
Ultrasensitive optical detection of anions by quantum dots
Yongbing
Lou
 *a,
Yixin
Zhao
b and
Jun-Jie
Zhu
*c
*a,
Yixin
Zhao
b and
Jun-Jie
Zhu
*c
aSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, P. R. China. E-mail: lou@seu.edu.cn; Fax: +86-025-52090620; Tel: +86-025-52090619
bSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China
cState Lab of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, P. R. China. E-mail: jjzhu@nju.edu.cn; Fax: +86-25-8357204; Tel: +86-25-8357204
First published on 12th October 2015
Abstract
Quantum dots (QDs) have received great interest for diverse applications over the past few decades due to their unique photophysical properties like their tunable band gap, facile solution processability and versatile surface functionalization with different ligands. Quantum dot based optical analysis techniques with high sensitivity and selectivity have been developed to detect anions in aqueous solution for environmental monitoring, medicinal diagnostics, and the analysis of biological samples and industrial processes. Here we review the latest research progress of semiconductor QDs for sensing of anions in aqueous solution or in vivo, and discuss the photophysical mechanisms and outlook for the potential development in QD based optical sensing for anions.
 Yongbing Lou | Professor Yongbing Lou received his BS degree in 1996 from Nankai University and received his MS degree in 1999 from Nanjing University in polymer materials. After he completed his PhD in chemistry from Case Western Reserve University in 2007, he joined the School of Chemistry and Chemical Engineering at Southeast University. His research interests include preparation of nanomaterials for energy conversion, nanoplasmonics, and nanosensing. Dr Lou has published more than 40 peer-reviewed papers. |
 Yixin Zhao | Professor Yixin Zhao received his BS and MS degree in chemistry from Shanghai Jiao Tong University in China in 2002 and 2005. He obtained his PhD from Case Western Reserve University in 2010, then he worked as a postdoctoral fellow at Pennsylvania State University and the National Renewable Energy Laboratory on high efficiency dye sensitized solar cells, organometal halide perovskite solar cells and photoelectrochemical water splitting cells. Dr Zhao has published more than 40 peer-reviewed papers in Chem. Rev., Proc. Natl. Acad. Sci. U. S. A., J. Am. Chem. Soc., Angew. Chem., Int. Ed., Energy Environ. Sci., Adv. Mater., Nano Lett. etc. |
 Jun-Jie Zhu | Professor Jun-Jie Zhu received his BS (1984) and PhD (1993) degrees from the Department of Chemistry, Nanjing University, China. Then, he began his academic career at the School of Chemistry and Chemical Engineering, Nanjing University. He entered Bar-Ilan University, Israel, as a postdoctoral researcher from 1998 to 1999. Since 2001, he has been a full professor at Nanjing University. His main research activities focus on the preparation and bioapplications of functional nanomaterials. |
1. Introduction
Selective detection of various anions in aqueous solution is a challenging area due to its importance in environmental monitoring, medicinal diagnostics, and the analysis of biological samples and industrial processes.1 For example, cyanides occur naturally in both the geological and biological world, where over 2000 plants including vegetables and fruits contain cyanogenic glycosides to release cyanide upon acid hydrolysis (e.g., as occurs when ingested).2 Cyanide is acutely toxic to mammals with a very steep and rate-dependent dose–response curve, which involves inhibition of cellular respiration and inactivation of cytochrome oxidase to lead to consequent cellular anoxia. Fluoride is commonly added to toothpaste or administered in the treatment of osteoporosis because of its beneficial effects in dental health, but the chronic exposure to high levels of fluoride could cause dental or even skeletal fluorosis.3 Sulfide anions are widespread in the environment from industrial processes and biological metabolism, which could cause gradual and cumulative damage to life, such as loss of consciousness and irritation of mucous membranes after over-exposure.4 Once protonated, they become even more toxic as they turn into HS− and H2S in an acidic environment. Many analytical methods have been developed to detect these anions, such as titration, chromatographic, fluorimetric, flow injection, electrochemistry, and luminescence analyses and combinations thereof.5–7 Generally, these probe techniques require complicated procedures, time-consuming analysis, large sample volumes, and specialized skills. Optical sensing approaches have attracted the attention of many researchers with respect to the cost, reliability, ease of preparation, reproducibility, accuracy, insensitivity to electrical interference and the possibility of remote sensing of hazardous areas using optical fibre techniques. Most fluorescent probes are abiotic supramolecular systems that commonly bind analytes by noncovalent interactions, such as hydrogen bonding, electrostatic attraction and coordination phenomena. The affinity between anion analytes and binding sites of organic fluorophores is not strong enough due to the lower charge density of anions, which causes the sensitivity for anion sensing to be commonly not ideal. In addition, these organic fluorophores usually have complicated synthetic procedures, poor chemical stability and photostability, low quantum yield, the requirement of specific wavelength excitation, and a tendency for photobleaching, which limit their practical applications.1,3,8 In the past few decades, a tremendous number of novel fluorescent nanomaterials have been developed, which hold great promise for optical sensing applications in terms of brightness, photostability with a relatively long fluorescence lifetime, a size-tunable fluorescence spectrum, and versatile surface modification compared to organic fluorophores.Semiconductor quantum dots (QDs), composed of an inorganic core and an organic outer layer of surfactant molecules (ligands) with physical dimensions smaller than the bulk-exciton Bohr radius, provide a new class of nanomaterials that reveal unique optical and electronic properties. The unique photophysical and chemical properties of semiconductor QDs have attracted substantial experimental and theoretical research efforts over the past 30 years.9–14 The size-dependent wavelength of their photoluminescence (PL) properties could be understood through the all spatial dimension confinement (quantum confinement) of the exciton. Compared with general organic fluorophores, QDs possess advantageous characteristics such as size-/shape-controlled absorption/luminescence properties, narrow emission bands with a large Stokes shift, broad absorbance bands, high fluorescence quantum yields, high stability, and increased photostability, which altogether make them interesting for optical sensing applications. The PL of QDs arises from the recombination of the exciton in two possible pathways: band gap recombination or trap state recombination. The ability to alter the electron transfer from/to the QDs and influence the radiative/non-radiative recombination of excited electrons in the conduction band and holes in the valence band, through analyte-specific interactions at the surface should offer considerable analytical capability. The changes of surface charges or ligand components of the QDs would greatly affect the efficiency of electron–hole recombination and consequently the luminescence efficiency. Subtle interactions between the QD surface ligands and the surrounding species (molecules/ions/biomass) could significantly change their PL characteristics, which is the main principle for them to be used for chemical/biological sensing.
Semiconductor QDs have been widely used for metal ion sensing, the sensing strategies originate from the interaction between analyte ions and QD surfaces/capping ligands during the photoexcitation of the QDs and the transfer of valence-band electrons to the conduction-band. As summarized in our previous paper,15 the sensing strategies for metal ions were divided into two main categories: (1) QD surface interaction based sensing, and (2) fluorescence resonance energy transfer (FRET) based sensing. As for the QD surface interaction, the QD fluorescence could be effectively quenched after analyte ion binding to the surface ligand, after ligand displacement by analyte ions, or quenched after enhancing the fluorescence in the first place. The QD fluorescence could also be enhanced after the passivation of surface trap states through the formation of a surface passivation layer by ligand analyte binding, or enhanced after quenching the fluorescence in the first place by “ion-imprinted” sites or functional quencher groups. As for FRET based sensing, the FRET between the QDs and the acceptors could be sensitively adjusted by the analyte ions. The analyte ions could bind to the QD surface or the acceptor to interrupt the FRET, and they can also disrupt the linkage between the QDs and the acceptor to turn off the FRET. In another way, the analyte ions could help build the linkage between the QDs and the acceptor to form the FRET. The broad diversity and high reactivity of metal ions have facilitated multiple strategies for QD based sensing. As for the anions, limited research on the interaction mechanism between QDs and anions has been conducted in the literature, which has confined the research progress of QD based anion sensing. Inspired by metal ion sensing, some sensing strategies have been applied for anion sensing in a similar way.
Highly sensitive determination of cyanide anions by using CdSe QDs surface-modified with tert-butyl-N-(2-mercaptoethyl)-carbamate groups in organic media was first reported by Sanz-Medel and coworkers.16 In order to use highly fluorescent QDs to sense anions optically in aqueous solution various techniques and strategies have been implemented for the production of water soluble QDs to be used in vivo and in vitro.14,17–19 Exchanging the surface organic ligands with water soluble ligands using electrostatic, hydrophobic interactions or host–guest interactions has been a general approach for many water soluble QDs. The common water soluble ligands are thiol-containing molecules, amino acids, amines, glycol, such as mercaptoacetic acid (MPA),5,20,21 2-mercaptoethane sulfonate (MES),22 glutathione (GSH),23 thioglycolic acid (TGA).24 Encapsulation of a layer of amphiphilic block copolymers25,26 or surface modification with hydrophilic functional groups is also commonly used to prepare water soluble QDs.16 In this review, we will comprehensively review the strategies and methods to apply semiconductor QDs as a highly sensitive optical probe for anion sensing in aqueous solution.
2. Anion sensing through specific binding to quench luminescence
Fundamental studies of the optoelectronic properties of QDs have revealed that the luminescence of a QD is very sensitive to the surface states. After photoexcitation of the QDs, the radiative recombination of the electron–hole pair leads to the luminescence of the QDs. After the target analyte binds to the QD surface, it will cause the excited electron–hole pair separation. Therefore it effectively leads to the quenching of the luminescence of the QDs, which is called photoinduced electron transfer (PET). This is the basic strategy to sense various anions by utilizing the specific binding of analyte anions to specially modified QDs. The high intrinsic affinity between the analyte ions and the surface ligands is the decisive factor for anion selectivity. Quenching mechanisms involve many different effects, including ion-induced surface states, inner filter effects, ion-induced surface charge changes and bond cleavage, ion binding interaction and the PET process. The literature reports utilizing this strategy for anion sensing are summarized in Table 1.| QDs | Capping ligandsa | Anions | Range (μM) | LOD (μM) | Ref. |
|---|---|---|---|---|---|
| a BMC = tert-butyl-N-(2-mercaptoethyl)-carbamate, MES = 2-mercaptoethane sulfonate, MPA = mercaptoacetic acid, GSH = glutathione, TGA = thioglycolic acid, CMC = carboxymethyl cellulose, PEG = polyethylene glycol, ME = 2-mercaptoethanol, HDA = hexadecylamine. | |||||
| CdSe | BMC | CN− | 0.1–10 | 0.11 | 16 |
| CdSe | MES | CN− | 1–250 | 1.1 | 22 |
| ZnS | PEG | CN− | 5.00–200 | 1.29 | 27 |
| ZnS | ME | CN− | 2.44–25.9 | 0.17 | 28 |
| ZnS:Mn | MPA | S2− | 2.5–38 | 0.15 | 21 |
| CdS | MPA | S2− | 3–56 | 6.5 | 5 |
| ZnS:Mn | ME | S2− | 1.2–26 | 0.33 | 29 |
| CdS | MPA | HSe− | 0.10–4.80 | 0.087 | 20 |
| CdSe–ZnS | Thiourea type | Cl−, F−, AcO− | — | — | 30 |
| CdSe | Pyridine type | I− | 0–50 | 0.0015 | 31 |
| CdTe | GSH | Cr2O72− | 0.05–4.6 | 0.04 | 23 |
| CdTe | TGA | VO3− | 0.10–2.02 | 0.021 | 24 |
| CdS | CMC | IO3− | 0.010–10 | 0.0060 | 32 |
| CdTe/ZnS | L-Cysteine | IO4− | 0.005–2.7 | 0.0036 | 33 |
| CdSe | HDA | SeO32− | 0.063–0.824 | 0.001 | 34 |
| CdSe/ZnS | HDA | SeO32− | 0.63–9.5 | 0.0063 | 35 |
Cyanide has been very common in industrial use, and it is one of the most investigated anions for QD based sensing. CdSe QDs with surface modification by tert-butyl-N-(2-mercaptoethyl)-carbamate (BMC) exhibited strong emission at 580 nm, which allowed a highly sensitive determination of free cyanide ions with a detection limit of 0.11 μM (Fig. 1).16 Cyanide would bring about a disruption of the hydrogen bonds between the BMC group, also the adsorption of negatively charged cyanide on the surface of CdSe QDs resulted in increased electron localization due to compression of the electron wave function in the dots which could account for the observed quenching of luminescence emission by cyanide. Water-soluble luminescent CdSe QDs capped with MES were synthesized for the selective determination of cyanide ions in aqueous solution with high sensitivity (detection limit of 1.1 μM), where both dynamic and static quenching acted together for the quenching process.22
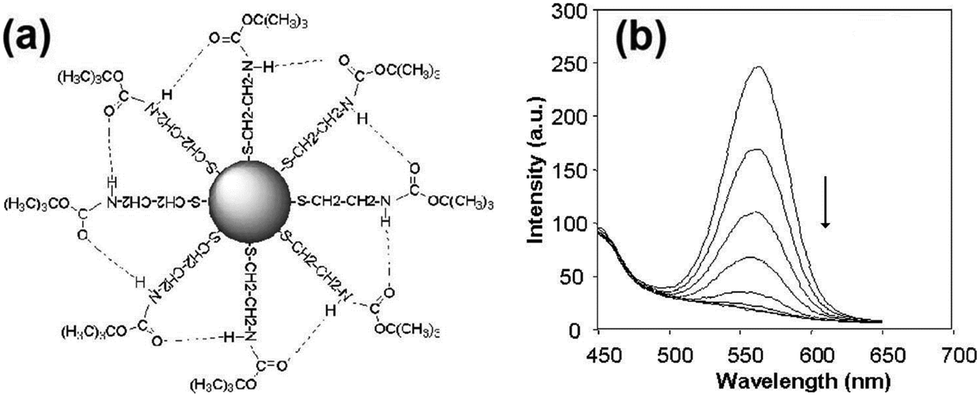 | ||
| Fig. 1 (a) Schematic diagram of a BMC–CdSe QD nanoparticle, and (b) effect of the addition of cyanide to the luminescence emission spectra of BMC–CdSe QDs. From top to bottom: addition of 0, 5, 10, 15, 20, 25, 30 μl of 0.05 M CN− ion to 3 ml QD solution after background was subtracted. Reprinted with permission from ref. 16, copyright 2004, Elsevier. | ||
The intrinsic toxicity of cadmium in common cadmium based QDs (CdS, CdSe, CdTe, etc.) casts doubt on the future applicability of these nanomaterials, especially for in vivo applications. Zinc sulfide and doped zinc sulfide with wide band gap energy are so far most studied, due to their low toxicity, tunable emission in the visible range and potential use for industrial and biomedical applications. Polyethylene glycol (PEG) coated ZnS QDs were fabricated by a microwave technique, which was used to develop a novel and highly sensitive luminescent sensor for cyanide ion detection in aqueous solution with a limit of detection of 1.29 μM.27 The negatively charged cyanide ions could bind to positively charged Zn2+ ions on the surface which led to quenching of the photoluminescence emission of ZnS QDs. 2-Mercaptoethanol (ME) capped ZnS QDs exhibited strong fluorescence emission at about 424 nm, where the intensity was linearly proportional to the cyanide ion concentration in the range 2.44 to 25.9 μM with a detection limit of 0.17 μM at pH 11.28 The quenching of the luminescence was attributed to anion binding followed by a redox reaction on the surface of the QDs and the effective electron transfer from the QDs to CN− ions. The high selectivity was assumed to be due to the fact that CN− could bind strongly with Zn2+, and form a layer of zinc complex on the QD surface.
Mn2+-doped ZnS QDs capped with MPA were prepared to selectively detect S2− ions.21 The linear range was from 2.5 to 38 μM with a detection limit of 0.15 μM. The adsorption of S onto QD surfaces would decrease the number of S2− vacancies to quench the QD emission, meanwhile it would increase the number of dangling bonds originating from the lone pairs on surface S2−, consequently the fluorescence of Mn2+ was also quenched. MPA functionalized CdS QDs were synthesized for the direct detection of S2− in aqueous solution.5 The adsorption of S2− on the CdS QD surface changed the surface state and thus facilitated non-radiative electron–hole recombination annihilation (Fig. 2a). Under optimum conditions, the method achieved a good linear relationship between the relative emission intensity ratio and the S2− concentration in the range from 3 to 56 μM and a detection limit of 6.5 μM (Fig. 2b). ZnS QDs doped with manganese (ZnS:Mn) were synthesized in aqueous solution with 2-mercaptoethanol (ME) as the capping agent, which could be used as a S2− sensor with a detection limit of 0.33 μM (Fig. 3).29 The emission of 594 nm is attributed to the 4T1–6A1 transition of Mn2+ impurities, which could be effectively quenched after S2− binding to the QD surface by an effective electron transfer mechanism.
