
Rationalizing the suitability of rhodamines as chromophores in dye-sensitized solar cells: a systematic molecular design study†
Giulio
Pepe
a,
Jacqueline M.
Cole
*abcd,
Paul G.
Waddell‡
ae and
James I.
Perry
a
aCavendish Laboratory, Department of Physics, University of Cambridge, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK. E-mail: jmc61@cam.ac.uk
bISIS Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0QX, UK
cArgonne National Laboratory, 9700 S. Cass Avenue, Argonne, IL 60439, USA
dDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Charles Babbage Road, Cambridge, CB3 0FS, UK
eAustralian Nuclear Science and Technology Organisation, Lucas Heights, NSW 2234, Australia
First published on 30th September 2016
Abstract
Rhodamines are chromophores that are employed in many dye applications. Their strong optical absorption in the visible region of the electromagnetic spectrum renders them attractive dye candidates for dye-sensitized solar cells (DSCs). However, they have not yet been systematically tested in DSCs as single- or co-sensitizers. Recent advances in concerted experimental and computational workflows involving molecular design protocols can afford a better understanding of the molecular origins of the optoelectronic properties in these sensitizers. Herein, we examine the suitability of rhodamines R560 (1), R575 (2), R590 (3), R610 (4), R620 (5), R640 (6), and R3B (7) as chromophores in co-sensitized DSCs. Our study follows a stepwise approach. Initially, structural and optical properties of the dyes are investigated by experimental and computational methods to reveal structure–property relationships and other useful features for DSC applications. Subsequently, 1–7 are investigated at the dye⋯TiO2 interface, both by calculations of dye-adsorption onto the surface of a modeled (TiO2)9 cluster, and by experimental studies of dye-adsorption on TiO2. For that purpose, a selection of rhodamine dyes are paired together, (1 and 5) and (1 and 7), for co-sensitization, among which 5 is also co-adsorbed with a fluorescein dye in order to explore chemical compatibility factors. The best dye candidates are identified from the findings of these adsorption studies in terms of dye aggregation, anchoring modes, and panchromatic response. Despite their promising dye⋯TiO2 adsorption and optical prospects, our results show that rhodamines lack a suitable intramolecular charge transfer pathway for dye-to-TiO2 electron injection to occur, thus precluding their photovoltaic function as DSC dyes. Our results are then assessed against ostensibly disparate reports of rhodamines performing successfully in DSC devices; this comparison necessitated the internal reproduction of previously reported co-sensitization experiments on 2 with the industrial standard reference dye, N3. We achieve reconciliation between our results and those in the literature by reasoning that while rhodamines cannot deliver photovoltaic function in DSCs in their own right, they can either act to facilitate or deplete the photovoltaic output of a DSC indirectly by affecting the TiO2 adsorption prospects of a photovoltaic-active dye with which a rhodamine is co-sensitized.
