
Advanced cathode materials for lithium-ion batteries using nanoarchitectonics
Renjie
Chen†
*ab,
Taolin
Zhao†
c,
Xiaoxiao
Zhang
a,
Li
Li
ab and
Feng
Wu
ab
aSchool of Material Science & Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: chenrj@bit.edu.cn
bCollaborative Innovation Center of Electric Vehicles in Beijing, Beijing 100081, China
cSchool of Materials Science and Engineering, Shijiazhuang Tiedao University, Shijiazhuang 050043, China
First published on 3rd May 2016
Abstract
In recent years, the global climate has further deteriorated because of the excessive consumption of traditional energy sources. The replacement of traditional fossil fuels with limited reserves by alternative energy sources has become one of the main strategies to alleviate the increasingly serious environmental issues. As a sustainable and promising store of renewable energy, lithium-ion batteries have replaced other types of batteries for many small-scale consumer devices. Notwithstanding their worldwide applications, it has become abundantly clear that the design and fabrication of electrode materials is urgently required to adapt to meet the growing global demand for energy and the power densities needed to make electric vehicles fully commercially viable. To dramatically enhance battery performance, further advances in materials chemistry are essential, especially in novel nanomaterials chemistry. The construction of nanostructured cathode materials by reducing particle size can boost electrochemical performance. The present review is intended to provide readers with a better understanding of the unique contribution of various nanoarchitectures to lithium-ion batteries over the last decade. Nanostructured cathode materials with different dimensions (0D, 1D, 2D, and 3D), morphologies (hollow, core–shell, etc.), and composites (mainly graphene-based composites) are highlighted, aiming to unravel the opportunities for the development of future-generation lithium-ion batteries. The advantages and challenges of nanomaterials are also addressed in this review. We hope to simulate many more extensive and insightful studies on nanoarchitectonic cathode materials for advanced lithium-ion batteries with desirable performance.
1. Introduction
Human health and social development are adversely impacted by the increasingly deteriorating environment resulting from excessive consumption of the traditional limited-reserve fossil fuels, including coal, oil, and natural gas. The development of clean and sustainable energy sources has been appealed for and pursued to replace these fossil fuels. Among the various energy storage devices, lithium-ion batteries (LIBs) have been recognized as the most promising candidate to date, and presently dominate the rechargeable battery market segment by virtue of their large specific capacity, high voltage, high power, high efficiency, and environmental friendliness.1,2 The most ubiquitous application of LIBs can be found in portable consumer electronics, including mobile phones, laptops, and cameras. Nonetheless, the gradually growing demand for the higher energy and power densities needed for electric vehicles (EVs) and hybrid electric vehicles (HEVs) can be hardly appeased by the present commercial LIBs.3Along with Tesla Motors' foray into the global EV market and local government policies regarding electric automobiles, the alternative energy automobile market boom has erupted. However, electric cars have not yet experienced development in leaps and bounds, mainly owing to the current level of battery technology. The specific energy, power, and safety of batteries directly affect the drive range, speed, and safety of electric vehicles. To date, the drive range of electric cars has been limited to be less than 500 km, and complete charging of the battery takes several hours. Increasing the energy density of batteries to 350 W h kg−1, which is twice the performance of existing batteries, would greatly improve the drive distance to an extent that would aid realizing the commercialization of electric cars. Owing to the limitations of current battery technology, the specific energy of even advanced LIBs is still limited. Increasing the quantity of batteries for longer running increases the weight of the vehicles, which then need an increased number of cells to sustain them, forming a vicious cycle. Moreover, as the number of cells is increased, heat dissipation becomes more difficult, which increases the risk of accidents. Thus, the way out of these issues is to develop advanced LIBs with superior electrochemical performance.
The electrochemical properties of LIBs largely depend on the active materials of the electrodes, which directly participate in electrochemical reactions and play a dominant role in improving the energy storage capability, the efficiency, and the energy conversion rate of the devices.4 The major obstacle hindering the extensive application of LIBs in the EV field is the cathode materials, which currently have a much lower capacity than that of the anode materials.5 Accordingly, it has become vital to design and fabricate cathode materials that will overcome the weak points of LIB systems.
As the development of nanoscience and nanotechnology has progressed, many researchers have devoted themselves to working on nanoarchitectonic cathode materials to pursue advanced materials with higher charge/discharge capacity, higher rate capability, lower cost, and longer cycle life. Great efforts have been made recently to employ nanostructured cathodes in LIBs.6–14 Previous studies have demonstrated that nanostructured materials are advantageous in improving the thermodynamics and kinetics of electrochemical reactions to some extent, as a result of shortening ionic/electronic diffusion distances and offering large surface areas for electrode reactions.
Thermodynamics reveals the optimum energy release or storage for an electrochemical reaction, which determines the travel distance of EVs. A high energy density is associated with high voltage and high capacity. Nanostructures can lead to higher capacity than micrometer-scale counterparts by increasing the utilization of the active materials and introducing new lithium storage space, and can also enhance the working voltage by alleviating electrochemical polarization. Kinetics determines the reaction rate, which is related to charge transport and transfer. Effective mass transport, including facile electronic conductivity and ionic diffusion, is helpful to achieve high power density. Nanosized materials can facilitate cathode kinetics through a reduced diffusion pathway for lithium insertion/extraction and an increased reaction interface.15
This paper reviews the recent advances in the nanotechnology applied in cathode materials for LIBs. The typical families of cathode nanomaterials with different crystal structures, morphologies, and synthetic routes are systematically described to allow readers to understand the merits and faults of different nanostructures. From nanostructured morphologies with different dimensions, including nanoparticles, nanotubes, nanowires, and nanosheets, to hierarchical nano/microstructures, core–shell structures and then nanocomposites, the electrochemical performance of typical cathode materials is significantly enhanced by such structures. Finally, we summarize the advantages and disadvantages of nanomaterials, and discuss the challenges and prospects to be faced in future LIB systems.
2. Principles of lithium-ion batteries
The commercialization of primary Li batteries was realized during the 1970s. With greater prospects, secondary Li batteries such as Li–TiS2,16 Li–MoS2,17 and Li–LixMnO2 systems appeared as a commercial reality during the early 1990s.18 To improve their safety problems, graphite replaced Li metal and was introduced as the anode material. With lithiated transition metal oxide LiMO2 as the cathode material, the LIB was successfully exploited and commercialized by Sony Corporation in 1991 and thereafter received tremendous research attention as a major energy storage device for portable electronic devices, transportation systems, and electrical power storage systems.19LIBs are concentration cells and are mainly composed of electrode materials (cathode and anode), an electrolyte, and a separator. Fig. 1 illustrates the reaction mechanism of the basic battery system. During charge and discharge processes, lithium ions shuttle reversibly between the two host structures of the cathode and anode, accompanied by redox reactions. On charging, lithium ions are extracted from the cathode to the anode through the electrolyte and separator, and electrons are released as the transition metal ions are oxidized. Thus, current flows from the anode to cathode in the external circuit. On discharging, the process is the opposite. The reversible electrochemical reaction that occurs in LIBs is a typical example of solid-state chemistry and reveals their charge-storage properties.
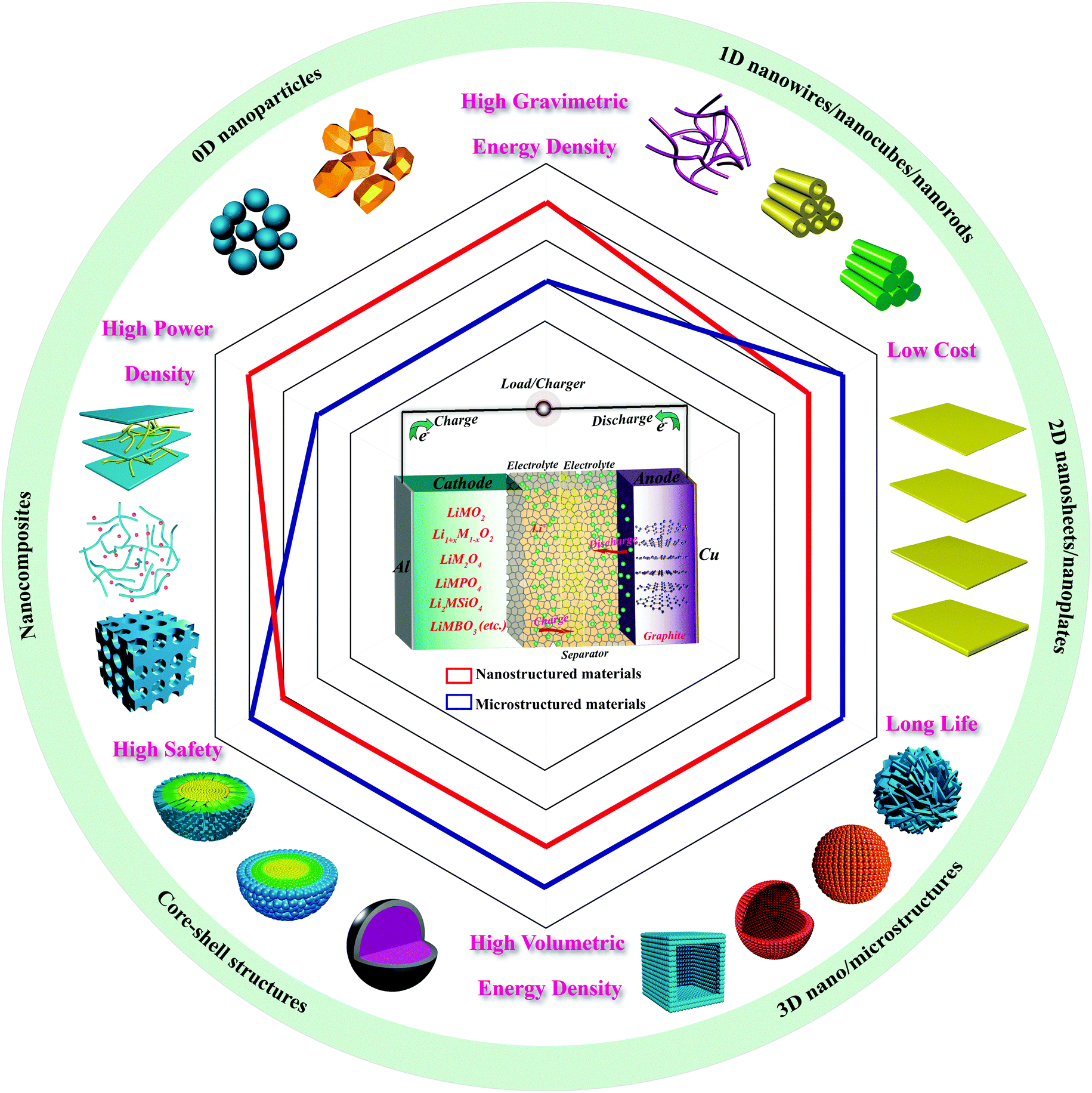 | ||
| Fig. 1 Illustration of the reaction mechanism and the typical nanotechnologies applied in various cathodes for achieving better LIB properties. | ||
Three reaction steps take place in LIBs: solid state diffusion of lithium ions, interfacial charge transfer between the electrode and electrolyte, and lithium ion transport in the electrolyte. The rate-controlled step is thought to be the solid state diffusion of Li+ in the electrode materials. The mean diffusion time (τ) is determined by the diffusion coefficient (D) and the diffusion length (L), according to the following formula: τ = L2/D. It is worth noting that the diffusion time decreases with the square of the diffusion length, which indicates that shortening the Li+ diffusion length is more effective when trying to enhance the rate capability of electrode materials. Accordingly, great efforts have been made to synthesize nanosized materials to reduce the diffusion length. Fig. 1 shows the typical nanotechnologies applied in various cathodes to obtain better LIBs with higher performance. The principal properties, including energy, power, life, cost, and safety, of these nanostructured materials and microstructured materials are also compared in Fig. 1. Obviously, morphology and shell control are vital to the construction of advanced cathodes for LIBs with high energy and power density. Moreover, the combination of various nanotechnology strategies is an effective route for achieving multiple functions. The following sections give a detailed discussion of this.
3. Typical families of cathode materials
For cathode materials, layered LiMO2, Li-rich layered Li1+xM1−xO2, spinel LiM2O4, olivine LiMPO4, and silicate compounds Li2MSiO4 and borate compounds LiMBO3 (where M is Fe, Co, Ni, Mn, Cu, Cr, etc.) have been extensively investigated. Beyond those materials, some other cathode materials like LiV3O8, fluorides (FeF3), and oxides (V2O5, MnO2) have also received much attention. All these cathode materials have unique and varied crystal structures (shown in Fig. 2) and show considerable capability and promise.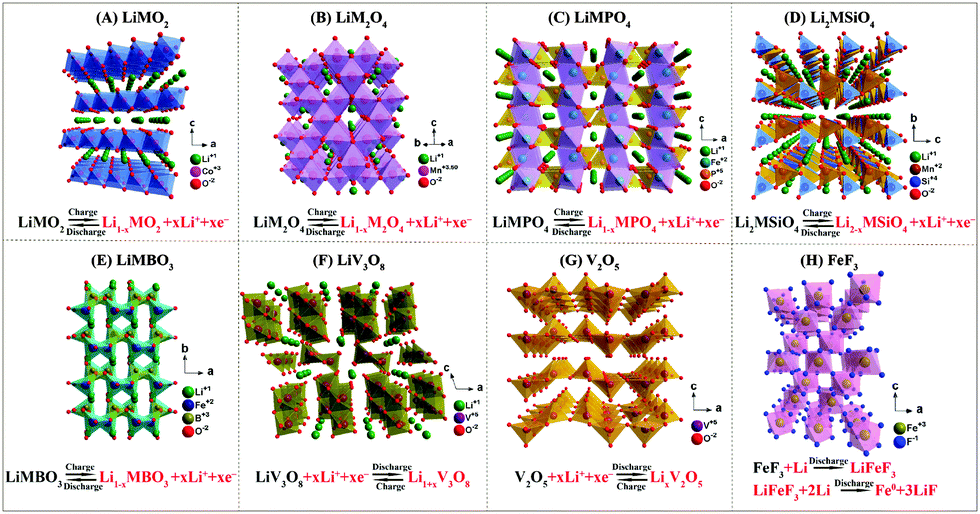 | ||
| Fig. 2 Crystal structures and electrochemical reactions of various cathodes: (A) layered LiMO2, (B) spinel LiM2O4, (C) olivine LiMPO4, (D) Li2MSiO4, (E) LiMBO3, (F) LiV3O8, (G) V2O5, (H) FeF3. | ||
3.1 Layered
The first commercial LIB cathode material, LiCoO2, possesses an ordered layered structure with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. Its practical capacity can only reach 140 mA h g−1, half of its high theoretical capacity of 274 mA h g−1, with a plateau potential of 3.9 V. Moreover, it suffers from high cost and poor safety. LiNiO2 is formed with a similar layered structure to that of LiCoO2 by the substitution of Co3+ with Ni3+. Its practical capacity is high, up to 190–210 mA h g−1. However, its commercialization is hindered by its difficult preparation process, poor thermal stability, and cation mixing of Li+ and Ni2+. The low-cost layered LiMnO2, which has a high practical capacity of ∼200 mA h g−1, is also plagued by Mn dissolution caused by the Jahn–Teller distortion of Mn3+ and oxygen evolution at a high charging potential, leading to the loss of the active material and serious safety issues.
m space group. Its practical capacity can only reach 140 mA h g−1, half of its high theoretical capacity of 274 mA h g−1, with a plateau potential of 3.9 V. Moreover, it suffers from high cost and poor safety. LiNiO2 is formed with a similar layered structure to that of LiCoO2 by the substitution of Co3+ with Ni3+. Its practical capacity is high, up to 190–210 mA h g−1. However, its commercialization is hindered by its difficult preparation process, poor thermal stability, and cation mixing of Li+ and Ni2+. The low-cost layered LiMnO2, which has a high practical capacity of ∼200 mA h g−1, is also plagued by Mn dissolution caused by the Jahn–Teller distortion of Mn3+ and oxygen evolution at a high charging potential, leading to the loss of the active material and serious safety issues.
Although LiNi0.5Mn0.5O2 can deliver a high discharge capacity of ∼200 mA h g−1 with a plateau potential of 3.8 V,20,21 its ionic diffusion is impeded by the cation mixing of the Ni and Li ions.22 The practical application of LiNi0.5Mn0.5O2 is limited by problems of capacity decay, poor cycling stability, and safety issues. To combine the advantages of LiCoO2, LiNiO2, and LiNi0.5Mn0.5O2, LiNi1−x−yMnxCoyO2 (NMC) was proposed and expected to replace both LiCoO2 and LiFePO4 as the cathode of choice in the near future. However, in spite of the promised theoretical capacity (∼280 mA h g−1), the practical capacity that can be accessed (∼170 mA h g−1) is still much lower. The sustained Mn dissolution from Mn-containing cathode materials into the electrolyte, accompanied by phase-transformation of the NMC structure, is believed to be responsible for the decay of its capacity during long-term cycling at high voltage (>4.2 V), high current density, or high temperature. Irreversible loss of the transition metal elements (Ni, Co, or Mn) in bulk NMC is also caused by side reactions with the electrolyte. Among the NMC cathode materials studied,23 LiNi1/3Mn1/3Co1/3O224–27 possesses the best compromise with a more stable structure, a moderate capacity, and lower cost. The Li+ ion intercalation–deintercalation process of LiNi1/3Mn1/3Co1/3O2 can be obviously facilitated through manufacturing the nanostructure, adjusting the surface and doping other cations.15 In addition, Ni-rich layered LiNi0.8Co0.1Mn0.1O2 has shown a fascinating capacity, but suffers from poor cycling stability caused by a structural transformation.28 LiNi0.80Co0.15Al0.05O2 (NCA) combines the attractive properties of LiNiO2 and LiCoO2 alongside a higher structural stability than LiNiO2 because the layered structure is maintained by Co doping, a larger practical specific capacity (∼200 mA h g−1), and lower cost than LiCoO2. The partial substitution of Ni3+ by the electrochemically inactive Al3+ ions plays a beneficial effect in improving the thermal stability of the material at high potentials by preventing overcharging.
The lack of high-capacity cathode materials has become the main obstacle to achieving advanced LIBs.28 The layered Li-rich cathode materials29,30xLi2MnO3·(1−x)LiMO2 (0 < x < 1, M = Ni, Mn, Co, etc.)31 have been considered to be one of the most promising cathode material series owing to their high specific capacity (>250 mA h g−1),32,33 low cost,33,34 and environmental benignity. Notably, the Li2MO3 component plays a vital role in storing the excess lithium and securing the high voltage and specific capacity. Nevertheless, the unavoidable rearrangement of surface and bulk structures leads to voltage fade and sluggish Li+ diffusion, resulting in a large initial irreversible capacity, poor cycling stability, and an intrinsic poor rate capability, which must be overcome before practical application is possible. Cationic doping to stabilize the crystal structure and minimize the voltage fade, using a stable surface coating layer to suppress side reactions, introducing Li-containing oxides with good conductivity to provide a Li+ diffusion tunnel, and synthesizing layered/spinel composites, are all effective and extensively studied approaches for improving the electrochemical performance of Li-rich cathodes. In addition, nanostructures, such as nanoparticles, nanorods, nanoplates, and hollow structures, can also improve the rate and cycling stability.15
Among all the potential vanadium oxide cathode materials, vanadium pentoxide (V2O5) with a layered structure can deliver excellent capacity by incorporating multiple lithium ions. A high capacity of 294 mA h g−1 can be obtained when two Li+ ions are intercalated. A comparable capacity can be still obtained even if only one Li+ is intercalated.35 Unfortunately, its poor structural stability, low Li+ diffusion coefficient (ca. 10−12 cm2 s−1), and moderate electrical conductivity (10−4–10−5 S cm−1)36 lead to a poor cycle life and low rate capability.
3.2 Spinel
Because of their robust host structure enabling 3D Li+ diffusion pathways, intrinsic low cost, and environmental friendliness, cubic spinel-structured cathodes with space symmetry Fd3m, mainly LiMn2O4 and LiMn1.5Ni0.5O2, have shown great potential for high-rate LIB applications.14,34,37 However, although spinel-type cathodes deliver a high discharge plateau, superior cycling, and outstanding rate capability, a capacity of less than 150 mA h g−1 is obtained during cycling above 3 V. The transition of the spinel phase to the rock salt phase38 occurs at around 3 V with further lithiation. Mn dissolution via disproportionation (2Mn3+ → Mn2+ + Mn4+) and Jahn–Teller distortion from the cubic to tetragonal phase39 hinder the practical application of spinel LiMn2O4. For EV applications, spinel LiMn2O4 can be preferable in cases where a long driving time without plug-in is the essential criterion. The substitution of Mn by Ni in LiMn1.5Ni0.5O2 can increase the mean oxidation state of the Mn ions for a more stable structure, and eliminate the Jahn–Teller distortion. Two crystal structures, namely the ordered P4332 phase and the disordered Fd3m phase, are present in spinel LiMn1.5Ni0.5O2.40,41 The oxidation potential for Ni2+ to Ni4+ can reach up to 4.7 V,42 making LiMn1.5Ni0.5O2 a promising high-voltage cathode material. Nevertheless, the stability of the present electrolytes and active materials makes it difficult to accommodate these high-voltage cathodes.3.3 Polyanion-type
Olivine-type LiMPO4 (M = Fe, Mn, Co, Ni) has attracted great interest as a cathode material for high-power LIBs. However, this family of compounds suffers from poor electronic conductivity and sluggish Li+ diffusion in the [010] direction. Nevertheless, olivine-type LiFePO4 has some obvious advantages, such as a very flat discharge potential around 3.4 V and excellent thermal and chemical stability. A carbon coating is often applied to improve its intrinsic low electronic conductivity and prevent particle growth. Despite its low cost and high safety level, its electrochemical performance at high or low temperature and high voltage are not ideal. The diffusion in large LiFePO4 crystals was found to be much slower than in nanoparticles because of the presence of a larger number of channel-blocking defects in the former.43Based on the Mn3+/Mn2+ redox couple, olivine LiMnPO4 has a high operating voltage of 4.1 V with a theoretical specific capacity of 170 mA h g−1. Notably, the (010) plane and the [010] direction are favorable for Li+ diffusion, which is vital for the good electrochemical performance of LiMnPO4.44 However, the low intrinsic electronic conductivity (<10−10 S cm−1) and large structure distortions induced by the Jahn–Teller effect of Mn3+ ions lead to a low practical capacity and poor rate performance for this material. Based on these issues, synthesized nanoparticles of LiMnPO4 can facilitate sufficient Li transport.
The possible two electron exchange reaction of Li2MnSiO4 based on the Mn3+/Mn2+ and Mn4+/Mn3+ redox couples is attractive, with a theoretical capacity of 333 mA h g−1.45,46 Low intrinsic electronic conductivity (<10−14 S cm−1), the Jahn–Teller distortion of Mn3+, and poor stability are also the obstacles for Li2MnSiO4.
The high theoretical energy density of LiMBO3 is ensured by its lightest polyanion group, BO3. In the case of LiFeBO3, a high capacity of 200 mA h g−1 was achieved in both computational and experimental investigations. Kinetic polarization and sensitivity to moisture are the main causes of the relatively poor performance of this material, and further optimization of the synthetic and operational conditions is essential.47
3.4 Other types of cathodes
To break the limits of the traditional cathodes, transition metal halides, especially fluoride-based materials with much higher gravimetric and volumetric capacities, have been developed as conversion-type cathode materials.As the most promising conversion cathode material, iron trifluoride (FeF3), which has a high theoretical capacity (712 mA h g−1), has good prospects for doubling the energy density of the current commercial cathodes.48 However, the non-optimal material properties and poor electrochemical kinetics of the conversion reaction are hindering its further development. Another promising cathode, FeF2, has been combined with carbon to form novel nanocomposites, enhancing the reversibility of electrochemical reactions between FeF2 and Li for FeF2 nano-confined within the conductive carbon.49
In addition to inorganic cathodes, organic cathode materials have also been studied as emerging materials for electrochemical energy storage,50,51 such as conductive polymers, sulphur compounds, nitrogen/oxygen free radical compounds and oxygen-containing conjugated compounds. To solve the serious dissolution into the electrolyte and promote electrical conductivity, conductive agents are preferred to be added, including metal nanoparticles and carbon nanomaterials.
Despite the rapid development of the present LIB cathodes, there are still many bottlenecks that restrict their further application, such as their structural instability, low discharge capacity and voltage, and poor rate capability. Various promising strategies have been proposed in this review to overcome these stubborn issues, including the reduction of particle size to the nanoscale, surface nanocoatings, construction of core–shell structures and nano/micro hierarchical structures, and combining two or more strategies to achieve complementarity.
4. Nanostructured cathode materials
4.1 Nanoparticles with different dimensions
As a prospective solution to improve on the inferiorities of cathode materials, nanoarchitectured cathodes with various morphologies, including zero dimension (0D; nanoparticles), one dimension (1D; nanotubes or nanowires), two dimension (2D; nanoplates or nanosheets), and three dimension (3D; hierarchical nanostructures), have been used. Such structures have been shown to possess unique lithium storage properties and exhibit improved electrochemical performance in terms of, for example, charge/discharge capacity, cycling stability, rate capability, and safety.Gaberscek et al. compared nine papers by different research groups using different synthesis methods and concluded that the capacity of LiFePO4 decreases linearly with increasing particle size.54 Reducing the particle size can also diminish the blocked capacity, and nanosized LiFePO4 is more tolerant to anti-site defects.55 LiFePO4 nanoparticles synthesized by a low-temperature precipitation process exhibited sloping voltage charge/discharge curves, characteristic of single-phase behavior. As shown in Fig. 3(a–c), monodisperse small platelet-shaped crystalline particles in the 15 to 100 nm range, with a very narrow percentage cumulative volumetric distribution peaking at 40 nm, showed excellent rate capability.56
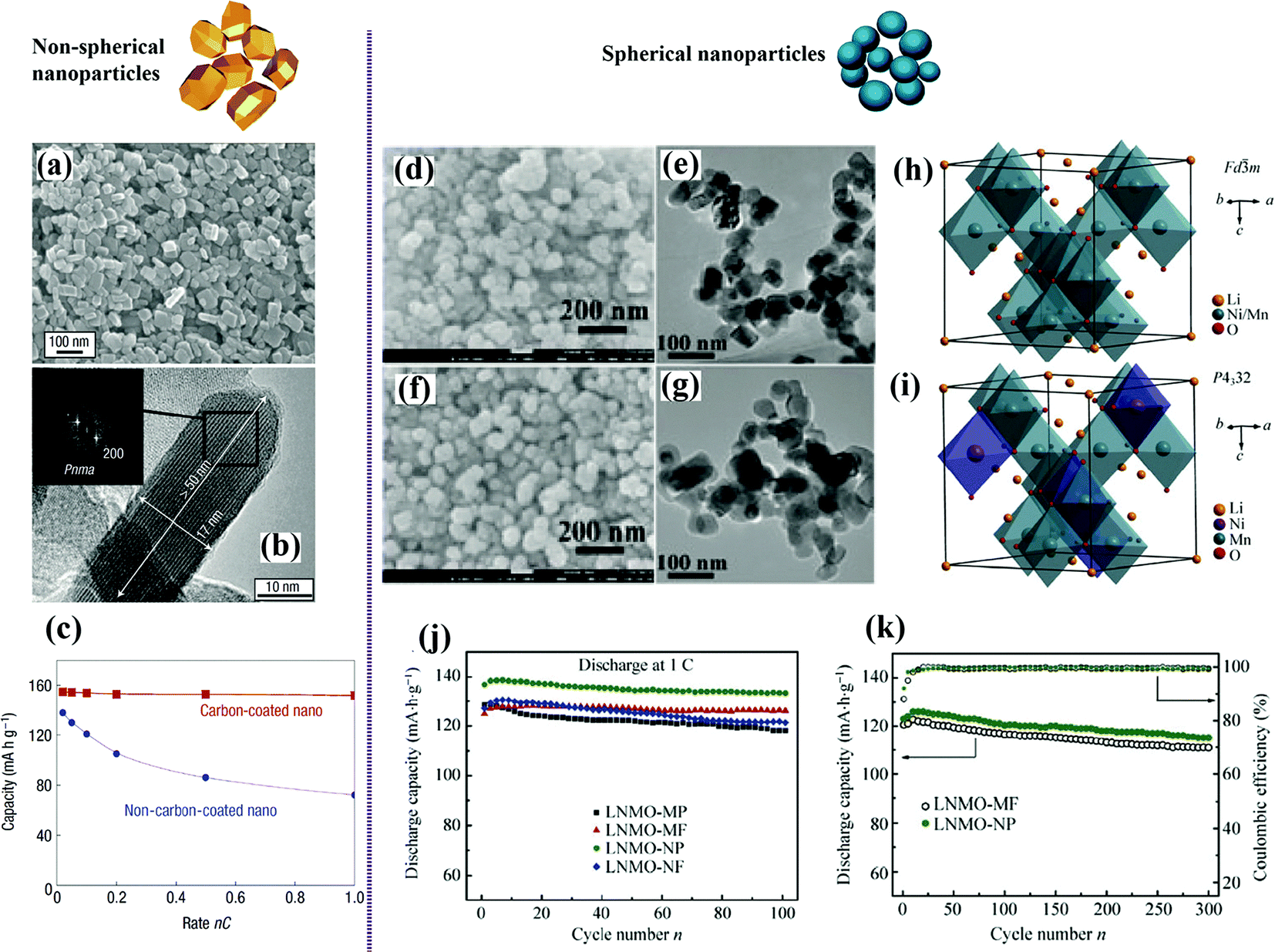 | ||
| Fig. 3 Non-spherical nanoparticles of LiFePO4:56 (a) SEM image, (b) RTEM image combined with the Fourier transform (inset) of one box showing the orientation of the crystallite, (c) electrochemical performance. Spherical nanoparticles of LiNi0.5Mn1.5O4:58 (d and f) SEM images, (e and g) TEM images, (h and i) crystal structures of LNMO with (h) Fd3m and (i) P4332 space groups, (j and k) electrochemical performance. | ||
Highly crystalline LiNi0.5Mn1.5O4 nanoparticles have been successfully synthesized through various methods.6,57 The combined influence of the crystal structure and particle morphology (size, shape, and surface area) on the electrochemical performance of LiNi0.5Mn1.5O4 is shown in Fig. 3(d–k). It has been confirmed that the ordered spinel phase favors cycle stability, while disordered spinel phase benefits the high-rate capability.58 The ordered spinel nanoparticles delivered a discharge capacity of nearly 130 mA h g−1 at 5C with a capacity retention exceeding 90% after 300 cycles. The improved rate capability was attributed to the high ionic conductivity caused by the reduced Li+ diffusion length and the increased surface area.59
Single crystalline nanowires are the most attractive morphology among all nanostructures because they resist aggregation and grain growth at high temperature and exhibit a negligible potential barrier among the nanosized grains. High-quality single crystalline cubic spinel LiMn2O4 nanowires synthesized using Na0.44MnO2 nanowires as a self-template have shown a relatively flat charge–discharge plateau even at 20 A g−1, excellent cycle stability, and high thermal stability.63Fig. 4(a–c) display the corresponding SEM, TEM, and electrochemical performance of the novel nanowires. The unique nanowire morphology decreased both the Li+ and electron diffusion lengths, and displayed industrial potential for an improved volumetric energy density and safety in practical LIBs. An inexpensive and scalable synthesis of porous FeF3 nanowires through temperature-controlled dehydration in an inert atmosphere has been reported. The first discharge capacity of the FeF3 nanowire cathode was up to 543 mA h g−1, and a capacity of 223 mA h g−1 was delivered after 50 cycles at 50 mA g−1, as a result of the large surface area, favorable morphology for accommodating transformation strains, and internal conductive pathway provided by the continuous network of Fe nanodomains.48 Li-deficient LixMn0.67Ni0.33O2 nanowires fabricated through a hydrothermal method using LiMn0.5Ni0.5O2 powder as a precursor delivered a high capacity of ∼230 mA h g−1 at 20 mA g−1, benefiting from the unique 1D nanowire structure.64 Li(Li0.15Ni0.25Mn0.6)O265 and Li0.88(Li0.18Co0.33Mn0.49)O266 nanowires prepared by a similar method also showed good cycling stability and excellent rate capability.
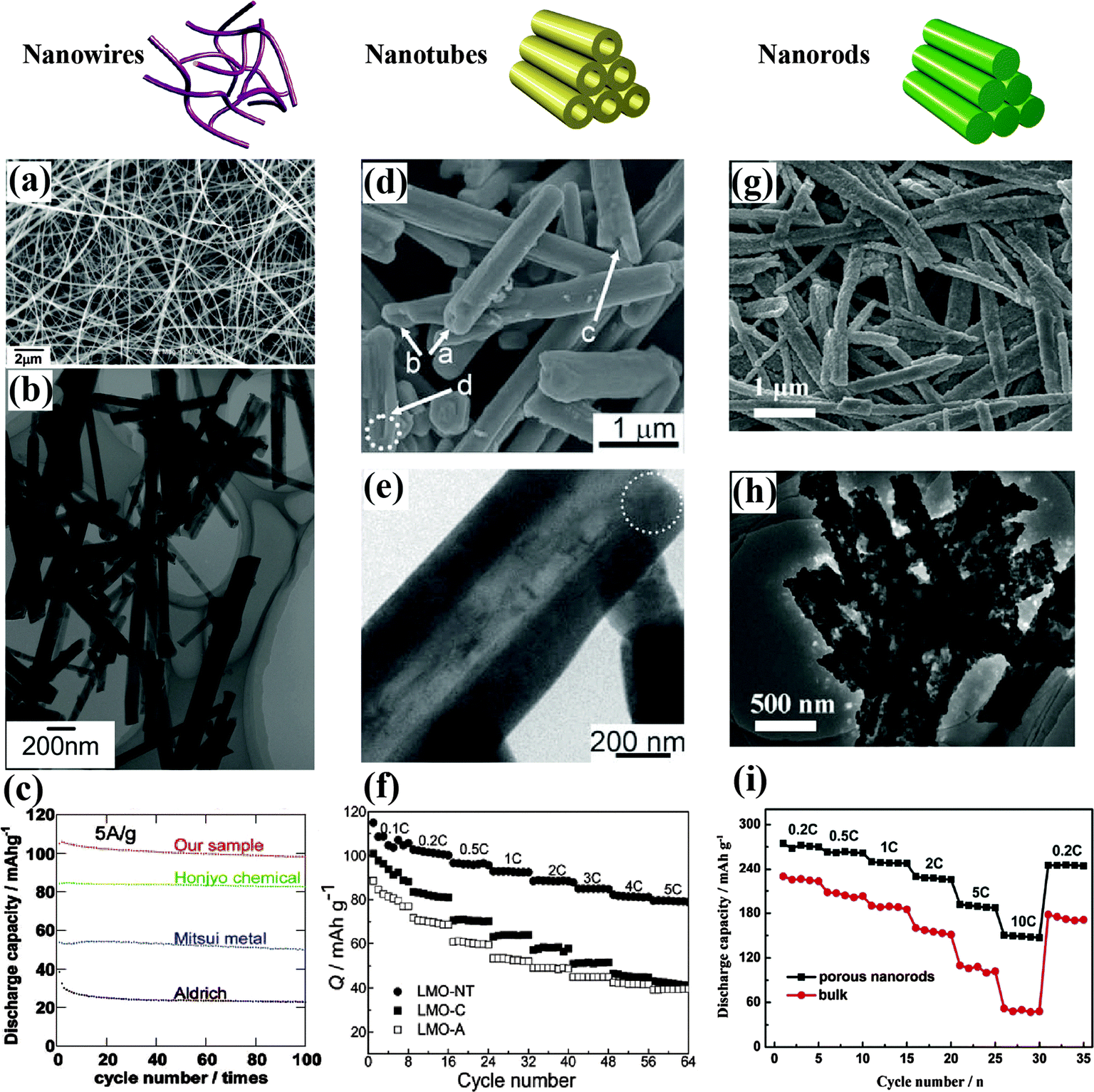 | ||
| Fig. 4 LiMn2O4 nanowires:63 (a) SEM image, (b) TEM image, (c) electrochemical performance. LiMn2O4 nanotubes:34 (d) SEM image, (e) TEM image, (f) electrochemical performance. 0.2Li2MnO3·0.8LiNi0.5Mn0.5O2 nanorods:68 (g) SEM image, (h) TEM image, (i) electrochemical performance. | ||
Channels for transfer are also a notable morphology of nanotubes that enhances the structure stability and rate capability. Single-crystalline nanotubes of spinel LiMn2O4 with a wall thickness of about 200 nm have been synthesized via a template-engaged reaction and exhibited remarkable high-rate performance, as displayed in Fig. 4(d–f).34 About 70% of their initial capacity was retained after 1500 cycles at a rate of 5C. Notably, the tubular LiMn2O4 retained its single-crystalline nature and preferential growth direction after long-term cycling at a relatively high rate, indicating its good structural stability during Li intercalation/deintercalation processes. LiMn2O4 nanotubes with a preferred orientation of (400) planes have been successfully prepared using multiwalled carbon nanotubes as a sacrificial template and reported to show a superfast second-level charge capability as a cathode for aqueous rechargeable lithium batteries.67 Their reversible capacity was 110 mA h g−1 and, even at the ultrahigh charging rate of 600C (6 s), a 53.9% capacity was obtained. This excellent cycling behavior was attributed to the porous tube structure buffering the strain and stress caused by Jahn–Teller effects.
Unique porous nanorods favor fast Li intercalation kinetics by shortening the Li+ diffusion distance and increasing the structural stability of the material by buffering the local volume change that occurs during the reversible insertion/extraction processes. As shown in Fig. 4(g–i), porous 0.2Li2MnO3·0.8LiNi0.5Mn0.5O2 nanorods fabricated using a morphology-inheritance strategy are composed of interconnected nanosized subunits with a highly porous structure, and displayed superior capacity and rate capability. The initial discharge capacity exceeded 275 mA h g−1 at 0.2C and the capacity retention approached 90% for up to 100 cycles.68 Porous LiMn2O4 nanorods consisting of aggregated nanoparticles have been synthesized by solid state lithiation of 1D nanoporous Mn2O3. A discharge capacity of 80 mA h g−1 at 30C was obtained, benefiting from the high diffusion coefficient of the porous nanorods, while the capacity retention approached 90% after 500 cycles at 2C. This durable high-rate capability was attributed to the unique porous 1D nanostructure that gave rise to fast Li-intercalation kinetics.9 Similarly, porous LiNi0.5Mn1.5O4 nanorods with a diameter of 100–400 nm constructed by the same method have exhibited a remarkable capacity retention of 91% after 500 cycles at 5C.69
Ultrathin V2O5 nanosheets with a thickness of 2.1–3.8 nm prepared via a simple and scalable liquid exfoliation technique have shown large reversible capacity, high Coulombic efficiency, stable cyclability, and ultrahigh rate capability. A capacity of 117 mA h g−1 (energy density: 158 W h kg−1) at a charge and discharge rate of 50C (power density: 20 kW kg−1) was obtained owing to the very short diffusion paths provided by this ultrathin thickness, as shown in Fig. 5A.70 Ultrathin LiMPO4 (M = Fe, Mn, Co, Ni) nanosheets (thickness: 3.7–4.6 nm) with exposed (010) surface facets have been fabricated by a liquid-phase exfoliation approach combined with a solvothermal lithiation process.71 As for LiFePO4, LiMnPO4, and LiCoPO4 nanosheets, the calculated time for Li+ diffusion over a [010]-thickness of <5 nm was less than 25, 2.5, and 250 μs, respectively, which were about 5 orders of magnitude lower than those for the corresponding bulk materials. The high energy and power densities and excellent rate capability (e.g., 18 kW kg−1 and 90 W h kg−1 at 80C rate) of LiFePO4 nanosheets have also been demonstrated.
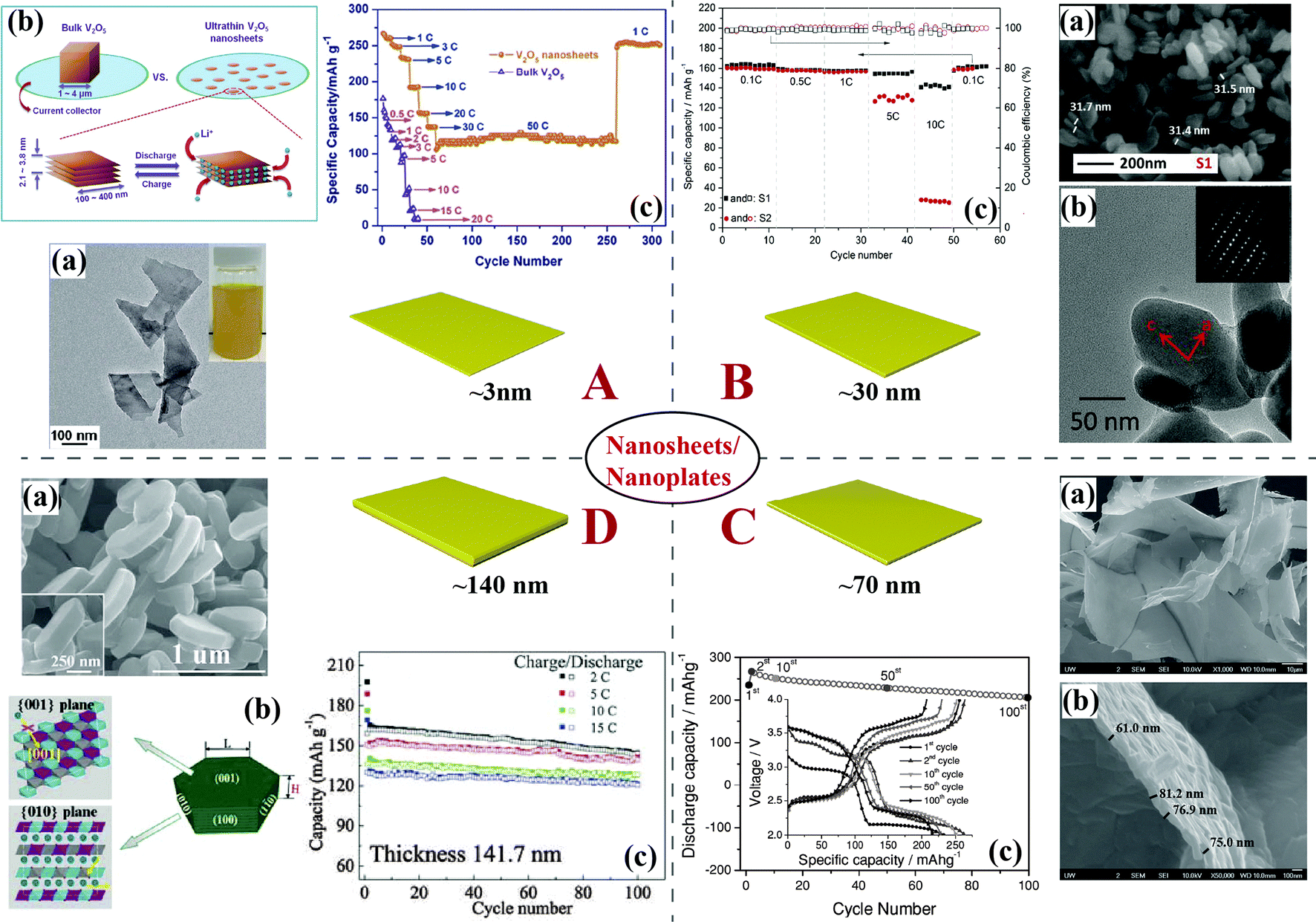 | ||
| Fig. 5 Typical 2D nanostructures and their corresponding morphology and electrochemical performance. (A) Ultrathin V2O5 nanosheets:70 (a) TEM image and the corresponding colloidal acetone dispersion (inset), (b) a schematic of lithiation processes for bulk V2O5versus {001}-oriented few-layer V2O5 nanosheets, (c) rate capability. (B) LiFePO4 nanoplates:72 (a) SEM images, (b) TEM images (SEAD inside), (c) discharge capacity and Coulombic efficiency vs. cycle number plots. (C) Leaf-like V2O5 nanosheets:73 (a) low- and (b) high-magnification FESEM images, (c) cycling performance at a current density of 500 mA g−1. Inset shows the charge/discharge curves. (D) LiNi1/3Co1/3Mn1/3O2 nanobricks:75 (a) SEM image of the lateral view of LNCM nanobricks, inset image is a magnified image of a single nanobrick, (b) schematic diagram of growth orientation, (c) cycling performance of LNCM at high rates. | ||
Olivine LiFePO4 nanoplates (Fig. 5B) with crystal orientation along the ac facet and bc facet, easily prepared by a glycol-based solvothermal process, have exhibited reversible capacities of around 160 mA h g−1 with a Coulombic efficiency of near 100%. Moreover, capacities of 156 mA h g−1 and 148 mA h g−1 were delivered at rates of 5C and 10C, respectively, demonstrating that the crystal orientation plays an important role in the superior electrochemical performance of the material.72 Porous LiMnPO4–C nanoplates with a thickness of ∼50 nm assembled by nanorods growing along the [010] direction in the (100) plane have been prepared via a solid-state reaction. The specific capacity of the porous nanoplates reached 168 mA h g−1 at 0.02C.10
Leaf-like V2O5 nanosheets (∼70 nm, shown in Fig. 5C) have been synthesized by a facile, green, and low-cost method. Their unique 2D nanoscale morphology, porous structure, and large specific surface area lend these materials a high specific capacity, good rate capability, and good cyclability. Their specific discharge capacity of 206 mA h g−1 was retained after 100 cycles at a current density of 500 mA g−1. Even at a very high current density of 5000 mA g−1, the 2D leaf-like V2O5 nanosheet electrode still delivered a high capacity of 104 mA h g−1.73 Cathodes based on LiPF6-etched LiFePO4@carbon nanocrystals shown to be nanoplates have delivered capacities of ∼155 mA h g−1, ∼135 mA h g−1, and ∼125 mA h g−1 at 1C, 5C, and 10C, respectively, with high capacity retention. A remarkable ultrahigh rate capability was also confirmed. At 61C (10.3 A g−1), a capacity of ∼70 mA h g−1 was obtained, while at 122C (20.7 A g−1) the capacity was ∼30 mA h g−1.74
Single-crystal LiNi1/3Co1/3Mn1/3O2 hexagonal nanobricks (thickness of around 141.7 nm) have been synthesized using Ni1/3Co1/3Mn1/3(OH)2 hexagonal nanosheets as both templates and precursors. Excellent cyclic stability at a high rate, ∼130 mA h g−1 at 15C, was obtained (Fig. 5D), which was attributed to the unique nanostructure with a high percentage of exposed {010} facets along the [001] direction, which ensured fast and efficient Li+ insertion and extraction.75
3D solid nanostructures. A hierarchical structured Li-rich Li1.2Mn0.6Ni0.2O2 cathode was proposed using a rational design that combined the advantages of a hierarchical architecture and the electrochemically active exposed {010} planes of the primary nanoplate structure.81 This unique hierarchical structure enables both efficient ion and electron transport for fast Li+ transport kinetics and leads to superior rate behavior and excellent capacity retention, as shown in Fig. 6(a–e). A hierarchical sphere-shaped Li1.2Ni0.13Mn0.54Co0.13O2 cathode with outstanding high-rate and cycling stability has been prepared using a versatile ionic interfusion method.82 The primary nanoplates assembled in this hierarchical cathode exhibited enhanced growth of the nanocrystal planes, such as the (010), (100), and (110) planes, which are favorable for Li+ insertion/extraction. High discharge capacities were realized and ultrafast charging to around 70% (175 mA h g−1) of the capacity at 0.1C within about 2.1 min was achieved.
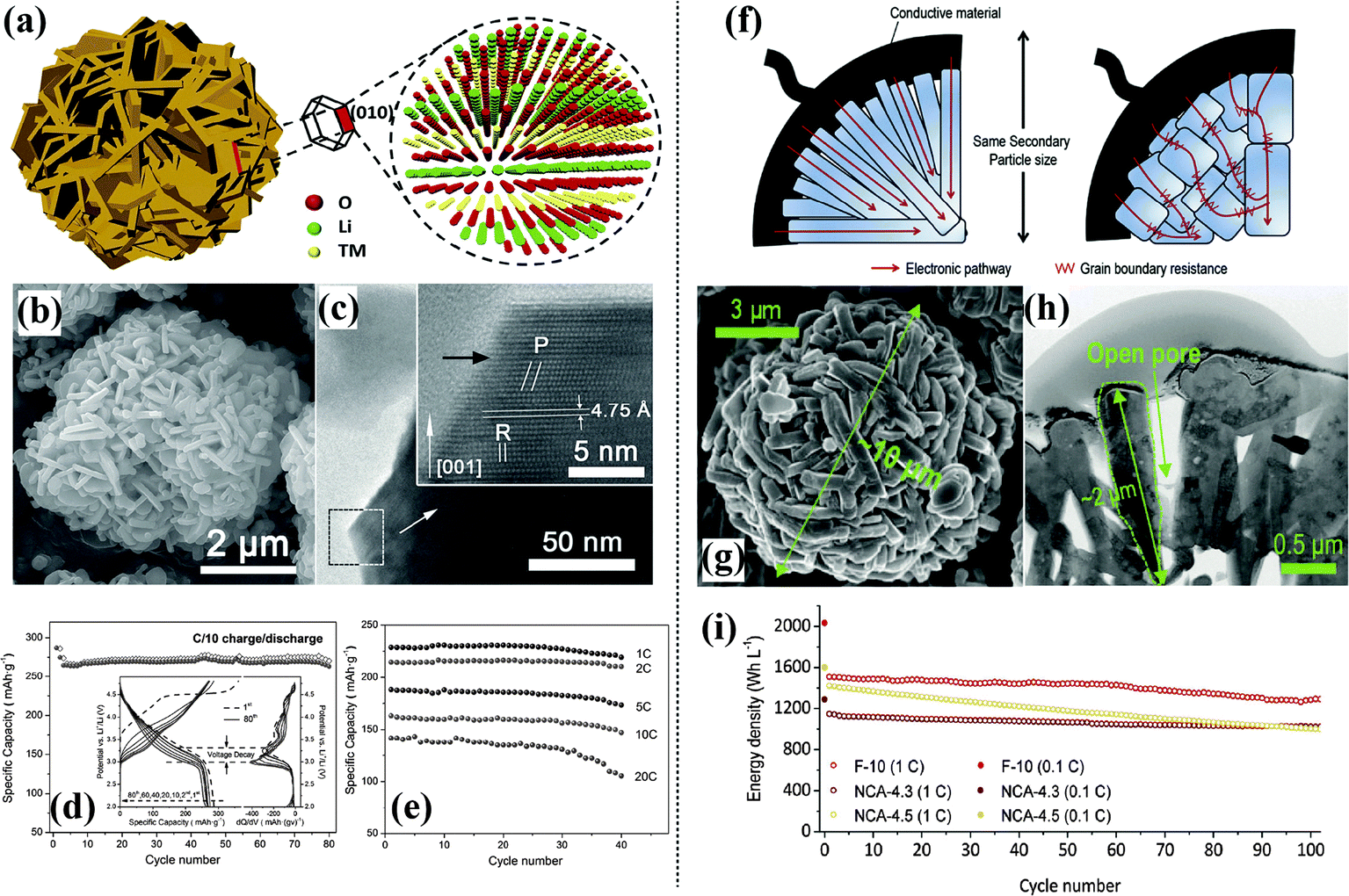 | ||
| Fig. 6 Hierarchical Li1.2Ni0.2Mn0.6O2 assembled in nanoplates:81 (a) schematic illustration, (b) SEM image, (c) HRTEM image of a lateral plane of a single nanoplate, inset is a magnified image of the frame, (d) cycling performance of HSLR materials at C/10 rate; inset is the corresponding voltage profile and dQ/dV plots, (e) electrochemical performance. Hierarchical 0.5Li2MnO3–0.5LiNi0.5Mn0.5O2:83 (f) schematic diagram of electron migration through secondary particles composed of primary particles with different morphologies; nanoflake-shaped (left) and nanoparticle-shaped (right), (g) SEM image, (h) TEM image, (i) electrochemical performance. | ||
A Li-rich cathode material with 10 μm-sized secondary particles composed of nanoscale flake-shaped primary particles designed using a unique engineering method exhibited a high average voltage, excellent long-term energy, and voltage retention of 93% over 600 cycles.83 The electron migration mechanism, SEM, TEM, and electrochemical performance of this material are shown in Fig. 6(f–i). Hierarchical nano/micro-structured LiNi0.5Mn1.5O4 with a high tap density of 1.7 g cc−1 has been prepared via a facile PEG-assisted co-precipitation method.39 A discharge capacity of more than 120 mA h g−1 at 40C and a capacity retention of 89% after 150 cycles at 5C was obtained. A facile solvothermal method combined with a calcination process was used to synthesize a novel peanut-like hierarchical Li-rich cathode material (Li1.2Mn0.54Ni0.18Co0.08O2) assembled by nanoparticles. A high reversible capacity, good cycle stability, and superior rate capability were obtained for this material. A discharge capacity of 229.9 mA h g−1 at a current density of 200 mA g−1 with a high capacity retention of 94.2% after 100 cycles was achieved.84
3D hollow nanostructures. In recent years, hollow cathodes composed of nanostructures have been considered to attempt to resolve the problem of rapid capacity fading over extended cycling. The large void space inside such hollow structures can provide more electrochemically active sites for lithium storage, facilitate the rapid transfer of electrons, and reversibly alleviate large volume changes and prevent pulverization during lithiation/delithiation. Their larger surface area, reduced diffusion length for Li+, and avoidance of high contact resistance generation also leads to better rate capabilities.
The preparation of novel hollow microspheres assembled from mesoporous single crystalline V2O5 nanorods has been successfully realized by a simple solvothermal treatment of NH4VO3 and ethylene glycol with subsequent annealing at 400 °C in air.85 Superior electrochemical performance, with high specific capacity, much improved capacity retention and long cycle life at various rates, was demonstrated. The SEM images and rate capability of this material are displayed in Fig. 7(a–c).
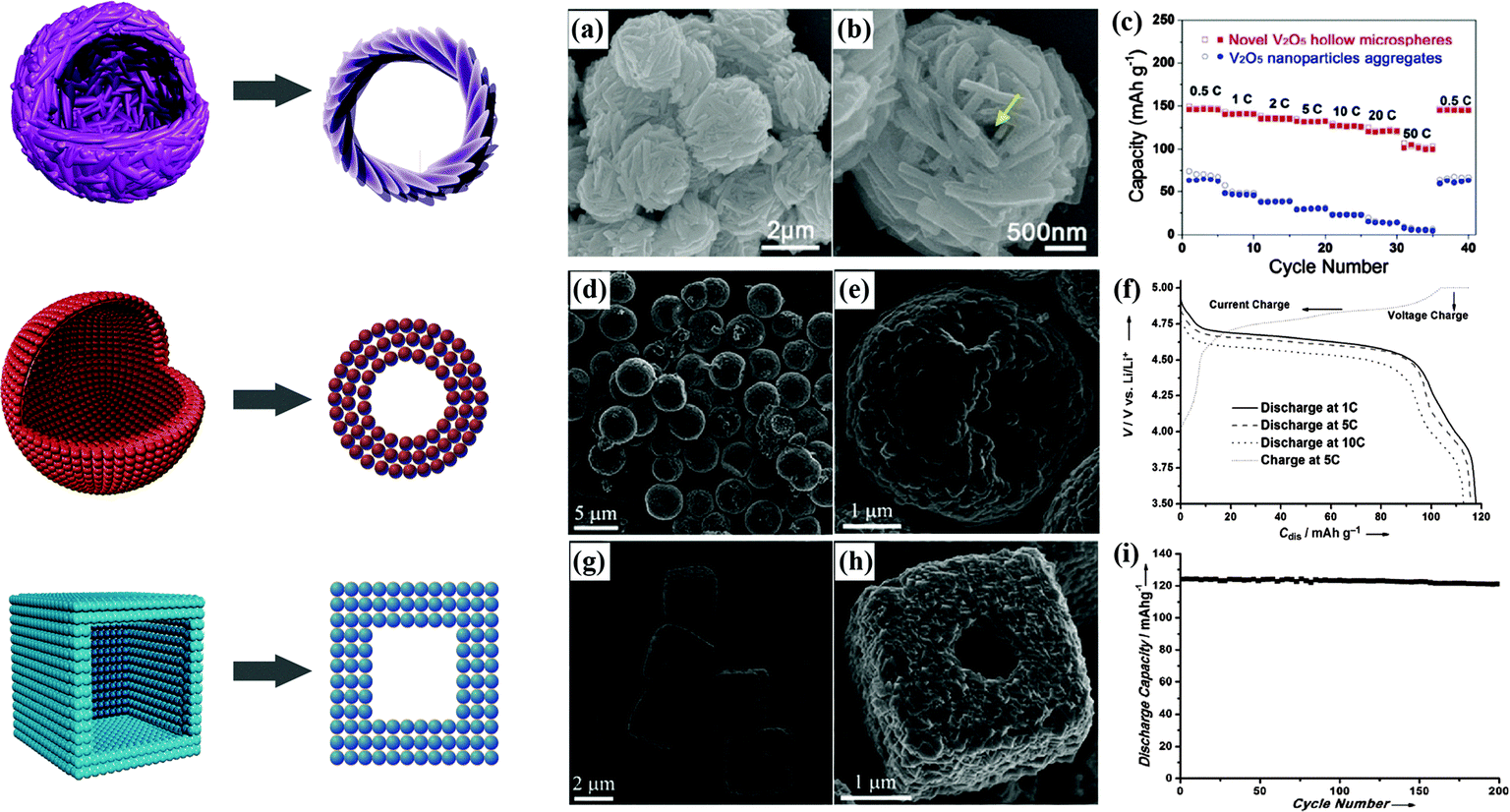 | ||
| Fig. 7 Hollow microspheres assembled by V2O5 nanorods:85 (a and b) SEM images, (c) rate capability. Hollow microspheres assembled by LiNi0.5Mn1.5O4 nanoparticles:14 (d and e) SEM images, (f) charge/discharge curves. Hollow microcubes assembled by LiNi0.5Mn1.5O4 nanoparticles:14 (g and h) SEM images, (i) cycling stability. | ||
Hollow LiNi0.5Mn1.5O4 microspheres and microcubes assembled by nanoparticles have been successfully synthesized using porous MnO2 spheres and cubes as precursors.14 The corresponding SEM images and electrochemical performance are shown in Fig. 7(d–i). The unique hollow structures delivered discharge capacities of 118 mA h g−1 at 1C and 104 mA h g−1 at 20C. Hollow LiMn2O4 nanostructures, such as the double-shelled hollow LiMn2O4 microspheres reported by Ding et al., are currently recognized to make Li+ insertion easier and can exhibit better rate capability than solid LiMn2O4 microspheres.86 Single-crystalline LiMn2O4 hollow nanocones synthesized using pre-synthesized MnO2 hollow nanocones as a self-sacrificial template have been reported to deliver a very high specific capacity, superior rate capability, and ultralong cycle life. A capacity of 100 mA h g−1 was obtained even at a very high rate of 50C. After 1000 cycles at 5C, almost 94.8% of the initial capacity was retained.87
Nanocrystal-assembled hollow 0.3Li2MnO3·0.7LiNi0.5Mn0.5O2 microspheres synthesized by a template route have delivered a high reversible capacity of 295 mA h g−1 after 100 cycles.88 The produced porous and hollow Li1.2Mn0.6Ni0.1Co0.1O2 microspheres were formed from the spontaneous aggregation of nanoparticles acting as building blocks. An appreciable discharge capacity of 242 mA h g−1 at 50 mA g−1 with an exceptional Coulombic efficiency of 97% were obtained, which was attributed to increased electrolyte penetration and reduced Li+ diffusion length originating from the hollow arrangement of interconnected nanocrystalline particles.89 Our group has also demonstrated that hollow spherical Li1.2Mn0.54Ni0.13Co0.13O2 structures assembled by nanoplate-shaped particles and quasi-sphere-shaped particles resulted in improved rate performance. The hollow cathode material consisting of assembled nanoplates showed better crystallinity and a layered structure, a higher surface area, and better electrochemical properties, especially at high discharge currents.90
The abovementioned studies clearly show the suitability of hollow nanostructured cathodes for LIB applications. Deploying new cathodes with hollow structures is the future direction of development.
3D core–shell nanostructured cathode materials. Because they have the best combination of utility and versatility through the regulation of different constituents, numerous core–shell structures have been successfully fabricated and applied in a wide range of fields.91–94 Core–shell nanostructured cathodes combine both the concept of “core–shell” and “nano” and have been extensively applied to LIBs. The main component of the core–shell structure, the inner core, plays a functional role in the lithium storage properties of the material. The inner core is surrounded by outer shells which can protect the inner core from outside side reactions or volume changes. Meanwhile, the shells can impart new physical or chemical properties and inhibit the agglomeration of internal particles, thus leading to superior electrochemical performance in terms of discharge capacity, cycle stability, and thermal stability. Additionally, the outer shell is generally designed to be nanoscale to allow ions to penetrate with negligible polarization. Such outer shells fall into two main categories, namely, inactive and active nanocoating shells.
Owing to their high electronic conductivity, considerable flexibility, low cost, and easy preparation, carbon materials are considered to be the most promising coating layer for effectively enhancing electronic transport and improving the electrochemical performance of various cathodes.
Carbon-coated LiMn2O4 clusters consisting of many single-crystal nanoparticles have shown ultrahigh rate capability. A gravimetric energy density of 300 W h kg−1 at a power density of 45 kW kg−1 and a volumetric energy density of 440 W h L−1 at a power density of 68 kW L−1 were obtained.37 Sphere-like Li3V2(PO4)3 nanoparticles have been prepared by synergistically using polyethylene glycol (PEG) and acetylene black for carbon coating and conductive network construction.95 The resulting material exhibited a high capacity retention of 83% even after 1000 cycles at 5C, and still delivered a considerable capacity after 5000 cycles. High rate capability without capacity fading at 30C was also demonstrated, resulting from the optimum design of the 3D continuous electron pathways.
Some other materials capable of forming a highly electrically conductive network, such as transition metals, conducting polymers, metal oxides, metal fluorides, and metal phosphates, have also been extensively investigated as nanocoating shells for modifying various cathodes. As HF scavengers, these thin protective shells with a size of several nanometers can react with the HF released from the electrolyte before the active core does.
AlF3-coated LiNi0.5Mn0.5O2 prepared by Sun et al. showed enhanced rate capability and thermal stability. The protective AlF3 shell, which had a thickness of ∼10 nm, effectively reduced Ni and Mn dissolution and inhibited the decomposition of the organic electrolyte.96 As illustrated in Fig. 8(e and i), the double-layer coated samples were proved to exhibit lower irreversible capacity loss and a higher discharge capacity than both the pristine and the single-layer coated samples, owing to the retention of a higher number of oxide ion vacancies in the layered lattice after the first charge.100 A series of functional surface modifications for Li-rich layered Li(Li1/3−2x/3NixMn2/3−x/3)O2, including Al2O3, RuO2, AlPO4 and CoPO4, were systematically investigated. The results of the study showed that 1 wt% Al2O3 and 1 wt% RuO2-coated Li(Li0.2Mn0.54Ni0.13Co0.13)O2 delivered a high capacity of 280 mA h g−1 at 0.05C with a capacity retention of 94.3% after 30 cycles.97,98
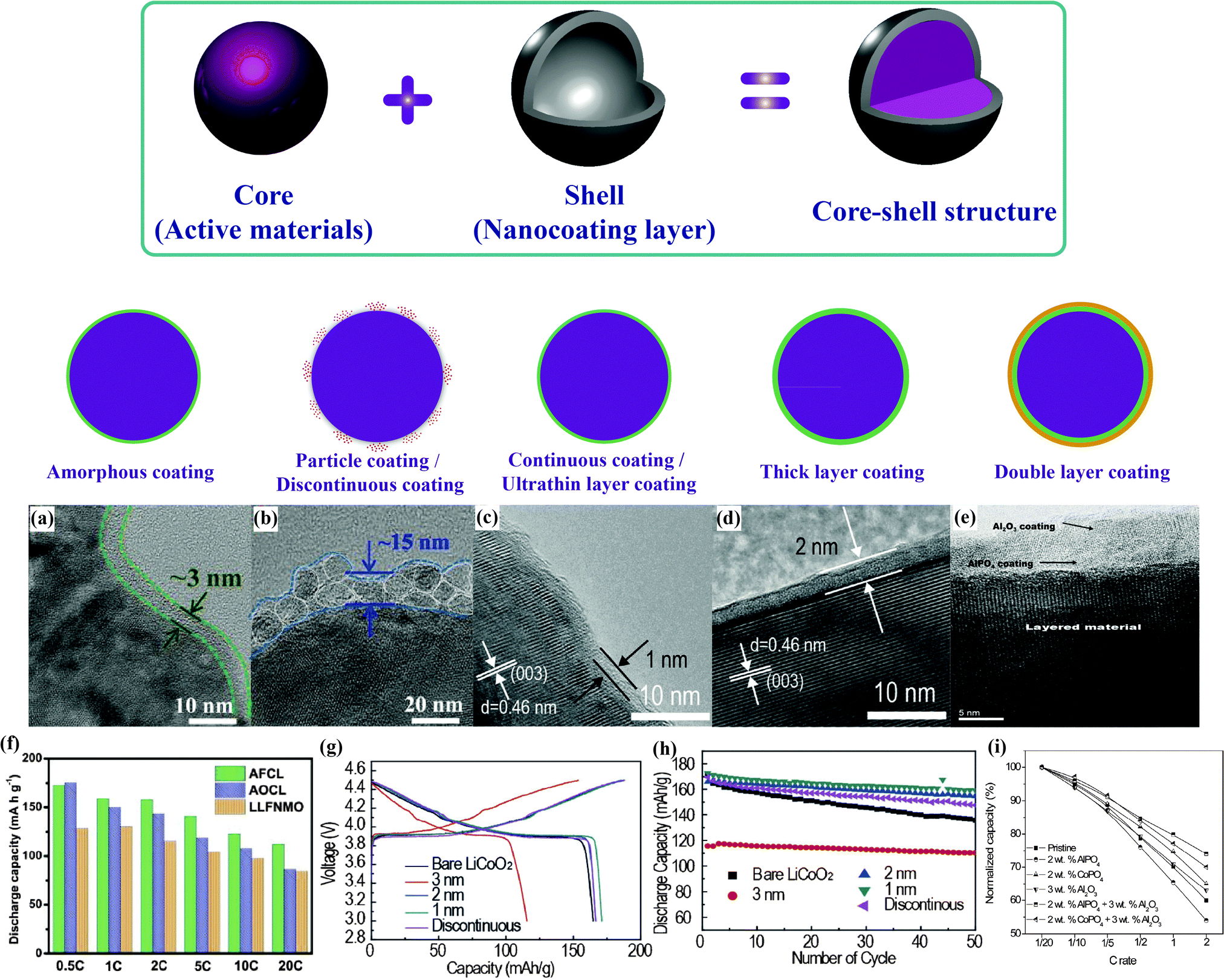 | ||
| Fig. 8 Morphology comparison:99 (a and b) TEM images of Li[Li0.2Fe0.1Ni0.15Mn0.55]O2 with different coating morphology, (f) electrochemical performance. Uniformity and thickness comparison:108 (c and d) TEM images of surface-modified LiCoO2 samples with different shell uniformity and thickness, (g and h) charge/discharge profiles and cycling performance. Multilayer comparison:98 (e) TEM images of double layer coating of Al2O3 and AlPO4, (i) comparison of normalized capacities. | ||
Recently, our group has mainly focused on the surface modification of Fe-based low-cost Li-rich cathode materials, which suffer from serious capacity fading and poor rate capability, through the introduction of several different inactive nanocoating shell materials with nanometer dimensions, such as Al2O3,99 AlF3,99 AlPO4,100 and FePO4.101Fig. 8(a, b and f) show TEM images of the obtained cathodes with different morphologies and their electrochemical performance. The reduced surface side reactions resulted from the isolation of the bulk cathode from the electrolyte, which led to enhanced electrochemical performance.
In conventional coating technologies, fast nucleation cannot be effectively avoided to prevent the formation of a partial coating layer. Accordingly, it is essential to precisely control the shell fabrication to ensure a uniform thickness for the designated function. Atomic layer deposition (ALD) techniques have been employed to construct uniform nanoshells typically thinner than 5 nm.102–106 Atomic-scale precision can be realized by depositing Al2O3 or TiO2 layers onto compressed cathode films. A higher first cycle Coulombic efficiency and improved cycling stability were obtained, which were directly attributable to the protective role of the surface films against electrode–electrolyte reactions.107 However, the high cost and laborious nature of the procedure limits the further application of ALD nanotechnology. A simple and economic synthetic strategy for growing uniform Al2O3 nanoshells on LiCoO2 on a larger scale has been developed to enable a large amount of powder to be handled with precise surface control.108 Perfect control of the Al2O3 nanoshells was realized using a suitable environment for manipulating the growth of aluminum hydroxide within a precision of one nanometer, as displayed in Fig. 8(c, d, g and h). Owing to the balance between polarization and surface protection, a uniform shell with a thickness of around 1–2 nm was found to show the best cyclability.
Besides the above familiar nanocoating layers, there are a number of more unique protective layers.109 A surface-Li2TiO3-rich coating layer was proved to be beneficial for the good cycle performance and thermal stability of 1D layered Mn-based oxide nanobelts.110 A mesoporous thin layer of the conducting polymer poly(3,4-ethylenedioxythiophene) (PEDOT) on the surface of V2O5 can decrease its polarization and increase the proportion of high-voltage capacity, leading to superior high-rate capability and cycling stability.111
Although inactive nanocoatings have been successfully applied to cathode materials, the addition of a non-electrochemically active coating layer decreases their gravimetric energy density. To solve this limitation of inactive nanocoating technology, researchers have begun to pursue the introduction of active outer shells with lithium storage properties and high stability to maximize charge/discharge capacities and optimize thermal stability.112,113
As shown in Fig. 9(a–i), the shell morphology of concentration gradient core–shell (CGCS) lithium oxides can be modified from nanoparticles to nanorods by varying the synthetic conditions.114,115 The presence of a dense nanorod-based shell led to lower cation mixing, higher electrical conductivity, smaller pore volume, higher tap density, and reduced surface area, resulting in a higher discharge capacity and a superior lithium intercalation stability.
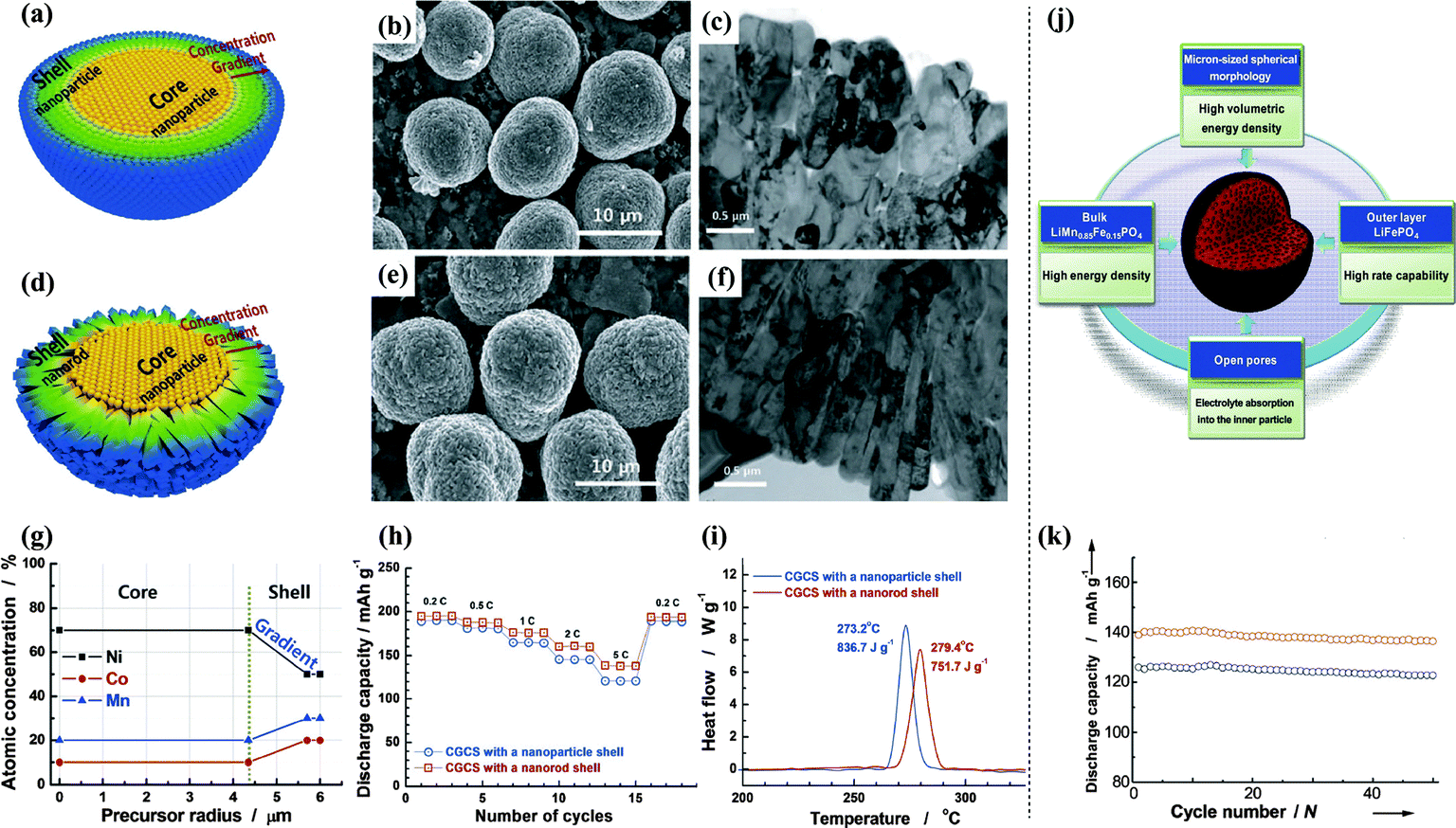 | ||
| Fig. 9 Schematics of concentration gradient core–shell particle with a (a) nanoparticle shell, (d) nanorod shell; (b) SEM image and (c) TEM image of Li[Ni0.60Co0.15Mn0.25]O2 with a nanoparticle shell; (e) SEM image and (f) TEM image of Li[Ni0.60Co0.15Mn0.25]O2 with a nanorod shell; (g) designed atomic concentration in the core and shell showing the concentration gradients of Ni, Co, and Mn; (h) capacity plots versus the C-rates; (i) DSC traces.114 (j) Double-structured micron-sized LiMn0.85Fe0.15PO4 modified with LiFePO4 and the resulting functions of each component, (k) cycling stability.116 | ||
LiFePO4 outer layers with different layer thickness were added to bulk LiMn0.85Fe0.15PO4, which allowed the fine-tuning of physical characteristics such as the tap density, porosity, and spherical morphology. The core–shell mechanism and electrochemical performance of the materials are shown in Fig. 9(j and k). The double-structured LiMn1−xFexPO4 material showed a specific discharge capacity approaching the theoretical limit, stable cyclability even at 60 °C, excellent performance at low temperatures, and an exceptionally high tap density.116
A spinel/layered core–shell heterostructured cathode was designed by our group. The nanosized spinel LiNixMn2−xO4 outer shell (∼10 nm thick), which provided a 3D Li+ insertion/extraction framework, protected the high Li+ storage capacity layered Li-rich inner core from erosion by the electrolyte and restrained the loss of bulk active-mass, resulting in a remarkable rate capability, extremely high capacity, and excellent cycling ability.117 A novel ultrathin spinel membrane (∼1–2 nm) was then encapsulated onto the layered Li-rich cathode material through a biomimetic technique. Superior high reversible capacity (over 290 mA h g−1), outstanding rate capability, and excellent cycling ability were obtained for the resulting cathode. The ultrathin 4 V spinel membrane, which acted as a Li+ pump, provided a Li+ diffusion highway between the electrolyte and the bulk layered mass. Meanwhile, voltage decay and thermal instability were also found to be alleviated.118 Additionally, the combination of an FeF3 electrochemical conversion reaction cathode and extraction/insertion cathode was realized and exhibited obvious improvements, including a high reversible capacity (260.1 mA h g−1 at 0.1C), superior rate capability (129.9 mA h g−1 at 20C) and good cycle stability.119
4.2 Nanocomposite cathodes
In addition to surface modification, the fabrication of nanocomposites has been reported to be an efficient strategy to obtain cathode materials with satisfactory electrochemical performance. The rational combination of promising cathode materials with conductive substrates such as graphene, carbon nanotubes, and carbon matrix is expected to enable the commercial availability of high density materials for LIBs.Li3V2(PO4)3/graphene nanocomposites prepared by a sol–gel process through the selective growth and/or enwrapping of Li3V2(PO4)3 nanoparticles on partially reduced graphene oxide have exhibited excellent high-rate and cycling stability, owing to the connection of the nanoparticles with the current collector through the conducting graphene network. The capacity of this material dropped only very slightly to 116 mA h g−1 (5C) and 108 mA h g−1 (20C) after 100 cycles, respectively, demonstrating a stable capacity retention with negligible capacity fading, as shown in Fig. 10(A).129
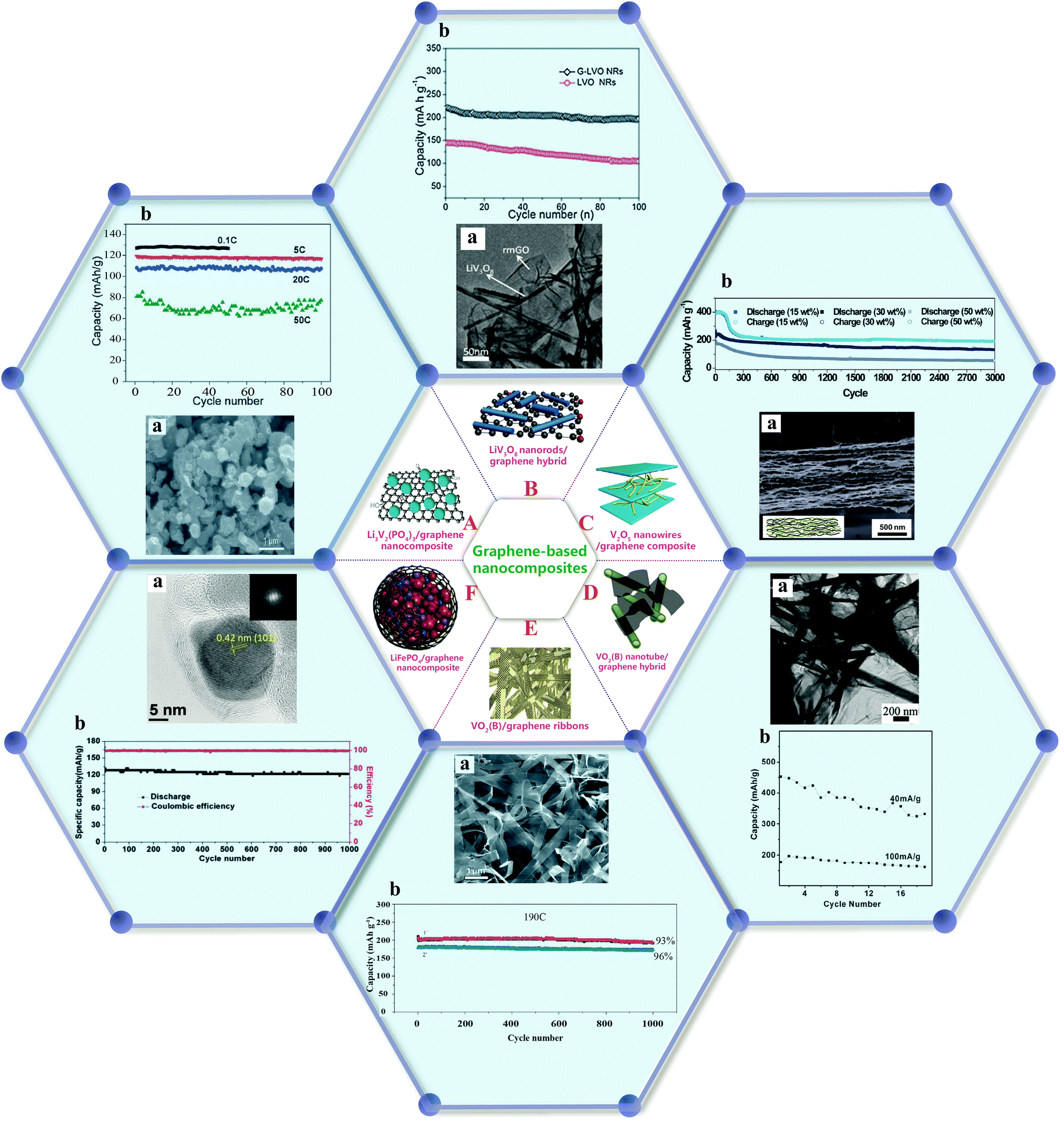 | ||
| Fig. 10 (A) Li3V2(PO4)3/graphene nanocomposite:129 (a) SEM image, (b) cycle performance at a cut-off voltage of 3–4.3 V (1C = 133 mA h g−1). (B) LiV3O8 nanorod/graphene hybrid:130 (a) TEM image, (b) cycling performance of the electrodes: G-LVO NRs and LVO NRs. (C) V2O5 nanowire/GO composite:131 (a) cross-sectional SEM image, (b) cycling tests for the composite paper with various V2O5 contents measured at 1000 mA g−1. (D) VO2(B) nanotube/graphene hybrid:132 (a) cycle life data of the hybrid, (b) TEM image. (E) VO2(B)/graphene ribbons:133 (a) SEM image, (b) capacity retentions at the highest rate of 190C (37.2 A g−1) for 1000 cycles. (F) LiFePO4@graphene nanocomposite: (a) high magnification TEM image, (b) cycling performance at a current density of 170 mA g−1 (1C) between 2.0 and 4.3 V.134 | ||
Compared with pure LiV3O8 nanorods, nanocomposites of LiV3O8 nanorods on graphene sheets (Fig. 10(B)) prepared by a facile in situ two-step solution-phase reaction route have shown high reversible capacity and good cyclic capacity retention after prolonged cycling. The enhanced lithium storage properties can be attributed to a high electrical conductivity and low ionic resistance.130
A nanostructured composite was designed through the incorporation of ultrathin V2O5 nanowires into graphene paper to improve the issues of capacity fading and poor cycle life caused by particle agglomeration.131 When cycled at 1000 mA g−1 for 3000 cycles, the nanocomposite paper with 15 wt% V2O5 exhibited extraordinary cycling performance and a reasonably high capacity of 191.5 mA h g−1, as displayed in Fig. 10(C). These results demonstrated that the integration of highly organized nanostructured cathodes with graphene could effectively enhance electronic and ionic conductivities, leading to good cycling stability.
VO2(B) nanotubes dispersed and trapped between graphene sheets have been realized by a hydrothermal reaction through the reduction of graphite oxide to graphene. As shown in Fig. 10(D), this nanocomposite exhibited a high capacity (∼450 mA h g−1) as well as cycling stability. The interaction between graphene sheets and nanotubes improved the charge-transfer process and suppressed the aggregation of the nanotubes.132
Single crystalline vanadium oxide (VO2) ribbons with graphene layers were also prepared by a simple bottom-up approach and achieved a high reversible capacity and ultrafast charging/discharging capability (20 s). As shown in Fig. 10(E), more than 90% of the initial capacity was retained after cycling more than 1000 times at an ultrahigh rate of 190C, demonstrating the excellent rate capability of the material.133
Highly crystalline LiFePO4 nanoparticles encapsulated by continuous and uniform graphene were synthesized by a solid state reaction between graphene-coated Fe nanoparticles and LiH2PO4.134 The specific capacity remained at 122 mA h g−1 (95.3% retention) after 1000 cycles at 170 mA g−1, as displayed in Fig. 10(F). Fast ionic diffusion and efficient electron transport arising from the highly conductive graphitic carbon network were believed to account for the improved cycle stability.
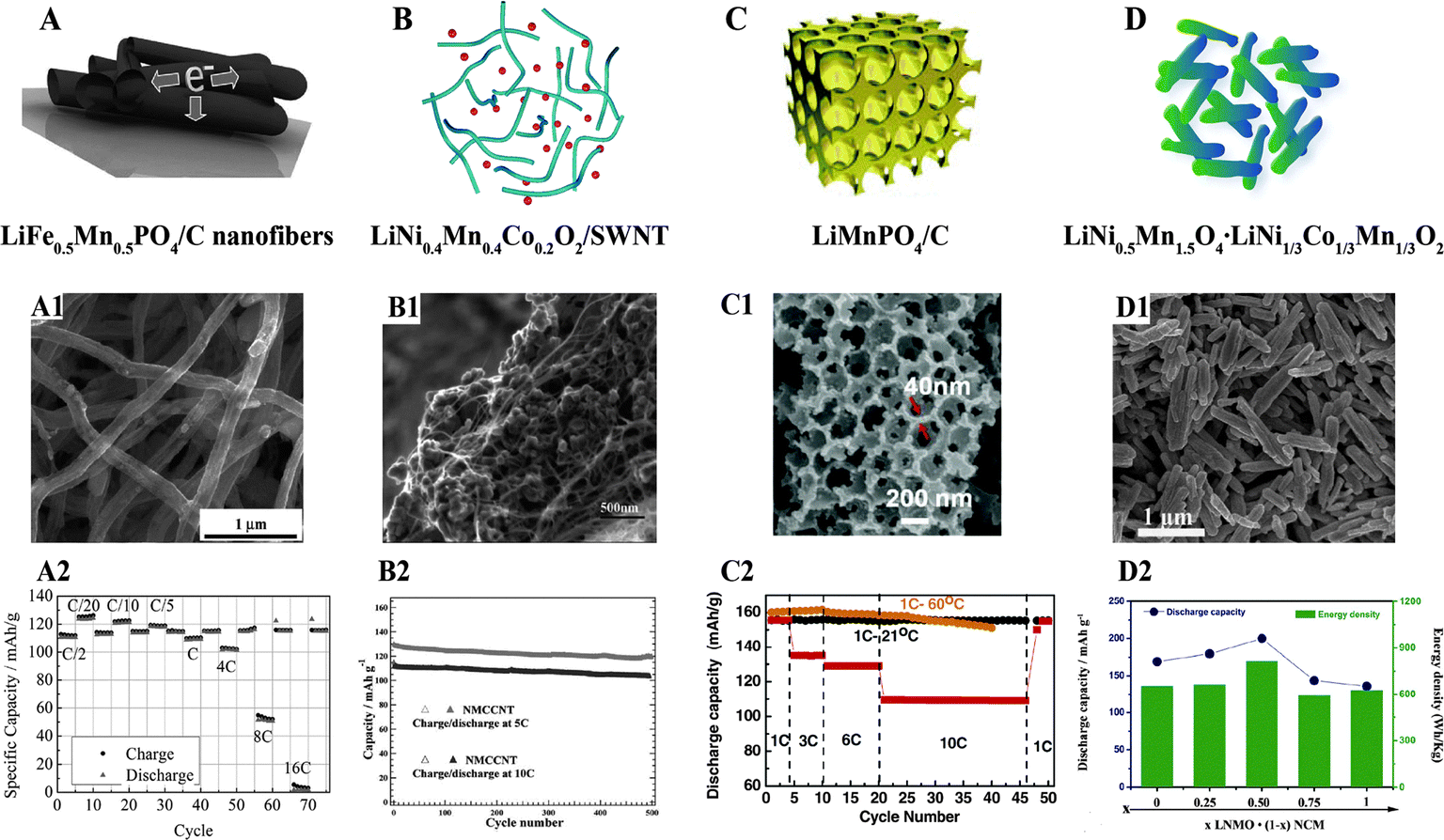 | ||
| Fig. 11 (A) LiFe0.5Mn0.5PO4/C nanofibers:136 (A1) SEM image, (A2) electrochemical performance; (B) LiNi0.4Mn0.4Co0.2O2/SWNT:137 (B1) SEM image, (B2) electrochemical performance; (C) LiMnPO4/C:139 (C1) SEM image, (C2) electrochemical performance; (D) LiNi0.5Mn1.5O4·LiNi1/3Co1/3Mn1/3O2:141 (D1) SEM image, (D2) electrochemical performance. | ||
Owing to their good electron conductivity, carbon nanotubes, including single-walled carbon nanotubes and multi-walled carbon nanotubes (MWCNTs), have been extensively used as electron conducting networks in various cathode materials. Ensuring homogeneous dispersion and good contact between CNTs and cathode materials are the key issues. A durable high-rate capability was achieved when LiNi0.4Mn0.4Co0.2O2 was mixed with single walled-carbon nanotubes, as displayed in Fig. 11(B–B2).137 The rate properties of Li1.2Co0.13Ni0.13Mn0.54O2/MWCNTs have been enhanced by a novel surface double phase network modification produced via flexible electrostatic heterocoagulation and thermal treatment.138
3D macroporous LiMnPO4–C flakes consisting of nanoparticles (∼6 nm) in a carbon matrix have been reported to deliver discharge capacities of 162 mA h g−1 at 0.1C and 110 mA h g−1 at 10C,139 as depicted in Fig. 11(C–C2). Similarly, a Li2MnSiO4/C nanocomposite with hierarchical and ordered macroporosity achieved a high reversible discharge capacity of 200 mA h g−1 at 0.1C (16 mA g−1) at 1.5–4.8 V at 45 °C. The high reversible capacity and good cycling stability came from the unique bimodal macro/mesoporous structure of the material, which facilitated electrical and ionic transport, fast electrolyte permeation within the cathode composite, and the suppression of volume change during cycling.140
Intergrown nanorod-shaped composites of spinel LiNi0.5Mn1.5O4 and layered LiNi1/3Co1/3Mn1/3O2 with a diameter of around 100 nm and length of 1–2 mm have been successfully synthesized by a simple self-supporting template method. The resulting LiNi0.5Mn1.5O4–LiNi1/3Co1/3Mn1/3O2 composite delivered a high discharge capacity of 200 mA h g−1 and a high energy density of 815 W h kg−1, as shown in Fig. 11(D–D2).141 The Li[Li0.2Mn0.54Ni0.13Co0.13]O2–5 wt% MoO3 composite prepared by our group exhibited a good cycling stability with a discharge capacity of 242.5 mA h g−1 after 50 cycles, and effectively decreased the irreversible first cycle capacity loss of the Li-rich cathode.142
5. Advantages and disadvantages of nanomaterials
Generally, phase purity and particle distribution and size significantly influence the electronic and ionic transport and then the electrochemical performance of cathode materials. Fig. 12 shows the differences in electronic and ionic transport within typical nanotechnologies applied in cathodes for LIBs. The several fascinating advantages of these nanotechnologies are accompanied by some challenges, which are derived from the nanostructure, and found to be associated with the enhanced electrochemical performance.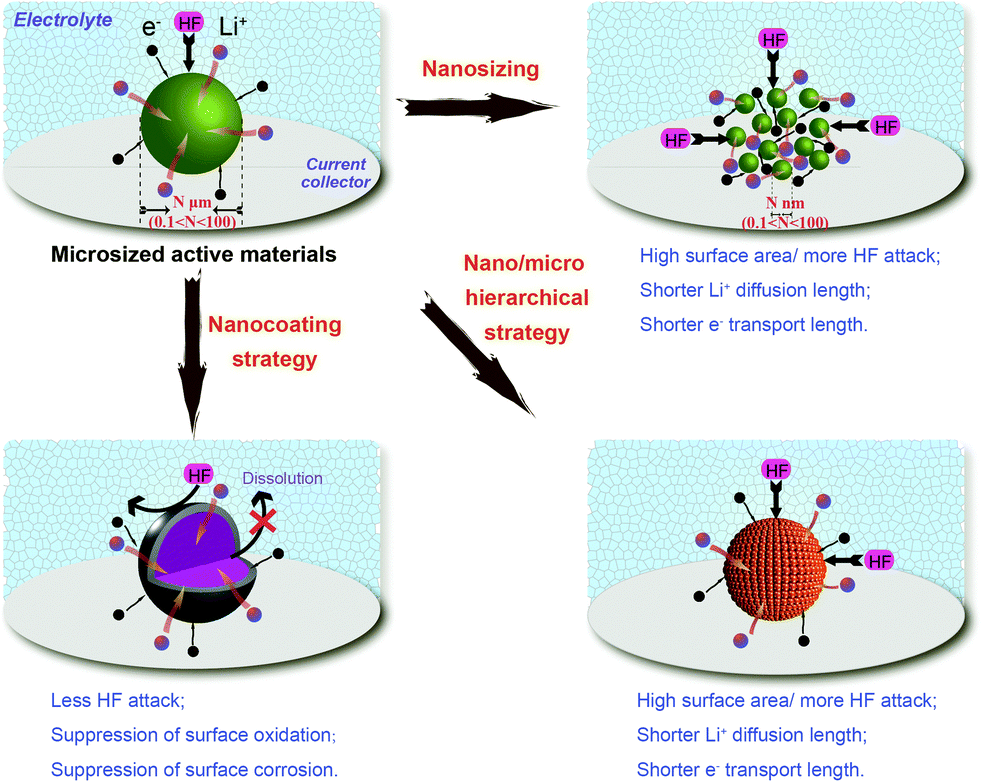 | ||
| Fig. 12 Impacts of typical nanotechnologies applied in cathodes for LIBs on electronic and ionic transport. | ||
Firstly, a high surface area can enrich the cathode with electrochemically active sites and increase the contact area between the active material and electrolyte. The increased contact area reduces the specific current density of the nano-active material and may also lead to improved charge/discharge rate capability, as listed in Table 1.59,70,143–159 However, when the nanomaterials are stored in air, their stability decreases with particle size owing to the large exposed area and they suffer significant surface oxidation, such as that seen for olivine LiFePO4.160 Undesirable surface side reactions induced by the highly reactive surface of nanomaterials are significantly intensified, resulting in a low Coulombic efficiency and poor cycle life. Meanwhile, it is difficult to disperse or mix nanoparticles with acetylene black and binders to prepare electrodes. Therefore, the increased contact resistance to some extent explains the performance fading observed in some cases. The volumetric capacities of nanomaterials are generally low, owing to their large surface area and porous structure, as shown in Fig. 13. A low thermodynamic stability can be caused by the presence of residual species such as organic surfactants on the surface of the nanomaterials, resulting in serious capacity fading and further serious safety problems at high temperature. The use of hierarchical nano/microstructures with the merits of both micromaterials and nanomaterials has been demonstrated to be an effective strategy for addressing these issues.
| Cathode materials | Particle size range | Average particle size (thickness)/nm | Surface area/m2 g−1 | Discharge capacity/mA h g−1 | Ref. |
|---|---|---|---|---|---|
| LiCoO2 | 4–8 nm | 6 | — | 68(1C) | 143 |
| 8–12 nm | 11 | — | 84(1C) | 143 | |
| 12–18 nm | 17 | — | 122(1C) | 143 | |
| 50 nm (200 °C) | — | 14 | 103(0.1C)/20(7C) | 144 | |
| 100 nm (500 °C) | — | 9 | 122(0.1C)/25(7C) | 144 | |
| 300 nm (700 °C) | — | 5 | 175(0.1C)/87(7C) | 144 | |
| 1000 nm (900 °C) | — | 3 | 182(0.1C)/18(7C) | 144 | |
| Li[Ni0.5Mn0.5]O2 | 3 μm | — | 0.778 | 148(0.14C)/48(10C) | 145 |
| 10 μm | — | 0.393 | 148(0.14C)/20(10C) | 145 | |
| LiCo1/3Mn1/3Ni1/3O2 | 56 nm (700 °C) | — | — | 174(0.1C)/98(30th) | 146 |
| 101 nm (800 °C) | — | — | 168(0.1C)/120(30th) | 146 | |
| LiFePO4 | 20–40 nm | 30 | — | 163 (1C) | 147 |
| 20–40 nm | 36 | — | 168 (0.6C) | 148 | |
| 60–100 nm | 70 | — | 145 (C/9) | 149 | |
| 100–200 nm | 140 | — | 158 (1C) | 150 | |
| 100–300 nm | 150 | — | 130 (1C) | 151 | |
| 200–400 nm | 300 | — | 120 (1C) | 152 | |
| ∼500 nm | 500 | — | 115(1C) | 153 | |
| 450–550 nm | 500 | — | 72 (1C) | 154 | |
| 600–1000 nm | 800 | — | 80 (1C) | 155 | |
| C–LiMnPO4 | 140 nm | — | — | 134(0.1C) | 156 |
| 160 nm | — | — | ∼127(0.1C) | 156 | |
| 200 nm | — | — | ∼100(0.1C) | 156 | |
| 270 nm | — | — | ∼92(0.1C) | 156 | |
| 830 nm | — | — | ∼67(0.1C) | 156 | |
| LiMn0.85Fe0.15PO4 | Nanosized | — | 53.5 | ∼163(0.05C)/108(3C) | 157 |
| 7 μm | — | 19.2 | ∼142(0.05C)/100(3C) | 157 | |
| LiNi0.5Mn1.5O4 | 16–28 nm | — | — | 100(1C) | 59 |
| 2–3 μm | — | — | 80(1C) | 59 | |
| 125 nm | — | — | ∼110(0.5C)/71(20C) | 158 | |
| 250 nm | — | — | ∼110(0.5C)/58(20C) | 158 | |
| 800 nm | — | — | ∼110(0.5C)/40(20C) | 158 | |
| V2O5 (nanosheet) | — | 3.5 | — | 42(10C) | 159 |
| — | 10 | — | 82(10C) | 159 | |
| — | 33 | — | 43(10C) | 159 | |
| — | 86 | — | 51(10C) | 159 | |
| — | 2.1–3.8 | 147.5 | 274(0.2C)/117(50C) | 70 | |
| — | 60–80 | 28 | 104(5 A g−1) | 73 | |
| LiFePO4/C | — | 4.3 | 128.5 | 163(0.2C)/139(10C) | 71 |
| LiMnPO4/C | — | 3.7 | 142.9 | 147(0.2C)/95(10C) | 71 |
| LiCoPO4/C (nanosheet) | — | 4.6 | 107.6 | 136(0.2C)/91(10C) | 71 |
| LiFePO4 (nanoplate) | — | 15 | — | 155(1C)/70(61C) | 74 |
| — | 30 | — | 160(0.1C)/156(5C) | 72 | |
| LiMnPO4 (nanoplate) | — | 50 | 37.3 | 168(0.02C)/117(1C) | 10 |
| LiCo1/3Mn1/3Ni1/3O2 (nanobrick) | — | 140 | — | 179.2(1C)/130(15C) | 75 |
| 0.2Li2MnO3·0.8LiNi0.5Mn0.5O2 (nanorod) | 50 nm × 5 μm | — | 7.3 | 275(0.2C)/192(5C) | 68 |
| LiNi0.5Mn1.5O4 (nanorod) | (100–400) nm × 10 μm | — | 6.9 | 140(1C)/109(20C) | 69 |
| LiMn2O4 (nanorod) | 250 nm × (3–6) μm | — | — | 105(10C)/90(500th) | 9 |
| LiMn2O4 (nanotube) | 600 nm × (1–4) μm | 200 | — | ∼96(5C)/67(1500th) | 34 |
| LiMn2O4 (nanowire) | (50–100) nm × 10 μm | — | — | 118(0.1 A g−1)/88(20 A g−1) | 63 |
| FeF3 (nanowire) | (30–70) nm × (2–15) μm | — | — | 543(1/14.2C)/223(50th) | 48 |
| LixNi0.33Mn0.67O2 (nanowire) | 20 nm × 1 μm | — | 27 | 300(0.1C) | 64 |
| Li1.15Ni0.25Mn0.6O2 (nanowire) | 30 nm × 1 μm | — | 65 | 311(4C)/256(7C) | 66 |
| Li1.06Co0.33Mn0.49O2 (nanowire) | (50–100) nm × 3 μm | — | 50 | 230(1C)/220(15C) | 65 |
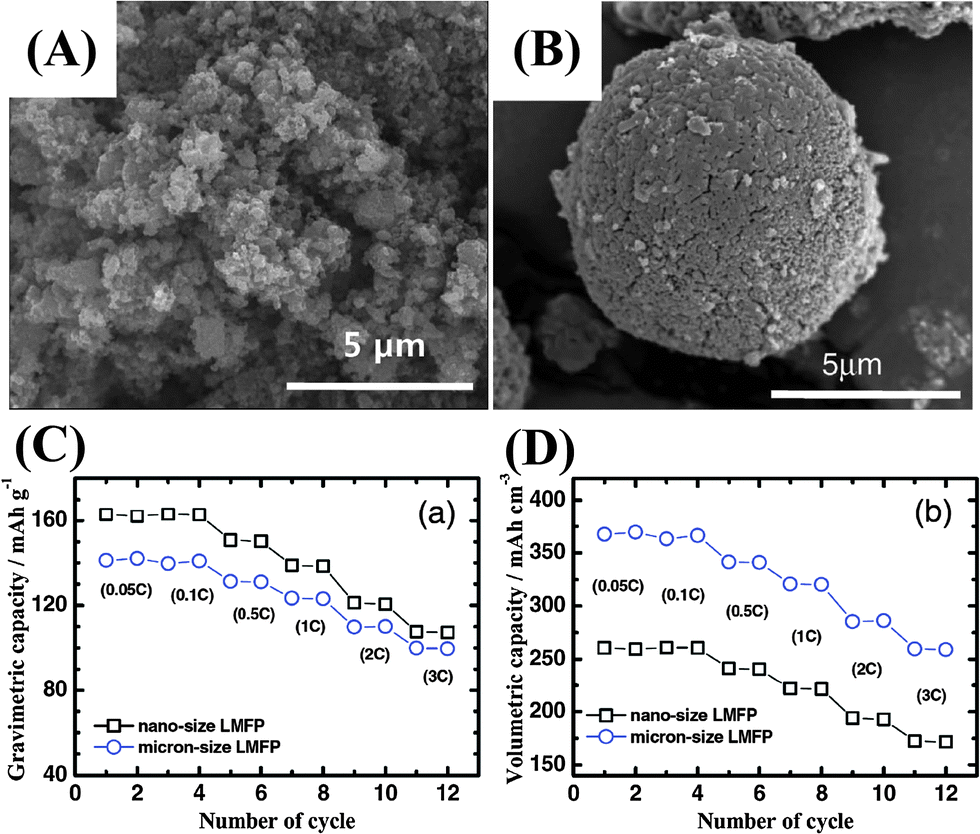 | ||
| Fig. 13 (A and B) SEM images of nano-sized and micro-sized LiMn0.85Fe0.15PO4 cathode material. Comparison of (C) gravimetric capacity and (D) volumetric capacity of nano-LiMn0.85Fe0.15PO4 and micro-LiMn0.85Fe0.15PO4.157 | ||
Secondly, when the size is decreased from micro to nano, the electrochemical kinetics of lithium storage might be significantly enhanced. The shorter Li+ diffusion pathways and faster Li+ diffusion kinetics arising from the reduction of particle size can lead to remarkable improvements in the rate capability, as illustrated in Fig. 12 and listed in Table 1. Electronic transport can also be promoted by combining nanosized particles with electronically conductive nanocoating shells.161 The chemical potentials of lithium ions and electrons may be changed, and thus the electrode potential as well as the work voltage of LIBs can be modified.162 New electrode reactions may occur in nanomaterials that are not seen in micrometer materials, such as the conversion mechanism. Additionally, when going to the nanoscale, materials inactive for Li storage have been observed to become active, such as LiFeO2.
Thirdly, nanoscale particles can more easily accommodate strains and structural changes during the cycling process. Therefore, structural transitions can be limited and the integrity of the electrode materials can be preserved. However, it is more difficult and complicated to synthesize nanomaterials and control their size and dimensions than bulk materials. Many of the synthesis routes of nanomaterials, for example, hydrothermal reactions and template methods, are complex and time consuming. Therefore, developing facile synthesis routes has become inevitably essential for the large-scale industrial production of nanomaterials.
6. Further directions: materials genome initiative
The Materials Genome Initiative (MGI), launched in 2011 in the United States, is a new and large-scale effort to develop a collaboration between materials scientists and computer scientists to accelerate advanced materials discovery and deployment, and specifically, to deploy proven computational methodologies to predict, screen, and optimize materials at an unparalleled scale and rate.163,164 The Materials Project, the core program of the MGI, combines high-throughput computation, web-based dissemination, and open-source analysis tools to compute the properties of all known materials.164,165 In the field of battery materials, high-throughput computation has already been applied to seek new materials with superior properties.166–168 Hautier et al. investigated a high-throughput ab initio analysis of phosphates as cathode materials.169 Various properties, including capacity, voltage, specific energy, energy density, and thermal stability were computationally evaluated for thousands of compounds. The computed results have been used to recommend specific chemistries within the phosphate class. Based on high-throughput computation, Ceder calculated the theoretical capacity of thousands of oxides, phosphates, borates, silicates, and sulfates at different voltages.170 Moreover, the oxygen chemical potential under which the charged state decomposes for oxides and phosphates was calculated for different voltages as well, as shown in Fig. 14(A and B). The data provide a first screening of materials with reasonable energy density and safety properties.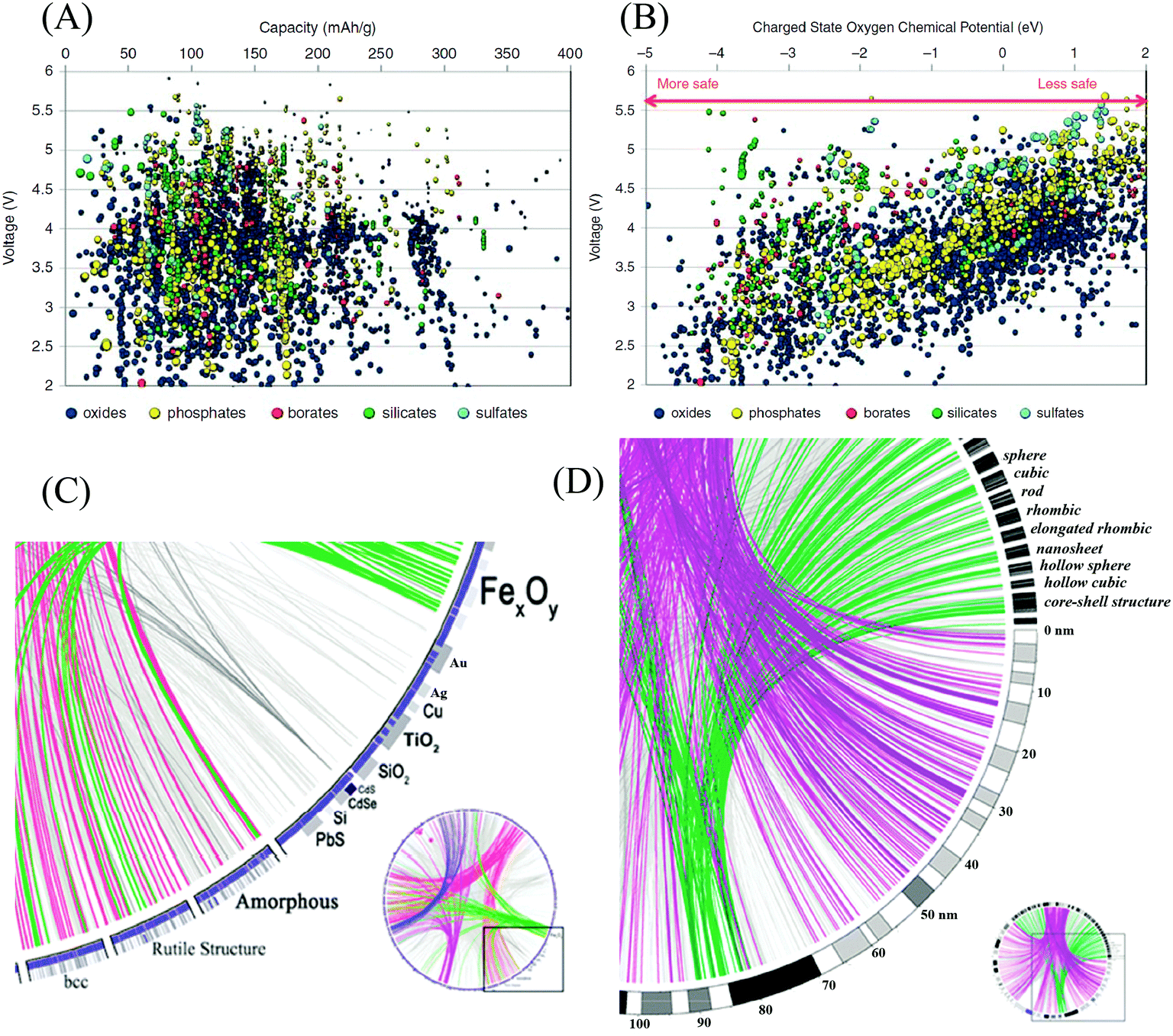 | ||
| Fig. 14 (A) Calculated lithium insertion voltage versus theoretical capacity for several thousand compounds.170 (B) Oxygen chemical potentials at which the charged state of thousands of cathode materials decompose versus their Li intercalation voltage.170 (C) Circos diagram of nanomaterials genome composition–structure relations.171 (D) Circos diagram of nanomaterials genome size–shape relations.171 | ||
Recently, inspired by the MGI discussed above and the Nanoinformatics Roadmap 2020 announced in 2010, Ozin et al. proposed the development of a Nanomaterials Genome Initiative (NMGI), which aims to build a central database of nanomaterials to serve the nanoscience and materials science communities with an opportunity to speed up the development continuum of nanomaterials.171 Like a human gene, the ‘gene’ of a nanomaterial is identified by descriptors including elemental composition, structure, size, shape, surface, degree of imperfection, self-assembly, and how these are connected to function and utility. The first step in the realization of this idea is the building of a database with a useful nomenclature system, in which each particular type of nanomaterial is assigned and described according to a common standard. Next, the data visualizing tool Circos was proposed by the authors as a way of presenting the nanomaterials genome. For example, the composition–structure pairs of a group of nanoparticles can be presented as shown in Fig. 14(C).171 Similarly, a Circos diagram for size–shape combinations is plotted in Fig. 14(D).171 Combining the database of nanomaterials and a multifunctional platform, the NMGI will facilitate and inspire researchers to investigate the best candidate nanomaterials for a vast number of research fields.
MGI combined with experiment design can offer a valuable route of exploring novel morphologies of particles and surface structures. The detailed analysis of MGI is ideally suited for assisting many researches, especially morphology control and structure design. A more clear understanding of the relationships between a nanomaterial and its microscopic states can simulate imaginative ideas and approaches to achieve improvements in the electrochemical properties. In addition, new materials with better compositional and structural harmony may also be discovered to be suitable for cathodes or anodes in LIBs. Future studies of the MGI on nanomaterials will play an increasing role in revealing the development rules and directions.
7. Summary and outlook
Various cathode materials, such as layered, spinel, and polyanion-type, with different crystal structures and features, have been extensively applied in LIBs. The prosperous field of nanostructured materials has inspired the exploration and development of many new electroactive materials for LIBs. As many properties highly depend on shape and size, morphological and structural control of nanomaterials are demonstrated to affect their performance by reducing transport path lengths. The influences of morphology and the synthetic route on the electrochemical performance of such materials have been widely studied. The present research efforts and progress in nanostructured cathode materials demonstrate that there are still great opportunities and huge challenges in the LIBs field. The strategies reported herein have not only realized the most promising cathodes in terms of capacity and cycling performance but also provide a convenient platform for future research. At the same time, these nanotechnologies could also be applied in the electrode materials of other energy storage applications, such as supercapacitors, lithium–sulfur batteries, and lithium–air batteries. Further investigation and development are essential to fulfill the future application of LIBs in electric vehicles.Generally, nanostructured materials are favorable for obtaining high energy and power density by means of shortening the ionic diffusion length and providing a large surface area for electrode reactions. However, the low volumetric energy density and the high porosity of nanostructured materials restrict its energy storage applications where space is a prime concern. Other than the development of novel advanced cathode materials, another suggestion is to design and exploit the combination of composites with hierarchical core–shell strategies and nanocoating or doping. Possible fruitful directions of future nanostructured cathode research should incorporate large-scale preparation, and high tap and volumetric energy density.
In conclusion, it is obvious that the main advances of LIBs for electronics, grid storage, and electric vehicles always depend on designing novel micro/nanostructures and exploring new cathode materials, which are based on the fundamental understanding of the microscale and nanoscale.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21373028), Major Achievements Transformation Project for Central University in Beijing, National Key Program for Basic Research of China (2015CB251106), Beijing Science and Technology Project (D151100003015001) and the Nature Science Foundation of Hebei Province (B2016210071).Notes and references
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- Y.-K. Sun, S.-T. Myung, B.-C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320–324 CrossRef CAS PubMed.
- K. G. Gallagher, S.-H. Kang, S. U. Park and S. Y. Han, J. Power Sources, 2011, 196, 9702–9707 CrossRef CAS.
- N. Liu, W. Li, M. Pasta and Y. Cui, Front. Phys., 2014, 9, 323–350 CrossRef.
- R. Pitchai, V. Thavasi, S. G. Mhaisalkar and S. Ramakrishna, J. Mater. Chem., 2011, 21, 11040–11051 RSC.
- J. C. Arrebola, A. Caballero, M. Cruz, L. Hernan, J. Morales and E. R. Castellon, Adv. Funct. Mater., 2006, 16, 1904–1912 CrossRef CAS.
- V. G. Kumar, J. S. Gnanaraj, S. Ben-David, D. M. Pickup, E. R. H. Van-Eck, A. Gedanken and D. Aurbach, Chem. Mater., 2003, 15, 4211–4216 CrossRef CAS.
- N. Twu, X. Li, A. Urban, M. Balasubramanian, J. Lee, L. Liu and G. Ceder, Nano Lett., 2015, 15, 596–602 CrossRef CAS PubMed.
- F. Cheng, H. Wang, Z. Zhu, Y. Wang, T. Zhang, Z. Tao and J. Chen, Energy Environ. Sci., 2011, 4, 3668–3675 CAS.
- D. Choi, D. Wang, I.-T. Bae, J. Xiao, Z. Nie, W. Wang, V. V. Viswanathan, Y. J. Lee, J.-G. Zhang, G. L. Graff, Z. Yang and J. Liu, Nano Lett., 2010, 10, 2799–2805 CrossRef CAS PubMed.
- X. Jie, Z. Jianming, L. Xiaolin, S. Yuyan and Z. Ji-Guang, Nanotechnology, 2013, 24, 424004 CrossRef PubMed.
- X. Zhang, R.-S. Kuehnel, H. Hu, D. Eder and A. Balducci, Nano Energy, 2015, 12, 207–214 CrossRef CAS.
- L. Su, Y. Jing and Z. Zhou, Nanoscale, 2011, 3, 3967–3983 RSC.
- L. Zhou, D. Zhao and X. Lou, Angew. Chem., Int. Ed., 2012, 51, 239–241 CrossRef CAS PubMed.
- K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- K. M. Abraham, D. M. Pasquariello and D. A. Schwartz, J. Power Sources, 1989, 26, 247–255 CrossRef CAS.
- K. Brandt and F. C. Laman, J. Power Sources, 1989, 25, 265–276 CrossRef CAS.
- P. Dan, E. Mengeritski, Y. Geronov, D. Aurbach and I. Weisman, J. Power Sources, 1995, 54, 143–145 CrossRef CAS.
- G. Jeong, Y.-U. Kim, H. Kim, Y.-J. Kim and H.-J. Sohn, Energy Environ. Sci., 2011, 4, 1986 CAS.
- Y. Makimura and T. Ohzuku, J. Power Sources, 2003, 119–121, 156–160 CrossRef CAS.
- M. S. Islam, R. A. Davies and J. D. Gale, Chem. Mater., 2003, 15, 4280–4286 CrossRef CAS.
- Y. Koyama, Y. Makimura, I. Tanaka, H. Adachi and T. Ohzuku, J. Electrochem. Soc., 2004, 151, A1499–A1506 CrossRef CAS.
- Z. Li, N. A. Chernova, M. Roppolo, S. Upreti, C. Petersburg, F. M. Alamgir and M. S. Whittingham, J. Electrochem. Soc., 2011, 158, A516–A522 CrossRef CAS.
- M. Wang, F. Wu, Y. Su and S. Chen, Sci. China, Ser. E: Technol. Sci., 2008, 52, 2737–2741 CrossRef.
- F. Wu, M. Wang, Y. Su, L. Bao and S. Chen, Electrochim. Acta, 2009, 54, 6803–6807 CrossRef CAS.
- F. Wu, M. Wang, Y. Su and S. Chen, J. Power Sources, 2009, 189, 743–747 CrossRef CAS.
- F. Wu, M. Wang, Y. Su, S. Chen and B. Xu, J. Power Sources, 2009, 191, 628–632 CrossRef CAS.
- M.-H. Kim, H.-S. Shin, D. Shin and Y.-K. Sun, J. Power Sources, 2006, 159, 1328–1333 CrossRef CAS.
- N. Yabuuchi, K. Yoshii, S.-T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS PubMed.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- Z. H. Lu, D. D. MacNeil and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A191–A194 CrossRef CAS.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815–A822 CrossRef CAS.
- C. S. Johnson, J. S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, Electrochem. Commun., 2004, 6, 1085–1091 CrossRef CAS.
- Y.-L. Ding, J. Xie, G.-S. Cao, T.-J. Zhu, H.-M. Yu and X.-B. Zhao, Adv. Funct. Mater., 2011, 21, 348–355 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- C. Wu, F. Feng and Y. Xie, Chem. Soc. Rev., 2013, 42, 5157–5183 RSC.
- S. Lee, Y. Cho, H.-K. Song, K. T. Lee and J. Cho, Angew. Chem., Int. Ed., 2012, 51, 8748–8752 CrossRef CAS PubMed.
- T. Ohzuku, M. Kitagawa and T. Hirai, J. Electrochem. Soc., 1990, 137, 769–775 CrossRef CAS.
- X. Zhang, F. Cheng, K. Zhang, Y. Liang, S. Yang, J. Liang and J. Chen, RSC Adv., 2012, 2, 5669–5675 RSC.
- J. H. Kim, S. T. Myung, C. S. Yoon, S. G. Kang and Y. K. Sun, Chem. Mater., 2004, 16, 906–914 CrossRef CAS.
- N. Amdouni, K. Zaghib, F. Gendron, A. Mauger and C. M. Julien, Ionics, 2006, 12, 117–126 CrossRef CAS.
- J. Cabana, M. Casas-Cabanas, F. O. Omenya, N. A. Chernova, D. Zeng, M. S. Whittingham and C. P. Grey, Chem. Mater., 2012, 24, 2952–2964 CrossRef CAS PubMed.
- R. Malik, A. Abdellahi and G. Ceder, J. Electrochem. Soc., 2013, 160, A3179–3197 CrossRef CAS.
- K. Zhang, Z. Hu, H. Gao, H. Feng, F. Cheng, Z. Tao and J. Chen, Sci. Adv. Mater., 2013, 5, 1676–1685 CrossRef CAS.
- Z. Gong and Y. Yang, Energy Environ. Sci., 2011, 4, 3223–3242 CAS.
- C. A. J. Fisher, N. Kuganathan and M. S. Islam, J. Mater. Chem. A, 2013, 1, 4207–4214 CAS.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Mater. Sci. Eng., R, 2012, 73, 51–65 CrossRef CAS.
- L. Li, F. Meng and S. Jin, Nano Lett., 2012, 12, 6030–6037 CrossRef CAS PubMed.
- W. T. Gu, A. Magasinski, B. Zdyrko and G. Yushin, Adv. Energy Mater., 2015, 5, 1401148 Search PubMed.
- X. Fan, C. Luo, J. Lamb, Y. Zhu, K. Xu and C. Wang, Nano Lett., 2015, 15, 7650–7656 CrossRef CAS PubMed.
- C. Luo, Y. J. Zhu, O. Borodin, T. Gao, X. L. Fan, Y. H. Xu, K. Xu and C. S. Wang, Adv. Funct. Mater., 2016, 26, 745–752 CrossRef CAS.
- H. K. Liu, G. X. Wang, Z. P. Guo, J. Z. Wang and K. Konstantinov, J. Nanosci. Nanotechnol., 2006, 6, 1–15 CrossRef CAS PubMed.
- S. H. Ye, J. Y. Lv, X. P. Gao, F. Wu and D. Y. Song, Electrochim. Acta, 2004, 49, 1623–1628 CrossRef CAS.
- M. Gaberscek, R. Dominko and J. Jamnik, Electrochem. Commun., 2007, 9, 2778–2783 CrossRef CAS.
- R. Malik, D. Burch, M. Bazant and G. Ceder, Nano Lett., 2010, 10, 4123–4127 CrossRef CAS PubMed.
- P. Gibot, M. Casas-Cabanas, L. Laffont, S. Levasseur, P. Carlach, S. Hamelet, J.-M. Tarascon and C. Masquelier, Nat. Mater., 2008, 7, 741–747 CrossRef CAS PubMed.
- K. M. Shaju and P. G. Bruce, Dalton Trans., 2008, 5471–5475 RSC.
- J. Yang, X. Han, X. Zhang, F. Cheng and J. Chen, Nano Res., 2013, 6, 679–687 CrossRef CAS.
- Y. Talyosef, B. Markovsky, R. Lavi, G. Salitra, D. Aurbach, D. Kovacheva, M. Gorova, E. Zhecheva and R. Stoyanova, J. Electrochem. Soc., 2007, 154, A682–A691 CrossRef CAS.
- J. Yang, L. Hu, J. Zheng, D. He, L. Tian, S. Mu and F. Pan, J. Mater. Chem. A, 2015, 3, 9601–9608 CAS.
- H. Song, Y. Liu, C. Zhang, C. Liu and G. Cao, J. Mater. Chem. A, 2015, 3, 3547–3558 CAS.
- S. Cavaliere, S. Subianto, I. Savych, D. J. Jones and J. Roziere, Energy Environ. Sci., 2011, 4, 4761–4785 CAS.
- E. Hosono, T. Kudo, I. Honma, H. Matsuda and H. Zhou, Nano Lett., 2009, 9, 1045–1051 CrossRef CAS PubMed.
- D. H. Park, S. T. Lim, S. J. Hwang, C. S. Yoon, Y. K. Sun and J. H. Choy, Adv. Mater., 2005, 17, 2834–2837 CrossRef CAS.
- Y. Lee, M. G. Kim and J. Cho, Nano Lett., 2008, 8, 957–961 CrossRef CAS PubMed.
- M. G. Kim, M. Jo, Y. S. Hong and J. Cho, Chem. Commun., 2009, 218–220 RSC.
- W. Tang, Y. Hou, F. Wang, L. Liu, Y. Wu and K. Zhu, Nano Lett., 2013, 13, 2036–2040 CrossRef CAS PubMed.
- J. Yang, F. Cheng, X. Zhang, H. Gao, Z. Tao and J. Chen, J. Mater. Chem. A, 2014, 2, 1636–1640 CAS.
- X. Zhang, F. Cheng, J. Yang and J. Chen, Nano Lett., 2013, 13, 2822–2825 CrossRef CAS PubMed.
- X. Rui, Z. Lu, H. Yu, D. Yang, H. H. Hng, T. M. Lim and Q. Yan, Nanoscale, 2013, 5, 556–560 RSC.
- X. Rui, X. Zhao, Z. Lu, H. Tan, D. Sim, H. H. Hng, R. Yazami, T. M. Lim and Q. Yan, ACS Nano, 2013, 7, 5637–5646 CrossRef CAS PubMed.
- L. Wang, X. He, W. Sun, J. Wang, Y. Li and S. Fan, Nano Lett., 2012, 12, 5632–5636 CrossRef CAS PubMed.
- Y. Li, J. Yao, E. Uchaker, J. Yang, Y. Huang, M. Zhang and G. Cao, Adv. Energy Mater., 2013, 3, 1171–1175 CrossRef CAS.
- A. Paolella, G. Bertoni, S. Marras, E. Dilena, M. Colombo, M. Prato, A. Riedinger, M. Povia, A. Ansaldo, K. Zaghib, L. Manna and C. George, Nano Lett., 2014, 14, 6828–6835 CrossRef CAS PubMed.
- F. Fu, G.-L. Xu, Q. Wang, Y.-P. Deng, X. Li, J.-T. Li, L. Huang and S.-G. Sun, J. Mater. Chem. A, 2013, 1, 3860–3864 CAS.
- Y. Wu, C. Cao, Y. Zhu, J. Li and L. Wang, J. Mater. Chem. A, 2015, 3, 15523–15528 CAS.
- W. Li, H. Zhang, Y. Mu, L. Liu and Y. Wang, J. Mater. Chem. A, 2015, 3, 15661–15667 CAS.
- R. Yi, F. Dai, M. L. Gordin, S. Chen and D. Wang, Adv. Energy Mater., 2013, 3, 295–300 CrossRef CAS.
- J. Xiao, L. Wan, S. Yang, F. Xiao and S. Wang, Nano Lett., 2014, 14, 831–838 CrossRef CAS PubMed.
- L. Xu, P. Hou, Y. Zhang, H. Zhang, D. Song, X. Shi, X. Wang and L. Zhang, J. Mater. Chem. A, 2015, 3, 21219–21226 CAS.
- L. Chen, Y. Su, S. Chen, N. Li, L. Bao, W. Li, Z. Wang, M. Wang and F. Wu, Adv. Mater., 2014, 26, 6756–6760 CrossRef CAS PubMed.
- L. Zhang, N. Li, B. Wu, H. Xu, L. Wang, X. Q. Yang and F. Wu, Nano Lett., 2015, 15, 656–661 CrossRef CAS PubMed.
- P. Oh, S. Myeong, W. Cho, M.-J. Lee, M. Ko, H. Y. Jeong and J. Cho, Nano Lett., 2014, 14, 5965–5972 CrossRef CAS PubMed.
- Y.-d. Zhang, Y. Li, X.-q. Niu, D.-h. Wang, D. Zhou, X.-l. Wang, C.-d. Gu and J.-p. Tu, J. Mater. Chem. A, 2015, 3, 14291–14297 CAS.
- Q. Yue, H. Jiang, Y. Hu, G. Jia and C. Li, Chem. Commun., 2014, 50, 13362–13365 RSC.
- Y.-L. Ding, X.-B. Zhao, J. Xie, G.-S. Cao, T.-J. Zhu, H.-M. Yu and C.-Y. Sun, J. Mater. Chem., 2011, 21, 9475–9479 RSC.
- H. Jiang, Y. Fu, Y. Hu, C. Yan, L. Zhang, P. S. Lee and C. Li, Small, 2014, 10, 1096–1100 CrossRef CAS PubMed.
- Y. Jiang, Z. Yang, W. Luo, X. Hu and Y. Huang, Phys. Chem. Chem. Phys., 2013, 15, 2954–2960 RSC.
- P. Remith and N. Kalaiselvi, Nanoscale, 2014, 6, 14724–14732 RSC.
- F. Wu, Z. Wang, Y. Su, Y. Guan, Y. Jin, N. Yan, J. Tian, L. Bao and S. Chen, J. Power Sources, 2014, 267, 337–346 CrossRef CAS.
- M. Cargnello, J. J. Delgado Jaen, J. C. Hernandez Garrido, K. Bakhmutsky, T. Montini, J. J. Calvino Gamez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- O. Chen, J. Zhao, V. P. Chauhan, J. Cui, C. Wong, D. K. Harris, H. Wei, H.-S. Han, D. Fukumura, R. K. Jain and M. G. Bawendi, Nat. Mater., 2013, 12, 445–451 CrossRef CAS PubMed.
- J. Li, R. Shunmugasundaram, R. Doig and J. R. Dahn, Chem. Mater., 2016, 28, 162–171 CrossRef CAS.
- J. Guo, Q. Wang, C. Qi, J. Jin, Y. Zhu and Z. Wen, Chem. Commun., 2016, 52, 5613–5616 RSC.
- L. Mai, S. Li, Y. Dong, Y. Zhao, Y. Luo and H. Xu, Nanoscale, 2013, 5, 4864–4869 RSC.
- Y.-K. Sun, S.-T. Myung, B.-C. Park and H. Yashiro, J. Electrochem. Soc., 2008, 155, A705–A710 CrossRef CAS.
- J. Liu and A. Manthiram, J. Mater. Chem., 2010, 20, 3961–3967 RSC.
- Q. Y. Wang, J. Liu, A. V. Murugan and A. Manthiram, J. Mater. Chem., 2009, 19, 4965–4972 RSC.
- T. Zhao, S. Chen, R. Chen, L. Li, X. Zhang, M. Xie and F. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 21711–21720 CAS.
- F. Wu, Q. Zhu, R. Chen, N. Chen, Y. Chen and L. Li, Nano Energy, 2015, 13, 546–553 CrossRef CAS.
- F. Wu, X. Zhang, T. Zhao, L. Li, M. Xie and R. Chen, J. Mater. Chem. A, 2015, 3, 9528–9537 CAS.
- Y. S. Jung, A. S. Cavanagh, L. A. Riley, S. H. Kang, A. C. Dillon, M. D. Groner, S. M. George and S. H. Lee, Adv. Mater., 2010, 22, 2172–2176 CrossRef CAS PubMed.
- Y. He, X. Yu, Y. Wang, H. Li and X. Huang, Adv. Mater., 2011, 23, 4938–4941 CrossRef CAS PubMed.
- X. Han, Y. Liu, Z. Jia, Y.-C. Chen, J. Wan, N. Weadock, K. J. Gaskell, T. Li and L. Hu, Nano Lett., 2014, 14, 139–147 CrossRef CAS PubMed.
- S. Sim, P. Oh, S. Park and J. Cho, Adv. Mater., 2013, 25, 4498–4503 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- X. Zhang, I. Belharouak, L. Li, Y. Lei, J. W. Elam, A. Nie, X. Chen, R. S. Yassar and R. L. Axelbaum, Adv. Energy Mater., 2013, 3, 1299–1307 CrossRef CAS.
- W. Zhang, Z.-X. Chi, W.-X. Mao, R.-W. Lv, A.-M. Cao and L.-J. Wan, Angew. Chem., Int. Ed., 2014, 53, 12776–12780 CrossRef CAS PubMed.
- X. Yang, R. Yu, L. Ge, D. Wang, Q. Zhao, X. Wang, Y. Bai, H. Yuan and H. Shu, J. Mater. Chem. A, 2014, 2, 8362–8368 CAS.
- J. Lu, Q. Peng, W. Wang, C. Nan, L. Li and Y. Li, J. Am. Chem. Soc., 2013, 135, 1649–1652 CrossRef CAS PubMed.
- D. Chao, X. Xia, J. Liu, Z. Fan, C. F. Ng, J. Lin, H. Zhang, Z. X. Shen and H. J. Fan, Adv. Mater., 2014, 26, 5794–5800 CrossRef CAS PubMed.
- C. Yang, Q. Zhang, W. Ding, J. Zang, M. Lei, M. Zheng and Q. Dong, J. Mater. Chem. A, 2015, 3, 7554–7559 CAS.
- F. Wang, Z. Chang, X. Wang, Y. Wang, B. Chen, Y. Zhu and Y. Wu, J. Mater. Chem. A, 2015, 3, 4840–4845 CAS.
- S.-J. Yoon, S.-T. Myung, H.-J. Noh, J. Lu, K. Amine and Y.-K. Sun, ChemSusChem, 2014, 7, 3295–3303 CrossRef CAS PubMed.
- H. J. Noh, J. W. Ju and Y. K. Sun, ChemSusChem, 2014, 7, 245–252 CrossRef CAS PubMed.
- S.-M. Oh, S.-T. Myung, J. B. Park, B. Scrosati, K. Amine and Y.-K. Sun, Angew. Chem., Int. Ed., 2012, 51, 1853–1856 CrossRef CAS PubMed.
- F. Wu, N. Li, Y. Su, H. Shou, L. Bao, W. Yang, L. Zhang, R. An and S. Chen, Adv. Mater., 2013, 25, 3722–3726 CrossRef CAS PubMed.
- F. Wu, N. Li, Y. Su, L. Zhang, L. Bao, J. Wang, L. Chen, Y. Zheng, L. Dai, J. Peng and S. Chen, Nano Lett., 2014, 14, 3550–3555 CrossRef CAS PubMed.
- T. Zhao, L. Li, R. Chen, H. Wu, X. Zhang, S. Chen, M. Xie, F. Wu, J. Lu and K. Amine, Nano Energy, 2015, 15, 164–176 CrossRef CAS.
- W. Fang, A. L. Hsu, Y. Song and J. Kong, Nanoscale, 2015, 7, 20335–20351 RSC.
- D. Higgins, P. Zamani, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 357–390 CAS.
- F. Fathollahi, M. Javanbakht, H. Omidvar and M. Ghaemi, J. Alloys Compd., 2015, 627, 146–152 CrossRef CAS.
- K. Chen, S. Song, F. Liu and D. Xue, Chem. Soc. Rev., 2015, 44, 6230–6257 RSC.
- A. R. Kamali, Green Chem., 2016, 18, 1952–1964 RSC.
- M. Liu, Y. Zhao, S. Gao, Y. Wang, Y. Duan, X. Han and Q. Dong, New J. Chem., 2015, 39, 1094–1100 RSC.
- Y. Zhao, Y. Wang, C. Ji, Z. Zhao and Z. Lv, RSC Adv., 2016, 6, 31762–31768 RSC.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664–2680 CrossRef CAS.
- G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS.
- H. Liu, P. Gao, J. Fang and G. Yang, Chem. Commun., 2011, 47, 9110–9112 RSC.
- R. Mo, Y. Du, N. Zhang, D. Rooney and K. Sun, Chem. Commun., 2013, 49, 9143–9145 RSC.
- J. W. Lee, S. Y. Lim, H. M. Jeong, T. H. Hwang, J. K. Kang and J. W. Choi, Energy Environ. Sci., 2012, 5, 9889–9894 CAS.
- C. Nethravathi, B. Viswanath, J. Michael and M. Rajamath, Carbon, 2012, 50, 4839–4846 CrossRef CAS.
- S. Yang, Y. Gong, Z. Liu, L. Zhan, D. P. Hashim, L. Ma, R. Vajtai and P. M. Ajayan, Nano Lett., 2013, 13, 1596–1601 CAS.
- H. Fei, Z. Peng, Y. Yang, L. Li, A.-R. O. Raji, E. L. G. Samuel and J. M. Tour, Chem. Commun., 2014, 50, 7117–7119 RSC.
- M. Pivko, M. Bele, E. Tchernychova, N. Z. Logar, R. Dominko and M. Gaberscek, Chem. Mater., 2012, 24, 1041–1047 CrossRef CAS.
- R. von Hagen, H. Lorrmann, K.-C. Möller and S. Mathur, Adv. Energy Mater., 2012, 2, 553–559 CrossRef CAS.
- C. Ban, Z. Li, Z. Wu, M. J. Kirkham, L. Chen, Y. S. Jung, E. A. Payzant, Y. Yan, M. S. Whittingham and A. C. Dillon, Adv. Energy Mater., 2011, 1, 58–62 CrossRef CAS.
- L. Guo, N. Zhao, J. Li, C. He, C. Shi and E. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 391–399 CAS.
- H. Yoo, M. Jo, B.-S. Jin, H.-S. Kim and J. Cho, Adv. Energy Mater., 2011, 1, 347–351 CrossRef CAS.
- G. He and A. Manthiram, Adv. Funct. Mater., 2014, 24, 5277–5283 CrossRef CAS.
- J. Yang, X. Zhang, X. Han, F. Cheng, Z. Tao and J. Chen, J. Mater. Chem. A, 2013, 1, 13742–13745 CAS.
- F. Wu, Z. Wang, Y. Su, N. Yan, L. Bao and S. Chen, J. Power Sources, 2014, 247, 20–25 CrossRef CAS.
- M. Okubo, E. Hosono, J. Kim, M. Enomoto, N. Kojima, T. Kudo, H. Zhou and I. Honma, J. Am. Chem. Soc., 2007, 129, 7444–7452 CrossRef CAS PubMed.
- M. Jo, Y.-S. Hong, J. Choo and J. Cho, J. Electrochem. Soc., 2009, 156, A430–A434 CrossRef CAS.
- K. S. Lee, S. T. Myung, J. S. Moon and Y. K. Sun, Electrochim. Acta, 2008, 53, 6033–6037 CrossRef CAS.
- J. H. Kim, J. H. Yi, Y. N. Ko and Y. C. Kang, Mater. Chem. Phys., 2012, 134, 254–259 CrossRef CAS.
- D.-H. Kim and J. Kim, Electrochem. Solid-State Lett., 2006, 9, A439–A442 CrossRef CAS.
- Y. Wang, Y. Wang, E. Hosono, K. Wang and H. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461–7465 CrossRef CAS PubMed.
- X.-L. Wu, L.-Y. Jiang, F.-F. Cao, Y.-G. Guo and L.-J. Wan, Adv. Mater., 2009, 21, 2710–2714 CrossRef CAS.
- C. Delacourt, P. Poizot, S. Levasseur and C. Masquelier, Electrochem. Solid-State Lett., 2006, 9, A352–A355 CrossRef CAS.
- H. C. Shin, W. I. Cho and H. Jang, Electrochim. Acta, 2006, 52, 1472–1476 CrossRef CAS.
- D. Y. W. Yu, C. Fietzek, W. Weydanz, K. Donoue, T. Inoue, H. Kurokawa and S. Fujitani, J. Electrochem. Soc., 2007, 154, A253–A257 CrossRef CAS.
- D. Wang, H. Li, Z. Wang, X. Wu, Y. Sun, X. Huang and L. Chen, J. Solid State Chem., 2004, 177, 4582–4587 CrossRef CAS.
- R. Dominko, M. Gaberscek, J. Drofenik, M. Bele, S. Pejovnik and J. Jamnik, J. Power Sources, 2003, 119–121, 770–773 CrossRef CAS.
- H. Liu, Q. Cao, L. J. Fu, C. Li, Y. P. Wu and H. Q. Wu, Electrochem. Commun., 2006, 8, 1553–1557 CrossRef CAS.
- T. Drezen, N.-H. Kwon, P. Bowen, I. Teerlinck, M. Isono and I. Exnar, J. Power Sources, 2007, 174, 949–953 CrossRef CAS.
- Y.-K. Sun, S.-M. Oh, H.-K. Park and B. Scrosati, Adv. Mater., 2011, 23, 5050–5054 CrossRef CAS PubMed.
- L. Xue, X. Li, Y. Liao, L. Xing, M. Xu and W. Li, J. Solid State Electrochem., 2015, 19, 569–576 CrossRef CAS.
- E. Ostreng, K. B. Gandrud, Y. Hu, O. Nilsen and H. Fjellvag, J. Mater. Chem. A, 2014, 2, 15044–15051 Search PubMed.
- G. Kobayashi, S.-i. Nishimura, M.-S. Park, R. Kanno, M. Yashima, T. Ida and A. Yamada, Adv. Funct. Mater., 2009, 19, 395–403 CrossRef CAS.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- P. Balaya, A. J. Bhattacharyya, J. Jamnik, Y. F. Zhukovskii, E. A. Kotomin and J. Maier, J. Power Sources, 2006, 159, 171–178 CrossRef CAS.
- Materials Genome Initiative for Global Competitiveness (June 2011) See: http://https://www.whitehouse.gov/sites/default/files/microsites/ostp/materials_genome_initiative-final.pdf.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- The Materials Project, See: http://https://www.materialsproject.org/.
- S. Curtarolo, G. L. W. Hart, M. B. Nardelli, N. Mingo, S. Sanvito and O. Levy, Nat. Mater., 2013, 12, 191–201 CrossRef CAS PubMed.
- T. Mueller, G. Hautier, A. Jain and G. Ceder, Chem. Mater., 2011, 23, 3854–3862 CrossRef CAS.
- G. Hautier, A. Jain, H. Chen, C. Moore, S. P. Ong and G. Ceder, J. Mater. Chem., 2011, 21, 17147–17153 RSC.
- G. Hautier, A. Jain, S. P. Ong, B. Kang, C. Moore, R. Doe and G. Ceder, Chem. Mater., 2011, 23, 3495–3508 CrossRef CAS.
- G. Ceder, MRS Bull., 2010, 35, 693–701 CrossRef CAS.
- C. Qian, T. Siler and G. A. Ozin, Small, 2015, 11, 64–69 CrossRef CAS PubMed.
Footnote |
| † R. C. and T. Z. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |