
Electrode materials with tailored facets for electrochemical energy storage
Faxing
Wang
ab,
Xiaowei
Wang
b,
Zheng
Chang
b,
Yusong
Zhu
*a,
Lijun
Fu
*a,
Xiang
Liu
a and
Yuping
Wu
*ab
aCollege of Energy and Institute for Electrochemical Energy Storage, Nanjing Tech University, Jiangsu Province, Nanjing 211816, China. E-mail: zhuys@njtech.edu.cn; l.fu@njtech.edu.cn
bNew Energy and Materials Laboratory (NEML), Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: wuyp@fudan.edu.cn
First published on 18th February 2016
Abstract
In recent years, the design and morphological control of crystals with tailored facets have become hot spots in the field of electrochemical energy storage devices. For electrode materials, morphologies play important roles in their activities because their shapes determine how many facets of specific orientation are exposed and therefore available for surface reactions. This review focuses on the strategies for crystal facet control and the unusual electrochemical properties of electrode materials bound by tailored facets. Here, electrode materials with tailored facets include transition metal oxides such as SnO2, Co3O4, NiO, Cu2O, and MnO2, elementary substances such as Si and Au, and intercalation compounds such as Li4Ti5O12, LiCoO2, LiMn2O4, LiFePO4, and Na0.7MnO2 for various applications of Li-ion batteries, aqueous rechargeable lithium batteries, Na-ion batteries, Li–O2 batteries and supercapacitors. How these electrode materials with tailored facets affect their electrochemical properties is discussed. Finally, research opportunities as well as the challenges in this emerging research frontier are highlighted.
1. Introduction
The specific exposed facets and their related surface energies exert significant influences on the kinetics and thermodynamics of heterogeneous reactions occurring at the interface.1–3 Thus, the design and morphological control of crystals with tailored facets are becoming hot spots in scientific research.4–9 In the case of anatase TiO2, for example, it is usually exposed with low index facets such as (001) and (101) as shown in Fig. 1. Specifically, its most common crystal shape is the truncated octahedral bipyramid comprising eight (101) facets (94%) on the sides and two (001) facets (6%) on the top and bottom truncated facets.5 However, theoretical studies indicate that the (001) facet of anatase TiO2 is much more reactive than its other surfaces.9 Surface energy is closely related to the density of undercoordinated Ti atoms. The right panel in Fig. 1 summarizes the possible shapes that may be produced. Synthesis of high energy facets of TiO2 has been achieved to improve its properties and extend its applications in the past decade.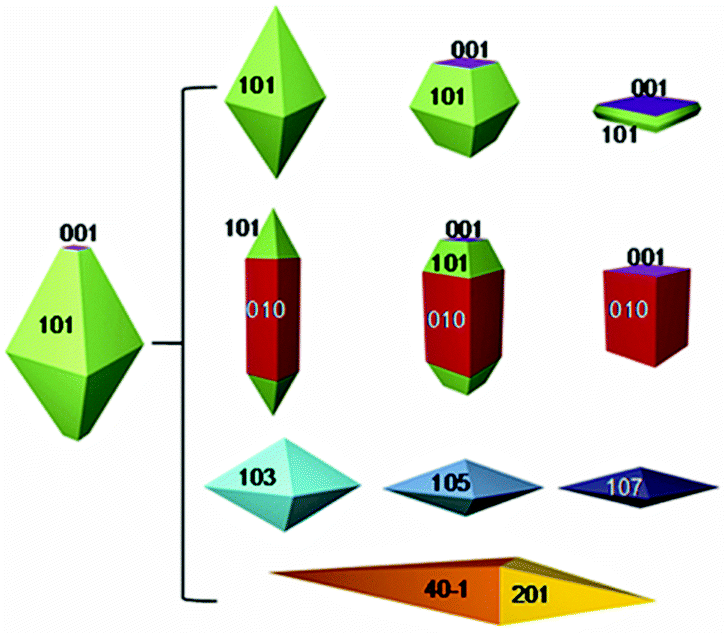 | ||
| Fig. 1 Equilibrium crystal shape of anatase TiO2 through the Wulff construction and the other evolved shapes (modified from ref. 5, copyright permission from American Chemical Society). | ||
At the same time, rechargeable batteries and supercapacitors are emerging as two important classes of electrochemical energy storage devices.10,11 Electrochemical energy storage involves physical interaction and/or chemical reaction at the surface or interface. The discharge/charge processes in rechargeable batteries and supercapacitors are accompanied by the transport of active ions across the surface of crystals. Thus, the interaction between active ions in the electrolyte and the surface of crystals is essential in the field of batteries and supercapacitors. To develop electrode materials with specific facets is of vital significance to control the interaction between active ions and surfaces of crystals. Different facets with different surface atomic structures may exhibit distinct abilities to host guest materials. In this regard, electrode materials with preferred orientation and exposure of facets have received widespread attention for electrochemical energy storage in recent years. For example, our group reported the synthesis of LiMn2O4 nanotubes with the exposed (400) plane,12 LiFePO4 nanoparticles with the major (010) exposed plane,13,14 and a hollow Li3VO4 microbox with the exposed (200) plane.15,16 Interestingly, based on density functional theory calculations and in situ mass measurements (electrochemical quartz crystal microbalance), we found that when the LiFePO4(010) surface is brought into contact with an aqueous solution, there is always a water molecule chemisorbed near the Fe site on the surface. This water molecule strengthens Li binding on surface sites and increases the binding energy, which lowers the energy barrier for Li diffusion from the subsurface to the (010) surface.13
However, most reviews on the unusual properties of crystals with exposed highly reactive facets mainly focus on the field of catalysis and gas adsorption.4–6 Those catalytic or adsorption processes critically depend on the surface atoms' arrangement and the number of dangling bonds on different crystal planes. Even though there are a few reviews dealing with crystals with tailored facets for electrochemical energy storage, they usually mainly focus on specific electrode materials such as TiO2,4,17,18 Co3O4,19 and positive electrode materials20 with predominantly exposed facets. Therefore, a comprehensive summary of crystals with tailored facets for electrochemical energy storage is lacking and highly desirable in order to rationally promote the further development of rechargeable batteries and supercapacitors.
In this review, we will summarize the latest developments in the applications of electrode materials with tailored facets for electrochemical energy storage in five fields, namely, Li-ion batteries, aqueous rechargeable lithium batteries, Na-ion batteries, Li–O2 batteries and supercapacitors. It will be shown that there is some relationship between crystals with exposed facets and their unusual electrochemical properties. The particular focus is directed to strategies for their shape control and the uniqueness of electrode materials with preferred orientation and facets in energy storage applications. To facilitate further research and development in this promising field, some future trends or directions are also discussed.
2. Electrode materials with tailored facets for Li ion batteries
Lithium is the third lightest element and has the lowest redox potential of all known elements (−3.05 V vs. the standard hydrogen potential). Though lithium has been incorporated into battery systems since the late 1960s, rechargeable lithium ion batteries were commercialized by Sony Corporation only in 1991.21,22 A Li-ion battery consists of three functional components (a negative electrode, a positive electrode and an electrolyte) and functions by converting the chemical potential into electrical energy via Faradaic reactions. This process includes heterogeneous charge transfer occurring at the surface of an electrode. Therefore, the crystal structure and morphology are critical to the reaction rate and transfer processes.23,242.1 Negative electrode materials with tailored facets
Negative electrode materials for Li-ion batteries can be classified into three different categories (1) intercalation compounds such as TiO2, Li4Ti5O12 and Li3VO4; (2) elementary substances based on alloy reactions, such as Si, Sn and Ge; and (3) metal oxides with conversion reactions, such as NiO, Co3O4 and Fe2O3. Some of their characteristics are summarized in Table 1.| Electrode | Tailored facets | Preparation method | Specific capacity/mA h g−1 | Rate/A g−1 | Cycle | Ref. |
|---|---|---|---|---|---|---|
| TiO2 | (001) | Solvothermal method | 200 | 3.4 | 100 | 25 |
| Li4Ti5O12 | (011) | Hydrothermal + calcination | 205 | 4 | 100 | 26 |
| SnO2 | (110) | Precipitation + calcination | 918 | 5 | 50 | 27 |
| NiO | (110) | Hydrothermal + calcination | 700 | 40C | 1000 | 28 |
| Cu2O | (001) | Hydrothermal method | 841 | 6.7 | 1000 | 29 |
| Co3O4 | (110) | Hydrothermal method | 946 | 2 | 50 | 30 |
Several years ago, anatase single crystal TiO2 with 47% highly reactive (001) facets was prepared by using hydrofluoric acid (HF) as a capping agent under hydrothermal conditions.1 The choice of capping agents is essential for controlling the facets grown in the TiO2 crystals. The adsorption of the capping agents reduces the surface free energy of materials with more active sites inhibiting the crystal growth along the corresponding direction. This breakthrough has attracted great interest in various synthesis methods to achieve TiO2 with specific facets, including hydrothermal,34–36 solvothermal,25,37,38 and template methods.39
For anatase TiO2, the Li+ ion diffusion coefficient is approximately 2.0 × 10−13 cm2 s−1 along the [001] orientation while it is only 7 × 10−14 cm2 s−1 along the [101] orientation.40 The energy barriers calculated based on density functional theory for Li+ ion insertion into (001) and (101) surfaces are 1.33 and 2.73 eV, respectively.41 Thus, the charge transfer and chemical diffusion coefficient for TiO2 are greatest along the (001) facet, and exposing this facet can result in a lower energy barrier for faster and more Li+ ion intercalation. As shown in Fig. 2, anatase TiO2 nanosheets composed of 80% exposed (001) facets demonstrate a high-rate of insertion/extraction of Li ions over extended cycling compared to anatase TiO2 with dominant (101) facets. These two samples have very different percentages of exposed (001) facets, about 80% for the nanosheets (Fig. 2a) and only about 2.2% for the octahedral sample dominated by (101) facets (Fig. 2b). At 10 C, anatase TiO2 nanosheets with 80% exposed (001) facets show a higher specific capacity compared to (101) dominated anatase TiO2 nano-octahedra (Fig. 2c). More Li+ ion insertion/extraction can occur through the less thermodynamically favored (001) surface, which agrees well with the theoretical prediction of a lower barrier for the surface transmission of Li+ ions across this facet compared with the (101) surface.
 | ||
| Fig. 2 TEM micrographs of the anatase TiO2 with dominant (a) (001) and (b) (101) surfaces (insets are corresponding geometrical models of the anatase single crystals), and (c) the cycling behaviors with dominant (001) (upper) and (101) (lower) facets at 10C (modified from ref. 41, copyright permission from The Royal Society of Chemistry). | ||
Ultrathin anatase TiO2 nanosheets with nearly 100% exposed (001) facets were also synthesized.25 The individual nanosheet adopts a random orientation and forms a three-dimensional highly nanoporous structure with a high specific surface area (170 m2 g−1). The short diffusion path length in these ultrathin TiO2 nanosheets leads to highly efficient solid-state diffusion of Li+ ions. So, even at a high current rate of 20 C (3.4 A g−1), a reversible capacity of 95 mA h g−1 could still be achieved. Similar results have also been demonstrated by sandwich-like TiO2/C composite hollow spheres34 and TiO2/graphene aerogel composites.35 Apart from the reactive (001) surface, the strong connection between TiO2 and conductive carbon also improves the electron transport efficiency. All these results clearly demonstrate the significance of tailoring the facets of TiO2 crystals for improving the lithium storage capability of negative electrode materials.
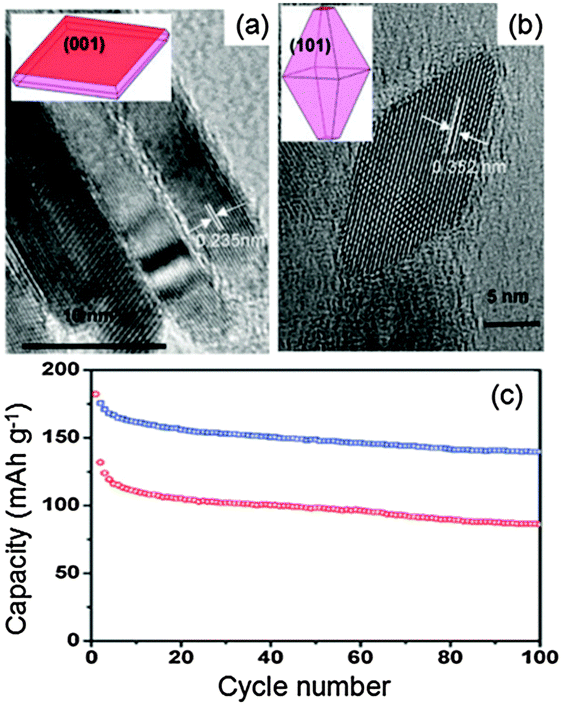
Spinel Li4Ti5O12 has plenty of porous channels along the [011] direction. These porous channels are favorable for fast Li ion diffusion. Therefore, the spinel Li4Ti5O12 negative electrode with the preferentially exposed (011) plane should be of benefit to high electrochemical performance due to the directly accessible channels for the intercalation of Li ions. With this consideration in mind, Li4Ti5O12 hollow spheres composed of nanoflakes with preferentially exposed (011) facets were fabricated via a facile hydrothermal process followed by calcination.26Fig. 3 shows the top view HRTEM micrographs of Li4Ti5O12 nanoflakes. Two sets of lattices form a dihedral angle of approximately 70.5° with each other with an equal inter-fringe spacing of 0.49 nm, corresponding to their (111) planes (Fig. 3a). The corresponding SAED pattern of the same region can be indexed to diffraction spots of the (011) zone, indicating that nearly 100% exposed surfaces are (011) planes. A more detailed crystal structure of the spinel Li4Ti5O12 with projection along the (011) direction can be found in partially enlarged HRTEM micrographs (Fig. 3b).
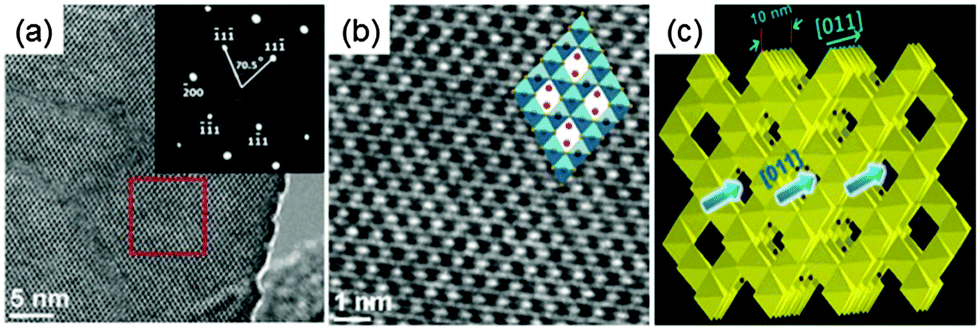 | ||
| Fig. 3 (a) The top view HRTEM micrographs of the nanoflake (inset is the corresponding SAED pattern); (b) partial enlarged details of the red box part in (a). The inset in b shows the corresponding crystal structure of spinel Li4Ti5O12 along (011); and (c) schematic illustration of Li+ ion diffusion in spinel structured Li4Ti5O12 (modified from ref. 26, copyright permission from American Chemical Society). | ||
Cycled at a high current density of 4 A g−1, Li4Ti5O12 nanoflakes show a high discharge capacity of 148 mA h g−1 in the first cycle, giving a retention rate of approximately 74% after 100 cycles.26 The preferentially exposed (011) planes of Li4Ti5O12 nanoflakes provide directly accessible channels for the intercalation/deintercalation of Li ions along the (011) direction (Fig. 3c). Besides, Li4Ti5O12 nanoflakes, with a thickness of approximately 10 nm, significantly decrease the diffusion distance for Li ions compared with the bulk materials. Additionally, the hollow structure of the Li4Ti5O12 spheres facilitates the permeation of the electrolyte within the electrode.
The intercalation of a Li atom into the surface and subsurface layers of Si(100) and Si(111) planes was studied by density functional theory calculations.47 It is found that once the Li atom is incorporated into the Si surface, Li diffuses faster by at least two orders of magnitude along the [100] direction than along the [111] direction. Similar results were also obtained through experimentally studying the shape and volume changes of Si with different orientations upon first lithiation.48 The Si nanopillars with axial orientations along (100), (110) and (111) planes were fabricated by etching a Si wafer surface with SiO2 nanospheres as the etch mask.
The nanopillars stand vertically on a fixed substrate as shown in Fig. 4. When they were discharged at 0.12 V (vs. Li+/Li) (partially lithiation), all of them show obvious anisotropic cross-sectional expansion. As shown in Fig. 4(a)–(f), the initially circular (100), (110), and (111) pillars transform into a cross, an ellipse, and a hexagon, respectively. With further discharge at 0.01 V (vs. Li+/Li) (full lithiation), the cross and elliptical cross-sectional shapes of the (100) and (110) nanopillars become more significant and the cross section of the 〈111〉 nanopillar is slightly hexagonal. In all three cases, the nanopillars expand most significantly along the (110) family of directions (Fig. 4d) because this channel is much larger than those along the (100) and (111) directions (Fig. 4e). Besides, the (111) and (100) nanopillars shrink in height after partial lithiation, while (110) nanopillars increase in height. It was suggested that Li enters the crystalline Si nanostructure through 〈110〉 ion channels and induces the collapse of some (111) facets by breaking Si–Si bonds. These findings provide guidelines for developing high power Si electrodes by increasing the prevalence of (110) facets to promote fast diffusion of Li ions. However, unfortunately few efforts have been put into the development of Si with tailored (110) facets.
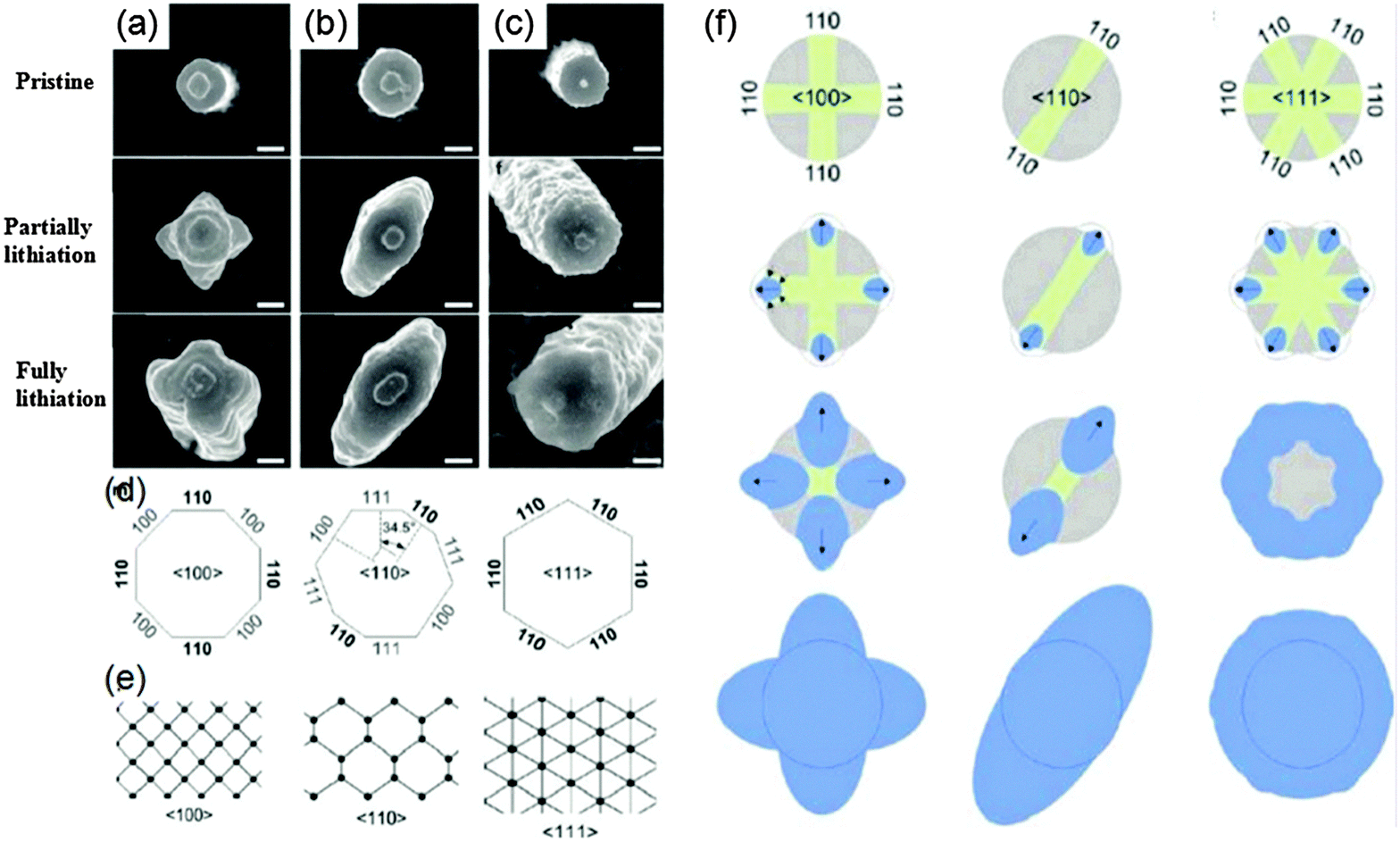 | ||
| Fig. 4 Top-view SEM micrographs of Si nanopillars with crystal orientation along (a) (100), (b) (110), and (c) (111) planes upon lithiation and each lithiation state, (d) schematic diagram of the crystallographic orientation of the facets on the sidewalls of each of the pillars, (e) view along three different directions of the diamond cubic lattice, and (f) schematic explaining anisotropic expansion of Si nanopillars (modified from ref. 48, copyright permission from American Chemical Society). | ||
Ultrathin SnO2 nanosheets along the [110] direction were assembled by oriented attachment of SnO2 nanoparticles with the help of additives (ethanol, NH3·H2O and urea).51 The –OH group of ethanol, the –NH2 group of urea, and the NH4+ group of aqueous ammonia play important roles as surface-modifying agents in the reactions. These functional groups can lead to the variation in the surface energy of SnO2 nanocrystals due to their different abilities to bind Sn ions or form hydrogen bonds. In another report, N-doped graphene–SnO2 sandwich films were fabricated via a three-step route.27 First, the 7,7,8,8-tetracyanoquinodimethane anion (TCNQ−) was adsorbed on the surface of graphene under electrostatic repulsion preventing the inter- or intra-π–π stacking of graphene. After addition of the Sn(II) salt, the graphene layers containing Sn–TCNQ self-assembled into a sandwich structure because of the strong electrostatic interactions between Sn2+ and TCNQ−. After that, the obtained precipitate was dried and annealed under an inert gas atmosphere to obtain the sandwich-type graphene–SnO2. During the synthesis process, the interlayer space of graphene acting as a nanoscale reactor effectively restricts the growth of the SnO2 nanoparticles and further drives the fusion of two or more nanoparticles into a 1D nanostructure within the lamellar superstructure. Most particles are epitaxially fused together towards [110] because of the continuity of the lattice (110) planes across the interface, suggesting that the oriented attachments occur along the (110) facets.
A much higher reversible capacity of 534 mA h g−1 was achieved for highly oriented SnO2 nanosheets along the (110) direction even after 50 cycles.27 The SnO2 nanosheets show a greatly enhanced lithium storage capability compared to commercial SnO2 nanoparticles. The specific capacity is maintained as high as 683 and 619 mA h g−1 at 1 and 2 A g−1, respectively. Even at a current density of 5 A g−1, they still deliver a capacity of 504 mA h g−1. As for the commercial SnO2 nanoparticles, the capacity rapidly decays to 153 mA h g−1 at 1 A g−1, and to 12 mA h g−1 at 2 A g−1. In the sandwich structure, the electronic transport length in graphene–SnO2 is effectively shortened to a level comparable to the particle size of the nanocrystals. Moreover, the ultra-small SnO2 nanocrystals (2–5 nm) with the preferred orientation of (110) planes render a very short transport length for Li ions during insertion/extraction processes.
The properties of transition metal oxide crystals are largely determined by exposed external surfaces. NiO crystals with dominantly exposed (110) reactive facets were obtained by the thermal conversion of hexagonal Ni(OH)2 nanoplatelets.28 The prepared NiO crystals preserve the single crystalline feature and hexagonal shape of the precursor Ni(OH)2 nanoplatelets. This may be because the (111) crystal facets of Ni(OH)2 and the (110) crystal facets of NiO have low crystal mismatch. The (110) lattice plane of Ni(OH)2 crystals has a d-spacing of 0.29 nm, which is very close to that of the (111) plane of NiO crystals (0.24 nm). The control of the crystal mismatch within a low range is highly favorable for monocrystallization.
CuO nanoplatelets with exposed (001) facets and hollow hierarchical Fe2O3 spheres self-organized from the ultrathin nanosheets of Fe2O3 were prepared by the hydrothermal process.29,52 The ultrathin Fe2O3 nanosheet subunits possess an average thickness of around 3.5 nm and show preferential exposure of (110) facets. The highly exposed (110) facets of Fe2O3 are largely dominated by the high density of Fe atoms. The crystal facet engineering in the formation of Fe2O3 mesocrystals was also studied in rhombic hematite mesocrystals by a facile solvothermal approach using N,N-dimethylformamide (DMF) and methanol as the mixed solvent.53 The reactants chemically transform into active particles to form the hematite crystals according to the following reactions:
| HCON(CH3)2 + H2O → NH(CH3)2 + HCOOH | (1) |
| HCOOH + CH3OH → HCOOCH3 + H2O | (2) |
| 2Fe3+ + 3H2O + 6NH(CH3)2 → Fe2O3 + 6NH2(CH3)2+ | (3) |
The shape control of Co3O4 with different well-defined crystal planar structures was facilely achieved by simply changing the content of NaOH and Co(NO3)2·6H2O without using a capping agent by a one-step hydrothermal method. Three kinds of Co3O4 crystals, a cube with the (001) plane, a truncated octahedron with (001) and (111) planes, and an octahedron with the (111) plane, were reported.30
NiO crystals retained a lithium storage capacity of 468 mA h g−1 at the 20C rate and 322 mA h g−1 at the 40C rate, respectively.28 The most stable crystal plane for NiO crystals is the (100) plane with the lowest surface energy of 0.958 J m−2. The (110) and (101) planes have relatively high values, larger than 1.47 J m−2. Because of the relatively high surface energy, the (110) crystal planes provide reactive sites for reaction with Li+ ions, which can facilitate fast conversion reaction during the charge and discharge process. Likely, CuO single crystals with exposed high-energy (001) facets also show high rate capability.29
A reversible discharge capacity as high as 815 mA h g−1 after the 200th cycle was delivered at a current density of 0.5 A g−1 when ultrathin nanosheets of Fe2O3 with preferentially exposed (110) facets were used as the negative electrode materials.52 The highly exposed (110) facets of Fe2O3, largely dominated by the high density of Fe atoms, play an important role in the Fe/Li2O interface. Fe2O3 mesocrystals obtained by crystal facet engineering also show improved cycling behavior.53
Electrochemical tests suggested that the electrochemical performance of Co3O4 is ranked as: octahedron > truncated octahedron > cube.30 The (111) plane is more beneficial to Li+ ion transport than the (001) plane. Co3O4 octahedra with (111) planes possessed the highest charge/discharge capacity and best cycling behavior. Through analyzing the surface atomic configurations in the (001), (111), and (110) planes of the Co3O4 unit cell, it can be clearly seen that the (001) plane contains only 2Co(II) but the (111) plane contains 3.75Co(III). There is a direct relationship between the electrochemical performance of Co3O4 and the redox reaction of Com+/Co. The Co3O4 octahedron has a larger area of exposed (111) planes and thus faster Co2+/Co redox reaction. The authors also predicted that Co3O4 with (110) planes would exhibit better electrochemical performance than Co3O4 with (001) and/or (111) planes because the (110) plane contains 5Co(II) and 4Co(III).
Hollow-structured Li3VO4 microboxes with the exposed (200) plane were synthesized via an in situ hydrothermal method reported by our group.16 We found that the hollow Li3VO4 microboxes were formed via an oxygen-engaged oxidation process, as well as by Ostwald ripening. The low ratio of vanadium shows an enhancement in the (200) plane, as we can see from the peak intensity of the XRD pattern of Li3VO4 (Fig. 5a). The trace oxygen dissolved in the solution may gradually oxidize the V2O3 surface to VO43−. Then reacting with LiOH, Li3VO4 precipitated on the surface could ensure that the surfaces of the V2O3 cubes are effectively covered, making the surface of V2O3 cubes less reactive than the freshly exposed interior. Actually, three lithium atoms and three oxygen atoms can form a radially arranged hexagon in the (100) plane (Fig. 5b), whereas two lithium atoms and two oxygen atoms can form a radially arranged rectangle in the (001) plane (Fig. 5c). Considering the diagonals of the hexagons and rectangles, the (100) plane has wider interatomic spacing for lithium. Therefore, lack of V cations could tend to increase the intensity of the (200) plane and decrease the intensity of the (002) plane. Similarly, the Ostwald ripening process was also reported to grow Ca2Ge7O16 nanowires with a preferred (001) growth direction.54
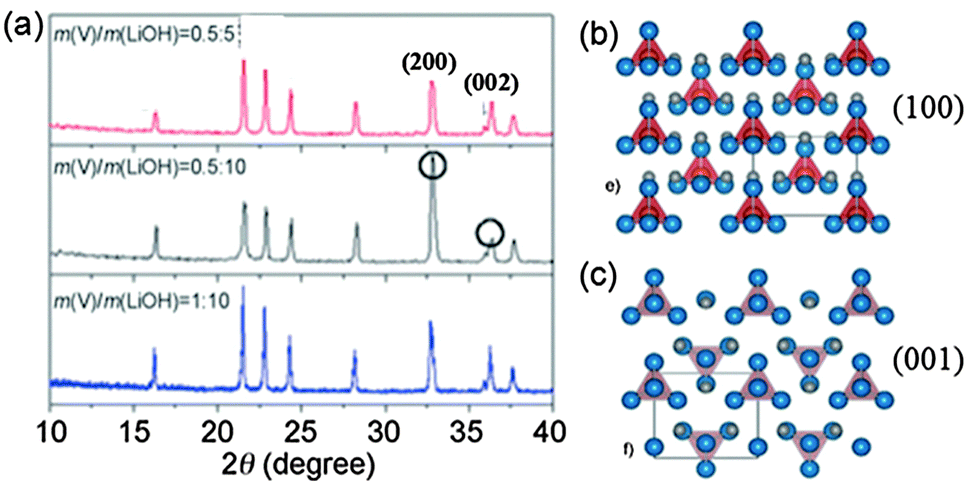 | ||
| Fig. 5 (a) XRD patterns of Li3VO4 synthesized from different ratios of Li and V, structure of the orthorhombic Li3VO4 viewed from the (b) (100) plane and (c) (001) plane. The blue atoms are O atoms and the grey atoms are Li atoms. The atoms in the red tetrahedron cages are V atoms (modified from ref. 16, copyright permission from Wiley). | ||
Li ions mainly intercalate into Li3VO4 in the potential range between 0.5 and 1.0 V (vs. Li+/Li), lower than the potential of Li4Ti5O12 and higher than that of graphite. Its theoretical capacity is 394 mA h g−1, in accordance with Li5VO4.55 The Li3VO4 electrode with the exposed (200) plane delivered a discharge capacity of 323 mA h g−1,16 close to that of commercial graphite56 and much higher than that of Li4Ti5O12 (in Section 2.1.2). It can also retain a reversible capacity of 83 mA h g−1 at 20 C. The Ca2Ge7O16 nanowires with a preferred (001) growth direction exhibit a reversible capacity of 420 mA h g−1.54 It also shows extremely long stable cycling. Actually, after the conversion reaction during the initial lithium uptake process, the in situ formed active Ge nanoparticles are highly dispersed within the mixed matrix of Li2O and CaO, which not only provides an elastic buffer to accommodate the volume changes, but also prevents the agglomeration of nanosized Ge particles.
2.2 Positive electrode materials with tailored facets
The insertion or intercalation compounds are among the most useful positive electrode materials for Li ion batteries, and some of their characteristics are summarized in Table 2.| Electrode | Tailored facets | Preparation method | Specific capacity/mA h g−1 | Rate/A g−1 | Cycle | Ref. |
|---|---|---|---|---|---|---|
| LiCoO2 | (100) | Hydrothermal + calcination | 189 | 1.4 | 200 | 57 |
| LiMn2O4 | (111) | Self-sacrifice template | 115 | 3 | 500 | 58 |
| LiNi1/3Co1/3Mn1/3O2 | (010) | Precipitation + calcination | 179 | 15C | 100 | 59 |
| LiFePO4 | (010) | Solvothermal method | 164 | 20C | 60 | 60 |
| Li1.2Ni0.2Mn0.6O2 | (010) | Precipitation + calcination | 230 | 20C | 80 | 61 |
| LiMnPO4 | (010) | Hydrothermal method | 130 | 0.5C | 20 | 62 |
LiCoO2 has a layered α-NaFeO2 type structure (Fig. 6a).65 By analyzing the crystal structure of LiCoO2, it is obvious to find that Li ions can fluently shuttle back and forth in the LiCoO2 crystal along the [100] or [010] directions (Fig. 6b and c). Li ions are hard to diffuse in the crystal along [001] (Fig. 6d) because a mass of atoms such as oxygen or cobalt obstruct the passing route. Thus, the (010) and (100) planes perpendicular to the (001) plane are favorable for the transportation of the Li+ ion.
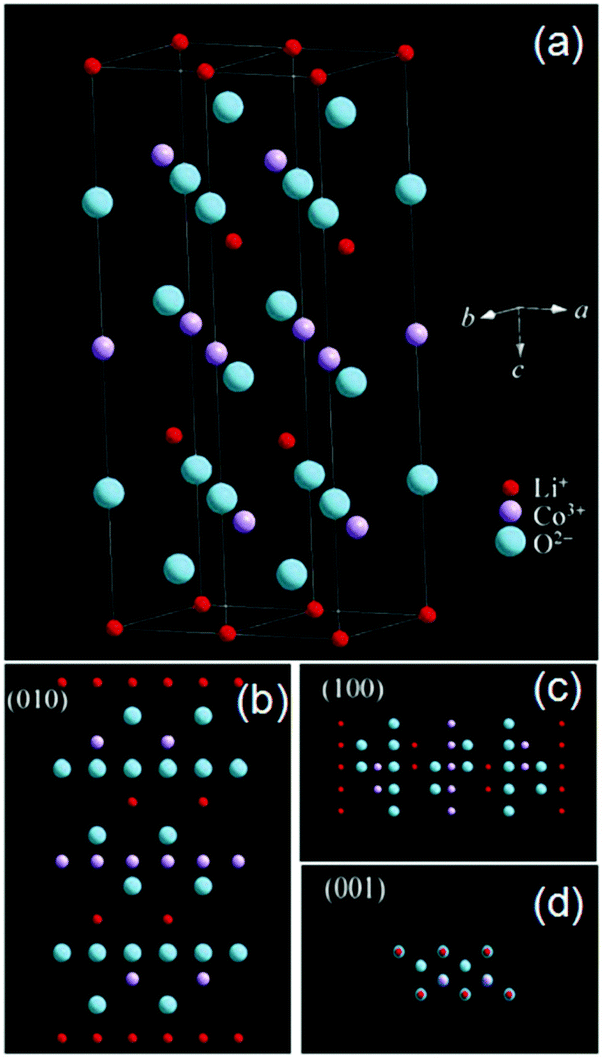 | ||
| Fig. 6 (a) The crystal structure of LiCoO2 and perspective views from (b) (001), (c) (100) and (d) (001) planes, respectively (modified from ref. 65, copyright permission from Springer). | ||
Uniform Co(OH)2 nanoplates were synthesized by co-precipitation and then transformed into LiCoO2 nanoplates by solid state reaction at 750 °C.65 Al doped LiCoO2 with the highly exposed (100) plane was synthesized via solid state reaction with Co6Al2CO(OH)16·H2O and LiOH powder.57
The initial discharge capacity of the Al doped LiCoO2 electrode is 189 mA h g−1 at 0.1C, higher than that of the conventional LiCoO2.57 After 200 cycles, it delivers a reversible capacity of 183 mA h g−1 (equal to 96.8% capacity retention). The rapid Li ion diffusion planes of (100) or their equivalent planes have a large exposure ratio up to 100%, which remarkably improves the cyclability and rate capability of Al doped LiCoO2. As discussed above, the (001) plane of LiCoO2 is not electrochemically active. However, the LiCoO2 nanoplates with the exposed (001) plane present a stable discharge capacity of 113 mA h g−1 at 1 A g−1 after 100 cycles.65 It was found that there are many cracks on the nanoplates which are perpendicular to the (001) plane and favor fast Li+ ion transportation.
LiMn2O4 epitaxial films with (111) and (110) orientations were synthesized by pulsed laser deposition using SrTiO3(111) and (110) substrates, respectively.69 LiMn2O4 nanosheets composed of single-crystals with exposed (111) facets were synthesized via a template-engaged reaction using ultrathin MnO2 nanosheets as a self-sacrificial template.58 Similarly, LiMn2O4 nanotubes,70 nanorods71 and nanowires72 with exposed (110) planes were also prepared using the corresponding shaped MnO2 as a self-sacrificial template.
Recently, structural changes of (111) and (110) surfaces for LiMn2O4 during the (de)intercalation process were reported.69 It is suggested that surface stability could be related to variations in the surface termination arrangements of oxygen ions and/or manganese ions with the valence changing during redox reactions. As shown in Fig. 7a, the (111) plane of LiMn2O4 is terminated by a cubic closed-packed oxygen arrangement in the spinel structure, while no closed-packed arrangement of oxygen appears in the (110) plane (Fig. 7b). The Mn ions are less densely arranged at the (110) surface and can easily come into close contact with the electrolyte, which makes Mn ions highly reactive towards solvents in the electrolyte. Therefore, the (110) plane in the spinel LiMn2O4 structure is less stable than the (111) surface. However, the orientation of the (110) plane is obviously aligned to Li+ ion diffusion, which can support fast Li ion insertion/extraction. So the above discussion may be the reason why all of the LiMn2O4 (nanotubes,70 nanorods71 and nanowires72) with (110) planes presents high-rate capabilities while LiMn2O4 nanosheets with exposed (111) facets exhibit good cycling behavior. For example, LiMn2O4 nanowires had a discharge capacity of about 80 mA h g−1 even at 150 C (22.2 A g−1),72 while nearly 100% of the initial capacity could be retained after 500 cycles using the LiMn2O4 nanosheets with exposed (111) facets.58
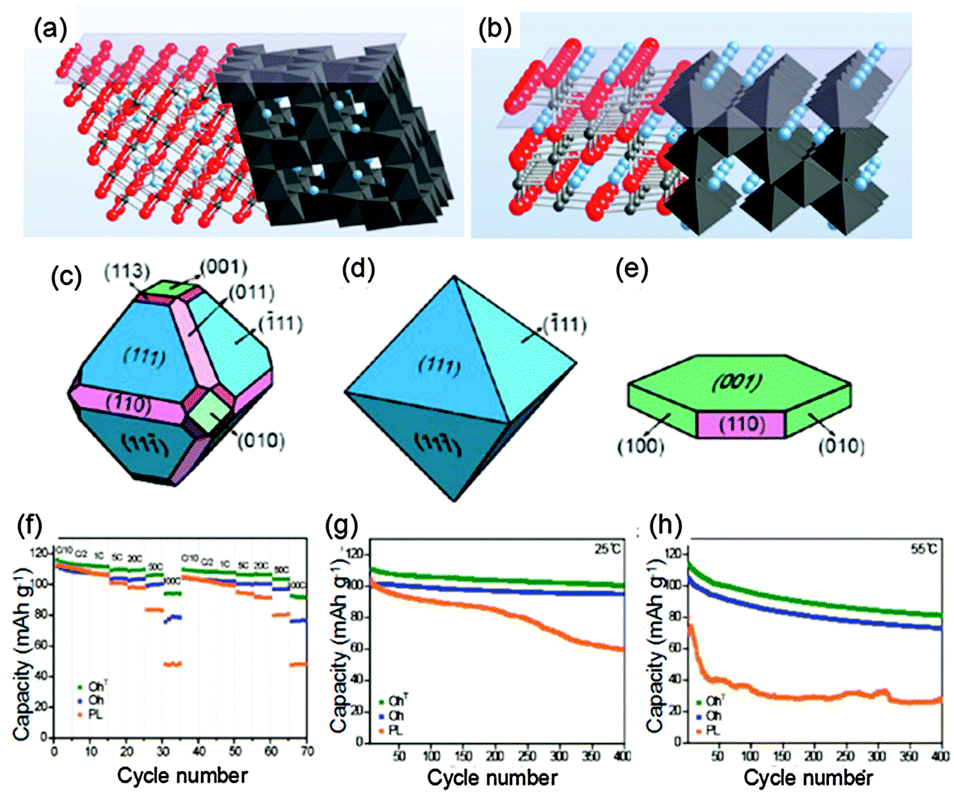 | ||
| Fig. 7 Atomic stacking of LiMn2O4 along (a) (111) and (b) (110) orientations (modified from ref. 69, copyright permission from American Chemical Society); schematic illustration of LiMn2O4 with various crystal shapes for (c) the truncated octahedron (OhT), (d) the bare octahedron (Oh), (e) platelets (Pl), and (f) their rate capability measured at various discharge C-rates, and cycling behaviors at 10C for discharge at (g) 25 and (h) 55 °C (modified from ref. 73, copyright permission from American Chemical Society). | ||
Following the above discussion, in order to obtain both excellent rate capability and cycling life, a truncated octahedral structure (Fig. 7c) was prepared through the solid state reaction of lithium hydroxide monohydrate with an Mn3O4 truncated octahedron.73 Its most surfaces are aligned along the (111) orientation activating minimal Mn dissolution, while a small portion of (110) planes is truncated along the directions that support Li ion diffusion. Octahedral structures enclosed by (111) planes (Fig. 7d) and nanoplates with even smaller dimensions (Fig. 7e) were also synthesized for comparison. Electrochemical test results (Fig. 7f–h) show that the truncated octahedral structure exhibits far better performance in both power density and cycle life compared to octahedral and nanoplate structures. Therefore, the concept of truncating a small portion of surfaces to support Li ion diffusion while leaving most remaining surfaces aligned along the crystalline orientations with minimal Mn dissolution enables excellent rate performance and cycle life simultaneously.