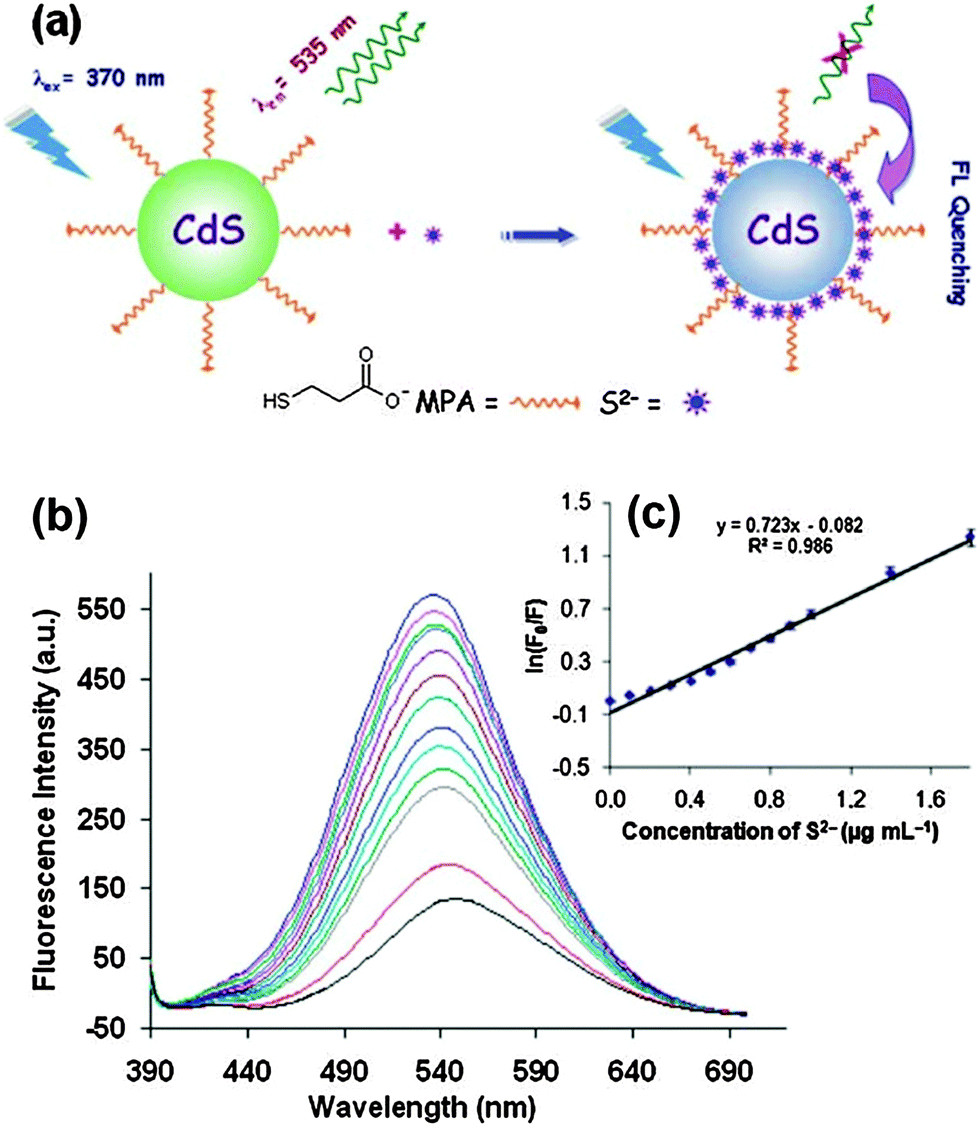 | ||
| Fig. 2 (a) Proposed graphic for the detection of S2− with MPA functionalized CdS QDs, (b) FL quenching of CdS QDs (0.0002 mol L−1) with the addition of different amounts of S2− (from top to bottom, 0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.4, 1.8 μg mL−1), and (c) the Stern–Volmer plots of S2− concentration dependence of the fluorescence intensity of CdS QDs. Reproduced from ref. 5. | ||
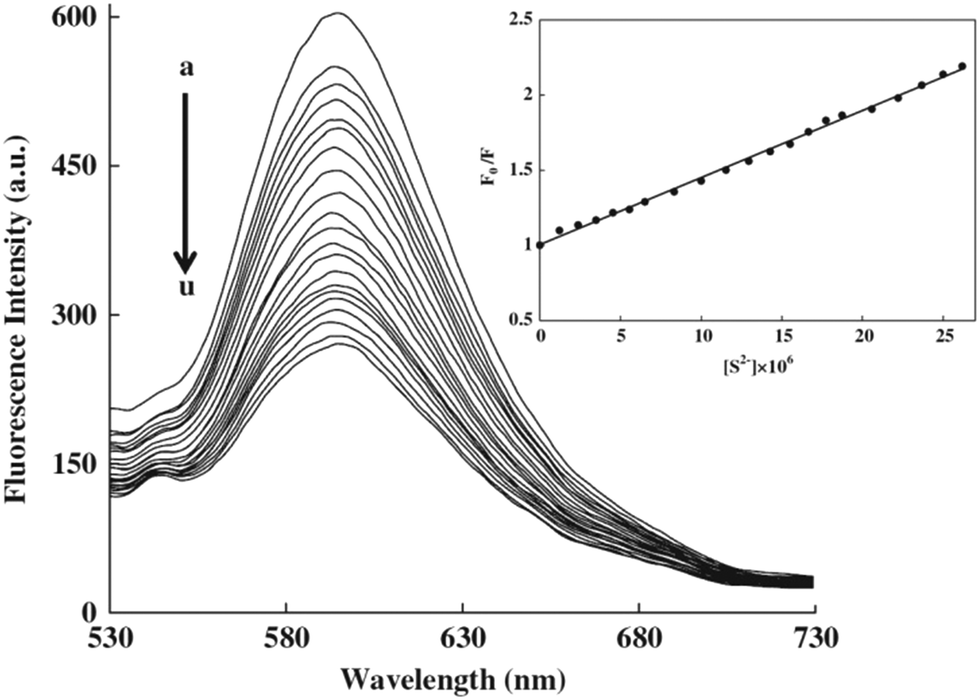 | ||
| Fig. 3 Effect of the sulfide concentration (0.00–2.62: 10−5 mol L−1; from (a) to (u)) on the fluorescence intensity of ZnS:Mn QDs. Inset: Stern–Volmer plot of sulfide concentration dependence of the fluorescence intensity of QDs with a 0.997 correlation coefficient. Reprinted with permission from ref. 29, copyright 2013, Elsevier. | ||
Water-soluble CdS QDs capped by MPA were synthesized by aqueous-phase arrested precipitation, which could be used for the selective detection of HSe− ions with a limit of detection of 0.087 μM.20 The quenching behavior after adding HSe− ions was due to the elimination of the S2− vacancies at the CdS QD surface and an outer semiconductor layer with a smaller band-gap provides an additional area of delocalization for electrons and holes. CdSe–ZnS QD functionalised with 1-(2-mercapto-ethyl)-3-phenyl-thiourea was prepared in the fluorophore–spacer–receptor format typical of PET based organic dye sensors as a modular system for anion sensing.30 The quenching effect by Cl−, F−, and AcO− is a result of hydrogen bonding interaction with the receptor molecule to enhance the rate of PET from the HOMO of the receptor to the QD.
CdSe QDs modified with 4-substituted pyridine type ligands containing thiourea groups (PyTU–CdSe QDs) were prepared through a ligand exchange process for the selective detection of I− ions with a limit of detection of 1.5 nM (Fig. 4).31 The preference for iodide suggests that the cavity formed by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N and CS⋯H–N of PyTUs on the surface of QDs is more complementary to the size of the iodide ion than to the size of other anions. The cavity formed by the thiourea group and the carbonyl group of the capping ligand group fitted well with the size of an iodide ion, which accounted for the high selectivity toward iodide ions.
O⋯H–N and CS⋯H–N of PyTUs on the surface of QDs is more complementary to the size of the iodide ion than to the size of other anions. The cavity formed by the thiourea group and the carbonyl group of the capping ligand group fitted well with the size of an iodide ion, which accounted for the high selectivity toward iodide ions.
 | ||
| Fig. 4 (a) Schematic diagram of the binding interaction between PyTU–CdSe QDs and iodide ions, (b) the effect of increasing concentration of iodide on the FL intensity of PyTU–CdSe QDs (from a to g: 0, 5 × 10−8, 5 × 10−7, 5 × 10−6, 1 × 10−5, 2.5 × 10−5, 5 × 10−5 M). Inset: A Stern–Volmer plot of the iodide concentration dependence of the FL intensity of PyTU–CdSe QDs with a 0.9982 correlation coefficient. Reproduced from ref. 31. | ||
The fluorescence intensity of GSH capped CdTe QDs was selectively reduced by Cr(VI) with a detection limit of 0.04 μM.23 The binding reaction of Cr2O72− and the surface GSH capping layer accounted for the fluorescence quenching. In a similar way, V(V) was sensitively detected by TGA capped CdTe QDs with a detection limit of 0.021 μM.24 Electron transfer made VO3− ions bind to the surface of the CdTe QDs, which facilitated nonradiative electron–hole recombination annihilation, leading to low PL quenching. The determination of iodate based on carboxymethyl cellulose (CMC) capped CdS QDs was reported by Zeng and coworkers.32 The relative fluorescence intensity of the CdS QDs was linearly proportional to IO3− over a concentration range from 0.010 to 10 μM with a detection limit of 0.0060 μM. The CdS QDs were directly oxidized by the iodate anion, resulting in the change in the surface structure of the CdS QDs and fluorescence quenching.
L-Cysteine-CdTe/ZnS core/shell QDs as pH-dependent fluorescent probes were proposed for the selective detection of IO4− ions, which exhibited a selective response for IO4− ions with a wide linear range from 50 nM to 2.7 μM and a detection limit of 3.6 nM at pH 3.5.33 Electron transfer between QDs and IO4− ions played the main role in the fluorescence quenching process, while collisions between QDs and IO4− ions could also lead to a decrease of the fluorescence intensity. CdSe QDs, which were stabilized by hexadecylamine (HDA) and confined in an organic droplet, were used in microfluorospectrometry as luminescent probes for the sensitive detection of Se(IV) with a detection limit of 0.63 μM.34 Derivatization by hydridation was used to convert SeO32− into volatile H2Se, which was rapidly transferred to the headspace of QD droplets. Aggregation was thought to occur as a result of binding between selenide trapped in the organic drop as selenium hydride and Cd2+ present on the surface of the QDs, which in turn, would result in the loss of stabilizing hexadecylamine groups. The same group also investigated a similar protocol with CdSe/ZnS QDs to detect Se(IV) and CH3Hg+ using the headspace single-drop microextraction (HS-SDME) approach along with a microvolume fluorospectrometer.35
3. Anion sensing based on the “off–on” strategy
Instead of directly quenching the QD fluorescence using analyte anions, binding a quencher group or a structure to quench fluorescence is another strategy to fabricate a new type of fluorescence sensor for anions. QD fluorescence is firstly quenched by surface state modification, by attaching a fluorescence quencher or a special structure coupled with a ligand through charge separation quenching or the FRET quenching mechanism. After adding the target anions, the QD fluorescence would be restored by passivating these defect sites, or blocking the charge separation and FRET between the QDs and the functional group. This kind of sensing design is called the “off–on” or “turn-on” strategy. The design or selection of a specific functional group or structure is the key to this strategy. The various functional groups or structures for sensing different anions are listed in Table 2.| QDs | Capping ligands or coatinga | Anions | Range (μM) | LOD (μM) | Ref. |
|---|---|---|---|---|---|
| a CTAB = cetyltrimethyl ammonium bromide, Cys = cysteamine, MSA = mercaptosuccinic acid, TEA = triethanolamine, PAA = polyacrylic acid. | |||||
| CdSe/ZnS | CTAB | F− | 0–300 | 0.68 | 36 |
| CdTe | Cys | F− | 0–60 | 5.0 | 37 |
| CdSe/ZnS | Ferrocenyl urea | F− | 300–5600 | 74.0 | 38 |
| CdTe | TGA–AuNPs | F− | 5–35 | 0.084 | 39 |
| CdSe@ZnS | Phthalocyanine | F− | 0.0015–0.12 | 0.000104 | 40 |
| CdTe | GSH–AuNPs | F− | 0.8–5.5 | 0.117 | 41 |
| CdSe | 2,2′-Bipyridine | CN− | 20–100 | 20 | 42 |
| CdTe | MSA–Cu2+ | CN− | 0.30–12 | 0.15 | 43 |
| CdS | Cys–Cu2+ | CN− | 2.5–20 | 1.13 | 44 |
| CdSe/ZnS | MPA–lucigenin | Cl− | 1000–250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
290 | 45 |
| CdTe@ZnS | GSH–phthalocyanine | Br− | 0.0010–0.048 | 0.00016 | 46 |
| CdSe | TEA–Hg2+ | I− | 4–32 | 0.28 | 47 |
| CdTe | TGA–Ca2+ | F−, CO32−, C2O42−, PO43− | 0–12000 |
440 | 48 |
| CdTe | TGA–Ni2+ | CO32−, C2O42−, PO43− | 0–1600 | 9.3 | 48 |
| CdTe | TGA–H2O2 | SO32− | 0–7600 | 6.8 | 48 |
| CdTe | PAA | CO32− | 0–14000 |
290 | 49 |
a. Fluorescence quenching by surface state modulation
The fluorescence of QDs (including emission intensity, emission wavelength, and emission lifetime) is sensitive to their surface states. In this strategy, the fluorescence of QDs is firstly quenched by modulation of the surface states with surface passivation or surface coating, while the fluorescence will be turned back on after interaction with analyte anions. A facile switchable fluorescent QD probe for F− ions based on hydrogen bonding-driven aggregation and analyte-triggered disaggregation was designed by Wang and coworkers.37 Cysteamine capped CdTe QDs underwent spontaneous self-aggregation via NH⋯N hydrogen bonds to quench the fluorescence, while the aggregates disassembled after F− ions formed NH⋯F hydrogen bonds resulting in fluorescence recovery of the QDs (Fig. 5a). Thus, the fluorescence off/on process enabled us to quantitate F− ions in aqueous media with a detection limit of 5.0 μM (Fig. 5b). A hybrid composite was formed from polyacrylic acid (PAA) and CdTe QDs through strong coordination interactions between the carboxyl groups of PAA and the Cd atoms on the surface of the QDs.49 The fluorescence of the QDs was completely quenched via the pull effect of PAA. Carbonate anions were hydrolyzed and partially protonated, which would react with the coordinated carboxyl groups of PAA, giving rise to the formation of hydrogen bonds. The fluorescence intensity would be gradually recovered after the addition of carbonate ions, showing a detection limit of 290 μM.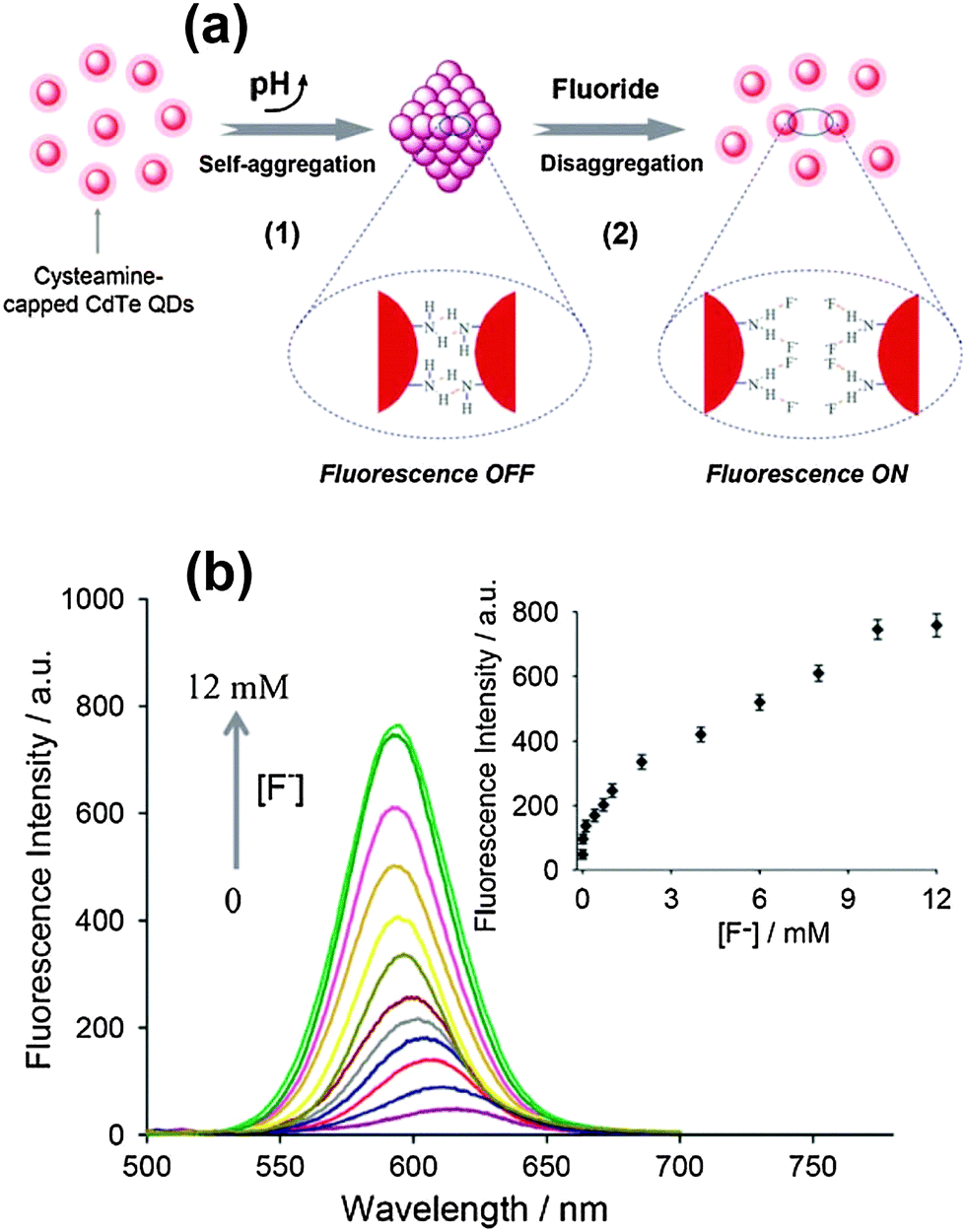 | ||
| Fig. 5 (a) A switchable fluorescent QD probe based on the aggregation/disaggregation mechanism, and (b) fluorescence spectra change of CdTe QD aggregate suspension (1.0 μM) upon addition of F− ions (0–12 mM), inset: The corresponding quantitative fluorescence intensity of QDs dependent on F− ions. Reproduced from ref. 37. | ||
CdSe/ZnS QDs coated with cetyltrimethyl ammonium bromide (CTAB) and a gemini surfactant [C12H25N+(CH3)2(CH2)4(CH3)2N+C12H25]·2Br− were reported for F− detection with a limit of detection of 0.68 μM.36 The fluorescence enhancement effect was due to the uniform surface arrangement after static interactions between the fluoride anion and the positively charged ammonium group of the surface of QD micelles (Fig. 6a). Such ordered orientation could suppress the quenching path to the medium by effective core protection and thus increases the luminescence intensity in the range of 0–300 μM (Fig. 6b).
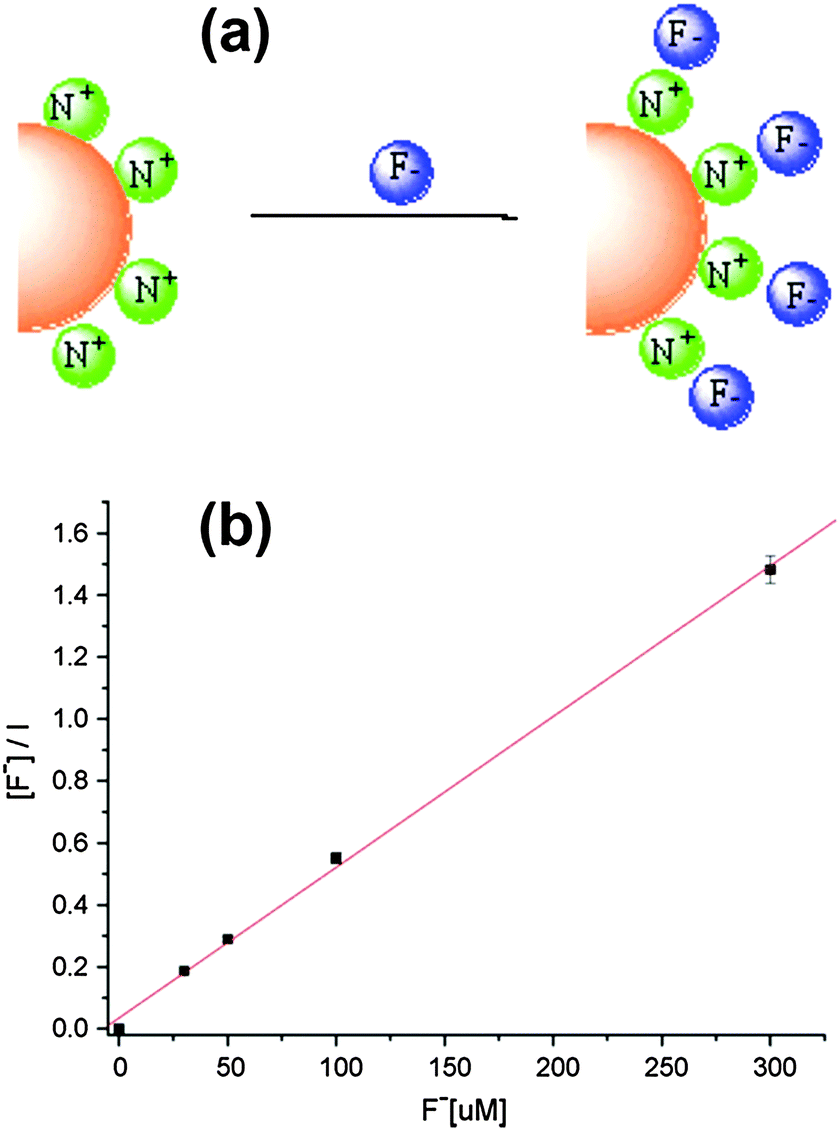 | ||
| Fig. 6 (a) Schematic illustration of the electrostatic interactions between single CdSe/ZnS QD micelles coated with a gemini surfactant and fluoride ions, and (b) Langmuir binding isotherm description of fluoride concentration dependence of the fluorescence intensity of surface modified CdSe/ZnS QDs. Reprinted with permission from ref. 36, copyright 2008, Elsevier. | ||
b. Fluorescence quenching by electron transfer
After adding metal cations or other electron acceptors to the QD system, the excited electrons will be transferred to the cation or the electron acceptor to quench the QD fluorescence effectively. After selectively adding analyte anions to break up the electron transfer path, the fluorescence will be turned back on. CdSe QDs conjugated with 2,2′-bipyridine-bound copper(II) ions were used to create a novel, selective turn-on fluorescence cyanide sensor in the range 20–100 μM.42 The ability of CuCl2:2,2′-bipyridine complex mixtures to quench the photoluminescence of CdSe QDs through the electron transfer mechanism was broken by the strong complexing ability of CN− to bind and remove copper ions (Fig. 7). CdSe/ZnS QDs attached with a ferrocenyl urea receptor were investigated for an “off–on” sensor for fluoride with a detection limit of 74.0 μM.38 The hydrogen bonding interaction between the urea protons and the fluoride ions would alter the rate of PET, resulting in the observed fluorescence enhancement upon fluoride addition. Since lucigenin is a broadly employed chloride ionophore, the CdSe/ZnS–MPA–lucigenin conjugate offered a good system to detect chloride ions with a detection limit of 0.29 mM.45 The quenching of QD fluorescence could be reversed after adding chloride ions, probably creating a competitive spin–orbit coupling pathway for lucigenin quenching, thus turning on the emission of the QDs.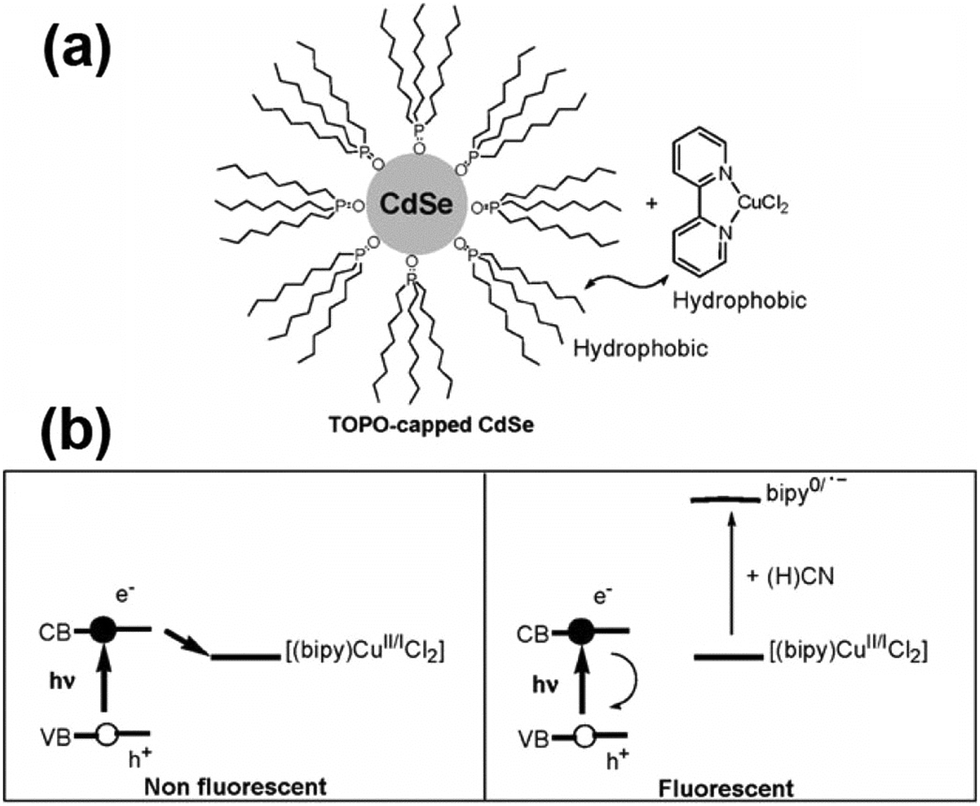 | ||
| Fig. 7 (a) Cyanide QD-based probe; and (b) fluorescence quenching and the cyanide sensing principle. Reproduced from ref. 42. | ||
The fluorescence of triethanolamine (TEA) coated CdSe QDs was greatly quenched only when Hg2+ and I− coexisted in solution, which could be used for the reciprocal recognition of Hg2+ ions and/or I− anions in aqueous solution.47 The detection limits of Hg2+ or I− ions were 0.19 μM or 0.28 μM, respectively. The fluorescence quenching of TEA coated QDs by the Hg2+·I− ion pair was not induced by the direct binding of Hg2+ with the TEA-QD surface, but by a diffusion double layer-type binding model of the soluble complex HgI42−. Dong and co-workers reported a “turn-on” fluorescent cyanide sensor based on copper ion-modified CdTe QDs with a low detection limit of 0.15 μM.43 The photoluminescence of the copper ion-modified CdTe QDs was firstly quenched due to the strong quenching ability of paramagnetic Cu2+, while in the presence of CN−, Cu2+ could be desorbed from the surface of the QDs because of the much higher stability constant of the complex of CN− and copper ions. The fluorescence of cysteamine (Cys) capped CdS QDs was effectively quenched by Cu2+, and could be recovered upon adding CN− to remove Cu2+ ions from the surface of QDs by forming a strong copper–cyanide complex.44 The proposed method showed good selectivity toward cyanide ions over other anions and could be efficiently applied to detect cyanide in drinking water samples with a detection limit of 1.13 μM. A flow injection chemiluminescence method was developed for the determination of cyanide CN− based on the recovered chemiluminescence signal by Cu2+ inhibiting a GSH capped CdTe QD and hydrogen peroxide system.50 Although this strategy seems promising for high selectivity with decreased background interference and various possible quenching structure designs, the reported sensitivity is somewhat below expectation compared to the previous strategy.
c. Fluorescence quenching by FRET
FRET involves the nonradiative energy transfer between proximal molecules or structures (0.5–10 nm), which occurs due to long-range dipole–dipole interactions between a donor (D) molecule in the excited state and an acceptor (A) molecule in the ground state.51 This strategy builds up an appropriate FRET system between QDs (D) and some specific acceptor groups (A) to quench the QD fluorescence first, while the analyte anions could either directly bind to the acceptor or break the linkage to interrupt the FRET. The high intrinsic sensitivity to small changes in the separation distance and orientation between the donor and acceptor dipoles will enable the ultrasensitive detection of trace amounts of analyte anions.The fluorescence of QDs was turned off by the efficient FRET between TGA modified CdTe QDs and citrate-capped Au NPs. Meanwhile, F−, a strongly nucleophilic anion, reacted with the H nucleus to break the hydrogen bonds and disassemble the AuNP segment, resulting in the fluorescence recovery of the quenched QDs. This new nano-assembly could be used for the sensitive detection of F− ions with a detection limit as low as 84 nM in aqueous solution (Fig. 8).39 GSH-capped CdTe@ZnS QDs were successfully conjugated to nickel tetraamino-phthalocyanine (NiTAPc) to form QD-NiTAPc nanocomplexes, in which GSH-CdTe@ZnS QDs transferred their energy to NiTAPc as a result of FRET and the fluorescence was quenched.46 Upon the interaction of NiTAPc with Br−, the fluorescence was turned on. The limit of detection of Br− could reach as low as 0.16 nM in aqueous solution. In a similar way, they also synthesized unsymmetrically substituted derivatives of aluminium amino phthalocyanines, which were conjugated to CdSe@ZnS QDs for sensitive F− ion detection.40 The detection limit could reach as low as 0.104 nM. Conjugation was formed through a H-bonding interaction between tripeptide GSH capped CdTe QDs and a bovine serum albumin (BSA) protein-conjugated Au25 nanocluster (NC) for the detection of both the Hg2+ ion and the F− ion.41 Strong fluorescence quenching was due to the energy transfer between QDs and the Au NC (quencher), which was restored in the presence of F− ions because the hydrogen bonds between GSH and BSA break down in the presence of F−. The limit of detection for the F− ion could reach 117 nM.
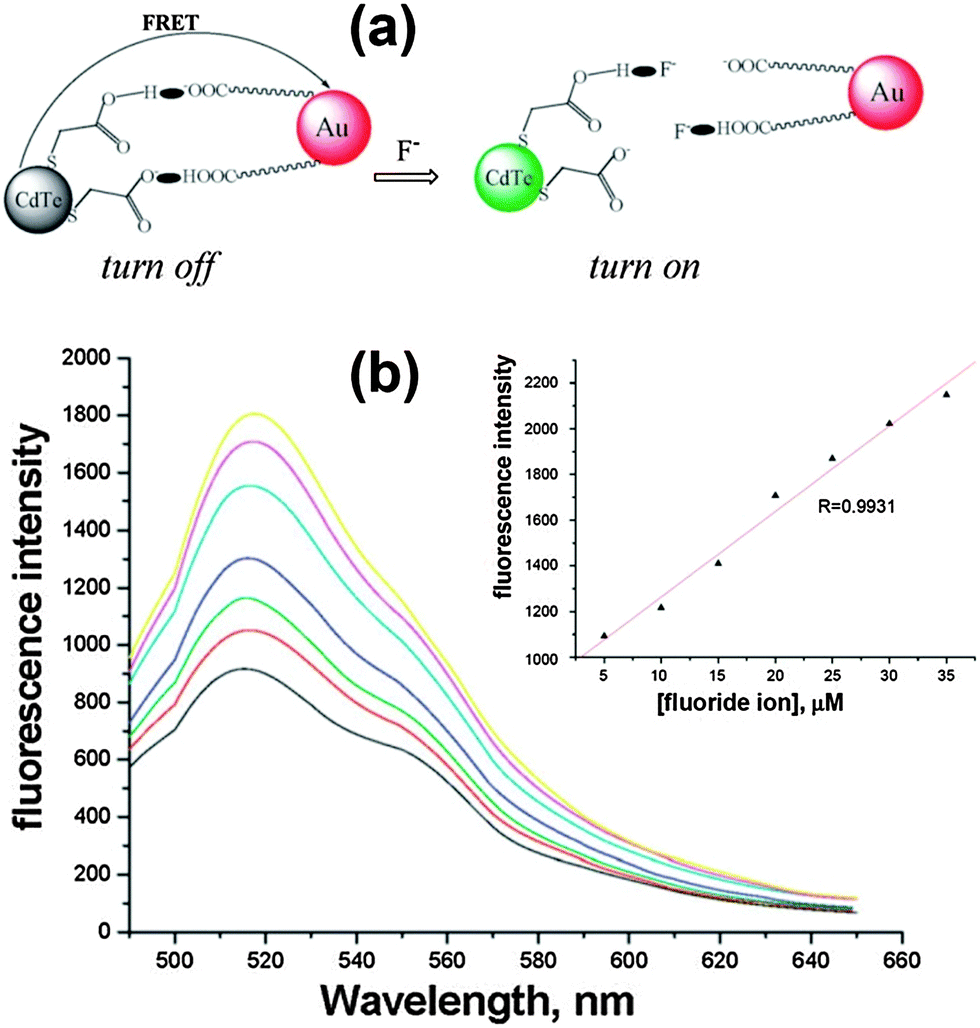 | ||
| Fig. 8 (a) Schematic illustration of FRET broken by adding F− anions, and (b) fluorescence response after different concentrations of F− were added to the analytical system under the optimal conditions (from bottom to top: 5, 10, 15, 20, 25, 30, 35 μM), inset: A linear correlation between the fluorescence intensity and the concentrations of F− in Tris-HAc buffer. Reproduced from ref. 39. | ||
Xia et al. designed three types of turn-on fluorescent method for anion sensing using TGA capped CdTe QDs.48 The fluorescence of the QDs is firstly quenched by three different mechanisms (fluorescence resonance energy transfer, electron transfer and surface state modulated fluorescence). The fluorescence recovered after adding various anions (F−, CO32−, C2O42−, PO43−, SO32−) due to modulating effects. These fluorescent probes gave moderate selectivity and the detection limit could reach the level of several μM.
4. In vivo anion sensing
Instead of monitoring the intensity changes after binding with the analyte anions, fluorescence-lifetime-based systems record the photoluminescence lifetime change after interacting with analytes. The fluorescence of QDs arises from the recombination of excitons, all changes in charge or composition on the surface and the nearby environment of the QDs could affect the efficiency of the electron–hole recombination. It further induces changes in the QD fluorescence efficiency, decays and dynamics. Wang and coworkers applied a nanosensor composed of a Cl− receptor (1-(2-mercapto-ethyl)-3-phenyl-thiourea) and CdSe/ZnS QDs to measure the dynamic Cl− responses in two human epithelial cell lines when their respective chloride channels were manipulated accordingly.53 Ruedas-Rama and coworkers designed Cl− sensitive CdSe/ZnS QD–lucigenin conjugates as time resolved fluorescence nanosensors, which could be expanded to in vivo cell imaging by taking advantage of fluorescence lifetime imaging techniques.52 The increase in the QD lifetime upon the addition of chloride ions is clearly visible in the arbitrary color scale image, as well as in the lifetime distributions (Fig. 9). The strong interaction between lucigenin and Cl− makes it a difficult task to reuse the QDs in subsequent analysis. Continuous exploration of sensitive and wide range detection of anions in vivo demands more efforts in the field.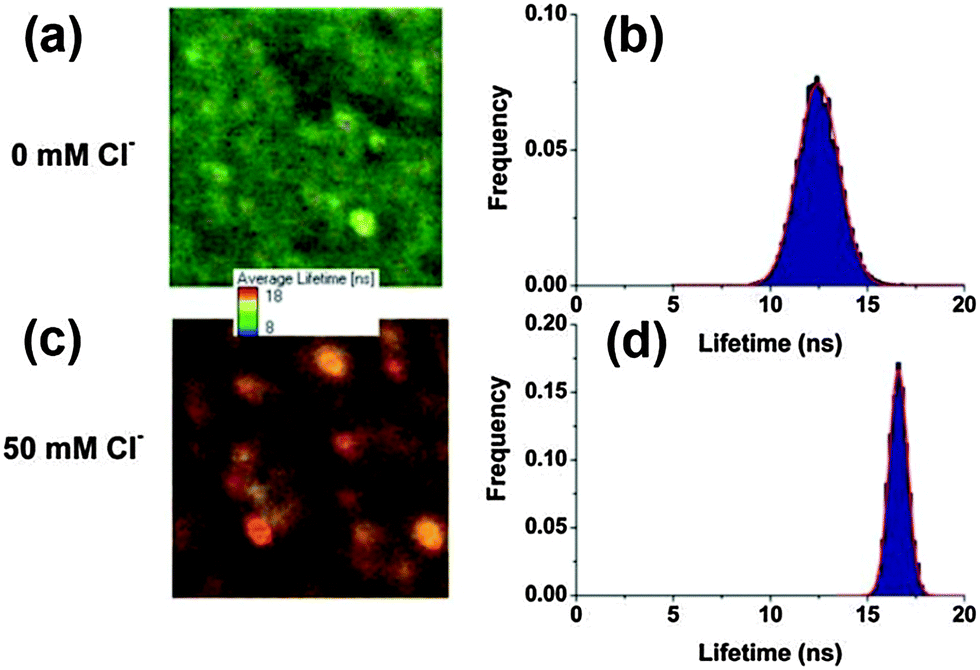 | ||
| Fig. 9 Fluorescence lifetime imaging and the corresponding lifetime distribution of QD–lucigenin conjugates in the presence of 0 mM Cl− (a and b) and 50 mM Cl− (c and d). Images are 9.80 × 9.80 mm (a) and 5.31 × 5.31 mm (c) represented in an arbitrary color scale between 8 (blue) and 18 ns (red), and modulated in intensity by the total number of photons per pixel. Reproduced from ref. 52. | ||
5. Future perspectives
Remarkable progress in the applications of semiconductor QDs as fluorescent sensors for anions has been made so far. Various hybrid nanostructure and sensing systems have made use of the unique photophysical properties of QDs (photostability, high quantum yields, tunable luminescence features) and the special interaction between the target analyte anions. Different sensing mechanisms including surface state modulation, photoinduced electron transfer, and fluorescence resonance energy transfer were implemented to develop various anion sensing systems. These analytical methods may find important applications in the analysis of industrial samples, environmental monitoring, and biosensing.Comparing the paramount work on the applications of QDs for heavy metal ions, small molecules, and biological sensing and bio-imaging (such as proteins, nucleic acids, and even whole cells),15,54–63 the application of QDs for anion sensing still has many aspects to be further explored in the future. The influence of experimental parameters including pH, QD concentration, interaction time, temperature and foreign ions on the fluorescence intensity has greatly affected the practical applications of QD anion sensors.29,52 It is critical to keep these parameters constant during anion sensing. CdSe QDs have showed good thermal sensitivity and surface modification played an important role in the enhancement of the characteristic thermal sensitivity,64–66 which would enable the possibility of building multi-functional QD sensors in combination with anion sensing. The design rules of the higher sensitivity of cation QD chemosensors will contribute to the further development of anion sensors with high sensitivity. FRET based anion sensing showed great potential for ultra-high sensitivity due to its intrinsic sensitivity to small changes in the separation distance and orientation. Special design of anion sensitive structures or effective organic fluorophores will find prospective applications in designing new FRET based sensing systems.1–4 FRET formation through analyte anion linkage or anion binding to the acceptor would also be the applicable strategy to design a new FRET sensing system. Multiplexed detection of analyte ions is also a challenge for numerous industrial and biochemical applications of fluorescence based sensors, which could be further developed based on common metal ion sensing systems.
Due to the high background and complexity of the intracellular environment, in vivo anion sensing is also one of the remaining scientific and technical challenges for further investigation. With the development of the synthesis of new types of QDs and nanomaterials, such as doped QDs, graphene QDs, graphene oxide, carbon dots and near-infrared QDs, and the elucidation of new optical properties of these nanomaterials, utilizing these new nanostructures for new sensing materials and innovative detection platforms could be envisaged.6,17,55,58,67,68 By combining with other techniques or materials, such as signal amplification techniques, flow injection system, charge coupled detectors, QD based fluorescence anion sensing could be even more powerful in terms of sensitivity and applicability.
Acknowledgements
This work was financially supported by the National Basic Research Program (2011CB933502) of China, the National Natural Science Foundation of China (21335004, 51372151, 21303103, 21475021 and 21427807), the Natural Science Foundation of Jiangsu Province (BK20141331), the State Key Laboratory of Analytical Chemistry for Life Science (SKLACLS1406), and the Fundamental Research Funds for the Central Universities.Notes and references
- J. Du, M. Hu, J. Fan and X. Peng, Chem. Soc. Rev., 2012, 41, 4511–4535 RSC.
- J. Ma and P. K. Dasgupta, Anal. Chim. Acta, 2010, 673, 117–125 CrossRef CAS PubMed.
- C. R. Wade, A. E. Broomsgrove, S. Aldridge and F. P. Gabbai, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- N. Lawrence, Talanta, 2000, 52, 771–784 CrossRef CAS.
- A. H. Gore, S. B. Vatre, P. V. Anbhule, S. H. Han, S. R. Patil and G. B. Kolekar, Analyst, 2013, 138, 1329–1333 RSC.
- X. Lou, D. Ou, Q. Li and Z. Li, Chem. Commun., 2012, 48, 8462–8477 RSC.
- C. Frigerio, D. S. Ribeiro, S. S. Rodrigues, V. L. Abreu, J. A. Barbosa, J. A. Prior, K. L. Marques and J. L. Santos, Anal. Chim. Acta, 2012, 735, 9–22 CrossRef CAS PubMed.
- H. J. Kim, M. H. Lee, L. Mutihac, J. Vicens and J. S. Kim, Chem. Soc. Rev., 2012, 41, 1173–1190 RSC.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- N. Gaponik, D. V. Talapin, A. L. Rogach, K. Hoppe, E. V. Shevchenko, A. Kornowski, A. Eychmüller and H. Weller, J. Phys. Chem. B, 2002, 106, 7177–7185 CrossRef CAS.
- P. Zrazhevskiy, M. Sena and X. H. Gao, Chem. Soc. Rev., 2010, 39, 4326–4354 RSC.
- N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2011, 44, 925–935 CrossRef CAS PubMed.
- Y. Lou, Y. Zhao, J. Chen and J.-J. Zhu, J. Mater. Chem. C, 2014, 2, 595–613 RSC.
- W. J. Jin, J. M. Costa-Fernández, R. Pereiro and A. Sanz-Medel, Anal. Chim. Acta, 2004, 522, 1–8 CrossRef CAS PubMed.
- C. M. Tyrakowski and P. T. Snee, Phys. Chem. Chem. Phys., 2014, 16, 837–855 RSC.
- H. Mattoussi, G. Palui and H. B. Na, Adv. Drug Delivery Rev., 2012, 64, 138–166 CrossRef CAS PubMed.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS PubMed.
- C. L. Wu and Y. B. Zhao, Anal. Bioanal. Chem., 2007, 388, 717–722 CrossRef CAS PubMed.
- B. H. Zhang, F. Y. Wu, Y. M. Wu and X. S. Zhan, J. Fluoresc., 2010, 20, 243–250 CrossRef CAS PubMed.
- W. J. Jin, M. T. Fernandez-Arguelles, J. M. Costa-Fernandez, R. Pereiro and A. Sanz-Medel, Chem. Commun., 2005, 883–885 RSC.
- L. Zhang, C. Xu and B. Li, Microchim. Acta, 2009, 166, 61–68 CrossRef CAS.
- M. Hou and J. Na, Anal. Bioanal. Chem., 2010, 397, 3589–3593 CrossRef CAS PubMed.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2002, 21, 41–46 CrossRef PubMed.
- X. Gao, Y. Cui, R. M. Levenson, L. W. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS PubMed.
- S. K. Mehta, K. Salaria and A. Umar, Spectrochim. Acta, Part A, 2013, 105, 516–521 CrossRef CAS PubMed.
- M. Shamsipur and H. R. Rajabi, Mater. Sci. Eng., C, 2014, 36, 139–145 CrossRef CAS PubMed.
- H. R. Rajabi, M. Shamsipur, A. A. Khosravi, O. Khani and M. H. Yousefi, Spectrochim. Acta, Part A, 2013, 107, 256–262 CrossRef CAS PubMed.
- J. F. Callan, R. C. Mulrooney, S. Kamila and B. McCaughan, J. Fluoresc., 2008, 18, 527–532 CrossRef CAS PubMed.
- H. Li, C. Han and L. Zhang, J. Mater. Chem., 2008, 18, 4543 RSC.
- C. R. Tang, Z. H. Su, B. G. Lin, H. W. Huang, Y. L. Zeng, S. Li, H. Huang, Y. J. Wang, C. X. Li, G. L. Shen and R. Q. Yu, Anal. Chim. Acta, 2010, 678, 203–207 CrossRef CAS PubMed.
- C.-X. Sui, Y.-F. Liu, P.-A. Li, D. Zhang and F. Xia, Anal. Methods, 2013, 5, 1695 RSC.
- I. Costas-Mora, V. Romero, F. Pena-Pereira, I. Lavilla and C. Bendicho, Anal. Chem., 2012, 84, 4452–4459 CrossRef CAS PubMed.
- I. Costas-Mora, V. Romero, F. Pena-Pereira, I. Lavilla and C. Bendicho, Anal. Chem., 2011, 83, 2388–2393 CrossRef CAS PubMed.
- H. Li and X. Wang, Sens. Actuators, B, 2008, 134, 238–244 CrossRef CAS PubMed.
- J. Liu, X. Yang, K. Wang, R. Yang, H. Ji, L. Yang and C. Wu, Chem. Commun., 2011, 47, 935–937 RSC.
- R. C. Mulrooney, N. Singh, N. Kaur and J. F. Callan, Chem. Commun., 2009, 686–688 RSC.
- M. Xue, X. Wang, H. Wang, D. Chen and B. Tang, Chem. Commun., 2011, 47, 4986–4988 RSC.
- O. Adegoke and T. Nyokong, Synth. Met., 2014, 188, 35–45 CrossRef CAS PubMed.
- B. Paramanik, S. Bhattacharyya and A. Patra, Chem. – Eur. J., 2013, 19, 5980–5987 CrossRef CAS PubMed.
- A. Touceda-Varela, E. I. Stevenson, J. A. Galve-Gasion, D. T. Dryden and J. C. Mareque-Rivas, Chem. Commun., 2008, 1998–2000 RSC.
- L. Shang, L. Zhang and S. Dong, Analyst, 2009, 134, 107–113 RSC.
- T. Noipa, T. Tuntulani and W. Ngeontae, Talanta, 2013, 105, 320–326 CrossRef CAS PubMed.
- M. J. Ruedas-Rama and E. A. Hall, Analyst, 2008, 133, 1556–1566 RSC.
- O. Adegoke and T. Nyokong, J. Photochem. Photobiol., A, 2013, 265, 58–66 CrossRef CAS PubMed.
- Z. B. Shang, Y. Wang and W. J. Jin, Talanta, 2009, 78, 364–369 CrossRef CAS PubMed.
- Y. Xia, J. Wang, Y. Zhang, L. Song, J. Ye, G. Yang and K. Tan, Nanoscale, 2012, 4, 5954–5959 RSC.
- G. Yang, P. Shen, K. Tan and Y. Xia, Microchim. Acta, 2014, 181, 607–613 CrossRef CAS.
- S. Han, J. Wang and S. Jia, Luminescence, 2015, 30, 38–43 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2009 Search PubMed.
- M. J. Ruedas-Rama, A. Orte, E. A. Hall, J. M. Alvarez-Pez and E. M. Talavera, Analyst, 2012, 137, 1500–1508 RSC.
- Y. Wang, H. Mao and L. B. Wong, Nanotechnology, 2010, 21, 055101 CrossRef PubMed.
- J. Li and J. J. Zhu, Analyst, 2013, 138, 2506–2515 RSC.
- R. Freeman, J. Girsh and I. Willner, ACS Appl. Mater. Interfaces, 2013, 5, 2815–2834 CAS.
- D. Jimenez de Aberasturi, J.-M. Montenegro, I. Ruiz de Larramendi, T. Rojo, T. A. Klar, R. Alvarez-Puebla, L. M. Liz-Marzán and W. J. Parak, Chem. Mater., 2012, 24, 738–745 CrossRef CAS.
- R. Freeman and I. Willner, Chem. Soc. Rev., 2012, 41, 4067–4085 RSC.
- F. Jiang, J. J. Zhang, J. R. Zhang and J. J. Zhu, Analyst, 2013, 138, 1962–1965 RSC.
- L. L. Li, Y. Chen, Q. Lu, J. Ji, Y. Y. Shen, M. Xu, R. Fei, G. H. Yang, K. Zhang, J. R. Zhang and J. J. Zhu, Sci. Rep., 2013, 3 Search PubMed.
- H. Y. Liu, S. M. Xu, Z. M. He, A. P. Deng and J. J. Zhu, Anal. Chem., 2013, 85, 3385–3392 CrossRef CAS PubMed.
- G. C. Fan, X. L. Ren, C. Zhu, J. R. Zhang and J. J. Zhu, Biosens. Bioelectron., 2014, 59, 45–53 CrossRef CAS PubMed.
- J. Ji, L. He, Y. Y. Shen, P. P. Hu, X. H. Li, L. P. Jiang, J. R. Zhang, L. L. Li and J. J. Zhu, Anal. Chem., 2014, 86, 3284–3290 CrossRef CAS PubMed.
- L. Bau, P. Tecilla and F. Mancin, Nanoscale, 2011, 3, 121–133 RSC.
- L. M. Maestro, E. M. n. Rodríguez, F. S. Rodríguez, M. C. I.-d. la Cruz, A. Juarranz, R. Naccache, F. Vetrone, D. Jaque, J. A. Capobianco and J. G. a. Solé, Nano Lett., 2010, 10, 5109–5115 CrossRef CAS PubMed.
- J.-M. Yang, H. Yang and L. Lin, ACS Nano, 2011, 5, 5067–5071 CrossRef CAS PubMed.
- D. Zhou, M. Lin, X. Liu, J. Li, Z. Chen, D. Yao, H. Sun, H. Zhang and B. Yang, ACS Nano, 2013, 7, 2273–2283 CrossRef CAS PubMed.
- P. Wu and X. P. Yan, Chem. Soc. Rev., 2013, 42, 5489–5521 RSC.
- L. L. Li, Y. Chen, Q. Lu, J. Ji, Y. Y. Shen, M. Xu, R. Fei, G. H. Yang, K. Zhang, J. R. Zhang and J. J. Zhu, Sci. Rep., 2013, 3, 1529 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |