
Recent advances in graphene-based hybrid nanostructures for electrochemical energy storage
Pan
Xiong
a,
Junwu
Zhu
*a,
Lili
Zhang
*b and
Xin
Wang
*a
aKey Laboratory for Soft Chemistry and Functional Materials of Ministry Education, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: wangx@njust.edu.cn; zhujw@njust.edu.cn; Fax: +86-25-8431-5054; Tel: +86-25-8430-5667
bJiangsu Key Laboratory for Chemistry of Low-Dimensional Materials, Huaiyin Normal University, Huai’an, Jiangsu 223300, P. R. China. E-mail: zll@hytc.edu.cn
First published on 16th March 2016
Abstract
In recent years, graphene has emerged as a promising candidate for electrochemical energy storage applications due to its large specific surface area, high electrical conductivity, good chemical stability, and strong mechanical flexibility. Moreover, its unique two-dimensional (2D) nanostructure can be used as an ideal building block for controllable functionalization with other active components and the resulting graphene-based hybrids exhibit desirable properties for improved energy storage capability. This review summarizes the most recent progress on graphene and graphene-based hybrid nanostructures for three frontier electrochemical energy storage device applications, i.e., lithium-ion batteries, lithium–sulfur batteries and supercapacitors. Finally, we outline the future perspectives and trends in this research field including several challenges and opportunities.
 Pan Xiong | Pan Xiong received his PhD degree in materials chemistry from the Nanjing University of Science and Technology in 2005. From December 2013 to December 2014, he worked in Professor Guihua Yu's group at the University of Texas at Austin as an exchange visiting scholar. His research interests focus on the synthesis of hybrid nanostructures for use in photocatalysis and energy storage applications. |
 Junwu Zhu | Junwu Zhu received his PhD degree in materials chemistry from the Nanjing University of Science and Technology in 2005. He is presently working as professor in the same university. His main research interests are focused on the preparation and application of functional materials based on carbon and nanostructured composites. |
 Lili Zhang | Lili Zhang is a professor of chemistry at Huaiyin Normal University. She received her PhD degree in materials chemistry from the Nanjing University of Science and Technology in 2006, followed by two year postdoctoral research with Professor Xin Wang at the same university. Her research interests are focused on nanostructured material systems for advanced energy and environmental technologies. |
 Xin Wang | Xin Wang received his PhD degree in inorganic chemistry at the Department of Chemistry, Nanjing University, in 1985. After that, he joined the Nanjing University of Science and Technology and was promoted to full professor in 1988. His research interests are focused on the design and synthesis of nanostructured materials for use in energy storage and conversion. |
1. Introduction
Due to the ever-increasing energy demand and consumption, along with the resulting global environmental problems and air pollution, the development of energy storage systems, especially high-performance, low-cost, and environmentally friendly electrochemical energy storage devices, has attracted tremendous attention around the world during the past several years.1–6 Generally, for electrochemical energy storage devices, the nature of electrode materials plays a vital role in improving the key electrochemical parameters, such as energy and power density (both gravimetric and volumetric capacities), cyclability, rate capability, safety and temperature dependence. For example, if the graphite anode having a theoretical capacity of 372 mA h g−1 in a standard Sony 18650 battery is replaced with an anode material having a capacity greater than 1000 mA h g−1, then more than a 20% increase in total capacity can be obtained.7 Therefore, for the purpose of pursuing higher performances for energy storage devices, rational design and facile synthesis of advanced electrode materials with higher energy storage capability, longer cycle life, better safety, and lower cost are of great importance.Since the two-dimensional (2D) single-atom-thick layer of carbon was isolated by Novoselov and co-workers in 20048 graphene and graphene-based composites have attracted increasing research interest in the past decade. The unique characteristics of graphene, such as large theoretical surface area (2630 m2 g−1), high charge carrier mobility (20 m2 V−1 s−1), mechanical flexibility, chemical stability, thermal conductivity (5000 W m−1 K−1), and optical transmittance (97.7%), make graphene an ideal alternative or a versatile additive for energy storage applications.1,2,4,9–11 For example, the first graphene-based supercapacitor was developed in 2008 with specific capacitances of 135 and 99 F g−1 in aqueous and organic electrolytes, respectively.12 The best graphene anode with a specific capacity of ∼1200 mA h g−1 at 100 mA g−1 was reported in 2010.13 A graphene-wrapped sulfur cathode for lithium–sulfur batteries with high and stable specific capacities of up to ∼600 mA h g−1 over more than 100 cycles was explored in 2011.14 Moreover, with regard to its unique 2D structural features, graphene has also been used as an ideal building block for controllable functionalization with other electroactive components, such as metal oxides/hydroxides and conducting polymers, and the resulting graphene-based hybrid nanostructures exhibit desirable properties for energy storage applications.9–11,15–18
In such a constantly developing area, a large number of fascinating research results related to graphene and graphene-based hybrids for energy storage applications have been presented in recent years. Therefore, it is of great importance to highlight the latest discoveries and achievements timely, and outline a roadmap for developing potential candidate materials. In this review, the recent advances in the applications of graphene and graphene-based hybrid nanostructures for three frontier electrochemical energy storage devices, namely lithium-ion batteries, lithium–sulfur batteries and supercapacitors, are summarized with a particular focus on the progress made over the last three years. The remaining challenges and future prospects are also discussed.
2. Graphene-based nanocomposites for lithium-ion batteries
Since first being introduced to the market in 1991 by Sony, lithium-ion batteries have become one of the main power sources for portable electronic devices, such as cell phones, laptop computers, and digital cameras, due to their high energy density (120–170 W h kg−1) and durable cycling life.19–22 However, lithium-ion batteries suffer from low power density, e.g., the complete charge time is usually about 6 h for a lithium-ion battery equipped mobile phone or laptop in the market at present. In principle, the charge/discharge process of lithium-ion batteries is realized through the migration/transport of electrons and Li+ between the anode and the cathode. Therefore, the key to developing a fast charge/discharge process (high power capability) is to design advanced electrode materials with high electrical conductivity for effective electron transport, large surface areas and well-organized nanostructures with a short diffusion length for Li+ transport. In this regard, 2D graphene with superior electrical conductivity (106 S cm−1) and a high surface area (2630 m2 g−1) is expected to be a promising candidate for high-performance lithium-ion batteries.1,2,9,10,16,23 Recently, various strategies based on graphene-based electrode materials have been developed, which will be summarized in the following sections.2.1 Graphene-based anode materials
A commercially used graphite anode has a theoretical capacity of 372 mA h g−1 by forming LiC6 upon Li-intercalation between stacked layers.19,24 While single-layer graphene sheets can theoretically host two times as many Li+ ions as conventional graphite through Li-adsorption on both sides.25 Yoo et al. first explored the Li storage capacity of graphene nanosheets prepared by a controlled exfoliation and reassembly process. The specific capacity of graphene nanosheets was found to be ∼540 mA h g−1, which is much larger than that of graphite.26 After that, a number of studies on graphene-based anodes have also been reported (Table 1).| Electrode materials | Capacity | Rate capability | Cycle life | Ref. |
|---|---|---|---|---|
| N-doped graphene | 740 mA h g−1 (100 mA g−1) | 300 mA h g−1 (3 A g−1) | 100 (96.5%) | 39 |
| S-doped graphene | 870 mA h g−1 (374 mA g−1) | 285 mA h g−1 (11.22 A g−1) | 40 | |
| P-doped graphene | 460 mA h g−1 (100 mA g−1) | 80 (100%) | 41 | |
| Edge-fluorinated graphene | 807.4 mA h g−1 (50 mA g−1) | 650.3 mA h g−1 (500 mA g−1) | 500 (76.6%) | 46 |
| Full fluorinated graphene | 843 mA h g−1 (50 mA g−1) | 336 mA h g−1 (500 mA g−1) | 50 (92.9%) | 48 |
| N,S-codoped graphene | 1090 mA h g−1 (200 mA g−1) | 297 mA h g−1 (5 A g−1) | 500 (66.6%) | 49 |
| Solvated graphene frameworks | 1158 mA h g−1 (100 mA g−1) | 472 mA h g−1 (5 A g−1) | 500 (93%) | 54 |
| N,S-codoped porous graphene | 860 mA h g−1 (0.5 A g−1) | 220 mA h g−1 (80 A g−1) | 3000 (100%) | 55 |
| N-doped graphene frameworks | 640 mA h g−1 (0.5 A g−1) | 210 mA h g−1 (10 A g−1) | 300 (100%) | 57 |
| MnO/graphene | 2014.1 mA h g−1 (200 mA g−1) | 625.8 mA h g−1 (3000 mA g−1) | 400 (96%) | 63 |
| MnO2/3D porous graphene | 836 mA h g−1 (100 mA g−1) | 433 mA h g−1 (1600 mA g−1) | 200 (75.6%) | 65 |
| MnO2/3D N-doped graphene | 909 mA h g−1 (400 mA g−1) | 636 mA h g−1 (1500 mA g−1) | 200 (>100%) | 66 |
| MnO2/N-doped graphene | 607.5 mA h g−1 (100 mA g−1) | 90 mA h g−1 (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mA g−1) 000 mA g−1) |
3000 (100%) | 67 |
| Mn3O4/graphene | 500 mA h g−1 (60 mA g−1) | 200 mA h g−1 (1500 mA g−1) | 68 | |
| Mn3O4/N-doped graphene | 800 mA h g−1 (200 mA g−1) | 400 mA h g−1 (2000 mA g−1) | 40 (∼100%) | 69 |
| Fe2O3/graphene aerogel | 995 mA h g−1 (100 mA g−1) | 372 mA h g−1 (5000 mA g−1) | 50 (95.2%) | 71 |
| Fe2O3/graphene sheet-on-sheet | 1074.9 mA h g−1 (100 mA g−1) | 622.4 mA h g−1 (1000 mA g−1) | 72 | |
| Hollow Fe3O4/graphene | 940 mA h g−1 (200 mA g−1) | 420 mA h g−1 (2000 mA g−1) | 50 (100%) | 73 |
| Fe3O4/graphene foam | 785 mA h g−1 (1C) | 190 mA h g−1 (60C) | 74 | |
| Fe3O4/graphene foam | 1200 mA h g−1 (924 mA g−1) | 300 mA h g−1 (18.48 A g−1) | 500 (>100%) | 75 |
| Fe3O4/graphene foam | 1200 mA h g−1 (200 mA g−1) | 444 mA h g−1 (5 A g−1) | 77 | |
| CoO/graphene | 640 mA h g−1 (100 mA g−1) | 170 mA h g−1 (20 A g−1) | 78 | |
| Co3O4 fiber/graphene | 1005.7 mA h g−1 (100 mA g−1) | 295 mA h g−1 (1000 mA g−1) | 40 (83.5%) | 82 |
| Co3O4 nanowall/graphene | 800 mA h g−1 (100 mA g−1) | 320 mA h g−1 (5000 mA g−1) | 500 (100%) | 83 |
| Porous Co3O4/graphene | 1236 mA h g−1 (89 mA g−1) | 371 mA h g−1 (4450 mA g−1) | 85 | |
| MoO2/3D graphene | 975.4 mA h g−1 (50 mA g−1) | 537.3 mA h g−1 (1000 mA g−1) | 86 | |
| Porous MoO2/graphene | 1300 mA h g−1 (83.8 mA g−1) | 390 mA h g−1 (4190 mA g−1) | 87 | |
| MoO2/N-doped graphene | 1138.5 mA h g−1 (100 mA g−1) | 873.7 mA h g−1 (1000 mA g−1) | 88 | |
| MoO3/graphene film | 291 mA h g−1 (100 mA g−1) | 151 mA h g−1 (2000 mA g−1) | 100 (59.1%) | 89 |
| MoO3/graphene | 1176 mA h g−1 (200 mA g−1) | 894 mA h g−1 (3000 mA g−1) | 100 (92%) | 90 |
| ZnFe2O4/graphene | 1181 mA h g−1 (100 mA g−1) | 713 mA h g−1 (2000 mA g−1) | 30 (101.6%) | 93 |
| ZnFe2O4/graphene | 1090 mA h g−1 (100 mA g−1) | 650 mA h g−1 (2000 mA g−1) | 200 (99%) | 95 |
| ZnMn2O4 nanorod/graphene | 707 mA h g−1 (100 mA g−1) | 440 mA h g−1 (2000 mA g−1) | 50 (73.6%) | 97 |
| Porous ZnMn2O4/graphene | 926.4 mA h g−1 (200 mA g−1) | 560.8 mA h g−1 (1200 mA g−1) | 98 | |
| ZnMn2O4@C/graphene | 705 mA h g−1 (232 mA g−1) | 403.5 mA h g−1 (4640 mA g−1) | 180 (99.4%) | 94 |
| ZnMn2O4/graphene | ∼860 mA h g−1 (200 mA g−1) | ∼568 mA h g−1 (3200 mA g−1) | 1500 (∼81%) | 96 |
| CoFe2O4@C/graphene | 925.6 mA h g−1 (100 mA g−1) | 305.3 mA h g−1 (1600 mA g−1) | 102 | |
| CoFe2O4/graphene | 916.8 mA h g−1 (500 mA g−1) | 556 mA h g−1 (3200 mA g−1) | 200 (99.2%) | 103 |
| CoFe2O4/graphene | 1300.7 mA h g−1 (200 mA g−1) | 574.9 mA h g−1 (1600 mA g−1) | 50 (69.5%) | 104 |
| CoFe2O4/graphene | 1091 mA h g−1 (100 mA g−1) | 636 mA h g−1 (3000 mA g−1) | 100 (102%) | 106 |
| NiFe2O4/graphene/C | 1150 mA h g−1 (100 mA g−1) | 520 mA h g−1 (1000 mA g−1) | 109 | |
| Porous NiCo2O4/graphene | 974 mA h g−1 (100 mA g−1) | 396 mA h g−1 (800 mA g−1) | 70 (80.1%) | 112 |
| Porous NiCo2O4/graphene | 1850 mA h g−1 (200 mA g−1) | 404 mA h g−1 (2000 mA g−1) | 113 | |
| TiO2/graphene | 189 mA h g−1 (100 mA g−1) | 94 mA h g−1 (10 A g−1) | 121 | |
| TiO2/graphene | 215 mA h g−1 (200 mA g−1) | 100 mA h g−1 (1600 mA g−1) | 100 (74%) | 123 |
| TiO2 nanosheet/graphene | 275 mA h g−1 (335 mA g−1) | 200 mA h g−1 (13400 mA g−1) |
1000 (80%) | 125 |
| Porous TiO2/graphene aerogel | 200 mA h g−1 (100 mA g−1) | 99 mA h g−1 (5000 mA g−1) | 126 | |
| TiO2/3D N-doped graphene | 165 mA h g−1 (168 mA g−1) | 96 mA h g−1 (3360 mA g−1) | 200 (83.3%) | 122 |
| Porous TiO2/graphene | 206 mA h g−1 (100 mA g−1) | 128 mA h g−1 (5 A g−1) | 127 | |
| Li4Ti5O12/graphene | 162 mA h g−1 (1750 mA g−1) | 126 mA h g−1 (17.5 A g−1) | 500 (96.2%) | 130 |
| Li4Ti5O12/graphene | 187 mA h g−1 (175 mA g−1) | 128 mA h g−1 (14 A g−1) | 2000 (63.2%) | 132 |
| Li4Ti5O12/graphene oxide | 201 mA h g−1 (87.5 mA g−1) | 162 mA h g−1 (5.25 A g−1) | 300 (97%) | 131 |
| MoS2/graphene | 912 mA h g−1 (100 mA g−1) | 571 mA h g−1 (1000 mA g−1) | 100 (88.6%) | 134 |
| MoS2/graphene | 1400 mA h g−1 (100 mA g−1) | 591 mA h g−1 (1000 mA g−1) | 200 (96.5%) | 136 |
| MoS2/3D graphene | 1020 mA h g−1 (100 mA g−1) | 466 mA h g−1 (4 A g−1) | 50 (86%) | 135 |
| MoS2/graphene paper | 1681 mA h g−1 (100 mA g−1) | 864 mA h g−1 (1000 mA g−1) | 100 (65.6%) | 137 |
| MoS2/graphene@carbon fiber | 1225 mA h g−1 (200 mA g−1) | 780 mA h g−1 (2.5 A g−1) | 250 (57.7%) | 138 |
| Si NW/graphene/RGO | 1600 mA h g−1 (2.1 A g−1) | 500 mA h g−1 (8.4 A g−1) | 100 (80%) | 156 |
| Si/graphene | 2250 mA h g−1 (100 mA g−1) | 1000 mA h g−1 (10 A g−1) | 120 (85%) | 151 |
| Si/graphene | 2699 mA h g−1 (0.56 A g−1) | 1148 mA h g−1 (28 A g−1) | 1000 (100%) | 152 |
| Si NW/graphene | 705 mA h g−1 (0.1 A g−1) | 550 mA h g−1 (6.8 A g−1) | 157 | |
| Porous Si/graphene | 1703 mA h g−1 (200 mA g−1) | 390 mA h g−1 (5 A g−1) | 160 (∼100%) | 153 |
| Multilayered Si/graphene | 2198 mA h g−1 (120 mA g−1) | 700 mA h g−1 (24 A g−1) | 150 (87%) | 154 |
| Sn@graphene/graphene | 1022 mA h g−1 (200 mA g−1) | 270 mA h g−1 (10 A g−1) | 1000 (96.3%) | 163 |
| SnO2/graphene | 1090 mA h g−1 (200 mA g−1) | 491 mA h g−1 (5 A g−1) | 200 (∼90%) | 164 |
| SnO2/graphene | 776 mA h g−1 (100 mA g−1) | 147 mA h g−1 (5 A g−1) | 1000 (∼100%) | 160 |
| SnO2/N-doped graphene | 1021 mA h g−1 (500 mA g−1) | 417 mA h g−1 (20 A g−1) | 500 (>100%) | 162 |
| SnO2/N-doped graphene | 1326 mA h g−1 (200 mA g−1) | 460 mA h g−1 (5 A g−1) | 100 (87.9%) | 165 |
| Ge/graphene | 915 mA h g−1 (100 mA g−1) | 273 mA h g−1 (5 A g−1) | 400 (84.9%) | 167 |
| Ge NW@graphene | 1059 mA h g−1 (4.8 A g−1) | 363 mA h g−1 (24 A g−1) | 200 (90%) | 168 |
| Ge NW@graphene | 463 mA h g−1 (1.6 A g−1) | 170 mA h g−1 (32 A g−1) | 50 (∼94.5%) | 169 |
| Ge/graphene/CNT | 1181.7 mA h g−1 (100 mA g−1) | 644.8 mA h g−1 (3200 mA g−1) | 100 (∼73%) | 170 |
| CNT/graphene | 900 mA h g−1 (100 mA g−1) | 370 mA h g−1 (1500 mA g−1) | 250 (98.92%) | 173 |
| CNT/graphene paper | 375 mA h g−1 (100 mA g−1) | 100 (88%) | 174 | |
| CNT/graphene paper | 303 mA h g−1 (30 mA g−1) | 50 (100%) | 175 | |
| CNT/graphene hybrid foam | 2640 mA h g−1 (186 mA g−1) | 236 mA h g−1 (27.9 A g−1) | 100 (100%) | 176 |
| MWCNT/graphene nanoribbon | 880 mA h g−1 (100 mA g−1) | 157 mA h g−1 (10 A g−1) | 177 | |
| Porous carbon/graphene | 988 mA h g−1 (0.1 A g−1) | 280 mA h g−1 (2 A g−1) | 100 (68.8%) | 182 |
| g-C3N4/graphene | 1705 mA h g−1 (100 mA g−1) | 934 mA h g−1 (1000 mA g−1) | 50 (89.4%) | 185 |
Doped graphenes, such as nitrogen (N)-doped,35–39 boron (B)-doped,36 sulfur (S)-doped,40 and phosphorus (P)-doped graphenes,41 have attracted great interest due to their improved electrochemical performance and electron transport ability, as shown both theoretically and experimentally.42–44 As the electronegativity of heteroatoms such as N (3.5) is higher than that of C (2.55), N-doped graphene is more favorable for the insertion of lithium. For example, Wu et al. reported that N-doped graphene and B-doped graphene could show a high reversible capacity of 1043 and 1540 mA h g−1 at a low rate of 50 mA g−1, respectively. Moreover, a high capacity of ∼199 and ∼235 mA h g−1 was still obtained for the N-doped graphene and B-doped graphene at a high rate of 25 A g−1 (about 30 s to fully charge), respectively. The high-rate performance of N- and B-doped graphenes is due to their disordered surface morphology, heteroatomic defects, enhanced surface wettability, increased interlayer distance, improved electrical conductivity, and thermal stability, which are beneficial for rapid surface Li+ absorption, ultrafast Li+ diffusion and electron transport.36 Cao's group used three different defect models (graphitic, pyridinic, and pyrrolic graphenes) to theoretically investigate the effects of the electron-deficiency of N-doped graphene on its Li storage ability based on first-principles calculations. The results showed that the pyridinic graphene is the most suitable for Li storage with a high storage capacity.45 Halogen atoms (F, Cl, Br, and I) with high electronegativity can also be incorporated into graphene for enhancing the electrochemical performance.46–48 Jeon et al. reported the synthesis of edge-selectively fluorinated graphene nanoplatelets by a mechanochemically driven reaction between active carbon species at the edges of graphene and fluorine gas (20 vol% in argon). Due to the large difference in electronegativity between C (χ = 2.55) and F (χ = 3.98), the edge-fluorinated graphene can provide enhanced chemical stability (a charge retention of 76.6% after 500 cycles).46 Feng's group reported a full fluorinated graphene nanosheet by liquid exfoliation of fluorinated graphite in 2-isopropanol (IPA) solvent. The resulting 2D amorphous-like microstructure with high fluorine content rendered such F-graphene nanosheets abundant F active sites for lithium storage and facilitated the rapid diffusion of lithium ions during charging/discharging processes, leading to a high reversible capacity of 780 mA h g−1 at 50 mA g−1 and excellent cycle performance.48 In addition to single heteroatom-doped graphene materials, co-doped graphene materials have also been developed to enhance the electrochemical performance. Recently, An et al. reported a N and S co-doped graphene through thermal treatment of covalently functionalized GO with 2-aminothiophenol (ATP) as a source of both N and S. Benefiting from the synergistic effects of N and S co-doping, the resulting N,S co-doped graphene anode showed super-high reversible capacity, excellent rate capability as well as long-term cycling performance for lithium-ion batteries (Fig. 1).49
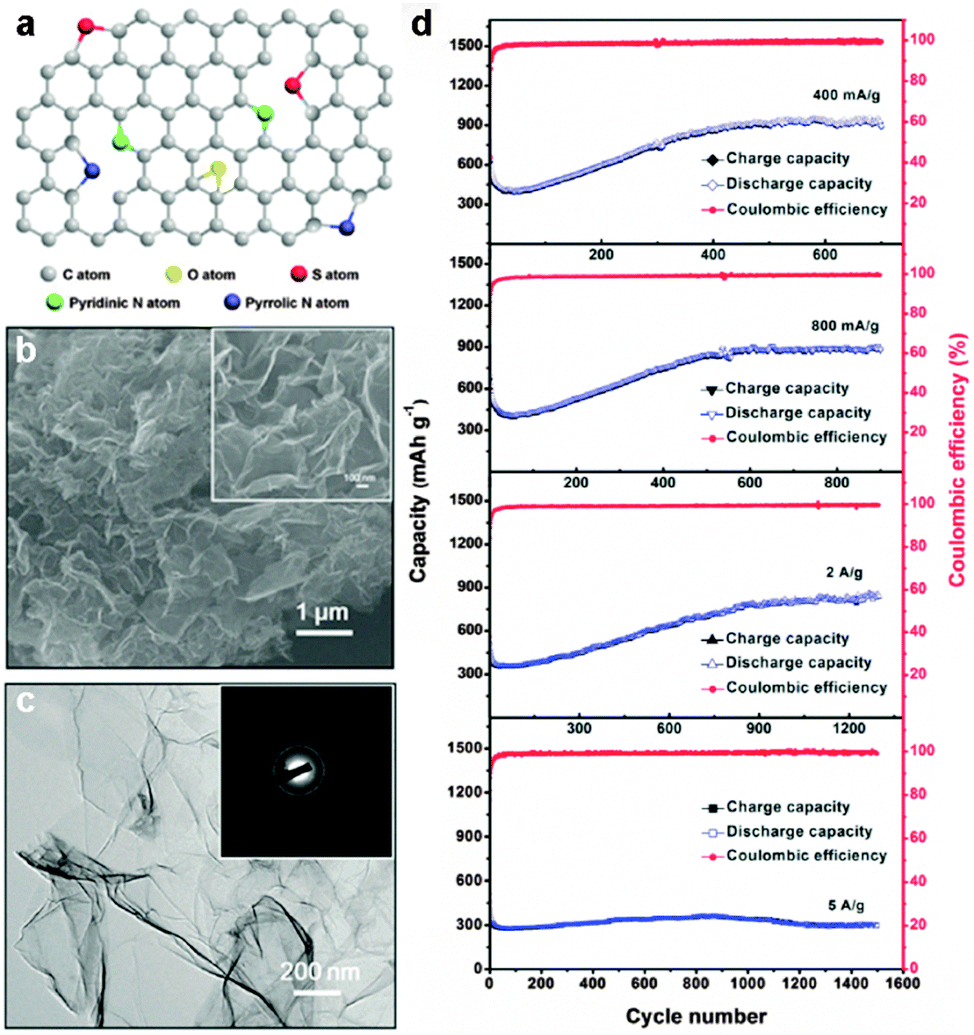 | ||
| Fig. 1 (a) Schematic illustration of the structure of N and S co-doped graphene. (b) SEM image of N and S co-doped graphene. Inset: Higher-magnification SEM image of N and S co-doped graphene. (c) TEM image of N and S co-doped graphene. Inset: Corresponding SAED pattern of N and S co-doped graphene. (d) Rate and cycling capability of the N and S co-doped graphene electrode at different rates. Reprinted with permission from ref. 49, copyright (2014), Wiley-VCH. | ||
Besides, three-dimensional (3D) graphene nanostructure-based anodes, such as graphene papers50,51 and graphene frameworks,52–55 have attracted great interest for lithium-ion batteries due to their large specific surface area, multidimensional continuous electron-transport pathways, and rapid ion-diffusion characteristics as well as excellent mechanical flexibility. In particular, Duan's group prepared solvated graphene frameworks (SGFs) through a convenient solvent-exchange approach. The as-obtained SGFs could be used directly as electrodes without adding any other binders or conductive additives and delivered a high reversible capacity of ∼1158 mA h g−1 at a charge/discharge rate of 0.1 A g−1, which was ∼2.6 times higher than that of unsolvated graphene frameworks (439 mA h g−1). Moreover, the SGFs showed excellent rate capability with a significantly large capacity of 472 mA h g−1 at a high charge/discharge rate of 5.0 A g−1 and superior cycling stabilities with a capacity retention of 93% over 500 charge/discharge cycles at 5.0 A g−1 (Fig. 2).54
 | ||
| Fig. 2 (a) Schematic representation of solvent exchange for the preparation of SGFs. (b) Preparation of a binder-free SGF electrode. A piece of SGF (left), its pressed-film electrode on copper foil placed in a coin cell case (middle), and SEM image of the cross-section of the pressed SGF film (right). Cycle performance and Coulombic efficiency of (c) SGF and (d) unsolvated graphene framework electrodes at a low current density of 0.1 A g−1. (e) Rate capabilities and cycle performance of SGF and unsolvated graphene framework electrodes over a wide range of current densities from 0.2 to 5.0 A g−1. (f) Cycling stability of the SGF electrode at a high current density of 5.0 A g−1 after a rate performance test. Reprinted with permission from ref. 54, copyright (2015), Wiley-VCH. | ||
Construction of a graphene nanostructure that combines a 3D highly conductive network, a hierarchically porous structure, and heteroatom doping together seems to be an ideal approach for achieving a high-performance electrode.55–59 Wang et al. reported the in situ fabrication of 3D doped hierarchically porous graphene. Due to the synergistic effect of the special 3D nanostructure and heteroatom doping, the as-prepared graphene electrode exhibited a high-power density of 116 kW kg−1, while the energy density remained as high as 322 W h kg−1 at 80 A g−1 (only 10 s to fully charge).55
Recently, graphene sheets combined with a number of transition metal oxides, such as MnO,62,63 MnO2,64–67 Mn3O4,68,69 Fe2O3,70–72 Fe3O4,73–77 CoO,78–80 Co3O4,81–85 MoO2,86–88 and MoO3,89,90 have been explored. For instance, Kan et al. prepared a Fe2O3/graphene sheet-on-sheet sandwich-like nanocomposite by a solvothermal method. Since Fe2O3 nanosheets have a better structure affinity with graphene nanosheets compared to Fe2O3 nanoparticles, which can more effectively prevent the agglomeration of graphene, larger intimate contact areas exist in sheet-on-sheet structure as compared to particle-on-sheet structure. The as-prepared Fe2O3/graphene sheet-on-sheet nanocomposite exhibited better specific capacities and cycling performances (a high capacity of 662.4 mA h g−1 was preserved after 100 cycles at 1000 mA g−1) compared with the conventional Fe2O3/graphene particle-on-sheet composite.72 A novel conversion mechanism between lithium and these transition metal oxides has been identified. The transition metal oxides are reduced to metal nanoparticles homogeneously embedded in a Li2O matrix. Simultaneously, Li2O can be reversibly decomposed in the presence of a transition metal as a catalyst.91 Sun and co-workers observed a reconstruction phenomenon of MnO/graphene electrodes due to the conversion reactions. During the lithiation and delithiation process, the initial MnO nanosheets were first reduced to Mn nanograins, which were then oxidized to Mn2+ and Mn4+, resulting in a thinner film composed of ultrafine MnOx nanoparticles that were uniformly loaded on or sandwiched between the graphene layers. Despite the initial morphology being lost, the close electrochemical connection between MnOx nanoparticles and the graphene matrix formed by the reconstruction process facilitated effective and rapid charge transfer, leading to high specific capacity and rate capability.63 Su et al. reported the direct observation of the conversion mechanism of Fe2O3/graphene anode by in situ transmission electron microscopy (TEM) technology. Upon first lithiation, single-crystalline Fe2O3 nanoparticles were transformed into multicrystalline Fe nanoparticles embedded in a Li2O matrix, which almost disappeared after delithiation. During the following delithiation process, FeO instead of Fe2O3 was detected. Therefore the eventual electrochemical processes of Fe2O3 anodes were revealed to be reversible conversion reactions between Fe nanograins and FeO nanograins.92
Recently, mixed transition metal oxides, such as ZnFe2O4,93–95 ZnMn2O4,96–98 CoFe2O4,99–106 NiFe2O4,107–109 NiCo2O4,110–113 and MnFe2O4,114–116 have been widely explored as upgraded anode materials owing to their higher electronic conductivity and larger specific capacities than those of simple transition metal oxides.117 One interesting example of these mixed transition metal oxides is ZnMn2O4, which can store Li+ through not only the conversion reaction, but also the alloying reaction between Zn and Li, leading to a high theoretical capacity of 784 mA h g−1. In order to resolve the intrinsically poor electronic/ionic conductivities of ZnMn2O4, Yu's group and our group designed a facile two-step synthesis approach for uniform growth of ultrafine ZnMn2O4 nanocrystals (4–5 nm) on RGO sheets to form a chemically integrated 2D ZnMn2O4/graphene nanostructure. A large specific capacity of ∼806 and 568 mA h g−1 was obtained at a low current density of 200 mA g−1 and a high current density of 3200 mA g−1, respectively, resulting in high rate capability with ∼71% capacity retention. A high long-term cyclability with a reversible capacity of ∼650 mA h g−1 over 1500 cycles at a current density of 2000 mA g−1 was obtained as well. The outstanding electrochemical performances of this 2D ZnMn2O4/graphene hybrid nanosheet can be largely attributed to the well-designed integrated 2D nanostructure, which not only provides a high surface area for Li storage, but also shortens diffusion pathways for Li+ shuttling in/out. Moreover, the possible covalent interaction between ZnMn2O4 and the graphene substrate not only contributes to the effective and rapid charge transfer between ZnMn2O4 and the current collector through the highly conductive graphene, but also plays an important role in maintaining the structural integrity upon cycling (Fig. 3).96 Spinel ferrites (MFe2O4, M = Co, Ni, Cu, Mn, and Zn), formed by replacement of one Fe in Fe3O4 by another metal cation, have been extensively regarded as promising anodes for lithium-ion batteries. Our group has explored a general one-step hydrothermal strategy for facile synthesis of a series of graphene–ferrite composite anodes, including ZnFe2O4/graphene,118 CoFe2O4/graphene,100 NiFe2O4/graphene,108 CuFe2O4/graphene,119 and MnFe2O4/graphene.120 By suitable tuning of the mass ratio between graphene and ferrite species for best synergetic effects, high reversible specific capacities of up to ∼950–1200 mA h g−1 as well as excellent cycling stability and rate capability can be achieved.
 | ||
| Fig. 3 (a) Schematic illustration of the 2D hybrid ZnMn2O4/graphene nanostructure and synergistic electrochemical characteristics of the 2D hybrid nanosheets. (b) SEM image of 2D ZnMn2O4/graphene hybrid nanosheets. (c) Rate performances of three different electrodes at various current densities of 200, 400, 800, 1600, and 3200 mA g−1. (d) Cycling performance and Coulombic efficiency of a 2D ZnMn2O4/graphene hybrid electrode at a current rate of 2000 mA g−1 for more than 1500 cycles. Reprinted with permission from ref. 96, copyright (2014), American Chemical Society. | ||
Different from the above transition metal oxides, TiO2 follows a lithium ion insertion/extraction mechanism, which possesses an advantage of negligible structure variation and consequently high cycling stability.121–127 Zhao's group reported a simple sol–gel strategy towards controllable manipulation of nucleation, growth, anchor, and crystallization of TiO2 nanoparticles on graphene, resulting in a TiO2 nanocrystal/RGO sheet hybrid with ultra-dispersed TiO2 nanoparticles (∼5 nm), ultrathin graphene sheets (≤3 layers), and a high surface area of ∼229 m2 g−1. Owing to the potential of strongly synergistic coupling effects, the resulting TiO2 nanocrystal/RGO sheet hybrid anode exhibited a high specific capacity of ∼94 mA h g−1 at ∼59C (10 A g−1), which was twice that of mechanically mixed composites (∼41 mA h g−1).121 Furthermore, Qiu and co-workers reported a simple one-step hydrothermal method toward the in situ growth of ultra-dispersed mesoporous TiO2 nanocrystals with (001) facets on 3D graphene aerogels (GAs), which could provide multidimensional electron transport pathways for Li storage. Due to the strong interaction between TiO2 and GAs, facet characteristics, high electrical conductivity, and the 3D hierarchically porous structure of the as-prepared TiO2/3D GA composites, a reversible capacity of 99 mA h g−1 could still be delivered at a very high rate of 5000 mA g−1.126
Similarly, spinel Li4Ti5O12 possessing reversible Li+ insertion/extraction properties together with negligible structural changes during the charge/discharge process has attracted considerable attention as an anode material for lithium-ion batteries.128–132 Kong et al. synthesized a 3D graphene-wrapped Li4Ti5O12 dandelion-like microsphere electrode by using a facile and scalable solution route. Due to the introduction of the graphene network which provides a fast and conductive electron transport path, Li4Ti5O12/graphene hybrid anodes displayed excellent rate performance and high cycling stability. An initial capacity of 261 mA h g−1 and a stable capability of 206 mA h g−1 after 500 cycles at 0.12 A g−1 were obtained.129
Among these metal sulfides, MoS2 is the most studied anode for Li storage. So far, a number of MoS2/graphene hybrids have been reported with significantly improved cycle life and rate performance. For example, Chen's group prepared MoS2/graphene composites by the L-cysteine-assisted hydrothermal method. The layered MoS2 crystals were anchored and grown on the surfaces of the RGO substrate. The incorporation of graphene can greatly enhance the whole conductivity of the MoS2/graphene composite electrode and provide rapid electron transport, resulting in a high specific capacity of ∼1100 mA h g−1 at a current density of 100 mA g−1, as well as excellent cycling stability and high-rate capability.133 Later, Zhang and co-workers deposited MoS2 on the surfaces of 3D graphene networks (3DGNs) by a facile chemical vapor deposition (CVD) method. Since the 3DGNs can provide highly efficient pathways for electronic and Li+ ion exchange during the charge/discharge cycles, the resulting MoS2/3DGN composites could be used as binder-free, conductive additive-free anodes for Li-ion batteries, exhibiting a high reversible capacity of 877 mA h g−1 at 100 mA g−1 and an excellent rate capability of 466 mA h g−1 at a high current density of 4 A g−1.135 Due to the strong covalent bonds within layers and weak van der Waals forces between layers, the layered MoS2 can be exfoliated to single-layer or few-layer sheets, showing different physical and chemical properties compared with its bulk counterparts.144–147 Recently, the exfoliated MoS2 nanosheets synthesized by the hydrolysis of lithiated MoS2 have been supported on the surfaces of the graphene substrate, resulting in a graphene-like MoS2/graphene nanocomposite. Due to the synergetic effect between highly conductive graphene nanosheets and graphene-like MoS2, the MoS2/graphene anodes exhibited a high reversible capacity of 1300–1400 mA h g−1, good cycling stability at 100 mA g−1, and excellent rate capability (Fig. 4).136
 | ||
| Fig. 4 (a) Schematic formation process of the MoS2/graphene nanocomposite. (b) Cycling behaviors of the synthesized samples: bulk MoS2, restacked MoS2, MoS2/graphene-15 and MoS2/graphene-30. (c) Rate capability and coulombic efficiency of the MoS2/graphene-15 electrode. Reprinted with permission from ref. 136, copyright (2014), Royal Society of Chemistry. | ||
Some graphene–Si nanocomposites with zero-dimensional (0D) Si nanoparticles150–155 or one-dimensional (1D) Si nanowires (NW)156,157 have been explored with enhanced lithium storage properties. For example, Cho's group reported a graphene–Si nanocomposite where island-shaped amorphous Si nanoparticles in the size range of 5–10 nm were strongly anchored on the graphene backbone. The as-prepared electrodes benefited from the synergetic effect between highly flexible graphene and critically sized Si nanoparticles for self-compacting behavior. The thickness of these loosely interconnected nanostructure electrodes decreased after cycling until reaching appropriate agglomeration without any cracks. Thus an excellent average charge capacity of ∼1103 mA h g−1 over 1000 cycles without capacity fading was achieved (Fig. 5a and b).152 Furthermore, a Si NW-based architecture via encapsulation of Si NWs with overlapped graphene sheets and RGO overcoats was explored by Wang and co-workers. On one hand, the overlapped graphene sheaths adapted to the volume change of embedded Si NWs by synergistic transformation. On the other hand, the RGO overcoats acted as mechanically robust and flexible matrices to accommodate the volume change of embedded Si NW/graphene nanocables. Thus a high reversible specific capacity of 1600 mA h g−1 at 2.1 A g−1 and high cycling stability of 80% capacity retention after 100 cycles were obtained due to the well-maintained structural and electrical integrity of the electrodes.156 Recently, Chen's group fabricated a 3D multilayered Si/RGO hybrid anode by assembly of alternating Si/RGO layers on porous Ni foams. The multilayered structures offering ample space for Si expansion were able to effectively mitigate detrimental volume expansion upon lithiation. As a result, high reversible specific capacity (2300 mA h g−1 at 0.05C), good rate capability (700 mA h g−1 at 10C), and superior cycling capability (87% capacity retention at 10C after 152 cycles) were obtained.154 Compared with silicon, silicon oxides (SiOx, 0 < x ≤ 2) with less volume change during lithiation usually exhibit better cycling stability. In order to improve the poor electronic conductivity of silicon oxides, Li et al. prepared a graphene nanoplatelet-supported SiOx-disordered carbon composite by self-assembly between graphene nanoplatelets and C2H5Si(OC2H5)3, followed by high-temperature treatment. A stable reversible capacity of about 630 mA h g−1 at a current density of 100 mA g−1 was achieved without obvious capacity fading after 250 cycles.158
 | ||
| Fig. 5 (a) Schematic view of elastic Si/graphene nanocomposites before and after electrochemical cycling. (b) Cycling performance of Si/graphene at a charge current density of 14 A g−1 and a discharge density of 2.8 A g−1 over 1000 cycles. Reprinted with permission from ref. 152, copyright (2014), American Chemical Society. (c) Schematic illustration of 3D Sn@graphene/graphene. (d) Cycle performance of the 3D Sn@graphene/graphene electrode at current densities of 0.2, 0.5, and 1 A g−1 for the initial six cycles and then 2 A g−1 for the subsequent 1000 cycles. Reprinted with permission from ref. 163, copyright (2014), American Chemical Society. TEM images of (e) as-grown Ge NWs, (f) a single layer of graphene on a Ge NW, and (g) a bilayer of graphene grown on a Ge NW. (h) Cycle performance of a graphene/Ge NW at each C-rate from 0.5 to 4.0C. Reprinted with permission from ref. 168, copyright (2013), Wiley-VCH. | ||
For Sn–graphene hybrid anodes, the incorporation of graphene159,160 and modified graphene (such as N-doped graphene,161,162 3D graphene,163,164 and 3D N-doped graphene165) also provides impressive advantages. Yang et al. described a facile solvothermal strategy for the in situ growth of mesoporous SnO2 on the graphene surface. The formation of mesoporous SnO2 crystals on the surface of graphene can avoid the aggregation of graphene, while the graphene substrate can restrain the collapse of the mesoporous structure. Therefore, the as-synthesized SnO2/graphene with large surface area, good electrical conductivity and stable porous structure exhibited greatly improved electrochemical performances as an anode material for Li-ion batteries.159 Later, SnO2 nanocrystals were hybridized with N-doped graphene sheets via Sn–N bonding as reported by Guo's group. The N-doped graphene sheets not only act as a flexible buffer to accommodate the huge volume change of SnO2 nanocrystals, but also provide enhanced lithium electrochemical activity compared with undoped graphene. The as-obtained SnO2/N-doped graphene hybrid showed a stable capacity of 1021 mA h g−1 under a current density of 0.5 A g−1. Moreover, a reversible charge capacity as high as 1346 mA h g−1 could be still achieved after 500 cycles.162 In another work, novel 3D porous graphene networks anchored with Sn nanoparticles (5–30 nm) encapsulated with graphene shells (∼1 nm) were synthesized using an in situ CVD technique using metal precursors as a catalyst and 3D self-assembled NaCl particles as a template. As a result, long-term cycling stability at high rates (approximately 96.3% capacity retention was maintained at 2 A g−1 after 1000 cycles) and superior rate capacity (a reversible capacity of 270 mA h g−1 at 10 A g−1) were achieved. The superior performances of this 3D hybrid anode can mainly be ascribed to the dual graphene encapsulation: flexible graphene shells suppress the aggregation of Sn nanoparticles and accommodate the volume expansion, leading to structural and interfacial stabilization of Sn nanoparticles; 3D porous graphene networks with superior electrical conductivity, high mechanical flexibility, and large surface area facilitate diffusion and transport of electrons and ions, leading to remarkably enhanced structural and electrical integrity (Fig. 5c and d).163
Ge also possesses a high theoretical capacity of 1624 mA h g−1. Recently, significant research efforts have been devoted to the graphene-supported Ge-based hybrid anodes.166–170 Lee's group used a low-pressure thermal evaporation approach to uniformly deposit crystalline Ge particles on graphene surfaces or embed into graphene sheets. A high initial coulombic efficiency of 80.4% in the first cycle and a capacity retention of 84.9% after 400 cycles were achieved.167 However, for the above deposit-type composite, it is difficult to provide a high-quality integrated protection layer for volume expansion, which frequently results in poor capacity retention during long cycling. An ideal approach to mitigate the volume expansion issue of Ge is to directly grow single to a few layers of graphene on Ge nanostructure surfaces. Cho and co-workers reported the direct growth of a single to a few (less than four) layers of graphene on Ge NWs. The Ge NW/graphene hybrid anode exhibited a high reversible specific capacity of 1059 mA h g−1 even after 200 cycles at a rate of 4.8 A g−1 (Fig. 5e–h).168 Then, Wang et al. reported a similar but completely catalyst-free route to prepare uniform, highly crystalline in situ graphene encapsulated Ge nanowires via the arc-discharge method. The graphene encapsulated Ge NW composites provided a reversible capacity of 430 mA h g−1 at a current density of 1600 mA g−1.169 The above two cases demonstrated that such a high-quality single- or few-layered graphene coating on Ge NWs not only would guarantee mechanically tight holding of the Ge NWs, but also would be suitable for Li ions to diffuse into and out of the Ge NWs throughout long cell cycles.
Honma's group first reported a graphene/CNT composite anode for Li-ion batteries. The interlayer distance of graphene was enhanced through CNT interaction. The graphene/CNT anode showed an increased specific capacity of 730 mA h g−1, which is much larger than that of graphite (540 mA h g−1).26 However, this graphene/CNT composite was prepared by a simple physical mixing of pre-fabricated CNTs and graphene, which may lead to poor interaction, and consequently unfavorable charge transport. To this point, Vinayan et al. reported the incorporation of multi-walled carbon nanotubes (MWCNTs) along with graphene sheets by strengthened electrostatic interaction between cationic polyelectrolyte modified graphene and acid treated MWCNTs. A discharge capacity of 768 mA h g−1 was obtained at a current density of 90 mA g−1 after 100 cycles.171 Wang's group prepared graphene/CNT composites by an in situ chemical vapor reduction and deposition process. CNTs with various lengths were uniformly grown on the surfaces of neighboring graphene to prevent restacking. Besides, the electrical conductivity of the graphene/CNT composite along the vertical direction was also improved compared with pure graphene. Thus, high reversible capacities of 573 mA h g−1 at a small current density of 74 mA g−1, and 520 mA h g−1 at a large current density of 744 mA g−1 were obtained, respectively.172 Similarly, Wang et al. reported a 3D graphene/CNT structure based on the optimized growth of hybrid CNT and graphene nanostructures on copper foil by a two-step CVD process. The seamlessly connected graphene/CNT hybrids provided a high reversible capacity of 900 mA h g−1 at a current density of 100 mA g−1 and a capacity retention of 98.92% at a current density of 600 mA g−1 for 250 cycles.173
2.2 Graphene-based materials for cathodes
The most common cathode materials for Li-ion batteries are lithium transition-metal oxides (such as LiCoO2 and LiMn2O4), lithium transition-metal phosphates LiMPO4 (M = Fe, Mn, Co, or Ni), and vanadium oxide (V2O5).9,10,186,187 Unfortunately, the inherently low ionic and electrical conductivities of these materials seriously limit Li+ insertion/extraction and charge transport rates. For example, the electronic conductivities of LiCoO2, LiMn2O4, and LiFePO4 are 10−4, 10−6, and 10−9 S cm−1, respectively, which are fairly low and can impair rate capability.10 Over the past decade, graphene and its derivatives have been extensively employed in the cathode system to enhance the electrochemical properties (Table 2).9,10| Electrode materials | Capacity | Rate capability | Cycle life | Ref. |
|---|---|---|---|---|
| LiMn2O4/graphene | 137 mA h g−1 (148 mA g−1) | 101 mA h g−1 (14.8 A g−1) | 100 (96%) | 188 |
| LiFePO4/graphene | 147.1 mA h g−1 (20 mA g−1) | 63 mA h g−1 (5000 mA g−1) | 200 (94.2%) | 195 |
| LiFePO4/graphene | 120 mA h g−1 (1C) | 85.1 mA h g−1 (50C) | 950 (91.4%) | 196 |
| C@LiFePO4/graphene | 164 mA h g−1 (0.2C) | 124 mA h g−1 (20C) | 1000 (95%) | 197 |
| LiFePO4/graphene | 168 mA h g−1 (0.5C) | 115 mA h g−1 (10C) | 198 | |
| LiFePO4/graphene | 208 mA h g−1 (0.1C) | 125 mA h g−1 (10C) | 199 | |
| LiFePO4/N-doped graphene | 166 mA h g−1 (0.5C) | 127 mA h g−1 (20C) | 200 | |
| Li3V2(PO4)3/graphene | 125.3 mA h g−1 (0.1C) | 105.7 mA h g−1 (20C) | 100 (96.7%) | 203 |
| Li3V2(PO4)3/graphene | 186 mA h g−1 (0.1C) | 135 mA h g−1 (10C) | 202 | |
| V2O5/graphene | 278 mA h g−1 (0.1C) | 165 mA h g−1 (2C) | 100 (78%) | 208 |
| V2O5 nanosheet/graphene | 223 mA h g−1 (0.2C) | 76 mA h g−1 (50C) | 160 (52%) | 207 |
The spinel LiMn2O4 cathode is being widely used due to its low production cost, environmental friendliness and good safety. In addition to its poor ionic and electronic conductivities, the dissolution of Mn2+ in the electrolyte is another severe obstacle that should be solved. With introduction of graphene which can serve as protection layer and provide large surface areas and improved Li diffusion kinetics, the cycling stability and rate capability of LiMn2O4/graphene hybrid cathodes have been remarkably improved compared to the pure LiMn2O4 cathode.188,189 Partially substituting Mn of LiMn2O4 with other metal ions such as Ni2+ afforded spinel LiNi0.5Mn1.5O4, which possesses fast 3D Li+ diffusion channels, a theoretical capacity of 146.7 mA h g−1, and a high operational voltage of ∼4.7 V. Similarly, in order to solve the poor conductivity of LiNi0.5Mn1.5O4, the GO-coated LiNi0.5Mn1.5O4 cathode190 and self-assembled LiNi0.5Mn1.5O4/graphene composites191 have been successfully developed for improving cycling stability and rate performance.
Olivine-structured LiFePO4 is one of the most popular commercial cathode materials for Li-ion batteries owing to its several advantageous features for Li storage such as low cost, environmental benignity, relatively large capacity, thermal stability, and excellent cycle life. However, the inherently low conductivity has hindered its practical application. Several reports have demonstrated the enhanced capacity of graphene/LiFePO4 composite cathodes, which were usually prepared by pyrolyzing GO or RGO together with either LiFePO4 precursors or LiFePO4 particles.192–198 Hu and co-workers reported the modification of LiFePO4 with few-layer graphene obtained by a scalable electrochemical exfoliation method. The as-obtained graphene-wrapped LiFePO4 hybrid cathode was able to deliver a capacity of 208 mA h g−1, which is beyond the theoretical capacity of LiFePO4 (170 mA h g−1). The excess capacity can be attributed to the reversible redox reaction between the Li ions of the electrolyte and the exfoliated graphene flakes, where the graphene flakes exhibited an ultrahigh capacity (>2000 mA h g−1).199 Recently, N-doped graphene prepared by using urea as a nitrogen source has also been employed to combine with LiFePO4. The resulting LiFePO4/N-doped graphene nanocomposite shows enhanced electrochemical performance in terms of high discharge capacities and voltages at high rates including sub-zero temperature conditions.200 Besides, considerable efforts have been devoted to the integration of graphene with other lithium transition-metal phosphate cathodes such as Li3V2(PO4)3, due to their higher discharge potential (e.g. 3.6–4.55 V) and higher theoretical capacity (e.g. 197 mA h g−1).201–204
V2O5 is another promising cathode candidate for Li-ion batteries because of its higher theoretical capacity (440 mA h g−1) as compared with those of the above cathodes. Besides the inferior electronic conductivity, the particle agglomeration and resultant short cycle life (typically less than a couple of hundred cycles) are more challenging issues for practical applications. Various graphene–V2O5 composites have been developed to resolve the sluggish electronic conductivity of V2O5 and increase Li+ transportation kinetics.205–208 However, the short cycle life of these V2O5/graphene hybrid cathodes has yet to be well resolved. Recently, Lee et al reported a hybrid structure composed of ultrathin V2O5 NWs homogeneously entangled with graphene sheets, where the agglomeration of the V2O5 NWs could be efficiently suppressed by the robust graphene sheets. As a result, a substantially improved cycle life over 100000 cycles at a high current density of 10000 mA g−1 was achieved.209
2.3 Graphene-based materials for lithium ion full batteries
The most common Li-ion batteries composed of a commercial Li-intercalated compound cathode (such as LiCoO2 or LiFePO4) and a graphitic anode have high energy densities of ∼120–150 W h kg−1, but suffer from low power densities compared to electrochemical capacitors.21 The direct method of increasing the rate capability is to replace the electrode materials with other nanomaterials having larger Li-storage capacity, a shortened Li+ diffusion length and better charge transport kinetics. To this end, a large fraction of current research is concentrated on assembly of Li-ion full batteries based on graphene210,211 and graphene-based hybrid212,213 electrodes, owing to their large specific capacity, high rate capability, and superior mechanical flexibility (Table 3).1,214| Anode materials | Cathode materials | Capacity | Rate capability | Cycle life | Ref. |
|---|---|---|---|---|---|
| a The discharge capacity of this full battery is based on the mass loading of anode materials. The discharge capacities of other full batteries shown in this table are based on the mass loading of cathode materials. | |||||
| Graphene | LiFePO4 | 165 mA h g−1 (1C) | 80 (87%) | 210 | |
| Graphene film | LiCoO2 | 127 mA h g−1 (0.2C) | 211 | ||
| Si/graphene | LiNi1/3Mn1/3Co1/3O2 | 137 mA h g−1 (C/15) | 15 (70%) | 212 | |
| Ge/graphene/C | LiCoO2 | 900 mA h g−1a (1C) |
100 (73%) | 213 | |
| Li4Ti5O12/3D graphene foam | LiFePO4/3D graphene foam | 143 mA h g−1 (0.2C) | 117 mA h g−1 (10C) | 100 (96%) | 215 |
| 2D ZnMn2O4/graphene | LiFePO4 nanosheets | 132 mA h g−1 (0.2C) | 58 mA h g−1 (10C) | 100 (94%) | 216 |
Hassoun and co-workers reported an advanced Li-ion full battery based on a graphene ink-based anode and a commercial LiFePO4 cathode. Due to the use of a high-performance graphene anode produced via drop casting graphene ink on a Cu substrate, the as-prepared full cell was operated for over 80 charge/discharge cycles at a 1C (170 mA g−1) rate with a reversible capacity of 165 mA h g−1, and displayed an estimated energy density of about 190 W h kg−1, exceeding ∼25–60% over the current lithium ion battery technology.210 Tuan's group assembled a lithium ion full battery by using a carbon-coated Ge nanoparticle/RGO anode and a commercial LiCoO2 cathode. The full cell displayed a stable capacity of ∼1000 mA h g−1 in 100 cycles and was able to power a wide range of electronic devices, including an light-emitting-diode (LED) array consisting of over 150 bulbs, blue LED arrays, a scrolling LED marquee, and an electric fan.213
Although the above full batteries using graphene or graphene hybrid anodes instead of conventional graphite exhibited much enhanced performances, the use of commercial micropowder cathodes had hindered the total charge transfer. In order to achieve higher performances, it is necessary to further replace the commercial micropowder cathodes with nanostructured alternatives for fabrication of full batteries, so that both the anode and the cathode belong to the nanostructure. For example, an all-3D graphene foam-based Li-ion full battery composed of Li4Ti5O12-loaded graphene foam anodes and LiFePO4-modified graphene foam cathodes was explored by Chen's group. By employing graphene foam, a 3D, flexible, and conductive interconnected network, as a current collector, the all-3D graphene foam-based lithium ion full battery showed promising rate capability, delivering a high specific capacity of 117 mA h g−1 at 10C with reserving 88% of the capacity at 1C. Moreover, a thin, lightweight, and flexible lithium ion full battery was also fabricated with a high-rate performance and energy density under repeated bending.215 Recently, Yu's group and our group reported an all-2D nanosheet-based Li-ion full battery with the 2D ZnMn2O4/graphene anode and the LiFePO4 nanosheet cathode. By taking advantage of these two unique 2D nanostructured electrodes, which can provide excellent electronic conductivity, more electrochemically active surfaces, and a reduced diffusion path for Li ion extraction/insertion, the as-assembled Li-ion full battery exhibited superior cycling performance and rate capability compared with those of control full batteries composed of the conventional graphite anode and the commercial LiFePO4 powder cathode. Further, a flexible pouch cell based on these two 2D nanostructures was also assembled. Even under various bending states including bent, folded and rolled, a stable reversible capacity could still be delivered (Fig. 6).216
 | ||
| Fig. 6 (a) Schematic of an all-nanosheet-based Li-ion full battery using 2D ZnMn2O4/graphene hybrid nanosheets and LiFePO4 nanosheets as the anode and the cathode, respectively. (b) Comparison of rate capabilities of an all-nanosheet-based full battery and a control full battery. (c) A flexible pouch cell based on the nanosheet anode and cathode can light a red LED logo (UT) at flat, 90° bent, 180° bent, and rolled states. (d) Cyclic performance of the pouch cell at flat, 90° bent, 180° bent, and rolled states for 20 cycles, respectively. Reprinted with permission from ref. 216, copyright (2015), Elsevier Ltd. | ||
3. Graphene-based nanocomposites for lithium–sulfur batteries
During the pursuing of even higher energy density for rechargeable batteries, alternative electrodes that possess different mechanisms other than the intercalation of Li ions have been extensively investigated. A lithium–sulfur (Li–S) battery that utilizes a Li anode and a S cathode can provide a high theoretical capacity of ∼1675 mA h gsulfur−1 upon the complete redox reaction (S8 + 16Li = 8Li2S), which corresponds to a high theoretical specific energy and volumetric energy density of ∼2572 W h kg−1 and ∼2835 W h L−1, respectively.6,217 Although tremendous progress in Li–S batteries has been made so far, some of the key challenges and critical issues still need to be addressed.5,218–223 Firstly, element S and its various lithiation products (Li2Sx, x = 1–8) are electrical and ionic insulating materials. For example, the electrical conductivity of S is 5 × 10−30 S cm−1 at 25 °C, which is so low that it will create a large internal resistance and block the charge transport. Thus, development of sulfur-based composites in which nanosized S particles are well embedded into conductive matrices such as conducting polymers or carbon materials has been a compelling research topic.218,224 Secondly, a large volume expansion of ∼80% will occur during lithiation due to the conversion of sulfur (∼2.03 g cm−3) to lithium sulfide (∼1.63 g cm−3). Repeated volume expansion/contraction during charge/discharge cycles may induce severe collapse of material's structure, resulting in rapidly declining capacity. A hollow structure or a buffering layer should be employed to provide sufficient space for accommodating volume changes.220,225,226 Thirdly, various soluble polysulfide intermediates (Li2Sx, x = 3–6) shuttle between the cathode and the anode, which lead to a low Coulombic efficiency and parasitic self-discharging. Finally, continuous deposition of the insulting Li2S layer onto the surface of the anode inhibits ion and electron transport, leading to degraded performances.11,227,228A number of approaches have been proposed to overcome the above obstacles.5,222,229,230 Among these strategies, introduction of carbon nanomaterials, especially graphene in Li–S batteries, has been widely investigated for the following reasons: (1) highly conductive graphene can provide a conductive framework for improved electrical properties and effective charge transfer; (2) flexible graphene can act as a 2D buffering layer and/or build a 3D porous network to accommodate the volume change of sulfur during repeated charge/discharge cycles; (3) functionalized graphene networks immobilize sulfur and lithium polysulfides, suppressing the shuttle phenomenon. The recent advances in graphene-based Li–S batteries are summarized in the following sections.
3.1 Graphene-based sulfur cathodes
Integrating sulfur with 2D conductive, flexible graphene is expected to afford improved performances for Li–S batteries. In this regard, various methods including ball-milling,231,232 melt-diffusion,233–237 self-assembly,14,238 chemical reaction approach,226,239 solution precipitation,240 electrodeposition,241 and electrochemical assembly242 have been explored to effectively combine sulfur with graphene-based or GO-based nanostructures for graphene-based sulfur cathodes (Table 4).| Cathode materials | S mass loading | Capacitya | Cycle life | Ref. |
|---|---|---|---|---|
| a The capacity is calculated based on the mass of sulfur. | ||||
| S/GO | 66 wt% | 1000 mA h g−1 (0.1C) | 50 (95.4%) | 226 |
| S/GO core–shell structure | 50 wt% | 900 mA h g−1 (1 A g−1) | 1000 (88.9%) | 240 |
| S/graphene | 70 wt% | 966.1 mA h g−1 (2C) | 500 (53%) | 232 |
| S/graphene | 70.2 wt% | 934 mA h g−1 (0.1 A g−1) | 60 (95.4%) | 241 |
| S/porous graphene | 67 wt% | 927 mA h g−1 (1C) | 100 (74%) | 234 |
| S/graphene nanoshell | 62 wt% | 1098 mA h g−1 (1C) | 1000 (38.2%) | 235 |
| S/templated graphene | 64 wt% | 734 mA h g−1 (10C) | 200 (85.5%) | 236 |
| S/graphene core–shell structure | 83.3 wt% | 514 mA h g−1 (3C) | 500 (94.2%) | 239 |
| S/graphene nanowalls | 66 wt% | 1261 mA h g−1 (209 mA g−1) | 120 (96%) | 242 |
| S/EDA–RGO | 60 wt% | 820 mA h g−1 (0.5C) | 350 (80%) | 243 |
| S/N-doped graphene | 60 wt% | 967 mA h g−1 (1C) | 500 (52.4%) | 245 |
| S/N-doped graphene | 80 wt% | 1356.8 mA h g−1 (0.1C) | 100 (62.5%) | 246 |
| S/3D graphene | 73 wt% | 1260 mA h g−1 (0.1C) | 100 (55.6%) | 237 |
| S/3D graphene | 63 wt% | 700 mA h g−1 (0.75 A g−1) | 100 (77.3%) | 238 |
| S/3D graphene | 80 wt%/12 mg cm−2 | 6.0 mA h cm−2/625 mA h g−1 (0.1C) | 300 (75.5%) | 248 |
| S/3D graphene foam | 52 wt% | 402.8 mA h g−1 (3.2 A g−1) | 400 (74.5%) | 249 |
| S/3D N-doped graphene | 87.6 wt% | 743 mA h g−1 (1.5 A g−1) | 200 (85.6%) | 250 |
| S/graphene-porous carbon | 68 wt%/0.74 mg cm−2 | 885.5 mA h g−1 (0.5C) | 100 (70%) | 252 |
| S-activated carbon/graphene | 1.2–1.5 mg cm−2 | 1113 mA h g−1 (0.2C) | 300 (88.9%) | 259 |
| S-MWCNT/graphene | 70 wt% | 1396 mA h g−1 (0.2C) | 100 (60.5%) | 254 |
| S-MWCNT/graphene | 68.93 wt% | 727.2 mA h g−1 (5C) | 200 (85.3%) | 255 |
| S/graphene/CNT | 50 wt% | 1048 mA h g−1 (1C) | 1000 (56.7%) | 256 |
| S/3D graphene-CNT | 1.0 mg cm−2 | 1179.6 mA h g−1 (0.2C) | 200 (83%) | 257 |
| S/3D graphene-CNT | 70 wt% | 946.2 mA h g−1 (0.3C) | 100 (74.4%) | 258 |
| S/graphene-SWCNT | 60 wt% | 822 mA h g−1 (5C) | 100 (80%) | 260 |
| S/graphene-CNT@porous carbon | 77 wt% | 914 mA h g−1 (1C) | 150 (72%) | 261 |
| S/N-doped CNT-graphene | 1.0 mg cm−2 | 1152 mA h g−1 (1C) | 80 (76.4%) | 262 |
| S/GO@PANI | 60.1 wt%/1.0 mg cm−2 | 1248 mA h g−1 (0.5C) | 100 (80.6%) | 263 |
| S/curved graphene@PANI | 55 wt% | 851 mA h g−1 (0.2C) | 100 (90%) | 264 |
| S/porous graphene@PEDOT:PSS | 60.1 wt%/1.0 mg cm−2 | 1198 mA h g−1 (0.1C) | 200 (70.5%) | 267 |
| S/N-doped graphene@PANI | 52.5 wt% | 1277.3 mA h g−1 (0.5C) | 100 (54.3%) | 265 |
| S/porous carbon/graphene@PPy | 64 wt% | 470 mA h g−1 (3C) | 400 (80%) | 266 |
 | ||
| Fig. 7 (a) Digital camera images of GO dispersed in different solutions at the beginning, after 12 h and after adding sulfur particles. (b) Galvanic charge/discharge performance and Coulombic efficiency of sulfur/GO at 1 A g−1 for 1000 cycles. Discharge specific capacities calculated based on the weight of sulfur only (green dotted line) and the total weight of sulfur/GO are plotted. Reprinted with permission from ref. 240, copyright (2014), American Chemical Society. | ||
Graphene with high conductivity and large surface areas is in favour of more effective charge transfer compared to GO. Wang et al. synthesized sulfur/graphene composites by heating a mixture of graphene nanosheets and elemental sulfur and the later was incorporated into the graphene aggregates. The sulfur/graphene composite showed improved electrical conductivity, while the cycling performance was not improved remarkably compared to pure sulfur.233 Li et al. explored an electrochemical assembly strategy to prepare a vertically aligned sulfur/graphene nanowall structure, in which sulfur nanoparticles were homogeneously embedded in the interlayers of graphene and the ordered graphene arrays were arranged perpendicularly to the electrically conductive substrates. Such a unique nanowall structure is favourable for fast lithium and electron transport in the electrode, leading to a high-rate performance (over 400 mA h g−1 at 8C, 13.36 A g−1) (Fig. 8). However, upon increasing sulfur content to 80.8%, the cycle performance decayed obviously.242
 | ||
| Fig. 8 (a) Schematic diagrams of vertically aligned sulfur–graphene nanowalls as a cathode for a Li–S battery, facilitating the fast diffusion of both lithium and electrons. (b and c) TEM images of the sectional slices of sulfur–graphene nanowalls with different magnifications, revealing that sulfur nanoparticles are anchored in between graphene layers and ordered graphene arrays in each sulfur–graphene nanowall. (d) Rate performance of sulfur–graphene nanowalls cycled at various current rates and long cycle performance at a constant current rate of 4C. Reprinted with permission from ref. 242, copyright (2015), American Chemical Society. | ||
In the above reports, the active sulfur was just impregnated between the interlayers of graphene nanosheets. Consequently, most of the active sulfur was exposed to the electrolyte and the formed polysulfides could still readily diffuse out of the graphene structure, which significantly undermined the cycling stability. To this point, Zhang's group confined active sulfur in the nanopores of highly porous activated graphene nanosheets (AGNs). The resultant AGN/S nanocomposites exhibited capacity retentions of 67% and 74% after 100 cycles at rates of 0.5C and 1C, respectively. The nanopores of AGNs could not only act as “microreactors” for the electrochemical reactions of sulfur, minimizing the dissolution and shuttling of polysulfide, but also ensure good electrolyte penetration for fast transport of lithium ions.234 Recently, Zhao et al. synthesized an intrinsically unstacked double-layer templated graphene (DTG) via a template-directed CVD method. The as-obtained DTG was composed of two unstacked graphene layers separated by a large quantity of protuberances. After incorporation of sulfur, this DTG/S composite exhibited high reversible capacities of ∼530 mA h g−1 and ∼380 mA h g−1 after 1000 cycles at 5C and 10C, respectively. The high performances of the DTG/S cathode were attributed to its excellent electrical conductivity, as well as to its unique porous structure, which allowed the effective storage of sulfur in the mesosized lamellar interlayer space, leading to an efficient connection between sulfur and graphene, which prevented the diffusion of polysulphides into the electrolyte.236 Besides, in order to better confine the sulfur nanocrystals in the electrode and impede polysulfide diffusion more effectively, graphene-wrapped sulfur core–shell nanocomposites have been explored.235,239,241 For example, Peng et al. employed hollow graphene nanoshells (HGNs) with a diameter of ca. 10–30 nm and a pore volume of 1.98 cm−3 g−1 as hosts to accommodate sulfur for Li–S batteries. The use of HGNs offered free space for sulfur expansion during discharge and prevented the diffusion of lithium polysulfide, resulting in an ultraslow decay rate of 0.06% per cycle during 1000 cycles. Besides, interconnected HGNs provided intercrossed ion channels for rapid diffusion of ions and excellent electron pathways, leading to a high retention of 70% when the current density increased from 0.1C to 2.0C (Fig. 9).235
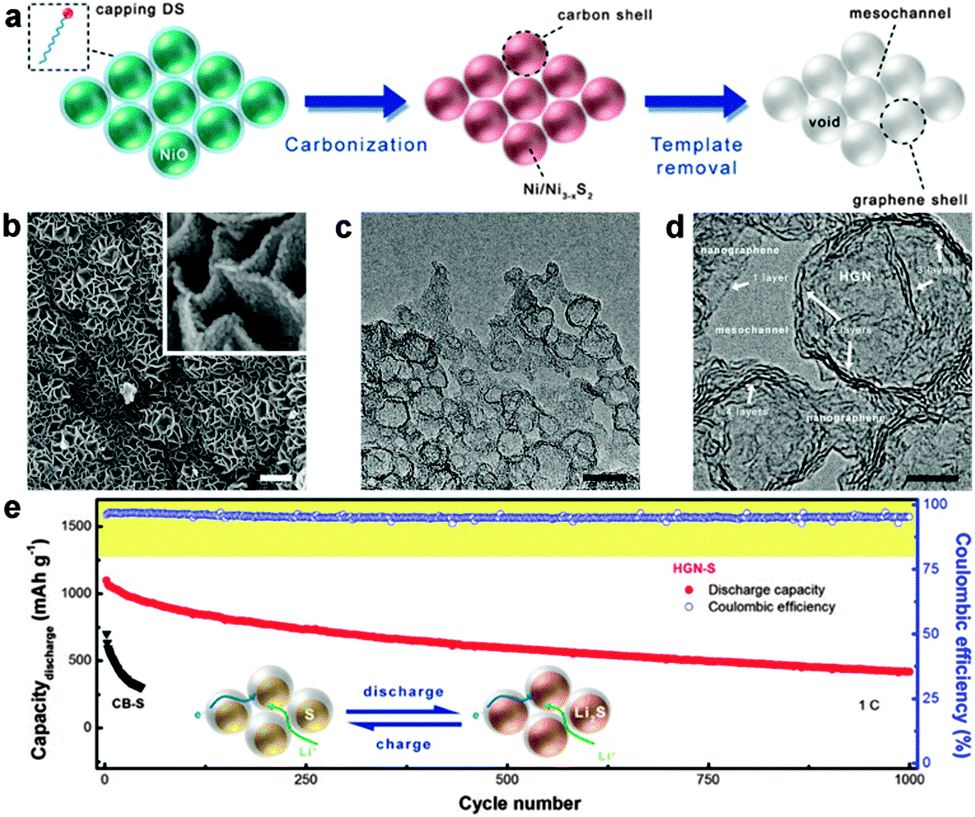 | ||
| Fig. 9 (a) Schematic illustration of the in situ catalytic self-limited assembly of HGNs. (b) SEM, (c) TEM, and (d) HR-TEM images of HGNs. The scale bars in b, inset of b, c, and d are 1 μm, 100 nm, 20 nm, and 5 nm, respectively. (e) Cycling performance at a current density of 1C and schematic illustration of HGN-S during discharge and charge (inset). Reprinted with permission from ref. 235, copyright (2014), American Chemical Society. | ||
As mentioned before, heteroatom doping can endow graphene with various new or improved properties, such as conductivity and lithium storage capacity.42,244–246 Zhang's group reported an N-doped graphene-wrapped sulfur cathode for Li–S batteries. The effects of N-doping are manifested not only in the significant improvement of the electronic conductivity of the overall S cathode, but also in the enhancement of ionic attractions between N and Li for effective lithium polysulfide trapping. Thus, the cycle life was prolonged to more than 2000 cycles with an extremely low capacity-decay rate (0.028% per cycle).245
 | ||
| Fig. 10 (a) Illustration of the formation process of the 3D graphene/S hybrid and schematic of fabrication of a self-supporting electrode. (b) SEM image of the interconnected fibrous microstructure of the graphene/S hybrid. (c) TEM and (d) HRTEM images of the graphene/S hybrid. (e) Capacity at different current densities of the graphene/S cathode. (f) Cycling performance and Coulombic efficiency of the graphene/S cathode at 0.75 A g−1 for 100 cycles after the high current density test. Reprinted with permission from ref. 238, copyright (2013), American Chemical Society. | ||
 | ||
| Fig. 11 (a) Schematic illustration for the synthesis process of the 3D-NGS composite: (a) GO suspension, (b) 3D-NG obtained from GO through a solvothermal process, and (c) the final 3D-NGS composite. (b) SEM images of the 3D-NG framework. (c) SEM image of the microstructure of the 3D-NGS composite. (d) Rate capability of 3D-NGS with current densities ranging from 100 mA g−1 to 1500 mA g−1 and the first discharge–charge profiles at different current densities. (e) Cycling stability and the corresponding coulombic efficiency of 3D-NGS at a current density of 100 mA g−1 for the first two cycles and 1500 mA g−1 for the following cycles. Reprinted with permission from ref. 250, copyright (2014), Royal Society of Chemistry. | ||
Chen's group improved the confinement of sulfur and polysulfides by uniformly coating a thin layer of porous carbon on both faces of graphene. In this architecture, graphene behaves as the electronic conductive channel, while the porous carbon layer provides high sulfur loading by impregnating sulfur in its pores and acts as a reservoir to alleviate the shuttle effect of polysulfide.252 Liu's group prepared a coaxial graphene–wrapped sulfur–carbon nanofiber (G–S–CNFs) nanocomposite for the cathode of Li–S batteries. However, it should be noted that graphene was wrapped around the S–CNF composites via electrostatic attraction. The obvious capacity decay in the first few cycles revealed that the polysulfides still partially diffused probably due to the loose cohesion between graphene and carbon nanofibers (Fig. 12).253 Compared with carbon fibers, carbon nanotubes have a much smaller diameter and a higher surface area, which is more beneficial for sulfur loading.254–256 Chen et al. sandwiched sulfur coated MWCNT composites in the interlayer of graphene sheets (GS). Due to the synergistic effects of GS and MWCNTs, much improved cycling stability and rate capability were achieved for the GS–MWCNT@S composite cathode compared with the composite lacking GS or MWCNTs.254 Besides, a higher sulfur loading of 49% was obtained compared with 33% in the case of carbon nanofibers.253 Furthermore, 3D graphene/CNT nanostructures with higher conductivity and better mesoporous structure were created as scaffolds for higher sulfur loading and superior electrochemical properties.257,258 For instance, He and co-workers synthesized a 3D CNT/graphene/sulfur (3DCGS) sponge with a high sulfur loading of 80.1%. The reversible discharge capacity for sulfur and the whole electrode was 1217 and 877.4 mA h g−1, respectively. A low capacity decay of 0.08% per cycle and a high-rate capacity of 653.4 mA h g−1 up to 4C were achieved as well (Fig. 13).257 Apart from the above carbon scaffolds, biomass-derived carbon was also composited with graphene to form a promising carbon hybrid substrate for loading sulfur in Li–S batteries.259
 | ||
| Fig. 12 (a) Schematic illustration of the assembled G–S–CNF multi-layered coaxial nanocomposites. TEM images of (b) pristine CNFs, (c) CNFs with sulfur coating, and (d) G–S–CNFs nanocomposite. (e) Capacity as a function of cycle numbers (50 cycles) of the S–CNF nanocomposite with (G–S–CNFs) and without (S–CNFs) graphene wrapping. (f) Cycling performances of the G–S–CNFs electrode at a high rate of 1C for 1500 cycles. Reprinted with permission from ref. 253, copyright (2013), American Chemical Society. | ||
 | ||
| Fig. 13 (a) Schematic of the synthesis procedure of the 3DCGS composite. (b) Cycling performance of the 3DGS and 3DCGS cathode at 0.2C for 200 cycles. (c) Rate performance at various C-rates of the 3DGS and 3DCGS cathode. Reprinted with permission from ref. 257, copyright (2015), Royal Society of Chemistry. | ||
The interactions between the components of most of the above-mentioned graphene/carbon hybrids are based on noncovalent effects such as electrostatic attraction, which may be unfavourable for polysulfide trapping and may increase contact resistance.253,255 Accordingly, covalently integrated graphene/CNT hierarchical architecture is highly desired with full inherited advantages of the component materials or even with unexpected properties.260–262 A graphene/single-walled carbon nanotube (G/SWCNT) hybrid was successfully fabricated by Wei's group via a facile template-assisted CVD process. The CNTs covalently anchored on graphene sheets enlarged internal spaces between the two stacked graphene layers for sulfur storage. A high electrical conductivity of 3130 S cm−1 enabled the as-obtained G/SWCNT–S cathode to exhibit a capacity as high as 650 mA h g−1 after 100 cycles even at a high current rate of 5C.260 Later, they explored an N-doped aligned CNT/graphene hybrid as a scaffold for sulfur. N-doping introduced more defects and active sites into the carbon framework, improving the electrochemical behaviours because of the enhanced affinity between sulfur and the scaffold. A high reversible capacity of ca. 880 mA h g−1 after 80 cycles could be achieved at 1.0C and a reversible capacity of ca. 770 mA h g−1 could be achieved even at a high current density of 5.0C.262 Furthermore, the same group coated a meso-/microporous carbon layer onto the sp2 interlinked graphene/CNT networks. In this graphene/CNT@porous carbon hybrid, the seamlessly linked graphene/CNT scaffold provides efficient pathways for electron transport, while the introduced porous carbon accommodates sulfur and polysulfides. An ultrahigh specific capacity of 1121 mA h g−1 at 0.5C and a favourable high-rate capability of 809 mA h g−1 at 10C were obtained. Moreover, the gravimetric energy density of the packaged cell was expected to be 400 W h kg−1 at a power density of 10000 W kg−1, approaching the level of engine driven systems (Fig. 14).261
 | ||
| Fig. 14 (a) Schematic illustration showing the nanoarchitectured graphene/CNT@porous carbon hybrid formation. (b) Rate performance compared with GSH–S, GSH@PC–S cathode materials in which the contents of S were ca. 50 wt%. (c) Ragone plot of the Li–S battery based on diverse sp2 nanocarbon cathode materials as well as other energy storage devices (all based on total mass of packaged device). Reprinted with permission from ref. 261, copyright (2014), Wiley-VCH. | ||
Some polymers have also been used to wrap the graphene/sulfur composites to prevent the diffusion of polysulfides. Conducting polymers, such as polyaniline (PANI),263–265 polypyrrole (PPy),266 and poly(3,4-ethylenedioxythiophene)–poly(styrene sulfonate) (PEDOT–PSS),267 with good compatibility with sulfur, are suitable for encapsulating sulfur. Dong et al. reported a rationally integrated sandwich-type hybrid nanosheet by conductive coating a PPy layer on sulfur-infiltrated graphene-backboned mesoporous carbon nanosheets. In this system, the 2D graphene backbone facilitates electron transport and ionic diffusion, the mesoporous carbon nanosheet reserves sulfur in its pores, and the outermost conducting PPy nanocoating prevents polysulfides from escaping and strengthens the entire structural integrity. The resulting sandwich-type hybrid nanosheet exhibited a high reversible capacity and a long lifespan of 400 cycles with an ultra-slow decay rate of 0.05% per cycle at the high rate of 1–3C (Fig. 15).266 Similarly, a conductive PEDOT-PSS layer was coated onto a sulfur impregnated porous graphene composite to facilitate charge transportation and prevent the dissolution of polysulfide. The sulfur composite cathode showed a reversible capacity of 718 mA h g−1 at 2C and a reversible capacity of 845 mA h g−1 at 0.1C after 200 cycles.267 Apart from the above conductive polymers, some natural polymers, such as amylopectin with hydroxyl groups, can also interact with GO to form a cross-linked 3D structure, helping to accommodate the volumetric expansion of sulfur. However, the relatively low Coulombic efficiency just stabilized around 90% and the capacity gradually faded during cycling, indicating that there was still some polysulfide dissolution and shuttling.268
 | ||
| Fig. 15 (a) Schematic illustration of the synthesis procedure of GCS@PPy hybrid nanosheets. (b) Schematic illustration of structural merits of the GCS@PPy hybrid nanosheets towards lithium storage. (c) Long-term cycling stability of the GCS@PPy electrodes at current rates of 0.5C, 1C and 3C. Cycling curve of the GS electrode at 0.5C is also displayed for comparison. Reprinted with permission from ref. 266, copyright (2015), Royal Society of Chemistry. | ||
Yushin's group reported a simple, low-cost, and scalable solution-based method to prepare nanostructured Li2S/graphene composites. The nanoparticles of Li2S precipitated heterogeneously on the defective graphene surface. There was a stable performance of Li2S/graphene composites without any Li SEI stabilizing electrolyte additive (such as LiNO3 or polysulfides) for over 200 cycles.271 Chen and co-workers reported in situ synthesis of a thermally exfoliated graphene/Li2S (TG/Li2S) nanocomposite via chemical reduction of TG/S composites by lithium triethylborohydride (LiEt3BH). A discharge capacity of 791 mA h g−1 based on the mass of Li2S after 100 cycles at 0.1C was retained. Moreover, a Li2S/Si full cell was produced by coupling the TG/Li2S cathode with the Si thin film anode. A high specific capacity of ∼900 mA h g−1 based on the mass of Li2S was delivered.272 Li et al. reported an in situ formed Li2S/graphene composite via one-pot pyrolysis of a mixture of graphene nanoplatelet aggregates (GNAs) and lithium sulfate (Li2SO4). The GNAs not only act as a reductant to reduce Li2SO4 and create Li2S at elevated temperature (Li2SO4 + 2C → 2CO2↑ + Li2S), but also are etched to thin graphene sheets, which are highly conductive 2D hosts for immobilization of Li2S.275 Recently, Wang et al. reported a Li2S/RGO cathode paper by simple drop-coating of the RGO paper with the Li2S anhydrous ethanol solution. This free-standing and binder-free Li2S/RGO paper could be directly used as a cathode without a metal substrate which showed a reversible discharge capacity of 816.1 mA h g−1 after 150 cycles at 0.1C, and 462.2 mA h g−1 after 200 cycles at 5C (Fig. 16).273 Furthermore, Hwa et al. reported the Li2S/GO nanospheres by coating a conformal carbon layer, which can prevent polysulfide dissolution and reduce the whole electrode resistance. Accordingly, the resulting core–shell Li2S/GO@C cathodes exhibited a high initial discharge capacity of 650 mA h g−1 of Li2S and a very low capacity decay rate of only 0.046% per cycle for 1500 cycles at 2C.274 Recently, Zhou et al. embedded dissolved lithium polysulphides (Li2S6) in a 3D N,S-codoped 3D graphene sponge. The N,S-codoped graphene conductive framework with high electrical conductivity and strong adsorption ability for polysulphides can provide Li ion rapid transport channels. As a result, a high-rate capacity of 430 mA h g−1 at 2C and excellent cycling stability with ∼0.078% capacity decay per cycle for 500 cycles were achieved (Fig. 17).276
 | ||
| Fig. 16 (a) Schematic illustration of the material preparation processes of the nano-Li2S/rGO paper and structural changes during cycling of nano-Li2S/RGO paper: (a) RGO paper. (b) Drop coating of the RGO paper by the solution of Li2S in anhydrous ethanol in the glovebox. (c) The final product, nano-Li2S/RGO paper. (d) The possible charge situation of the nano-Li2S/RGO paper. (b) Rate performance of the nano-Li2S/RGO paper electrode with a current rate ranging from 0.05 to 7C. (c) Cycling performance of the nano-Li2S/RGO paper electrode at a high current rate of 5C. Reprinted with permission from ref. 273, copyright (2015), American Chemical Society. | ||
 | ||
| Fig. 17 (a) A lightweight N,S-codoped graphene sponge standing on a dandelion. (b) Illustration of the formation process of the N,S-codoped graphene electrode and schematic of the fabrication of a Li/dissolved polysulphide cell with the N,S-codoped graphene electrode after adding the polysulphide catholyte. (c) SEM image of the N,S-codoped graphene sponge. (d) Rate performance of the N,S-codoped graphene electrode with a graphene-coated separator at different current densities. (e) Cycling performance and Coulombic efficiency of the Li polysulphide batteries with the N,S-codoped graphene electrodes and the graphene-coated separator at a 0.5C rate for 200 cycles. Reprinted with permission from ref. 276, copyright (2015), Nature Publishing Group. | ||
3.2 Graphene-based interlayers
As discussed above, much attention has been focused on the ‘inside’ modification of the cathodes, namely employing sulfur–graphene composites to enhance the electrical conductivity and suppress the loss of soluble polysulphide intermediates for high capacity and long cycle life. Modification of the ‘outside’ of the cathodes, such as cell configuration, may be a new strategy for improving the performance of Li–S batteries. Recent studies illustrate that adding a suitable interlayer between the sulfur cathode and separator has shown great promise in preventing the diffusion of soluble polysulphide intermediates for enhanced cyclability.230 An ideal interlayer is expected to selectively control the shuttling of soluble polysulphides anions via strong interactions, while not disturbing the rapid charge transport at the same time. Therefore, various carbon-based interlayers including microporous carbon,277 CNTs,278 GO,279 RGO,280 and graphene227,281 have been developed. Especially, Chen's group designed a unique sandwich structure with pure sulfur between two graphene membranes. One sulfur coated graphene membrane was used as a current collector to provide excellent electric conductivity and the other graphene membrane coated on a commercial polymer separator acted as an interlayer to preserve lithium polysulfides localized in the cathode side. Besides, the sandwich structure could accommodate the large volumetric expansion of sulfur during lithiation. These effects could improve the cyclic stability and rate capability of the Li–S battery.227 Later, the same group used a graphene coated commercial polypropylene (PP) separator as an internal current collector to support the sulfur cathode, resulting in a flexible integrated structure of sulfur and graphene on a PP separator for Li–S batteries. A high capacity of 663 mA h g−1 after cycling over 500 cycles at 1.5 A g−1 and a very small capacity decay of 0.064% per cycle were achieved. A high capacity of 522 mA h g−1 was still maintained at 3 A g−1 corresponding to a capacity retention of 71.1% of its initial value at 1.5 A g−1. Moreover, a flexible Li–S battery with a retained capacity of 722 mA h g−1 and a Coulombic efficiency of ∼98% at 0.75 A g−1 after 30 cycles was obtained (Fig. 18).281 | ||
| Fig. 18 (a) Schematic of the electrode configuration using an integrated structure of sulfur and the G@PP separator and the corresponding battery assembly. (b) Cycling stability of the S–G@PP separator cell at 1.5 and 3 A g−1 for 500 cycles after the high-current-density test. (c) The optical images show a red LED logo lighted by a bent Li–S battery. (d) Cycling performance of the battery under the bent state for 30 cycles at 0.75 A g−1. Reprinted with permission from ref. 281, copyright (2015), Wiley-VCH. | ||
As discussed above, the nonpolar graphene shows weak interaction with polar polysulfide anions, thus functionalized graphene with controllable surface modification has been developed for effective confinement of polysulfides. For example, Zhang and co-workers reported a permselective GO membrane for high-performance Li–S batteries. The GO membrane with negatively charged oxygen groups was permeable for positively charged lithium ions but rejected the transportation of negatively charged polysulfide ions (Sn2−) due to the electrostatic interactions. Consequently, an improved Coulombic efficiency from 67–75% to over 95–98% at 0.1C, and a reduced cyclic capacity decay rate from 0.49% per cycle to 0.23% per cycle were obtained.279 Vizintin et al. explored a fluorinated reduced graphene oxide (F-RGO) deposited on the glass fiber sheets as a separator for Li–S batteries. Better capacity retention upon cycling proved the superior polysulfide confinement efficiency of this F-RGO separator interlayer compared to the pure glass fiber separator.282 Recently, Huang et al. applied a TiO2/graphene hybrid film as a promising interlayer for Li–S batteries. The interconnected graphene provided rapid electron transfer paths and physically entrapped S and polysulfides. Meanwhile, TiO2 further chemically suppressed the dissolution of polysulfides based on electrostatic attraction (S–Ti–O). The sulfur composite cathode modified with the TiO2/graphene interlayer exhibited a high reversible capacity of 1040 mA h g−1 over 300 cycles at 0.5C (Fig. 19).283
 | ||
| Fig. 19 (a) Schematic of electrode configuration for the Li–S battery with a graphene/TiO2 coating film. (b) Typical photographs of the cathode coated with graphene/TiO2 film. Typical (c) cross-sectional SEM image and (d) front-view SEM image of the cathode with graphene/TiO2 coating film. (e) Cycling stability of PCNTs–S, PCNTs–S@G, PCNTs–S@G/3%T cathodes at 0.5C. Reprinted with permission from ref. 283, copyright (2015), Wiley-VCH. | ||
3.3 Graphene-based anodes
A Li–S battery usually contains a metallic lithium-based anode. Although lithium metal has an extremely high specific capacity (∼3860 mA h g−1), its drawbacks, especially safety issues, restrict its practical application. Uncontrolled dendrite lithium arising from unstable SEI formed on the surfaces of the Li anode may pierce the separator and cause short circuit. In addition, shuttle effects may gradually thicken the Li2S2/Li2S passivation layer on lithium anode surfaces, increasing the resistance and decreasing the Coulombic efficiency.5,11 Therefore, developing attractive anode candidates to replace metallic lithium in Li–S batteries is necessary. In addition to employing the lithium sulfide (Li2S) cathode, which can be combined with safer Li-free anodes (e.g., Si, Sn, C) to avoid potential safety vulnerabilities as mentioned above,272 recent studies demonstrate that the lithiated graphene anode may be a promising candidate for safe Li–S batteries.284–286Jang's group reported a safe graphene-supported Li metal anode to solve the above issues. Graphene sheets with a high specific surface area could significantly reduce the anode current density to prolong the dendrite initiation time and restrain the growth of dendrites.284 Mukherjee et al. reported a defect-induced plating of metallic lithium into the interior of a porous graphene network. The network structure could act as a cage to effectively entrap lithium metal and suppress the lithium dendritic growth.285 Recently, Zhang and co-workers proposed an in situ formed SEI coated graphene framework with Li deposition as a high-efficiency and high-stability Li metal anode for Li–S batteries. The graphene framework could not only inhibit the growth of Li dendrites, but also sustain the rapid Li ion diffusion. Consequently, a stable Li metal anode with a cycling Coulombic efficiency of ∼97% and dendrite-free morphology after more than 100 cycles at 1.0C was obtained.286
4. Graphene-based nanocomposites for supercapacitors
As another promising class of alternative energy storage systems for future sustainable energy supply, supercapacitors, also known as electrochemical capacitors, have attracted significant interest during the past few decades due to their long cycle life (>100000 cycles), rapid charging/discharging rate, especially ultrahigh power density (at least 10 kW kg−1).287–290 However, the commercialized supercapacitors can only deliver a limited energy density of 5–10 W h kg−1, which is much lower than that of batteries (such as 120–170 W h kg−1 for Li-ion batteries).
Generally, supercapacitors can be divided into two classes: porous carbon nanomaterial-based electrochemical double-layer capacitors (EDLCs) and conducting polymer- or transition metal oxide/hydroxide-based pseudocapacitors.
4.1 Graphene-based composites for electrochemical double-layer capacitors
In EDLCs, charge can be stored by reversible adsorption of electrolyte ions on electrode material surfaces, without involving Faradaic processes during charging/discharging processes. Theoretically, the amount of stored charge is directly proportional to the specific surface areas (SSAs) of electrode materials.12 In this regard, graphene and graphene-based nanocomposites with large SSAs and high electrical conductivity would be a perfect candidate for boosting the capacity of EDLCs (Table 5).291,292| Electrode materials | SSA (m2 g−1) | Capacitya | Energy density | Cycle life | Ref. |
|---|---|---|---|---|---|
| a The numbers 2 and 3 refer to two- and three-electrode test systems, respectively. | |||||
| Activated graphene | 3100 | 165 F g−1 (2) | 70 W h kg−1 | 10000 (∼97%) |
296 |
| Activated RGO | 2400 | 120 F g−1 (2) | 26 W h kg−1 | 2000 (∼95%) | 298 |
| Activated graphene | 3290 | 174 F g−1 (2) | 74 W h kg−1 | 1000 (∼94%) | 299 |
| 100 F cm−3 (2) | 44 W h L−1 | ||||
| Graphene hydrogel | 175 F g−1 (2) | 302 | |||
| Graphene hydrogel | 951 | 220 F g−1 (2) | 5.7 W h kg−1 | 2000 (∼92%) | 303 |
| 3D porous graphene film | 71.0 mF cm−2 (2) | 9.8 μW h cm−2 | 5000 (∼98.3%) | 304 | |
| 3D graphene | 1005 | 250 F g−1 (2) | 305 | ||
| Graphene films | 215 F g−1 (2) | 150.9 W h kg−1 | 10000 (∼97%) |
306 | |
| 273.1 F g−1 (2) | |||||
| 3D porous graphene | 835 | 169 F g−1 (2) | 3.76 W h kg−1 | 5000 (∼92%) | 307 |
| Compact graphene films | 255.5 F cm−3 (2) | 59.9 W h L−1 | 309 | ||
| Holy graphene framework | 1560 | 298 F g−1 (2) | 35 W h kg−1 | 10000 (∼91%) |
310 |
| 212 F cm−3 (2) | 49 W h L−1 | ||||
| N-doped graphene | 280 F g−1 (2) | 10000 (∼99.8%) |
315 | ||
| N-doped graphene | 465.0 | 248.4 F g−1 (2) | 5000 (∼96.1%) | 314 | |
| 3D N-doped graphene | 280 | 484 F g−1 (3) | 1000 (∼100%) | 317 | |
| N-doped graphene hydrogels | 1521 | 326 F g−1 (3) | 1200 (∼92%) | 316 | |
| 3D N,B-doped graphene | 249.0 | 239 F g−1 (3) | 8.65 W h kg−1 | 1000 (∼100%) | 318 |
| 62 F g−1 (2) | |||||
| Graphene/carbon black | 112 F g−1 (2) | 3000 (∼94%) | 322 | ||
| Graphene/CNT | 331 F g−1 (3) | 2000 (∼80%) | 323 | ||
| 3D graphene/CNT | 903 | 413 F g−1 (3) | 5000 (∼115%) | 321 | |
| 3D graphene/CNT | 237 | 318 F g−1 (2) | 11.1 W h kg−1 | 1000 (∼95%) | 320 |
| 3D graphene/CNT | 2285 | 60 W h kg−1 | 5000 (∼100%) | 325 | |
| N-doped RGO/SWCNT | 396 | 305 F cm−3 (3) | 6.3 mW h cm−3 | 10000 (∼93%) |
324 |
| 300 F cm−3 (2) | |||||
293 and a capacitance of 550 F g−1 would be obtained if the surface area could be completely utilized294 However, the capacitances of graphene-based supercapacitors obtained so far are far below the theoretical value. Ruoff et al initiated the exploration of graphene for supercapacitors.12,295–298 A chemically modified graphene (CMG) with an SSA of ∼705 m2 g−1 was synthesized by reduction of GO aqueous dispersion with hydrazine hydrate and delivered specific capacitances of 135 and 99 F g−1 in aqueous and organic electrolytes, respectively.12 It should be noted that the relatively small SSA may result in the decrease of capacity, which is mainly attributed to the aggregation and restacking of graphene sheets during electrode preparation.
In order to avoid the restacking and increase the SSA of graphene, several strategies have been developed. A chemical activation method was explored to create a 3D meso- and microporous graphene network with an ultrahigh SSA of up to ∼3290 m2 g−1.296,299 An enhanced capacitance of 165 or 200 F g−1 was obtained in organic or ionic liquid electrolytes, respectively. Although a higher SSA was obtained, the accessible surface area to electrolytes was still limited. Besides, the continuous charge transport pathway was suppressed due to the poor connections between isolated graphene sheets. To solve these problems, an effective strategy is to construct 3D graphene-based frameworks with continuously interconnected macroporous structure for the integration of perfect sheet-to-sheet connectivity, large surface area, and high electrical conductivity towards high performance.300,301 Shi's group reported the synthesis of a self-assembled graphene hydrogel (GH) via a simple hydrothermal reduction.302,303 The GH has a well-defined and crosslinked 3D porous structure, with a large SSA of ∼964 m2 g−1, exhibiting a specific capacitance as high as 220 F g−1.303 Recently, Xiong and co-workers fabricated a flexible large-area hierarchical porous graphene film by blade-casting of GO hydrogel and post-casting reduction. Such films possess interpenetrating 3D hierarchical porous structures, high strength and modulus, large specific area, and good electrical conductivity. A flexible supercapacitor fabricated with these porous graphene films exhibited high areal specific capacitance (71.0 mF cm−2 at 1 mA cm−2), excellent rate performance (79.0% retention at 100 mA cm−2), long cycle life (98.3% retention after 5000 cycles), as well as almost negligible capacity loss at different bending states (Fig. 20).304 Wang et al. developed a 3D strutted graphene with a large SSA of 1005 m2 g−1 and a high conductivity of 20000 S m−1 by using a sugar-blowing approach. The as-obtained 3D graphene nanostructures exhibited a high capacitance of 250 F g−1 at 1 A g−1.305 Li's group developed a flow-directed self-assembly method to prepare self-stacked solvated graphene films. A large amount of water trapped in the solvated graphene film could act as a “spacer” to prevent the restacking of CMG sheets. The obtained solvated graphene film displayed greatly improved performance compared to its freeze-dried or thermal-annealed forms. The maximum capacitance reached 215 F g−1 in an aqueous electrolyte and a capacitance of 156.5 F g−1 could be retained even at an ultrahigh current density of 1080 A g−1.306 Recently, Zou and co-workers reported a mild and environmentally friendly method to fabricate a 3D interconnected graphene framework, which has hierarchical macropores and in-plane nanopores. Different from the well-known KOH-activation process which can produce in-plane nanopores on graphene sheets but possibly destroy the original 2D continuity, such a 3D graphene framework was prepared by a novel NaI-activation process. As a result, the 3D hierarchically porous graphene framework with original continuity, a shortened mass diffusion length, and suppressed interlayer restacking delivered a specific capacitance of up to 169 F g−1 and surface-normalized capacitance close to 21 μF cm−2.307 Although the above reported 3D graphene nanostructures could give a high gravimetric capacitance of over 200 F g−1, a large number of void spaces (macropores) decreased the packing density (∼0.069 g cm−3) of final devices, thus leading to a low volumetric capacitance (∼18 F cm−3).2,306,308 To this point, Li's group further prepared a highly compact graphene film with a packing density of up to ∼1.33 g cm−3. This liquid-mediated densely compacted graphene film exhibited both a high gravimetric capacitance of over 200 F g−1 and volumetric capacitance exceeding 200 F cm−3. An ultrahigh energy density of 59.9 W h L−1, comparable to lead–acid batteries (50–90 W h L−1), was also obtained.309 Duan et al. explored a highly dense holey graphene framework (HGF) through a mild defect-etching reaction.310,311 The in-plane nanopores can function as the ion diffusion shortcuts between different layers of graphene to greatly speed up the ion transport across the entire film and facilitate ion access to the entire surface area, which is impossible to be realized in non-holey GF.311 In this way, both high gravimetric capacitance (298 F g−1) and volumetric capacitance (212 F cm−3) could be achieved simultaneously.310
 | ||
| Fig. 20 (a) Schematic illustration of the fabrication process of the porous graphene film. (b) Photographs of the graphene film. (c) SEM and (d) TEM images and electron diffraction patterns of the porous graphene film. (e) Photographs of the corresponding flexible supercapacitor. (f) Areal specific capacitance and (g) cyclic life at the normal and 180° bending states. Reprinted with permission from ref. 304, copyright (2015), Wiley-VCH. | ||
Doped graphene sheets are also promising electrode materials for supercapacitors because of their better conductivity and stability as well as improved wettability and the existence of pseudocapacitance.42,312,313 Chen's group developed an efficient and facile strategy to fabricate highly crumpled N-doped graphene nanosheets by thermal treatment of cyanamide modified GO. The resulting crumpled N-doped graphene nanosheets with a pore volume as high as 3.42 cm3 g−1 exhibited a high specific capacity of 248 F g−1.314 Jeong et al. developed N-doped graphene using a simple plasma doping process. The as-prepared N-doped graphene exhibited 4 times larger specific capacitance compared to that of pristine graphene.315 Guo et al. synthesized N-doped graphene hydrogels through a one-pot hydrothermal route with GO as raw material and urea as reducing-doping agent, which had a large specific surface area of ∼1500 m2 g−1 and yielded a high specific capacitance of 308 F g−1.316 The effects of N doping content and doping configurations on the capacitive performance of N-doped graphene were further studied. The results showed that graphitic N can facilitate electron transfer, thereby improving the capacitive behaviour by lowering the charge-transfer resistance of the electrode at a high current density,315 while pyridinic and pyrrolic N can offer high pseudocapacitance in an alkaline aqueous solution, consequently leading to large specific capacitance.316 Besides, a 3D N-doped graphene framework (NGF) was developed by a hydrothermal method of GO aqueous suspension mixed with pyrrole. Based on the synergetic effect of 3D open-pore structure and N doping, the 3D NGF-based supercapacitor displayed a high specific capacitance of 484 and 415 F g−1 at 1 and 100 A g−1, respectively (Fig. 21).317 A 3D B, N co-doped graphene was explored by hydrothermal reaction and freeze-drying processes using GO and ammonia boron trifluoride (NH3BF3) as precursors. The 3D B, N co-doped graphene electrodes exhibited a high capacitance of 239 F g−1 because of the synergetic effects between the two co-dopants, outperforming the control samples without doping or doped with only B or N.318
 | ||
| Fig. 21 (a and b) Photographs of an as-prepared superlight NGF and one with a piece of NGF size of 1.8 cm × 1.1 cm × 1.2 cm standing on a dandelion. (c and d) SEM images of the NGF. (e) The specific capacitances calculated from the discharge curves under different current densities. (f) Cyclic stability of the NGF-based capacitor with a current density of 100 A g−1. Reprinted with permission from ref. 317, copyright (2012), Wiley-VCH. | ||
4.2 Graphene-based composites for pseudocapacitors
Typical active pseudocapacitive materials such as conducting polymers or transition metal oxides/hydroxides possessing larger specific capacitance enable higher energy densities compared with carbon nanomaterials in EDLCs.289,327,328 However, they usually suffer from relatively poor conductivity and limited stability, inferior to EDLCs in terms of power density and cycling life.15 In this regard, multi-component electrode materials composed of graphene and pseudocapacitive electrode materials could be viable candidates for improving performances of pseudocapacitors (Table 6). Usually, in these multi-component electrode materials, metal oxides/hydroxides or conducting polymers can enhance the capacitance remarkably, while graphene, as a support, can not only increase the effective utilization of active materials, but also improve the electrical conductivity and structural integrity of the composite electrodes.329| Electrode materials | Capacitya | Energy density | Cycle life | Ref. |
|---|---|---|---|---|
| a The numbers 2 and 3 refer to two- and three-electrode test systems, respectively. | ||||
| MnO2/graphene | 280 F g−1 (3) | 1000 (93%) | 330 | |
| MnO2/graphene | 411 F g−1 (3) | 10000 (85%) |
331 | |
| Co3O4/graphene | 472 F g−1 (2) | 39.0 W h kg−1 | 1000 (95.6%) | 332 |
| Co3O4/graphene | 580 F g−1 (2) | 80 W h kg−1 | 20000 (86.2%) |
333 |
| Ni(OH)2/N-doped graphene | 1151 F g−1 (3) | 2500 (∼100%) | 334 | |
| Ni(OH)2/graphene | 1672 F g−1 (3) | 2000 (81%) | 335 | |
| NiO dot/graphene | 1181.1 F g−1 (3) | 1000 (83.5%) | 336 | |
| Fe3O4/graphene | 368 F g−1 (3) | 1000 (∼100%) | 337 | |
| V2O5/graphene | 218 F g−1 (3) | 22 W h kg−1 | 338 | |
| TiO2/graphene | 225 F g−1 (2) | 20.1 W h kg−1 | 2000 (86.5%) | 340 |
| TiO2/N-doped graphene | 385.2 F g−1 (3) | 50000 (98.8%) |
341 | |
| NiCo2O4/graphene | 618 F g−1 (3) | 342 | ||
| ZnMn2O4/graphene | 618 F g−1 (2) | 37 W h kg−1 | 4000 (91%) | 343 |
| Ni3S2/graphene@Ni | 987.8 F g−1 (3) | 3000 (97.9%) | 344 | |
| Co3S4/graphene@Ni | 1369 F g−1 (3) | 3000 (96.6%) | 344 | |
| CoNi2S4/graphene | 2009.1 F g−1 (3) | 345 | ||
| Ni(OH)2/MnO2/graphene | 1985 F g−1 (3) | 54 W h kg−1 | 347 | |
| Ni(OH)2/low-defect graphene | 1162.7 F g−1 (3) | 2000 (>90%) | 348 | |
| 3D MnO2/graphene | 29.8 F g−1 (2) | 6.8 W h kg−1 | 500 (93.3%) | 350 |
| 3D MnO2/graphene | 389 F g−1 (3) | 351 | ||
| 3D Ni(OH)2/graphene | 532.5 F g−1 (3) | 14.9 W h kg−1 | 2000 (∼100%) | 352 |
| 3D Co3O4/graphene | 757.5 F g−1 (3) | 500 (94.5%) | 353 | |
| 3D Co3O4/graphene | 1390 F g−1 (3) | 35.14 W h kg−1 | 800 (90%) | 357 |
| 3D Co3O4/graphene | 1100 F g−1 (3) | 354 | ||
| 3D NiO/graphene | 425 F g−1 (3) | 2000 (79%) | 356 | |
| 3D CoMoO4/graphene | 2741 F g−1 (3) | 100000 (96.36%) |
355 | |
| 3D NiCo2O4/graphene | 1402 F g−1 (3) | 5000 (∼76.6%) | 358 | |
| Graphene/PANI | 863.2 F g−1 (3) | 11.3 W h kg−1 | 1000 (85.6%) | 360 |
| Graphene/PANI | 1170 F g−1 (3) | 1000 (85.7%) | 361 | |
| Graphene/PANI | 23 mF cm−2 (2) | 2000 (100%) | 362 | |
| Graphene/PANI | 286 F g−1 (3) | 19.02 W h kg−1 | 2000 (94%) | 363 |
| Graphene/PANI | 431 F g−1 (3) | 500 (74%) | 371 | |
| Graphene/PPy | 350 F g−1 (2) | 1000 (∼100%) | 364 | |
| Graphene/PPy | 351 F g−1 (3) | 82.4 W h kg−1 | 1000 (82%) | 365 |
| Graphene/PANI/graphene | 581 F g−1 (3) | 10.79 W h kg−1 | 10000 (85%) |
377 |
| 3D graphene/PANI | 1024 F g−1 (3) | 120 W h kg−1 | 5000 (86.5%) | 372 |
| 3D graphene/PANI | 751.3 F g−1 (3) | 1000 (93.2%) | 373 | |
| 3D graphene/PANI | 790 F g−1 (2) | 17.6 W h kg−1 | 5000 (80%) | 374 |
| 3D graphene/PANI | 530 F g−1 (2) | 10000 (93%) |
375 | |
| 3D graphene/PANI | 380 F g−1 (2) | 5000 (90%) | 376 | |
| Coupled graphene/PANI | 590 F g−1 (3) | 1000 (91%) | 370 | |
| Coupled graphene/PANI | 422 F g−1 (3) | 1000 (76%) | 369 | |
| Graphene/MnO2/PEDOT:PSS | 380 F g−1 (3) | 3000 (95%) | 378 | |
| Graphene/MnO2/PANI | 636.5 F g−1 (3) | 10000 (85%) |
385 | |
| Graphene/Fe2O3/PANI | 638 F g−1 (3) | 107 W h kg−1 | 5000 (92%) | 379 |
| Graphene/SnO2/PPy | 616 F g−1 (3) | 19.4 W h kg−1 | 1000 (100%) | 380 |
| Graphene/SnO2/PEDOT | 184 F g−1 (3) | 22 W h kg−1 | 5000 (100%) | 381 |
| Graphene/CoFe2O4/PANI | 767.7 F g−1 (3) | 79.7 W h kg−1 | 5000 (96%) | 382 |
| Graphene/NiFe2O4/PANI | 667 F g−1 (3) | 92.7 W h kg−1 | 10000 (90%) |
383 |
| Graphene/MnFe2O4/PANI | 307.2 F g−1 (2) | 13.5 W h kg−1 | 2000 (74%) | 384 |
 | ||
| Fig. 22 (a) Possible formation mechanism of Ni(OH)2/MnO2/RGO hybrid spheres. (b) Typical SEM image of the Ni(OH)2/MnO2/RGO hybrid spheres. (c) Typical TEM images of the hybrid sphere (the inset is the SAED pattern of the whole hybrid sphere). (d) Comparison of specific capacitances of Ni(OH)2/MnO2/RGO, Ni(OH)2/MnO2/G12 (C12H25N(CH3)2(CH2)6(CH3)2NC12H25Br2), and Ni(OH)2/RGO electrodes. Reprinted with permission from ref. 347, copyright (2014), American Chemical Society. | ||
It should be noted that, for the above graphene–metal oxide/hydroxide hybrids, the noncovalent interaction between metal oxide/hydroxide and graphene limits the charge transfer at the interfaces and it is incapable of sustaining structural integrity during long-term cycling. To this end, Zhou and co-workers reported the self-assembly of MoO3 nanocrystal on graphene nanosheets by oxygen-bonding interactions at the interface. The resulting MoO3/graphene hybrids possessing abundant exposed active sites, rapid ion diffusion and electron transport exhibited high specific capacitances and excellent cycling stability in both aqueous (527 F g−1 at the current density of 1.0 A g−1, 100% retention after 10000 cycles) and solid electrolytes (373 F g−1 at 1.0 A g−1, 100% retention after 5000 cycles).349
Besides, 3D graphene nanostructures such as hydrogels, foams, and films have also been employed to prepare the graphene–metal oxide/hydroxide 3D hybrid nanostructures.350–358 For example, Choi et al. reported a 3D macroporous nanohybrid by depositing a thin layer of MnO2 onto the CMG framework. The 3D porous graphene with a large surface area allowed fast ionic and electronic transport, and thus endowed 3D MnO2/CMG hybrid electrodes with high electrochemical performances, such as a high specific capacitance of 389 F g−1 at 1 A g−1 and 97.7% capacitance retention upon a current increase to 35 A g−1.351 Our group reported a facile heterogeneous chemical deposition method to confine Ni(OH)2 particles in a graphene film, resulting in a sandwich-like 3D graphene/Ni(OH)2 film. Due to the simultaneously ordered ion diffusion channels and high electrical conductivity, the 3D hybrid film showed a high capacitance of 532.5 F g−1 and no capacitance loss after 2000 cycles.352 Recently, Yu et al. prepared a novel honeycomb-like strongly coupled CoMoO4/3D graphene hybrid by hydrothermal deposition of CoMoO4 on 3D graphene foam. This 3D CoMoO4/graphene hybrid electrode exhibited excellent specific capacitance as high as 2741 and 1101 F g−1 at a current density of 1.43 and 85.71 A g−1, respectively. Moreover, an outstanding cycling stability with 96.36% retention of its initial specific capacitance was observed after 100000 cycles at a current density of 400 A g−1 (Fig. 23).355
 | ||
| Fig. 23 (a) The typical synthesis procedure of honeycomb-like CoMoO4/3D graphene hybrid electrodes. (b) Cross-sectional SEM image of a honeycomb-like CoMoO4/3D graphene hybrid. (c) Typical TEM images of honeycomb-like CoMoO4. (d) Cycling performance of a honeycomb-like CoMoO4/3D graphene hybrid electrode for 100000 cycles at a current density of 400 A g−1. (bottom) The initial profiles for the first ten charge/discharge cycles, ten charge/discharges profiles after 50000 cycles, and ten charge/discharge profiles after 100000 cycles, respectively. Reprinted with permission from ref. 355, copyright (2014), Wiley-VCH. | ||
For these graphene-conducting polymer composites, taking graphene/PANI as an example, graphene and PANI may interact via electrostatic attraction, hydrogen bonding and π–π stacking, and the resulting synergistic effects can lead to higher electrochemical capacitance and better stability.368 However, it is still worth pointing out that the noncovalent interaction between graphene and PANI is helpless for the charge transfer at the graphene/PANI interface, and incapable of accommodating the volumetric changes of PANI during repeated charging/discharging cycles. Intimately coupled interaction, such as covalent bonding, is expected to decrease interfacial resistance and endure volumetric changes upon long-term cycling. To this end, a covalently grafted graphene/PANI composite was synthesized by Gao and co-workers. The graphene substrates were first modified with p-aniline groups, which further served as initiation sites for the chemical grafting and polymerization of aniline. The as-prepared chemically grafted graphene/PANI composite delivered a high electrochemical capacitance of 422 F g−1 at a discharge rate of 1 A g−1, which was dramatically higher than that of graphene or pure PANI.369 Similarly, Wang et al. chemically grafted the PANI chains onto surfaces of graphene modified with the aminophenyl group, which acted as initiation sites for aniline polymerization.370 Recently, Kim and co-workers reported a novel fabrication method of highly flexible and scalable graphene/PANI hybrid film by the solution process. The as-prepared redoped graphene/PANI m-cresol/chloroform suspension was cast onto a large-sized glass substrate (23 cm × 23 cm, 13 inch) and then placed on a heat source to evaporate any residual solvent. The large-scale graphene/PANI film (23 cm × 23 cm, 13 inch) was obtained after the annealing process. Additionally, due to the great advantage of the solution process, scalable films with different sizes were fabricated easily by changing the size of the glass substrate. As electrode materials for supercapacitors, these scalable and flexible films exhibited a high capacitance of 431 F g−1 and stable performances even under 180° bending (Fig. 24).371
 | ||
| Fig. 24 (a) Schematic illustration of the sequential steps for fabricating a large-scale graphene/PANI film. (b) Digital photographs of a flexible graphene/PANI film. The top image shows the folding characteristics of the film. The bottom images illustrate the high flexibility (bending and twisting) of the film. (c) CV curves of graphene/PANI film electrodes with two different bending angles of 0° and 180° at a scan rate of 5 mV s−1. Reprinted with permission from ref. 371, copyright (2014), Wiley-VCH. | ||
Moreover, 3D graphene nanostructures such as 3D graphene frameworks372–374 and 3D porous graphene films375,376 have also been used as substrates to produce 3D graphene/PANI composites. Especially, Xiao et al. reported a scalable and modular method to prepare a unique sandwich-structured RGO/PANI/RGO nanohybrid paper. The freestanding and flexible RGO paper was firstly fabricated by a facile and scalable printing technique and the bubbling delamination method. Then, a PANI layer was uniformly coated on the RGO paper by in situ electropolymerization, followed by wrapping an ultrathin RGO layer on its surface. A symmetric supercapacitor based on this RGO/PANI/RGO nanohybrid paper and the polymer gel electrolyte exhibited a large areal capacitance of 107 mF cm−2 at a current density of 2 mA cm−2, a high energy density of 5.4 mW h cm−3 (10.79 W h kg−1), and excellent cycling stability (Fig. 25).377
 | ||
| Fig. 25 (a) Schematic image illustrating the formation of a freestanding RGO paper. (b) Fabrication process of a GO film by the printing method. (c and d) Peeling GO film from the printing paper using the bubbling delamination method in 1 M HCl (since HI is dark colored, HCl was used instead to illustrate this process). (e) Photograph of a large-scale RGO paper. (f) Photograph of the RGO paper with excellent flexibility. (g) Photograph of RGO, PANI/RGO and RGO/PANI/RGO papers. (h) Areal capacitance versus discharge current for the solid-state symmetric device. The inset shows the photograph of a red LED lit by in-series solid-state SCs with three units. (i) Ragone plot of energy density versus power density for different solid-state device. Reprinted with permission from ref. 377, copyright (2015), Nature Publishing Group. | ||
The most popular strategy to prepare the ternary graphene–metal oxide-conducting polymer ternary composites includes two steps: synthesis of graphene/metal oxide binary hybrids first and then coating the conducting polymers onto the surfaces of binary hybrids. Based on this strategy, Yu et al. developed a ternary graphene/MnO2/PEDOT:PSS composite through 3D conductive wrapping of graphene/MnO2 nanostructures with PEDOT:PSS. As a result, the specific capacitance of the electrode had substantially increased to ∼380 F g−1, outperforming the graphene/MnO2 nanostructures (∼260 F g−1) without conductive wrapping. Additionally, more than 95% capacitance retention was obtained after 3000 cycles.378 Our group reported series work on graphene–metal oxide-conducting polymer ternary composites, such as graphene/Fe2O3/PANI,379 graphene/SnO2/PPy,380 and graphene/SnO2/PEDOT,381 and demonstrated their improved electrochemical performances. Further considering that mixed transition metal oxides such as ferrites exhibit unique electrochemical attributes such as different redox states and good electrochemical stability, a series of graphene–ferrite-conducting polymer ternary hybrids, including graphene/CoFe2O4/PANI,382 graphene/NiFe2O4/PANI383 and graphene/MnFe2O4/PANI384 have also been explored as upgraded electrode materials for supercapacitors by our group. These ternary graphene–metal oxide-conducting polymer hybrids exhibited large specific capacitance, good rate capability and high cycling stability, superior to those of individual components and corresponding binary hybrids. Recently, Li et al. reported a facile two-step strategy to fabricate a free-standing MnO2 nanoflake/PANI nanorod/RGO ternary composite paper. MnO2 nanoflakes were first electrodeposited on RGO paper, followed by assembly of PANI nanorods between MnO2 nanoflakes by in situ polymerization. The flexible MnO2/PANI/RGO ternary hybrid paper was directly used as the working electrode and showed a large specific capacitance (636.5 F g−1 at 1.0 A g−1), high rate capacity (53.8% capacitance retention at 50 A g−1), and excellent cycling stability (85% capacitance retention after 104 cycles) (Fig. 26).385
 | ||
| Fig. 26 (a) Schematic illustration of the preparation process of the RGO/MnO2/PANI nanocomposite. (b) Digital images of the RGO paper (the inset shows the digital image of RGO/MnO2/PANI). (c) SEM image of the cross-sectional view of RGO/MnO2/PANI. (d) Specific capacitances of RGO/PANI, RGO/MnO2 and RGO/MnO2/PANI at different current densities. (e) Cycling stability of RGO/MnO2/PANI at a current density of 20 A g−1, the inset shows the last 10 cycles of the charge/discharge curves. Reprinted with permission from ref. 385, copyright (2015), Royal Society of Chemistry. | ||
4.3 Graphene-based composites for asymmetric supercapacitors
In order to achieve high energy density without sacrificing power density, asymmetric supercapacitors (ASCs) that integrate the Faradic electrode (as an energy source) and the capacitive electrode (as a power source) in one configuration have been extensively explored. Usually, carbon-based materials, such as graphene with high surface area and conductivity, which facilitates fast charge/discharge, can be utilized for the negative electrode (capacitive electrode), while transition metal oxides and conducting polymers, which provide reversible redox reactions, are suitable for the positive electrode (Faradic electrode). Accordingly, there is no doubt that graphene/transition metal oxide composites and graphene/conducting polymer hybrids with improved specific capacitance and cycling stability can also be used as positive electrodes. In other words, graphene-based nanostructures can be employed in both negative and positive electrodes for ASCs with high power and energy density (Table 7).386–395| Electrode materials | Capacitya | Energy densityb | Power densityc | Cycle life | Ref. |
|---|---|---|---|---|---|
| a Specific capacitance of asymmetric supercapacitors. b Maximum energy density of asymmetric supercapacitors. c Maximum power density of asymmetric supercapacitors. | |||||
| IL–CMG//RuO2/IL–CMG | 175 F g−1 | 19.7 W h kg−1 | 6.8 kW g−1 | 2000 (∼95%) | 386 |
| Porous graphene//Ni(OH)2/graphene | 218.4 F g−1 | 77.8 W h kg−1 | 15.2 kW kg−1 | 3000 (∼94.3%) | 387 |
| Activated graphene//PANI/graphene/CNT | 107 F g−1 | 41.5 W h kg−1 | 25.0 kW kg−1 | 5000 (∼91.4%) | 389 |
| RGO paper//MnO2/RGO paper | 113 mF cm−2 | 35.1 μW h cm−2 | 3.8 mW cm−2 | 3600 (∼84%) | 388 |
| Fe3O4/graphene//NiOOH/Ni3S2/3D graphene | 233 F g−1 | 82.5 W h kg−1 | 930 W kg−1 | 392 | |
| RGO//Ni(OH)2/RGO | 75 W h kg−1 | 40000 W kg−1 |
10000 (∼89%) |
394 | |
| CNT/graphene//Mn3O4/graphene | 72.6 F g−1 | 32.7 W h kg−1 | 9.0 kW kg−1 | 10000 (∼86%) |
396 |
Yan et al. fabricated a high-voltage ASC using porous graphene as a negative electrode and Ni(OH)2/graphene as a positive electrode, respectively. The optimized ASC could be cycled reversibly in a high-voltage region of 0–1.6 V and displayed a maximum specific capacitance of 218.4 F g−1 and a high energy density of 77.8 W h kg−1.387 Sharma's group reported an ASC using graphene nanoribbons (GNRs) as a negative electrode and metal oxide modified GNRs as a positive electrode, respectively.393,395 For example, an ASC using GNR and CoMn2O4/GNR composites as negative and positive electrodes, respectively, demonstrated a high energy density of 84.69 W h kg−1 and long term stable cycling performance with ∼96% capacitance retention and ∼97.5% columbic efficiency.393 In addition, metal oxide or carbon nanomaterial modified graphene can also be used as negative electrode for ASCs.390–392 For example, an all-solid-state ASC with MnO2/graphene foam as the positive electrode and CNT/graphene as the negative electrode exhibited high energy density, remarkable rate capability, and reasonable cycling performance in a high-voltage region of 0 to 1.8 V.391 In some cases, graphene-based ternary hybrids have been exploited as negative or positive electrodes for ASCs. Liu et al. deposited MnO2 or PPy onto 3D graphene foam/carbon nanotube (GF/CNT) films and then fabricated an ASC based on the resulting GF/CNT/MnO2 and GF/CNT/PPy ternary hybrid films. The ASC was able to provide an output voltage of 1.6 V and deliver a high energy density of 22.8 W h kg−1.390
The development of smart wearable or rolling-up electronic devices has prompted the demand for flexible energy storage devices, including flexible ASCs that can function under considerable physical deformation. Carbonaceous nanomaterials, especially graphene, has been widely used to fabricate flexible ASCs because of its unprecedented combination of large surface area, superior electrical conductivity, and excellent mechanical flexibility.386,390,391,396,397 Choi et al. fabricated a solid-state flexible ASC based on an ionic liquid functionalized-chemically modified graphene (IL–CMG) film as the negative electrode and a hydrous RuO2/IL–CMG composite film as the positive electrode. The as-prepared all-graphene film-based ASCs not only showed a high energy density (19.7 W h kg−1) and power density (6.8 kW g−1), but also could operate even under harsh mechanical conditions including twisted and bent states (Fig. 27).386 Gao et al. reported an all-solid-state ASC based on free-standing CNT/graphene paper and Mn3O4/graphene paper electrodes. The resultant all-graphene paper-based ASC demonstrated an increased cell voltage of 1.8 V, stable cycling performance (capacitance retention of 86.0% after 10000 cycles), an improved energy density of 32.7 W h kg−1, and more importantly, a distinguished mechanical flexibility (Fig. 28).396
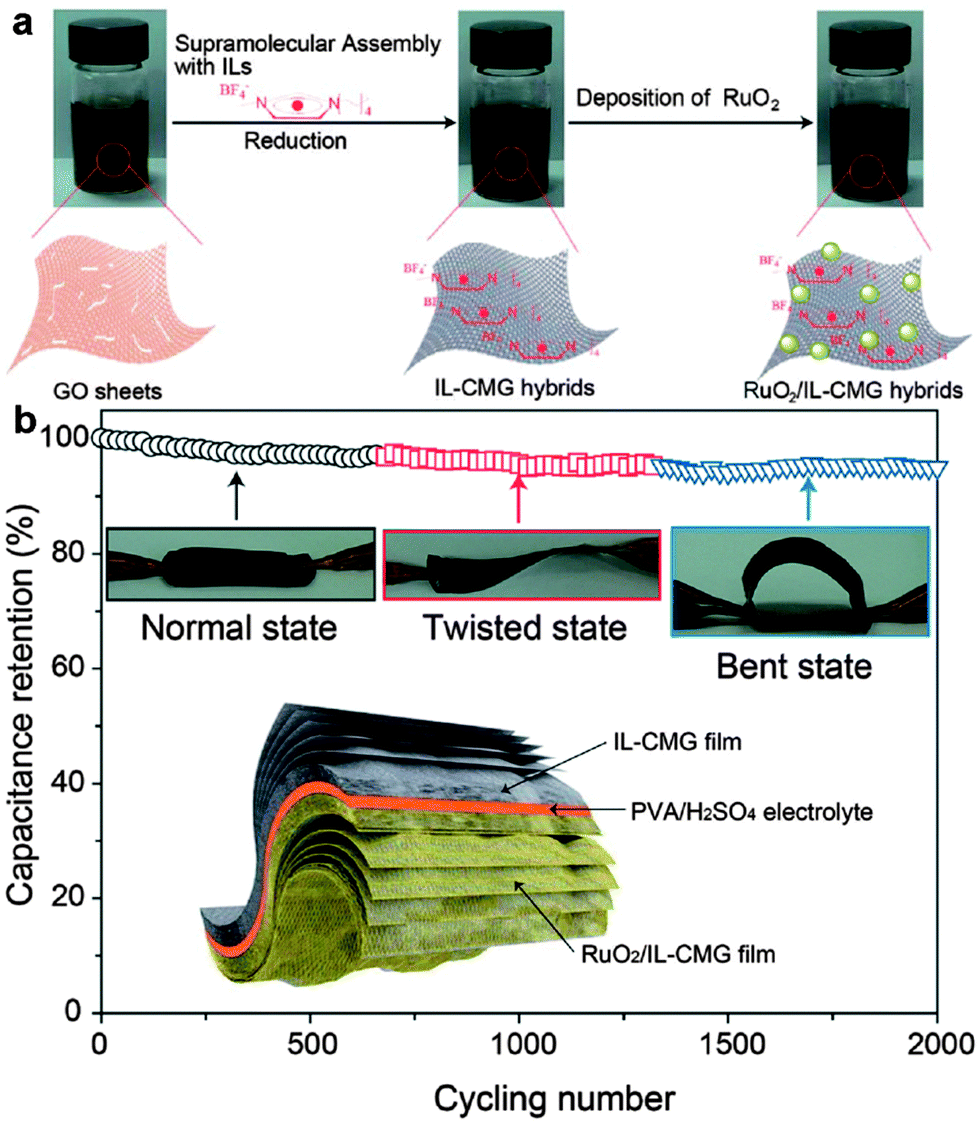 | ||
| Fig. 27 (a) Experimental procedure of the synthesis for water-soluble IL–CMG and RuO2/IL–CMG hybrids. (b) Long-term cycling stability of ASC devices at normal, twisted, and bent states with a constant current density of 1 A g−1 over 2000 cycles. The inset shows the schematic diagram of an all-solid-state flexible thin ASC. Reprinted with permission from ref. 386, copyright (2012), Royal Society of Chemistry. | ||
 | ||
| Fig. 28 (a) Illustration of the fabrication process of a flexible all-solid-state ASC based on a polymer gel electrolyte and free-standing CNT/graphene and Mn3O4/graphene paper electrodes. (b) Cross-sectional SEM image of the CNT/graphene paper. (c) Photograph shows that the free-standing CNT/graphene paper is flexible enough to be folded up. (d) Cross-section SEM image of the Mn3O4/graphene paper. (e) Photograph shows that the free-standing Mn3O4/graphene paper is flexible enough to be folded up. (f) Ragone plots of ASC (line a) and two corresponding symmetric supercapacitors (line b and c). (g) Cycling behavior of ASC with a cell voltage of 1.8 V at a scan rate of 50 mV s−1. (h) A green light-emitting diode (LED) lighted by two ASCs connected in series. (i) Specific capacitance retention ratio of the ASC at normal (n), bending (b), and twisting (t) states and after being bent repeatedly (number of times indicated). The inset shows the photograph of the ASC at normal, bending, and twisting states. Reprinted with permission from ref. 396, copyright (2012), American Chemical Society. | ||
5. Conclusions, challenges and perspectives
Owing to its unique properties such as 2D structure, large specific surface area, excellent electrical conductivity, together with strong mechanical flexibility, graphene has shown great potential not only in its direct use as an electrode material, but also in acting as a versatile building block for synthesis of graphene-based hybrid nanostructures with improved energy storage capability. This review summarized the recent progress on graphene and graphene-based hybrid nanostructures for three frontier energy storage systems, that is, Li-ion batteries, Li–S batteries and supercapacitors. Graphene and its derivatives, such as doped graphene and 3D graphene frameworks, as well as graphene-based multicomponent hybrids with tunable pore structure and morphology, large surface area, and an interconnected charge transport pathway, have shown improved performances in terms of specific capacity, rate capability, cycling stability, and power/energy densities. These exciting developments have opened up new pathways for high-performance graphene-based materials for energy storage.Despite the advances, great challenges urgently need to be addressed before future commercialization. Firstly, similar to many other novel materials in the past, the main important thing is to shorten the distance between laboratory-scale research and practical applications of graphene-based hybrids for energy storage. Nowadays, most graphene-based materials are produced by the reduction of GO to RGO, which has the potential for scalability but may introduce intrinsic and extrinsic defects. These defects can strongly affect electrochemical properties. For example, more defects on graphene are propitious to enhance the Li storage capability, but obviously reduce the conductivity of graphene. As a result, RGO-based anodes exhibit an attractive initial capacity but often suffer a low first-cycle Coulombic efficiency and fast capacity fading. Therefore, how to manufacture large-scale and high-quality graphene and graphene-based materials in a cost-effective way is the most essential issue. Secondly, the mechanisms for the improved performances of graphene and graphene-based hybrids are still not completely clear. A better understanding of the charge transfer and storage mechanisms on single-layer graphene and graphene-based hybrids, as well as the relationship between charge storage and defects, layer number, size, and surface state of graphene could provide a direction for the nanostructure design and property optimization. To this end, in situ characterization techniques combined with theoretical calculations are required to reveal the complicated mechanisms. Thirdly, the highly porous nature of graphene offers a large surface area and favourable ion diffusion for high gravimetric capacitance but results in low volumetric energy density due to the low packing density. Graphene-based composites containing a large portion of graphene usually show a low packing density. While a high loading of nanoparticles on graphene usually results in poor cycle stability because of their weak interaction and the severe aggregation of nanoparticles on graphene sheets. Therefore, to improve the volumetric energy density and maintain high gravimetric capacitance simultaneously, the mass ratio of graphene to nanoparticles should be appropriately controlled. Finally, it is crucial to achieve the desirable controllability of interface interactions among individual components inside of graphene-based hybrids, especially for energy storage applications that involve charge transfer processes. A well-integrated hybrid with intimate interactions such as chemical covalent bonding will be able to offer effective charge transport for high rate capability as well as well-sustained structural integrity for long cycle life. Unfortunately, most graphene-based composites reported so far are synthesized simply by mixing or dispersing all components, which leads to a relatively poor interfacial interaction. There is little reason to believe that a breakthrough in energy storage can be achieved by such cursory blend.
In addition to the above mentioned systems, graphene and graphene-based composites have also shown outstanding performances in sodium-ion batteries,398–402 lithium–air batteries,403–406 and sodium–air batteries.407,408 More encouragingly, increasing efforts has been devoted to the development of graphene-based energy storage devices with small, thin, lightweight and even flexible properties for advanced wearable electronics. Although many challenges still need to be overcome, the past few years have witnessed the exciting advances in the research on graphene and graphene-based hybrids for energy storage. In the future, it may be possible that low-cost graphene-based supercapacitors exhibit a power density of ∼100 kW kg−1, batteries show an energy density of at least 250 W h kg−1 and stable cyclability up to 5000 cycles. Overall, with the continuous efforts of many scientists and engineers around the world, it is believed that these interesting targets will be realized in the future.
Acknowledgements
This work was supported by National High Technology Research and Development Program of China (863 Program, No. 2013AA050905), NSF of China (No. 51572125, 51472101, 21206075) and PAPD of Jiangsu.Notes and references
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271 CrossRef PubMed.
- D. P. Dubal, O. Ayyad, V. Ruiz and P. Gomez-Romero, Chem. Soc. Rev., 2015, 44, 1777 RSC.
- K. Chen, S. Song, F. Liu and D. Xue, Chem. Soc. Rev., 2015, 44, 6230 RSC.
- X. Fang and H. Peng, Small, 2015, 11, 1488 CrossRef PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef PubMed.
- A. M. Chockla, K. C. Klavetter, C. B. Mullins and B. A. Korgel, ACS Appl. Mater. Interfaces, 2012, 4, 4658 Search PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef PubMed.
- J. Zhu, D. Yang, Z. Yin, Q. Yan and H. Zhang, Small, 2014, 10, 3480 CrossRef PubMed.
- G. Kucinskis, G. Bajars and J. Kleperis, J. Power Sources, 2013, 240, 66 CrossRef.
- M. Yu, R. Li, M. Wu and G. Shi, Energy Storage Mater., 2015, 1, 51 CrossRef.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef PubMed.
- P. Lian, X. Zhu, S. Liang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2010, 55, 3909 CrossRef.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644 CrossRef PubMed.
- Y. Huang, J. Liang and Y. Chen, Small, 2012, 8, 1805 CrossRef PubMed.
- N. C. Zhang, H. Yin and Y. Hou, J. Mater. Chem. A, 2014, 2, 15 Search PubMed.
- M. Srivastava, J. Singh, T. Kuila, R. K. Layek, N. H. Kim and J. H. Lee, Nanoscale, 2015, 7, 4820 RSC.
- W. Sun and Y. Wang, Nanoscale, 2014, 6, 11528 RSC.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef PubMed.
- Y.-K. Sun, S.-T. Myung, B.-C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320 CrossRef PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS PubMed.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novák, Adv. Mater., 1998, 10, 725 CrossRef CAS.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Science, 1995, 270, 590 CAS.
- E. Yoo, J. Kim, E. Hosono, H.-S. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277 CrossRef CAS PubMed.
- G. Wang, X. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049 CrossRef CAS.
- D. Pan, S. Wang, B. Zhao, M. Wu, H. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136 CrossRef CAS.
- L.-Z. Bai, D.-L. Zhao, T.-M. Zhang, W.-G. Xie, J.-M. Zhang and Z.-M. Shen, Electrochim. Acta, 2013, 107, 555 CrossRef CAS.
- R. Mukherjee, A. V. Thomas, A. Krishnamurthy and N. Koratkar, ACS Nano, 2012, 6, 7867 CrossRef CAS PubMed.
- X. Li, Y. Hu, J. Liu, A. Lushington, R. Li and X. Sun, Nanoscale, 2013, 5, 12607 RSC.
- D. Yoon, K. Y. Chung, W. Chang, S. M. Kim, M. J. Lee, Z. Lee and J. Kim, Chem. Mater., 2015, 27, 266 CrossRef CAS.
- S.-L. Kuo, W.-R. Liu, C.-P. Kuo, N.-L. Wu and H.-C. Wu, J. Power Sources, 2013, 244, 552 CrossRef CAS.
- O. A. Vargas C, A. Caballero and J. Morales, Nanoscale, 2012, 4, 2083 RSC.
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4, 6337 CrossRef CAS PubMed.
- Z.-S. Wu, W. Ren, L. Xu, F. Li and H.-M. Cheng, ACS Nano, 2011, 5, 5463 CrossRef CAS PubMed.
- H. Wang, C. Zhang, Z. Liu, L. Wang, P. Han, H. Xu, K. Zhang, S. Dong, J. Yao and G. Cui, J. Mater. Chem., 2011, 21, 5430 RSC.
- C. He, R. Wang, H. Fu and P. K. Shen, J. Mater. Chem. A, 2013, 1, 14586 CAS.
- Y. Liu, X. Wang, Y. Dong, Z. Wang, Z. Zhao and J. Qiu, J. Mater. Chem. A, 2014, 2, 16832 CAS.
- Y. S. Yun, V.-D. Le, H. Kim, S.-J. Chang, S. J. Baek, S. Park, B. H. Kim, Y.-H. Kim, K. Kang and H.-J. Jin, J. Power Sources, 2014, 262, 79 CrossRef CAS.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932 CrossRef CAS PubMed.
- X. Wang, G. Sun, P. Routh, D.-H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067 RSC.
- C. N. R. Rao, K. Gopalakrishnan and A. Govindaraj, Nano Today, 2014, 9, 324 CrossRef CAS.
- Y. Xue, B. Wu, Q. Bao and Y. Liu, Small, 2014, 10, 2975 CrossRef CAS PubMed.
- C. Ma, X. Shao and D. Cao, J. Mater. Chem., 2012, 22, 8911 RSC.
- I.-Y. Jeon, M. J. Ju, J. Xu, H.-J. Choi, J.-M. Seo, M.-J. Kim, I. T. Choi, H. M. Kim, J. C. Kim, J.-J. Lee, H. K. Liu, H. K. Kim, S. Dou, L. Dai and J.-B. Baek, Adv. Funct. Mater., 2015, 25, 1170 CrossRef CAS.
- J. Xu, I.-Y. Jeon, J.-M. Seo, S. Dou, L. Dai and J.-B. Baek, Adv. Mater., 2014, 26, 7317 CrossRef CAS PubMed.
- L. Zhan, S. Yang, Y. Wang, Y. Wang, L. Ling and X. Feng, Adv. Mater. Interfaces, 2014, 1, 1300149 Search PubMed.
- W. Ai, Z. Luo, J. Jiang, J. Zhu, Z. Du, Z. Fan, L. Xie, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2014, 26, 6186 CrossRef CAS PubMed.
- C. Wang, D. Li, C. O. Too and G. G. Wallace, Chem. Mater., 2009, 21, 2604 CrossRef CAS.
- G. Ning, C. Xu, Y. Cao, X. Zhu, Z. Jiang, Z. Fan, W. Qian, F. Wei and J. Gao, J. Mater. Chem. A, 2013, 1, 408 CAS.
- M. Ye, Z. Dong, C. Hu, H. Cheng, H. Shao, N. Chen and L. Qu, Small, 2014, 10, 5035 CAS.
- S. Y. Jeong, S. Yang, S. Jeong, I. J. Kim, H. J. Jeong, J. T. Han, K.-J. Baeg and G.-W. Lee, Small, 2015, 11, 2774 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Zhong, B. Papandrea, Y. Huang and X. Duan, Angew. Chem., Int. Ed., 2015, 54, 5345 CrossRef CAS PubMed.
- Z.-L. Wang, D. Xu, H.-G. Wang, Z. Wu and X.-B. Zhang, ACS Nano, 2013, 7, 2422 CrossRef CAS PubMed.
- Z.-J. Jiang and Z. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 19082 CAS.
- X. Jia, G. Zhang, T. Wang, X. Zhu, F. Yang, Y. Li, Y. Lu and F. Wei, J. Mater. Chem. A, 2015, 3, 15738 CAS.
- X. Liu, Y. Wu, Z. Yang, F. Pan, X. Zhong, J. Wang, L. Gu and Y. Yu, J. Power Sources, 2015, 293, 799 CrossRef CAS.
- Z.-Y. Sui, C. Wang, Q.-S. Yang, K. Shu, Y.-W. Liu, B.-H. Han and G. G. Wallace, J. Mater. Chem. A, 2015, 3, 18229 CAS.
- Z.-S. Wu, G. Zhou, L.-C. Yin, W. Ren, F. Li and H.-M. Cheng, Nano Energy, 2012, 1, 107 CrossRef CAS.
- X. Huang, Z. Zeng, Z. Fan, J. Liu and H. Zhang, Adv. Mater., 2012, 24, 5979 CrossRef CAS PubMed.
- Y. J. Mai, D. Zhang, Y. Q. Qiao, C. D. Gu, X. L. Wang and J. P. Tu, J. Power Sources, 2012, 216, 201 CrossRef CAS.
- Y. Sun, X. Hu, W. Luo, F. Xia and Y. Huang, Adv. Funct. Mater., 2013, 23, 2436 CrossRef CAS.
- L. Li, A.-R. O. Raji and J. M. Tour, Adv. Mater., 2013, 25, 6298 CrossRef CAS PubMed.
- Y. Li, Q. Zhang, J. Zhu, X.-L. Wei and P. K. Shen, J. Mater. Chem. A, 2014, 2, 3163 CAS.
- Z.-Y. Sui, C. Wang, K. Shu, Q.-S. Yang, Y. Ge, G. G. Wallace and B.-H. Han, J. Mater. Chem. A, 2015, 3, 10403 CAS.
- T. Yang, T. Qian, M. Wang, J. Liu, J. Zhou, Z. Sun, M. Chen and C. Yan, J. Mater. Chem. A, 2015, 3, 6291 CAS.
- I. Nam, N. D. Kim, G.-P. Kim, J. Park and J. Yi, J. Power Sources, 2013, 244, 56 CrossRef CAS.
- S.-K. Park, A. Jin, S.-H. Yu, J. Ha, B. Jang, S. Bong, S. Woo, Y.-E. Sung and Y. Piao, Electrochim. Acta, 2014, 120, 452 CrossRef CAS.
- G. Wang, H. Wang, S. Cai, J. Bai, Z. Ren and J. Bai, J. Power Sources, 2013, 239, 37 CrossRef CAS.
- L. Xiao, D. Wu, S. Han, Y. Huang, S. Li, M. He, F. Zhang and X. Feng, ACS Appl. Mater. Interfaces, 2013, 5, 3764 CAS.
- J. Kan and Y. Wang, Sci. Rep., 2013, 3, 3502 Search PubMed.
- R. Wang, C. Xu, J. Sun, L. Gao and C. Lin, J. Mater. Chem. A, 2013, 1, 1794 CAS.
- J. Luo, J. Liu, Z. Zeng, C. F. Ng, L. Ma, H. Zhang, J. Lin, Z. Shen and H. J. Fan, Nano Lett., 2013, 13, 6136 CrossRef CAS PubMed.
- X. Hu, M. Ma, M. Zeng, Y. Sun, L. Chen, Y. Xue, T. Zhang, X. Ai, R. G. Mendes, M. H. Rümmeli and L. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 22527 CAS.
- L. Zhao, M. Gao, W. Yue, Y. Jiang, Y. Wang, Y. Ren and F. Hu, ACS Appl. Mater. Interfaces, 2015, 7, 9709 CAS.
- Z. Zhang, F. Wang, Q. An, W. Li and P. Wu, J. Mater. Chem. A, 2015, 3, 7036 CAS.
- X.-l. Huang, R.-Z. Wang, D. Xu, Z.-l. Wang, H.-G. Wang, J.-J. Xu, Z. Wu, Q.-C. Liu, Y. Zhang and X.-B. Zhang, Adv. Funct. Mater., 2013, 23, 4345 CrossRef CAS.
- M. Zhang, F. Yan, X. Tang, Q. Li, T. Wang and G. Cao, J. Mater. Chem. A, 2014, 2, 5890 CAS.
- S. J. R. Prabakar, R. S. Babu, M. Oh, M. S. Lah, S. C. Han, J. Jeong and M. Pyo, J. Power Sources, 2014, 272, 1037 CrossRef CAS.
- B. G. Choi, S.-J. Chang, Y. B. Lee, J. S. Bae, H. J. Kim and Y. S. Huh, Nanoscale, 2012, 4, 5924 RSC.
- X. Yang, K. Fan, Y. Zhu, J. Shen, X. Jiang, P. Zhao, S. Luan and C. Li, ACS Appl. Mater. Interfaces, 2013, 5, 997 CAS.
- L. Li, G. Zhou, X.-Y. Shan, S. Pei, F. Li and H.-M. Cheng, J. Power Sources, 2014, 255, 52 CrossRef CAS.
- J. Xu, J. Wu, L. Luo, X. Chen, H. Qin, V. Dravid, S. Mi and C. Jia, J. Power Sources, 2015, 274, 816 CrossRef CAS.
- M. Zhang, R. Li, X. Chang, C. Xue and X. Gou, J. Power Sources, 2015, 290, 25 CrossRef CAS.
- Z. X. Huang, Y. Wang, Y. G. Zhu, Y. Shi, J. I. Wong and H. Y. Yang, Nanoscale, 2014, 6, 9839 RSC.
- K. Palanisamy, Y. Kim, H. Kim, J. M. Kim and W.-S. Yoon, J. Power Sources, 2015, 275, 351 CrossRef CAS.
- X. Wang, Y. Xiao, J. Wang, L. Sun and M. Cao, J. Power Sources, 2015, 274, 142 CrossRef CAS.
- L. Noerochim, J.-Z. Wang, D. Wexler, Z. Chao and H.-K. Liu, J. Power Sources, 2013, 228, 198 CrossRef CAS.
- G. D. Park, J. H. Kim, Y. J. Choi and Y. C. Kang, Electrochim. Acta, 2015, 173, 581 CrossRef CAS.
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef CAS PubMed.
- Q. Su, D. Xie, J. Zhang, G. Du and B. Xu, ACS Nano, 2013, 7, 9115 CrossRef CAS PubMed.
- X. Yao, J. Kong, D. Zhou, C. Zhao, R. Zhou and X. Lu, Carbon, 2014, 79, 493 CrossRef CAS.
- L. Lin and Q. Pan, J. Mater. Chem. A, 2015, 3, 1724 CAS.
- Y. Dong, Y. Xia, Y.-S. Chui, C. Cao and J. A. Zapien, J. Power Sources, 2015, 275, 769 CrossRef CAS.
- P. Xiong, B. Liu, V. Teran, Y. Zhao, L. Peng, X. Wang and G. Yu, ACS Nano, 2014, 8, 8610 CrossRef CAS PubMed.
- Z. Zheng, Y. Cheng, X. Yan, R. Wang and P. Zhang, J. Mater. Chem. A, 2014, 2, 149 CAS.
- D. Cai, D. Wang, H. Huang, X. Duan, B. Liu, L. Wang, Y. Liu, Q. Li and T. Wang, J. Mater. Chem. A, 2015, 3, 11430 CAS.
- Y. Dong, Y.-S. Chui, X. Yang, R. Ma, J.-M. Lee and J. A. Zapien, ChemElectroChem, 2015, 2, 1010 CrossRef CAS.
- H. Xia, D. Zhu, Y. Fu and X. Wang, Electrochim. Acta, 2012, 83, 166 CrossRef CAS.
- S. Liu, J. Xie, C. Fang, G. Cao, T. Zhu and X. Zhao, J. Mater. Chem., 2012, 22, 19738 RSC.
- M. Zhang, X. Yang, X. Kan, X. Wang, L. Ma and M. Jia, Electrochim. Acta, 2013, 112, 727 CrossRef CAS.
- B. Wang, S. Li, J. Liu, M. Yu, B. Li and X. Wu, Electrochim. Acta, 2014, 146, 679 CrossRef CAS.
- S. Li, B. Wang, J. Liu and M. Yu, Electrochim. Acta, 2014, 129, 33 CrossRef CAS.
- G. Zeng, N. Shi, M. Hess, X. Chen, W. Cheng, T. Fan and M. Niederberger, ACS Nano, 2015, 9, 4227 CrossRef CAS PubMed.
- L. Wang, L. Zhuo, C. Zhang and F. Zhao, J. Power Sources, 2015, 275, 650 CrossRef CAS.
- Y. Xiao, J. Zai, X. Li, Y. Gong, B. Li, Q. Han and X. Qian, Nano Energy, 2014, 6, 51 CrossRef CAS.
- Y. Fu, Y. Wan, H. Xia and X. Wang, J. Power Sources, 2012, 213, 338 CrossRef CAS.
- E. K. Heidari, B. Zhang, M. H. Sohi, A. Ataie and J.-K. Kim, J. Mater. Chem. A, 2014, 2, 8314 CAS.
- G. Gao, H. B. Wu and X. W. Lou, Adv. Energy Mater., 2014, 4, 1400422 Search PubMed.
- Y. Chen, J. Zhu, B. Qu, B. Lu and Z. Xu, Nano Energy, 2014, 3, 88 CrossRef CAS.
- Y. Chen, M. Zhuo, J. Deng, Z. Xu, Q. Li and T. Wang, J. Mater. Chem. A, 2014, 2, 4449 CAS.
- S. Liu, J. Wu, J. Zhou, G. Fang and S. Liang, Electrochim. Acta, 2015, 176, 1 CrossRef CAS.
- S. Liu, J. Xie, Q. Su, G. Du, S. Zhang, G. Cao, T. Zhu and X. Zhao, Nano Energy, 2014, 8, 84 CrossRef CAS.
- H. Tang, P. Gao, A. Xing, S. Tian and Z. Bao, RSC Adv., 2014, 4, 28421 RSC.
- Y. Xiao, J. Zai, L. Tao, B. Li, Q. Han, C. Yu and X. Qian, Phys. Chem. Chem. Phys., 2013, 15, 3939 RSC.
- C. Yuan, H. B. Wu, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488 CrossRef CAS PubMed.
- H. Xia, Y. Qian, Y. Fu and X. Wang, Solid State Sci., 2013, 17, 67 CrossRef CAS.
- Y. Fu, Q. Chen, M. He, Y. Wan, X. Sun, H. Xia and X. Wang, Ind. Eng. Chem. Res., 2012, 51, 11700 CrossRef CAS.
- G. Zeng, J. Zhang, Y. Fu, F. Nie and X. Wang, Int. J. Mater. Res., 2015, 106, 915 CrossRef CAS.
- W. Li, F. Wang, S. Feng, J. Wang, Z. Sun, B. Li, Y. Li, J. Yang, A. A. Elzatahry, Y. Xia and D. Zhao, J. Am. Chem. Soc., 2013, 135, 18300 CrossRef CAS PubMed.
- X. Jiang, X. Yang, Y. Zhu, H. Jiang, Y. Yao, P. Zhao and C. Li, J. Mater. Chem. A, 2014, 2, 11124 CAS.
- J. Qiu, P. Zhang, M. Ling, S. Li, P. Liu, H. Zhao and S. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 3636 CAS.
- E. Liu, J. Wang, C. Shi, N. Zhao, C. He, J. Li and J.-Z. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 18147 CAS.
- V. Etacheri, J. E. Yourey and B. M. Bartlett, ACS Nano, 2014, 8, 1491 CrossRef CAS PubMed.
- B. Qiu, M. Xing and J. Zhang, J. Am. Chem. Soc., 2014, 136, 5852 CrossRef CAS PubMed.
- S. Yang, C. Cao, P. Huang, L. Peng, Y. Sun, F. Wei and W. Song, J. Mater. Chem. A, 2015, 3, 8701 CAS.
- Y. Shi, L. Wen, F. Li and H.-M. Cheng, J. Power Sources, 2011, 196, 8610 CrossRef CAS.
- D. Kong, W. Ren, Y. Luo, Y. Yang and C. Cheng, J. Mater. Chem. A, 2014, 2, 20221 CAS.
- Y. Yang, B. Qiao, X. Yang, L. Fang, C. Pan, W. Song, H. Hou and X. Ji, Adv. Funct. Mater., 2014, 24, 4349 CrossRef CAS.
- Z. Xie, X. Li, W. Li, M. Chen and M. Qu, J. Power Sources, 2015, 273, 754 CrossRef CAS.
- C. Chen, Y. Huang, H. Zhang, X. Wang, G. Li, Y. Wang, L. Jiao and H. Yuan, J. Power Sources, 2015, 278, 693 CrossRef CAS.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720 CrossRef CAS PubMed.
- Z. Wang, T. Chen, W. Chen, K. Chang, L. Ma, G. Huang, D. Chen and J. Y. Lee, J. Mater. Chem. A, 2013, 1, 2202 CAS.
- X. Cao, Y. Shi, W. Shi, X. Rui, Q. Yan, J. Kong and H. Zhang, Small, 2013, 9, 3433 CrossRef CAS PubMed.
- Y. Liu, Y. Zhao, L. Jiao and J. Chen, J. Mater. Chem. A, 2014, 2, 13109 CAS.
- M. Yang, S. Ko, J. S. Im and B. G. Choi, J. Power Sources, 2015, 288, 76 CrossRef CAS.
- F. Xiong, Z. Cai, L. Qu, P. Zhang, Z. Yuan, O. K. Asare, W. Xu, C. Lin and L. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 12625 CAS.
- X. Jiang, X. Yang, Y. Zhu, J. Shen, K. Fan and C. Li, J. Power Sources, 2013, 237, 178 CrossRef CAS.
- Z. X. Huang, Y. Wang, J. I. Wong and H. Y. Yang, 2D Mater., 2015, 2, 024010 CrossRef.
- K. Chang, Z. Wang, G. Huang, H. Li, W. Chen and J. Y. Lee, J. Power Sources, 2012, 201, 259 CrossRef CAS.
- J. Yin, H. Cao, Z. Zhou, J. Zhang and M. Qu, J. Mater. Chem., 2012, 22, 23963 RSC.
- R. Chen, T. Zhao, W. Wu, F. Wu, L. Li, J. Qian, R. Xu, H. Wu, H. M. Albishri, A. S. Al-Bogami, D. A. El-Hady, J. Lu and K. Amine, Nano Lett., 2014, 14, 5899 CrossRef CAS PubMed.
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766 CrossRef CAS PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- X. Huang, Z. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934 RSC.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- H. Tian, F. Xin, X. Wang, W. He and W. Han, J. Materiomics, 2015, 1, 153 CrossRef.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31 CrossRef CAS PubMed.
- J. Ji, H. Ji, L. L. Zhang, X. Zhao, X. Bai, X. Fan, F. Zhang and R. S. Ruoff, Adv. Mater., 2013, 25, 4673 CrossRef CAS PubMed.
- Y. Wen, Y. Zhu, A. Langrock, A. Manivannan, S. H. Ehrman and C. Wang, Small, 2013, 9, 2810 CrossRef CAS PubMed.
- M. Ko, S. Chae, S. Jeong, P. Oh and J. Cho, ACS Nano, 2014, 8, 8591 CrossRef CAS PubMed.
- S. Jing, H. Jiang, Y. Hu and C. Li, J. Mater. Chem. A, 2014, 2, 16360 CAS.
- J. Chang, X. Huang, G. Zhou, S. Cui, P. B. Hallac, J. Jiang, P. T. Hurley and J. Chen, Adv. Mater., 2014, 26, 758 CrossRef CAS PubMed.
- I. H. Son, J. Hwan Park, S. Kwon, S. Park, M. H. Rummeli, A. Bachmatiuk, H. J. Song, J. Ku, J. W. Choi, J.-M. Choi, S.-G. Doo and H. Chang, Nat. Commun., 2015, 6, 7393 CrossRef CAS PubMed.
- B. Wang, X. Li, X. Zhang, B. Luo, M. Jin, M. Liang, S. A. Dayeh, S. T. Picraux and L. Zhi, ACS Nano, 2013, 7, 1437 CrossRef CAS PubMed.
- F. M. Hassan, A. R. Elsayed, V. Chabot, R. Batmaz, X. Xiao and Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 13757 CAS.
- M. Li, Y. Yu, J. Li, B. Chen, A. Konarov and P. Chen, J. Power Sources, 2015, 293, 976 CrossRef CAS.
- S. Yang, W. Yue, J. Zhu, Y. Ren and X. Yang, Adv. Funct. Mater., 2013, 23, 3570 CrossRef CAS.
- W. Li, D. Yoon, J. Hwang, W. Chang and J. Kim, J. Power Sources, 2015, 293, 1024 CrossRef CAS.
- X. Wang, X. Cao, L. Bourgeois, H. Guan, S. Chen, Y. Zhong, D.-M. Tang, H. Li, T. Zhai, L. Li, Y. Bando and D. Golberg, Adv. Funct. Mater., 2012, 22, 2682 CrossRef CAS.
- X. Zhou, L.-J. Wan and Y.-G. Guo, Adv. Mater., 2013, 25, 2152 CrossRef CAS PubMed.
- J. Qin, C. He, N. Zhao, Z. Wang, C. Shi, E.-Z. Liu and J. Li, ACS Nano, 2014, 8, 1728 CrossRef CAS PubMed.
- Y. Wan, Y. Sha, S. Luo, W. Deng, X. Wang, G. Xue and D. Zhou, J. Power Sources, 2015, 295, 41 CrossRef CAS.
- C. Tan, J. Cao, A. M. Khattak, F. Cai, B. Jiang, G. Yang and S. Hu, J. Power Sources, 2014, 270, 28 CrossRef CAS.
- D.-J. Xue, S. Xin, Y. Yan, K.-C. Jiang, Y.-X. Yin, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2012, 134, 2512 CrossRef CAS PubMed.
- J.-G. Ren, Q.-H. Wu, H. Tang, G. Hong, W. Zhang and S.-T. Lee, J. Mater. Chem. A, 2013, 1, 1821 CAS.
- H. Kim, Y. Son, C. Park, J. Cho and H. C. Choi, Angew. Chem., Int. Ed., 2013, 52, 5997 CrossRef CAS PubMed.
- C. Wang, J. Ju, Y. Yang, Y. Tang, J. Lin, Z. Shi, R. P. S. Han and F. Huang, J. Mater. Chem. A, 2013, 1, 8897 CAS.
- S. Fang, L. Shen, H. Zheng and X. Zhang, J. Mater. Chem. A, 2015, 3, 1498 CAS.
- B. P. Vinayan, R. Nagar, V. Raman, N. Rajalakshmi, K. S. Dhathathreyan and S. Ramaprabhu, J. Mater. Chem., 2012, 22, 9949 RSC.
- S. Chen, P. Chen and Y. Wang, Nanoscale, 2011, 3, 4323 RSC.
- W. Wang, I. Ruiz, S. Guo, Z. Favors, H. H. Bay, M. Ozkan and C. S. Ozkan, Nano Energy, 2014, 3, 113 CrossRef CAS.
- Y. Hu, X. Li, J. Wang, R. Li and X. Sun, J. Power Sources, 2013, 237, 41 CrossRef CAS.
- C. Zhong, J.-Z. Wang, D. Wexler and H.-K. Liu, Carbon, 2014, 66, 637 CrossRef CAS.
- A. P. Cohn, L. Oakes, R. Carter, S. Chatterjee, A. S. Westover, K. Share and C. L. Pint, Nanoscale, 2014, 6, 4669 RSC.
- M. Sahoo and S. Ramaprabhu, Nanoscale, 2015, 7, 13379 RSC.
- Z.-J. Fan, J. Yan, T. Wei, G.-Q. Ning, L.-J. Zhi, J.-C. Liu, D.-X. Cao, G.-L. Wang and F. Wei, ACS Nano, 2011, 5, 2787 CrossRef CAS PubMed.
- H. Kim, X. Huang, X. Guo, Z. Wen, S. Cui and J. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 18590 CAS.
- Y. Yang, R. Pang, X. Zhou, Y. Zhang, H. Wu and S. Guo, J. Mater. Chem., 2012, 22, 23194 RSC.
- Y. Yan, Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Chem. Commun., 2012, 48, 10663 RSC.
- R. Guo, L. Zhao and W. Yue, Electrochim. Acta, 2015, 152, 338 CrossRef CAS.
- Y. Zhao, J. Zhang and L. Qu, ChemNanoMat, 2015, 1, 298 CrossRef CAS.
- Y. Gong, M. Li and Y. Wang, ChemSusChem, 2015, 8, 931 CrossRef CAS PubMed.
- Y. Fu, J. Zhu, C. Hu, X. Wu and X. Wang, Nanoscale, 2014, 6, 12555 RSC.
- J. W. Fergus, J. Power Sources, 2010, 195, 939 CrossRef CAS.
- Y. Wang and G. Cao, Adv. Mater., 2008, 20, 2251 CrossRef CAS.
- S.-M. Bak, K.-W. Nam, C.-W. Lee, K.-H. Kim, H.-C. Jung, X.-Q. Yang and K.-B. Kim, J. Mater. Chem., 2011, 21, 17309 RSC.
- X. Zhao, C. M. Hayner and H. H. Kung, J. Mater. Chem., 2011, 21, 17297 RSC.
- X. Fang, M. Ge, J. Rong and C. Zhou, J. Mater. Chem. A, 2013, 1, 4083 CAS.
- S. J. R. Prabakar, Y.-H. Hwang, B. Lee, K.-S. Sohn and M. Pyo, J. Electrochem. Soc., 2013, 160, A832 CrossRef CAS.
- J. Yang, J. Wang, D. Wang, X. Li, D. Geng, G. Liang, M. Gauthier, R. Li and X. Sun, J. Power Sources, 2012, 208, 340 CrossRef CAS.
- Y. Shi, S.-L. Chou, J.-Z. Wang, D. Wexler, H.-J. Li, H.-K. Liu and Y. Wu, J. Mater. Chem., 2012, 22, 16465 RSC.
- X. Zhou, F. Wang, Y. Zhu and Z. Liu, J. Mater. Chem., 2011, 21, 3353 RSC.
- J. Ha, S.-K. Park, S.-H. Yu, A. Jin, B. Jang, S. Bong, I. Kim, Y.-E. Sung and Y. Piao, Nanoscale, 2013, 5, 8647 RSC.
- W.-B. Luo, S.-L. Chou, Y.-C. Zhai and H.-K. Liu, J. Mater. Chem. A, 2014, 2, 4927 CAS.
- B. Wang, B. Xu, T. Liu, P. Liu, C. Guo, S. Wang, Q. Wang, Z. Xiong, D. Wang and X. S. Zhao, Nanoscale, 2014, 6, 986 RSC.
- X. Guo, Q. Fan, L. Yu, J. Liang, W. Ji, L. Peng, X. Guo, W. Ding and Y. Chen, J. Mater. Chem. A, 2013, 1, 11534 CAS.
- B. Lung-Hao Hu, F.-Y. Wu, C.-T. Lin, A. N. Khlobystov and L.-J. Li, Nat. Commun., 2013, 4, 1687 CrossRef PubMed.
- J.-P. Jegal, K.-C. Kim, M. S. Kim and K.-B. Kim, J. Mater. Chem. A, 2014, 2, 9594 CAS.
- X. Rui, D. Sim, K. Wong, J. Zhu, W. Liu, C. Xu, H. Tan, N. Xiao, H. H. Hng, T. M. Lim and Q. Yan, J. Power Sources, 2012, 214, 171 CrossRef CAS.
- B. Pei, Z. Jiang, W. Zhang, Z. Yang and A. Manthiram, J. Power Sources, 2013, 239, 475 CrossRef CAS.
- B. Cheng, X.-D. Zhang, X.-H. Ma, J.-W. Wen, Y. Yu and C.-H. Chen, J. Power Sources, 2014, 265, 104 CrossRef CAS.
- Y. Jiang, W. Xu, D. Chen, Z. Jiao, H. Zhang, Q. Ma, X. Cai, B. Zhao and Y. Chu, Electrochim. Acta, 2012, 85, 377 CrossRef CAS.
- X. Rui, J. Zhu, D. Sim, C. Xu, Y. Zeng, H. H. Hng, T. M. Lim and Q. Yan, Nanoscale, 2011, 3, 4752 RSC.
- Y. Qian, A. Vu, W. Smyrl and A. Stein, J. Electrochem. Soc., 2012, 159, A1135 CrossRef CAS.
- J. Cheng, B. Wang, H. L. Xin, G. Yang, H. Cai, F. Nie and H. Huang, J. Mater. Chem. A, 2013, 1, 10814 CAS.
- Y. Yang, L. Li, H. Fei, Z. Peng, G. Ruan and J. M. Tour, ACS Appl. Mater. Interfaces, 2014, 6, 9590 CAS.
- J. W. Lee, S. Y. Lim, H. M. Jeong, T. H. Hwang, J. K. Kang and J. W. Choi, Energy Environ. Sci., 2012, 5, 9889 CAS.
- J. Hassoun, F. Bonaccorso, M. Agostini, M. Angelucci, M. G. Betti, R. Cingolani, M. Gemmi, C. Mariani, S. Panero, V. Pellegrini and B. Scrosati, Nano Lett., 2014, 14, 4901 CrossRef CAS PubMed.
- K. Rana, S. D. Kim and J.-H. Ahn, Nanoscale, 2015, 7, 7065 RSC.
- L. Ji, H. Zheng, A. Ismach, Z. Tan, S. Xun, E. Lin, V. Battaglia, V. Srinivasan and Y. Zhang, Nano Energy, 2012, 1, 164 CrossRef CAS.
- F.-W. Yuan and H.-Y. Tuan, Chem. Mater., 2014, 26, 2172 CrossRef CAS.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113 CAS.
- N. Li, Z. Chen, W. Ren, F. Li and H.-M. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17360 CrossRef CAS PubMed.
- P. Xiong, L. Peng, D. Chen, Y. Zhao, X. Wang and G. Yu, Nano Energy, 2015, 12, 816 CrossRef CAS.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018 RSC.
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821 RSC.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125 CrossRef CAS PubMed.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186 CrossRef CAS PubMed.
- L. Chen and L. L. Shaw, J. Power Sources, 2014, 267, 770 CrossRef CAS.
- J. Scheers, S. Fantini and P. Johansson, J. Power Sources, 2014, 255, 204 CrossRef CAS.
- S. Xin, L. Gu, N.-H. Zhao, Y.-X. Yin, L.-J. Zhou, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2012, 134, 18510 CrossRef CAS PubMed.
- W. Li, G. Zheng, Y. Yang, Z. W. Seh, N. Liu and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7148 CrossRef CAS PubMed.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522 CrossRef CAS PubMed.
- G. Zhou, S. Pei, L. Li, D.-W. Wang, S. Wang, K. Huang, L.-C. Yin, F. Li and H.-M. Cheng, Adv. Mater., 2014, 26, 625 CrossRef CAS PubMed.
- H. Al Salem, G. Babu, C. V. Rao and L. M. R. Arava, J. Am. Chem. Soc., 2015, 137, 11542 CrossRef CAS PubMed.
- D.-W. Wang, Q. Zeng, G. Zhou, L. Yin, F. Li, H.-M. Cheng, I. R. Gentle and G. Q. M. Lu, J. Mater. Chem. A, 2013, 1, 9382 CAS.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751 CrossRef CAS PubMed.
- T. Lin, Y. Tang, Y. Wang, H. Bi, Z. Liu, F. Huang, X. Xie and M. Jiang, Energy Environ. Sci., 2013, 6, 1283 CAS.
- J. Xu, J. Shui, J. Wang, M. Wang, H.-K. Liu, S. X. Dou, I.-Y. Jeon, J.-M. Seo, J.-B. Baek and L. Dai, ACS Nano, 2014, 8, 10920 CrossRef CAS PubMed.
- J.-Z. Wang, L. Lu, M. Choucair, J. A. Stride, X. Xu and H.-K. Liu, J. Power Sources, 2011, 196, 7030 CrossRef CAS.
- B. Ding, C. Yuan, L. Shen, G. Xu, P. Nie, Q. Lai and X. Zhang, J. Mater. Chem. A, 2013, 1, 1096 CAS.
- H.-J. Peng, J. Liang, L. Zhu, J.-Q. Huang, X.-B. Cheng, X. Guo, W. Ding, W. Zhu and Q. Zhang, ACS Nano, 2014, 8, 11280 CrossRef CAS PubMed.
- M.-Q. Zhao, Q. Zhang, J.-Q. Huang, G.-L. Tian, J.-Q. Nie, H.-J. Peng and F. Wei, Nat. Commun., 2014, 5, 3410 Search PubMed.
- C. Xu, Y. Wu, X. Zhao, X. Wang, G. Du, J. Zhang and J. Tu, J. Power Sources, 2015, 275, 22 CrossRef CAS.
- G. Zhou, L.-C. Yin, D.-W. Wang, L. Li, S. Pei, I. R. Gentle, F. Li and H.-M. Cheng, ACS Nano, 2013, 7, 5367 CrossRef CAS PubMed.
- H. Xu, Y. Deng, Z. Shi, Y. Qian, Y. Meng and G. Chen, J. Mater. Chem. A, 2013, 1, 15142 CAS.
- J. Rong, M. Ge, X. Fang and C. Zhou, Nano Lett., 2014, 14, 473 CrossRef CAS PubMed.
- L. Zhang, H. Huang, H. Yin, Y. Xia, J. Luo, C. Liang, Y. Gan, X. Tao and W. Zhang, J. Mater. Chem. A, 2015, 3, 16513 CAS.
- B. Li, S. Li, J. Liu, B. Wang and S. Yang, Nano Lett., 2015, 15, 3073 CrossRef CAS PubMed.
- Z. Wang, Y. Dong, H. Li, Z. Zhao, H. Bin Wu, C. Hao, S. Liu, J. Qiu and X. W. Lou, Nat. Commun., 2014, 5, 5002 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS.
- Y. Qiu, W. Li, W. Zhao, G. Li, Y. Hou, M. Liu, L. Zhou, F. Ye, H. Li, Z. Wei, S. Yang, W. Duan, Y. Ye, J. Guo and Y. Zhang, Nano Lett., 2014, 14, 4821 CrossRef CAS PubMed.
- X. Wang, Z. Zhang, Y. Qu, Y. Lai and J. Li, J. Power Sources, 2014, 256, 361 CrossRef CAS.
- C. Li and G. Shi, Adv. Mater., 2014, 26, 3992 CrossRef CAS PubMed.
- S. Lu, Y. Chen, X. Wu, Z. Wang and Y. Li, Sci. Rep., 2014, 4, 4629 Search PubMed.
- K. Xi, P. R. Kidambi, R. Chen, C. Gao, X. Peng, C. Ducati, S. Hofmann and R. V. Kumar, Nanoscale, 2014, 6, 5746 RSC.
- C. Wang, K. Su, W. Wan, H. Guo, H. Zhou, J. Chen, X. Zhang and Y. Huang, J. Mater. Chem. A, 2014, 2, 5018 CAS.
- G. Zhou, L. Li, C. Ma, S. Wang, Y. Shi, N. Koratkar, W. Ren, F. Li and H.-M. Cheng, Nano Energy, 2015, 11, 356 CrossRef CAS.
- X. Yang, L. Zhang, F. Zhang, Y. Huang and Y. Chen, ACS Nano, 2014, 8, 5208 CrossRef CAS PubMed.
- S. Lu, Y. Cheng, X. Wu and J. Liu, Nano Lett., 2013, 13, 2485 CrossRef CAS PubMed.
- R. Chen, T. Zhao, J. Lu, F. Wu, L. Li, J. Chen, G. Tan, Y. Ye and K. Amine, Nano Lett., 2013, 13, 4642 CrossRef CAS PubMed.
- J. Xie, J. Yang, X. Zhou, Y. Zou, J. Tang, S. Wang and F. Chen, J. Power Sources, 2014, 253, 55 CrossRef CAS.
- L. Sun, W. Kong, Y. Jiang, H. Wu, K. Jiang, J. Wang and S. Fan, J. Mater. Chem. A, 2015, 3, 5305 CAS.
- J. He, Y. Chen, P. Li, F. Fu, Z. Wang and W. Zhang, J. Mater. Chem. A, 2015, 3, 18605 CAS.
- S. Niu, W. Lv, C. Zhang, Y. Shi, J. Zhao, B. Li, Q.-H. Yang and F. Kang, J. Power Sources, 2015, 295, 182 CrossRef CAS.
- M. Yu, R. Li, Y. Tong, Y. Li, C. Li, J.-D. Hong and G. Shi, J. Mater. Chem. A, 2015, 3, 9609 CAS.
- M.-Q. Zhao, X.-F. Liu, Q. Zhang, G.-L. Tian, J.-Q. Huang, W. Zhu and F. Wei, ACS Nano, 2012, 6, 10759 CrossRef CAS PubMed.
- H.-J. Peng, J.-Q. Huang, M.-Q. Zhao, Q. Zhang, X.-B. Cheng, X.-Y. Liu, W.-Z. Qian and F. Wei, Adv. Funct. Mater., 2014, 24, 2772 CrossRef CAS.
- C. Tang, Q. Zhang, M.-Q. Zhao, J.-Q. Huang, X.-B. Cheng, G.-L. Tian, H.-J. Peng and F. Wei, Adv. Mater., 2014, 26, 6100 CrossRef CAS PubMed.
- S. Moon, Y. H. Jung and D. K. Kim, J. Power Sources, 2015, 294, 386 CrossRef CAS.
- X. Li, M. Rao, H. Lin, D. Chen, Y. Liu, S. Liu, Y. Liao, L. Xing, M. Xu and W. Li, J. Mater. Chem. A, 2015, 3, 18098 CAS.
- K. Ding, Y. Bu, Q. Liu, T. Li, K. Meng and Y. Wang, J. Mater. Chem. A, 2015, 3, 8022 CAS.
- Y. Dong, S. Liu, Z. Wang, Y. Liu, Z. Zhao and J. Qiu, Nanoscale, 2015, 7, 7569 RSC.
- H. Li, M. Sun, T. Zhang, Y. Fang and G. Wang, J. Mater. Chem. A, 2014, 2, 18345 CAS.
- W. Zhou, H. Chen, Y. Yu, D. Wang, Z. Cui, F. J. DiSalvo and H. D. Abruña, ACS Nano, 2013, 7, 8801 CrossRef CAS PubMed.
- Z. W. Seh, H. Wang, N. Liu, G. Zheng, W. Li, H. Yao and Y. Cui, Chem. Sci., 2014, 5, 1396 RSC.
- K. Han, J. Shen, C. M. Hayner, H. Ye, M. C. Kung and H. H. Kung, J. Power Sources, 2014, 251, 331 CrossRef CAS.
- F. Wu, J. T. Lee, A. Magasinski, H. Kim and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 639 CrossRef CAS.
- K. Zhang, L. Wang, Z. Hu, F. Cheng and J. Chen, Sci. Rep., 2014, 4, 6467 CrossRef CAS PubMed.
- C. Wang, X. Wang, Y. Yang, A. Kushima, J. Chen, Y. Huang and J. Li, Nano Lett., 2015, 15, 1796 CrossRef CAS PubMed.
- Y. Hwa, J. Zhao and E. J. Cairns, Nano Lett., 2015, 15, 3479 CrossRef CAS PubMed.
- Z. Li, S. Zhang, C. Zhang, K. Ueno, T. Yasuda, R. Tatara, K. Dokko and M. Watanabe, Nanoscale, 2015, 7, 14385 RSC.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS PubMed.
- Y.-S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- Y.-S. Su and A. Manthiram, Chem. Commun., 2012, 48, 8817 RSC.
- J.-Q. Huang, T.-Z. Zhuang, Q. Zhang, H.-J. Peng, C.-M. Chen and F. Wei, ACS Nano, 2015, 9, 3002 CrossRef CAS PubMed.
- X. Wang, Z. Wang and L. Chen, J. Power Sources, 2013, 242, 65 CrossRef CAS.
- G. Zhou, L. Li, D.-W. Wang, X.-Y. Shan, S. Pei, F. Li and H.-M. Cheng, Adv. Mater., 2015, 27, 641 CrossRef CAS PubMed.
- A. Vizintin, M. Lozinšek, R. K. Chellappan, D. Foix, A. Krajnc, G. Mali, G. Drazic, B. Genorio, R. Dedryvère and R. Dominko, Chem. Mater., 2015, 27, 7070 CrossRef CAS.
- Z. Xiao, Z. Yang, L. Wang, H. Nie, M. E. Zhong, Q. Lai, X. Xu, L. Zhang and S. Huang, Adv. Mater., 2015, 27, 2891 CrossRef CAS PubMed.
- A. Zhamu, G. Chen, C. Liu, D. Neff, Q. Fang, Z. Yu, W. Xiong, Y. Wang, X. Wang and B. Z. Jang, Energy Environ. Sci., 2012, 5, 5701 CAS.
- R. Mukherjee, A. V. Thomas, D. Datta, E. Singh, J. Li, O. Eksik, V. B. Shenoy and N. Koratkar, Nat. Commun., 2014, 5, 3710 CAS.
- X.-B. Cheng, H.-J. Peng, J.-Q. Huang, R. Zhang, C.-Z. Zhao and Q. Zhang, ACS Nano, 2015, 9, 6373 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797 RSC.
- G. Yu, X. Xie, L. Pan, Z. Bao and Y. Cui, Nano Energy, 2013, 2, 213 CrossRef CAS.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828 CrossRef CAS PubMed.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41 CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906 CrossRef CAS PubMed.
- J. Xia, F. Chen, J. Li and N. Tao, Nat. Nanotechnol., 2009, 4, 505 CrossRef CAS PubMed.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294 CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS PubMed.
- M. D. Stoller, C. W. Magnuson, Y. Zhu, S. Murali, J. W. Suk, R. Piner and R. S. Ruoff, Energy Environ. Sci., 2011, 4, 4685 CAS.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806 CrossRef CAS PubMed.
- T. Kim, G. Jung, S. Yoo, K. S. Suh and R. S. Ruoff, ACS Nano, 2013, 7, 6899 CrossRef CAS PubMed.
- S. He and W. Chen, Nanoscale, 2015, 7, 6957 RSC.
- Y. Xu, G. Shi and X. Duan, Acc. Chem. Res., 2015, 48, 1666 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324 CrossRef CAS PubMed.
- L. Zhang and G. Shi, J. Phys. Chem. C, 2011, 115, 17206 CAS.
- Z. Xiong, C. Liao, W. Han and X. Wang, Adv. Mater., 2015, 27, 4469 CrossRef CAS PubMed.
- X. Wang, Y. Zhang, C. Zhi, X. Wang, D. Tang, Y. Xu, Q. Weng, X. Jiang, M. Mitome, D. Golberg and Y. Bando, Nat. Commun., 2013, 4, 2905 Search PubMed.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833 CrossRef CAS PubMed.
- Z. Zuo, T. Y. Kim, I. Kholmanov, H. Li, H. Chou and Y. Li, Small, 2015, 11, 4922 CrossRef CAS PubMed.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917 CrossRef CAS PubMed.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CAS.
- Y. Xu, C.-Y. Chen, Z. Zhao, Z. Lin, C. Lee, X. Xu, C. Wang, Y. Huang, M. I. Shakir and X. Duan, Nano Lett., 2015, 15, 4605 CrossRef CAS PubMed.
- J. Zhao, H. Lai, Z. Lyu, Y. Jiang, K. Xie, X. Wang, Q. Wu, L. Yang, Z. Jin, Y. Ma, J. Liu and Z. Hu, Adv. Mater., 2015, 27, 3541 CrossRef CAS PubMed.
- J. Han, L. L. Zhang, S. Lee, J. Oh, K.-S. Lee, J. R. Potts, J. Ji, X. Zhao, R. S. Ruoff and S. Park, ACS Nano, 2013, 7, 19 CrossRef CAS PubMed.
- Z. Wen, X. Wang, S. Mao, Z. Bo, H. Kim, S. Cui, G. Lu, X. Feng and J. Chen, Adv. Mater., 2012, 24, 5610 CrossRef CAS PubMed.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472 CrossRef CAS PubMed.
- H.-L. Guo, P. Su, X. Kang and S.-K. Ning, J. Mater. Chem. A, 2013, 1, 2248 CAS.
- Y. Zhao, C. Hu, Y. Hu, H. Cheng, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2012, 51, 11371 CrossRef CAS PubMed.
- Z.-S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2012, 24, 5130 CrossRef CAS PubMed.
- C. X. Guo and C. M. Li, Energy Environ. Sci., 2011, 4, 4504 CAS.
- Y. Wang, Y. Wu, Y. Huang, F. Zhang, X. Yang, Y. Ma and Y. Chen, J. Phys. Chem. C, 2011, 115, 23192 CAS.
- Z.-Y. Yang, Y.-F. Zhao, Q.-Q. Xiao, Y.-X. Zhang, L. Jing, Y.-M. Yan and K.-N. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 8497 CAS.
- Y. Wang, J. Chen, J. Cao, Y. Liu, Y. Zhou, J.-H. Ouyang and D. Jia, J. Power Sources, 2014, 271, 269 CrossRef CAS.
- X. Cui, R. Lv, R. U. R. Sagar, C. Liu and Z. Zhang, Electrochim. Acta, 2015, 169, 342 CrossRef CAS.
- D. Yu, K. Goh, H. Wang, L. Wei, W. Jiang, Q. Zhang, L. Dai and Y. Chen, Nat. Nanotechnol., 2014, 9, 555 CrossRef CAS PubMed.
- Y. Zhu, L. Li, C. Zhang, G. Casillas, Z. Sun, Z. Yan, G. Ruan, Z. Peng, A.-R. O. Raji, C. Kittrell, R. H. Hauge and J. M. Tour, Nat. Commun., 2012, 3, 1225 CrossRef PubMed.
- J. Lin, C. Zhang, Z. Yan, Y. Zhu, Z. Peng, R. H. Hauge, D. Natelson and J. M. Tour, Nano Lett., 2012, 13, 72 CrossRef PubMed.
- J. Chen, C. Li and G. Shi, J. Phys. Chem. Lett., 2013, 4, 1244 CrossRef CAS PubMed.
- S. Bose, T. Kuila, A. K. Mishra, R. Rajasekar, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 767 RSC.
- P. Xiong, J. Zhu and X. Wang, J. Power Sources, 2015, 294, 31 CrossRef CAS.
- L. Mao, K. Zhang, H. S. On Chan and J. Wu, J. Mater. Chem., 2012, 22, 1845 RSC.
- Y. Sun, Z. Fang, C. Wang, K. R. R. M. Ariyawansha, A. Zhou and H. Duan, Nanoscale, 2015, 7, 7790 RSC.
- C. Xiang, M. Li, M. Zhi, A. Manivannan and N. Wu, J. Power Sources, 2013, 226, 65 CrossRef CAS.
- Q. Liao, N. Li, S. Jin, G. Yang and C. Wang, ACS Nano, 2015, 9, 5310 CrossRef CAS PubMed.
- H. Chang, J. Kang, L. Chen, J. Wang, K. Ohmura, N. Chen, T. Fujita, H. Wu and M. Chen, Nanoscale, 2014, 6, 5960 RSC.
- S. Bag and C. R. Raj, J. Mater. Chem. A, 2014, 2, 17848 CAS.
- M. Jing, C. Wang, H. Hou, Z. Wu, Y. Zhu, Y. Yang, X. Jia, Y. Zhang and X. Ji, J. Power Sources, 2015, 298, 241 CrossRef CAS.
- M. Liu and J. Sun, J. Mater. Chem. A, 2014, 2, 12068 CAS.
- A. Choudhury, J. S. Bonso, M. Wunch, K. S. Yang, J. P. Ferraris and D. J. Yang, J. Power Sources, 2015, 287, 283 CrossRef CAS.
- M. Lee, B.-H. Wee and J.-D. Hong, Adv. Energy Mater., 2015, 5, 1401890 Search PubMed.
- C. Xiang, M. Li, M. Zhi, A. Manivannan and N. Wu, J. Mater. Chem., 2012, 22, 19161 RSC.
- S. Yang, Y. Lin, X. Song, P. Zhang and L. Gao, ACS Appl. Mater. Interfaces, 2015, 7, 17884 CAS.
- H. Wang, C. B. Holt, Z. Li, X. Tan, B. Amirkhiz, Z. Xu, B. Olsen, T. Stephenson and D. Mitlin, Nano Res., 2012, 5, 605 CrossRef CAS.
- P. Ahuja, R. K. Sharma and G. Singh, J. Mater. Chem. A, 2015, 3, 4931 CAS.
- D. Ghosh and C. K. Das, ACS Appl. Mater. Interfaces, 2015, 7, 1122 CAS.
- W. Du, Z. Wang, Z. Zhu, S. Hu, X. Zhu, Y. Shi, H. Pang and X. Qian, J. Mater. Chem. A, 2014, 2, 9613 CAS.
- X. Rui, H. Tan and Q. Yan, Nanoscale, 2014, 6, 9889 RSC.
- H. Chen, S. Zhou and L. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 8621 CAS.
- J. Zhu, S. Chen, H. Zhou and X. Wang, Nano Res., 2012, 5, 11 CrossRef CAS.
- K. Zhou, W. Zhou, X. Liu, Y. Sang, S. Ji, W. Li, J. Lu, L. Li, W. Niu, H. Liu and S. Chen, Nano Energy, 2015, 12, 510 CrossRef CAS.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2012, 7, 174 CrossRef PubMed.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020 CrossRef CAS PubMed.
- S. Chen, J. Zhu, L. Qiu, D. Li and X. Wang, Chem. – Eur. J., 2013, 19, 7631 CrossRef CAS PubMed.
- J. Yuan, J. Zhu, H. Bi, X. Meng, S. Liang, L. Zhang and X. Wang, Phys. Chem. Chem. Phys., 2013, 15, 12940 RSC.
- X.-C. Dong, H. Xu, X.-W. Wang, Y.-X. Huang, M. B. Chan-Park, H. Zhang, L.-H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206 CrossRef CAS PubMed.
- X. Yu, B. Lu and Z. Xu, Adv. Mater., 2014, 26, 1044 CrossRef CAS PubMed.
- C. Wu, S. Deng, H. Wang, Y. Sun, J. Liu and H. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 1106 CAS.
- C. Wu, Q. Shen, R. Mi, S. Deng, Y. Shu, H. Wang, J. Liu and H. Yan, J. Mater. Chem. A, 2014, 2, 15987 CAS.
- C. Zhang, T. Kuila, N. H. Kim, S. H. Lee and J. H. Lee, Carbon, 2015, 89, 328 CrossRef CAS.
- Q. Zhang, E. Uchaker, S. L. Candelaria and G. Cao, Chem. Soc. Rev., 2013, 42, 3127 RSC.
- X. Liu, Y. Zheng and X. Wang, Chem. – Eur. J., 2015, 21, 10408 CrossRef CAS PubMed.
- H.-B. Zhao, J. Yang, T.-T. Lin, Q.-F. Lü and G. Chen, Chem. – Eur. J., 2015, 21, 682 CrossRef CAS PubMed.
- X. Zang, X. Li, M. Zhu, X. Li, Z. Zhen, Y. He, K. Wang, J. Wei, F. Kang and H. Zhu, Nanoscale, 2015, 7, 7318 RSC.
- R. R. Salunkhe, S.-H. Hsu, K. C. W. Wu and Y. Yamauchi, ChemSusChem, 2014, 7, 1551 CrossRef CAS PubMed.
- Y. Zhao, J. Liu, Y. Hu, H. Cheng, C. Hu, C. Jiang, L. Jiang, A. Cao and L. Qu, Adv. Mater., 2013, 25, 591 CrossRef CAS PubMed.
- Y. Song, J.-L. Xu and X.-X. Liu, J. Power Sources, 2014, 249, 48 CrossRef CAS.
- J. Zhang and X. S. Zhao, J. Phys. Chem. C, 2012, 116, 5420 CAS.
- D. Sun, L. Jin, Y. Chen, J.-R. Zhang and J.-J. Zhu, ChemPlusChem, 2013, 78, 227 CrossRef CAS.
- H. Wang, Q. Hao, X. Yang, L. Lu and X. Wang, ACS Appl. Mater. Interfaces, 2010, 2, 821 CAS.
- Z. Gao, F. Wang, J. Chang, D. Wu, X. Wang, X. Wang, F. Xu, S. Gao and K. Jiang, Electrochim. Acta, 2014, 133, 325 CrossRef CAS.
- L. Wang, Y. Ye, X. Lu, Z. Wen, Z. Li, H. Hou and Y. Song, Sci. Rep., 2013, 3, 3568 Search PubMed.
- M. Kim, C. Lee and J. Jang, Adv. Funct. Mater., 2014, 24, 2489 CrossRef CAS.
- S. B. Kulkarni, U. M. Patil, I. Shackery, J. S. Sohn, S. Lee, B. Park and S. Jun, J. Mater. Chem. A, 2014, 2, 4989 CAS.
- M. Yu, Y. Ma, J. Liu and S. Li, Carbon, 2015, 87, 98 CrossRef CAS.
- P. Yu, X. Zhao, Z. Huang, Y. Li and Q. Zhang, J. Mater. Chem. A, 2014, 2, 14413 CAS.
- Y. Wang, X. Yang, L. Qiu and D. Li, Energy Environ. Sci., 2013, 6, 477 CAS.
- Y. Meng, K. Wang, Y. Zhang and Z. Wei, Adv. Mater., 2013, 25, 6985 CrossRef CAS PubMed.
- F. Xiao, S. Yang, Z. Zhang, H. Liu, J. Xiao, L. Wan, J. Luo, S. Wang and Y. Liu, Sci. Rep., 2015, 5, 9359 CrossRef CAS PubMed.
- G. Yu, L. Hu, N. Liu, H. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 4438 CrossRef CAS PubMed.
- X. Xia, Q. Hao, W. Lei, W. Wang, D. Sun and X. Wang, J. Mater. Chem., 2012, 22, 16844 RSC.
- W. Wang, Q. Hao, W. Lei, X. Xia and X. Wang, RSC Adv., 2012, 2, 10268 RSC.
- W. Wang, W. Lei, T. Yao, X. Xia, W. Huang, Q. Hao and X. Wang, Electrochim. Acta, 2013, 108, 118 CrossRef CAS.
- P. Xiong, H. Huang and X. Wang, J. Power Sources, 2014, 245, 937 CrossRef CAS.
- W. Wang, Q. Hao, W. Lei, X. Xia and X. Wang, J. Power Sources, 2014, 269, 250 CrossRef CAS.
- P. Xiong, C. Hu, Y. Fan, W. Zhang, J. Zhu and X. Wang, J. Power Sources, 2014, 266, 384 CrossRef CAS.
- H. Li, Y. He, V. Pavlinek, Q. Cheng, P. Saha and C. Li, J. Mater. Chem. A, 2015, 3, 17165 CAS.
- B. G. Choi, S.-J. Chang, H.-W. Kang, C. P. Park, H. J. Kim, W. H. Hong, S. Lee and Y. S. Huh, Nanoscale, 2012, 4, 4983 RSC.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632 CrossRef CAS.
- A. Sumboja, C. Y. Foo, X. Wang and P. S. Lee, Adv. Mater., 2013, 25, 2809 CrossRef CAS PubMed.
- J. Shen, C. Yang, X. Li and G. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 8467 CAS.
- J. Liu, L. Zhang, H. B. Wu, J. Lin, Z. Shen and X. W. Lou, Energy Environ. Sci., 2014, 7, 3709 CAS.
- Z. Zhang, F. Xiao, L. Qian, J. Xiao, S. Wang and Y. Liu, Adv. Energy Mater., 2014, 4, 1400064 Search PubMed.
- T.-W. Lin, C.-S. Dai and K.-C. Hung, Sci. Rep., 2014, 4, 7274 CrossRef CAS PubMed.
- P. Ahuja, V. Sahu, S. K. Ujjain, R. K. Sharma and G. Singh, Electrochim. Acta, 2014, 146, 429 CrossRef CAS.
- Y. Liu, R. Wang and X. Yan, Sci. Rep., 2015, 5, 11095 CrossRef PubMed.
- S. K. Ujjain, G. Singh and R. K. Sharma, Electrochim. Acta, 2015, 169, 276 CrossRef CAS.
- H. Gao, F. Xiao, C. B. Ching and H. Duan, ACS Appl. Mater. Interfaces, 2012, 4, 7020 CAS.
- M. Li, Z. Tang, M. Leng and J. Xue, Adv. Funct. Mater., 2014, 24, 7495 CrossRef CAS.
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980 CrossRef CAS PubMed.
- D. Y. W. Yu, P. V. Prikhodchenko, C. W. Mason, S. K. Batabyal, J. Gun, S. Sladkevich, A. G. Medvedev and O. Lev, Nat. Commun., 2013, 4, 2922 Search PubMed.
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759 CrossRef CAS PubMed.
- B. Qu, C. Ma, G. Ji, C. Xu, J. Xu, Y. S. Meng, T. Wang and J. Y. Lee, Adv. Mater., 2014, 26, 3854 CrossRef CAS PubMed.
- X. Rui, W. Sun, C. Wu, Y. Yu and Q. Yan, Adv. Mater., 2015, 27, 6670 CrossRef CAS PubMed.
- J. Xiao, D. Mei, X. Li, W. Xu, D. Wang, G. L. Graff, W. D. Bennett, Z. Nie, L. V. Saraf, I. A. Aksay, J. Liu and J.-G. Zhang, Nano Lett., 2011, 11, 5071 CrossRef CAS PubMed.
- E. Yoo and H. Zhou, ACS Nano, 2011, 5, 3020 CrossRef CAS PubMed.
- H.-G. Jung, Y. S. Jeong, J.-B. Park, Y.-K. Sun, B. Scrosati and Y. J. Lee, ACS Nano, 2013, 7, 3532 CrossRef CAS PubMed.
- H. Kim, H.-D. Lim, J. Kim and K. Kang, J. Mater. Chem. A, 2014, 2, 33 CAS.
- W. Liu, Q. Sun, Y. Yang, J.-Y. Xie and Z.-W. Fu, Chem. Commun., 2013, 49, 1951 RSC.
- Y. Li, H. Yadegari, X. Li, M. N. Banis, R. Li and X. Sun, Chem. Commun., 2013, 49, 11731 RSC.
| This journal is © The Royal Society of Chemistry 2016 |