
Realizing the full potential of conjugated polymers: innovation in polymer synthesis†
Pierre-Olivier
Morin
,
Thomas
Bura
and
Mario
Leclerc
*
Department of Chemistry, Université Laval, Quebec City, Qc, Canada G1V 0A6. E-mail: mario.leclerc@chm.ulaval.ca
First published on 3rd September 2015
Abstract
Plastic electronics is closely linked to advances in polymer synthesis. Based on conjugated polymers, this technology aims to exploit the features of metals and inorganic semi-conductors while preserving mechanical properties unique to polymers. In the first part of this review, we present a retrospective study of the development of different polymerization protocols with their respective key polymers. We report different methods, starting with Ziegler–Natta followed by electro-polymerization, metathesis, Kumada, Negishi, GRIM, Stille, and Suzuki. In the second part of this review, we outline the recent advances made in direct (hetero)arylation polymerization (DHAP) which is particularly promising for the future development of efficient, greener and low-cost electronic devices.
 Pierre-Olivier Morin | Pierre-Olivier Morin received his BS degree in chemistry in 2010 from Université Laval (Quebec, Canada). Currently, he is a PhD candidate at the Department of Chemistry at Université Laval. His research interests include the design, synthesis, and characterization of conjugated polymers for plastic electronics, more especially for transistor and solar cell applications. During his doctoral research, he also focused his effort on expanding the scope of the novel DHAP polymerization reaction. |
 Thomas Bura | Thomas Bura obtained his PhD degree in chemistry from Université de Strasbourg (France) in 2013. He received the prize for the best thesis in chemistry from Université de Strasbourg. During his PhD studies, he developed new BODIPY synthesis for several applications such as optoelectronics, biomedical sensing, and energy transfer. He then started postdoctoral research in the group of Prof. Mario Leclerc (Université Laval, Québec, Canada). His research interests lie in the development of polymeric optoelectronic materials, especially photovoltaic materials synthesized by the novel DHAP polymerization reaction. |
 Mario Leclerc | Mario Leclerc was awarded a PhD degree in chemistry from Université Laval, Quebec City, Canada, in 1987, under the guidance of Prof. R. E. Prud'homme. After a short post-doctoral stay at INRS-Energie et Matériaux near Montréal with Prof. L. H. Dao, he joined the Max-Planck-Institute for Polymer Research, in Mainz, Germany, as a post-doctoral fellow in the research group of Prof. G. Wegner. In 1989, he accepted a position of professor in the department of chemistry at the Université de Montréal. He returned to Université Laval in 1998. Since 2001, he has held a Canada Research Chair on Electroactive and Photoactive Polymers. His current research activities include the synthesis and characterization of new conjugated oligomers and polymers for applications in micro- and nano-electronics, electro-optics, and genomics. |
1 Introduction
Over the last few decades, conjugated polymers have attracted a lot of attention from both academia and industrial laboratories since they can combine the best features of metals or inorganic semiconducting materials (excellent electrical and optical properties) with those of synthetic polymers (mechanical flexibility, simple processing, and low cost). These materials make now possible the fabrication of electronic devices printed on different substrates. Such printed devices include, for instance, sensors, light-emitting diodes, transistors, and photovoltaic cells. It is important to note that the development of the so-called plastic electronics was and still is strongly linked to the availability of reliable and versatile polymerization methods (e.g. Ziegler–Natta, metathesis, Kumada, Negishi, Stille, Suzuki, etc.) to afford well-defined and reproducible conjugated polymers. This review aims to establish the links between the development of novel polymerization methods and advances in the field of conjugated polymers. In particular, the recent direct (hetero)arylation polymerization (DHAP) is particularly promising for the future development of efficient and low-cost printed electronic devices.2 First generation of conjugated and conducting polymers
Ironically, the first report of a highly conducting polymer was obtained from an ill-defined Ziegler–Natta polymerization of acetylene (Scheme 1). Indeed, by accident, a student of Professor H. Shirakawa (Tokyo Institute of Technology) prepared a “many molar” instead of the usual “millimolar” concentration of the catalyst and obtained a polyacetylene thin film which looked like a metallic foil instead of the usual dark, powdery, polymeric material obtained before (the first example of doped polyacetylene was made a decade earlier by Berets and Smith1 with compressed pellets). During a visit to Japan, Professor A. G. MacDiarmid invited Professor H. Shirakawa to come to the University of Pennsylvania to investigate in more detail this new form of polyacetylene. In collaboration with Professor A. J. Heeger, this team reported in 1977 that upon partial oxidation or reduction (the so-called doping reaction), the conductivity of intrinsically semiconducting polyacetylene increases over 100 S cm−1.2 This trio eventually received the 2000 Nobel Prize in Chemistry for this discovery.3–5 Unfortunately, this first conducting polymer is not processable and unstable in air.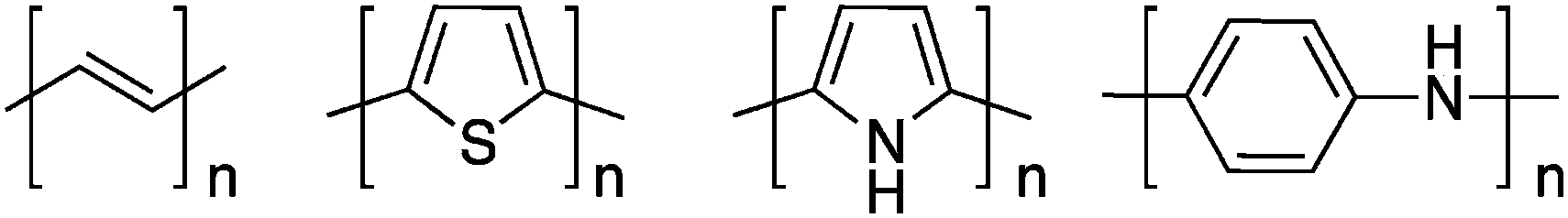 | ||
| Scheme 1 First generation of conjugated polymers. | ||
Rapidly, many scientists investigated more stable aromatic conjugated polymers. For instance, in the beginning of the 80's, a lot of studies were devoted to electropolymerized polythiophene,6 polypyrrole,6,7 and polyaniline8 (Scheme 1). This oxidative electrochemical method has the advantage of being able to prepare thin films of these infusible and insoluble rigid-rod conjugated polymers in one step. However, the ultimate goal is to develop polymeric materials that combine the electrical and optical features of metals or semiconductors with the processing advantages and mechanical properties of traditional polymers.
3 Design and synthesis of processable conjugated polymers
Attempts to achieve these requirements led to the development of a second generation of conjugated polymers with much better processability. A first example of “processable” polyacetylene was demonstrated using a two-step procedure by Edwards and Feast in 1980.9 Using a ring-opening metathesis polymerization (ROMP), a soluble polymeric non-conjugated precursor could be converted upon heating into a polyacetylene thin film and a volatile by-product (Scheme 2).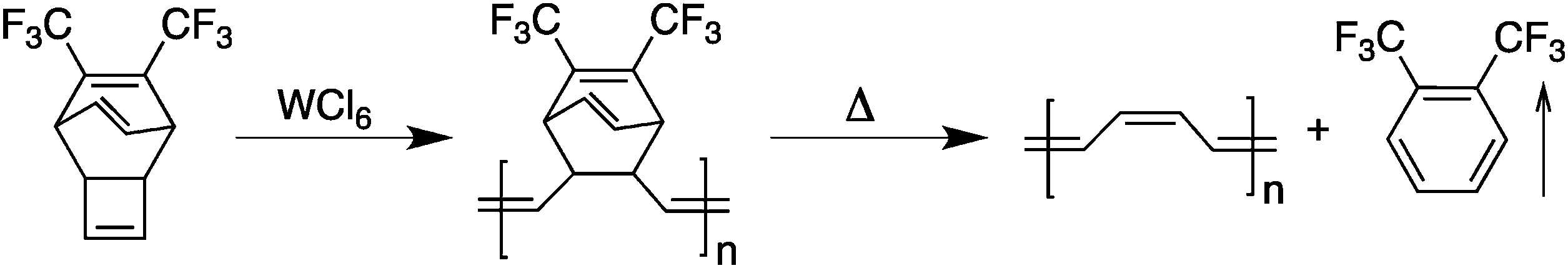 | ||
| Scheme 2 Synthesis of polyacetylene from a processable precursor. | ||
A major breakthrough occurred in 1986–1987 with the first syntheses of highly conjugated and processable poly(3-alkylthiophene)s10–12 by a simple one-step oxidation reaction (Scheme 3). The fact that these five-membered rings can exhibit an anti co-planar conformation reduces the steric hindrance developed by the presence of the solubilizing side-chains. Following the first studies on poly(3-alkylthiophene)s, it became quite clear that the synthesis of well-defined head-to-tail coupled (which should yield the lowest steric hindrance from the side chains and possibly a more efficient three-dimensional packing) poly(3-alkylthiophene)s would lead to a significant improvement in the performance of these polymeric materials.13 Therefore, in an attempt to bring more reliable synthetic procedures towards the field of electronic materials, a variety of synthetic strategies were utilized and allowed significant advances in this research field. For instance, these investigations led to the synthesis of processable, air-stable, semi-transparent, highly conducting poly(3,4-ethylenedioxythiophene) (PEDOT, Scheme 3).14
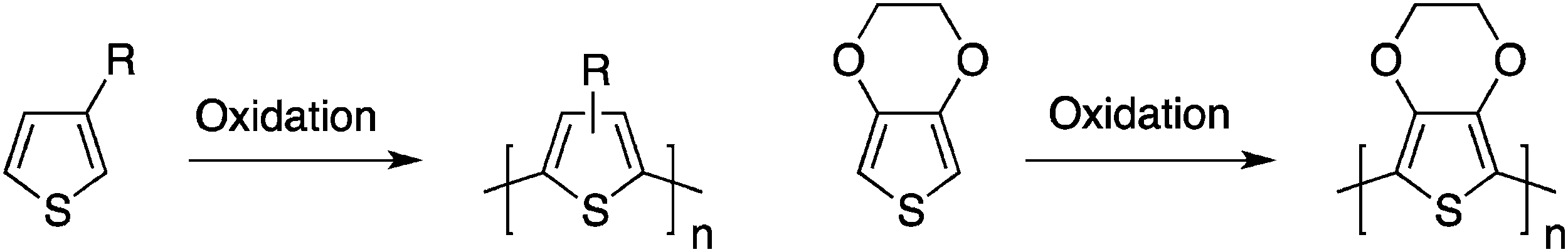 | ||
| Scheme 3 Synthesis of processable poly(3-alkylthiophene)s and poly(3,4-ethylenedioxythiophene). | ||
In parallel, McCullough15 and Rieke16 independently reported the first preparations of well-defined regioregular poly(3-alkylthiophene)s by metal-catalyzed polymerization methods (Scheme 4). These relatively complicated polymerization procedures have been optimized and simplified, leading to the Grignard metathesis method (GRIM).17 For instance, this existing polymerization method eliminates the need for highly reactive metals and can even lead to well-defined block copolymers. Among all poly(3-alkylthiophene)s investigated, regioregular poly(3-hexylthiophene) (rr-P3HT) has shown the best electrical properties together with adequate processability. Both rr-P3HT and PEDOT are currently the most utilized conjugated polymers and are commercially available from various commercial sources.
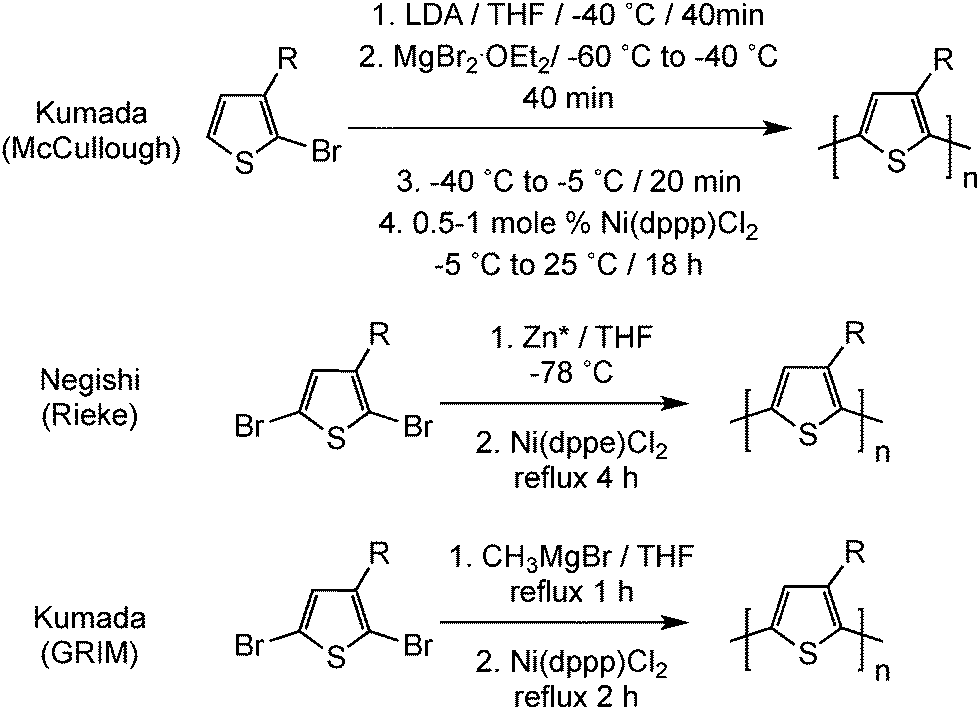 | ||
| Scheme 4 Synthesis of processable regioregular poly(3-alkylthiophene)s (Zn* = Rieke zinc). | ||
Rapidly, the focus shifted from the synthesis of highly conducting polymers to the design of stable semiconducting polymers with tunable electronic and optical properties. This shift was mainly driven by the development of polymeric light-emitting diodes and photovoltaic cells in the 90's. For this purpose, the rigidification of the conjugated backbone, the introduction of electron-withdrawing or electron-donating side groups, and the increase of the quinoid (versus aromatic) character of the main chain have been widely utilized to modulate the bandgap of such conjugated polymers.18 However, one of the most efficient approaches involves the alternation of electron-rich and electron-poor units leading to a push–pull architecture.19
Over the last few years, a third generation of semiconducting copolymers was therefore developed to satisfy the needs for these electronic applications.20 Among them, a large number of conjugated alternating copolymers based on phenylenes, fluorenes, carbazoles, and thiophenes were synthesized. There are relatively few synthetic methods allowing an efficient preparation of such alternating copolymers; most of these copolymers are obtained via the well-known Suzuki21,22 or Stille23,24 cross-coupling polymerization reactions (Scheme 5). We should mention the importance of these Pd-catalyzed cross-coupling reactions for the formation of a carbon–carbon single bond allowing the polymerization of conjugated units. Professors R. F. Heck, E.-i. Negishi and A. Suzuki shared the 2010 Nobel Prize in Chemistry for the development of those synthetic tools.25–27 However, these state-of-the-art methods generally involve numerous synthetic steps and organometallic reagents that give rise to metal waste and various by-products. Low cost, environmentally friendly, and more efficient synthetic procedures would therefore be a great asset to the sustainable preparation of affordable and efficient conjugated polymers.
 | ||
| Scheme 5 Typical polymerization reactions for conjugated alternating push–pull copolymers. | ||
4 Towards greener and cheaper polymerization methods
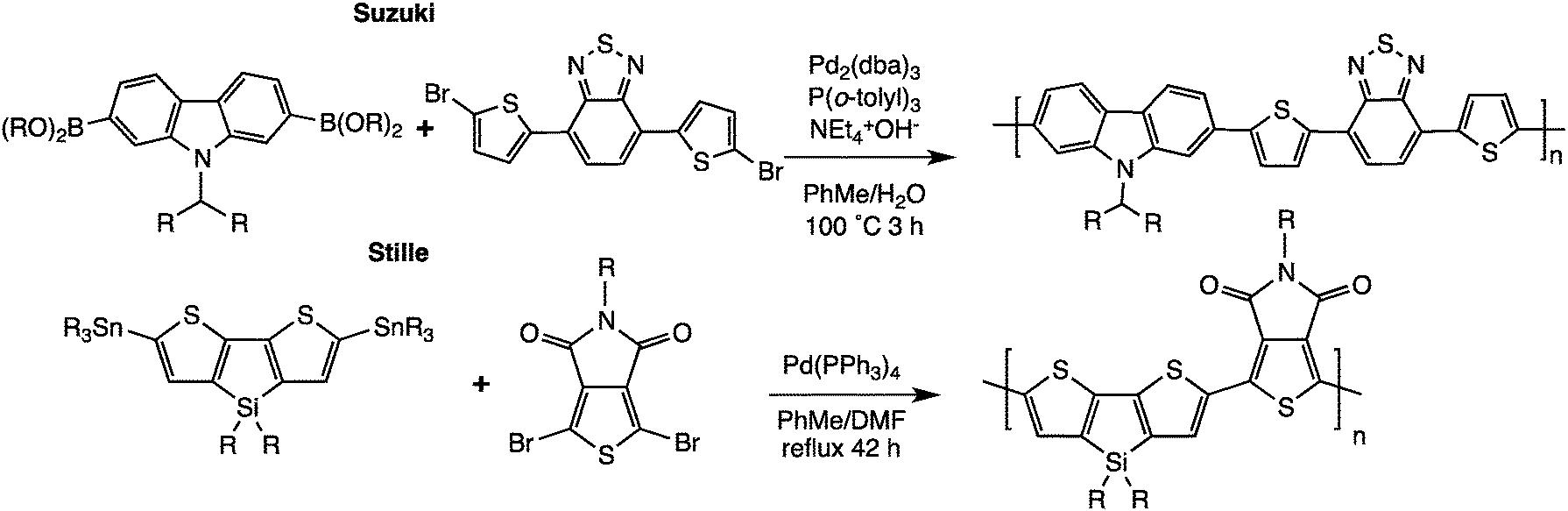
As mentioned above, simple, reproducible, environmentally benign, and low-cost polymerization methods can be seen as the Holy-Grail for plastic electronics. Interestingly, metal-catalyzed direct arylation of aromatic compounds has been recently developed. When first used for the synthesis of pharmaceutical small molecules,28 this reaction exhibits several key benefits: fewer reaction steps, easier purification and only acidic by-products. Indeed, as described in Scheme 6, these new reactions allow the formation of carbon–carbon bonds between (hetero)arenes and (hetero)aryl halides, which do not require organometallic intermediates, thereby significantly reducing both synthetic steps and cost. From an industrial perspective, these advantages can be particularly important for the large-scale development of plastic electronics.29 This new technique has already demonstrated the successful incorporation of materials into highly performing polymeric solar cells30–32 and field-effect transistors.33–35 This novel polymerization method has also made possible the preparation of well-defined polymers for electrochromic windows,36 chemical sensors,37 memory devices,38 and gas storage39 that would have been difficult to obtain using traditional polymerization methods.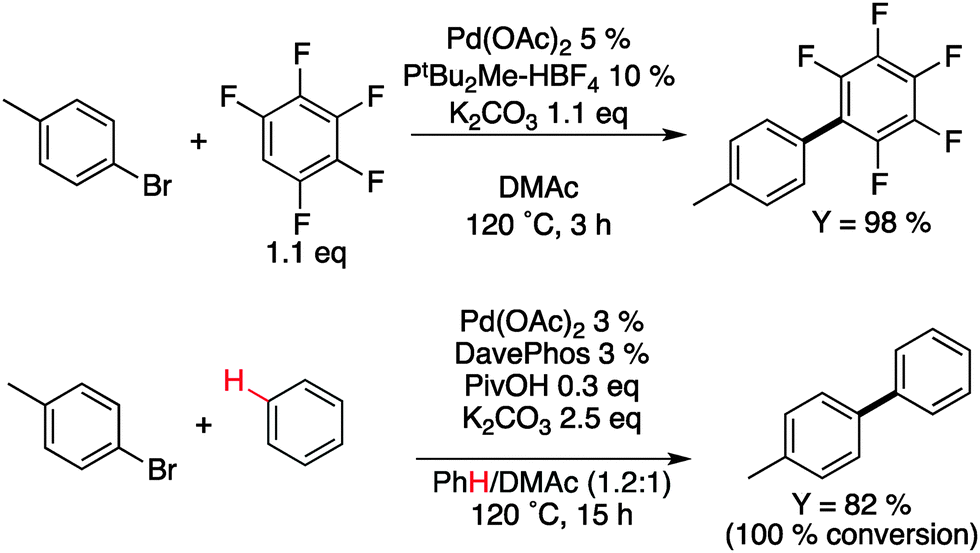 | ||
| Scheme 6 Examples of direct arylation on benzene.40,41 | ||
The first example of a direct (hetero)arylation polymerisation (DHAP) was reported in 1999 by Lemaire and collaborators.42 They polymerized 2-iodo-3-octylthiophene using Heck-type conditions to obtain poly(3-octylthiophene) with a number-average molecular weight of 3 kDa and a regioregularity of 90% (Scheme 7).43
 | ||
| Scheme 7 Poly(3-octylthiophene) polymerized by DHAP. | ||
Although this approach was innovative, the low molecular weight and relatively poor regioregularity limited the interest of the polymer chemists. A decade later, helped by a better understanding of the mechanisms involved in direct arylation reactions40,41,44–47 the group of Ozawa successfully obtained P3HT with molecular weight and regioregularity comparable to those obtained using either McCullough, Rieke or GRIM methods (Mn = 31 kDa, rr > 98%) (Scheme 8).48
 | ||
| Scheme 8 Poly(3-hexylthiophene) obtained by DHAP. | ||
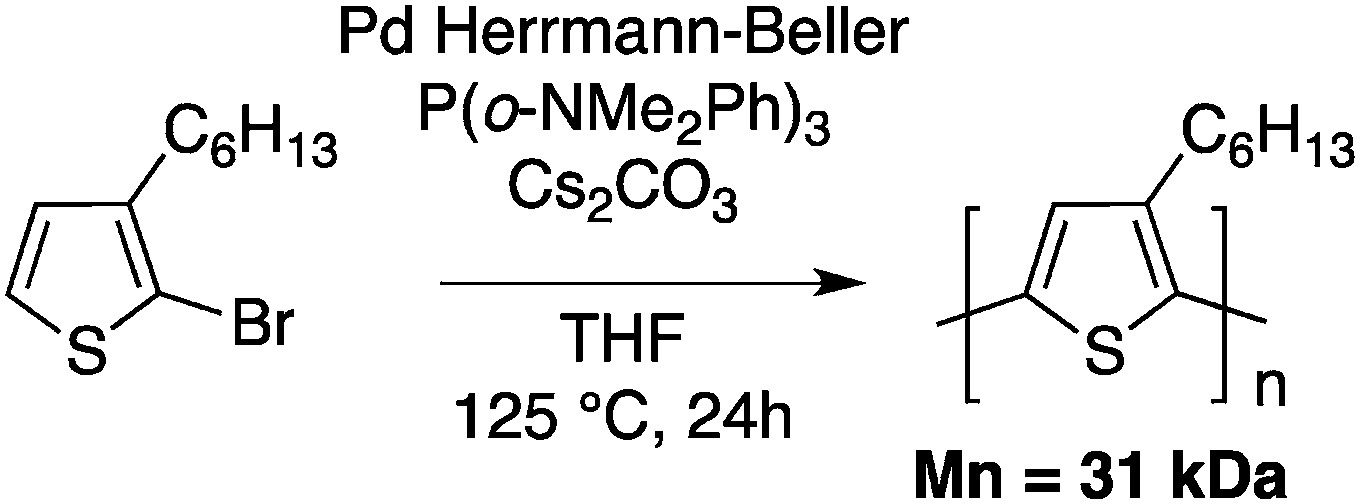
Few months prior to the work reported by Ozawa, Kumar and Kumar reported the synthesis of conjugated copolymers based on 3,4-alkylenedioxythiophenes (ProDOT/EDOT) using, what was called at that time, a single step reductive polymerization catalyzed by palladium acetate (Scheme 9).49
 | ||
| Scheme 9 Poly(3,4-alkylenedioxythiophene) obtained by DHAP. | ||
The main goal of Kumar and Kumar was the development of a new polymerization method, which is economically viable, inert to the presence of functional groups and uses less stringent polymerization conditions (compared to Grignard metathesis (GRIM), for example). Few months later, Kanbara and co-workers50 reported a push–pull copolymer made of 2,7-dibromofluorene and tetrafluorobenzene (Scheme 10).
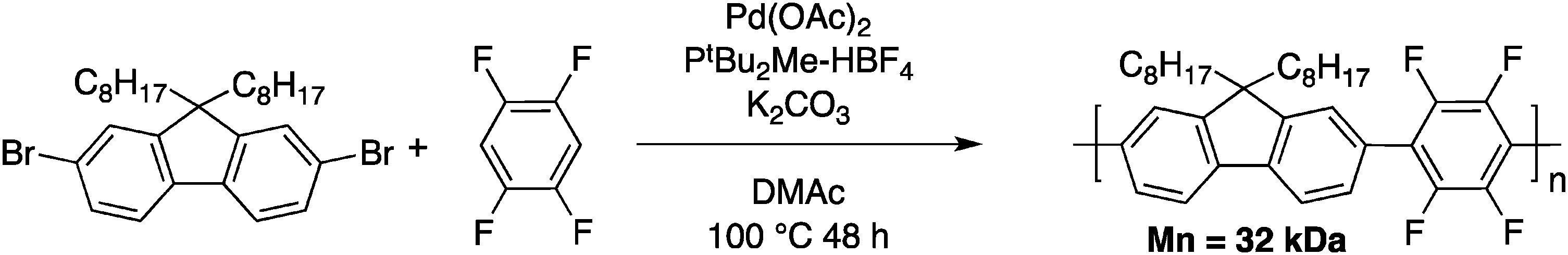 | ||
| Scheme 10 Poly(2,7-fluorene-alt-tetrafluorobenzene) obtained by DHAP. | ||
Tetrafluorobenzene was chosen as a model compound since the acidic C–H bonds, owing to the fluorine substituents, enable direct arylation with good selectivity and yields.40,51 Upon optimization of the DHAP conditions (ligand, solvent, concentration and reaction time), they successfully obtained a high molecular weight polymer (Mn = 32 kDa). They also investigated these newly developed DHAP conditions for the synthesis of 3,6-carbazole-alt-tetrafluorobenzene. Moderate molecular weights were obtained due to limited solubility of both comonomers. More importantly, Kanbara and co-workers underlined the fact that unfavourable coupling reactions can occur with DHAP. Indeed, using model compounds, they found that direct heteroarylation polymerization can lead to unexpected C–H bond activation, resulting in branched structures.50 Soon after, Leclerc and coworkers published a copolymer made from a bithiophene and a thieno[3,4-c]pyrrole-4,6-dione (TPD) derivative (Scheme 11).52
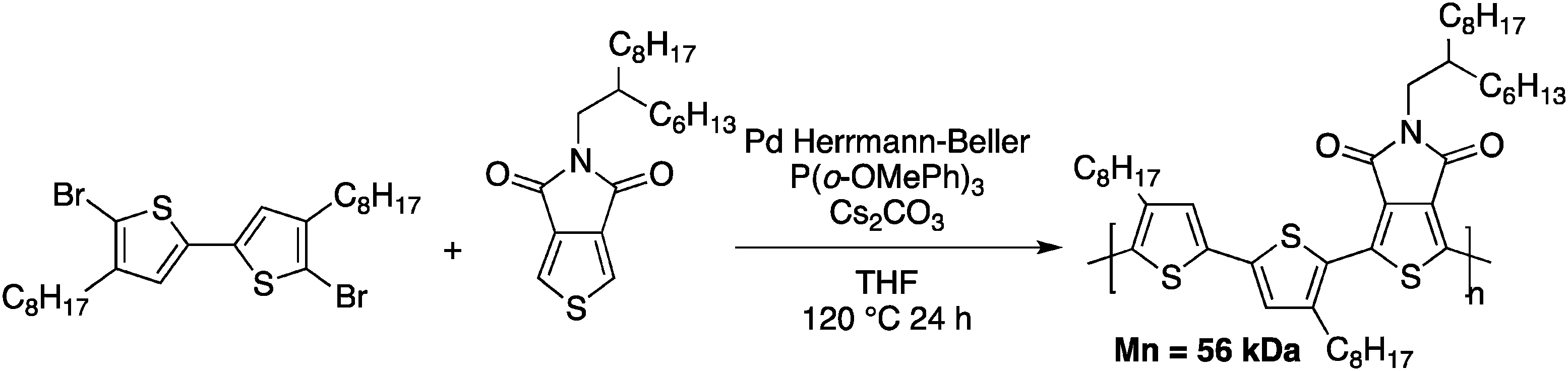 | ||
| Scheme 11 Poly(bithiophene-alt-thienopyrroledione) obtained by DHAP. | ||
TPD-based copolymers are usually obtained using Stille cross-coupling reactions, which utilize thermally unstable organometallic reagents and produce stoichiometric amounts of toxic tin by-products. It is worth noting that TPD-based copolymers are interesting polymeric materials for organic electronic devices such as plastic solar cells and field-effect transistors.53–57
This TPD-based copolymer obtained by DHAP exhibits a higher molecular weight than its analog obtained using Stille cross-coupling polymerization (Mn = 56 kDa vs. Mn = 9 kDa) with comparable, if not slightly better physical properties. These results have catalyzed the interest in direct heteroarylation polymerization. Indeed, since 2012, more than 50 papers related to direct heteroarylation polymerization have been published and have been the subject of at least five reviews.58–62
These first studies were based on two general polymerization methods. The first method is directly inspired by the work done on small molecules where the reaction takes place in polar solvents with good coordination ability (e.g. DMAc, DMF, HMPA, and NMP), combined with a base (M2CO3) and the utilization of an additive (proton shuttle; carboxylic acid). Solvents with good coordination capability are beneficial since the DHAP can proceed without phosphine ligands which can be a source of impurities.32,63 Indeed, it has been reported that a small amount of phosphine can be introduced into the polymer backbone during polymerization reaction.64 The second approach for direct heteroarylation polymerization is based on the utilization of less polar solvents (e.g. tetrahydrofurane, toluene, and dioxane) which promote polymer solubility and often lead to high molecular weights.65 To increase the reactivity of the catalyst in those solvents, a phosphine ligand is usually needed (e.g. P(o-OMePh)3, P(o-NMe2Ph)3, P(Cy)3, and P(t-Bu)3).
However, the main drawback of this new polymerization method was the lack of selectivity with monomers having multiple available protons which can lead to structural defects (branching and cross-linking). Since such defects in a polymeric structure cannot be removed by purification, a better understanding of the possible side reactions became an important subject of the study. These structural defects change the electrical and optical properties of the resulting polymers and eventually lead to a loss of solubility due to cross-linking as reported by Kanbara and co-workers.50
5 Towards defect-free conjugated polymers
While a minimum of defects might be tolerable,66,67 their elimination is clearly the final goal. The first strategy used to prevent such unwanted side-reactions was the design of monomers bearing blocking groups.68 For instance, this first approach has been used on substrates like 3,4-disubstituted hetero(arene) derivatives (Scheme 12). More precisely, different copolymers and homopolymers have been made using 3,4-ethylenedioxythiophene (EDOT),69 3,4-propylenedioxythiophene (ProDOT),63 thieno[3,4-c]pyrrole-4,6-dione (TPD),70,71 dialkyl-bithiazole,72,73 3,4-dicyanothiophene70 and 3,3′,4,4′-tetramethyl-2,2′-bithiophene68,74 derivatives. For example, TPD monomers have been polymerized by Berrouard and co-workers who demonstrated that by using DHAP they could obtain new polymers with a better conjugation length compared to previous polymerization methods.75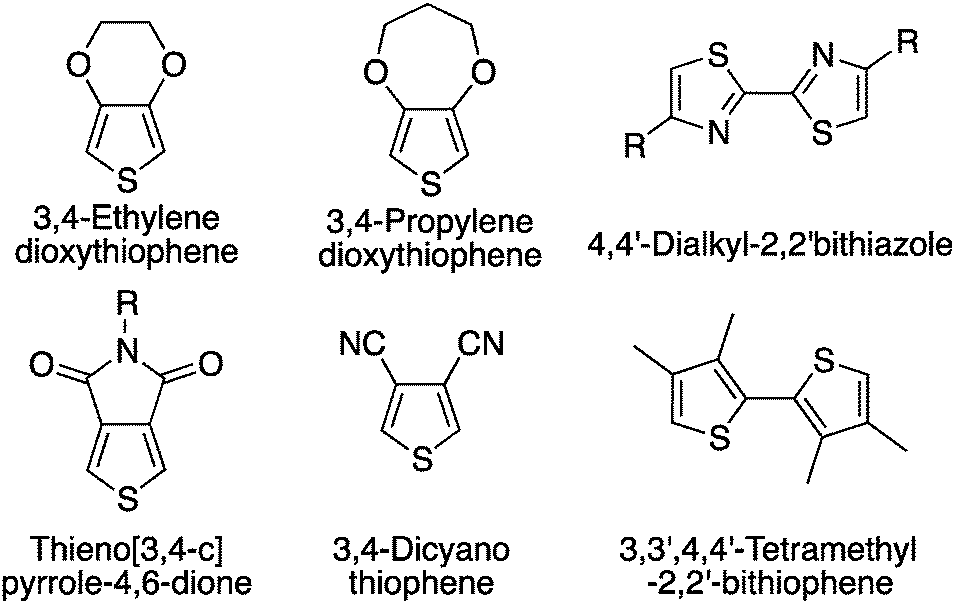 | ||
| Scheme 12 Protected (hetero)arene monomers used in DHAP. | ||
Since then, the scientific community has not only revisited the synthesis by DHAP of the known conjugated polymers (obtained by classical polymerization methods) but has also designed new polymers with β-blocking groups. For example, bithiophene and terthiophene based polymers with β-substituted thiophene (n-octyl or n-dodecyl chain; Scheme 13) have allowed the preparation of well-defined high molecular weight materials.52 In this series, the alkyl chains act as effective blocking groups and one can think that steric hindrance coming from the alkyl chains limits the reactivity of the remaining protons on the thiophene derivatives.30
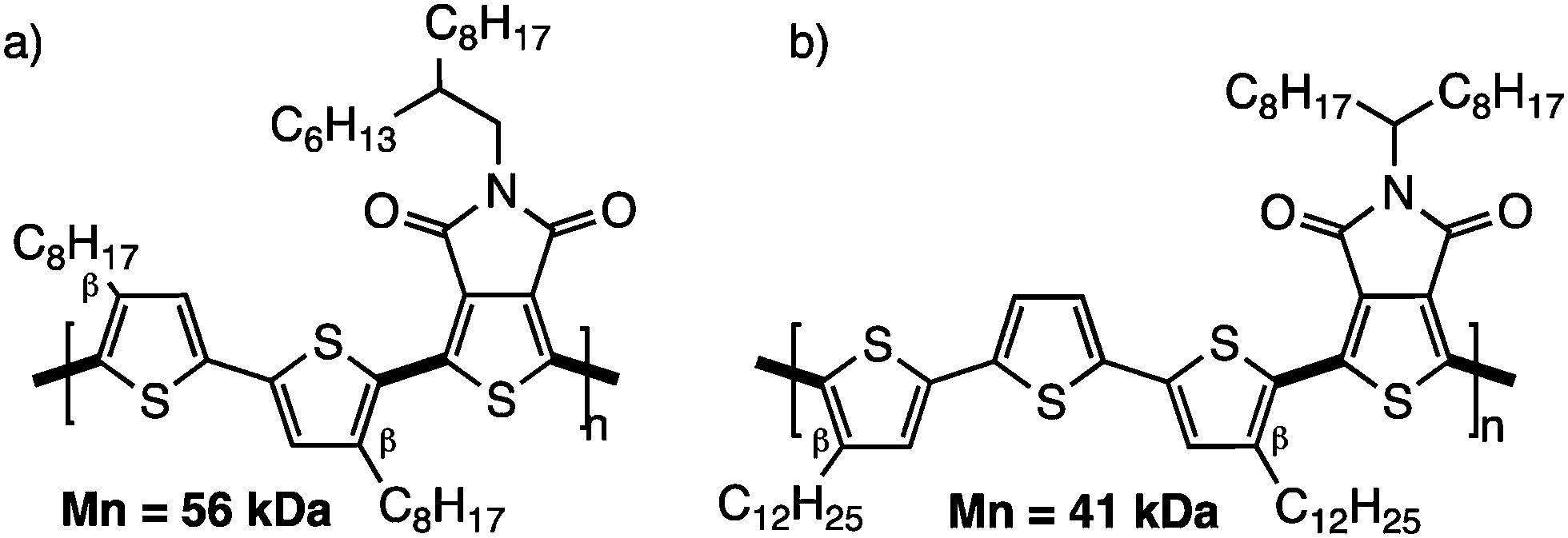 | ||
| Scheme 13 4,4′-Dioctylbithiophene (a) and 4,4′′-didodecylterthiophene (b) copolymerized with a thienopyrroledione. | ||
Unfortunately, in some cases, the β-blocking strategy can have a dramatic effect on the properties of the resulting material. Indeed, Leclerc et al. have revisited the synthesis of poly(dithienosilole-alt-thienopyrroledione) (P(DTS-TPD), Scheme 14a; an efficient polymer used in polymeric solar cells55,57) by using DHAP. Suspecting selectivity issues, they modified the chemical structure of dithienosilole by adding alkyl chains at the β-positions. This strategy led to new well defined polymers with β-protected positions (Scheme 14b) but with poor electro-optical properties. Indeed, a higher bandgap attributed to a significant torsion angle between units, lower mobilities and poor packing was observed for the new β-protected dithienosilole-based copolymers.76
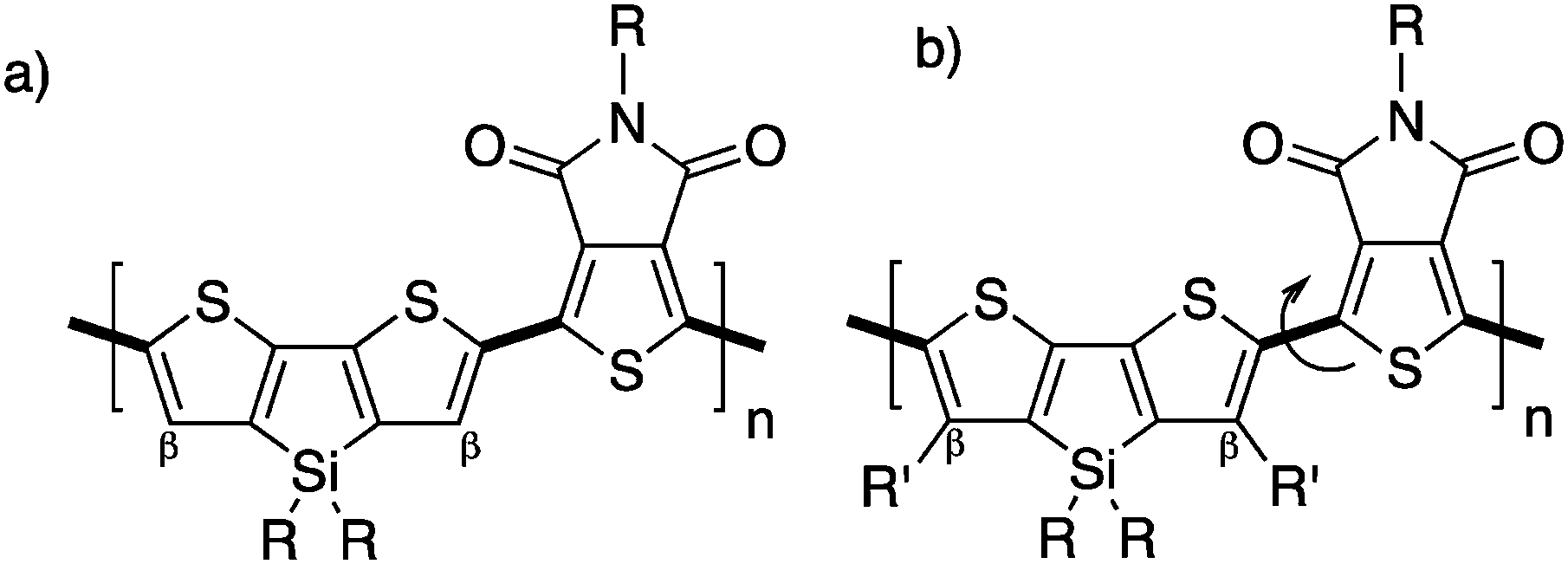 | ||
| Scheme 14 Copolymers of (a) dithienosilole-alt-thienopyrroledione and (b) β-protected dithienosilole-alt-thienopyrroledione. | ||
In parallel, Sommer et al.77 have investigated the direct heteroarylation polymerization between 2,7-dibromo-9-(N-heptadecanyl)-carbazole and 4,7-bis(4-hexyl-2-thienyl)-2,1,3-benzothiadiazole, focusing on the side reactions as a function of polymerization conditions (Scheme 15). They chose this system because both monomers exhibit three different C–H bonds. Moreover, the targeted copolymer is an analog of PCDTBT, a well-known, efficient, and stable material for polymeric solar cells.78 Upon careful 1H NMR analyses, they concluded that homocoupling was the main source of defects and did not find any evidence of β-branching. They also suggested that the shape of the size exclusion chromatography (SEC) curve could be a simple indicator of homocoupling.
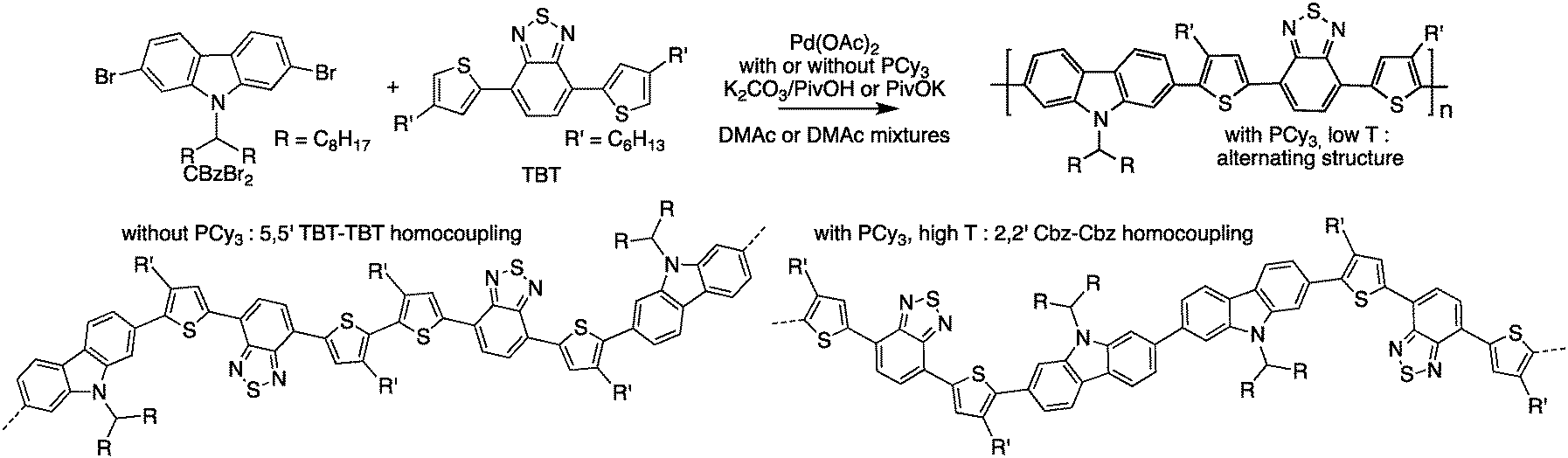 | ||
| Scheme 15 Polymerization of 2,7-dibromo-9-(N-heptadecanyl)-carbazole and 4,7-bis(4-hexyl-2-thienyl)-2,1,3-benzothiadiazole and homocoupling defects. | ||
Despite the fact that α–β selectivity and homocoupling have been observed by different research groups,48,67,68 we have recently reported that some time-controlled DHAP reactions can yield well-defined and processable polymers.79 Indeed, by using an appropriate catalytic system and particularly a close monitoring of polymerization time, various bromo(arene)s and unprotected thiophene derivatives have been successfully polymerized without any evidence of unwanted side reactions (branching, crosslinking and homocoupling) (Scheme 16). More precisely, bithiophene, dithienyl-difluorobenzothiadiazole and diketopyrropyrole derivatives were successfully polymerized by DHAP with 2,7-dibromofluorenes, 2,7-dibromocarbazoles and 1,4-dibromobenzenes.
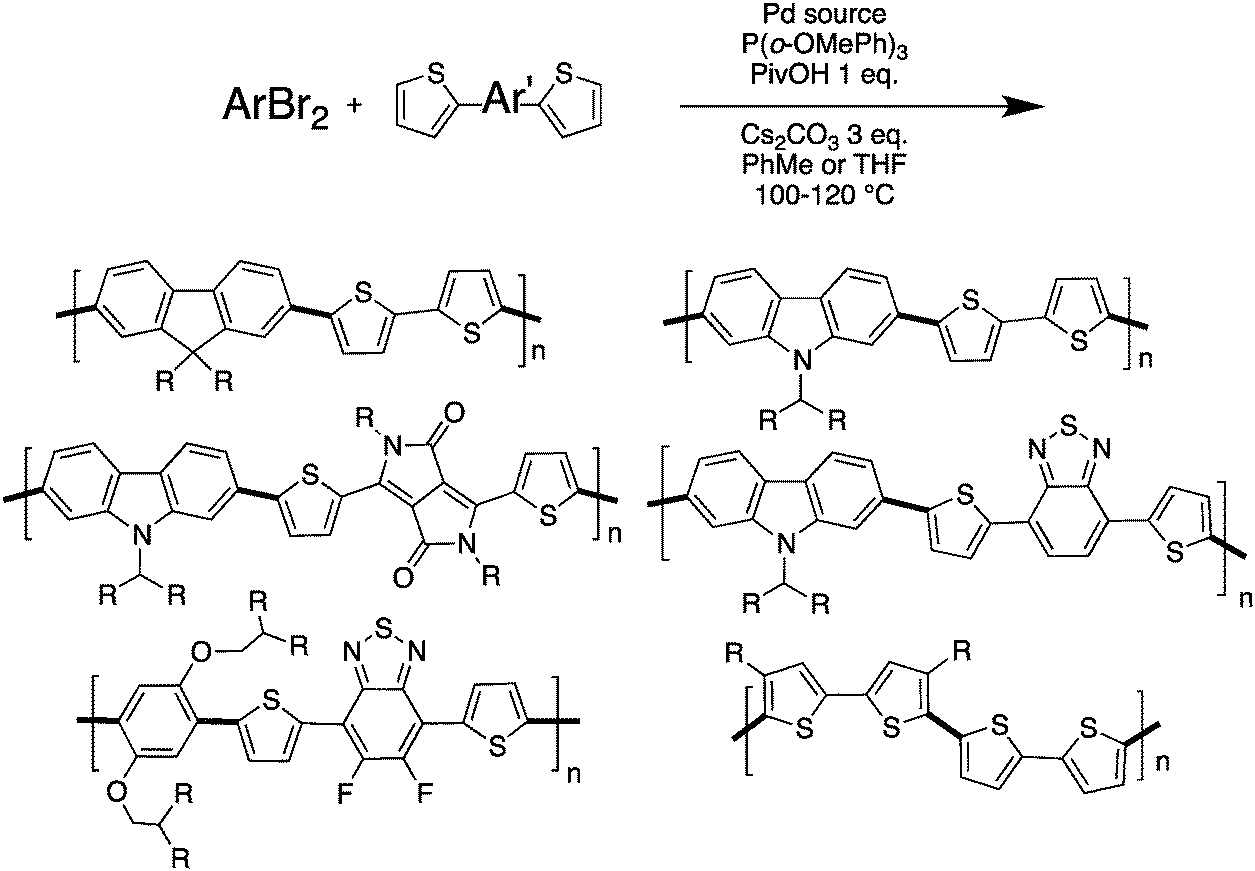 | ||
| Scheme 16 (Hetero)arene units bearing unprotected thiophene with α–β protons successfully polymerized by DHAP.79 | ||
While in this work we were able to polymerize protected bromo-thiophenes (Scheme 17a), these sets of conditions did not allow the synthesis of polymers from unprotected bromo-thiophenes (Scheme 17b). Most common procedures DHAP with unprotected bromo-thiophenes always led to polymers having a higher bandgap than polymers obtained using classical methods, which means that defects are probably introduced into the conjugated backbone.
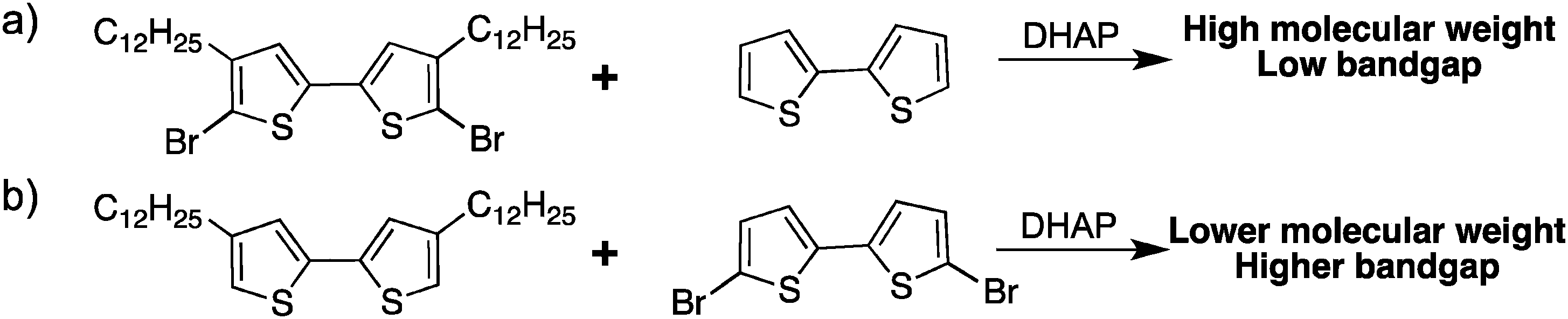 | ||
| Scheme 17 Influence of the brominated monomer with DHAP. | ||
In parallel, while studying β-branching in P3HT, Rudenko and coworkers have found that additives can also control the α–β selectivity.80 Indeed, neodecanoic acid (NDA) efficiently suppressed the β-branching often observed during the polymerization of thiophene derivatives. Based on these promising results, we have successfully polymerized unprotected bromo-thiophenes. Indeed, these new polymerization conditions allowed the synthesis of poly(quaterthiophene) with properties better than oxidative coupling and equal or similar to those obtained from Stille coupling.81 We also highlighted the importance of increasing the steric hindrance for the polymerization of unprotected bromo-thiophene to prevent β-branching (Scheme 18). The steric hindrance could either come from an alkyl in the γ position (a) or by adding the NDA as a bulky additive (b).
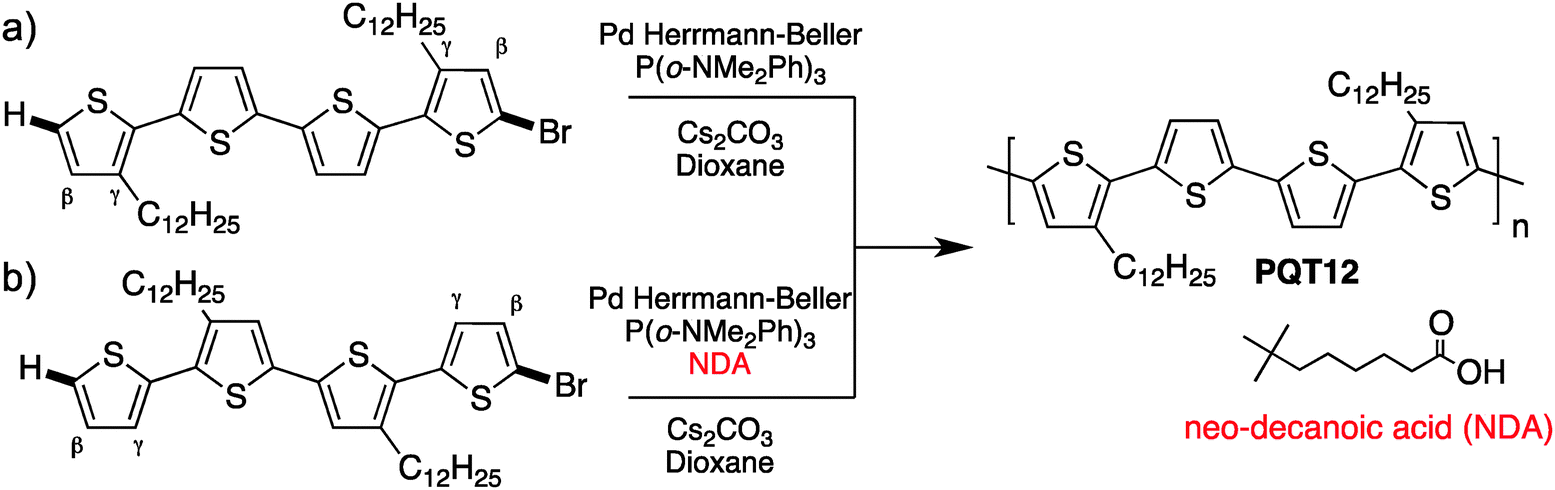 | ||
| Scheme 18 Polymerization of poly(quaterthiophene) from two different monomers. | ||
The scope of DHAP has therefore been expanded since its introduction. First, DHAP has been performed using bromo(arene)s or β-protected bromo-hetero(arene)s with β-blocked comonomers (Scheme 19a). Subsequently, DHAP has been successfully applied to bromo(arene)s or β-protected bromo-hetero(arene)s with unprotected comonomers (Scheme 19b). Finally, monomers made of unprotected bromo-thiophenes (Scheme 19c) have been polymerized but they still represent a challenge.
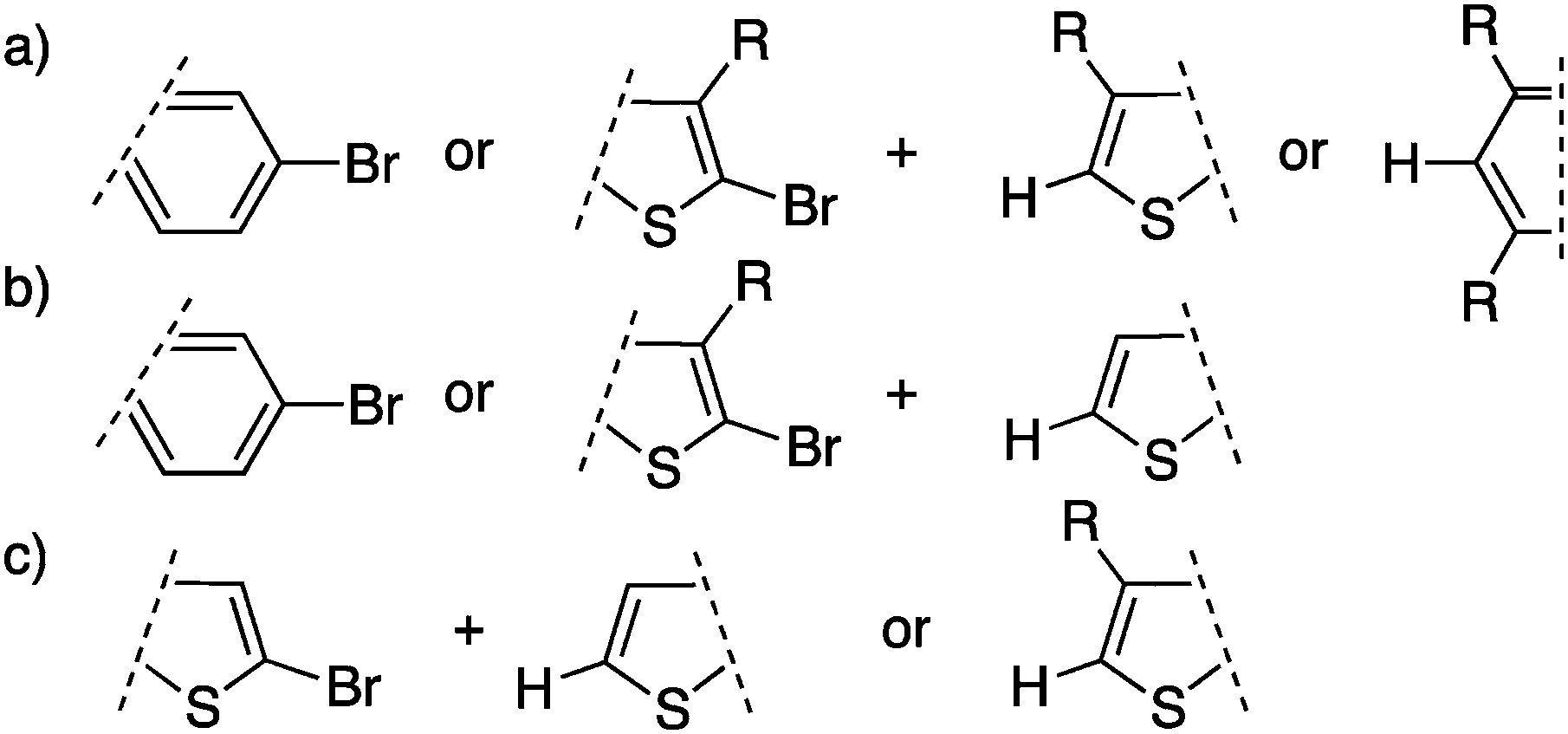 | ||
| Scheme 19 Different types of cross-coupling reactions. | ||
In particular and as discussed earlier, homocoupling (which most probably results from dehalogenation) can prevent the synthesis of defect-free polymers. This phenomenon is not unique to DHAP and has been observed before for typical palladium-catalyzed polymerization methods.82 However, due to the nature of its catalytic system, an undesired dehalogenation process is more likely to occur with DHAP.41 Additionally, this issue is not the same in all cases, and depends on the type of monomer bearing the bromine atoms (Scheme 20). If dehalogenation takes place on a bromo(arene), the dehalogenated product should not be reactive enough to continue the polymerization. In the other cases, particularly with unprotected bromo-thiophenes, the dehalogenated product becomes active, leading to homocoupling reactions.
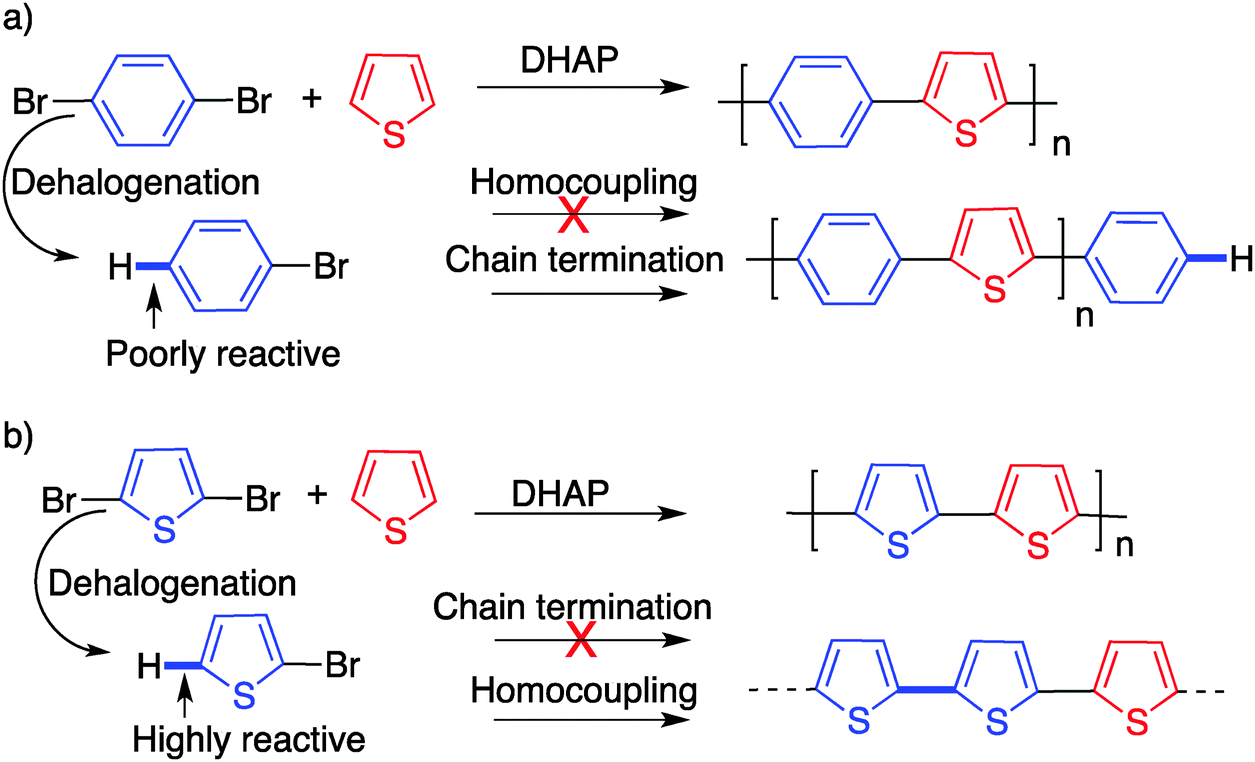 | ||
| Scheme 20 Dehalogenation and the possible defect occurring with a bromo arene (a) and with a bromo (hetero)arene (b). | ||
In order to prevent dehalogenated-induced side-reactions (particularly for thiophene-based materials), optimized catalytic systems should be developed. Up to now, selectivity issues and homocoupling have been addressed for many electro-rich moieties; however, polymerization of electron poor unprotected halogenated-thiophenes is still problematic. One can think that the use of a bulky electron rich ligand (usually used in Pd assisted polymerization) should be useful to prevent this undesired side-reaction. Moreover, to limit or prevent the dehalogenation process, the halogen should be placed on the more electron-rich moiety unlike other palladium assisted cross-coupling reactions such as Stille and Suzuki.79,83
6 Outlook
From the research performed up to now, DHAP can be much more selective than first anticipated. Careful NMR and physical analyses have revealed polymeric materials that are comparable if not better, to those obtained from conventional cross-coupling procedures. Even on substrates having different C–H bonds, new catalytic systems with additives, ligands and Pd precatalysts have solved some selectivity issues.79,84,85 For instance, well defined copolymers such as poly(dithienosilole-alt-thienopyrroledione) P(DTS-TPD),84 poly(cyclopentadithiophene-alt-benzothiadiazole) P(CPDT-BT)86 and poly(naphthalenebisdicarboximide-alt-bithiophene) P(NDI-2T)35 can now be obtained (Scheme 21).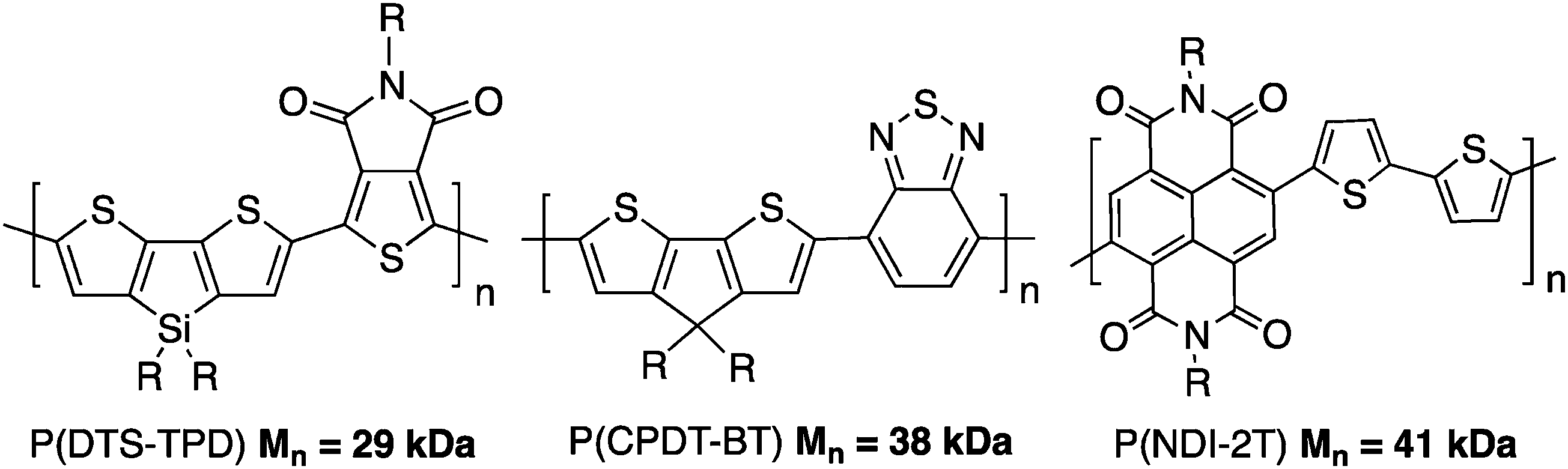 | ||
| Scheme 21 Three well known polymers obtained by DHAP. | ||
However, the polymerization seems to be substrate dependent and it is not possible to predict with accuracy which system will work on a new set of monomers. During the next few years, we expect this community to continue to expand the scope of DHAP by finding novel conditions to achieve high degrees of polymerization and good selectivity. Optimization is a multi-parameter process where the nature and amount of metallic source, ligand, additive, base and variables like solvent, concentration, temperature and time need to be taken into consideration. Nevertheless, when successfully applied, DHAP will always provide simple, low-cost, and environmentally benign syntheses of conjugated polymers. Furthermore, recent advances have been made vis-à-vis quasi-living polymerization of conjugated polymers via Pd-catalyzed coupling.87–89 Such tools have gained interest for the preparation of well defined polymers with low polydispersity or block copolymers. In the future, it will be interesting to apply these strategies to the DHAP system to expand the scope of feasible architectures. Finally, it is interesting to note that modern synthetic tools such as continuous-flow synthesis can also applied to DHAP.90 All these synthetic advances should help paving the way for the future implementation of plastic electronics.
Funding sources
No competing financial interests have been declared.Acknowledgements
This work was supported by both discovery and strategic grants from NSERC. M.L. thanks the Killam Foundation for a fellowship.References
- D. J. Berets and D. S. Smith, Trans. Faraday Soc., 1968, 64, 823–828 RSC.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- H. Shirakawa, Angew. Chem., Int. Ed., 2001, 40, 2574–2580 CrossRef.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581–2590 CrossRef CAS.
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591–2611 CrossRef CAS.
- G. Tourillon and F. Garnier, J. Electroanal. Chem., 1982, 135, 173–178 CrossRef CAS.
- A. F. Diaz, K. K. Kanazawa and G. P. Gardini, J. Chem. Soc., Chem. Commun., 1979, 635–636 RSC.
- A. F. Diaz and J. A. Logan, J. Electroanal. Chem., 1980, 111, 111–114 CrossRef CAS.
- J. H. Edwards and W. J. Feast, Polymer, 1980, 21, 595–596 CrossRef CAS.
- K.-Y. Jen, G. G. Miller and R. L. Elsenbaumer, J. Chem. Soc., Chem. Commun., 1986, 1346–1347 RSC.
- M. Sato, S. Tanaka and K. Kaeriyama, J. Chem. Soc., Chem. Commun., 1986, 873–874 RSC.
- S. Hotta, S. D. D. V. Rughooputh, A. J. Heeger and F. Wudl, Macromolecules, 1987, 20, 212–215 CrossRef CAS.
- M. Leclerc, F. M. Diaz and G. Wegner, Makromol. Chem., 1989, 190, 3105–3116 CrossRef CAS.
- S. Kirchmeyer and K. Reuter, J. Mater. Chem., 2005, 15, 2077–2088 RSC.
- R. D. McCullough and R. D. Lowe, J. Chem. Soc., Chem. Commun., 1992, 70–72 RSC.
- T. A. Chen and R. D. Rieke, J. Am. Chem. Soc., 1992, 114, 10087–10088 CrossRef CAS.
- R. S. Loewe, S. M. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250–253 CrossRef CAS.
- J. Roncali, Chem. Rev., 1997, 97, 173–206 CrossRef CAS PubMed.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Polym. Bull., 1992, 29, 119–126 CrossRef CAS.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- M. Rehahn, A.-D. Schlüter, G. Wegner and W. J. Feast, Polymer, 1989, 30, 1060–1062 CrossRef CAS.
- J. Sakamoto, M. Rehahn, G. Wegner and A. D. Schlüter, Macromol. Rapid Commun., 2009, 30, 653–687 CrossRef CAS PubMed.
- Z. Bao, W. Chan and L. Yu, Chem. Mater., 1993, 5, 2–3 CrossRef CAS.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- E.-i. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
- R. Po, A. Bernardi, A. Calabrese, C. Carbonera, G. Corso and A. Pellegrino, Energy Environ. Sci., 2014, 7, 925–943 CAS.
- J. Jo, A. Pron, P. Berrouard, W. L. Leong, J. D. Yuen, J. S. Moon, M. Leclerc and A. J. Heeger, Adv. Energy Mater., 2012, 2, 1397–1403 CrossRef CAS.
- D. H. Wang, A. Pron, M. Leclerc and A. J. Heeger, Adv. Funct. Mater., 2013, 23, 1297–1304 CrossRef CAS.
- J. Kuwabara, T. Yasuda, S. J. Choi, W. Lu, K. Yamazaki, S. Kagaya, L. Han and T. Kanbara, Adv. Funct. Mater., 2014, 24, 3226–3233 CrossRef CAS.
- A. Luzio, D. Fazzi, F. Nübling, R. Matsidik, A. Straub, H. Komber, E. Giussani, S. E. Watkins, M. Barbatti, W. Thiel, E. Gann, L. Thomsen, C. R. McNeill, M. Caironi and M. Sommer, Chem. Mater., 2014, 26, 6233–6240 CrossRef CAS.
- J.-R. Pouliot, B. Sun, M. Leduc, A. Najari, Y. Li and M. Leclerc, Polym. Chem., 2015, 6, 278–282 RSC.
- R. Matsidik, H. Komber, A. Luzio, M. Caironi and M. Sommer, J. Am. Chem. Soc., 2015, 137, 6705–6711 CrossRef CAS PubMed.
- J. A. Kerszulis, K. E. Johnson, M. Kuepfert, D. Khoshabo, A. L. Dyer and J. R. Reynolds, J. Mater. Chem. C, 2015, 3, 3211–3218 RSC.
- J. G. Weis and T. M. Swager, ACS Macro Lett., 2015, 4, 138–142 CrossRef CAS.
- W. Elsawy, M. Son, J. Jang, M. J. Kim, Y. Ji, T.-W. Kim, H. C. Ko, A. Elbarbary, M.-H. Ham and J.-S. Lee, ACS Macro Lett., 2015, 4, 322–326 CrossRef CAS.
- D.-P. Liu, Q. Chen, Y.-C. Zhao, L.-M. Zhang, A.-D. Qi and B.-H. Han, ACS Macro Lett., 2013, 2, 522–526 CrossRef CAS.
- M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS PubMed.
- M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed.
- M. Sévignon, J. Papillon, E. Schulz and M. Lemaire, Tetrahedron Lett., 1999, 40, 5873–5876 CrossRef.
- J. Hassan, E. Schulz, C. Gozzi and M. Lemaire, J. Mol. Catal. A: Chem., 2003, 195, 125–131 CrossRef CAS.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
- B. Liégault, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826–1834 CrossRef PubMed.
- A. Battace, M. Lemhadri, T. Zair, H. Doucet and M. Santelli, Adv. Synth. Catal., 2007, 349, 2507–2516 CrossRef CAS.
- A. Battace, M. Lemhadri, T. Zair, H. Doucet and M. Santelli, Organometallics, 2007, 26, 472–474 CrossRef CAS.
- Q. Wang, R. Takita, Y. Kikuzaki and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 11420–11421 CrossRef CAS PubMed.
- A. Kumar and A. Kumar, Polym. Chem., 2010, 1, 286–288 RSC.
- W. Lu, J. Kuwabara and T. Kanbara, Macromolecules, 2011, 44, 1252–1255 CrossRef CAS.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2011, 77, 658–668 CrossRef PubMed.
- P. Berrouard, A. Najari, A. Pron, D. Gendron, P.-O. Morin, J.-R. Pouliot, J. Veilleux and M. Leclerc, Angew. Chem., Int. Ed., 2012, 51, 2068–2071 CrossRef CAS PubMed.
- Y. Zou, A. Najari, P. Berrouard, S. Beaupré, B. Réda Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS PubMed.
- X. Guo, R. P. Ortiz, Y. Zheng, M.-G. Kim, S. Zhang, Y. Hu, G. Lu, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 13685–13697 CrossRef CAS PubMed.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed.
- C. Cabanetos, A. El Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. J. Fréchet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656–4659 CrossRef CAS PubMed.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S.-W. Tsang, T.-H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115–120 CrossRef CAS.
- A. Facchetti, L. Vaccaro and A. Marrocchi, Angew. Chem., Int. Ed., 2012, 51, 3520–3523 CrossRef CAS PubMed.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed.
- K. Okamoto, J. Zhang, J. B. Housekeeper, S. R. Marder and C. K. Luscombe, Macromolecules, 2013, 46, 8059–8078 CrossRef CAS.
- K. Wang and M. Wang, Curr. Org. Chem., 2013, 17, 999–1012 CrossRef CAS.
- S. Kowalski, S. Allard, K. Zilberberg, T. Riedl and U. Scherf, Prog. Polym. Sci., 2013, 38, 1805–1814 CrossRef CAS.
- L. A. Estrada, J. J. Deininger, G. D. Kamenov and J. R. Reynolds, ACS Macro Lett., 2013, 2, 869–873 CrossRef CAS.
- S. A. Macgregor, Chem. Soc. Rev., 2007, 36, 67–76 RSC.
- M. Wakioka, Y. Kitano and F. Ozawa, Macromolecules, 2013, 46, 370–374 CrossRef CAS.
- S.-W. Chang, H. Waters, J. Kettle, Z.-R. Kuo, C.-H. Li, C.-Y. Yu and M. Horie, Macromol. Rapid Commun., 2012, 33, 1927–1932 CrossRef CAS PubMed.
- A. E. Rudenko, A. A. Latif and B. C. Thompson, Nanotechnology, 2014, 25, 014005 CrossRef PubMed.
- Y. Fujinami, J. Kuwabara, W. Lu, H. Hayashi and T. Kanbara, ACS Macro Lett., 2012, 1, 67–70 CrossRef CAS.
- H. Zhao, C.-Y. Liu, S.-C. Luo, B. Zhu, T.-H. Wang, H.-F. Hsu and H.-h. Yu, Macromolecules, 2012, 45, 7783–7790 CrossRef CAS.
- J.-R. Pouliot, L. G. Mercier, S. Caron and M. Leclerc, Macromol. Chem. Phys., 2013, 214, 453–457 CrossRef CAS.
- F. Grenier, P. Berrouard, J.-R. Pouliot, H.-R. Tseng, A. J. Heeger and M. Leclerc, Polym. Chem., 2013, 4, 1836–1841 RSC.
- W. Lu, J. Kuwabara and T. Kanbara, Polym. Chem., 2012, 3, 3217–3219 RSC.
- M. Kuramochi, J. Kuwabara, W. Lu and T. Kanbara, Macromolecules, 2014, 47, 7378–7385 CrossRef CAS.
- J. Kuwabara, Y. Nohara, S. J. Choi, Y. Fujinami, W. Lu, K. Yoshimura, J. Oguma, K. Suenobu and T. Kanbara, Polym. Chem., 2013, 4, 947–953 RSC.
- P. Berrouard, S. Dufresne, A. Pron, J. Veilleux and M. Leclerc, J. Org. Chem., 2012, 77, 8167–8173 CrossRef CAS PubMed.
- L. G. Mercier, B. R. Aich, A. Najari, S. Beaupre, P. Berrouard, A. Pron, A. Robitaille, Y. Tao and M. Leclerc, Polym. Chem., 2013, 4, 5252–5260 RSC.
- F. Lombeck, H. Komber, S. I. Gorelsky and M. Sommer, ACS Macro Lett., 2014, 3, 819–823 CrossRef CAS.
- S. Beaupre and M. Leclerc, J. Mater. Chem. A, 2013, 1, 11097–11105 CAS.
- P.-O. Morin, T. Bura, B. Sun, S. I. Gorelsky, Y. Li and M. Leclerc, ACS Macro Lett., 2015, 4, 21–24 CrossRef CAS.
- A. E. Rudenko and B. C. Thompson, Macromolecules, 2015, 48, 569–575 CrossRef CAS.
- T. Bura, P.-O. Morin and M. Leclerc, Macromolecules, 2015, 48, 5614–5620 CrossRef CAS.
- R. C. Chiechi and J. C. Hummelen, ACS Macro Lett., 2012, 1, 1180–1183 CrossRef CAS.
- F. Livi, N. S. Gobalasingham, E. Bundgaard and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2015 DOI:10.1002/pola.27728.
- E. Iizuka, M. Wakioka and F. Ozawa, Macromolecules, 2015, 48, 2989–2993 CrossRef CAS.
- S. Hayashi and T. Koizumi, Polym. Chem., 2015, 6, 5036–5039 RSC.
- S. Kowalski, S. Allard and U. Scherf, Macromol. Rapid Commun., 2015, 36, 1061–1068 CrossRef CAS PubMed.
- T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595–5619 CrossRef CAS PubMed.
- L. Ying, P. Zalar, S. D. Collins, Z. Chen, A. A. Mikhailovsky, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 6496–6501 CrossRef CAS PubMed.
- E. Elmalem, A. Kiriy and W. T. S. Huck, Macromolecules, 2011, 44, 9057–9061 CrossRef CAS.
- F. Grenier, B. R. Aïch, Y.-Y. Lai, M. Guérette, A. B. Holmes, Y. Tao, W. W. H. Wong and M. Leclerc, Chem. Mater., 2015, 27, 2137–2143 CrossRef CAS.
Footnote |
| † This work is dedicated to Professor Dr Gerhard Wegner for his 75th anniversary. |
| This journal is © The Royal Society of Chemistry 2016 |