In contrast to the results of LiMn2O4 with the (110) plane,70–72 the truncated LiNi0.5Mn1.5O4 sample with the presence of (110) planes exhibits poor electrochemical performance while the octahedral LiNi0.5Mn1.5O4 material with high exposure of (111) planes demonstrates the highest reversible capacity and the best rate capability.76 From the analysis in the above sections, we know that the (111) plane in the spinel structure is more stable than the (110) planes, while (110) planes can support fast Li ion insertion/extraction. So the electrochemical properties of LiNi0.5Mn1.5O4 are very sensitive to the dissolution of Mn(III). Actually, most prepared LiNi0.5Mn1.5O4 still contains a small amount of Mn(III), which can be shown by a small redox peak at 4.0 V (vs. Li+/Li). The Mn ions are less densely arranged at the (110) surface, which can easily come into close contact with the electrolyte, making the Mn ions highly reactive towards solvents in the electrolyte.
LiNi1/3Co1/3Mn1/3O2 with a hexagonal α-NaFeO2 layer structure, which is similar to that of LiCoO2, is made up of MO2 oxygen layers perpendicular to the c axis, indexed to (001) facets that include the (001) and (001) planes at the top and bottom.59 The close packed (001) facets are therefore not electrochemically active for Li+ ion transportation due to their close-packed structure. The (010) facets are perpendicular to the (001) facets and have an open structure with a wide window between the layers for Li+ ion migration. Therefore, producing LiNi1/3Co1/3Mn1/3O2 crystals with a high percentage of exposed (010) facets will facilitate the fast and efficient transportation of Li+ ions.
Single crystalline LiNi1/3Co1/3Mn1/3O2 nanobricks with a high percentage of exposed (010) facets were synthesized by a solid-state reaction of LiOH·H2O with nickel–cobalt–manganese hydroxide precursors.59 When a plate-like structure is formed, PVP molecules immediately adsorb on its negatively charged (001) surfaces via the amine groups, thus reducing the growth rate along the (001) direction and leading to the formation of (001)-plane-dominated nanoplates. Then, the produced precursor Ni1/3Co1/3Mn1/3(OH)2 was mixed with a lithium salt and calcined at high temperature. The Ni1/3Co1/3Mn1/3(OH)2 hexagonal nanosheets with the reaction of the Li salt along the (010) direction result in a significant increase in the percentage of lateral (010) facets. Besides, a polyol medium (ethylene glycol) can also be used to control the preparation of the LiNi1/3Co1/3Mn1/3O2 nanoplates with exposed (010) active facets.79 Hierarchical cubed LiNi1/3Co1/3Mn1/3O2 with the enhanced growth of (010) facets was synthesized by using cube structured MnCO3 as a self-sacrificial template.80
In case of single crystalline LiNi1/3Co1/3Mn1/3O2 nanobricks with a high percentage of exposed (010) facets, the initial discharge capacities are 159, 151, 136 and 130 mA h g−1 at 2, 5, 10 and 15C rates, respectively.59 Both LiNi1/3Co1/3Mn1/3O2 nanoplates and microcubes with exposed (010) active facets display a high initial discharge capacity of above 200 mA h g−1 at a low current density.79,80 All of them exhibited excellent cycling behaviors. Moreover, the LiNi1/3Co1/3Mn1/3O2 crystal with a high percentage of exposed (010) facets also ensured an ordered atomic arrangement, which improves the stability of the crystallographic structure upon cycling.59
Many theoretical calculations and atomistic models were applied to study the surface energy of LiFePO4 structure. Through studying Li+ ion transportation in olivine LiFePO4 by first-principles calculations, it was found that the (010) plane possesses a lower Li ion migration energy and a higher Li ion diffusion coefficient, up to several orders of magnitude, than that of the (001) plane.86 Therefore, considerable attempts have been made to enhance their electrochemical performances by construction of LiFePO4 materials with maximal exposure of the (010) plane over other planes. Solvothermal and hydrothermal reactions are two effective strategies to prepare LiFePO4 with an increased percentage of (010) planes.60,87–90 The selection of chelating agent, reaction temperature and time, composition and concentration of surfactant plays critical roles in the growth of LiFePO4 nanocrystals.
LiFePO4 nanoplates with exposure of different crystal planes of (010) and (100) demonstrate similar discharge capacities at low current densities but quite different ones at high current densities (5 and 10C-rates).60 For example, LiFePO4 nanoplates with (010) planes delivered 156 and 148 mA h g−1 at 5 and 10C-rates, respectively, while the latter delivered 132 and only 28 mA h g−1 at the 5C-rate and the 10C-rate, respectively. It was reported that the crystallographic plane of LiFePO4 nanoplates was controlled by the mixing procedure of the starting materials. In addition, the b-axis thickness plays a critical role in the percentage of (010) planes and the electrochemical performance of LiFePO4. With a decreased b-axis thickness, LiFePO4/C nanoplates present increasingly improved electrochemical properties in comparison with those at larger b-axis thickness, owing to the higher percentage of (010) planes than that at smaller b-axis thickness.88 The key point is to allow LiFePO4 to grow along the ac plane and decrease the thickness of the b-axis as much as possible, leading to more exposure of (010) planes.
The benefits of faceting have also been observed for lithium-rich layered oxide materials. In this positive electrode material with α-NaFeO2 structure, it was widely reported that Li+ ions prefer to intercalate along the direction parallel to the Li+ ion layers, as discussed in Sections 2.2.1 and 2.2.4. The (010) facets can facilitate the migration of Li+ ions between the MO6 octahedron interlayers. Two different synthetic approaches were employed to synthesize lithium-rich layered oxide materials with deliberately (010) planes.62,95 It has been demonstrated that a different precursor and a shorter hydrothermal time both enable the Li1.17Ni0.25Mn0.58O2 nanoplates to grow simultaneously along the (010) and (001) directions, leading to the formation of (010)-facet-dominated nanoplates.95 Another layered lithium-rich material, hierarchical Li1.2Mn0.6Ni0.2O2 quasi-spheres, whose surface is constructed with (010) planes, was prepared by using the Ni0.2Mn0.6(OH)1.6 precursor as a self-sacrificial template.61 The hierarchical nanostructured Ni0.2Mn0.6(OH)1.6 precursors were synthesized through a co-precipitation reaction. After heat-treatment with the lithium salts at 900 °C, the nanoplates are developed with round edges, shrunk lateral dimensions, and expanded thickness, leading to a decrease in the non-electrochemically active (001) facets.
At a 6C-rate, the reversible capacity of Li1.17Ni0.25Mn0.58O2 nanoplates could reach around 200 mA h g−1 and 186 mA h g−1 after 50 cycles.95 In comparison with that in the conventional thermodynamic equilibrium nanoplate material of lithium-rich layered oxides, the active surface area in this habit-tuned nanoplate material of the Li1.17Ni0.25Mn0.58O2 sample is increased by about 50% although the proportion of the (010) nanoplates is only about 1/7. The hierarchical structured Li1.2Mn0.6Ni0.2O2 yields high initial specific discharge capacities of 230.8 and 141 mA h g−1 at the 1C and 20C rate, respectively, demonstrating an outstanding high-rate performance,61 which is attributed to the increased active surface area for Li+ ion transportation in the lithium-rich layered oxide. This unique hierarchical structure combines the advantages of a hierarchical architecture with electrochemically active (010) planes. The special directional alignment of nanoplates provides paths for Li+ ion rapid insertion/extraction, while the hierarchical structure gives an efficient 3D electron transport network, which enables both efficient ion and electron transport for fast Li+ ion transport kinetics.
LiMnPO4 microspheres with different crystallographic orientations were assembled via a facile hydrothermal route.62 Na2S·9H2O is employed as a sole additive for controlling the phase, shape and crystallographic orientation of LiMnPO4 microspheres. The Na2S·9H2O additive can be rapidly hydrolyzed into sulphions and hydroxyl ions, which selectively adsorb on different crystallographic facets of a LiMnPO4 crystal in aqueous solution. Li2FeSiO4 and Li2MnSiO4 nanosheets with growth oriented along the a-axis were prepared by a rapid one pot supercritical fluid synthesis method.99 The surface tension of supercritical fluids completely vanishes above the critical point of the fluid, which is of particular utility in controlling the surface and interface chemistries of the nanostructured materials.
The synthesized LiMnPO4 microspheres assembled with nanoplates exhibit discharge capacities of 130 mA h g−1 at 0.05C and 76.8 mA h g−1 at 0.5C, respectively, which showed a superior electrochemical performance over microspheres assembled with edges and prisms.62 This is due to the exposure of (010) facets. The (010) direction is the thinnest part of the crystal allowing for fast Li+ ion diffusion. Ultrathin Li2MnSiO4 nanosheets show a discharge capacity of 340 mA h g−1.99 For the first time, two Li ions were successfully extracted/inserted using the Li2MnSiO4 nanosheets. The sheet-like morphology oriented along the a-axis plays a significant role in achieving the two Li ion insertion/extraction of Li2MnSiO4. Both the orthorhombic and monoclinic structures of the Li2MSiO4 family are based on the slightly distorted hexagonal close packing of oxygen ions with all cations in the tetrahedral voids and a pseudohexagonal plane parallel to (001).
3 Electrode materials with tailored facets for aqueous rechargeable lithium batteries
Research on Li-ion battery technology shows that the use of flammable organic electrolytes might lead to thermal run-away and safety accidents. Moreover, the cost of a Li-ion battery is relatively high due to the organic electrolytes and special cell assembly technology such as the requirement of a strictly dry environment during manufacturing processes. In this regard, aqueous rechargeable lithium batteries (ARLBs) were proposed in 1994.100 However, the inferior capacity retention and low energy density limit their practical application. In 2007, ARLBs using various electrode materials based on redox reactions were reported by our groups and have attracted wide attention as promising systems in the past several years.101 ARLBs can overcome some disadvantages of Li-ion batteries. They are inherently safe by avoiding the use of flammable organic electrolytes. Moreover, the cost of the aqueous electrolyte is low because expensive salts can be replaced by cheap salts. In addition, the ionic conductivity of aqueous electrolytes is high, about two orders of magnitudes higher than that of organic electrolytes, which ensures high rate capability and thus high specific power.102–1043.1 Negative electrodes with tailored facets
As for the negative electrode materials of ARLBs, vanadium oxides, molybdenum oxides and some lithium intercalation compounds have been reported. Among them, LiV3O8,105 H2V3O4106 and VO2107 with preferred orientation and exposure of facets were studied in the past several years.Hierarchical LiV3O8 nanofibers, assembled from nanosheets that have exposed (100) facets, were fabricated by using electrospinning combined with calcination.105 During calcination, PVA was used and decomposed to release CO2, whilst NH4VO3 reacted with Li(CH3COO)·H2O to produce LiV3O8 nanoparticles. Besides acting as the template for forming the fibers, PVA could prevent LiV3O8 nanoparticles from aggregating into larger ones, making them grow into small nanosheets with exposed (100) facets owing to the self-limitation properties of LiV3O8. Single-crystal H2V3O8 nanowires along the [001] growth direction were obtained through a facile method by one-step hydrothermal treatment of commercial V2O5 powder.106 Flowerlike VO2 (B) micro-nanostructures assembled by single-crystalline nanosheets have been successfully synthesized via a hydrothermal route using polyvinyl pyrrolidone (PVP) as a capping agent.107
Compared with the other LiV3O8 micro/nanostructures, the hierarchical LiV3O8 with exposed (100) facets clearly shows better electrochemical performance in ARLBs. When the pH value of the electrolyte was adjusted to below 5, it was found that nanosheets with exposed (100) facets could effectively alleviate proton co-intercalation into the LiV3O8 electrode. In the crystal structure of LiV3O8, the (010) and (001) facets provide a lot of channels while the (100) facet has fewer and smaller channels, which makes it more difficult for H+ ions to intercalate into this material. A small amount of H+ ions intercalating into these channels instead of Li+ ions will lead to capacity fading. The pH value of the electrolyte has little influence on the cycling performance of the hierarchical LiV3O8 nanofibers with exposed (100) facets.105
The post-treated flowerlike VO2 (B) electrode shows an initial discharge capacity reaching 81 mA h g−1, which is higher than that of the flowerlike VO2 (B) sample before annealing.107 Compared to V2O5, H2V3O8 (or V3O7·H2O) has a higher electronic conductivity arising from a mixed-valence of V4+/V5+. The H2V3O8 nanowires allow a full intercalation of Li+ ions in preference to hydrogen evolution, and thus can deliver a specific capacity of 234 mA h g−1,106 much higher than that of any other vanadium oxides (VO2107 and V2O5108) in aqueous electrolytes. In H2V3O8, the V3O8 layers are held together by van der Waals interactions together with hydrogen bonding, in which this weakly bonded layer structure can favor the mobility of Li+ ions between the layers.
3.2 Positive electrodes with exposed facets
As discussed in Section 2.2, intercalation compounds (LiCoO2, LiNi1/3Co1/3Mn1/3O2, LiMn2O4 and LiFePO4) are used as positive electrode materials for lithium ion batteries. In aqueous electrolytes, lithium intercalation and de-intercalation can also occur in/from these materials as in organic electrolytes.LiMn2O4 nanotubes with a preferred orientation of (400) planes were prepared by using multiwalled carbon nanotubes as sacrificial templates.12 The oriented MnO2 was deposited on the CNTs. Then the as-prepared LiMn2O4 showed a typical crystal orientation after reacting with a Li salt at 700 °C. In the standard LiMn2O4, the intensity of (111) planes is much stronger than that of (400) planes.66,68 For the prepared LiMn2O4 nanotubes, their intensities are almost the same, suggesting that LiMn2O4 has a crystal orientation. LiFePO4 crystals with major (010) exposed facets were prepared by the reflux route in ethylene glycol solution under atmospheric pressure.13
Both LiMn2O4 with a preferred orientation of (400) facets and LiFePO4 crystals with major (010) exposed facets present superfast second-level charge capability (up to 1000C).13,65 In the case of the crystal orientation of the LiMn2O4 nanotube, there are more (001) or (010) planes on the edges of these planes or vertical to (400) planes, which is similar to the LiCoO2 nanoplates with exposed (001) planes.65 The 8a sites for lithium intercalation and deintercalation are situated at the (001) or (010) planes. Therefore, more Li sites are exposed to the aqueous electrolyte due to the preferred growth of (400) planes.
As for LiFePO4 with exposure of (010) facets, the reason for fast charge in an aqueous electrolyte is a little relatively complicated. H2O adsorption at different atomic sites on the LiFePO4(010) surface was calculated and the most stable structure was identified when three H2O molecules adsorb at three different sites, as illustrated in Fig. 8a and b (the 1, 2, 3 sites). These sites are the exact locations of the O vacancies at the corners of FeO6 and LiO6 octahedra in a stoichiometric (010) LiFePO4 surface. Such unique arrangement of water molecules at those LiFePO4(010) sites was verified using an accurate in situ mass measurement (EQCM) and Fourier transform infrared spectroscopy (FTIR). Each Li+ ion in the aqueous electrolyte is always coordinated by 4 water molecules in its primary solvation sheath. Two water molecules will be stripped away from this complex in order for a Li+ ion to intercalate into the nanoparticle (i to ii, and to iii in Fig. 8c and d). Then the resultant Li+(H2O)2 can approach the LiFePO4 surface with almost no additional barrier and docks at the site (iii to iv in Fig. 8c and d) to form a structure similar to the scenario of 3H2O on top of LiFePO4 in Fig. 9b. Such interfaces (HSLE) are effective in promoting fast mass transfer, which results in high rate capability (Fig. 8e). After this Li+ ion diffuses into the LiFePO4 bulk along the Li channel, these two H2O molecules will desorb from the surface.
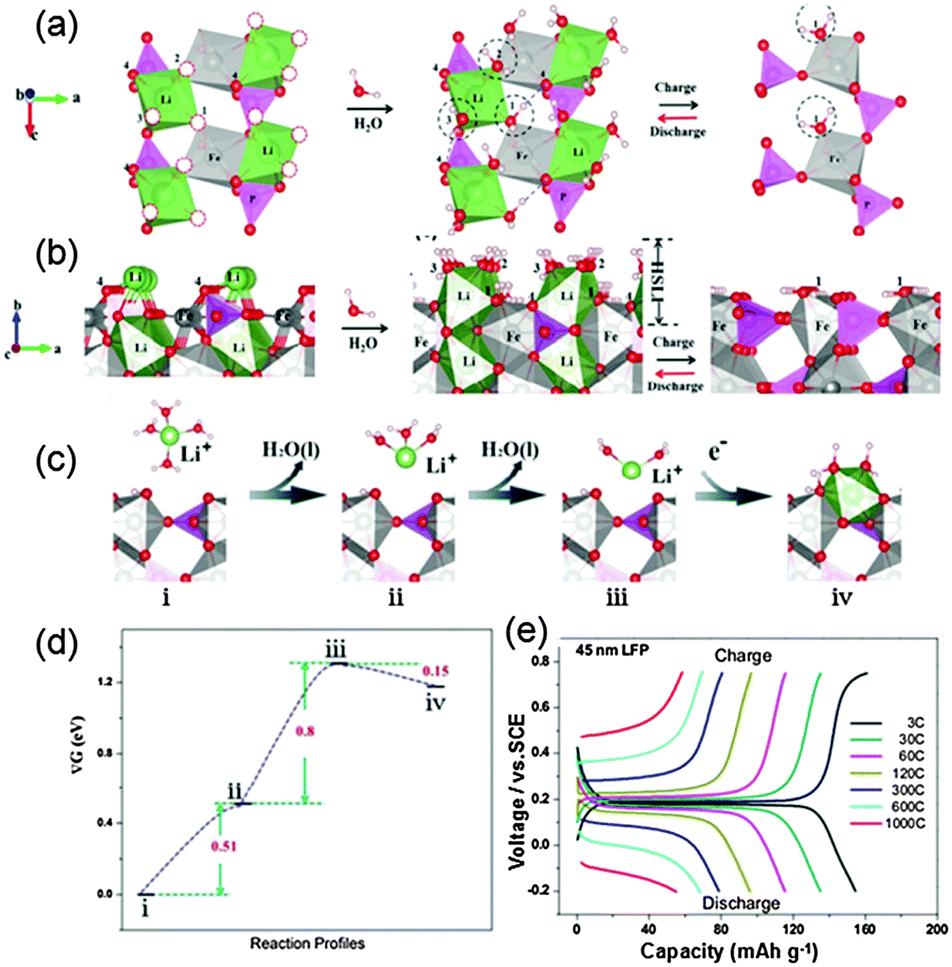 | ||
| Fig. 8 (a) Top and (b) side views of LiFePO4/vacuum, LiFePO4/H2O, and FePO4/H2O along the (010) direction. The four LiO6 octahedra correspond to four Li diffusion channels. The dashed red circles denote the O vacancies at the surface. (c) The reaction profiles for Li+ ion transport across the FePO4/water interface in the discharge process and (d) their energies at each step (right hand panels). (e) Charge and discharge curves at different current densities (1C = 170 mA g−1) between 0.2 and 0.75 V vs. SCE in a 0.5 M Li2SO4 aqueous electrolyte (modified from ref. 13, copyright permission from American Chemical Society). | ||
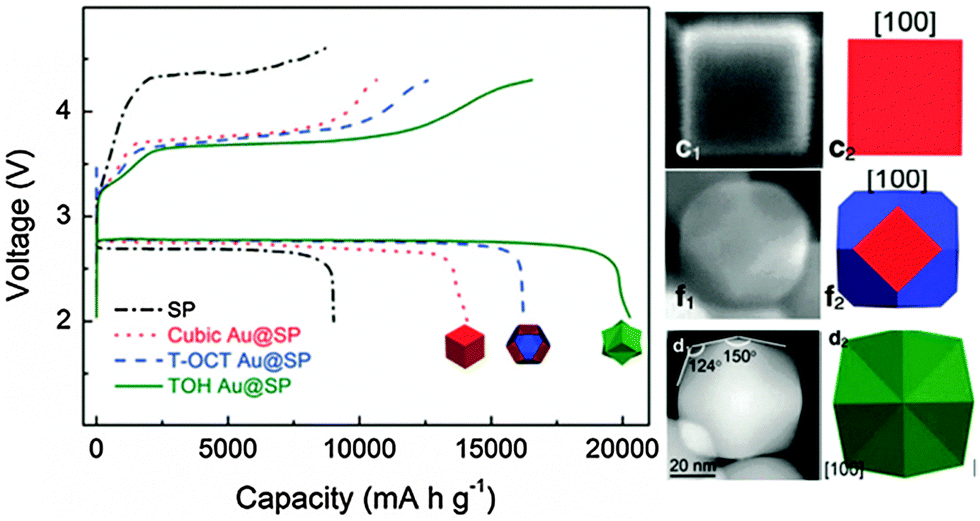 | ||
| Fig. 9 Charge–discharge curves of the cubic Au NCs@SP (red line), T-OCT Au NCs@S P (blue line), TOH Au NCs@SP (green line) and bare SP electrodes (black line) at 100 mA g−1 in the first cycle. The right images are their FEM or TEM micrographs and structure models (modified from ref. 122, copyright permission from Nature Publishing Group). | ||
4 Electrode materials with tailored facets for Na-ion batteries
Historically, progress in Na-ion batteries is parallel to that of lithium ion batteries.109 Unfortunately, Na-based systems rapidly fell into oblivion because Na+ ions are much larger in radius than Li+ ions and it is more difficult to find a suitable host material to accommodate Na+ ions. However, for sustainability reasons, Na-ion batteries have recaptured the scientific community's attention in recent years. The breakthroughs in materials science open new horizons for new energy storage and conversion devices.1104.1 Negative electrodes with exposed tailored facets
In this section, recent research progress on negative electrodes with tailored facets for Na ion batteries is summarized, such as SnO2 octahedral nanocrystals consisting of dominantly exposed (221) high energy facets111 and SnO nanocrystals with exposed (001) facets.112SnO2 with exposed (221) high energy facets and SnO with exposed (001) facets were synthesized by a hydrothermal method using PVP and Na2SO4 as a morphology directing agent, respectively.111,112 Sulfate ions are most strongly adsorbed onto faces perpendicular to the c-axis of the crystal through bridging-bidentate adsorption, leading to the retarded growth along the c-axis and the formation of the facet crystals.112
SnO2 nanocrystals with exposed (221) facets demonstrated a good high rate performance.111 It was found that Na ions first insert into SnO2 crystals at the voltage range from 3 to 0.8 V (vs. Na+/Na), and that the exposed (1 × 1) tunnel-structure could facilitate the initial insertion of Na ions. Then, Na ions react with SnO2 to form NaxSn alloys and Na2O in the low voltage range from 0.8 to 0.01 V (vs. Na+/Na). The SnO with exposed (001) facets delivered specific capacities of 525, 438, and 421 mA h g−1 at current densities of 40, 80, and 160 mA g−1, respectively.112 Similar to SnO2, SnO nanocrystals are dominated by (001) facets, exposing the 2D diffusion pathways for the insertion of Na+ ions into SnO crystals and the facilitated reaction towards sodium at high current densities.
4.2 Positive electrodes with tailored facets
Positive electrode materials are considered in this section including V2O5 hollow nanospheres constructed from hierarchical nanocrystals with predominantly exposed (110) crystal facets.113 Na0.54Mn0.50Ti0.51O2 with the growth direction perpendicular to the c-axis,114 Na0.7MnO2 single crystals with predominantly exposed (100) facets115 and β-MnO2 nanorods with exposed (111) crystal facets116 have been reported.V2O5 with predominantly exposed (110) crystal planes was synthesized via a polyol-induced solvothermal process.113 The preferred orientation of V2O5 nanocrystals is not triggered by the preparation conditions, but influenced by the vanadyl ethylene glycolate precursor's crystal structure. Tunnel-structured Na0.54Mn0.50Ti0.51O2 nanorods were synthesized by a facile molten salt method.114 These nanorods are grown in the direction normal to the Na-ion tunnels, which could greatly shorten the diffusion distance of Na ions and benefit the transfer kinetics. Na0.7MnO2 nanoplates with exposed (100) crystal planes and β-MnO2 nanorods with exposed (111) crystal planes were synthesized by a hydrothermal method.115,116 The individual Na0.7MnO2 nanoplate shows a perfect rhombus shape. Its corresponding SAED spot pattern could be well-indexed to the orthorhombic crystal structure along the (100) zone axis. And the facet vertical to the incident electron beam is the (100) crystal plane. The two basal planes of the rhombus nanoplates are the (100) facets, which are the predominantly exposed facets. For Na0.7MnO2 with layered structure, each layer perpendicular to the c axis is indexed to the (001) crystal plane. The (001) crystal plane is not electrochemically active for Na+ ion transport while the (100) crystal plane is an active plane for Na+ ion insertion/extraction, owing to the existence of a 2D channel for Na+ ion transport. So Na0.7MnO2 nanoplates with predominantly exposed (100) facets could easily facilitate the insertion/extraction of Na+ into/from the crystal structure. At a current density of 0.18 A g−1, the Na0.7MnO2 nanoplate electrode still delivers an initial capacity of 125 mA h g−1.115
Beta-MnO2 nanorods deliver a high initial discharge capacity of 350 mA h g−1. Although the discharge capacity decreases gradually upon cycling, it still maintains a high specific capacity of 200 mA h g−1 after 100 cycles.116 These β-MnO2 nanorods have exposed (111) crystal planes with a high density of (1 × 1) tunnels. The (1 × 1) tunnel not only provides facile transport for Na-ion insertion and extraction but also accommodates Na-ions.
5 Electrode materials with tailored facets for Li–O2 batteries
The first Li–O2 battery was introduced in 1996, and it was composed of lithium, an organic-impregnated polyacrylonitrile electrolyte, and a carbon electrode.117 A decade later, the Li–O2 battery using organic carbonates as electrolytes was demonstrated.118 After that, research on Li–O2 batteries quickly became a hot topic.119–121 A typical rechargeable non-aqueous Li–O2 battery is comprised of a Li metal as the negative electrode, a Li conducting organic electrolyte and a catalyst with oxygen as a positive electrode. Catalytic processes critically depend on the surface atoms' arrangement and the number of dangling bonds on different crystal planes.The catalytic properties of polyhedral Au nanocrystals (NC) with different index facets were studied in Li–O2 batteries, including cubic gold (Au) NCs enclosed by (100) facets, truncated octahedral Au NCs enclosed by (100) and (110) facets, and trisoctahedral (TOH) Au NCs enclosed by 24 high-index (441) facets.122 This preparation system has three species including a reducing agent (ascorbic acid), a gold precursor (HAuCl4) and a capping agent (CTAB or CTAC surfactant). It has been found that CTAB molecules can bind more strongly to the (100) than the (111) facets.123 Different CTAB concentrations can produce shapes with (100) or (111) facets. When CTAB is replaced by CTAC at the same concentration for the synthesis, TOH Au NCs bound by (441) facets dominate in the final product. The controlled synthesis of Co3O4 with different shapes and crystal planes and their catalytic properties for Li–O2 batteries have also been systematically studied, including nanocubes, pseudo octahedra, nanosheets, hexagonal nanoplatelets and nano-laminar.124 They are exposed with (100), (110), (111), and (112) crystal facets, respectively.
Compared to the carbon black (Super-P, SP), all these Au NCs significantly reduce the charge potential and show high reversible capacities (Fig. 9). Particularly, TOH Au NC catalysts demonstrate the lowest charge/discharge overpotential and the highest capacity of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 298 mA h g−1. Density functional theory calculations on the different Au crystal planes and their interaction with the Li and O atoms show that the interaction energy between the Au and the Li and O atoms decreases as the surface energy of the Au crystal planes increases. The oxygen adsorption energy on the surface of the (441) crystal planes is lower than those of the (100) and (111) crystal planes, which makes TOH Au NCs more active toward the oxygen evolution reaction (OER), thus leading to an enhanced electrochemical performance.122
298 mA h g−1. Density functional theory calculations on the different Au crystal planes and their interaction with the Li and O atoms show that the interaction energy between the Au and the Li and O atoms decreases as the surface energy of the Au crystal planes increases. The oxygen adsorption energy on the surface of the (441) crystal planes is lower than those of the (100) and (111) crystal planes, which makes TOH Au NCs more active toward the oxygen evolution reaction (OER), thus leading to an enhanced electrochemical performance.122
For the Co3O4 catalyst, the essential factor promoting the OER is its surface crystal planes.124 The correlation between different Co3O4 crystal planes and their effect on reducing charge–discharge over-potential is established in the following order: (111) > (112) > (110) > (100). Similar to the above Au crystal planes, Co3O4(111) crystal planes with highest surface energy have the largest interaction with Li and O atoms, leading to the highest catalytic properties for the Li and O reaction.
6 Electrode materials with exposed facets for supercapacitors
Supercapacitors have some advantages over batteries such as short charge–discharge time, high power density, high charge–discharge efficiency and long cycling life. According to different charge-storage mechanisms of electrode materials, supercapacitors can be categorized into electrochemical double layer capacitors (EDLCs) and Faradaic pseudocapacitors.125,126 In terms of the electrode configuration, supercapacitors can be fabricated as symmetric and asymmetric supercapacitors. The well-studied materials for EDLCs are usually based on nanostructured carbon materials. Pseudocapacitive materials cover transition metal oxides, conducting polymers and intercalation compounds. Among them, carbon materials and conducting polymers are amorphous, so these electrode materials are not included in this section.6.1 Negative electrodes with tailored facets
Hierarchical nanotubular titanium nitride (TiN) was fabricated by magnesiothermic reduction of titania replicas of ordinary filter paper (cellulose substance).127 The preferred growth direction is perpendicular to [200]. The Fe3O4@SnO2 core/shell nanorod film was achieved by fabricating Fe3O4 pristine nanorod film from FeOOH followed by the hydrothermal coating with SnO2 nanoparticles.128 Fe3O4 nanorods possess single-crystalline nature and grow along the [110] direction. MoO3 nanoplates with a crystal orientation along [001] were prepared via a sol–gel method by our group.129 The prepared MoO3 consists of nanoplates which are on average about 1 μm × 1 μm × 100 nm. A core/shell structure of PPy grown on V2O5 nanoribbons was fabricated by using SDB-(dodecylbenzenesulfonate) as a surfactant by our group.130 V2O5 nanoribbons were prepared by the hydrothermal treatment of NH4VO3 and the poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) copolymer in acid solution at 120 °C.TiN possesses attractive properties such as thermal conductivity, corrosion resistance and chemical stability. The shape of its CV curve is approximated to rectangular in a 1 M KOH electrolyte, showing an ideal double layer capacitive behavior.127 The specific capacitance of TiN is very low, only 74.2 F g−1 at 0.16 A g−1. When Fe3O4@SnO2 core/shell nanorod film is tested in the potential range of −0.8 to 0.2 V (vs. Ag/AgCl) in 1 M Na2SO3 solution, 82.8% of the initial capacitance could be stabilized after 2000 cycles.128 It is claimed that the capacitance of magnetite Fe3O4 originates from the surface redox reaction of sulfur in the form of sulfite anions, as well as the redox reactions between Fe2+ and Fe3+ accompanied by intercalation of sulfite ions to balance the extra charge with the iron oxide layers. Besides, SnO2 absorbs solvated cations (Na+) on the electrode surface from the electrolyte.
MoO3 nanoplates with a crystal orientation along [001] have a specific capacitance of 280 F g−1 in the Li2SO4 aqueous electrolyte, which is higher than that of the bulk MoO3 (208 F g−1).129 The nanoplate structure makes the solvated Li+ ions reach the MoO3 surface more easily than the bulk MoO3. Our group also investigated the insertion/extraction of K+ ions into/from V2O5 occurring in the (001)-plane-constituted interlayer space.130 V2O5 retains a layered structure with distinct diffraction peaks of (001), (003), and (004) planes. When K+ ions insert into the V2O5 electrodes possessing a K/V ratio of 0.35, the calculated interlayer space of the (001) plane decreases to 9.45 Å in comparison with the original value of 10.5 Å. This probably results from the reinforced interaction between K+ and the V2O5 skeleton. At the end of charge and discharge, the interlayer space expands slightly at the end of the charge accompanied by K+ ion extraction. The specific capacitance of V2O5 nanoribbons is 162 F g−1 at 100 mA g−1. The PPy shell coated on the V2O5 core further improves the charge transfer process and prevents vanadium dissolution into the aqueous electrolyte.
6.2 Positive electrodes with tailored facets
The capacitance of Co3O4 is directly linked to surface properties and its electrochemical performance is greatly influenced by any change related to the surface morphology of this electroactive material.131 A Co3O4@CNT hybrid was prepared by depositing Co3O4 on CNTs through a simple hydrothermal method. The size of the Co3O4 nanoparticles with preferentially exposed (220) planes is 10–15 nm.132 Co3O4 nanomesh with large-scale exposed (112) crystal planes is obtained based on the formation of single-crystal (NH4)2Co8(CO3)6(OH)6·4H2O nanosheets as the precursor and their subsequent conversion upon heating in air.133 The monocrystallization and conversion process from (NH4)2Co8 (CO3)6(OH)6·4H2O to Co3O4 is considered as thermodynamically supported recrystallization. The exposed crystal plane of Co3O4 on the largest scale is changed from (110) to (112) with the precursors changing from Co(CO3)0.5(OH)0.11·H2O to (NH4)2Co8(CO3)6(OH)6·4H2O, which is convinced by XRD measurements.NiO hexagons with exposed (110) facets on metallic Ni backbones were prepared via a simple hydrothermal method followed by annealing at 300 °C for 1 h.134 The presence of SO42− anions in the solution is primarily responsible for the restricted crystal growth in the perpendicular direction as they are strongly adsorbed onto the surfaces perpendicular to the c-axis through bridging-bidentate adsorption, thereby resulting in oriented hexagonal nanoplatelets of Ni(OH)2 on the surface. Upon annealing, the hexagon shapes are preserved and the exposed facets are the (110) facets which allow a lower lattice mismatch with Ni(OH)2. The NiO crystals have dominantly exposed (110) facets on both the hexagonal surfaces together with (002) and (111) facets as edges, forming a close-packed hexagonal nanoplatelet structure.
Alpha-MnO2 nanowires grown on flexible carbon fabric were synthesized by a hydrothermal approach.135 The growth direction of these MnO2 nanowires is very close to the normal direction of the (112) plane. Mn3O4 octahedral nanoparticles with (101) facets were prepared by a simple controlled oxidation method,136 similar to the growth mechanism of Li3VO4.15,16 Mn3O4 exists naturally as hausmannite, a distorted spinel in which Mn(II) and Mn(III) occupy the tetrahedral and octahedral sites, respectively. The most stable phase of Mn3O4 is a truncated tetragonal bipyramidal structure in which the (101) facets are primarily exposed with a small percentage of (001) and (100) facets. Growth of the (101) plane depends on the availability of Mn(III), which is formed by the oxidation of Mn(II), because of the greater density of Mn(III) in the (101) plane than that of the (001) plane.
The hybrid of Co3O4 nanocrystals coupled with CNTs can cycle over 9000 cycles in 1 M KOH aqueous solution.132 When assembled into an asymmetric supercapacitor by using activated carbon as the negative electrode, the hybrid capacitor shows excellent cycling performance between 0 and 1.8 V with an energy density of 31 W h kg−1 and a power density of 3 kW kg−1. Co3O4 nanomesh shows a capacitance of 297 F g−1 when scanned at 2 A g−1 in 3 M KOH aqueous solution though the voltage window is only in the range from 0.25 to 0.5 V (vs. Ag/AgCl),133 and 288 F g−1 could be achieved even at 10 A g−1. However, Co3O4 nanostructures with dominant (111) or (100) crystal planes do not have highly expected capacitive performance, and their specific capacitances are below 20 F g−1 and degrade rapidly upon increasing the scan rates. Actually, the dominant (112) crystal plane in the Co3O4 nanomesh has much higher surface energy than the conventional (111) and (100) crystal planes, leading to higher activity in supercapacitors.133 The Ni/NiO composite electrode with exposed high surface energy facets exhibits a specific capacitance as high as 2100 F g−1.134 When assembled as an asymmetric supercapacitor with mesoporous carbon as the negative electrode, the energy and power densities are calculated to be 17 W h kg−1 and 3.5 kW kg−1, respectively. Besides, this asymmetric supercapacitor also delivers good cycling performance over 2000 cycles, as the mesoporous wire-like network structure can uniformly distribute the stress across the electrode.
The areal capacitance of the MnO2 nanowire electrode can be 150 mF cm−2 at a current density of 1 mA cm−2.135 The specific capacitance corresponds to 197.4 F g−1 at an equivalent current density of 1.3 A g−1. A solid-state flexible asymmetric supercapacitor was assembled with MnO2 nanowires and Fe2O3 nanotubes as the electrodes using a gel electrolyte. It demonstrates excellent stability in a large potential window of 1.6 V and exhibits an excellent energy density of 0.55 mW h cm−3. The Mn3O4 octahedral nanoparticles show a high capacitance of 260 F g−1 at a scan rate of 1 mV s−1.136 Based on density functional theory calculations, Na preferentially binds to the (101) surface with a binding energy of 2.04 eV, compared to that of 1.4 eV on the (001) surface.
7 Conclusion and outlook
Over several years, the increasingly large amount of efforts devoted to electrode materials with tailored facets has resulted in a rich database for their synthesis, characterization and wide range of applications. This review has summarized recent advances in the synthesis chemistries and distinctive electrochemical properties of electrode materials with tailored facets for Li-ion batteries, aqueous rechargeable lithium batteries, Na-ion batteries, Li–O2 batteries as well as supercapacitors. The relationship between the crystal facets and their electrochemical activities has been analyzed and discussed.Generally, the synthesis can be achieved by a wet-chemistry route with addition of a surfactant. More specifically, through preferential binding of the surfactants with certain crystal planes, the shape of the crystals can be finely tuned. A surfactant is the most important preparation parameter as a morphology-directing agent (or a template, or a capping agent) in the formation of the desired electrode materials with tailored facets. Besides, the exposure of crystal facets can be controlled to some extent by selecting the appropriate precursor, crystal structure and ratio of precursors, and other reaction conditions such as temperature. The precursors can control both the chemical compositions and morphologies of the crystals while temperature can significantly control the speed of crystal growth in some cases.
On the other hand, the relationship between the crystal facets and their electrochemical activities can be summarized as follows:
(1) The charge transfer resistance and chemical diffusion coefficient are high along some facets, and exposing these facets can result in a lower energy barrier for faster transport of active ions across the surface of crystals such as TiO2 as the negative electrodes for Li-ion batteries.40,41
(2) Some facets have relatively high surface energy and these crystal facets provide reactive sites for fast redox reaction during the charge and discharge process, such as transition metal oxides based on conversion reaction for Li-ion batteries,28–30,52,53 catalysts for Li–O2 batteries,122–124 and electrode materials for supercapacitors.136
(3) Active ions can quickly shuttle back and forth in crystals along some directions while they cannot diffuse in the crystal along other directions because a mass of other atoms obstruct their passage. Thus, those facets, perpendicular to the directions that allow transportation of active ions, render a very short transport length for ions during insertion/extraction, such as most intercalation compounds for Li-ion batteries and Na-ion batteries.57,115
(4) There are many cracks on the exposed crystal planes and these cracks can favor fast ion transportation, such as LiCoO2 with exposed (001) planes and LiMn2O4 with exposed (400) planes.12,65
(5) A small amount of non-active ions intercalating into the channels of electrode materials will lead to capacity fading. Some exposed facets can effectively alleviate non-active ion co-intercalation into the electrode materials because these facets have fewer and smaller channels which only allow active ion intercalation into the material such as LiV3O18 with exposed (100) planes for aqueous rechargeable lithium batteries.105
(6) There will be a water molecule chemisorbed near some sites of some facets. This water molecule strengthens the active ion binding on surface sites and increases the binding energy, which lowers the energy barrier for active ion diffusion from the subsurface to this facet, such as LiFePO4 with exposed (010) planes for aqueous rechargeable lithium batteries.13
(7) Some ions in the crystals are less densely arranged at some facets and can easily come into close contact with the electrolyte, which makes these ions highly reactive towards solvents in the electrolyte, such as LiMn2O4 with exposed (111) planes for Li-ion batteries.73
The mechanism for the enhancement of electrochemical performances varies with materials and applications. In the case of most electrode materials for batteries, some planes possessing relatively low migration energy for active-ions like Li+ and Na+ are needed. To predict the electrochemical properties, the active-ion transport in some complex structures should be confirmed at first. Thus some well-established atomic modeling techniques are of utmost significance for electrode material optimization. The arrangement of surface atoms and surface energy also mean a lot especially for those electrode materials used in Li–O2 batteries and supercapacitors. For example, the surface energy of some planes of electrode materials can be calculated with the help of the Vienna ab initio simulation package (VASP).19 Currently, despite these significant advances achieved in this field, research on electrode materials with exposed tailored facets is still in its preliminary stage. Great opportunities and huge challenges coexist in this field. Throughout this review, we hope to generate more interest in them and boost extensive investigation in related areas. Thus, new vital interest and some challenges listed below are expected to motivate future studies.
To begin with, the technology for the synthesis of electrode materials with tailored facets is facing a challenge in establishing some theories that enable us to predict the type of surface or plane produced by a certain method. In fact, the selection of surfactant still remains empirical currently. Even with the use of same adsorbent as the surface controller, it is possible to prepare nanocrystals with different morphologies. Therefore, the surface binding structures of molecular adsorbents need to be well characterized at the molecular level. Besides, most synthesis strategies involve the use of morphology-controlling agents that must eventually be removed in order to obtain clean facets. This process might lead to some uncontrollable changes in the surface atomic structure of the crystal.
In addition, the integration of several instruments based on in situ characterization techniques is necessary. Obviously, in situ observations are essential to acquire a true understanding of the electrode surfaces. For example, in situ X-ray diffraction137 and in situ TEM techniques138 are strongly recommended to investigate the mechanism and structural evaluation of electrode materials.
Last but not least, developing new materials and structures is always expected, especially for Na-ion batteries and Li–O2 batteries. Exploring reliable electrode materials with suitable structures that can allow the intercalation/deintercalation of Na ions with high efficiency and meanwhile possessing excellent cycling stability is needed for the further development of Na-ion batteries. Designing different structured and efficient catalysts towards both oxygen reduction and/or evolution reactions is one of key strategies to improve Li–O2 battery performance. Meanwhile, many new electrochemical energy storage devices have emerged in recent years, including Mg-ion batteries,139 Al-ion batteries,140 Zn-ion batteries,141 F- and Cl-ion batteries,142,143 Na- and K–O2 batteries,144,145 Li–CO2 batteries,146 Li–Br2 batteries,147 metal ion capacitors148 and so on. However, there are few reports on their electrode materials with tailored facets. Therefore, seeking new electrode materials with tailored facets will become a hot spot in the field of electrochemical energy storage devices.
Acknowledgements
Financial support from the Distinguished Young Scientists Program of the National Natural Science Foundation of China (NSFC51425301) is gratefully appreciated.Notes and references
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- E. M. Larsson, C. Langhammer, I. Zoric and B. Kasemo, Science, 2009, 326, 1091–1094 CrossRef CAS PubMed.
- Y. G. Sun and Y. N. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- C. L. Pang, R. Lindsay and G. Thornton, Chem. Soc. Rev., 2008, 37, 2328–2353 RSC.
- G. Liu, H. G. Yang, J. Pan, Y. Q. Yang, G. Q. Lu and H. M. Cheng, Chem. Rev., 2014, 114, 9559–9612 CrossRef CAS PubMed.
- M. Chen, B. Wu, J. Yang and N. Zheng, Adv. Mater., 2012, 24, 862–879 CrossRef CAS PubMed.
- S. Liu, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef CAS PubMed.
- L. Zhang, W. Niu and G. Xu, Nano Today, 2012, 7, 586–605 CrossRef CAS.
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Gratzel, Phys. Rev. Lett., 1998, 81, 2954–2957 CrossRef CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. D. Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- W. Tang, Y. Y. Hou, F. X. Wang, L. L. Liu, Y. P. Wu and K. Zhu, Nano Lett., 2013, 13, 2036–2040 CrossRef CAS PubMed.
- J. X. Zheng, Y. Y. Hou, Y. D. Duan, X. H. Song, Y. Wei, T. C. Liu, J. T. Hu, H. Guo, Z. Q. Zhuo, L. L. Liu, Z. L. Chang, X. W. Wang, D. Zherebetskyy, Y. Fang, Y. Lin, K. Xu, L. W. Wang, Y. P. Wu and F. Pan, Nano Lett., 2015, 15, 6102–6109 CrossRef CAS PubMed.
- Y. Shi, S. L. Chou, J. Z. Wang, D. Wexler, H. J. Li, H. K. Liu and Y. P. Wu, J. Mater. Chem., 2012, 22, 16465–16470 RSC.
- Y. Shi, J. Z. Wang, S. L. Chou, D. Wexler, H. J. Li, K. Ozawa, H. K. Liu and Y. P. Wu, Nano Lett., 2013, 13, 4715–4720 CrossRef CAS PubMed.
- Y. Shi, J. Gao, H. D. Abruna, H. J. Li, H. K. Liu, D. Wexler, J. Z. Wang and Y. P. Wu, Chem. – Eur. J., 2014, 20, 5608–5612 CrossRef CAS PubMed.
- W. Q. Fang, X. Q. Gong and H. G. Yang, J. Phys. Chem. Lett., 2011, 2, 725–734 CrossRef CAS.
- W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946–2008 RSC.
- H. Sun, H. M. Ang, M. O. Tade and S. Wang, J. Mater. Chem. A, 2013, 1, 14427–14442 CAS.
- G. L. Xu, Q. Wang, J. C. Fang, Y. F. Xu, J. T. Li, L. Huang and S. G. Sun, J. Mater. Chem. A, 2014, 2, 19941–19962 CAS.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 CAS.
- Y. P. Wu, E. Rahm and R. Holze, Electrochim. Acta, 2002, 47, 3491–3507 CrossRef CAS.
- C. Li, H. P. Zhang, L. J. Fu, H. Liu, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, Electrochim. Acta, 2006, 51, 3872–3883 CrossRef CAS.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124–6130 CrossRef CAS PubMed.
- Y. M. Jiang, K. X. Wang, X. Y. Wu, H. J. Zhang, B. M. Bartlett and J. S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19791–19796 CAS.
- X. Wang, X. Cao, L. Bourgeois, H. Guan, S. Chen, Y. Zhong, D. M. Tang, H. Li, T. Zhai, L. Li, Y. Bando and D. Golberg, Adv. Funct. Mater., 2012, 22, 2682–2690 CrossRef CAS.
- D. Su, M. Ford and G. Wang, Sci. Rep., 2012, 2, 924–930 Search PubMed.
- D. Su, X. Xie, S. Dou and G. Wang, Sci. Rep., 2014, 4, 5753–5761 CrossRef CAS PubMed.
- X. Xiao, X. Liu, H. Zhao, D. Chen, F. Liu, J. Xiang, Z. Hu and Y. Li, Adv. Mater., 2012, 24, 5762–5766 CrossRef CAS PubMed.
- L. J. Fu, H. Liu, H. P. Zhang, C. Li, T. Zhang, Y. P. Wu, R. Holze and H. Q. Wu, Electrochem. Commun., 2006, 8, 1–4 CrossRef CAS.
- L. J. Fu, T. Zhang, Q. Cao, H. P. Zhang and Y. P. Wu, Electrochem. Commun., 2007, 9, 2140–2144 CrossRef CAS.
- C. Wang, F. Wang, Y. Zhao, Y. Li, Q. Yue, Y. Liu, Y. Liu, A. A. Elzatahry, A. Al-Enizi, Y. Wu, Y. Deng and D. Zhao, Nano Res., 2016, 9, 165–173 CrossRef CAS.
- L. Liu, Q. Fan, C. Sun, X. Gu, H. Li, F. Gao, Y. Chen and L. Dong, J. Power Sources, 2013, 221, 141–148 CrossRef CAS.
- B. Qiu, M. Xing and J. Zhang, J. Am. Chem. Soc., 2014, 136, 5852–5855 CrossRef CAS PubMed.
- L. Ren, Y. Liu, X. Qi, K. S. Hui, K. N. Hui, Z. Huang, J. Li, K. Huang and J. Zhong, J. Mater. Chem., 2012, 22, 21513–21518 RSC.
- S. Liu, H. Jia, L. Han, J. Wang, P. Ga, D. Xu, J. Yang and S. Che, Adv. Mater., 2012, 24, 3201–3204 CrossRef CAS PubMed.
- J. Liu, J. S. Chen, X. Wei, X. W. Lou and X. W. Liu, Adv. Mater., 2011, 23, 998–1002 CrossRef CAS PubMed.
- S. Ding, J. S. Chen, Z. Wang, Y. L. Cheah, S. Madhavi, X. Hu and X. W. Lou, J. Mater. Chem., 2011, 21, 1677–1680 RSC.
- R. Hengerer, L. Kavan, P. Krtil and M. Gratzel, J. Electrochem. Soc., 2000, 147, 1467–1472 CrossRef CAS.
- C. H. Sun, X. H. Yang, J. S. Chen, Z. Li, X. W. Lou, C. Li, S. C. Smith, G. Q. Lu and H. G. Yang, Chem. Commun., 2010, 46, 6129–6131 RSC.
- G. J. Wang, J. Gao, L. J. Fu, N. H. Zhao, Y. P. Wu and T. Takamura, J. Power Sources, 2007, 174, 1109–1112 CrossRef CAS.
- Y. Shi, J. Gao, H. D. Abruña, H. K. Liu, H. J. Li, J. Z. Wang and Y. P. Wu, Nano Energy, 2014, 8, 297–304 CrossRef CAS.
- T. Zhang, H. P. Zhang, L. C. Yang, B. Wang, Y. P. Wu and T. Takamura, Electrochim. Acta, 2008, 53, 5660–5664 CrossRef CAS.
- T. Zhang, L. Fu, J. Gao, L. Yang, Y. Wu and H. Wu, Pure Appl. Chem., 2006, 78, 1889–1896 CrossRef CAS.
- T. Zhang, J. Gao, L. J. Fu, L. C. Yang, Y. P. Wu and H. Q. Wu, J. Mater. Chem., 2007, 17, 1321–1325 RSC.
- S. C. Jung and Y. K. Han, Phys. Chem. Chem. Phys., 2011, 13, 21282–21287 RSC.
- S. W. Lee, M. T. McDowell, J. W. Choi and Y. Cui, Nano Lett., 2011, 11, 3034–3039 CrossRef CAS PubMed.
- N. Zhao, G. Wang, Y. Huang, B. Wang, B. Yao and Y. Wu, Chem. Mater., 2008, 20, 2612–2614 CrossRef CAS.
- N. H. Zhao, L. C. Yang, P. Zhang, G. J. Wang, B. Wang, B. D. Yao and Y. P. Wu, Mater. Lett., 2010, 64, 972–975 CrossRef CAS.
- C. Wang, G. Du, K. Stahl, H. Huang, Y. Zhong and J. Z. Jiang, J. Phys. Chem. C, 2012, 116, 4000–4011 CAS.
- J. Zhu, Z. Yin, D. Yang, T. Sun, H. Yu, H. E. Hoster, H. H. Hng, H. Zhang and Q. Yan, Energy Environ. Sci., 2013, 6, 987–993 CAS.
- X. Duan, L. Mei, J. Ma, Q. Li, T. Wang and W. Zheng, Chem. Commun., 2012, 48, 12204–12206 RSC.
- W. Li, Y. X. Yin, S. Xin, W. G. Song and Y. G. Guo, Energy Environ. Sci., 2012, 5, 8007–8013 CAS.
- H. Li, X. Liu, T. Zhai, D. Li and H. Zhou, Adv. Energy Mater., 2013, 3, 428–432 CrossRef CAS.
- Y. P. Wu, E. Rahm and R. Holze, J. Power Sources, 2003, 114, 228–236 CrossRef CAS.
- H. T. Xu, H. Zhang, L. Liu, Y. Feng and Y. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 20979–20986 CAS.
- W. Sun, F. Cao, Y. Liu, X. Zhao, X. Liu and J. Yuan, J. Mater. Chem., 2012, 22, 20952–20957 RSC.
- F. Fu, G. L. Xu, Q. Wang, Y. P. Deng, X. Li, J. T. Li, L. Huang and S. G. Sun, J. Mater. Chem. A, 2013, 1, 3860–3864 CAS.
- L. Wang, X. He, W. Sun, J. Wang, Y. Li and S. Fan, Nano Lett., 2012, 12, 5632–5636 CrossRef CAS PubMed.
- L. Chen, Y. Su, S. Chen, N. Li, L. Bao, W. Li, Z. Wang, M. Wang and F. Wu, Adv. Mater., 2014, 26, 6756–6760 CrossRef CAS PubMed.
- X. L. Pan, C. Y. Xu and L. Zhen, CrystEngComm, 2012, 14, 6412–6418 RSC.
- X. Wang, Q. Qu, Y. Hou, F. Wang and Y. Wu, Chem. Commun., 2013, 49, 6179–6181 RSC.
- Q. Cao, H. P. Zhang, G. J. Wang, Q. Xia, Y. P. Wu and H. Q. Wu, Electrochem. Commun., 2007, 9, 1228–1232 CrossRef CAS.
- X. Xiao, X. Liu, L. Wang, H. Zhao, Z. Hu, X. He and Y. Li, Nano Res., 2012, 5, 395–401 CrossRef CAS.
- F. X. Wang, S. Y. Xiao, Y. Shi, L. L. Liu, Y. S. Zhu, Y. P. Wu, J. Z. Wang and R. Holze, Electrochim. Acta, 2013, 93, 301–306 CrossRef CAS.
- Y. Liu, B. Zhang, F. Wang, Z. Wen and Y. Wu, Pure Appl. Chem., 2014, 86, 593–609 CAS.
- F. X. Wang, S. Y. Xiao, X. W. Gao, Y. S. Zhu, H. P. Zhang, Y. P. Wu and R. Holze, J. Power Sources, 2013, 242, 560–565 CrossRef CAS.
- M. Hirayama, H. Ido, K. S. Kim, W. Cho, K. Tamura, J. Mizuki and R. Kanno, J. Am. Chem. Soc., 2010, 132, 15268–15276 CrossRef CAS PubMed.
- Y. L. Ding, J. Xie, G. S. Cao, T. J. Zhu, H. M. Yu and X. B. Zhao, Adv. Funct. Mater., 2011, 21, 348–355 CrossRef CAS.
- D. K. Kim, P. Muralidharan, H. W. Lee, R. Ruffo, Y. Yang, C. K. Chan, H. Peng, R. A. Huggin and Y. Cui, Nano Lett., 2008, 8, 3948–3952 CrossRef CAS PubMed.
- H. W. Lee, P. Muralidharan, R. Ruffo, C. M. Mari, Y. Cui and D. K. Kim, Nano Lett., 2010, 10, 3852–3856 CrossRef CAS PubMed.
- J. S. Kim, K. S. Kim, W. Cho, W. H. Shin, R. Kanno and J. W. Choi, Nano Lett., 2012, 12, 6358–6365 CrossRef CAS PubMed.
- H. Kawaura, D. Takamatsu, S. Mori, Y. Orikasa, H. Sugaya, H. Murayama, K. Nakanishi, H. Tanida, Y. Koyama, H. Arai, Y. Uchimoto and Z. Ogumi, J. Power Sources, 2014, 245, 816–821 CrossRef CAS.
- J. Rana, S. Glatthaar, H. Gesswein, N. Sharma, J. R. Binder, R. Chernikov, G. Schumacher and J. Banhart, J. Power Sources, 2014, 255, 439–449 CrossRef CAS.
- K. R. Chemelewski, E. S. Lee, W. Li and A. Manthiram, Chem. Mater., 2013, 25, 2890–2897 CrossRef CAS.
- F. Wang, S. Xiao, Z. Chang, Y. Yang and Y. Wu, Chem. Commun., 2013, 49, 9209–9211 RSC.
- F. Wang, Y. Liu, X. Wang, Z. Chang, Y. Wu and R. Holze, ChemElectroChem, 2015, 2, 1024–1030 CrossRef CAS.
- J. Li, R. Yao and C. Cao, ACS Appl. Mater. Interfaces, 2014, 6, 5075–5082 CAS.
- Y. Wu, C. Cao, Y. Zhu, J. Li and L. Wang, J. Mater. Chem. A, 2015, 3, 15523–15528 CAS.
- H. Liu, C. Li, H. P. Zhang, L. J. Fu, Y. P. Wu and H. Q. Wu, J. Power Sources, 2006, 159, 717–720 CrossRef CAS.
- H. Liu, P. Zhang, G. C. Li, Q. Wu and Y. P. Wu, J. Solid State Electrochem., 2008, 12, 1011–1015 CrossRef CAS.
- S. Xiao, F. Wang, Y. Yang, Z. Chang and Y. Wu, RSC Adv., 2014, 4, 76–81 RSC.
- S. Y. Xiao, Y. Q. Yang, M. X. Li, F. X. Wang, Z. Chang, Y. P. Wu and X. Liu, J. Power Sources, 2014, 270, 53–58 CrossRef CAS.
- Y. S. Zhu, S. Y. Xiao, M. X. Li, Z. Chang, F. X. Wang, J. Gao and Y. P. Wu, J. Power Sources, 2015, 288, 368–375 CrossRef CAS.
- D. Morgan, A. V. Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30–A32 CrossRef CAS.
- C. Nan, J. Lu, C. Chen, Q. Peng and Y. Li, J. Mater. Chem., 2011, 21, 9994–9996 RSC.
- B. Pei, H. Yao, W. Zhang and Z. Yang, J. Power Sources, 2012, 220, 317–323 CrossRef CAS.
- Y. Liu, J. Gu, J. Zhang, F. Yu, J. Wang, N. Nie and W. Li, RSC Adv., 2015, 5, 9745–9751 RSC.
- R. Mei, X. Song, Y. Yang, Z. An and J. Zhang, RSC Adv., 2014, 4, 5746–5752 RSC.
- F. Wang, S. Xiao, M. Li, X. Wang, Y. Zhu, Y. Wu, A. Shirakawa and J. Peng, J. Power Sources, 2015, 287, 416–421 CrossRef CAS.
- F. X. Wang, S. Y. Xiao, Z. Chang, M. X. Li, Y. P. Wu and R. Holze, Int. J. Electrochem. Sci., 2014, 9, 6182–6190 Search PubMed.
- H. J. Yu and H. S. Zhou, J. Phys. Chem. Lett., 2013, 4, 1268–1280 CrossRef CAS PubMed.
- F. Wang, Z. Chang, X. Wang, Y. Wang, B. Chen, Y. Zhu and Y. Wu, J. Mater. Chem. A, 2015, 3, 4840–4845 CAS.
- G. Z. Wei, X. Lu, F. S. Ke, L. Huang, J. T. Li, Z. X. Wang, Z. Y. Zhou and S. G. Sun, Adv. Mater., 2010, 22, 4364–4367 CrossRef CAS PubMed.
- C. L. Hu, H. H. Yi, F. X. Wang, S. Y. Xiao, Y. P. Wu, D. Wang and D. L. He, J. Power Sources, 2014, 255, 355–359 CrossRef CAS.
- W. Chen, M. Lan, D. Zhu, C. Ji, X. Feng, C. Yang, J. Zhang and L. Mi, J. Mater. Chem. A, 2013, 1, 10912–10917 CAS.
- R. J. Gummow and Y. He, J. Power Sources, 2014, 253, 315–331 CrossRef CAS.
- D. Rangappa, K. D. Murukanahally, T. Tomai, A. Unemoto and I. Honma, Nano Lett., 2012, 12, 1146–1151 CrossRef CAS PubMed.
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CAS.
- G. Wang, L. Fu, N. Zhao, L. Yang, Y. Wu and H. Wu, Angew. Chem., Int. Ed., 2007, 46, 295–297 CrossRef CAS PubMed.
- W. Tang, Y. Zhu, Y. Hou, L. Liu, Y. Wu, K. P. Loh, H. Zhan and K. Zhu, Energy Environ. Sci., 2013, 6, 2093–2104 CAS.
- Z. Chang, Y. Yang, M. Li, X. Wang and Y. Wu, J. Mater. Chem. A, 2014, 2, 10739–10755 CAS.
- H. Kim, J. Hong, K. Y. Park, H. Kim, S. W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- L. Liang, M. Zhou and Y. Xie, Chem. – Asian J., 2012, 7, 565–571 CrossRef CAS PubMed.
- H. Li, T. Zhai, P. He, Y. Wang, E. Hosono and H. Zhou, J. Mater. Chem., 2011, 21, 1780–1787 RSC.
- S. Zhang, Y. Li, C. Wu, F. Zheng and Y. Xie, J. Phys. Chem. C, 2009, 113, 15058–15067 CAS.
- W. Tang, X. W. Gao, Y. S. Zhu, Y. B. Yue, Y. Shi, Y. P. Wu and K. Zhu, J. Mater. Chem., 2012, 22, 20143–20145 RSC.
- G. H. Newman and L. P. Klemann, J. Electrochem. Soc., 1980, 127, 2097–2099 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- D. Su, C. Wang, H. Ahn and G. Wang, Phys. Chem. Chem. Phys., 2013, 15, 12543–12550 RSC.
- D. Su, X. Xie and G. Wang, Chem. – Eur. J., 2014, 20, 3192–3197 CrossRef CAS PubMed.
- D. W. Su, S. X. Dou and G. X. Wang, J. Mater. Chem. A, 2014, 2, 11185–11194 CAS.
- X. Jiang, S. Liu, H. Xu, L. Chen, J. Yang and Y. Qian, Chem. Commun., 2015, 51, 8480–8483 RSC.
- D. Su, C. Wang, H. J. Ahn and G. Wang, Chem. – Eur. J., 2013, 19, 10884–10889 CrossRef CAS PubMed.
- D. Su, H. J. Ahn and G. Wang, NPG Asia Mater., 2013, 5, e70 CrossRef CAS.
- K. M. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1–5 CrossRef CAS.
- T. Ogasawara, A. D'ebart, M. Holzapfel, P. Novak and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390–1393 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- Z. Guo, X. Dong, Y. Wang and Y. Xia, Chem. Commun., 2015, 51, 676–678 RSC.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- D. Su, S. Dou and G. Wang, NPG Asia Mater., 2015, 7, e155 CrossRef CAS.
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12, 1765–1770 RSC.
- D. Su, S. Dou and G. Wang, Sci. Rep., 2014, 4, 5767–5775 CrossRef PubMed.
- F. Wang, S. Xiao, Y. Hou, C. Hu, L. Liu and Y. Wu, RSC Adv., 2013, 3, 13059–13084 RSC.
- F. Yu, L. Zhu, T. You, F. Wang and Z. Wen, RSC Adv., 2015, 5, 96165–96169 RSC.
- X. Liu, Y. Zhang, T. Wu and J. Huang, Chem. Commun., 2012, 48, 9992–9994 RSC.
- R. Li, X. Ren, F. Zhang, C. Du and J. Liu, Chem. Commun., 2012, 48, 5010–5012 RSC.
- W. Tang, L. Liu, S. Tian, L. Li, Y. Yue, Y. Wu and K. Zhu, Chem. Commun., 2011, 47, 10058–10060 RSC.
- Q. Qu, Y. Zhu, X. Gao and Y. Wu, Adv. Energy Mater., 2012, 2, 950–955 CrossRef CAS.
- X. Wang, M. Li, Z. Chang, Y. Yang, Y. Wu and X. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 2280–2285 CAS.
- X. Wang, M. Li, Z. Chang, Y. Wang, B. Chen, L. Zhang and Y. Wu, J. Electrochem. Soc., 2015, 162, A1966–A1971 CrossRef CAS.
- Y. Wang, H. J. Zhang, J. Wei, C. C. Wong, J. Lin and A. Borgna, Energy Environ. Sci., 2011, 4, 1845–1854 CAS.
- A. Paravannoor, T. S. Sonia, S. V. Nair and A. Balakrishnan, Mater. Lett., 2014, 135, 180–183 CrossRef CAS.
- P. Yang, Y. Ding, Z. Lin, Z. Chen, Y. Li, P. Qiang, M. Ebrahimi, W. Mai, C. P. Wong and Z. L. Wang, Nano Lett., 2014, 14, 731–736 CrossRef CAS PubMed.
- M. P. Yeager, W. Du, Q. Wang, N. A. Deskins, M. Sullivan, B. Bishop, D. Su, W. Xu, S. D. Senanayake, R. Si, J. Hanson and X. Teng, ChemSusChem, 2013, 6, 1983–1992 CrossRef CAS PubMed.
- S. Misra, N. Liu, J. Nelson, S. S. Hong, Y. Cui and M. F. Toney, ACS Nano, 2012, 6, 5465–5473 CrossRef CAS PubMed.
- J. Y. Huang, L. Zhong, C. M. Wang, J. P. Sullivan, W. Xu, L. Q. Zhang, S. X. Mao, N. S. Hudak, X. H. Liu, A. Subramanian, H. Fan, L. Qi, A. Kushima and J. Li, Science, 2010, 330, 1515–1520 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- F. Wang, F. Yu, X. Wang, Z. Chang, L. Fu, Y. Zhu, Z. Wen, Y. Wu and W. Huang, ACS Appl. Mater. Interfaces, 2016 DOI:10.1021/acsami.5b06142.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 CrossRef CAS PubMed.
- X. Zhao, S. Ren, M. Bruns and M. Fichtner, J. Power Sources, 2014, 245, 706–711 CrossRef CAS.
- X. Zhao, Z. Z. Karger, D. Wang and M. Fichtner, Angew. Chem., Int. Ed., 2013, 52, 13621–13624 CrossRef CAS PubMed.
- P. Hartmann, C. L. Bender, M. Vracar, A. K. Dürr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228–232 CrossRef CAS PubMed.
- X. Ren and Y. Wu, J. Am. Chem. Soc., 2013, 135, 2923–2926 CrossRef CAS PubMed.
- Y. Liu, R. Wang, Y. Lyu, H. Li and L. Chen, Energy Environ. Sci., 2014, 7, 677–681 CAS.
- Z. Chang, X. Wang, Y. Yang, J. Gao, M. Li, L. Liu and Y. Wu, J. Mater. Chem. A, 2014, 2, 19444–19450 CAS.
- F. Wang, X. Wang, Z. Chang, X. Wu, X. Liu, L. Fu, Y. Zhu, Y. Wu and W. Huang, Adv. Mater., 2015, 27, 6962–6968 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |