared to those using an HPLC method,42 and these are tabulated in Table 2. The results show that the two methods are in good agreement and the proposed method can be used for effective determination of COP in urine samples.
| Experiment no. | Spiked value (ng mL−1) | Found (ng mL−1) | Recovery percent | HPLC method |
|---|---|---|---|---|
| *N = 3. | ||||
| 1 | — | 12.01 ± 0.04* | — | 11.71 ± 0.07 |
| 2 | 10.00 | 21.80 ± 0.03 | 98.1 | 23.41 ± 0.06 |
| 3 | 20.00 | 31.43 ± 0.01 | 97.3 | 32.04 ± 0.02 |
Table 3 shows a comparison between the results obtained by the present method with those obtained by some other methods reported for the determination of COP. This comparison shows that the proposed method analytical performance is comparable with some sensitive instrumental methods such as HPLC, and in some cases the method is superior. Other advantages of the proposed method are its simplicity, lower cost, low LOD, wide linear range, easy adsorbent separation and high removal capacity of the adsorbent.
| Method | LOD (ng mL−1) | Sample | Ref. |
|---|---|---|---|
| a Headspace solid-phase microextraction. b Reverse-phase high-performance liquid chromatography. c Liquid–liquid microextraction followed by high-performance liquid chromatography. d Liquid chromatography tandem mass spectrometry. | |||
| HS-SPMEa | 1.0 | Human hair | 43 |
| RPHPLCb | 3.0 | Human blood plasma | 42 |
| LC–MS | 10.0 | Human hair | 44 |
| LLME–HPLCc | 0.6 | Human urine | 9 |
| LC-MS/MSd | 1.0 | Human urine | 45 |
| MIP/SPE/FL | 0.67 | Human urine | This work |
Conclusions
The novel molecularly imprinted nanoadsorbent, used for recognizing codeine, was prepared using magnetite nanospheres as the magnetic core, a synthesized aminoimide monomer as the functional monomer, ethylene glycol dimethacrylate as the cross-linker, ammonium persulfate as the initiator and codeine as the template. The synthesized core–shell adsorbent has been characterized using XRD, FTIR and TEM measurements. The recognition properties and applications based on the solid-phase extraction of the synthesized nanoadsorbent were evaluated. The resulting nanoadsorbent showed a relatively large adsorption capacity and fast binding kinetics for recognizing the drug, and the method showed high recovery, good accuracy and repeatability in determining the drug in human urine samples.Acknowledgements
This study was financially supported by the Bu-Ali Sina University Research Council and the Center of Excellence in Development of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS), and the authors acknowledge the support provided for this study.References
- Y. Li, K. Li, G. Song, J. Liu, K. Zhang and B. Ye, Sens. Actuators, B, 2013, 182, 401 CrossRef CAS.
- D. T. Gimenes, R. R. Cunha, M. M. A. d. C. Ribeiro, P. F. Pereira, R. A. A. Muñoz and E. M. Richter, Talanta, 2013, 116, 1026 CrossRef CAS PubMed.
- B. A. Sproule, U. E. Busto, G. Somer, M. K. Romach and E. M. Sellers, J. Clin. Psychopharmacol., 1999, 19, 367 CrossRef CAS PubMed.
- M. Hakkinen, T. Launiainen, E. Vuori and I. Ojanpera, Forensic Sci. Int., 2012, 222, 327 CrossRef CAS PubMed.
- M. Krogh, A. S. Christophersen and K. E. Rasmussen, J. Chromatogr., 1993, 621, 41 CrossRef CAS PubMed.
- K. A. Rees, P. A. McLaughlin and M. D. Osselton, J. Anal. Toxicol., 2012, 36, 1 CrossRef CAS PubMed.
- U. Hofmann, S. Seefried, E. Schweizer, T. Ebner, G. Mikus and M. Eichelbaum, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 727, 81 CrossRef CAS.
- H. He, S. D. Shay, Y. Caraco, M. Wood and A. J. Wood, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 708, 185 CrossRef CAS.
- M. Shamsipur and N. Fattahi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 2978 CrossRef CAS PubMed.
- Z. Hu, Q. Zou, J. Tian, L. Sun and Z. Zhang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 3937 CrossRef CAS PubMed.
- G. M. Greenway, L. J. Nelstrop and S. N. Port, Anal. Chim. Acta, 2000, 405, 43 CrossRef CAS.
- B. Qiu, X. Chen, H. L. Chen and G. N. Chen, Luminescence, 2007, 22, 189 CrossRef CAS PubMed.
- M. E. Capella-Peiro, D. Bose, M. F. Rubert and J. Esteve-Romero, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 839, 95 CrossRef CAS PubMed.
- T. Zhou, H. Yu, Q. Hu and Y. Fang, J. Pharm. Biomed. Anal., 2002, 30, 13 CrossRef CAS PubMed.
- D. S. Popa, R. Oprean, E. Curea and N. Preda, J. Pharm. Biomed. Anal., 1998, 18, 645 CrossRef CAS PubMed.
- B. Weingarten, H. Y. Wang and D. M. Roberts, J. Chromatogr. A, 1995, 696, 83 CrossRef CAS PubMed.
- S. Gandhi, P. Sharma, N. Capalash, R. S. Verma and C. R. Suri, Anal. Bioanal. Chem., 2008, 392, 215 CrossRef CAS PubMed.
- A. Afkhami, H. Khoshsafar, H. Bagheri and T. Madrakian, Sens. Actuators, B, 2014, 203, 909 CrossRef CAS.
- A. Afkhami, M. Abbasi-Tarighat, M. Bahram and H. Abdollahi, Anal. Chim. Acta, 2008, 613, 144 CrossRef CAS PubMed.
- T. Madrakian, A. Afkhami, M. A. Zolfigol, M. Ahmadi and N. Koukabi, Nano-Micro Lett., 2012, 4, 57 CrossRef CAS.
- T. Madrakian, A. Afkhami, M. Rahimi, M. Ahmadi and M. Soleimani, Talanta, 2013, 115, 468 CrossRef CAS PubMed.
- T. Madrakian, A. Afkhami, H. Mahmood-Kashani and M. Ahmadi, J. Iran. Chem. Soc., 2013, 10, 481 CrossRef CAS.
- T. Madrakian, A. Afkhami and M. Ahmadi, Chemosphere, 2013, 90, 542 CrossRef CAS PubMed.
- A. Afkhami, R. Moosavi and T. Madrakian, Talanta, 2010, 82, 785 CrossRef CAS PubMed.
- A. Afkhami, H. Bagheri and T. Madrakian, Desalination, 2011, 281, 151 CrossRef CAS.
- T. Madrakian, A. Afkhami, H. Mahmood-Kashani and M. Ahmadi, Talanta, 2013, 105, 255 CrossRef CAS PubMed.
- B. Sellergren, Molecularly Imprinted Polymers: Man-Made Mimics of Antibodies and their Application in Analytical Chemistry, Elsevier Science, 2000 Search PubMed.
- T. Madrakian, M. Ahmadi, A. Afkhami and M. Soleimani, Analyst, 2013, 138, 4542 RSC.
- Z. Lin, W. Cheng, Y. Li, Z. Liu, X. Chen and C. Huang, Anal. Chim. Acta, 2012, 720, 71 CrossRef CAS PubMed.
- X. Mao, H. Sun, X. He, L. Chen and Y. Zhang, Anal. Methods, 2015, 7, 4708 RSC.
- Y. Ji, J. Yin, Z. Xu, C. Zhao, H. Huang, H. Zhang and C. Wang, Anal. Bioanal. Chem., 2009, 395, 1125 CrossRef CAS PubMed.
- W. Rao, R. Cai, Y. Yin, F. Long and Z. Zhang, Talanta, 2014, 128, 170 CrossRef CAS PubMed.
- H. He, D. Xiao, J. He, H. Li, H. He, H. Dai and J. Peng, Analyst, 2014, 139, 2459 RSC.
- T. Madrakian, A. Afkhami, B. Zadpour and M. Ahmadi, J. Ind. Eng. Chem., 2015, 21, 1160 CrossRef CAS.
- X. Wang, L. Wang, X. He, Y. Zhang and L. Chen, Talanta, 2009, 78, 327 CrossRef CAS PubMed.
- X. Kan, Q. Zhao, D. Shao, Z. Geng, Z. Wang and J.-J. Zhu, J. Phys. Chem. B, 2010, 114, 3999 CrossRef CAS PubMed.
- M. Ahmadi, T. Madrakian and A. Afkhami, Anal. Chim. Acta, 2014, 852, 250 CrossRef CAS PubMed.
- M. Ahmadi, T. Madrakian and A. Afkhami, New J. Chem., 2015, 39, 163 RSC.
- H. Freundlich and W. Heller, J. Am. Chem. Soc., 1939, 61, 2228 CrossRef CAS.
- I. Langmuir, J. Am. Chem. Soc., 1916, 38, 2221 CrossRef CAS.
- E. Fuguet, M. Reta, C. Gibert, M. Rosés, E. Bosch and C. Ràfols, Electrophoresis, 2008, 29, 2841 CAS.
- V. Nitsche and H. Mascher, J. Pharm. Sci., 1984, 73, 1556 CrossRef CAS PubMed.
- F. Sporkert and F. Pragst, Forensic Sci. Int., 2000, 107, 129 CrossRef CAS PubMed.
- D. K. Huang, C. Liu, M. K. Huang and C. S. Chien, Rapid Commun. Mass Spectrom., 2009, 23, 957 CrossRef CAS PubMed.
- T. Nema, E. C. Chan and P. C. Ho, J. Mass Spectrom., 2011, 46, 891 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |