
Synthesis of novel TEMPO stable free (poly)radical derivatives and their host–guest interaction with cucurbit[6]uril†
Gabriela
Ionita
*a,
Augustin M.
Madalan
b,
Ana Maria
Ariciu
ac,
Andrei
Medvedovici
d and
Petre
Ionita
*ac
a“Ilie Murgulescu” Institute of Physical Chemistry of the Romanian Academy, Bucharest, 060021, Romania. E-mail: ige@icf.ro; pionita@icf.ro; Fax: +40 213121147
bInorganic Chemistry Department, Faculty of Chemistry, University of Bucharest, Str. Dumbrava Rosie nr. 23, 020464-Bucharest, Romania
cDepartment of Organic Chemistry, Biochemistry and Catalysis, University of Bucharest, 90-92 Panduri, Bucharest, Romania
dDepartment of Analytical Chemistry, University of Bucharest, 90-92 Panduri, Bucharest, Romania
First published on 10th November 2015
Abstract
A number of ten stable free mono-, di- and tri-radicals of the TEMPO nitroxide type were synthesized and characterized via physico-chemical methods (elemental analysis, MS, UV-Vis, IR, ESR, and X-ray, where appropriate). The design of the compounds was chosen such that supramolecular interaction could be gained via hydrogen-bonding or π–π interactions; therefore the compounds should contain amino- or urea-moieties, (nitro)aromatic rings, or a crown ether residue. The formation of inclusion complexes between these (poly)radicals and cucurbit[6]uril was studied via Electron Spin Resonance (ESR) spectroscopy. The binding constants were evaluated from the analysis of the rotational correlation time dependence on the concentration of cucurbit[6]uril. These constants are an order of magnitude lower than the values reported before for complexes of TEMPO derivatives with β-cyclodextrin. The complexation behaviour of cucurbit[6]uril was revealed in the ESR spectra of the nitroxides in the presence and in the absence of the host molecules, recorded at low temperatures. The experiments revealed that the mobilities of the nitroxides investigated are higher in the absence of the host molecule, a different behaviour compared with complexes with cyclodextrins. This behavior can be understood by taking into account the role played by the presence of salts necessary to ensure the solubility of cucurbit[6]uril in water.
Introduction
Nitroxides are a family of N,N-disubstituted NO˙ free radicals with the unpaired electron delocalised between the nitrogen and oxygen atom. The shielding substituents, usually alkyl substituents,1,2 prevent radical–radical dismutation and also limit the access of reactive substances which can quench the radical species. Tetramethylpiperidine-N-oxyl (TEMPO) based radicals represent a well known class of stable nitroxide which has found applications in various fields of chemistry. Often, these radicals are used as spin labels or probes to report on microenvironment and molecular interactions in various assemblies,1 but they can be used as spin traps,3 antioxidants,4 mediators in free radical polymerisation5 or as building blocks for molecular based magnetic materials.6The presence of two or more TEMPO like moieties in a molecule may lead to intramolecular spin exchange interactions quantified by the exchange interaction constant, J.7 The ESR spectrum of a poly-nitroxide depends on the relation between J and the hyperfine coupling constant, aN. The exchange coupling in biradicals or polyradicals can be exploited to report on the microenvironmental changes occurring in a particular system.8–10
A considerable number of studies addressing the use of ESR spectroscopy in the investigation of various types of host–guest interactions have been reported in the last four decades.11 The advantages of this method include its high sensitivity to the molecular environment in the immediate vicinity of the spin label and the ability to probe interspin distances and molecular dynamics.1,11 Most of these studies regarding the interaction between host molecules and monoradicals proved the changes in aN and the rotational correlation time, τc. Although in this field the most prominent class of host molecules is represented by the cyclodextrin family, which is considered to be a natural compound family (as it results from the enzymatic degradation of starch), the studies on synthetic cyclic compounds with defined hydrophobic cavities (cucurbituril,12 calixarenes13,14) have been expanding in the last decades.12 The recognition properties of different molecular hosts are exploited in already found applications in developing pseudorotaxanes and separation systems, and drug formulation.14–16
In this paper, we report the synthesis of a series of new mono-, di, and tri-nitroxide type radicals (Chart 1), except compound 2a which has been reported by others,17,18 and their recognition properties in the presence of cucurbit[6]uril (Q[6]), evaluated via analysis of the ESR spectra.
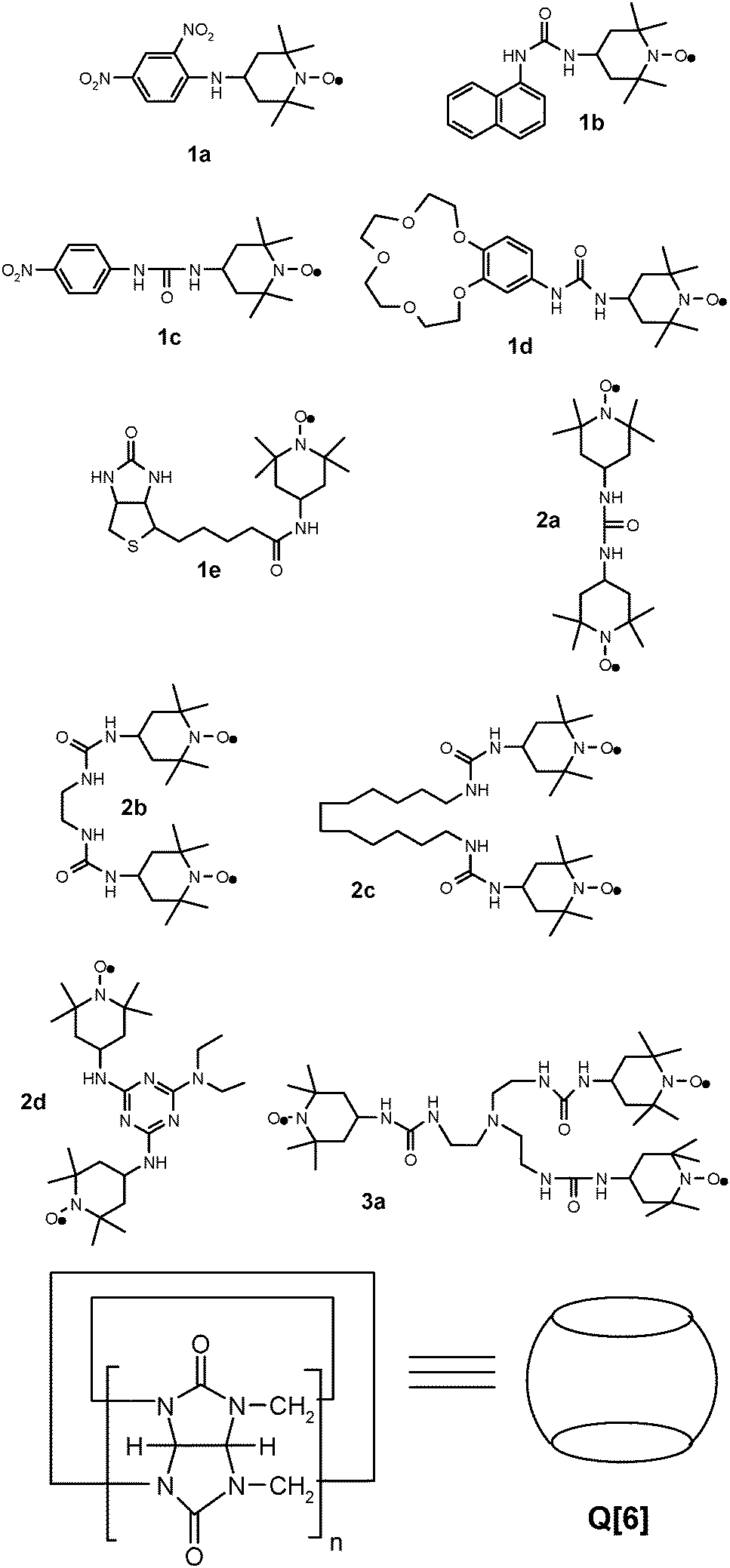 | ||
| Chart 1 The structure of the nitroxide radicals used in this study and of cucurbit[6]uril ([Q6]). | ||
Cucurbiturils are synthetic macrocyclic compounds containing methylene-linked glycoluril fragments, obtained upon condensation of formaldehyde with glycoluril (the condensation product of urea and glyoxal) in acidic medium. Cucurbiturils were suggestively named after the pumpkin like shape, having hydrophobic cavities marked by methylene groups and hydrophilic portals represented by the carbonyl groups, so they can easily act as a versatile host, forming a large number of possible inclusion complexes.12,19
Compared with the other well recognised classes of guest molecules, cucurbiturils are hosts proficient at binding a range of guests into their rigid cavities, like organic molecules through hydrophobic interactions, or metal cations and protonated alkyl and aryl amines via ion–dipole interactions involving their carbonyl portal ends.19 Quite a few studies regarding the inclusion complexes of cucurbiturils with monoradical20–24 or with biradical16,25 nitroxide type radicals have been reported.
In our attempt to study the inclusion complexes of the radicals presented in Chart 1 with Q[6], we have started to evaluate the binding constant considering the variation of the rotational correlation time, τc, with the concentration of the guest molecule. The ESR parameters in the temperature range 293–333 K have been used to evaluate the thermodynamic parameters of the inclusion complexes between the radicals and Q[6]. Moreover, being inspired by the results of a previously reported study,26 regarding the complexes between cyclodextrins with TEMPO type radicals at temperatures below 273 K, we investigated the behaviour of these new radicals with Q[6] at low temperatures.
Experimental
Materials
All chemicals and materials were supplied by Sigma-Aldrich and used as received. 4-Isocyanato-TEMPO was synthesized as reported.27 UV-Vis spectra were recorded using a UVD-3500 spectrometer, in methanol and at ambient temperature. IR spectra were recorded using a Bruker Vertex 70 spectrometer (as solid samples, ATR). ESI-MS spectra were recorded using Agilent 1200 series modules (quaternary pump G4204A, automated injector/thermostat G1367E/G1330B, ESI source G1948B, triple quadrupole MS G6410B). ESR spectra were recorded using a Jeol JES-FA 100 spectrometer, in dichloromethane (DCM) as the solvent and at room temperature; for the variable temperature spectra, see discussion.X-ray crystallography
X-ray diffraction measurements were performed using a STOE IPDS II diffractometer, operating with a Mo-Kα (λ = 0.71073 Å) X-ray tube with a graphite monochromator. The structures were solved using direct methods and refined using full-matrix least squares techniques based on F2. The non-H atoms were refined with anisotropic displacement parameters. Calculations were performed using the SHELX-2014 crystallographic software package. A summary of the crystallographic data and the structure refinement for crystals 1a and 1c are given in Table 1. CCDC 1402759 (1a) and 1402760 (1b).| Compound | 1a | 1c |
|---|---|---|
| Chemical formula | C15H21N4O5 | C16H25N4O5 |
| M (g mol−1) | 337.36 | 353.40 |
| Temperature, (K) | 293(2) | 293(2) |
| Wavelength, (Å) | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n |
| a (Å) | 7.8007(6) | 7.0768(7) |
| b (Å) | 10.7877(5) | 25.637(4) |
| c (Å) | 20.2847(16) | 10.7812(11) |
| α (°) | 90 | 90 |
| β (°) | 91.562(7) | 101.712(8) |
| γ (°) | 90 | 90 |
| V (Å3) | 1706.4(2) | 1915.3(4) |
| Z | 4 | 4 |
| D c (g cm−3) | 1.313 | 1.226 |
| μ (mm−1) | 0.100 | 0.092 |
| F(000) | 716 | 756 |
| Goodness-of-fit on F2 | 0.973 | 0.724 |
| Final R1, wR2 [I > 2σ(I)] | 0.0479, 0.0985 | 0.0451, 0.0432 |
| R 1, wR2 (all data) | 0.1063, 0.1162 | 0.2069, 0.0687 |
| Largest diff. peak and hole (e Å−3) | 0.120, −0.158 | 0.105, −0.083 |
ESR measurements
ESR spectra were recorded on a Jeol JES FA 100 spectrometer, equipped with a temperature variable controller. The general settings used were as follows: centre field 3356 G, sweep field 80 G, frequency 100 kHz, gain in the range 100–200, sweep time 480 s, time constant 0.3 s, modulation width 1 G, and microwave power 1 mW. The ESR spectra were recorded either in the temperature range 293–323 K or 273–100 K. Each sample was left in the resonator cavity at a particular temperature for 5 minutes to equilibrate. The rotational correlation times were determined using the following equation: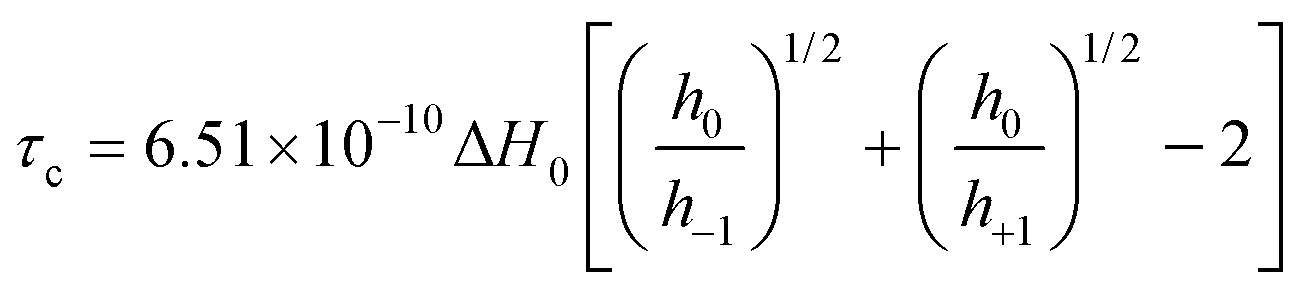 | (1) |
Sample preparation
A stock solution of Q[6] × 10−2 M was prepared in HCl (20 mM)/NaCl (2 M). From this solution, a series of solutions with a concentration of Q[6] in the range 2 × 10−4–10−2 M were prepared. Solutions of TEMPO-derivatives at a concentration of 10−2 M in ethanol were prepared. For ESR measurements, the solvent of an appropriate volume of this solution was evaporated and a new solution was obtained by adding a solution of HCl (20 mM)/NaCl (2 M) or a solution of Q[6] in order to obtain a concentration of 10−4 M of the radical, in each case. To prepare the samples for ESR measurements at temperatures below 273 K, an aliquot of the ethanol solutions of the nitroxides was evaporated. Each residue was redissolved in a 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (w/w) water solution of (HCl/NaCl):glycerol to result in a 4 × 10−4 M concentration for the spin probe.
:2 (w/w) water solution of (HCl/NaCl):glycerol to result in a 4 × 10−4 M concentration for the spin probe.
Results and discussion
The synthesis of all compounds used in this work, namely stable monoradicals 1a–e, diradicals 2a–d, and triradical 3a (Chart 1), was performed practically in one or two steps, starting from commercially available compounds. Thus, 4-amino-TEMPO reacts with the selected halogen-reactive derivatives (via nucleophilic substitution) to yield compounds 1a, 1e and 2d. A key intermediate in the synthesis of the other compounds was 4-isocyanato-TEMPO, obtained from 4-amino-TEMPO, as the literature data showed.27 4-Isocyanato-TEMPO easily reacts with the selected amine yielding urea derivatives 1b–d and 2a–c. Both types of reactions occur with good yields.All of these TEMPO derivatives were characterized via elemental analysis, ESI-MS, UV-Vis and IR spectrometry, and ESR. For the derivatives 1a and 1c, obtained as crystals, X-ray diffraction was also employed to characterize their structures. All analyses confirmed the purity and the structure of the obtained radicals.
The UV-Vis spectra of the TEMPO radicals may differ due to the presence of other moieties covalently attached. Thus, in the UV-Vis spectra, the most bathochromic shift was noticed for compound 1a, which contains a dinitrophenyl ring. In the IR spectra, nitro groups are present at about 1320 and 1590 cm−1, the aromatic rings at about 3000 cm−1, the amino groups at about 3300 cm−1, and the carbonyl groups at about 1600–1700 cm−1, while the nitroxide moiety appears at about 1300 cm−1. In the ESR spectra, the three lines due to the interaction of the unpaired electron with the nitrogen nucleus have hyperfine coupling constants of about 15.5 Gauss (for the specific values, see the Experimental part). The ESR spectra of the radicals presented in Chart 1 are shown in the ESI,† Table S1.
Description of the X-ray structures of 1a and 1c
Crystals suitable for X-ray diffraction analysis were obtained for compounds 1a and 1c from a DCM/ethyl acetate solution upon slow evaporation of the solvent.The molecular structure of compound 1a determined via X-ray diffraction on a single crystal is depicted in Fig. 1 along with the atom labelling scheme. The 2,4-dinitrophenylamino fragment is placed in the equatorial position of C5 belonging to the piperidine ring. The two nitro groups are co-planar with the phenyl ring and the nitro group from the ortho position is involved in the intramolecular hydrogen interaction with the amino group (N2–H2⋯O2 = 1.97 Å).
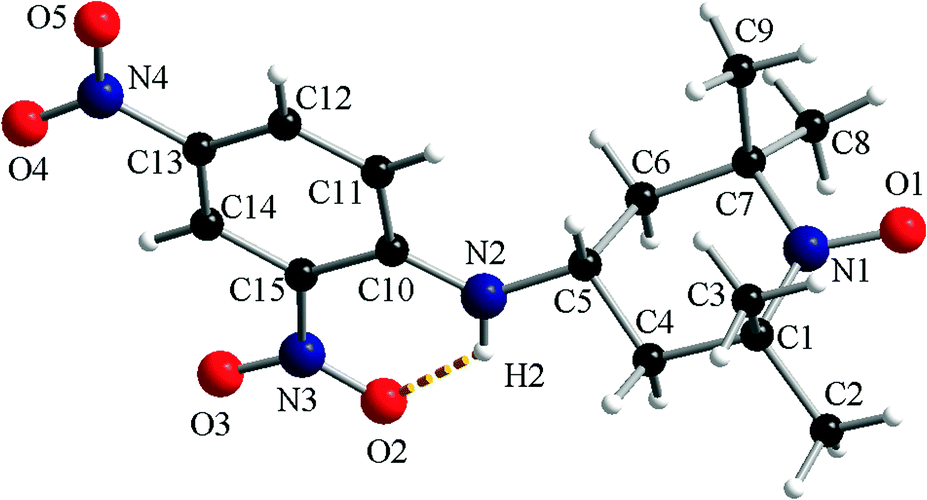 | ||
| Fig. 1 A perspective view of the molecular structure of 1a. | ||
In the crystal, for compound 1a, the molecular units are organized in supramolecular columns running along the crystallographic a axis (ESI,† Fig. S1a). The supramolecular columns are generated by the π–π stacking interactions established between the neighbouring dinitrophenyl fragments (A ≈ 3.16–3.19 Å; B ≈ 3.39–3.45 Å). The analysis of the packing diagram of 1a reveals that the supramolecular pillars are arranged in a herringbone fashion (ESI,† Fig. S1b).
The crystal structure of compound 1c consists of organic radicals and crystallization water molecules (Fig. 2). The urea and 4-nitrophenyl fragments are almost co-planar with a dihedral angle between their mean planes of 7.8°. The urea fragment is located in the equatorial position of C8 from the piperidine ring.
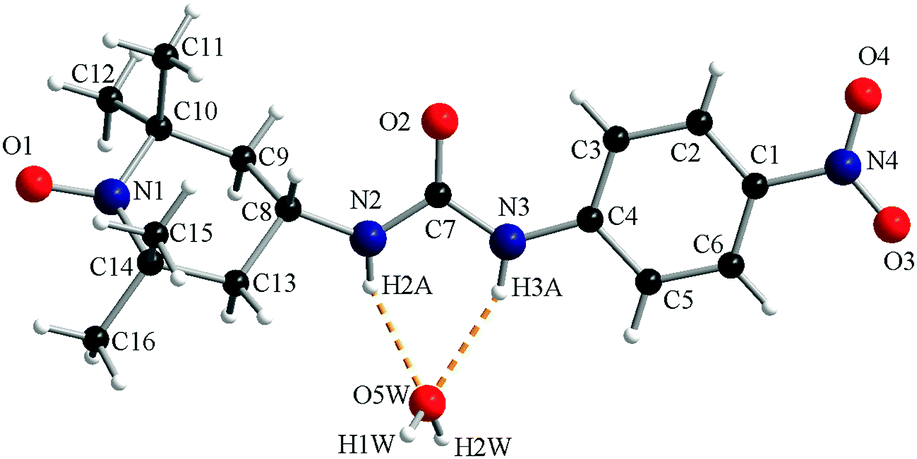 | ||
| Fig. 2 A perspective view of the asymmetric unit of 1c. | ||
The N–H groups of the urea fragment act as hydrogen bond donors toward the crystallization water molecule (N2–H2A⋯O5W = 2.12 Å and N3–H3A⋯O5W = 2.16 Å). The water molecule is a hydrogen bond donor toward two oxygen atoms, one belonging to the urea fragment of a neighbouring organic molecule (O5W–H2W⋯O2′ = 1.87 Å, ′ = −1 + x, y, z) and one oxyl atom from another radical (O5W–H1W⋯O1′′ = 1.90 Å, ′′ = −0.5 + x, 0.5 − y, 0.5 + z; Fig. 3).
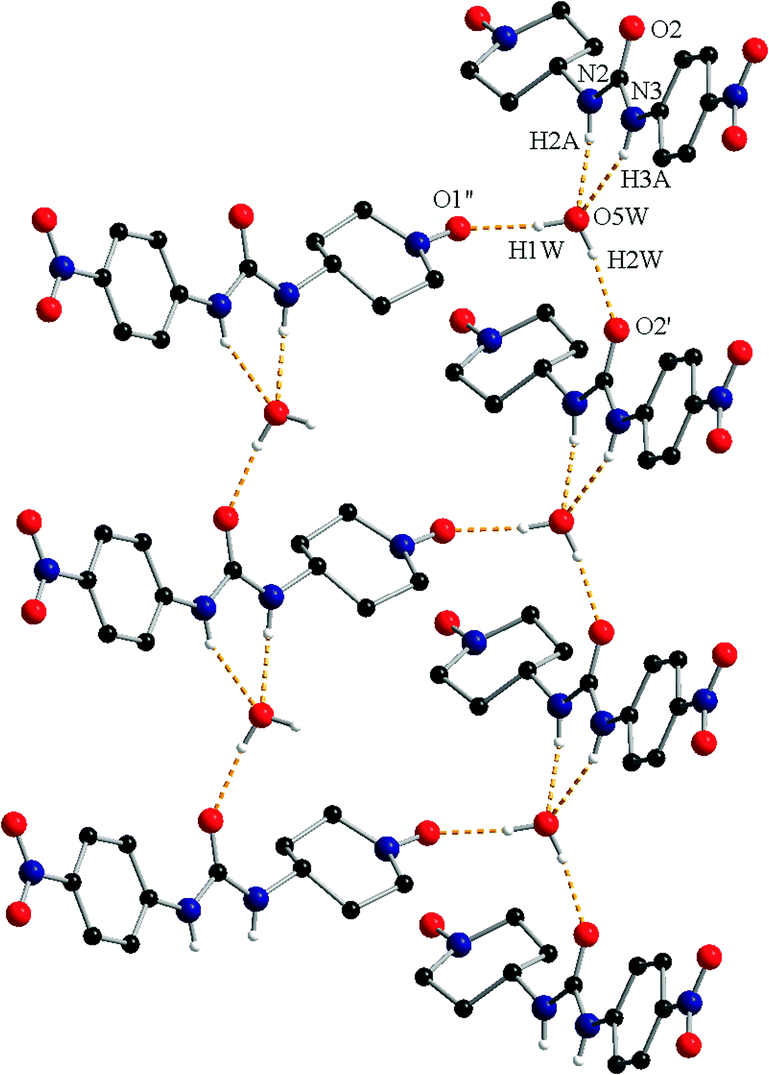 | ||
| Fig. 3 View of the hydrogen interaction pattern in crystal 1c (symmetry codes: ′ = −1 + x, y, z; ′′ = −0.5 + x, 0.5 − y, 0.5 + z). The methyl groups and the hydrogen atoms not involved in hydrogen bonding are omitted. | ||
The hydrogen bonds generate a supramolecular two dimensional (2D) architecture (ESI,† Fig. S2 – in orange) which further develops into a 3D architecture through the π–π stacking interactions (3.47–3.60 Å) established between the 4-nitrophenyl-urea moieties.
Interaction of nitroxide radicals with Q[6]
The ESR spectra of the mono-nitroxides presented in Chart 1, recorded in DCM, are characterised by the values of hyperfine splitting, aN, in the range of 15.4–15.6 G. Similar values for aN were calculated for the bi- and tri-radicals, considering the lines which appear at low and at high magnetic fields in the ESR spectra. Like the commercially available TEMPO derivatives, the radicals presented in Chart 1 can find applications as spin probes for various systems. For instance, here we analysed the host–guest complexation of radicals with Q[6] using ESR measurements, considering the sensitivity of this method to changes in the dynamics of spin probes and in the polarity of the microenvironment sensed.11 The suitability of this method to describe the behaviour of TEMPO type radicals in host–guest complexes formed with cyclodextrins and cucurbiturils or calixarenes has been already demonstrated in an appreciable number of studies.11,20–25,29 The portal diameter of Q[6] is comparable to that of α-cyclodextrin, while the interior diameter has an intermediary value between those corresponding to α- and β-cyclodextrin.30,31 However, due to the rigidity of the Q[6] molecules31 we can expect similarities in the binding abilities of Q[6] and α-cyclodextrin to nitroxides. The evidence for the formation of the Q[6] complexes with the radicals presented in Chart 1 is the decrease of the aN values (Fig. 4) and the slower tumbling of the radicals. In Table 2, the differences in aN values are summarised for three commercially available TEMPO radicals and for the radicals presented in Chart 1, corresponding to the ESR spectra of their solutions at the maximum concentration and in the absence of the host (Q[6]).32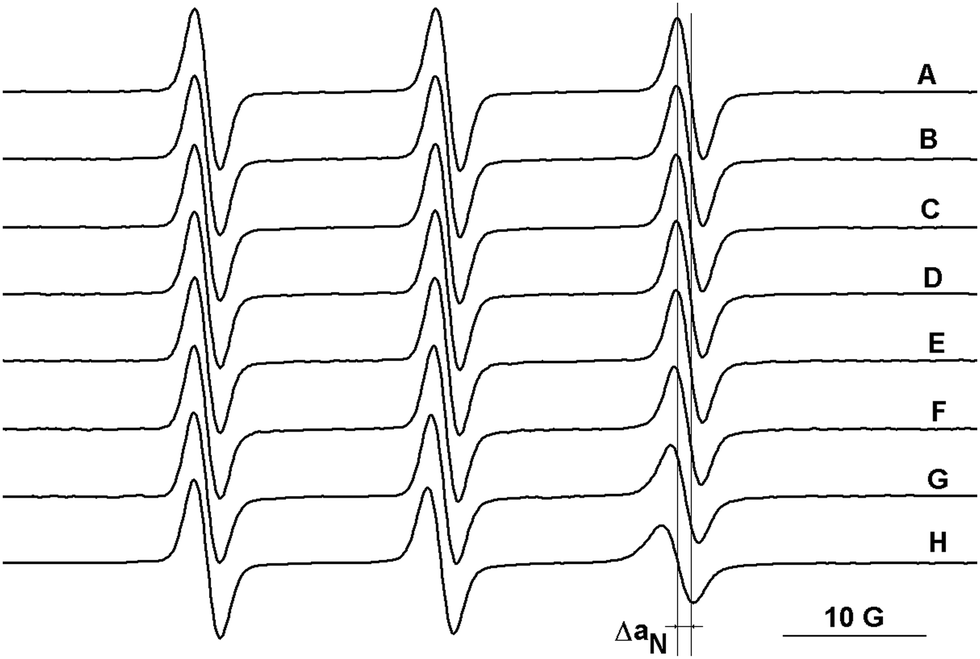 | ||
| Fig. 4 The ESR spectra of 1a in: (A) 0.2 M HCl and in solutions of Q[6] at (B) 10−4 M, (C) 5 × 10−4 M, (D) 7 × 10−4 M, (E) 10−3 M, (F) 2.5 × 10−3 M, (G) 5 × 10−3 M, and (H) 10−2 M. | ||
| Radical | ΔaN (G) | Radical | ΔaN (G) | Radical | ΔaN (G) |
|---|---|---|---|---|---|
| TEMPO | 0.27 | 1a | 0.44 | 2a | 0 |
| 1b | 0.45 | 2b | 0.22 | ||
| 4-Carboxy-TEMPO | 0.30 | 1c | 0.20 | 2c | 0.47 |
| 4-Amino-TEMPO | 0.60 | 1d | 0.37 | 2d | 0.56 |
| 1e | 0.27 | 3a | 0.07 | ||
The ESR parameters of the radicals are sensitive to the presence of Q[6], except for radicals 2a and 3a which probably do not form complexes, most likely due to geometrical restrictions. The values presented in Table 2 show that the largest variation of aN is observed in the case of the protonated form of 4-amino-TEMPO. In this case, it can be considered that ΔaN = 0.6 G describes the deepest inclusion of the nitroxide group in the cavity of Q[6] and the formation of a stable complex. The values of ΔaN observed for the TEMPO and 4-carboxy TEMPO radicals reveal a moderate exchange between the complexed and uncomplexed radical species. A similar decrease in aN with 4-amino-TEMPO is noticed for 2d due to the protonation of the amino group. The amino groups from the structures of 1a and 1b are less protonated than those of 4-amino-TEMPO due to the electrophilic character of the aromatic fragments from these compounds. The variation of aN observed for radicals 1a, 1b and 1d suggests that the nitroxide moiety is partly included or deeply included with a moderate rate of exchange. In contrast, a faster exchange between the complexed and uncomplexed nitroxide moieties of 1c and 1e can be considered as the values of ΔaN are similar to those observed for the TEMPO and 4-carboxy TEMPO radicals. The ESR spectra of biradicals 2a–2b and tri-radical 3a show the lines attributed to spin–spin interactions. Depending on the structural features, such as the flexibility of the linkage between the paramagnetic moieties and the distance between them, theoretically, it is possible to prove the complexation with Q[6] by monitoring the changes in the intensity of these lines as the concentration of the host molecule increases. The intensities of the lines attributed to spin–spin interactions are also dependent on the solvent nature. For all of the poly-nitroxides, these lines are less intense in water solutions, indicating a lower frequency of the collisions between the paramagnetic moieties linked in a given compound. The complexation of 2a by the rigid molecule Q[6] is somehow restricted due to the short distance between the paramagnetic moieties. For the other poly-nitroxides, the intensities of the lines attributed to spin–spin interactions decrease as the concentration of Q[6] increases (ESI,† Fig. S3). In the case of compound 2c, the linker between the paramagnetic moieties is flexible but the length of the alkyl chain is long enough to determine weak exchange interactions. The complexation of the radicals with Q[6] shields the exchange interaction in the polyradicals. As is the case for the monoradicals, the variation of aN for the biradicals determined by the presence of Q[6] provides qualitative information on the complexation process. Thus, in the case of 2b, a fast exchange between the complexed and uncomplexed species can be considered while in the case of the biradical 2d, the nitroxide moieties are deeply included. The value of ΔaN observed for 2c indicates a faster exchange between the complexed/uncomplexed species compared with 2d. The literature describes two models for the interaction between biradicals linked by flexible structures16 with Q[n], where n is 6, 7 or 8. One refers to the formation of pseudorotaxane like complexes (Fig. 5a), which is more probable for interactions with Q[7] and Q[8], but this model is debatable for the systems analysed in this study, considering the structural characteristics of the host and the guest molecules. In the case of Q[6], the complexation of the nitroxide moieties is seen as an insertion among the carbonyl moieties16 and this model seems to be suitable for understanding the experimental EPR data obtained in the present study (Fig. 5b and c).
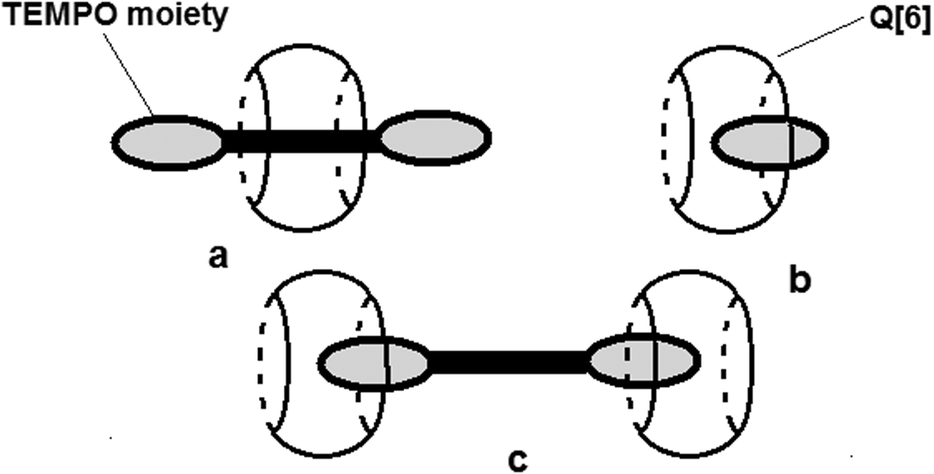 | ||
| Fig. 5 Schematic representation of the different complexation modes of a radical with cucurbiturils: (a) pseudorotaxane like complex for a biradical, (b) 1:1 complexation for a monoradical, and (c) 1:2 complexation for biradical/Q[6]. | ||
The fact that both aN and the intensities of the lines attributed to spin–spin interactions for compound 2a are insensitive to the presence of Q[6] might be an indication that the formation of complexes with low stoichiometry (1:1 ratio, Fig. 5b) is favoured.
The thermodynamic parameters of the host–guest systems can be obtained via the analysis of the changes of the rotational correlation time, τc, calculated from the ESR spectra of the paramagnetic guest using eqn (1), as a function of the host concentration.
The binding constants were estimated from the dependence of τc on the concentration of Q[6], assuming a 1:1 complexation ratio for all radicals. Although a higher stoichiometry is possible (e.g. 1:2 for radical/Q[6]), especially at a high concentration of the host, we considered that the complex with the 1:1 ratio is the prevailing species in each case. τc is proportional to the molecular size of the paramagnetic molecule or paramagnetic complex. The ratio between the values of τc for a radical in solution in the presence and in the absence of Q[6] indicates that the formation of a 1:1 complex is more probable. A linear relation has been assumed between τc and the concentration of the complex in solution (see ESI†), in order to estimate the binding constants. This procedure has been previously applied for the evaluation of the binding constants between the cyclodextrin and nitroxide type radicals.33,34 In Table 3, the values of the binding constants for those radicals that showed a dependence of τc on the concentration of Q[6] at 293 K are summarised. The data presented in Table 3 reveal that the binding constants for these complexes are in general an order of magnitude lower than those observed for the interaction between TEMPO type radicals and β-cyclodextrin.33–37 They are similar to the binding constants observed for the complexes between nitroxide type radicals and α-cyclodextrin.38
| Radical | 1a | 1b | 1d | 1e | 2b | 2c | 2d |
|---|---|---|---|---|---|---|---|
| K (l × mol−1) | 70 | 110 | 175 | 99 | 124 | 165 | 170 |
Aiming to obtain the thermodynamic parameters corresponding to the host–guest complexation of these guests molecules with Q[6], the ESR spectra of radicals 1b and 2c were recorded in the temperature range of 293–323 K with a step of 5 K. For each temperature the binding constants were calculated in the same manner as for the data obtained at 293 K, from the dependence of the rotational correlation times on the concentration of Q[6]. By combining the Gibbs free energy equation with the Gibbs free energy isotherm equation, it was possible to determine, from the van't Hoff plots, the thermodynamic parameters associated with the complexation process (ESI,† Fig. S4). These parameters obtained for compounds 1b and 2c are as follows: ΔS = −32.9 (1b) and −65.2 (2c) J mol−1 K−1 and ΔH = −21.2 (1b) and −31.7 (2c) kJ mol−1. The thermodynamic values obtained for the complexes of these two nitroxides with Q[6] are of the same order of magnitude but lower than similar values obtained for β-cyclodextrin/nitroxide complexes.39–41 Evidently, the variation of the entropy is negative due to a restricted motion of the radicals (paramagnetic moiety) in the corresponding complexes with Q[6]. The formation of the cucurbituril complexes represents enthalpy driven processes, since the variations of enthalpy are negative values. Compared with similar studies regarding the thermodynamics of β-cyclodextrin inclusion complexes,39–41 the values of enthalpy obtained for radicals 1b and 2c are smaller. On the other hand, the enthalpy values found for these two radicals are similar to those reported for the interaction of proxyl-type radicals with α-cyclodextrin.38
Information on the steric orientation of the host and guest molecules in the complex can be indirectly provided by the analysis of the ESR features of the spectra recorded below room temperature in the absence and in the presence of Q[6] at the highest concentration of the host. This approach has been used in previous studies reporting the behaviour of some commercially available nitroxide radicals and cyclodextrin complexes in frozen solutions or in the solid state.26,42 These studies showed that the spin probe mobilities are higher in the complexes with cyclodextrins compared with the mobilities in the bulky solute.
In contrast to the data observed for TEMPO26 or DOXYL42 radicals/cyclodextrin complexes at low temperature, in this study it was noticed that the motions of the radicals presented here freeze at higher temperatures in the presence of Q[6] than in the solvent. As an illustration, Fig. 6 shows the spectra of the radical 1b in NaCl/HCl water solution/glycerol (8:2) in the absence and in the presence of Q[6] (10−2 M). In the temperature range 253–273 K, the ESR spectra indicate relatively fast dynamics, although a progressive reduction of the tumbling can be observed both in the absence and in the presence of Q[6] as the temperature decreases.
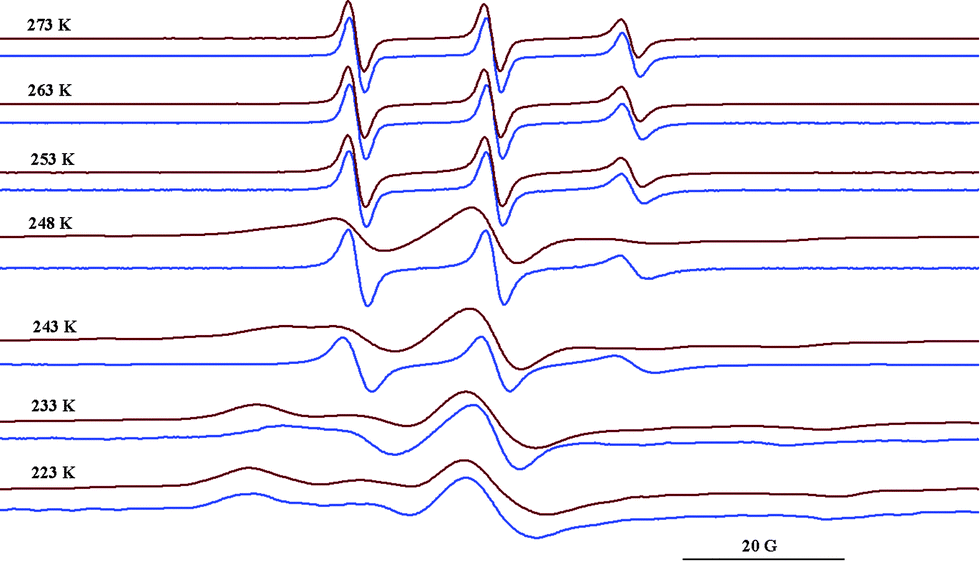 | ||
| Fig. 6 ESR spectra of 1b in NaCl/HCl water solution/glycerol (8:2) at different temperatures in the absence (blue) and in the presence of Q[6] (red). | ||
It was observed that the motions of the radical freeze at temperatures below 223 K in NaCl/HCl water solution/glycerol (8:2). In a similar solution, the tumbling of the radical is considerably slowed down in the presence of Q[6] at 248 K, while at 233 K the EPR spectrum shows a powder pattern. This behaviour can be explained by the fact that the presence of ions at a high concentration avoids the vitrification of the solution at higher temperatures and thus the probe has relatively fast dynamics even at lower values of the temperature. In the presence of Q[6], the paramagnetic moieties are probably capping the host cavity rather than being deeply included in the cucurbituril cavity. Due to the urea moiety being close to the paramagnetic group, it is likely that the N–H groups from the radical molecule will interact with the carbonyl group from Q[6] via hydrogen bonding. This is expressed in the ESR spectra which reveal the radical immobilisation at temperature values 20 K higher than those in the absence of the host. Fig. 7 illustrates schematically the spin probe surroundings in a HCl/NaCl/glycerol solution in the absence and in the presence of Q[6] assuming a 1:1 inclusion complex.
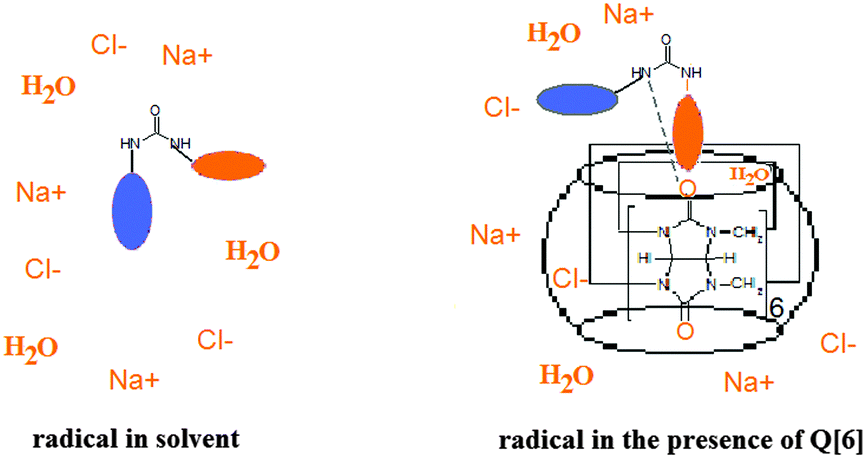 | ||
| Fig. 7 Schematic representation of the radical environments. | ||
These results together with those previously reported26 indicate that the behaviour of the nitroxides in the presence of host molecules in vitrified solutions can provide information on the complex geometry.
Conclusions
To summarise, we have prepared a series of new TEMPO based (poly)radicals. Due to their structural features with fragments able to generate hydrogen bond networks, it was possible to characterise two of the new compounds via X-ray diffraction. All of these radicals form complexes with cucurbit[6]uril and the analysis of the spectral parameters as a function of the Q[6] concentration provided the thermodynamic parameters of complexation. Owing to the geometrical features of Q[6], we showed that the formation of the complexes took place in such a manner that the paramagnetic moiety covered the entrance of Q[6] rather than being deeply included in the cavity. This conclusion is sustained by the thermodynamic parameters of complexation and behaviour of the radicals at low temperature. Moreover, the spectra recorded at temperatures below 273 K support the model of capping one side of the host. The stability of such complexes is probably determined by the hydrogen bonds established between the NH group from the radical structure and the CO group from the glycoluril groups. This ESR study on the interaction of TEMPO derivatives with Q[6] can be extended to investigate similar systems involving superior terms of the cucurbituril family at variable temperatures. Such studies will provide a general picture on the host–guest complexes of cucurbiturils with paramagnetic compounds.Acknowledgements
This work was supported by a grant from the Romanian National Authority for Scientific Research, CNCS – UEFISCDI, project numbers PN-II-ID-PCE-2011-3-0408 and PN-II-ID-PCE-2011-3-0328.Notes and references
- Advanced ESR Methods in Polymer research, ed. S. Schlick, Wiley Interscience, 2006 Search PubMed.
- T. Vogler and A. Studer, Synthesis, 2008, 1979 CAS.
- R. P. Mason, Free Radical Biol. Med., 2004, 36, 1214 CrossRef CAS PubMed.
- J. B. Mitchell, M. C. Krishna, P. Kuppusamy, J. A. Cook and A. Russo, Exp. Biol. Med., 2001, 226, 620 CAS.
- A. Studer, Chem. – Eur. J., 2001, 7, 1159 CrossRef CAS.
- D. B. Amabilino and J. Veciana, in Magnetism, Molecules to Materials II, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2001, vol. II, pp. 1–60 Search PubMed.
- M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed.
- K. C. Ko, D. Cho and J. Y. Lee, J. Phys. Chem. A, 2013, 117, 3561 CrossRef CAS PubMed.
- E. Coulaud, D. Hagebaum-Reigner, D. Siri, P. Tordo and N. Ferre, Phys. Chem. Chem. Phys., 2012, 14, 5504 RSC.
- I. Krstic, R. Hansel, O. Romainczyk, J. W. Engels, V. Dotsch and T. F. Prisner, Angew. Chem., Int. Ed., 2011, 50, 5070 CrossRef CAS PubMed.
- M. Lucarini and E. Mezzina, Electron Paramagn. Reson., 2011, 22, 41 CAS.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394 RSC.
- M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479 CrossRef CAS PubMed.
- S. B. Nimsea and T. Kim, Chem. Soc. Rev., 2013, 42, 366 RSC.
- X.-L. Ni, X. Xiao, H. Cong, L.-L. Liang, K. Cheng, X.-J. Cheng, N.-N. Ji, Q.-J. Zhu, S.-F. Xue and Z. Tao, Chem. Soc. Rev., 2013, 42, 9480 RSC.
- E. Mileo, C. Casati, P. Franchi, E. Mezzina and M. Lucarini, Org. Biomol. Chem., 2011, 9, 2920 CAS.
- Y. Basel and A. Hassner, J. Org. Chem., 2000, 65, 6368 CrossRef CAS PubMed.
- M. Kavala, V. Brezova, L. Svorc, Z. Vihonska, P. Olejnikova, J. Moncol, J. Kozisek, P. Herich and P. Szolcsanyi, Org. Biomol. Chem., 2014, 12, 4491 CAS.
- A. C. Bhasikuttan, H. Pal and J. Mohanty, Chem. Commun., 2011, 47, 9959 RSC.
- E. Mezzina, F. Cruciani, G. F. Pedulli and M. Lucarini, Chem. – Eur. J., 2007, 13, 7223 CrossRef CAS PubMed.
- E. Mileo, E. Mezzina, F. Grepioni, G. F. Pedulli and M. Lucarini, Chem. – Eur. J., 2009, 15, 7859 CrossRef CAS.
- D. Bardelang, K. Banaszak, H. Karoui, A. Rockenbauer, M. Waite, K. Udachin, J. A. Ripmeester, C. I. Ratcliffe, O. Ouari and P. Tordo, J. Am. Chem. Soc., 2009, 131, 5402 CrossRef CAS PubMed.
- I. Kirilyuk, D. Polovyanenko, S. Semenov, I. Grigor’ev, O. Gerasko, V. Fedin and E. Bagryanskaya, J. Phys. Chem. B, 2010, 114, 1719 CrossRef CAS PubMed.
- N. Jayaraj, M. Porel, M. F. Ottaviani, M. V. S. N. Maddipatla, A. Modelli, J. P. Da Silva, B. R. Bhogala, B. Captain, S. Jockusch, N. J. Turro and V. Ramamurthy, Langmuir, 2009, 25, 13820 CrossRef CAS PubMed.
- S. Yi, B. Captain, M. F. Ottaviani and A. E. Kaifer, Langmuir, 2011, 27, 5624 CrossRef CAS PubMed.
- G. Ionita and V. Chechik, Phys. Chem. Chem. Phys., 2010, 12, 6956 RSC.
- T. E. Edwards, T. M. Okonogi, B. H. Robinson and S. T. Sigurdsson, J. Am. Chem. Soc., 2001, 123, 1527 CrossRef CAS PubMed.
- T. J. Stones, T. Buckman, P. L. Nordio and H. M. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1965, 54, 1010 CrossRef.
- M. Porel, M. F. Ottaviani, S. Jockusch, N. J. Turro and V. Ramamurthy, RSC Adv., 2013, 3, 427 RSC.
- J. Szejtli, Chem. Rev., 1998, 98, 1743 CrossRef CAS PubMed.
- L. Isaacs, Chem. Commun., 2009, 619 RSC.
- The authors thank the referee for suggesting the analysis of aN changes of the radicals in the presence of Q[6].
- G. Ionita and V. Chechik, Org. Biomol. Chem., 2005, 3, 3096 CAS.
- V. Chechik and G. Ionita, Org. Biomol. Chem., 2006, 4, 3505 CAS.
- G. Ionita, A. Caragheorgheopol, H. Caldararu, L. Jones and V. Chechik, Org. Biomol. Chem., 2009, 7, 598 CAS.
- Y. Kotake and E. G. Janzen, J. Am. Chem. Soc., 1989, 111, 2066 CrossRef CAS.
- M. Okazakiand and K. Kuwata, J. Phys. Chem., 1984, 88, 3163 CrossRef.
- M. P. Eastman, B. Freiha, C. C. Hsu, K. C. Lurn and A. Allen Chang, J. Phys. Chem., 1987, 91, 1953 CrossRef CAS.
- V. Chechik and G. Ionita, New J. Chem., 2007, 31, 1726 RSC.
- G. Fukuhara, T. Mori, T. Wada and Y. Inow, J. Org. Chem., 2000, 65, 8041 CrossRef.
- Y. Inoue, T. Wada, N. Sugahara, R. Yamamoto, K. Kimura, L. H. Tong, X. M. Gao and Z. J. Hou, J. Org. Chem., 2000, 65, 8041 CrossRef CAS PubMed.
- B. Dzikovski, D. Tipikin, V. Livshits, K. Earle and J. Freed, Phys. Chem. Chem. Phys., 2009, 11, 6676 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: More ESR and X-ray data/pictures. CCDC 1402759 and 1402760. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj01518a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |