
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen reduction reaction catalysts used in microbial fuel cells for energy-efficient wastewater treatment: a review
Heyang
Yuan
a,
Yang
Hou
*b,
Ibrahim M.
Abu-Reesh
c,
Junhong
Chen
*d and
Zhen
He
*a
aDepartment of Civil and Environmental Engineering, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA. E-mail: zhenhe@vt.edu
bDepartment of Chemistry and Food Chemistry & Center for Advancing Electronics Dresden (CFAED), Technische Universitaet Dresden, 01062 Dresden, Germany. E-mail: houyang1213@hotmail.com
cDepartment of Chemical Engineering, College of Engineering, Qatar University, P.O. Box 2713, Doha, Qatar
dDepartment of Mechanical Engineering, University of Wisconsin-Milwaukee, 3200 North Cramer Street, Milwaukee, Wisconsin 53211, USA. E-mail: jhchen@uwm.edu
First published on 2nd May 2016
Abstract
Microbial fuel cells (MFCs) as an energy-efficient wastewater treatment technology have attracted increasing interest in the past decade. Cathode catalysts for the oxygen reduction reaction (ORR) present a major challenge for the practical applications of MFCs. An ideal cathode catalyst should be scalable, durable, and cost-effective. A variety of non-precious metal catalysts have been developed for MFC applications, including carbon-based catalysts, metal-based catalysts, metal–carbon hybrids, metal–nitrogen–carbon complexes, and biocatalysts. This paper comprehensively reviews these materials with emphasis on their synthesis, performance, durability, and cost. It is anticipated that insights offered in this review could facilitate the development of ORR catalysts for MFC applications towards energy-efficient wastewater treatment.
 Heyang Yuan | Mr Heyang Yuan is now a PhD student advised by Dr Zhen He in the Via Department of Civil and Environmental Engineering at Virginia Polytechnic Institute and State University (Virginia Tech). He received his BS from Tongji University and MS from Technische Universitaet München, where he has conducted research in microbial communities in activated sludge. At Virginia Tech, his research focuses on fundamentals and optimization of microbial desalination cells and nano-structured materials for electrode/catalyst application in bioelectrochemical systems. |
 Yang Hou | Dr Yang Hou received his PhD degree from the School of Environmental Science & Technology of Dalian University of Technology in 2011, and then did 3.5 years of postdoctoral research in the Department of Chemistry at the University of California, Riverside, and Department of Mechanical Engineering, University of Wisconsin–Milwaukee. Now he is a research associate working with Professor Xinliang Feng in the Center for Advancing Electronics Dresden, Technische Universitaet Dresden. His current research focuses on the design and synthesis of low-dimensional nanomaterials for energy and environmental applications. He has published nearly 60 papers and is an editorial board member at Scientific Reports. |
 Ibrahim M. Abu-Reesh | Professor Ibrahim M. Abu-Reesh received his master's and doctorate degrees from the Chemical Engineering Department at Washington University, St. Louis, Missouri, USA, and his BSc degree in petroleum refining engineering from Suez Canal University, Egypt. Dr Abu-Reesh has taught in chemical engineering departments for about 20 years before joining Qatar University (Fall 2011) at institutions such as the University of Jordan, King Fahd University of Petroleum and Minerals, and the American University of Sharjah, as a visiting professor. Dr Abu-Reesh's research publications are in the area of biochemical engineering and environmental biotechnology. |
 Junhong Chen | Junhong Chen is a Distinguished Professor of Mechanical Engineering and Materials Science and Engineering at the University of Wisconsin–Milwaukee, and a Fellow of American Society for Mechanical Engineers (ASME). He is also the Director of the Industry-University Cooperative Research Center (I/UCRC) on Water Equipment and Policy, supported by the U.S. National Science Foundation (NSF) and water-based industrial partners. His research interests lie in nanostructure-based sensors and nanocarbon-based hybrid nanomaterials for sustainable energy and environment (http://www.uwm.edu/nsee/). |
 Zhen He | Dr Zhen He is an Associate Professor in the Via Department of Civil and Environmental Engineering at Virginia Tech. Prior to this position, he was an Assistant Professor of Civil Engineering and Mechanics at the University of Wisconsin–Milwaukee. He received his BS from Tongji University, MS from the Technical University of Denmark, and PhD from Washington University in St. Louis, and all of them were in environmental engineering. His research focuses on the development of biotechnologies for sustainable water and wastewater treatment. He has published more than 100 journal papers and is an associate editor for Water Environment Research. |
1. Introduction
The growing demand for water and energy has become a critical issue for sustainable societal development in the 21st Century. Globally there are 783 million people lacking access to clean water and 2.5 billion people lacking adequate water sanitation.1 Meanwhile, electricity is unavailable to more than 1.3 billion people.1 There is a strong nexus between water and energy. For example, water and wastewater utilities consume 3% of the electricity in the USA, which accounts for 35% of the total municipal energy budgets.2 On the other hand, wastewater is increasingly considered as an energy resource, and the potential energy that can be extracted from domestic wastewater is estimated to be 3.8 kW h g−1 COD (chemical oxygen demand).3 This energy can be recovered as methane by anaerobic digestion, which is mainly applied to treat concentrated wastes such as sludge; however, inefficient collection of methane gas could result in the release of methane into the atmosphere as a greenhouse gas.4 Therefore, there is an urgent need to develop energy-efficient and environmentally-friendly methods for sustainable wastewater treatment.Microbial fuel cells (MFCs) are green technologies that can directly convert the organic energy in wastewater into electricity.5 Similar to chemical fuel cells, MFCs are composed of an anode and a cathode (Fig. 1). The electrochemically active microorganisms (i.e., exoelectrogens) that grow in the anode are fed by the organic matter in wastewater and respire extracellularly by transferring electrons to the anode electrode.6 When an appropriate electron acceptor (e.g., oxygen) is present in the cathode and the overall thermodynamics of the cell is favourable, the electrons flow spontaneously through the external circuit to the cathode for reduction reactions. As such, electrical energy is produced and wastewater is treated. Compared to conventional activated sludge processes, MFCs can theoretically achieve a positive energy balance and produce significantly less waste sludge, which further enhances energy efficiency.7 Moreover, the electricity produced by MFCs is cleaner than CH4 in terms of greenhouse effects.
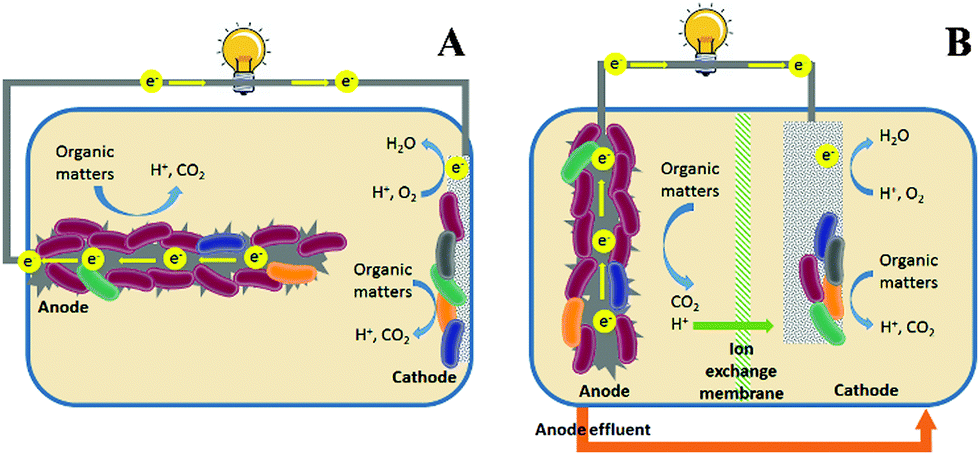 | ||
| Fig. 1 Schematic of (A) single-chamber MFCs and (B) two-chamber MFCs. | ||
One of the main challenges is the development of efficient and stable cathode catalysts for MFCs.8 Oxygen is an ideal electron acceptor for MFCs because of its high redox potential, availability, and sustainability. However, the oxygen reduction reaction (ORR) is kinetically sluggish, resulting in a large proportion of potential loss.9 Although platinum (Pt) shows a high ORR catalytic activity, a Pt-based cathode could account for more than 50% of the total capital cost of lab-scale MFCs, and thus it is not economically viable for large-scale wastewater treatment.10 A number of materials have been studied as alternative ORR catalysts for MFCs. Recent publications have reviewed these catalysts from the perspectives of their structures and the ORR mechanisms,11–14 but the unique features of the ORR catalysts for MFC applications are not well demonstrated. Furthermore, the role of MFCs as a research platform in evaluating the activity and durability of catalysts remains to be explained.
This review comprehensively summarizes the ORR catalysts used in MFCs with a focus on their synthesis/modification procedure, durability, performance, stability, and economics. The criteria that ORR catalysts should meet for MFC applications and the evaluation methods based on MFC experiments are demonstrated. The cathode catalysts are categorized into carbon-based catalysts, metal-based catalysts, carbon–metal hybrids, metal–nitrogen–carbon complexes and biocatalysts. The synthesis/modification, the consequently enhanced performance, and the stability of the catalysts are discussed in detail. The costeffectiveness of the catalysts is interpreted as the maximum power density (MPD) normalized to the cost for comparison. This review is expected to provide insights into the development of ORR catalysts for MFC-based wastewater treatment.
2. ORR catalysts used in MFCs
The ideal ORR catalysts used in MFCs are expected to be cost-effective and have high catalytic activity, because MFCs are engineered primarily for wastewater treatment, and thus the capital and maintenance costs should be comparable to conventional treatment technologies.7 At this stage of development, the energy generated by MFCs is mainly used to balance the energy consumption rather than to generate additional economic benefits,15 further highlighting the importance of economic feasibility of the cathode catalysts. Hence, properties associated with practicality, including simple and large-scale synthesis, low cost, and high durability should be given priority when developing ORR catalysts for MFC applications.Besides their cost effectiveness, the durability of the ORR catalysts in MFCs is another major challenge, because the cathode is constantly exposed to wastewater containing organic matter, contaminants and microorganisms. In single-chamber MFCs (Fig. 1A), the catalysts directly contact the wastewater and may be poisoned by the intermediate products such as methanol, chloride, sulfide, etc.16–18 Furthermore, organisms can form a biofilm on the cathode surface and degenerate catalytic performance by blocking the O2 transport.19,20 Although in some circumstances the biofilm may serve as a biocatalyst,21 the interaction between the organisms and the catalyst remains unknown and warrants further studies. Similar problems are also found in two-chamber MFCs (Fig. 1B), where the anode effluent is commonly introduced into the cathode for post-treatment.22 Poisoning of the catalysts by intermediates leads to high potential loss and reduced power production. Previous studies showed an increased CH4 production at a lower current, indicating the competition for substrates between exoelectrogens and methanogens.23 To make MFCs for practical applications, the ORR catalysts must be tolerant to poisoning, resistant to biofouling and able to restore catalytic activity after cleaning.
Whilst ORR catalysts are developed to enhance MFC performance, MFCs also present a powerful tool to study the durability and activity of the catalysts. The assessment using MFCs provides insightful information that cannot be obtained by ex situ electrochemical characterization methods. For example, multi-cycle cyclic voltammetry (CV) as a method to measure durability is typically accomplished with 6000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in ∼48 h depending on the scan rate, which is much shorter than the operation period of MFCs (i.e., several weeks to months). Similarly, the chronoamperometric curve for assessing catalytic stability lasts only a few hours, and the typical operation potential (e.g., −0.4 V vs. Ag/AgCl) is much more negative than that in MFCs (i.e., ∼0 V vs. Ag/AgCl). Electrochemical impedance spectrometry (EIS) can be used to measure the distribution of internal resistance,24 but the overall internal resistance for calculating the MPD cannot be obtained. In contrast, the measurement of the polarization curve and cathode potential in MFCs not only yields the overall internal resistance, but can also be used for comparison of the concentration overpotential at high current output.25 Moreover, long-term MFC experiments help gain a better understanding of the complicated effects of poisoning and fouling caused by real wastewater.26–28
000 cycles in ∼48 h depending on the scan rate, which is much shorter than the operation period of MFCs (i.e., several weeks to months). Similarly, the chronoamperometric curve for assessing catalytic stability lasts only a few hours, and the typical operation potential (e.g., −0.4 V vs. Ag/AgCl) is much more negative than that in MFCs (i.e., ∼0 V vs. Ag/AgCl). Electrochemical impedance spectrometry (EIS) can be used to measure the distribution of internal resistance,24 but the overall internal resistance for calculating the MPD cannot be obtained. In contrast, the measurement of the polarization curve and cathode potential in MFCs not only yields the overall internal resistance, but can also be used for comparison of the concentration overpotential at high current output.25 Moreover, long-term MFC experiments help gain a better understanding of the complicated effects of poisoning and fouling caused by real wastewater.26–28
It should be noted that the choice of external resistance is critical when performing MFC experiments. The difference in current production between the catalysts of interest and the control Pt/C is artificially diminished by using a large resistance (Fig. 2). As aforementioned, the current will determine the anode potential and will consequently affect the microbial activity, metabolite production and fouling. It is thus recommended that, when evaluating the long-term stability, MFCs should be operated at a high current mode (i.e., low external resistance) and/or a high power mode (i.e., external resistance equals the internal resistance), which are the typical operational conditions in real applications.
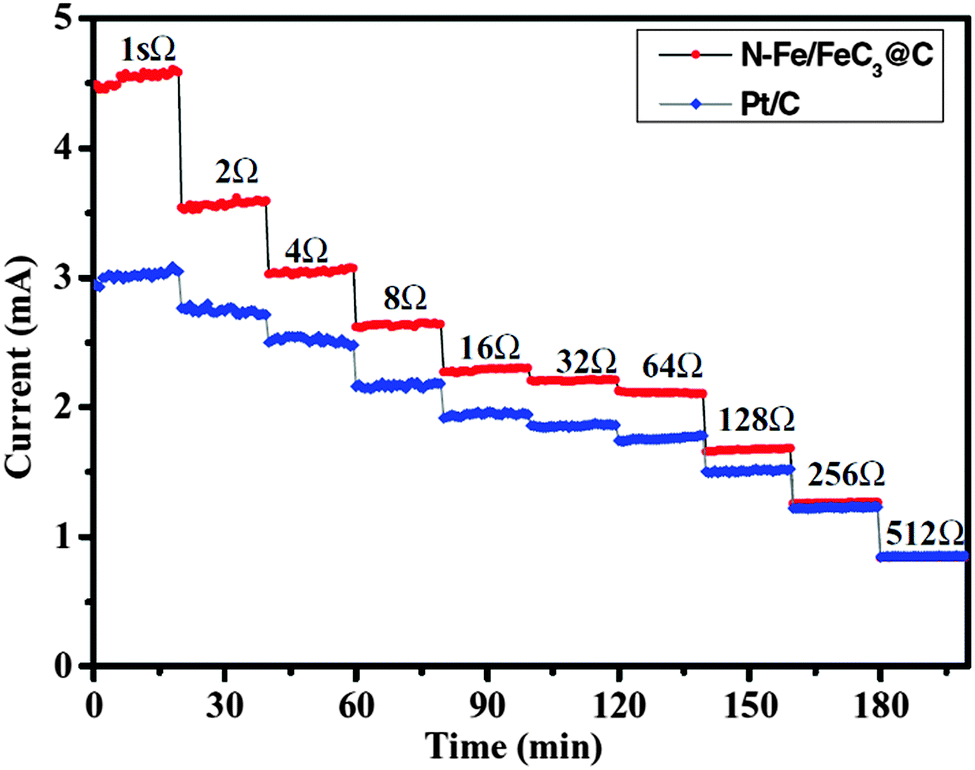 | ||
| Fig. 2 Current production as a function of external resistance. Reproduction with permission from ref. 29. Copyright (2012) John Wiley & Sons. | ||
3. Carbon-based catalysts
3.1 Carbon black
Carbon black (CB) is a product from incomplete combustion or thermal decomposition of hydrocarbons.30 Due to its high stability and large specific surface area, CB is widely used as the support material for metal catalysts.31 However, simple chemical modification and/or the introduction of functional groups can create active sites that make CB itself a metal-free ORR catalyst.In a study treating CB with nitric acid, the MPD of the MFC equipped with the modified CB was 3.3 times that with pristine CB and was 78% of that with Pt/C.32 A similar enhancement in MPD (71% of that with Pt/C) was reported in another study by using nitric acid and ammonia gas as treatment reagents, which was likely attributed to the successful introduction of oxygen and nitrogen atoms on the CB surface.33 Pyrolyzing CB and polytetrafluoroethylene (PTFE) under an ammonium atmosphere resulted in the co-doping of nitrogen and fluorine atoms.34 As a consequence, the electron transfer number increased from 2.7 for the un-doped CB to 3.8 for the co-doped one, and the MPD of the MFC with the treated CB reached 672 mW m−2, which was 1.2 times that with Pt/C.
CB as a cathode catalyst shows high economic viability. For example, polypyrrole/carbon black (PPy/C) yielded a MPD that is 70% of that with Pt/C. When the MPD was normalized to the material cost, the composite was 15 times more efficient than Pt/C.35 Despite the excellent cost effectiveness, the durability of CB catalysts in MFCs remains unknown. Furthermore, systematic doping of heteroatoms (N, O, S, P, etc.) in CB for improving catalytic activity warrants further studies.
3.2 Activated carbon
Activated carbon (AC) refers to porous carbon materials (surface area > 1000 m2 g−1) that are produced by the thermal or chemical activation of a wide range of carbonaceous precursors.36 The preparation of AC from silk fibroin was the first study showing that AC could catalyse ORR, which was attributed by the authors to the intrinsic nitrogen atoms.37 Later, peat-based AC was used as the MFC catalyst and achieved a MPD of 1220 mW m−2, 1.2 times that with Pt/C, indicating that AC might hold great promise for MFC applications.38 The results of the two studies also suggest that AC produced from different precursors will possess different chemical functional groups, BET specific surface areas and active sites, which may synergistically affect ORR catalysis. AC made from five precursors, including peat, coconut shell, hardwood carbon, phenolic resin and bituminous coal, were examined as the ORR catalysts in MFCs and yielded varied performance.39 The AC derived from bituminous coal performed relatively poorly in terms of onset potential (0.09 V vs. Ag/AgCl) and electron transfer number (2.4), but generated the highest MPD (77% of Pt/C) among those AC. It was found that the content of strong acid functional groups in the AC negatively correlated with the onset potential, while the BET specific surface area could not be used to predict the catalytic performance. A similar conclusion on the surface area was drawn by another study on AC produced from six different precursors.40 Interestingly, both studies observed high oxygen contents (4–10%), but negligible-to-no nitrogen in the AC, leaving the ORR catalysis mechanisms to be further understood.A number of modifications of AC have been carried out to overcome the limitations caused by the precursors. For instance, simple physical modification by blending CB with AC presents an effective method to reduce both the ohmic and charge transfer resistances, thereby increasing the MPD by 16% compared to bare AC.41 Chemical treatment with alkali or acid has also been reported to enhance ORR performance via the formation of possible chemical bonds. The AC pre-treated with potassium hydroxide reduced the internal resistance and yielded a MPD 16% higher than the untreated AC, possibly because of the increased electrolyte–catalyst affinity caused by the adsorbed OH−.42 Meanwhile, the pre-treatment of AC with phosphoric acid at 80 and 400 °C showed 35% and 55% increase in the MPD, respectively.43 The introduction of non-acidic oxygen content and P in the form of C–O–P bonding might account for the improved catalysis.44,45 On the other hand, the acidic functional groups resulting from H3PO4 treatment were speculated to be detrimental to ORR catalysis,44 consistent with the findings in those studies using AC from different precursors.39
Treatment using nitrogen-containing chemicals is one of the most effective methods for improving the ORR catalysis of AC as it can achieve multiple benefits. Treating AC in ammonia gas at 700 °C not only removed oxygen functional groups, but also introduced nitrogen atoms, leading to a MPD (2450 mW m−2) 28% higher than untreated AC and 16% higher than Pt/C, respectively.46 Nitrogen doping has been demonstrated to be an effective strategy to enhance catalytic activity.47 Three different types of doped nitrogen may play important roles in ORR catalysis: graphitic-N may favor the reduction of O2 to H2O2via the 2e− pathway, whereas pyridinic- and pyrrolic-N are likely to contribute to the 4e− pathway.48,49 A recent study reported an N-doped AC catalyst with a remarkably high nitrogen content (8.65% total N and 5.56% pyridinic-N) by acid/alkaline pre-treatment and using cyanamide as the nitrogen precursor (Fig. 3).50 The modified AC achieved an electron transfer number of 3.99 and a MPD (650 mW m−2) 44% higher than Pt/C. In addition to nitrogen-doping, nitrogen-containing chemicals such as ammonium bicarbonate could serve as a pore former to increase porosity and alter pore size distribution, which could reduce charge transfer resistance and consequently enhance the MFC performance.51
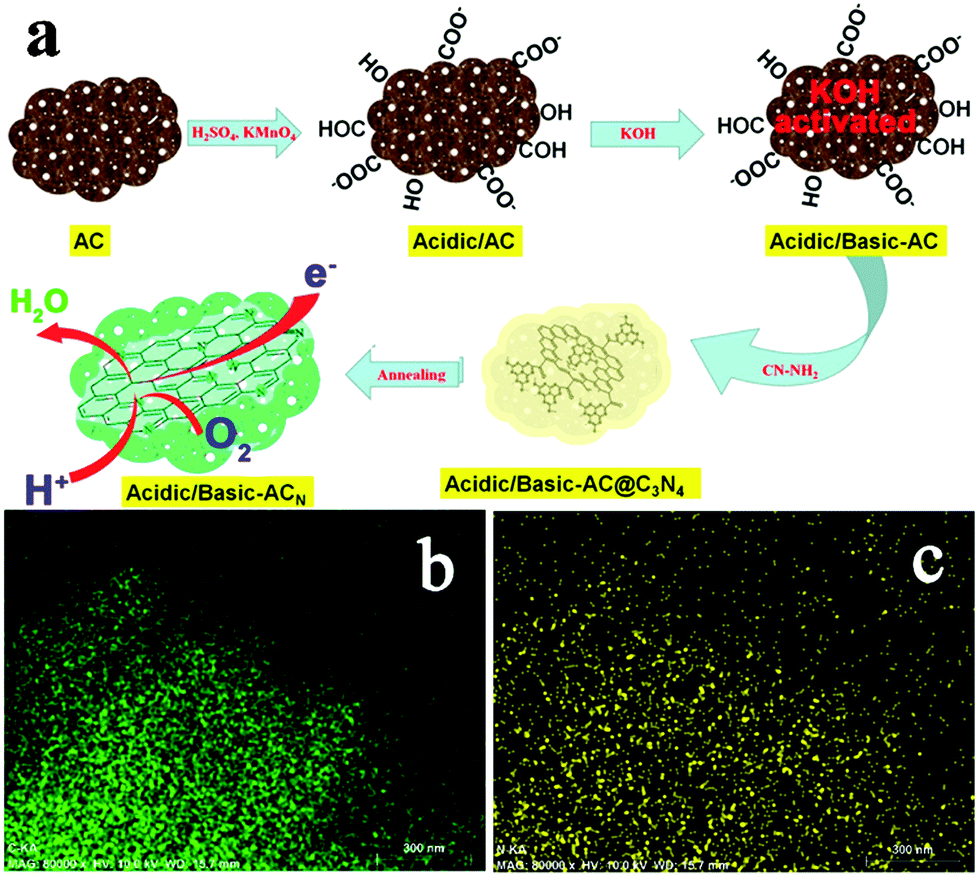 | ||
| Fig. 3 (a) The synthesis of the acidic/basic-ACN, (b) the carbon element mapping, and (c) the nitrogen element mapping. Reproduction with permission from ref. 50. Copyright (2014) American Chemical Society. | ||
AC as an ORR catalyst exhibits excellent electrochemical durability. While the current density of a Pt/C cathode dropped by 73% after 7 h of the chronoamperometry test, a nitrogen-doped AC cathode showed only 30% decrease.50 However, biofouling on the AC cathode and degenerated MFC performance were still observed.40 The disinfectant quaternary ammonium compound was added in AC to inhibit biofilm growth.52 After 2 months of operation, the protein content on the modified AC cathode was 26 times lower than that on the control electrode, leading to a lower charge transfer resistance (Fig. 4) and a more stable MPD. Long-term studies suggested that the MPD of the MFCs using an AC–CB mixture as the cathode catalysts decreased by only 1–7% after 5 months of operation.41,53 After 16 months of operation, the performance of AC–CB dropped to 79% of its original level, but could be recovered to 90% using a simple acid washing (60 mM HCl solution).53 On the other hand, acid cleaning did not noticeably recover the performance of Pt/C cathodes, which generated a MPD only 21% of its initial value.
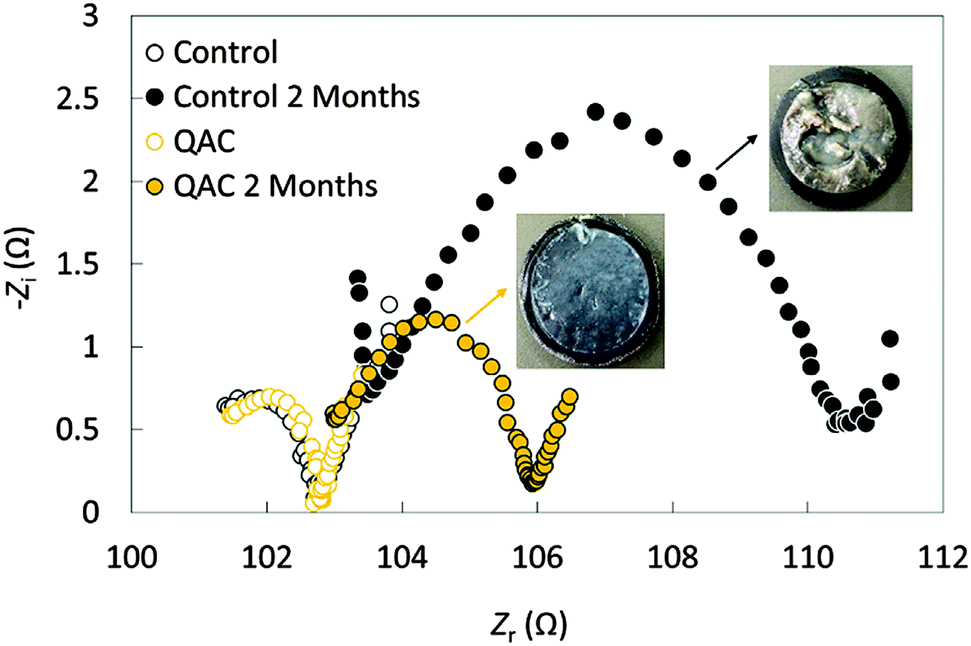 | ||
| Fig. 4 Nyquist plots of the control and QAC before and after 2 months of operation. Reproduction with permission from ref. 52. Copyright (2014) Elsevier. | ||
AC has attracted much attention due to its high cost effectiveness. Compared to commercial Pt/C, whose price is $28 g−1 (10% Pt on Vulcan XC 72, Premetek Co., Wilmington, DE, USA) and the normalized MPD is only ∼1 mW $−1 (with a typical loading rate of 5 mg cm−2), AC costs less than $0.002 g−1. The unit price of the AC blended with CB was lower than $1 m−2, together with the high power production leading to a normalized MPD of 1210 mW $−1.53 It was also estimated that a complete AC cathode, including the AC catalyst, the PTFE binder and the metal support, costs $30–60 m−2, which resulted in a normalized MPD of 22–41 mW $−1 depending on the MFC performance.54,55 The normalized MPD could be further improved to 94–98 mW $−1 by pressing AC–PTFE on a stainless steel mesh,56 or by phase inversion of an AC/CB/PVDF (poly(vinylidene fluoride)) mixture,57 demonstrating its potential for practical applications.
3.3 Carbon nanofibers/nanotubes
Carbon nanofibers (CNFs) are composed of stacked corn-shaped graphene sheets and show high electrical conductivity and a high BET specific surface area.58,59 Similar to the treatment of AC, alkaline/acid activation and nitrogen doping are common strategies to improve the ORR catalysis of CNFs. Immersing CNFs into 8 M KOH solution increased its surface area from 275 m2 g−1 to 2100 m2 g−1, and consequently enhanced the MPD by 79% with respect to the untreated CNFs.60 By comparison, HNO3 treatment of CNFs did not significantly change the BET surface area, but shifted the pore size distribution, which might favor ORR catalysis.61 Nitrogen-doped CNFs via the pyrolysis of pyridine showed high catalytic activity and obtained a MPD comparable to that with Pt/C.62 The combination of nitrogen doping and chemical activation with KOH yielded a CNF material with a large BET surface area (1984 m2 g−1) and high catalytic activity (electron transfer number 3.6), and the MFC equipped with the modified CNFs generated a MPD (1377 mW m−2) similar to that of the Pt/C-based MFC.63 Recently, heteroatom-doped porous CNFs were obtained via the pyrolysis of natural spider silk (Fig. 5).64 Owing to the abundant electronegative N and S atoms within the carbon lattice and the high BET surface area (721.6 m2 g−1), the CNFs achieved a MPD of 1800 mW m−2, 1.6 times higher than Pt/C.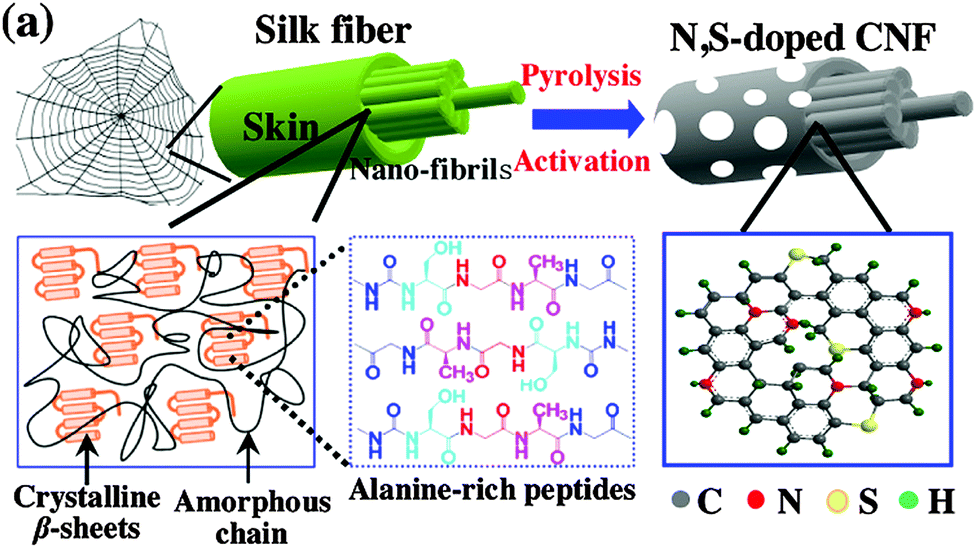 | ||
| Fig. 5 The synthesis of heteroatom-doped CNFs derived from spider silk. Reproduction with permission from ref. 64. Copyright (2016) Elsevier. | ||
Carbon nanotubes (CNTs) are one or multiple layers of graphene sheets wrapped in a concentric manner,65,66 whose catalytic activity can be tuned by heteroatom doping.67 It has been reported that vertically aligned nitrogen-doped CNTs catalyse ORR mainly via the 4e− pathway.68 The N-doped CNTs showed lower internal resistance and a more positive onset potential in cyclic voltammetry tests compared to Pt/C.69 The MFCs with the N-doped CNTs thus outperformed the Pt/C-MFCs in terms of power production. Quantum mechanics calculations suggested that nitrogen dopants increased the positive charge density of carbon atoms and induced charge delocalization, which could enhance parallel diatomic adsorption of O2 and weaken O–O bonds during ORR catalysis.70 In addition to pre-treatment and nitrogen doping, mixing CNTs with a conductive polymer, such as polyaniline (PANI) or polypyrrole (PPy), presents a simple method to enhance the cathode performance.71,72 Although the ORR catalysis of those CNT/polymer composites was slightly inferior to that of N-doped CNTs and Pt/C, the simple and large-scale production makes them competitive catalysts for practical applications.
Both CNFs and CNTs as cathode catalysts are more durable than Pt/C. Activated N-doped CNFs showed less attenuation than Pt/C in chronoamperometry tests, likely because the graphitic-N in the carbon plane was less susceptible to protonation.63 With respect to long-term stability, the Pt/C cathode severely deteriorated due to sulfide poisoning and biofilm growth, but the activated CNF cathode showed only a slight decrease in the MPD.61 The CNF synthesized using spider silk was not affected by the crossover effects of various chemicals (e.g. formate, ethanol, lactate, methanol, acetate, sulfide, and ascorbate), and constantly produced high MPD in 3 months of continuous operation.64
The cost of CNFs prepared by the electrospinning of polyacrylonitrile was estimated to be $7.5 g−1, and increased slightly to $9.6 g−1 after KOH treatment; but the normalized MPD of the modified CNFs was still 2.6 times that with Pt/C.60 The normalized MPD of the polyaniline/multi-walled CNT composite reached 31 mW $−1 owing to the low cost of commercial multi-walled CNTs.71 However, CNFs and CNTs were still not competitive to AC. The CNFs obtained from spider silk used free natural materials and might provide insight into the cost-effective fabrication of sustainable cathode catalysts.
3.4 Graphite/graphene
Graphite is multiple layers of carbon sheets bonded through weak van der Waals interaction. Due to its high electrical conductivity and high stability, graphite is commonly used as a fuel cell electrode.73 Exfoliation of graphite can form single-layer carbon nanosheets known as graphene.74 The discovery of graphene was awarded the 2010 Nobel Prize for Physics and has drawn extensive attention in the past decade.75,76 With high electrical conductivity and a high BET surface area, graphene-based cathode catalysts have been demonstrated to effectively catalyse the ORR in MFCs.14Pristine graphite is not considered catalytic toward the ORR because of the lack of active sites. However, the MFC filled with granular graphite generated a stable MPD of 50 W m−3, comparable to many common catalysts.77 Furthermore, the COD removal (1.46 kg m−3 d−1) by the graphite-based MFC was higher than that of conventional aerobic processes, which was of practical significance for wastewater treatment. Activation of graphite with HNO3 and H3PO4 could enhance the MPD by 2 and 2.4 times, respectively.78,79 In another study, the graphite treated with HNO3 achieved a MPD similar to that with Pt/C, which could be attributed to the high BET surface area of the modified graphite and the introduction of nitrogen and oxygen functional groups.80
The modification of graphene has mainly focused on the doping of heteroatoms. Detonation of cyanuric chloride and trinitrophenol could effectively incorporate three different nitrogen species (i.e., pyridinic-N, pyrrolic-N, and graphitic-N) in graphene, leading to a high electron transfer number of 3.7.81 Implantation of mesoporous graphitic carbon nitride in N-doped graphene further enhanced the catalytic activity (complete 4e− ORR pathway), and yielded a MPD of 1618 mW m−2, 14% higher than Pt/C.82 The co-doping of nitrogen and sulfur in carbon could provide dual active sites for ORR catalysis.83 The N/S co-doped carbon nanosheets produced a lower MPD but a comparable current density to Pt/C, suggesting that this catalyst could be used in bioelectrochemical systems that require high current output.84 Graphene and N-doped graphene generally have a relatively low surface area, which can be increased by using KOH activation.85 A more direct way to increase the BET surface area is to prepare crumpled graphene particles by capillary compression in rapidly evaporating aerosol droplets.86,87
Similar to other carbon catalysts, both graphite and graphene catalysts exhibit high stability. In the presence of sulfide, the MPD using N-doped graphite as the cathode catalyst was not affected and became similar to that using Pt (Fig. 6a).26 Compared to the 17% decrease in the MPD of Pt/C-MFC after 35 cycles, the MPD of the MFC with N-doped graphene decreased by only 9%.81 It was further reduced to 4% by implanting C3N4 in the N-doped graphene.82 It was proposed that the oxygen species introduced during the synthesis of N-doped graphene could protect catalytic C–N groups from being attacked by protons, which might account for the superior stability (Fig. 6b).88
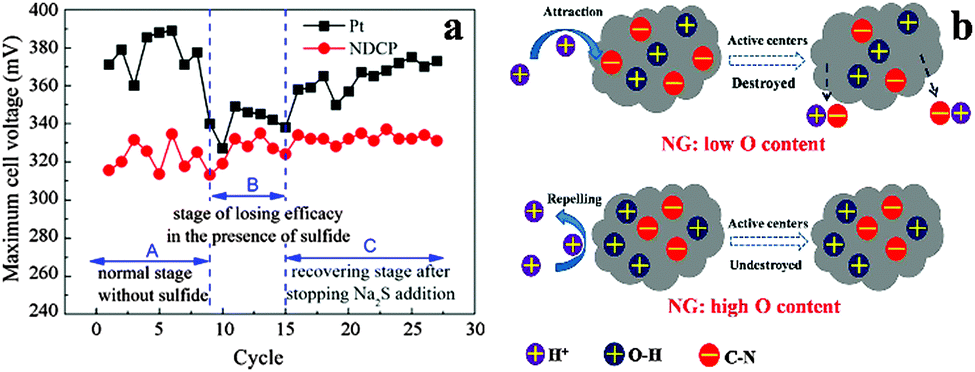 | ||
| Fig. 6 (a) The maximum cell voltage evolution in a typical three-stage process. Reproduction with permission from ref. 25 and 26. Copyright (2012) Elsevier. (b) Proposed mechanism for the improvement of long-time activity of N-doped graphene by O–H groups. Reproduction with permission from ref. 88. Copyright (2013) American Chemical Society. | ||
The cost effectiveness of graphite and graphene is comparable to that of CNFs/CNTs, but still less competitive to AC. Based on the literatures, the normalized MPD is approximately 12 mW $−1 for granular graphite,77 and 10 mW $−1 for N/S co-doped carbon nanosheets.84 The major problem of the heteroatom-doped graphene lies in its complicated synthesis process and low yields. In the typical procedures, graphene oxide is first prepared, reduced to graphene and then subjected to modification.89 Other synthesis methods such as chemical vapor deposition, unzipping of MWCNTs and detonation, etc. are either energy/time-consuming or involve hazardous chemicals.90,91 Therefore, a facile and efficient approach to creating graphene-based catalysts is highly desired and should be the focus of future studies.
3.5 Carbon synthesized from sustainable precursors
In addition to the well-defined categories, many other carbon materials have been developed using a variety of sustainable precursors, including biochar obtained from the pyrolysis of sewage sludge;92,93 the heteroatom-doped carbon derived from cellulose,94–96 dopamine,97 straw,98 chitin,99 and petroleum coke;100 and the carbon nanoparticle-coated porous biocarbon prepared from plant moss.101 The MFCs equipped with these carbon catalysts have shown a high electron transfer number and comparable or superior catalytic performance to Pt/C. For example, three cellulose-based catalysts generated MPDs ranging from 1041 to 2293 mW m−2 due to the different MFC configurations, but all outperformed their control MFCs using Pt/C.94–96This group of carbon cathodes is of particular interest, because they meet the requirement of durability and economy besides their high activity. Chitin-based carbon sheets were tolerant to methanol crossover, and the voltage output remained 97% of its original value after 60 days of operation.99 The moss-based biocarbon was also tolerant to methanol and showed considerable stability in chronoamperometric tests.101 The normalized MPD of the moss-based biocarbon may even be competitive to AC, as its precursor is free and readily available. According to the estimated price, loading rate and power production, the normalized MPD of N/P co-doped cellulose carbon could reach up to 143 mW $−1,95 highlighting its feasibility for MFC practical applications.
4. Metal-based catalysts
4.1 Metals and alloys
Many pure metals exhibit strong ORR catalytic activity. For example, noble metal Pt is considered to be the most active catalyst using the theoretical calculation of O2-, O-, and OH-binding energy,102 and it is proposed to catalyse the ORR via a dissociative mechanism at a low current density and an associative mechanism at a high current density, both of which are 4e− pathways.103 Many efforts have been devoted to reduce the amount of Pt loading on the MFC cathode without compromising the catalytic performance. The deposition of Pt on carbon paper by electron beam evaporation reduced the thickness of the Pt layer and minimized Pt loading, but significantly increased the current output of the MFC.104 A more practical way of reducing the loading is to alloy Pt with inexpensive transition metals, such as Fe,105 Co and Ni,106 which has been studied to further reduce the oxygen binding energies and enhance the catalytic activity.107 With appropriate Pt:metal ratios, Pt-based alloys could generate higher MPDs than commercial Pt/C.108,109
The stability of the metal catalysts presents the most challenging issue that hinders their commercial applications. Leaching of metallic Co from Pt–Co alloy was observed under an acidic electrochemical environment.108 Similar degeneration occurred for a Pt–Fe alloy, although its MPD in MFCs under neutral conditions was more stable than that with Pt/C.109 To cope with the possible biofouling, silver nanoparticles were used as the cathode catalysts and antimicrobial agents.110 It was observed that the biomass attached on the AgNP-coated cathode after 50 days was 44% less than that on plain graphite and 25% less than that on Pt/C (Fig. 7). The cost of metal-based catalysts is also a major concern. The use of scrap metals recycled from electronics and automobiles was sustainable and could produce a satisfactory MPD (422 mW m−2), but the normalized MPD was only 0.05 mW $−1 due to the large quantity of materials used in the cathode.111
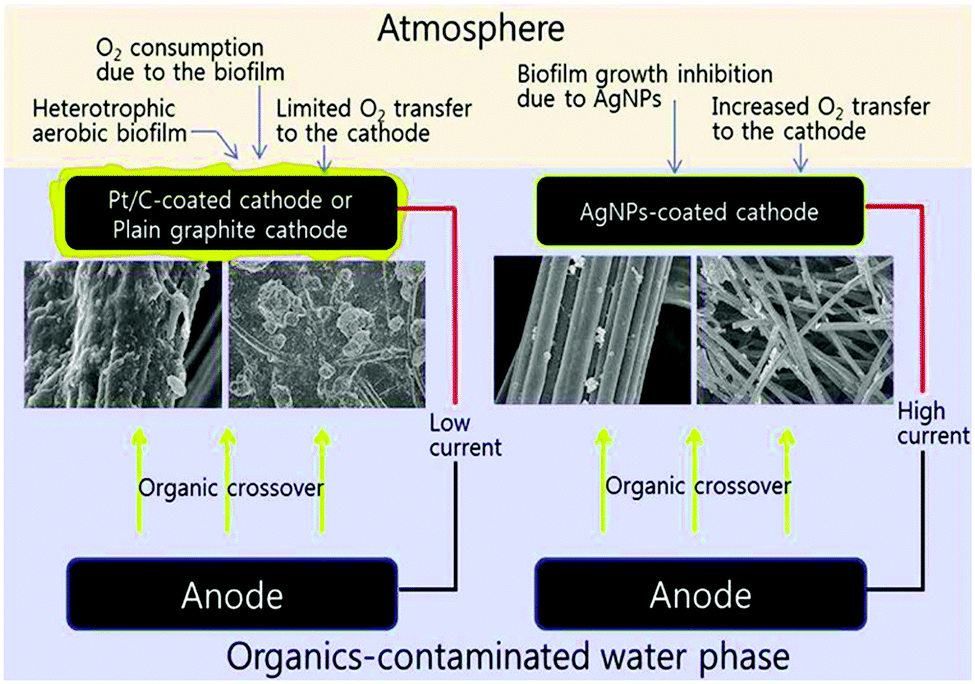 | ||
| Fig. 7 Biofouling on the cathode with Pt/C, plain graphite and Ag nanoparticles. Reproduction with permission from ref. 110. Copyright (2011) American Chemical Society. | ||
4.2 Metal oxides
Manganese dioxide has been successfully used as the cathode catalyst in aqueous and non-aqueous fuel cells for many decades.112 It is thus not surprising to find extensive studies on MnO2 in MFCs. However, most of the MnO2 achieved MPDs only half of that with Pt/C, regardless of structure modifications or doping of other transition metals.113–115 A change of the oxidation state of manganese oxides may affect the catalytic activity. Different components in MnOx with varied manganese valences, including MnO2, Mn2O3, Mn3O4, Mn5O8 and MnOOH, may coexist and contribute differently to ORR catalysis.116 Electrochemical deposition methods could synthesize MnOx with a lower oxidation state, thereby increasing the electron transfer number up to 3.5.117 Meanwhile, core–shell–shell MnO2 nanowires could facilitate electron transfer due to the interactions between the conductive middle layer and MnO2.118 A simple synthesis strategy using a hydrothermal method and in situ chemical polymerization was developed to prepare MnO2/PPy/MnO2 multi-walled nanotubes.119 The modified MnO2 catalysts showed much lower ohmic and charge transfer resistance than the pristine MnO2 nanotube and produced a MPD close to that of Pt/C.Spinel manganese–cobaltite, a mixed valence transition metal oxide that can be obtained using facile synthesis methods, is a promising catalyst for the ORR under alkaline conditions.120 This is particularly attractive for MFC applications, because it can reduce the investment on buffer solution, and consequently improve the cost effectiveness. The manganese cobaltite/PPy nanocomposite showed an electron transfer number of 3.9 in KOH electrolyte and produced a MPD 91% of that with Pt/C under neutral conditions.121 Doping copper in spinel manganese–cobaltite could affect both the BET surface area and the ORR electron-transfer pathway, and thus could achieve an improved MFC performance.122 Another type of spinel, manganese–ferrite, showed the complete 4e− pathway after the incorporation of PANI.123
Many other metal oxides, such as lead dioxide,124 perovskite oxides,125 vanadium oxides,126,127 cobalt oxide and zirconium oxide,128,129 have been studied as the cathode catalysts in MFCs. Perovskite oxides produced the highest normalized MPD of 116 mW $−1 among those metal oxides due to their low cost ($0.041 g−1).125 However, the MPD of perovskite oxides decreased by 15% after 15 cycles, indicating that the stability of the material needed to be improved. The stability and cost of other metal oxides remain to be investigated, and the possible leaching of the metals to the treated wastewater, the toxic effects and environmental impacts should be taken into consideration.
5. Metal–carbon hybrids
5.1 Metal–AC
AC with superior electrochemical properties has increasingly been used to replace CB as the support material for metal catalysts. Mechanical mixing of MnO2 and AC led to a reduced internal resistance, and the weight ratio was optimized to be 1:1.130 The addition of ceria, an oxygen storage material, with a fractional change between Ce3+ and Ce4+, could further improve the cathode performance.131 Similarly, mixing Co3O4 or NiCo2O4 with AC resulted in an increased MPD by maintaining the high BET surface area and reducing the charge transfer resistance.132,133 To achieve more homogeneous mixing, the non-stoichiometric Fe3O4 and AC were sonicated and an MPD 83% higher than the pristine AC was produced.134
Electrochemical deposition of MnO2 on AC can form closer interactions between the materials, as evidenced by the varied surface area and pore size distribution of MnO2–AC hybrids, which can be a key factor for cathode performance.135 The same method was adopted to deposit silver on AC, and a MPD 1.7 times higher than that with bare AC was achieved.136 The mixed components of zero-valent, monovalent and divalent Ag were hypothesized to transform mutually, thereby contributing to ORR catalysis. In another study of depositing CuxO, the lattice defects and stacking faults of the Cu crystal were shown to affect the electrochemical characteristics.137 It was also found that electrodepositing Cu2O on AC can change the surface roughness and the pore structure, and create lattice (111) planes and surface oxygen defects in n-type Cu2O, all of which might account for the improved electro-transfer kinetics.138 Similar to the electrodeposition strategy, chemical synthesis can incorporate metals in carbon structures and yield synergetic effects. Pyrolyzing the AC with Fe–ethylenediaminetetra acetic acid systematically decreased the surface area with increased Fe–EDTA content, but introduced N and Fe atoms into the surface structure as the active sites, resulting in a MPD 93% of that with Pt/C.139
The information on the stability and cost of the metal–AC catalysts in MFCs is limited. The Fe–EDTA/AC cathode with a Fe–EDTA-to-AC ratio of 0.2:1 did not show an appreciable change in the MPD after 4.5 months of operation, whereas the Fe–EDTA/AC cathodes with higher ratios degenerated as the Pt/C cathode did,139 which could be an implication that the structural change by pyrolysis might adversely affect the catalyst durability. In order to better understand the application niches of metal–AC hybrids, future work should focus on the long-term performance and biofouling in real wastewater.
5.2 Metal–CNTs
The feasibility of CNTs as an alternative support of Pt for MFC applications has been demonstrated using sonication or a simple mixing method.140,141 In addition, in situ reduction of Pt salts on the CNT surface could reduce Pt loading and slightly increase the MPD compared to Pt/C.142 The Pt loading on the CNT textile by electrochemical deposition was further reduced to one fifth of that on a carbon cloth, but the CNT-textile–Pt cathode achieved twice higher MPD than the carbon cloth–Pt cathode.143 The distinction of CNT–Pt from commercial Pt/C lies in the porous structure of the CNT-textile and the facilitated mass transfer of oxygen to the catalysts (Fig. 8).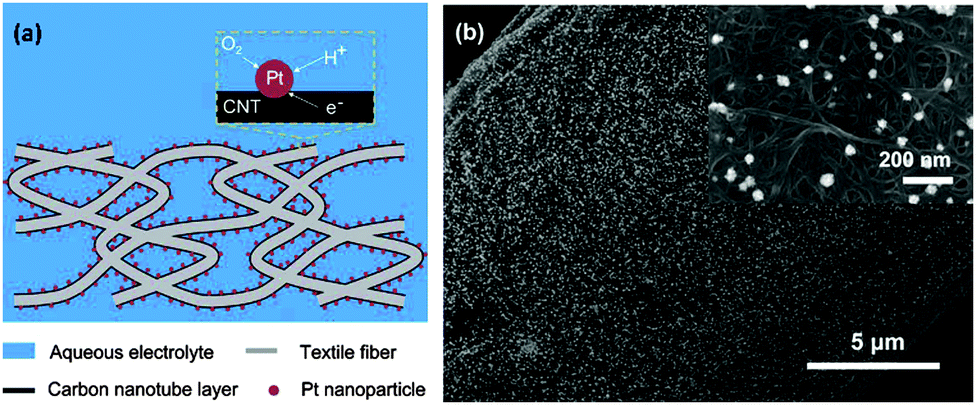 | ||
| Fig. 8 (a) Schematic of the CNT-textile–Pt composite in aqueous electrolyte and (b) SEM image of the CNT-textile–Pt showing the uniform distribution of Pt nanoparticles. Reproduction with permission from ref. 143. Copyright (2011) The Royal Society of Chemistry. | ||
As discussed previously, MnO2 is a promising non-precious metal catalyst and has been hybridized with CNTs in several studies. When mixed with CNTs using sonication, β-MnO2 outperformed α-MnO2 and γ-MnO2 in terms of power production, but was slightly inferior to Pt/C.144 Hydrothermal reduction of KMnO4 on CNTs achieved a MPD much higher than physical mixing and comparable to that with Pt/C.145 In another study, coating CNTs with PPy doubled the conductivity of the Mn–CNT composites.146 Furthermore, treating the CNTs with acid could introduce functional groups that might serve as the additional actives sites in the MnO2–CNT hybrids.147 It was observed that both the MPD and electron transfer number were systematically improved with increased MnO2 content in the CNTs, which could be easily tuned by varying the initial KMnO4 concentration.148 The MPD of the MnO2–CNT hybrids exceeded that of the control Pt/C when the MnO2 content was higher than 60%.
An in situ reduction method was also used to hybridize cobalt oxides or nickel oxides in CNTs, usually followed by calcination at 400–500 °C.149,150 The power production of the Co3O4–CNT cathode remained stable in 16 cycles. Meanwhile, the cost of NiO–CNTs was estimated to be $0.3 g−1, leading to a normalized MPD of 45 mW $−1. The CNFs dispersed with alumina–nickel and doped with nitrogen could achieve an MPD of up to 1850 mW m−2,151,152 but the complicated synthesis, which was composed of impregnating, calcination, reduction and chemical vapor deposition, may not be suitable for mass production. In addition, the long-term stability of these novel metal–CNT catalysts has not been demonstrated yet.
5.3 Metal–graphite/graphene
As previously mentioned, both pristine graphite and graphene do not possess defects in the carbon structures and thus are not catalytically active. Thermal deposition of Fe(CO)5 on graphite felt could introduce Fe2O3 and FeOOH as the active sites, and thereby achieving a high MPD comparable to that with Pt/C.153 To closely incorporate iron in carbon, cornstalks or pomelo skins were impregnated with an FeCl3 solution and graphitized at 1000 °C.154 The resulting iron oxide/partly graphitized carbon composite showed the lowest ohmic and charge transfer resistance in comparison with bare partly graphitized carbon and Pt/C, and consequently produced the highest MPD. Later, Ag+ and Fe3+ were used for impregnation to form dual-metal modified graphitic carbon composites.155 Both the Ag nanoparticles and the Fe3O4 acted as the active sites, exhibiting a 4e− ORR pathway. In a recent work graphitic carbon was synthesized from a PANI precursor, and the dual active centers were replaced with N-doped carbon and cobalt sulfide.156 The multi-crystalline Co/Co9S8 heterojunction formed at high temperature, together with the nitrogen species originating from the PANI precursor, was speculated to contribute to the improved ORR kinetics and cathode performance.Graphene has been demonstrated to be an effective support material for metal catalysts such as Pt–Pd and MnO2.157,158In situ reduction of MnO2 on graphene using a microwave method significantly increased the MPD from 1470 mW m−2 (pure MnO2) to 2083 mW m−2, which was also 22% higher than that with Pt/C.159 It is worth noting that the MnO2 hydrothermally reduced on graphite oxide, which is considered non-conductive,76 has achieved a high MPD of 3359 mW m−2,160 and the detailed catalytic mechanisms remain to be studied. The synthesis of metal–graphene catalysts is typically a two-step procedure consisting of the reduction of graphene oxide and the embedding of metals, which may not be practical for MFC applications. To address this issue, SnO2–graphene catalysts were prepared through microwave-assisted simultaneous reduction of graphite oxide and oxidation of tin salts.161 A one-step strategy was also adopted to prepare a novel N-doped graphene/CoNi-alloy encased within bamboo-like carbon nanotube hybrids.162 The CoNi alloy particles were encapsulated at the end of the CNTs, whilst the N-doped graphene filled the inner cavities of the CNTs (Fig. 9). This is the first report that N-doped graphene acts as the active sites instead of the support to enhance ORR catalysis. Moreover, the MFC equipped with the N–G@CoNi/BCNT produced the same current density as that with Pt/C under alkaline conditions (pH = 10, Fig. 9g), highlighting the attractive synergetic effects of the individual components and their potential in buffer-free MFCs.
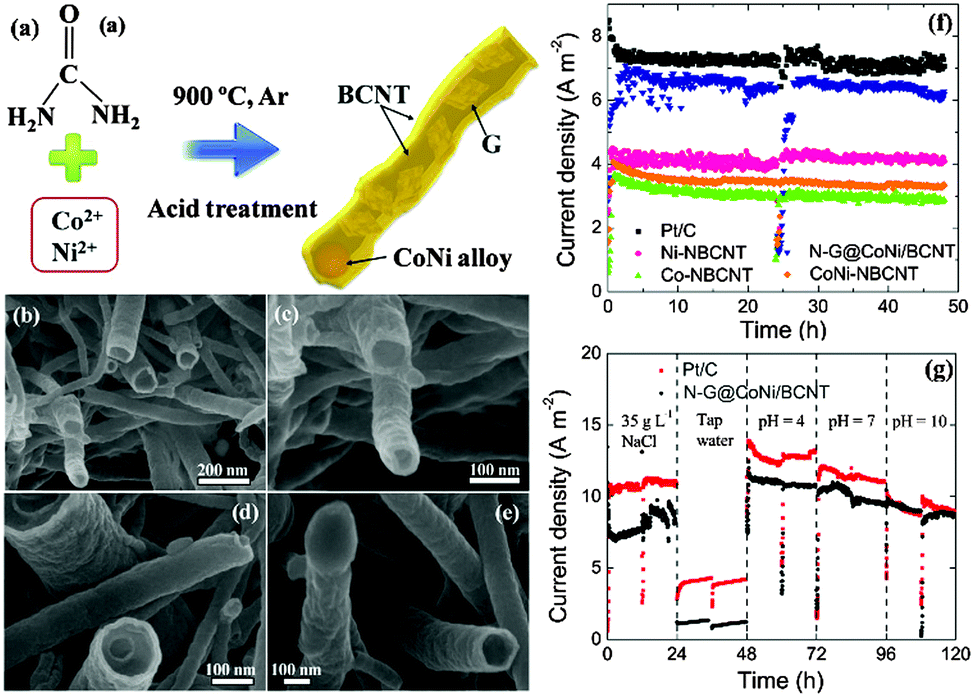 | ||
| Fig. 9 (a) The synthesis of the N–G@CoNi/BCNT, (b–e) FESEM images of the N–G@CoNi/BCNT at different magnifications, (f) current density of the MFC equipped with different cathode electrodes, and (g) current density of Pt/C and N–G@CoNi/BCNT in different catholytes. Reproduction with permission from ref. 162. Copyright (2016) Elsevier. | ||
Because the transition metals are relatively unstable and graphite/graphene are considered biocompatible,163,164 the durability of metal–graphite/graphene catalysts will be a critical issue. The power production of the graphitic carbon derived from cornstalks and pomelo skins decreased by 17% and 10% after 18 cycles of operation, respectively, but the decrease was alleviated to 7% by mixing those two types of graphitic carbons.154 The degeneration was likely attributed to the loose structure and the invasion of biofilms. Replacing the active sites with antibacterial Ag nanoparticles could reduce biofouling and consequently lead to only 4% decrease in the MPD after 17 cycles.155
The cost of metal–graphite/graphene catalysts mainly depends on their precursors and synthesis procedures. For instance, the one-step synthesis of N–G@CoNi/BCNTs used 5 g of urea as the carbon and nitrogen source to prepare 1 g of the product (Fig. 9a), whose normalized MPD reached 150 mW $−1.162 The graphitic carbon produced from renewable resources such as cornstalks or pomelo skins could be even more cost-effective and may meet the requirement of sustainability and economy.
6. Metal–nitrogen–carbon complexes
6.1 Metal macrocycles
Unlike the previously discussed Ni/N–CNFs or N–G@CoNi/BCNTs, in which the metal and nitrogen species do not form chemical bonding, metal–nitrogen–carbon (M–N–C) complexes refer to a wide range of materials with the metal cations coordinated with nitrogen functional groups in carbonaceous matrices.165 Metal macrocycles, a class of well-defined M–N–C complexes whose ORR catalytic activity has been studied for fifty years,166 are among the first alternative cathode catalysts examined in MFCs.167 The major issue of metal macrocycles is their instability in an acid medium.168 This is of critical concern particularly for two-chamber MFCs with the anode effluent being further treated in the cathode, where a high concentration of protons is present due to the microbial oxidation of the organic matter (Fig. 1B). Pyrolysis of metal macrocycles at 400–1000 °C can significantly improve both stability and activity, and thus makes them feasible for MFC applications169With the same loading rate of 2 mg cm−2, pyrolyzed iron phthalocyanine (FePc, 700 °C) could achieve a current density comparable to that with Pt/C in the acid medium.170 Another iron macrocyclic complex, Cl–Fe tetramethoxyphenyl porphyrin, was heat-treated at 800 °C and produced a MPD 80% of that with Pt/C.171 Although the pyrolyzed iron macrocycles are stable in the acid medium, they still suffer from deterioration under alkaline conditions, which possibly involves deactivation by H2O2 generated from the 2e− ORR pathway.172,173 MnOx was therefore hybridized with FePc to degrade the accumulated H2O2 (Scheme 1), and an improved cathode performance was observed.174 Whilst the active metal–N center is playing a key role in ORR catalysis, the interaction between the carbon matrix and the carbon support may also affect catalytic kinetics. It has been reported that replacing CB with Ketjen black carbon can increase the power production by 20%.175 The FePc supported by multi-walled CNTs or PANI, and the iron tetrasulfophthalocyanine mixed with graphene have also shown enhanced MPDs comparable to or even higher than Pt/C.176–178 A composite of FePc, polyindole and CNTs was obtained without thermal treatment, but showed an electron transfer number of 3.9 and a MPD (799 mW m−2) 1.2 times that with Pt/C,179 which might be practical for MFC-based wastewater treatment due to the simple fabrication procedures.
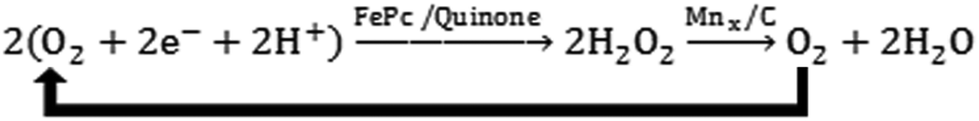 | ||
| Scheme 1 Reaction scheme for synergetic ORR catalysis by FePc and Mn. Reproduction with permission from ref. 174. | ||
The feasibility of Co macrocycles as an alternative cathode catalyst in MFCs was first demonstrated together with FePc a decade ago.167 Satisfactory MPDs were generated by pyrolyzed cobalt tetramethylphenylporphyrin (CoTMPP) and cobalt naphthalocyanine (∼75% relative to that with Pt/C).180,181 Later, several modifications on cobalt macrocycles were conducted to further promote the catalytic performance. Depositing CoTMPP and FePc on CNTs produced a MPD of 751 mW m−2, 1.5 times that with Pt/C.182 Cobalt oxide was mixed with cobalt phthalocyanine (CoPc) to serve as the downstream active sites to reduce HO2− production and minimize the possible inhibitory effects on the metal–N center.183 As a consequence, the electron transfer number was increased from ∼3.5 to 4 with 20% less production of HO2−. The addition of CoO and NiO in non-pyrolyzed binuclear CoPc could increase the MPD by 14% and 23%, respectively.184 Meanwhile, pyrolyzing binuclear CoPc at different temperatures could increase the nitrogen abundance, particularly that of pyrrolic-N,185 which might be beneficial for improving the ORR performance.
Both pyrolyzed FePc and CoTMPP were demonstrated to be durable in long-term MFC experiments.171,180 However, the open circuit potential of FePc distinctly decreased when sulfide was added.17 This may pose a potential challenge for MFC applications as sulfate in wastewater is readily reduced to sulfide by sulfate-reducing bacteria under anaerobic conditions.186 Encouragingly, in the same study, FePc was not affected in the presence of various organic metabolites, such as formate, ethanol, lactate and methanol. When exposed to 40 mM methanol, the single-chamber MFC equipped with CoTMPP was not affected, whereas that with Pt/C produced an open circuit potential only half of that in the absence of methanol.18 The tolerance to the organic matter is of practical relevance for the cathode catalysts in MFCs, as organic matter is constantly added as a carbon source to facilitate power generation.187
While technologically promising, metal macrocycles do not seem to be economically feasible for wastewater treatment. The binuclear CoPc-based catalysts were estimated to cost over $100 m−2,184,185 making the normalized MPD only 4–6 mW $−1. The normalized MPD was increased to 11 mW $−1 using the CoO–FePc mixture,188 but was still not competitive with carbon-based catalysts.
6.2 M–N–C complexes synthesized from other precursors
During heat treatment, the atomic configuration of metal macrocycles is likely to decompose and form core–shell or substrate–anchor structures, which has been suggested to be the actual active sites of ORR catalysis.165 It was therefore proposed and verified that precursors other than metal macrocycles could be used to form the metal–Nx/C active sites.189 Since then, many efforts have been made to construct M–N–C catalysts from a variety of precursors, including polymers, simple organic compounds and metal–organic frameworks (MOFs).The typical synthesis procedures with polymers start from the polymerization of monomers followed by coordination with metal salts and the final pyrolysis. For example, poly(2,6-diaminopyridine) (PDAP) was impregnated with Co and Fe salts and pyrolyzed under an NH3 atmosphere at 700 °C (Fig. 10).190 The obtained products showed an electron transfer number of 3.96, and a MPD (1.2 W m−2) 2.2 times that with Pt/C. The results suggested that the abundance of the Fe–Nx and Co–Nx structures exerted strong influence on ORR catalysis. Similar power production was obtained when melamine-formaldehyde resin (MFR) was used as the precursor and the same procedures were followed.191 The polymerization step could also be performed after mixing the monomers with the metal salts.192 Regardless of the synthesis procedures, the pyrolysis temperature is considered the major factor that affects the structure and the consequent catalytic activity. The electron transfer number of a Fe–N–C catalyst increased from 3.48 to 3.86 when the temperature was increased from 800 °C to 900 °C, but decreased to 3.69 when it was further increased to 1000 °C.193 The M–N–C complex derived from PANI at 700 °C and 900 °C showed different BET surface areas and varied content of N species, which might explain the different cathode performances.194
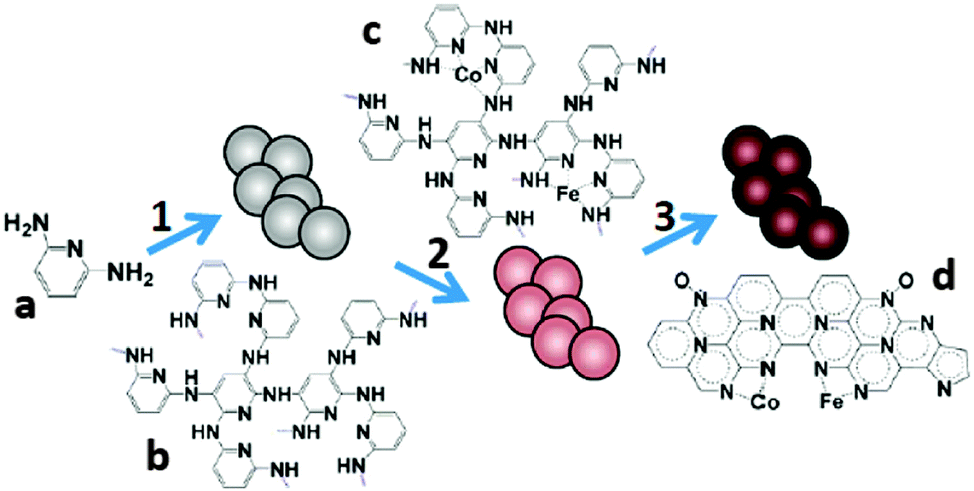 | ||
| Fig. 10 The synthesis of Co/Fe–PDAP: (1) polymerization, (2) metal coordination and (3) pyrolysis. Reproduction with permission from ref. 190. Copyright (2012) American Chemical Society. | ||
The strategy using simple organic compounds as the precursor also involves multi-step synthesis and high temperature treatment. For the preparation of Fe- and N-functionalized graphene, graphite oxide was first chemically reduced at 100 °C in the presence of FeCl3 and graphitic C3N4, dried at 130 °C and finally pyrolyzed at 800 °C.195 Although the Fe–N–graphene showed the same onset potential as Pt/C and produced a MPD (1149.8 mW m−2) 2.1 times that with Pt/C, the complicated synthesis may not be applicable for large-scale wastewater treatment. To simplify the procedures, FeCl3 was directly pyrolyzed with cyanamide to form core–shell structured Fe–N–C nanorods, leading to a MPD of 4.3 W m−3, slightly higher than that with Pt/C.29 FeCl3 was also pyrolyzed with other nitrogen-containing organic compounds such as melamine and ethylenediamine, and the MPDs produced by those Fe–N–C catalysts were both higher than that by Pt/C.196,197 A recent study prepared Fe–N–C complexes using mebendazole and aminoantipyrine as precursors and observed an attractive phenomenon: the power density of the MFCs equipped with those two catalysts increased when the catholyte pH was increased from 7 to 11.198 While the detailed catalysis mechanisms remain to be understood, the effective ORR catalysis at high pH is favorable for MFC applications as it can reduce the cost of buffer solutions.
MOFs are highly defined 3D-structured materials composed of metal centers and organic ligands. Owing to the high BET surface area (theoretically up to 10000 m2 g−1), uniform open cavities and tunable microstructures, MOFs have gained growing interest in a variety of applications.199 The abundant M–N–C sites have also made MOFs an ideal candidate for ORR catalysis. There have been a few pioneering studies using MOFs as the precursor to synthesize ORR catalysts in the past five years.200–204 The application of MOF-based ORR catalysts in MFCs has been reported by two recent studies. Pyrolyzing a Co-MOF (ZIF-67) in the presence of NiCl2 at 800 °C resulted in the formation of N species, Co–N bonding and Ni species in the composite.205 On the other hand, the BET surface area was reduced from 555 m2 g−1 for the precursor Co-MOF to 194 m2 g−1 for the Ni/Co-MOF. Due to the synergetic effects, the Ni/Co-MOF showed an onset potential more positive than Pt/C, indicating higher ORR catalytic activity. Subsequently, the MFC equipped with the Ni/Co-MOF produced a high MPD of 4336 mW m−2, 1.7 times that with Pt/C. The direct pyrolysis of the same Co-MOF could also yield high catalytic performance, which could be tuned by the heat treatment temperature.28 It was observed that increasing the pyrolysis temperature could systematically change the BET surface area, shift the pore size distribution and tune the content of carbon and nitrogen.
The configuration change caused by pyrolysis has endowed M–N–C complexes with superior durability.165 The performance of the Co/Fe–N–C catalysts prepared from polymers such as PDAP and MFR was not affected by methanol crossover,190,191 and the catalyst from PANI showed only 5% decrease in the MPD after 6 months of operation.192 In terms of current output, the MFCs equipped with the Fe–N–C nanorods remained unchanged for 6 months when operated with an external resistance of 20 Ω.29 The study on Fe–AAPyr has delivered more insights into the effects of pollutants.206 This Fe–AAPyr catalyst generated much more stable electricity than Pt/C in the presence of sulfide and sulfate with concentrations up to 20 mM (Fig. 11). Meanwhile, a thick biofilm was observed on the Fe–AAPyr surface, but did not affect the cathode performance. Biofouling has also been reported to exert a negligible influence on the cathode performance of the pyrolyzed Co-MOF.28
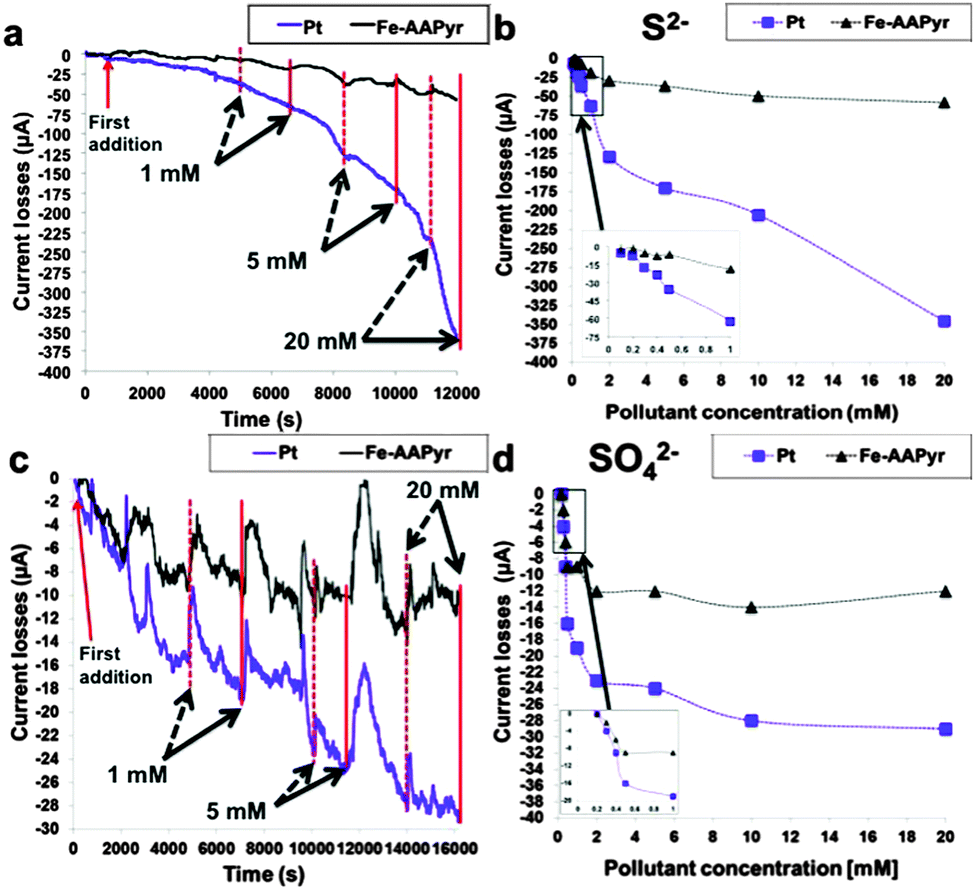 | ||
| Fig. 11 (a) Chronoamperometry study with the addition of S2−, (b) current losses as a function of the S2− concentration, (c) chronoamperometry study with the addition of SO42− and (d) current losses as a function of SO42− concentration. Reproduction with permission from ref. 206. Copyright (2015) Nature Publishing Group. | ||
The cost effectiveness of M–N–C complexes largely depends on the precursor. Take Fe–AAPyr and Fe–MBZ as examples, the normalized MPD of those catalysts was less than 5 mW $−1 due to the high cost of AAPyr and MBZ ($3.2–3.6 g−1).198 The price could be reduced to 5% of that of Pt by using cyanamide as the precursor.29 Furthermore, the Fe–N–C catalysts obtained from the inexpensive aniline was estimated to cost only $0.08 g−1,194 making them even competitive to AC-based catalysts.
7. Biocatalysts
Microorganisms can take up electrons from a cathode electrode to reduce oxygen, thereby acting as the ORR catalysts in MFCs.207 It has been observed that the anode biofilm and several electrochemically active pure cultures (e.g., Acidithiobacillus ferrooxidans and Pseudomonas aeruginosa) are able to facilitate ORR kinetics.208–210 Moreover, seven isolates from seawater and one green alga have also been reported to catalyze the ORR,211,212 indicating that extracellular electron uptake is a common strategy for aerobic respiration. Fundamental studies have suggested that the electron transfer of the microbes on biocathodes involves one or multiple pathways that are commonly found in exoelectrogens.12,213,214The uniqueness of biocatalysts is that the catalytic efficiency is not only determined by the activity of the individual cells, but also affected by the microbial interactions in the community. For instance, the power density produced by the MFCs with pure-culture biocathodes was an order of magnitude lower than that with mixed culture,215 a phenomenon consistently observed in the studies on the MFC anode.216 The better performance could be ascribed to the mutualistic interactions between electrochemically active microorganisms and other organisms during metabolism and respiration.217–219 An example of such mutualism is the integrated algae-MFC, in which the algae provide dissolved oxygen, and the ORR in return buffers the pH of the algal growth medium (i.e., catholyte).220,221 The synergetic cooperation of algae and biocathodes is of particular interest in sustainable wastewater treatment, as it can further polish the anode effluent by the removal of nitrogen and phosphate and can simultaneously produce electricity and algal biomass as energy.
The fact that the microorganisms in the MFC anodes and biocathodes share similar electron transfer pathways implies that the features of an effective anode electrode, such as good biocompatibility, high surface area and high conductivity,14 should be considered when developing the electrodes of biocathodes. Several carbon materials, including graphite, activated carbon and semicoke were examined as the support of biocathodes and produced satisfactory power densities of up to 100 W m−3 or 700 mW m−2.222–226 Modifying carbon materials with conductive polymers could introduce functional groups that help alleviate the change in dissolved oxygen and pH, and encourage biofilm attachment, both of which could improve the cathode performance.227,228 Nano-structured carbon materials such as CNTs and graphene with a high surface area and high conductivity have also been shown to facilitate the electron transfer of biocathodes.229–231
In addition to microorganisms, redox active enzymes have also been used in MFC cathodes as the biocatalysts. Laccase is a copper containing oxidoreductase that can effectively catalyse the ORR with the assistance of redox mediators (e.g., (2,2′-azino-bis(3-ethylbenzo-thiazoline-6-sulfonic acid)diammonium salt)).232,233 Immobilizing laccase on an air-breathing cathode could avoid the use of the mediator and enhance the mass transfer.234 To further overcome the limitations caused by enzyme purification, white-rot fungus was inoculated in the cathode to directly excrete laccase for ORR catalysis.235 Another enzyme, bilirubin oxidase, was immobilized on a yeast surface or immobilized on an air cathode and was proved to be feasible as the cathode catalyst.236,237
In terms of cost, microorganisms and enzymes excreted by yeast or fungi may be ideal ORR catalysts for MFC applications. The cathode ORR catalysts can affect wastewater treatment efficiency through current generation, which can stimulate the removal of organic contaminants. Moreover, biocatalysts can be used to remove the excess organic matters from the anode effluent, thereby producing high-quality water and further enhancing the cost effectiveness of wastewater treatment. With different carbon materials as the support, the COD removal by biocatalysts could reach up to 90%.225,226 Biocatalysts could also facilitate the removal of toxic and refractory pollutants such as p-nitrophenol (PNP).238 The MFC biocathode removed 100% of the PNP in 50 h, while the abiotic cathode achieved only 70.2% removal. The enzymatic cathode with immobilized laccase has been reported to simultaneously generate power and decolorize azo dye, a primary pollutant in industrial wastewater that affects the clarity and oxygen solubility.239
Biocatalysts suffer from stability issues, which are largely attributed to the microbial community dynamics. It has been reported that the biocathode with the potential fixed at 250 mV vs. Ag/AgCl, at which MPD is typically obtained, shows a long start-up period and negligible current generation.240 Further experiments using phylogenetic analysis revealed that the biocathode poised at higher potential had a higher diversity, indicating a stronger competition between the electrochemically active microorganisms and other organisms.241 The heterotrophs are likely to outcompete the biocathode community for oxygen, leading to decreased power production after long-term operation.242 For the enzyme-based MFCs, bilirubin oxidase was immediately deactivated in wastewater. The restoration of the catalytic activity of biocatalysts may be more difficult than that of abiotic catalysts, as the non-functional biofilm needs to be removed and the time-consuming acclimation needs again to be performed. Hence, the application of biocatalysts requires advances in engineering to maintain a stable environment for the organisms and enzymes.
8. Perspectives
The goal of MFC development is to achieve energy-efficient wastewater treatment, and accordingly the ideal cathode catalysts should be simple to synthesize, durable after long-term operation, stable in wastewater and cost-effective (Table 1). In this regard, carbon-based catalysts, particularly N-doped AC, may be the most promising candidate for practical applications. The normalized MPD of 1210 mW $−1 achieved by the AC mixed with CB is by far the highest reported in the literature. Further studies are needed to understand the roles of AC precursors, elucidate the mechanisms of heteroatom doping and develop a simple treatment for mass production. Metal-based catalysts do not seem competitive compared with carbon materials in terms of durability and cost, but the hybridization of metal and carbon can combine the advantages of the individual components and thus greatly enhance catalytic performance. M–N–C complexes present a new group of catalysts with excellent catalytic activity and high stability, and are particularly attractive with the effective ORR catalysis under alkaline conditions. Finally, biocatalysts are sustainable and free, but the resilience towards fluctuation remains to be improved.| Category | Catalysts/synthesis | n | MPDb | Stability | Cost effectiveness | Ref. | |
|---|---|---|---|---|---|---|---|
| a n, electron transfer number. b MPD, maximum power density. | |||||||
| Carbon-based catalysts | Carbon black | Polypyrrole/carbon black composite | 401.8 mW m−2 | Biofilm formation | 15 times that with Pt/C | 35 | |
| Activated carbon | Activated carbon/carbon black mixture | 1210 mW m−2 | 15% decrease in MPD after 16 months | 1210 mW $−1 | 53 | ||
| Carbon nanofibers | Alkaline treatment | 61.3 mW m−2 | 2.6 times that with Pt/C | 60 | |||
| Carbon nanotubes | Polyaniline/multi-walled CNTs composite | 488 mW m−2 | 31 mW $−1 | 71 | |||
| Graphene | N/S co-doped carbon nanosheets | 3.5 | 1.5 W m−2 | Stable after 6000 cycles of CV | 10 mW $−1 | 84 | |
| Carbon-based catalysts synthesized from biochar, cellulose, dopamine, straw, chitin, petroleum coke and moss | 3.5 | 2293 mW m−2 | Stable in long-term experiments, tolerant to crossover | >143 mW $−1 (free precursors) | 95 | ||
| Metal-based catalysts | Platinum | Electron beam evaporation | Reduced Pt loading | 104 | |||
| Silver nanoparticles | Inhibit bacterial growth | 110 | |||||
| Perovskite oxides | 405 mW m−2 | 15% decrease in MPD after 15 cycles | 116 mW $−1 | 125 | |||
| Metal–carbon hybrids | Metal–AC | Pyrolysis of Fe–EDTA and AC | 1580 mW m−2 | No change in MPD after 4.5 months | 139 | ||
| Metal–CNT | NiO/CNT with in situ reduction and calcination | 3.5 | 670 mW m−2 | 45 mW $−1 | 149 | ||
| Metal–graphite | Carbonized AgNPs/Fe3O4/GC | 3.98 | 1712 mW m−2 | Inhibit bacterial growth | 155 | ||
| Metal–graphene | N-Doped graphene/CoNi alloy encased within bamboo-like CNT | 3.63 | 2.0 mW m−2 | 150 mW $−1 | 162 | ||
| Metal–nitrogen–carbon complexes | Metalmacrocycles | CoO–FePc mixture | 654 mW m−2 | 85% decrease in MPD after 60 days | 11 mW $−1 | 188 | |
| N–Fe/Fe3C@C | Pyrolysis of cyanamide and FeCl3 | 3.98 | 4.2 W m−3 | Stable current output over 6 months | 5% of that of Pt | 29 | |
| Biocatalysts | Microorganisms | Mixed culture | 554 mW m−2 | 64% decrease in MPD after 9 cycles | Free | 242 | |
| Enzymes | Laccase | 26 W m−3 | 4% loss in potential after 5 days | 234 | |||
One of the advantages of biocatalysts over abiotic catalysts is the additional treatment of wastewater by microbes. However, recent studies have demonstrated that integrated catalyst-membrane assembly can also produce effluent with high quality.243 Coating multiwalled CNTs on nonwoven polyester formed an ultrafiltration membrane with a pore size of ∼65 nm that could simultaneously produce energy and filtrate wastewater in a single-chamber MFC.244 The MFC using the membrane modified with a C–Mn–Fe–O catalyst could remove up to 90% COD and 80% NH4+–N.245 In addition to the good performance, the integration of ORR catalysts with membranes can improve system stability. Fouling mitigation was observed when the membrane was coated with different catalysts,246,247 possibly because the electric field reduced the deposition of sludge on the membrane surface, and the H2O2 produced by the cathode removed the foulants.248 With stable energy and water production, catalyst–membrane assembly may be more cost-effective than individual MFCs and membrane bioreactors. It was estimated that the cathode membrane prepared from carbon foam and transition metals cost only 10% of the total price of Pt/C and commercial filtration membranes.249 Such a combination may provide a new direction in the study of abiotic ORR catalysts for MFC-based wastewater treatment.
Regardless of the catalyst materials, biofouling is almost inevitable when the cathode electrode is exposed to wastewater. The inefficient ORR catalysis, i.e., the 2e− pathway with the production of H2O2, may provide a solution for the fouling issue. Most of the ORR catalysts discussed in this review produce a considerable amount of H2O2 under normal MFC operation. For example, at the MPD, C–CoPc generated a current density of 2.5 A m−2 (green triangle, Fig. 12A) and the corresponding cathode potential of −0.2 V vs. SCE (Fig. 12B). The electron transfer number at this cathode potential was measured to be 3, with 50% of the ORR product being hydrogen peroxide ions (Fig. 12C).183 The high H2O2 production, as discussed in the catalyst–membrane assembly, may contribute to fouling mitigation. The study using graphite particles as the cathode electrode could produce 196.5 mg L−1 H2O2 in 24 h, but did not further investigate the effects on fouling.250 Therefore, the relation between H2O2 production and biofilm formation can be a focus for future research.
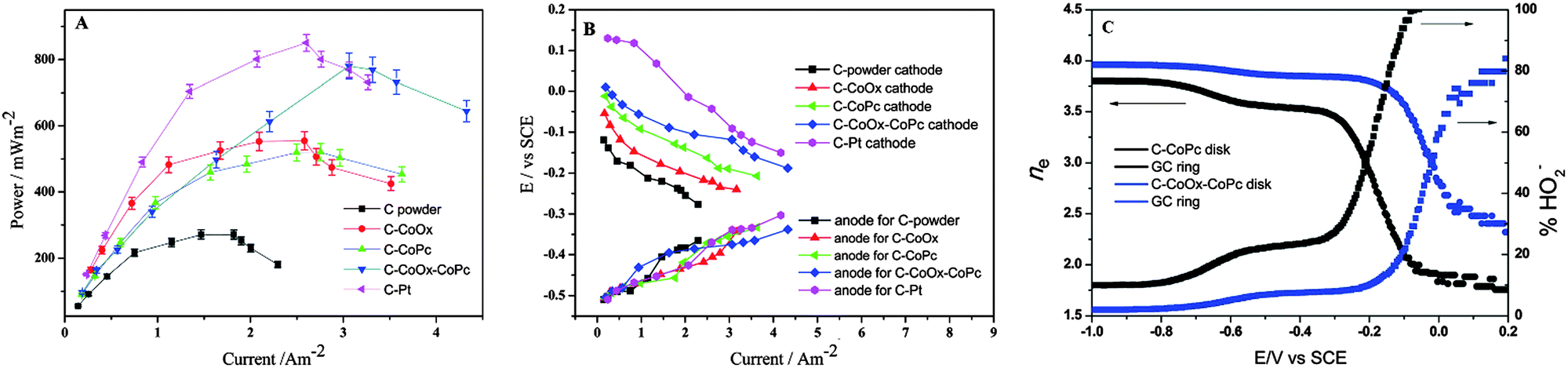 | ||
| Fig. 12 (A) Plot of power density vs. current density for MFCs constructed with carbon power, C–CoOx, C–CoPc, C–CoOx–CoPc and C–Pt; (B) individual electrode potential measured with respect to SCE as a function of current density and (C) plots of the number of electrons transferred (left) and the percent yield of hydrogen peroxide (right) during the oxygen reduction as a function of potential. Reproduction with permission from ref. 183. Copyright (2014) The Royal Society of Chemistry. | ||
Depending on the pH conditions, the overall equation of ORR can either be proton consuming (acid medium) or hydroxide producing (alkaline medium).47 Experiments and modeling suggest that the ORR in the neutral catholyte proceeds dominantly via the OH−-producing pathway.251 The OH− accumulated at the catalytic sites could lead to a potential loss over 0.3 V at high current density. Such a potential loss may explain the finding that power density negatively correlates with the micropore volume of the AC.39 The small pore hinders OH− transportation, thereby resulting in large concentration overpotential. Moreover, the commonly used Nafion binder, which was not anion-conducting, was speculated to further contribute to OH− build-up.251 The functional groups on the catalyst surface may act in a similar way and significantly affect the OH− transportation and ORR kinetics. In addition to potential loss, the elevated pH may affect the stability of the catalysts. For example, iron macrocycles lose catalytic activity under alkaline conditions.173 The effects of high pH on the stability of other catalysts is of practical significance and thus warrant further studies.
The development of ORR catalysts can greatly benefit bioelectrochemical systems (BES) other than MFCs, such as microbial desalination cells (MDCs) and microbial electrolysis cells (MECs). MDCs also use oxygen as the cathode electron acceptor and can achieve simultaneous wastewater treatment and desalination at low energy consumption.252,253 While the unit price of the ORR catalysts in MDCs remains the same as in MFCs, the cost effectiveness may be significantly increased when fresh water production and net energy balance are considered. MECs can produce hydrogen gas by using protons as the electron acceptor.254,255 Similar to ORR, hydrogen evolution reaction (HER) is sluggish and needs the presence of catalysts.256,257 Multi-functional catalysts for ORR, OER (oxygen evolution reaction) and HER have recently been reported,258–262 which can make BES a more versatile technology that can be easily switched between MFCs and MECs to meet the special requirement of applications. It can be expected that ORR catalysts will play an important role in energy-efficient water and wastewater treatment in the future, and there will be an increasing need for identifying their appropriate application niches.
Acknowledgements
This work was made possible by NPRP grant # 6-289-2-125 from the Qatar National Research Fund (a member of Qatar Foundation). The statements made herein are solely the responsibility of the authors.Notes and references
- WWAP (United Nations World Water Assessment Programme), The United Nations World Water Development Report 2014: Water and Energy, Paris, UNESCO, 2014, ISBN 978-92-3-104259-1 Search PubMed.
- R. Goldstein and W. Smith, Water & Sustainability, US Electricity Consumption for Water Supply & Treatment-The Next Half Century, Electric Power Research Institute, vol. 4, 2002 Search PubMed.
- P. L. McCarty, J. Bae and J. Kim, Environ. Sci. Technol., 2011, 45, 7100–7106 CrossRef CAS PubMed.
- A. L. Smith, S. J. Skerlos and L. Raskin, Water Res., 2013, 47, 1655–1665 CrossRef CAS PubMed.
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5192 CrossRef CAS PubMed.
- D. R. Lovley, Annu. Rev. Microbiol., 2012, 66, 391–409 CrossRef CAS PubMed.
- W.-W. Li, H.-Q. Yu and Z. He, Energy Environ. Sci., 2014, 7, 911–924 CAS.
- B. E. Logan, Appl. Microbiol. Biotechnol., 2010, 85, 1665–1671 CrossRef CAS PubMed.
- Y. Fan, E. Sharbrough and H. Liu, Environ. Sci. Technol., 2008, 42, 8101–8107 CrossRef CAS PubMed.
- R. A. Rozendal, H. V. Hamelers, K. Rabaey, J. Keller and C. J. Buisman, Trends Biotechnol., 2008, 26, 450–459 CrossRef CAS PubMed.
- Z. Wang, C. Cao, Y. Zheng, S. Chen and F. Zhao, ChemElectroChem, 2014, 1, 1813–1821 CrossRef CAS.
- X.-W. Liu, W.-W. Li and H.-Q. Yu, Chem. Soc. Rev., 2014, 43, 7718–7745 RSC.
- K. Ben Liew, W. R. W. Daud, M. Ghasemi, J. X. Leong, S. Su Lim and M. Ismail, Int. J. Hydrogen Energy, 2014, 39, 4870–4883 CrossRef CAS.
- H. Yuan and Z. He, Nanoscale, 2015, 7, 7022–7029 RSC.
- Z. Ge, J. Li, L. Xiao, Y. Tong and Z. He, Environ. Sci. Technol. Lett., 2014, 1, 137–141 CrossRef CAS.
- T. Schmidt, U. Paulus, H. Gasteiger and R. Behm, J. Electroanal. Chem., 2001, 508, 41–47 CrossRef CAS.
- F. Harnisch, S. Wirth and U. Schröder, Electrochem. Commun., 2009, 11, 2253–2256 CrossRef CAS.
- B. Liu, C. Brückner, Y. Lei, Y. Cheng, C. Santoro and B. Li, J. Power Sources, 2014, 257, 246–253 CrossRef CAS.
- P. D. Kiely, G. Rader, J. M. Regan and B. E. Logan, Bioresour. Technol., 2011, 102, 361–366 CrossRef CAS PubMed.
- Y. Yuan, S. Zhou and J. Tang, Environ. Sci. Technol., 2013, 47, 4911–4917 CrossRef CAS PubMed.
- Z. He and L. T. Angenent, Electroanalysis, 2006, 18, 2009–2015 CrossRef CAS.
- F. Zhang, Z. Ge, J. Grimaud, J. Hurst and Z. He, Environ. Sci. Technol., 2013, 47, 4941–4948 CrossRef CAS PubMed.
- S. Jung and J. M. Regan, Appl. Environ. Microbiol., 2011, 77, 564–571 CrossRef CAS PubMed.
- Z. He and F. Mansfeld, Energy Environ. Sci., 2009, 2, 215 CAS.
- F. Zhao, R. C. Slade and J. R. Varcoe, Chem. Soc. Rev., 2009, 38, 1926–1939 RSC.
- Y. Feng, X. Shi, X. Wang, H. Lee, J. Liu, Y. Qu, W. He, S. M. S. Kumar, B. H. Kim and N. Ren, Biosens. Bioelectron., 2012, 35, 413–415 CrossRef CAS PubMed.
- X.-B. Gong, S.-J. You, X.-H. Wang, Y. Gan, R.-N. Zhang and N.-Q. Ren, J. Power Sources, 2013, 225, 330–337 CrossRef CAS.
- S. You, X. Gong, W. Wang, D. Qi, X. Wang, X. Chen and N. Ren, Adv. Energy Mater., 2016, 6 DOI:10.1002/aenm.201501497.
- Z. Wen, S. Ci, F. Zhang, X. Feng, S. Cui, S. Mao, S. Luo, Z. He and J. Chen, Adv. Mater., 2012, 24, 1399–1404 CrossRef CAS PubMed.
- J.-B. Donnet, Carbon black: science and technology, CRC Press, 1993 Search PubMed.
- PEM fuel cell electrocatalysts and catalyst layers: fundamentals and applications, ed. J. Zhang, Springer Science & Business Media, 2008 Search PubMed.
- N. Duteanu, B. Erable, S. M. Senthil Kumar, M. M. Ghangrekar and K. Scott, Bioresour. Technol., 2010, 101, 5250–5255 CrossRef CAS PubMed.
- G. Yang, Y. Sun, Z. Yuan, P. Lü, X. Kong, L. Li, G. Chen and T. Lu, Chin. J. Catal., 2014, 35, 770–775 CrossRef CAS.
- K. Meng, Q. Liu, Y. Huang and Y. Wang, J. Mater. Chem. A, 2015, 3, 6873–6877 CAS.
- Y. Yuan, S. Zhou and L. Zhuang, J. Power Sources, 2010, 195, 3490–3493 CrossRef CAS.
- H. Marsh and F. R. Reinoso, Activated carbon, Elsevier, 2006 Search PubMed.
- T. Iwazaki, R. Obinata, W. Sugimoto and Y. Takasu, Electrochem. Commun., 2009, 11, 376–378 CrossRef CAS.
- F. Zhang, S. Cheng, D. Pant, G. V. Bogaert and B. E. Logan, Electrochem. Commun., 2009, 11, 2177–2179 CrossRef CAS.
- V. J. Watson, C. Nieto Delgado and B. E. Logan, Environ. Sci. Technol., 2013, 47, 6704–6710 CAS.
- A. Janicek, N. Gao, Y. Fan and H. Liu, Fuel Cells, 2015, 15, 855–861 CrossRef CAS.
- X. Zhang, X. Xia, I. Ivanov, X. Huang and B. E. Logan, Environ. Sci. Technol., 2014, 48, 2075–2081 CrossRef CAS PubMed.
- X. Wang, N. Gao, Q. Zhou, H. Dong, H. Yu and Y. Feng, Bioresour. Technol., 2013, 144, 632–636 CrossRef CAS PubMed.
- Z. Chen, K. Li and L. Pu, Bioresour. Technol., 2014, 170, 379–384 CrossRef CAS PubMed.
- Z. Chen, K. Li, P. Zhang, L. Pu, X. Zhang and Z. Fu, Chem. Eng. J., 2015, 259, 820–826 CrossRef CAS.
- Y. Liu, K. Li, Y. Liu, L. Pu, Z. Chen and S. Deng, J. Mater. Chem. A, 2015, 3, 21149–21158 CAS.
- V. J. Watson, C. Nieto Delgado and B. E. Logan, J. Power Sources, 2013, 242, 756–761 CrossRef CAS.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- B. Zhang, Z. Wen, S. Ci, S. Mao, J. Chen and Z. He, ACS Appl. Mater. Interfaces, 2014, 6, 7464–7470 CAS.
- D. Li, Y. Qu, J. Liu, W. He, H. Wang and Y. Feng, J. Power Sources, 2014, 272, 909–914 CrossRef CAS.
- N. Li, Y. Liu, J. An, C. Feng and X. Wang, J. Power Sources, 2014, 272, 895–899 CrossRef CAS.
- X. Zhang, D. Pant, F. Zhang, J. Liu, W. He and B. E. Logan, ChemElectroChem, 2014, 1, 1859–1866 CrossRef CAS.
- H. Dong, H. Yu and X. Wang, Environ. Sci. Technol., 2012, 46, 13009–13015 CrossRef CAS PubMed.
- S. Cheng and J. Wu, Bioelectrochemistry, 2013, 92, 22–26 CrossRef CAS PubMed.
- B. Wei, J. C. Tokash, G. Chen, M. A. Hickner and B. E. Logan, RSC Adv., 2012, 2, 12751–12758 RSC.
- W. Yang, W. He, F. Zhang, M. A. Hickner and B. E. Logan, Environ. Sci. Technol. Lett., 2014, 1, 416–420 CrossRef CAS.
- J. Huang, Y. Liu and T. You, Anal. Methods, 2010, 2, 202–211 RSC.
- L. Feng, N. Xie and J. Zhong, Materials, 2014, 7, 3919–3945 CrossRef CAS.
- M. Ghasemi, S. Shahgaldi, M. Ismail, B. H. Kim, Z. Yaakob and W. R. Wan Daud, Int. J. Hydrogen Energy, 2011, 36, 13746–13752 CrossRef CAS.
- C. Santoro, A. Stadlhofer, V. Hacker, G. Squadrito, U. Schröder and B. Li, J. Power Sources, 2013, 243, 499–507 CrossRef CAS.
- S. Chen, Y. Chen, G. He, S. He, U. Schröder and H. Hou, Biosens. Bioelectron., 2012, 34, 282–285 CrossRef CAS PubMed.
- X. Yang, W. Zou, Y. Su, Y. Zhu, H. Jiang, J. Shen and C. Li, J. Power Sources, 2014, 266, 36–42 CrossRef CAS.
- L. Zhou, P. Fu, X. Cai, S. Zhou and Y. Yuan, Appl. Catal., B, 2016, 188, 31–38 CrossRef CAS.
- S. van Dommele, Nitrogen doped carbon nanotubes: synthesis, characterization and catalysis, Utrecht University, 2008, ISBN 978-90-6464-263-0 Search PubMed.
- M. F. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 CAS.
- L. Feng, Y. Yan, Y. Chen and L. Wang, Energy Environ. Sci., 2011, 4, 1892–1899 CAS.
- Y.-R. He, F. Du, Y.-X. Huang, L.-M. Dai, W.-W. Li and H.-Q. Yu, J. Mater. Chem. A, 2016, 4, 1632–1636 CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- Y. Jiang, Y. Xu, Q. Yang, Y. Chen, S. Zhu and S. Shen, Int. J. Energy Res., 2014, 38, 1416–1423 CrossRef CAS.
- M. Ghasemi, W. R. Wan Daud, S. H. A. Hassan, T. Jafary, M. Rahimnejad, A. Ahmad and M. H. Yazdi, Int. J. Hydrogen Energy, 2015, 41, 4872–4878 CrossRef.
- D. Chung, J. Mater. Sci., 2002, 37, 1475–1489 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. Morozov, D. Jiang, Y. Zhang, S. Dubonos, I. Grigorieva and A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- R. S. Edwards and K. S. Coleman, Nanoscale, 2013, 5, 38–51 RSC.
- F. Perreault, A. Fonseca de Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC.
- S. Freguia, K. Rabaey, Z. Yuan and J. Keller, Electrochim. Acta, 2007, 53, 598–603 CrossRef CAS.
- B. Erable, N. Duteanu, S. M. S. Kumar, Y. Feng, M. M. Ghangrekar and K. Scott, Electrochem. Commun., 2009, 11, 1547–1549 CrossRef CAS.
- L. Zhang, Z. Lu, D. Li, J. Ma, P. Song, G. Huang, Y. Liu and L. Cai, J. Cleaner Prod., 2016, 115, 332–336 CrossRef CAS.
- X. Shi, Y. Feng, X. Wang, H. Lee, J. Liu, Y. Qu, W. He, S. M. S. Kumar and N. Ren, Bioresour. Technol., 2012, 108, 89–93 CrossRef CAS PubMed.
- L. Feng, Y. Chen and L. Chen, ACS Nano, 2011, 5, 9611–9618 CrossRef CAS PubMed.
- L. Feng, L. Yang, Z. Huang, J. Luo, M. Li, D. Wang and Y. Chen, Sci. Rep., 2013, 3 DOI:10.1038/srep03306.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed.
- H. Yuan, Y. Hou, Z. Wen, X. Guo, J. Chen and Z. He, ACS Appl. Mater. Interfaces, 2015, 7, 18672–18678 CAS.
- Q. Wen, S. Wang, J. Yan, L. Cong, Y. Chen and H. Xi, Bioelectrochemistry, 2014, 95, 23–28 CrossRef CAS PubMed.
- J. Luo, H. D. Jang, T. Sun, L. Xiao, Z. He, A. P. Katsoulidis, M. G. Kanatzidis, J. M. Gibson and J. Huang, ACS Nano, 2011, 5, 8943–8949 CrossRef CAS PubMed.
- L. Xiao, J. Damien, J. Luo, H. D. Jang, J. Huang and Z. He, J. Power Sources, 2012, 208, 187–192 CrossRef CAS.
- Y. Liu, H. Liu, C. Wang, S.-X. Hou and N. Yang, Environ. Sci. Technol., 2013, 47, 13889–13895 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- Y.-X. Huang, X.-W. Liu, J.-F. Xie, G.-P. Sheng, G.-Y. Wang, Y.-Y. Zhang, A.-W. Xu and H.-Q. Yu, Chem. Commun., 2011, 47, 5795–5797 RSC.
- Y.-C. Yong, X.-C. Dong, M. B. Chan-Park, H. Song and P. Chen, ACS Nano, 2012, 6, 2394–2400 CrossRef CAS PubMed.
- Y. Yuan, T. Yuan, D. Wang, J. Tang and S. Zhou, Bioresour. Technol., 2013, 144, 115–120 CrossRef CAS PubMed.
- Y. Yuan, T. Liu, P. Fu, J. Tang and S. Zhou, J. Mater. Chem. A, 2015, 3, 8475–8482 CAS.
- Q. Liu, S. Chen, Y. Zhou, S. Zheng, H. Hou and F. Zhao, J. Power Sources, 2014, 261, 245–248 CrossRef CAS.
- Q. Liu, Y. Zhou, S. Chen, Z. Wang, H. Hou and F. Zhao, J. Power Sources, 2015, 273, 1189–1193 CrossRef CAS.
- G. Yue, K. Meng and Q. Liu, ChemPlusChem, 2015, 80, 1133–1138 CrossRef CAS.
- S. Zhong, L. Zhou, L. Wu, L. Tang, Q. He and J. Ahmed, J. Power Sources, 2014, 272, 344–350 CrossRef CAS.
- L. Liu, Q. Xiong, C. Li, Y. Feng and S. Chen, RSC Adv., 2015, 5, 89771–89776 RSC.
- H. Yuan, L. Deng, X. Cai, S. Zhou, Y. Chen and Y. Yuan, RSC Adv., 2015, 5, 56121–56129 RSC.
- P. Zhang, X.-H. Liu, K.-X. Li and Y.-R. Lu, Int. J. Hydrogen Energy, 2015, 40, 13530–13537 CrossRef CAS.
- L. Zhou, P. Fu, D. Wen, Y. Yuan and S. Zhou, Appl. Catal., B, 2016, 181, 635–643 CrossRef CAS.
- F. H. B. Lima, J. Zhang, M. H. Shao, K. Sasaki, M. B. Vukmirovic, E. A. Ticianelli and R. R. Adzic, J. Phys. Chem. C, 2007, 111, 404–410 CAS.
- V. P. Zhdanov and B. Kasemo, Electrochem. Commun., 2006, 8, 1132–1136 CrossRef CAS.
- H. I. Park, U. Mushtaq, D. Perello, I. Lee, S. K. Cho, A. Star and M. Yun, Energy Fuels, 2007, 21, 2984–2990 CrossRef CAS.
- J.-N. Zhang, S.-J. You, Y.-X. Yuan, Q.-L. Zhao and G.-D. Zhang, Electrochem. Commun., 2011, 13, 903–905 CrossRef CAS.
- Y.-Y. Chang, H.-Z. Zhao, C. Zhong and A. Xue, Russ. J. Electrochem., 2014, 50, 885–890 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- Z. Yan, M. Wang, Y. Lu, R. Liu and J. Zhao, J. Solid State Electrochem., 2013, 18, 1087–1097 CrossRef.
- Z. Yan, M. Wang, J. Liu, R. Liu and J. Zhao, Electrochim. Acta, 2014, 141, 331–339 CrossRef CAS.
- J. An, H. Jeon, J. Lee and I. S. Chang, Environ. Sci. Technol., 2011, 45, 5441–5446 CrossRef CAS PubMed.
- O. Lefebvre, Z. Tan, Y. Shen and H. Y. Ng, Bioresour. Technol., 2013, 127, 158–164 CrossRef CAS PubMed.
- M. M. Thackeray, M. H. Rossouw, A. de Kock, A. P. de la Harpe, R. J. Gummow, K. Pearce and D. C. Liles, J. Power Sources, 1993, 43, 289–300 CrossRef CAS.
- I. Roche, K. Katuri and K. Scott, J. Appl. Electrochem., 2009, 40, 13–21 CrossRef.
- L. Zhang, C. Liu, L. Zhuang, W. Li, S. Zhou and J. Zhang, Biosens. Bioelectron., 2009, 24, 2825–2829 CrossRef CAS PubMed.
- X. Li, B. Hu, S. Suib, Y. Lei and B. Li, J. Power Sources, 2010, 195, 2586–2591 CrossRef CAS.
- K. A. Stoerzinger, M. Risch, B. Han and Y. Shao-Horn, ACS Catal., 2015, 5, 6021–6031 CrossRef CAS.
- X.-W. Liu, X.-F. Sun, Y.-X. Huang, G.-P. Sheng, K. Zhou, R. J. Zeng, F. Dong, S.-G. Wang, A.-W. Xu, Z.-H. Tong and H.-Q. Yu, Water Res., 2010, 44, 5298–5305 CrossRef CAS PubMed.
- J.-Y. Liao, D. Higgins, G. Lui, V. Chabot, X. Xiao and Z. Chen, Nano Lett., 2013, 13, 5467–5473 CrossRef CAS PubMed.
- H. Yuan, L. Deng, J. Tang, S. Zhou, Y. Chen and Y. Yuan, ChemElectroChem, 2015, 2, 1152–1158 CrossRef CAS.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79–84 CrossRef CAS PubMed.
- S. Khilari, S. Pandit, D. Das and D. Pradhan, Biosens. Bioelectron., 2014, 54, 534–540 CrossRef CAS PubMed.
- D. Hu, H. Wang, J. Wang and Q. Zhong, Energy Technol., 2015, 3, 48–54 CrossRef CAS.
- S. Khilari, S. Pandit, J. L. Varanasi, D. Das and D. Pradhan, ACS Appl. Mater. Interfaces, 2015, 7, 20657–20666 CAS.
- J. M. Morris, S. Jin, J. Wang, C. Zhu and M. A. Urynowicz, Electrochem. Commun., 2007, 9, 1730–1734 CrossRef CAS.
- H. Dong, H. Yu, X. Wang, Q. Zhou and J. Sun, J. Chem. Technol. Biotechnol., 2013, 88, 774–778 CrossRef CAS.
- K. B. Ghoreishi, M. Ghasemi, M. Rahimnejad, M. A. Yarmo, W. R. W. Daud, N. Asim and M. Ismail, Int. J. Energy Res., 2014, 38, 70–77 CrossRef CAS.
- M. T. Noori, M. M. Ghangrekar and C. K. Mukherjee, Int. J. Hydrogen Energy, 2016, 41, 3638–3645 CrossRef CAS.
- X.-B. Gong, S.-J. You, X.-H. Wang, J.-N. Zhang, Y. Gan and N.-Q. Ren, Biosens. Bioelectron., 2014, 55, 237–241 CrossRef CAS PubMed.
- B. Mecheri, A. Iannaci, A. D’Epifanio, A. Mauri and S. Licoccia, ChemPlusChem, 2016, 81, 80–85 CrossRef CAS.
- I. Singh and A. Chandra, Bioresour. Technol., 2013, 142, 77–81 CrossRef CAS PubMed.
- I. Singh and A. Chandra, Int. J. Hydrogen Energy, 2016, 41, 1913–1920 CrossRef CAS.
- B. Ge, K. Li, Z. Fu, L. Pu and X. Zhang, Bioresour. Technol., 2015, 195, 180–187 CrossRef CAS PubMed.
- B. Ge, K. Li, Z. Fu, L. Pu, X. Zhang, Z. Liu and K. Huang, J. Power Sources, 2016, 303, 325–332 CrossRef CAS.
- Z. Fu, L. Yan, K. Li, B. Ge, L. Pu and X. Zhang, Biosens. Bioelectron., 2015, 74, 989–995 CrossRef CAS PubMed.
- P. Zhang, K. Li and X. Liu, J. Power Sources, 2014, 264, 248–253 CrossRef CAS.
- L. Pu, K. Li, Z. Chen, P. Zhang, X. Zhang and Z. Fu, J. Power Sources, 2014, 268, 476–481 CrossRef CAS.
- Z. Liu, K. Li, X. Zhang, B. Ge and L. Pu, Bioresour. Technol., 2015, 195, 154–161 CrossRef CAS PubMed.
- X. Zhang, K. Li, P. Yan, Z. Liu and L. Pu, Bioresour. Technol., 2015, 187, 299–304 CrossRef CAS PubMed.
- X. Xia, F. Zhang, X. Zhang, P. Liang, X. Huang and B. E. Logan, ACS Appl. Mater. Interfaces, 2013, 5, 7862–7866 CAS.
- D. V. Sanchez, P. Huynh, M. E. Kozlov, R. H. Baughman, R. D. Vidic and M. Yun, Energy Fuels, 2010, 24, 5897–5902 CrossRef CAS.
- H. Wang, Z. Wu, A. Plaseied, P. Jenkins, L. Simpson, C. Engtrakul and Z. Ren, J. Power Sources, 2011, 196, 7465–7469 CrossRef CAS.
- M. Ghasemi, M. Ismail, S. K. Kamarudin, K. Saeedfar, W. R. W. Daud, S. H. A. Hassan, L. Y. Heng, J. Alam and S.-E. Oh, Appl. Energy, 2013, 102, 1050–1056 CrossRef CAS.
- X. Xie, M. Pasta, L. Hu, Y. Yang, J. McDonough, J. Cha, C. S. Criddle and Y. Cui, Energy Environ. Sci., 2011, 4, 1293–1297 CAS.
- M. Lu, S. Kharkwal, H. Y. Ng and S. F. Y. Li, Biosens. Bioelectron., 2011, 26, 4728–4732 CrossRef CAS PubMed.
- Y. Zhang, Y. Hu, S. Li, J. Sun and B. Hou, J. Power Sources, 2011, 196, 9284–9289 CrossRef CAS.
- M. Lu, L. Guo, S. Kharkwal, H. N. Wu, H. Y. Ng and S. F. Y. Li, J. Power Sources, 2013, 221, 381–386 CrossRef CAS.
- K. B. Liew, W. R. Wan Daud, M. Ghasemi, K. S. Loh, M. Ismail, S. S. Lim and J. X. Leong, Int. J. Hydrogen Energy, 2015, 40, 11625–11632 CrossRef CAS.
- Y. Chen, Z. Lv, J. Xu, D. Peng, Y. Liu, J. Chen, X. Sun, C. Feng and C. Wei, J. Power Sources, 2012, 201, 136–141 CrossRef CAS.
- J. Huang, N. Zhu, T. Yang, T. Zhang, P. Wu and Z. Dang, Biosens. Bioelectron., 2015, 72, 332–339 CrossRef CAS PubMed.
- T.-S. Song, D.-B. Wang, H. Wang, X. Li, Y. Liang and J. Xie, Int. J. Hydrogen Energy, 2015, 40, 3868–3874 CrossRef CAS.
- S. Singh and N. Verma, Int. J. Hydrogen Energy, 2015, 40, 5928–5938 CrossRef CAS.
- A. Modi, S. Singh and N. Verma, Electrochim. Acta, 2016, 190, 620–627 CrossRef CAS.
- P. Wang, B. Lai, H. Li and Z. Du, Bioresour. Technol., 2013, 134, 30–35 CrossRef CAS PubMed.
- M. Ma, Y. Dai, J.-L. Zou, L. Wang, K. Pan and H.-G. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 13438–13447 CAS.
- M. Ma, S. You, X. Gong, Y. Dai, J. Zou and H. Fu, J. Power Sources, 2015, 283, 74–83 CrossRef CAS.
- R. Li, Y. Dai, B. Chen, J. Zou, B. Jiang and H. Fu, J. Power Sources, 2016, 307, 1–10 CrossRef CAS.
- S. Khilari, S. Pandit, M. M. Ghangrekar, D. Das and D. Pradhan, RSC Adv., 2013, 3, 7902–7911 RSC.
- X. Quan, Y. Mei, H. Xu, B. Sun and X. Zhang, Electrochim. Acta, 2015, 165, 72–77 CrossRef CAS.
- Q. Wen, S. Wang, J. Yan, L. Cong, Z. Pan, Y. Ren and Z. Fan, J. Power Sources, 2012, 216, 187–191 CrossRef CAS.
- G. Gnana kumar, Z. Awan, K. Suk Nahm and J. Stanley Xavier, Biosens. Bioelectron., 2014, 53, 528–534 CrossRef CAS PubMed.
- N. Garino, A. Sacco, M. Castellino, J. A. Muñoz-Tabares, A. Chiodoni, V. Agostino, V. Margaria, M. Gerosa, G. Massaglia and M. Quaglio, ACS Appl. Mater. Interfaces, 2016, 8, 4633–4643 CAS.
- Y. Hou, H. Yuan, Z. Wen, S. Cui, X. Guo, Z. He and J. Chen, J. Power Sources, 2016, 307, 561–568 CrossRef CAS.
- X. Xie, G. Yu, N. Liu, Z. Bao, C. S. Criddle and Y. Cui, Energy Environ. Sci., 2012, 5, 6862–6866 CAS.
- Z. He, J. Liu, Y. Qiao, C. M. Li and T. T. Y. Tan, Nano Lett., 2012, 12, 4738–4741 CrossRef CAS PubMed.
- C. Tang and Q. Zhang, J. Mater. Chem. A, 2016, 4, 4998–5001 CAS.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- F. Zhao, F. Harnisch, U. Schröder, F. Scholz, P. Bogdanoff and I. Herrmann, Electrochem. Commun., 2005, 7, 1405–1410 CrossRef CAS.
- E. H. Yu, S. Cheng, B. E. Logan and K. Scott, J. Appl. Electrochem., 2008, 39, 705–711 CrossRef.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 CAS.
- F. Zhao, F. Harnisch, U. Schröder, F. Scholz, P. Bogdanoff and I. Herrmann, Environ. Sci. Technol., 2006, 40, 5193–5199 CrossRef CAS PubMed.
- L. Birry, P. Mehta, F. Jaouen, J. P. Dodelet, S. R. Guiot and B. Tartakovsky, Electrochim. Acta, 2011, 56, 1505–1511 CrossRef CAS.
- H. Schulenburg, S. Stankov, V. Schünemann, J. Radnik, I. Dorbandt, S. Fiechter, P. Bogdanoff and H. Tributsch, J. Phys. Chem. B, 2003, 107, 9034–9041 CrossRef CAS.
- R. Chen, H. Li, D. Chu and G. Wang, J. Phys. Chem. C, 2009, 113, 20689–20697 CAS.
- R. Burkitt, T. R. Whiffen and E. H. Yu, Appl. Catal., B, 2016, 181, 279–288 CrossRef CAS.
- E. HaoYu, S. Cheng, K. Scott and B. Logan, J. Power Sources, 2007, 171, 275–281 CrossRef CAS.
- Y. Yuan, B. Zhao, Y. Jeon, S. Zhong, S. Zhou and S. Kim, Bioresour. Technol., 2011, 102, 5849–5854 CrossRef CAS PubMed.
- Y. Yuan, J. Ahmed and S. Kim, J. Power Sources, 2011, 196, 1103–1106 CrossRef CAS.
- Y. Zhang, G. Mo, X. Li and J. Ye, J. Power Sources, 2012, 197, 93–96 CrossRef CAS.
- M.-T. Nguyen, B. Mecheri, A. Iannaci, A. D’Epifanio and S. Licoccia, Electrochim. Acta, 2016, 190, 388–395 CrossRef CAS.
- S. Cheng, H. Liu and B. E. Logan, Environ. Sci. Technol., 2006, 40, 364–369 CrossRef CAS PubMed.
- J. R. Kim, J.-Y. Kim, S.-B. Han, K.-W. Park, G. D. Saratale and S.-E. Oh, Bioresour. Technol., 2011, 102, 342–347 CrossRef CAS PubMed.
- L. Deng, M. Zhou, C. Liu, L. Liu, C. Liu and S. Dong, Talanta, 2010, 81, 444–448 CrossRef CAS PubMed.
- J. Ahmed, H. J. Kim and S. Kim, RSC Adv., 2014, 4, 44065–44072 RSC.
- B. Li, X. Zhou, X. Wang, B. Liu and B. Li, J. Power Sources, 2014, 272, 320–327 CrossRef CAS.
- B. Li, M. Wang, X. Zhou, X. Wang, B. Liu and B. Li, Bioresour. Technol., 2015, 193, 545–548 CrossRef CAS PubMed.
- M. H. Gerardi, The microbiology of anaerobic digesters, John Wiley & Sons, 2003 Search PubMed.
- H. Yuan, I. M. Abu-Reesh and Z. He, J. Membr. Sci., 2016, 502, 116–123 CrossRef CAS.
- J. Ahmed, Y. Yuan, L. Zhou and S. Kim, J. Power Sources, 2012, 208, 170–175 CrossRef CAS.
- S. Gupta, D. Tryk, I. Bae, W. Aldred and E. Yeager, J. Appl. Electrochem., 1989, 19, 19–27 CrossRef CAS.
- Y. Zhao, K. Watanabe and K. Hashimoto, J. Am. Chem. Soc., 2012, 134, 19528–19531 CrossRef CAS PubMed.
- Y. Zhao, K. Watanabe and K. Hashimoto, J. Mater. Chem. A, 2013, 1, 1450–1456 CAS.
- X. Tang, H. Li, W. Wang, Z. Du and H. Y. Ng, RSC Adv., 2014, 4, 12789–12794 RSC.
- Y. Su, H. Jiang, Y. Zhu, W. Zou, X. Yang, J. Chen and C. Li, J. Power Sources, 2014, 265, 246–253 CrossRef CAS.
- X. Tang, H. Li, Z. Du and H. Y. Ng, RSC Adv., 2015, 5, 79348–79354 RSC.
- S. Li, Y. Hu, Q. Xu, J. Sun, B. Hou and Y. Zhang, J. Power Sources, 2012, 213, 265–269 CrossRef CAS.
- Y.-Z. Chan, Y. Dai, R. Li, J.-L. Zou, G.-H. Tian and H.-G. Fu, Carbon, 2015, 89, 8–19 CrossRef CAS.
- Y. Pan, X. Mo, K. Li, L. Pu, D. Liu and T. Yang, Bioresour. Technol., 2016, 206, 285–289 CrossRef CAS PubMed.
- C. Santoro, A. Serov, C. W. Narvaez Villarrubia, S. Stariha, S. Babanova, A. J. Schuler, K. Artyushkova and P. Atanassov, ChemSusChem, 2015, 8, 828–834 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- G. Goenaga, S. Ma, S. Yuan and D.-J. Liu, ECS Trans., 2010, 33, 579–586 CAS.
- S. Ma, G. A. Goenaga, A. V. Call and D.-J. Liu, Chem. – Eur. J., 2011, 17, 2063–2067 CrossRef CAS PubMed.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- A. Morozan and F. Jaouen, Energy Environ. Sci., 2012, 5, 9269–9290 CAS.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 CAS.
- H. Tang, S. Cai, S. Xie, Z. Wang, Y. Tong, M. Pan and X. Lu, Adv. Sci., 2016, 3, 1500265 Search PubMed.
- C. Santoro, A. Serov, C. W. N. Villarrubia, S. Stariha, S. Babanova, K. Artyushkova, A. J. Schuler and P. Atanassov, Sci. Rep., 2015, 5 DOI:10.1038/srep16596.
- P. Clauwaert, D. van der Ha, N. Boon, K. Verbeken, M. Verhaege, K. Rabaey and W. Verstraete, Environ. Sci. Technol., 2007, 41, 7564–7569 CrossRef CAS PubMed.
- K. Y. Cheng, G. Ho and R. Cord-Ruwisch, Environ. Sci. Technol., 2010, 44, 518–525 CrossRef CAS PubMed.
- S. Carbajosa, M. Malki, R. Caillard, M. F. Lopez, F. J. Palomares, J. A. Martín-Gago, N. Rodríguez, R. Amils, V. M. Fernández and A. L. De Lacey, Biosens. Bioelectron., 2010, 26, 877–880 CrossRef CAS PubMed.
- A. Cournet, M. Bergé, C. Roques, A. Bergel and M.-L. Délia, Electrochim. Acta, 2010, 55, 4902–4908 CrossRef CAS.
- S. Parot, I. Vandecandelaere, A. Cournet, M.-L. Délia, P. Vandamme, M. Bergé, C. Roques and A. Bergel, Bioresour. Technol., 2011, 102, 304–311 CrossRef CAS PubMed.
- X.-W. Liu, X.-F. Sun, Y.-X. Huang, D.-B. Li, R. J. Zeng, L. Xiong, G.-P. Sheng, W.-W. Li, Y.-Y. Cheng, S.-G. Wang and H.-Q. Yu, Biotechnol. Bioeng., 2013, 110, 173–179 CrossRef CAS PubMed.
- S. Freguia, S. Tsujimura and K. Kano, Electrochim. Acta, 2010, 55, 813–818 CrossRef CAS.
- M. Rosenbaum, F. Aulenta, M. Villano and L. T. Angenent, Bioresour. Technol., 2011, 102, 324–333 CrossRef CAS PubMed.
- K. Rabaey, S. T. Read, P. Clauwaert, S. Freguia, P. L. Bond, L. L. Blackall and J. Keller, ISME J., 2008, 2, 519–527 CrossRef CAS PubMed.
- B. E. Logan, Nat. Rev. Microbiol., 2009, 7, 375–381 CrossRef CAS PubMed.
- A. Venkataraman, M. A. Rosenbaum, S. D. Perkins, J. J. Werner and L. T. Angenent, Energy Environ. Sci., 2011, 4, 4550 CAS.
- B. E. Morris, R. Henneberger, H. Huber and C. Moissl-Eichinger, FEMS Microbiol. Rev., 2013, 37, 384–406 CrossRef CAS PubMed.
- V. B. Wang, K. Sivakumar, L. Yang, Q. Zhang, S. Kjelleberg, S. C. J. Loo and B. Cao, Sci. Rep., 2015, 5 DOI:10.1038/srep11222.
- Z. He, J. Kan, F. Mansfeld, L. T. Angenent and K. H. Nealson, Environ. Sci. Technol., 2009, 43, 1648–1654 CrossRef CAS PubMed.
- L. Xiao, E. B. Young, J. A. Berges and Z. He, Environ. Sci. Technol., 2012, 46, 11459–11466 CrossRef CAS PubMed.
- S. J. You, N. Q. Ren, Q. L. Zhao, J. Y. Wang and F. L. Yang, Fuel Cells, 2009, 9, 588–596 CrossRef CAS.
- J. Wei, P. Liang, X. Cao and X. Huang, Bioresour. Technol., 2011, 102, 10431–10435 CrossRef CAS PubMed.
- G.-D. Zhang, Q.-L. Zhao, Y. Jiao, J.-N. Zhang, J.-Q. Jiang, N. Ren and B. H. Kim, J. Power Sources, 2011, 196, 6036–6041 CrossRef CAS.
- Y. Zhang, J. Sun, Y. Hu, S. Li and Q. Xu, Int. J. Hydrogen Energy, 2012, 37, 16935–16942 CrossRef CAS.
- H. Tursun, R. Liu, J. Li, R. A. Abro, X. Wang, Y. Gao and Y. Li, Front. Microbiol., 2016, 7, 6 Search PubMed.
- C. Li, L. Ding, H. Cui, L. Zhang, K. Xu and H. Ren, Bioresour. Technol., 2012, 116, 459–465 CrossRef CAS PubMed.
- H. Zhang, R. Zhang, G. Zhang, F. Yang and F. Gao, Int. J. Hydrogen Energy, 2014, 39, 11250–11257 CrossRef CAS.
- X.-W. Liu, X.-F. Sun, Y.-X. Huang, G.-P. Sheng, S.-G. Wang and H.-Q. Yu, Energy Environ. Sci., 2011, 4, 1422–1427 CAS.
- L. Zhuang, Y. Yuan, G. Yang and S. Zhou, Electrochem. Commun., 2012, 21, 69–72 CrossRef CAS.
- Y. Zhang, J. Sun, Y. Hu, S. Li and Q. Xu, J. Power Sources, 2013, 239, 169–174 CrossRef CAS.
- O. Schaetzle, F. Barriere and U. Schroder, Energy Environ. Sci., 2009, 2, 96–99 CAS.
- H. Luo, S. Jin, P. H. Fallgren, H. J. Park and P. A. Johnson, Chem. Eng. J., 2010, 165, 524–528 CrossRef CAS.
- S. R. Higgins, C. Lau, P. Atanassov, S. D. Minteer and M. J. Cooney, ACS Catal., 2011, 1, 994–997 CrossRef CAS.
- C. Wu, X.-W. Liu, W.-W. Li, G.-P. Sheng, G.-L. Zang, Y.-Y. Cheng, N. Shen, Y.-P. Yang and H.-Q. Yu, Appl. Energy, 2012, 98, 594–596 CrossRef CAS.
- A. Szczupak, D. Kol-Kalman and L. Alfonta, Chem. Commun., 2012, 48, 49–51 RSC.
- C. Santoro, S. Babanova, P. Atanassov, B. Li, I. Ieropoulos and P. Cristiani, J. Electrochem. Soc., 2013, 160, H720–H726 CrossRef CAS.
- L. Zhang, X. Jiang, J. Shen, K. Xu, J. Li, X. Sun, W. Han and L. Wang, RSC Adv., 2016, 6, 29072–29079 RSC.
- I. S. P. Savizi, H.-R. Kariminia and S. Bakhshian, Environ. Sci. Technol., 2012, 46, 6584–6593 CrossRef CAS PubMed.
- A. Ter Heijne, D. P. Strik, H. V. Hamelers and C. J. Buisman, Environ. Sci. Technol., 2010, 44, 7151–7156 CrossRef CAS PubMed.
- X. Xia, Y. Sun, P. Liang and X. Huang, Bioresour. Technol., 2012, 120, 26–33 CrossRef CAS PubMed.
- X. Xia, J. C. Tokash, F. Zhang, P. Liang, X. Huang and B. E. Logan, Environ. Sci. Technol., 2013, 47, 2085–2091 CrossRef CAS PubMed.
- H. Yuan and Z. He, Bioresour. Technol., 2015, 195, 202–209 CrossRef CAS PubMed.
- L. Malaeb, K. P. Katuri, B. E. Logan, H. Maab, S. P. Nunes and P. E. Saikaly, Environ. Sci. Technol., 2013, 47, 11821–11828 CrossRef CAS PubMed.
- Y. Li, L. Liu, F. Yang and N. Ren, J. Membr. Sci., 2015, 484, 27–34 CrossRef CAS.
- J. Liu, L. Liu, B. Gao, F. Yang, J. Crittenden and N. Ren, Int. J. Hydrogen Energy, 2014, 39, 17865–17872 CrossRef CAS.
- Y. Li, L. Liu and F. Yang, J. Membr. Sci., 2016, 505, 130–137 CrossRef CAS.
- Y.-K. Wang, W.-W. Li, G.-P. Sheng, B.-J. Shi and H.-Q. Yu, Water Res., 2013, 47, 5794–5800 CrossRef CAS PubMed.
- T. Yu, L. Liu, Q. Yang, J. Song and F. Yang, RSC Adv., 2015, 5, 48946–48953 RSC.
- J.-Y. Chen, N. Li and L. Zhao, J. Power Sources, 2014, 254, 316–322 CrossRef CAS.
- S. C. Popat, D. Ki, B. E. Rittmann and C. I. Torres, ChemSusChem, 2012, 5, 1071–1079 CrossRef CAS PubMed.
- X. Cao, X. Huang, P. Liang, K. Xiao, Y. Zhou, X. Zhang and B. E. Logan, Environ. Sci. Technol., 2009, 43, 7148–7152 CrossRef CAS PubMed.
- Y. Kim and B. E. Logan, Desalination, 2013, 308, 122–130 CrossRef CAS.
- H. Liu, S. Grot and B. E. Logan, Environ. Sci. Technol., 2005, 39, 4317–4320 CrossRef CAS PubMed.
- Y. Zhang and I. Angelidaki, Water Res., 2014, 56, 11–25 CrossRef CAS PubMed.
- H. Yuan, J. Li, C. Yuan and Z. He, ChemElectroChem, 2014, 1, 1828–1833 CrossRef CAS.
- Y. Hou, B. Zhang, Z. Wen, S. Cui, X. Guo, Z. He and J. Chen, J. Mater. Chem. A, 2014, 2, 13795–13800 CAS.
- X. Fan, Z. Peng, R. Ye, H. Zhou and X. Guo, ACS Nano, 2015, 9, 7407–7418 CrossRef CAS PubMed.
- Y. Hou, S. Cui, Z. Wen, X. Guo, X. Feng and J. Chen, Small, 2015, 11, 5940–5948 CrossRef CAS PubMed.
- Y. Hou, Z. Wen, S. Cui, S. Ci, S. Mao and J. Chen, Adv. Funct. Mater., 2015, 25, 872–882 CrossRef CAS.
- J. Lu, W. Zhou, L. Wang, J. Jia, Y. Ke, L. Yang, K. Zhou, X. Liu, Z. Tang, L. Li and S. Chen, ACS Catal., 2016, 6, 1045–1053 CrossRef CAS.
- Y. Hou, M. R. Lohe, J. Zhang, S. Liu, X. Zhuang and X. Feng, Energy Environ. Sci., 2016, 9, 478–483 CAS.
| This journal is © The Royal Society of Chemistry 2016 |