Design, System, ApplicationDye-sensitized solar cells (DSCs) would highly benefit from a rational design of their dye molecules. This is because DSC function is controlled by the way in which dye molecules absorb light from the sun, and initiate photovoltaic current by injecting electrons into the conduction band of TiO2 whose surface contains adsorbed dye molecules. This dye⋯TiO2 interface represents the DSC working electrode; yet its structure and optical properties are poorly understood for so many dyes, despite being at the core of DSC function. Rhodamine dyes are a case in point. Their use has been demonstrated in co-sensitized DSCs, where multiple dyes adsorb onto TiO2. This experimental and computational study unravels the molecular structures and optical properties of seven rhodamine dyes, and interprets them in terms of DSC molecular design. Considerations focus on dye intramolecular charge transfer pathways, dye-to-TiO2 binding (anchoring) ability, dye interoperability with other device components, co-sensitization fabrication approaches, chemical compatibility with other dyes during co-sensitization, dye aggregation, solvent effects. This analysis reveals the extent by which rhodamines can themselves deliver photovoltaic output, and how co-sensitization influences DSC performance. We discover that rhodamines are not DSC-active per se, but they can facilitate DSC-active dyes in device function via co-sensitization. |
1. Introduction
The development of renewable energy technologies is a prime concern of the scientific community, given the accelerating depletion of fossil fuels1 and the detrimental environmental effects associated with their combustion,2 combined with an increasing global demand for energy. Dye-sensitized solar cells (DSCs) represent a promising photovoltaic solution to this problem. The fabrication costs of DSCs are low compared to their silicon counterparts, they exhibit excellent indoor performance,3 and they potentially offer applications as solar-powered ‘smart windows’.4–6 However, since their invention,7 DSC power conversion efficiencies (PCE) have remained lower than those of other types of photovoltaic devices, so their innovation has been limited to niche, rather than generic, applications.That said, recent efforts in exploiting dye co-sensitization, to augment PCE by the few percent needed to make DSCs more competitive in the general photovoltaic market, have been met with good success. For example, co-sensitization was used to elevate the PCE of a zinc-based porphyrin dye (∼9.5% when singly-sensitized) by about 3% to produce the world-record DSC performance in 2011.8 Last year, a new world record in DSC performance was made using, for the first time, a combination of metal-free organic dyes, where co-sensitization yielded 14.3% efficiency under one sun illumination.9
Despite these successes, rational molecular design strategies which would enable the systematic engineering of DSCs that employ co-sensitization (hereafter called co-DSCs) still remain elusive. The fact that the dye is one of the most important components of a DSC, whose function is controlled at the molecular level, makes this dearth in molecular engineering particularly startling. A dye has a dual function in the DSC device, acting as the light harvester and the driver in electrical current initiation, via the dye⋯TiO2 electron injection process. The optical absorption profile of a dye and its molecular structure thus determine the performance of the cell.
The optical absorption profile of a dye should be as broad as possible in order to harness the maximum range of the solar spectrum. Since UV/vis absorption spectra of Ru-based dyes exhibit good breadth, they feature historically as the most commonly used DSC sensitizers.10 However, metal-free organic dyes have recently received more attention,11 on account of their high molar extinction coefficients (ε) and low toxicity. Moreover, they are easily available and cost effective.3 A major drawback of organic dyes, though, is their typically narrow optical absorption spectra, relative to metal–organic dyes, which restricts the maximum short-circuit current density (JSC) attainable.12 That said, this problem can be tackled by employing co-sensitization. One of the key assets of co-DSCs is that the multiple dyes contained within the same DSC absorb light at complementary wavelengths, such that their combined use results in a device that exhibits a broad optical absorption spectrum (Fig. 1(a)).13
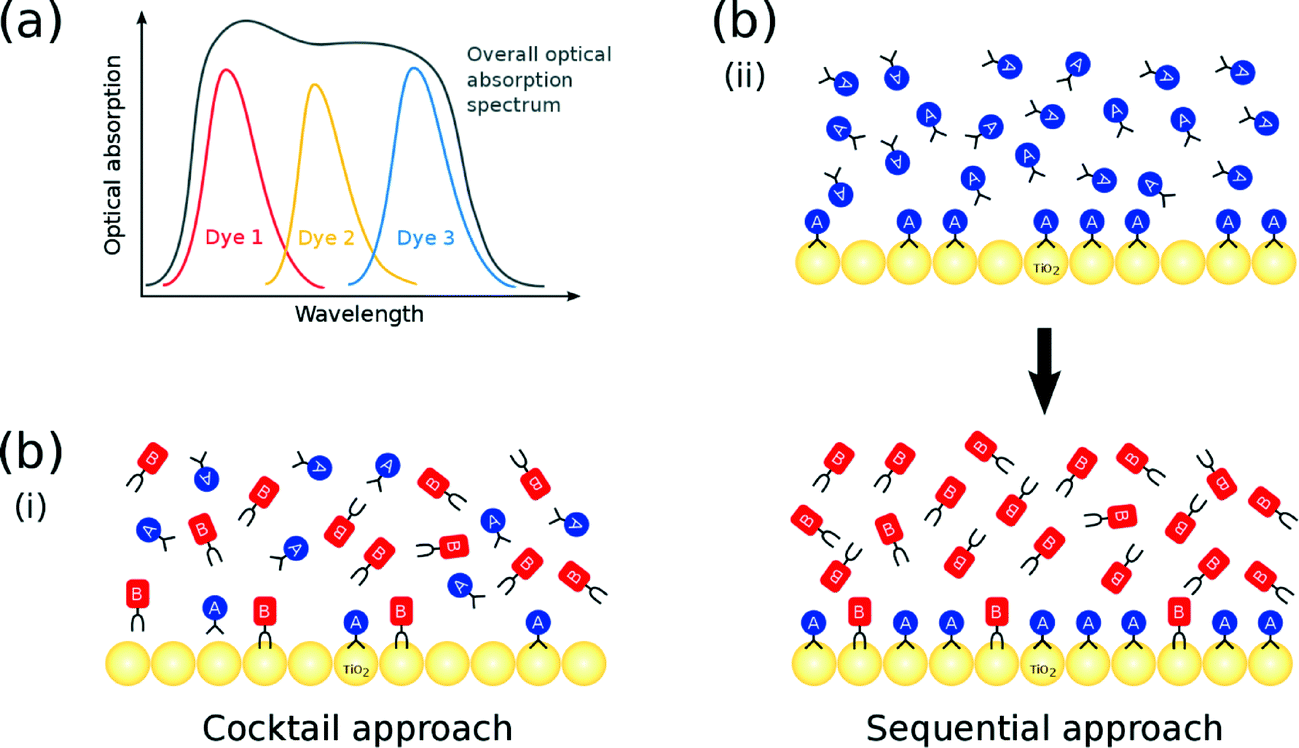 | ||
| Fig. 1 (a) Complementary absorption of multiple co-sensitized dyes; (b) (i) cocktail and (ii) sequential co-sensitization approach using two dyes A and B. | ||
The molecular structure of a dye affects the photovoltaic output of a DSC device since it prescribes the manner by which it binds onto the surface of the TiO2 semiconductor, to form the dye⋯TiO2 interface which operates as the DSC working electrode. Dye co-sensitization could also have an auxiliary role here, since it offers an opportunity to tailor this dye⋯TiO2 interfacial structure to afford optimal DSC function, by exploiting the many more dye adsorption control parameters that are available in the co-DSC device fabrication process, relative to single dye sensitization. Co-DSCs are fabricated predominantly from one of two methods: the (i) cocktail or (ii) sequential approach. In the former, TiO2 is sensitized by a solution that contains a mixture of two or more dyes; as such, each dye competes simultaneously for an adsorption site on the TiO2 surface (Fig. 1(b)(i)). In the latter, the TiO2 substrate is consecutively soaked in solutions of the respective dyes. The earlier a particular dye is employed in the sensitization process, the higher is its chance to encounter available adsorption sites. Consequently, dyes that are employed early in this sequence are usually responsible for the largest share of the dye coverage in the resulting co-DSC working electrodes; in contrast, dyes that feature at later stages in this sequence often merely fill remaining gaps on the TiO2 surface (Fig. 1(b)(ii)). Ultimately, the possible structural outcomes associated with either of these two co-sensitization fabrication methods are numerous and diverse in terms of dye⋯TiO2 interfacial adsorption dynamics, dye distribution, and dye coverage. Accordingly, it would prove very helpful to establish a molecular model that successfully predicts the dye⋯TiO2 interfacial structure, which results from applying a given co-sensitization fabrication method to a certain combination of dyes. A design strategy could then be used to engineer the dye structure and its distribution across the TiO2 surface. In turn, this would enable the function of the working electrodes to be manipulated with molecular precision.
Given the current knowledge base of co-sensitization studies available in the literature, a model that encompasses all control parameters does not seem to be tractable. Indeed, such a model would need to account for a multitude of factors that influence the resulting dye⋯TiO2 interfacial structure, such as differences in molecular size and shape of the co-sensitizing dyes, the nature of their chemical interactions (e.g. intermolecular forces, dye aggregation, or chemical reactions), their relative concentrations within a given dye sensitizing solution, and the manner by which dye adsorption dynamics manifest on the TiO2 surface. So far, very little is known about these factors in the context of DSC co-sensitization. However, a first step to overcome this dearth of knowledge can be achieved via the use of case studies, which explore the molecular structure and optical properties of specific families of dyes, whose DSC device function could be enabled, or improved, by co-sensitization.
To this end, we have recently reported on three case studies, each prospecting co-DSC applications for a chemical family of dyes: 4-(1,3-pentadienyl)-N,N-dimethylanilines,14 oxazines15 and cyanines.16 These studies used a combined experimental and computational approach to unravel relationships between dye structure and optical function in a chemical family of molecular chromophores that were a priori well known as dyes for other applications. The structure–function relationships, so formed for the dyes in those studies, were thence used as reference dyes in a molecular engineering workflow that was designed to generate new, hypothetical chemical derivatives, via in silico calculations, which carried suitable DSC dye attributes. Such attributes included suitable dye⋯TiO2 anchoring groups, and optical absorption bands that complement those of other dyes within the hypothetical series of chemical derivatives to the extent that the co-sensitization of complementary pairs of these in silico-generated dyes were predicted to afford an overall panchromatic optical response in a co-DSC device (cf.Fig. 1(a)).
The study conducted herein similarly employs a combined experimental and computational approach to uncover relationships between molecular structure and optical function, in a specific chemical family of dyes. In this case, a series of rhodamine chromophores was investigated, which are generally known according to their trade names (herein numerically labeled): R560 (1), R575 (2), R590 (3), R610 (4), R620 (5), R640 (6), and R3B (7) (Fig. 2). Dyes 1–7 were selected for this study since they exhibit photophysical properties, commonly exploited in biochemistry as fluorescent probes, whereby their optical absorption and emission spectra span the visible spectrum and thus allow multicolor labeling.17,18 The same photophysical properties also render them attractive as prospective DSC sensitizers. Indeed, rhodamines have already been investigated in the context of DSC applications. However, their potential has not been investigated systematically. For example, several rhodamines have been used for single sensitization purposes in DSCs, but in this context, attention was focused predominantly on the DSC fabrication technology, rather than on the performance of the rhodamines.19–21 Rhodamines have also been used for co-sensitization purposes, e.g. in conjunction with a metal-free porphyrin dye22 or in a supporting capacity for a Ru-based dye.23 Even though isolated positive results emerged from these studies, the performance of these dyes remains to be rationalized or improved in order to generate competitive DSCs. Moreover, the studies so far reported are far from systematic or exhaustive, thus precluding the ability to draw helpful conclusions from them with respect to correlations between molecular structure and DSC performance.
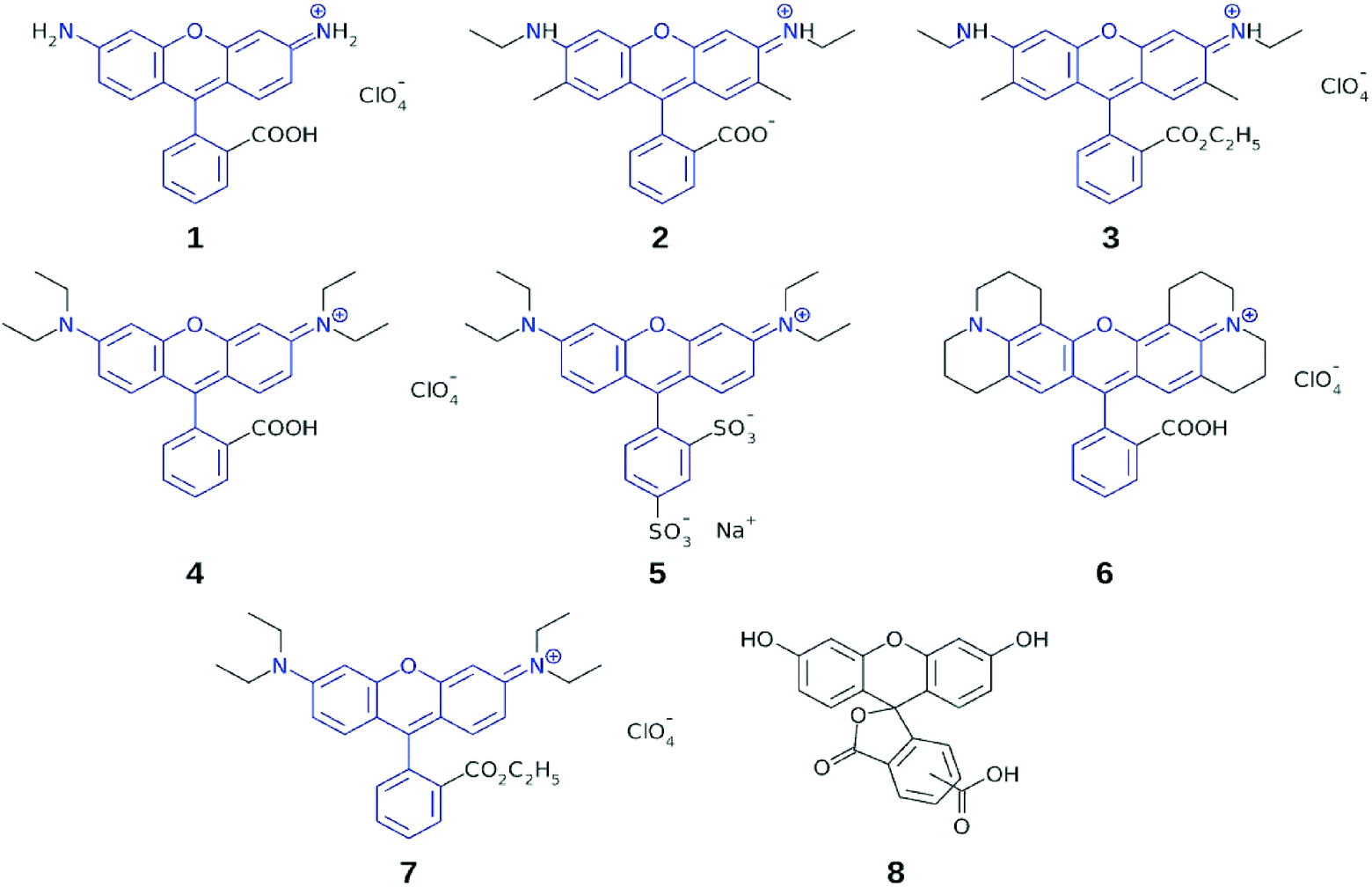 | ||
| Fig. 2 Subject dyes 1–7; common molecular fragments between the dyes are highlighted in blue; chemically related fluorescein dye, 8, was used with 5 in a co-sensitization test. | ||
Dyes 1–7 share a common molecular fragment, which is a xanthene heterocycle with terminal nitrogen atoms connected to a substituted phenyl ring. The nitrogen atoms may accommodate additional substituents, while adsorption onto the TiO2 surface occurs via the anchoring group on the substituted phenyl ring. Dyes 1–7 differ in the number and type of alkyl groups that they contain, as well as in the chemical nature of the anchoring moiety.
In one key respect, this study contrasts with our three previous case studies on prospective DSC co-sensitizers, where the subject dyes in previous work were treated as references in the creation of hypothetical chemical derivatives which might be suitable for co-DSC applications. In this work, the structure–function relationships herein established for the subject dyes are used to short-list several of them to go forward for actual co-sensitization experiments. The molecular engineering workflow presented in this work (Fig. 3) therefore allows an internal experimental validation of the predicted co-DSC attributes of the short-listed dyes, through their subsequent co-sensitization and DSC fabrication. The nature of this workflow also enables possible practical issues such as dye aggregation, chemical compatibility, solvent compatibility, and optimal sensitization sequences to be assessed.
 | ||
| Fig. 3 Molecular engineering workflow employed in this study. | ||
The outline of this molecular engineering workflow, shown in Fig. 3, discriminates where the use of experimental and computational tools are used to extract certain types of information via the yellow and green box classification, respectively; the interplay between this acquired information is described by annotated arrows. Firstly, the dye portion of the DSC was investigated exclusively in terms of the structural and optoelectronic properties of 1–7, which revealed their molecular structure, molar extinction coefficients, highest occupied and lowest unoccupied molecular orbital (HOMO and LUMO) energies, and lowest-lying vertical excitation energies (EV). On the basis of these results, relationships between the molecular structure of the dyes and their optical properties were elicited. Secondly, the dye⋯TiO2 interfacial structure that comprises the DSC working electrode was examined for 1–7, by determining their dye⋯TiO2 adsorption properties. Experimentally, this entailed adsorbing each dye, 1–7, onto TiO2 films. For the dyes that possess a classical carboxylate anchoring group3,24 (1, 2, 4, and 6), these experimental studies were complemented by a computational examination of the adsorbed dye at the dye⋯TiO2 interface.
Several of the subject dyes, or pairs of dyes, were then identified as prospective for co-DSC applications, according to a collective assessment of the structure and optical function of these dyes, when in their isolated form or when incorporated as a dye⋯TiO2 interface. To this end, the relative merits of 1–7 were judged against important co-DSC dye molecular design criteria, such as panchromatic optical absorption, efficient anchoring, and low molecular dye aggregation. These identified dyes, or pairs of dyes, were short-listed for experimental validation, i.e. they were taken forward for co-sensitization tests. This experimental work is depicted in the grey box in the lower part of Fig. 3, and involved either the co-sensitization of a short-listed pair of subject dyes (1 and 5, or 1 and 7), or the co-sensitization of one of the selected subject dyes (5) with a chemically related fluorescein dye (8; see Fig. 2). The use of two co-sensitization methods, the cocktail and the sequential approach, was explored, and the light-harvesting prospects of the resultant thin-film co-DSC working electrodes were assessed using solid-state UV/vis absorption spectroscopy. In order to understand the results of these co-sensitization studies within the broader context of related work, 1 and 2 were further selected for co-DSC device fabrication and testing, using the industrial standard dye, N3, as the co-adsorbant.
An auxiliary diagram (Fig. 4) summarizes which of the individual dyes engages with each of these characterization tools. In many cases, complementary dye characterization tools are used to acquire the same information, as denoted by the visual overlap of various bounded regions; where present, these complementary efforts substantiate the results. In other cases, only computational results relay structural or optical property information. This situation may denote computation being brought in to deal with the practical problem of unavailable experimental data, that is often encountered in chemical investigations, e.g. due to limited sample quantities, experimental difficulties such as poor crystallization, or solubility issues. One salient advantage of a concerted experimental and computational approach is this ability to verify results by information duality, or to fill in gaps in information where needed. This is much aligned with the spirit of the Materials Genome Initiative, where the time trajectory of ‘molecule-to-market’ needs to be accelerated;25 one needs computation to support experiments since one cannot afford to wait for all relevant experimental information to be realized before innovating a new device technology.
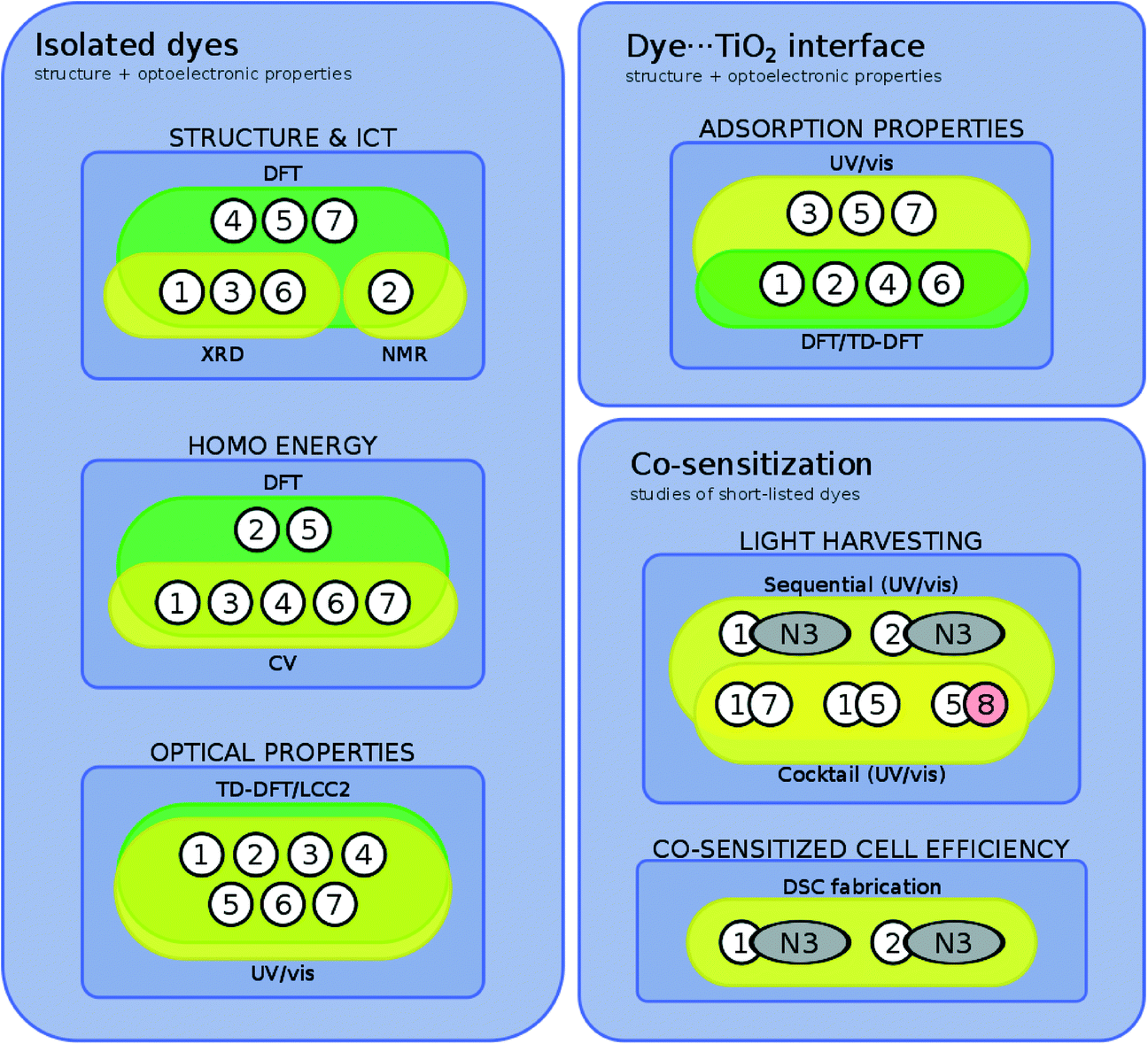 | ||
| Fig. 4 Summary of categorizing dyes/dye pairs by their experimental and/or theoretical characterization tools. | ||
2. Experimental and computational methods
2.1 Materials
The rhodamine dyes R560 ([C20H15N2O3][ClO4]; 1), R575 ([C26H26N2O3]; 2), R590 ([C27H29N2O3][ClO4]; 3), R610 ([C28H31N2O3][ClO4]; 4), R620 ([C27H29N2O7S2][Na]; 5), R640 ([C32H31N2O3][ClO4]; 6), R3B ([C29H33N2O3][ClO4]; 7), and the fluorescein dye C21H12O7 (8) were purchased in powdered form from Exciton. Standard grade solvents (methanol, ethanol, isopropanol, acetone, acetonitrile, valeronitrile, dimethyl sulfoxide, and dichloromethane) were purchased from common commercial suppliers and used without further purifications. TiO2 (DSL 18NR-T) particles (diameter: ∼20 nm), fluorine-doped tin oxide (FTO)-coated glass (TEC15), and the I−/I3− electrolyte solution (EL-HPE) were obtained from Dyesol. H2PtCl6·6H2O was purchased from SigmaAldrich.2.2 Single-crystal X-ray diffraction
Single crystals of 1, 3, and 6, suitable for X-ray diffraction analysis, were obtained from the slow evaporation of saturated ethanol solutions of the respective compounds at room temperature. Diffraction data were collected at 120 K on a Rigaku Saturn 724+ CCD diffractometer (λMo-Kα = 0.71073 Å) equipped with SHINE Optics focusing and an Oxford Cryosystems CryostreamPlus open-flow N2 cooling device. Cell refinement, data collection and integration were carried out using the Rigaku CrystalClear-SM Expert 2.0 software package,26 while absorption correction was implemented using ABSCOR.27 Structures were solved by direct methods and refined by full-matrix least-squares methods on F2. All refinements were performed using SHELXS,28 whereby hydrogen atoms were positioned geometrically and refined as riding on their parent atoms. A full summary of the crystal data, as well as crystallographic collection and refinement details is provided in the associated deposition material (ESI† S.1).2.3 Nuclear magnetic resonance spectroscopy
All NMR measurements were carried out by the NMR service facility of the Department of Chemistry, University of Cambridge (UK). 1H and 13C{1H} NMR spectra of 2 in DMSO-d6 and CD3OD were collected on a Bruker Avance 500 Cryo Ultrashield at room temperature. Chemical shifts (δ) are expressed in ppm referenced with respect to residual solvent peaks (δ = 0 ppm) as the internal standard. All NMR spectra are shown in the ESI† (S.2).2.4 UV/vis absorption and fluorescence spectroscopy
UV/vis absorption spectra of methanolic solutions of 1–8 (0.01 mM) were recorded on an Agilent Cary 300 UV-vis spectrophotometer with uncertainties of ±1 nm. Molar extinction coefficients (ε) for individual peaks were calculated using the slope of the linear regression line obtained from the absorbance of five different dye concentrations. Individual absorbance values for each concentration were measured five times in order to exclude potential systematic errors and obtain a standard uncertainty on the measurement. For 4 and 5, the dye aggregation behavior was examined, as their spectra contained shoulder peaks that could potentially be attributed to the formation of dimers and trimers in solution. In order to control the relative changes in peak intensity, dye concentrations and the solvent were varied. The absorption spectra of methanolic solutions of 4 and 5 were measured for 15 concentrations (5 × 10−4–3 × 10−8 M) and measured at 1.5 × 10−5 M in methanol, acetone, dimethyl sulfoxide, acetonitrile, and dichloromethane (see ESI† S.3). Fluorescence spectra of 1–8 were recorded on an Agilent Cary Eclipse Fluorescence spectrophotometer using a xenon flash lamp. The excitation wavelength was changed for each compound and the emission spectrum was recorded in methanol for each dye. A summary of the salient spectral information for all dyes is shown in Table 2.2.5 Cyclic voltammetry
Cyclic voltammograms of 1–7 in acetonitrile solutions (0.5 mM) were recorded on an Autolab PGSTAT101 (working electrode: carbon; counter electrode: platinum; reference electrode: Ag/AgCl in KCl) at scan rates of 25, 50, 75, and 100 mV s−1. Analyte solutions were deoxygenated by nitrogen sparging immediately prior to each measurement. The Ag/AgCl reference electrode was calibrated using the ferrocene/ferrocenium (Fc/Fc+) redox couple as an external standard. HOMO energies were calculated from the voltammograms by extracting the mean value between the oxidation and reduction peaks. Energies were referenced to vacuum conditions by subtracting 4.398 eV from the Ag/AgCl-referenced values (see ESI† S.4).2.6 Preparation of TiO2 films for adsorption and desorption studies
TiO2 films were deposited on transparent glass slides using the doctor-blade technique,29 to create a single layer of TiO2 nanopores with a thickness of ∼4 μm. Subsequently, these TiO2 films were sintered (T = 500 °C; 30 min) and, unless specified otherwise, sensitized by dipping in dye solution (0.5 mM in acetone) for 8 hours at room temperature. Excess dye was removed from these films by rinsing with acetone. Dye desorption, for quantification of dye-adsorption, was achieved by dipping the sensitized films in a NaOH solution (0.1 M in H2O/ethanol, 1/1, v/v) at room temperature (see ESI† S.6).2.7 Fabrication of DSCs and photovoltaic characterization
TiO2 films were deposited on fluorine-doped tin oxide (FTO)-coated glass as described in the previous section. Counter electrodes were generated by doctor blading a solution of H2PtCl6·6H2O (52 mM in isopropanol) onto these films, followed by sintering (T = 385 °C; 30 min). Cells were not sealed and the circuit was closed by adding 0.1 mL of an I−/I3− electrolyte solution (50 mM in acetonitrile/valeronitrile) and holding in place the two electrodes using a paper clip. For each dye, five cells were fabricated and I–V curves were recorded by measuring the current and the voltage for each cell under AM1.5 illumination at 100 mW cm−2, using an ABET Sun 2000 solar simulator. A mask was employed in order to limit the functional area of the tested cells to 0.16 cm2.2.8 Computational studies
Electronic structure calculations were carried out for 1–7 in order to examine their optoelectronic properties. These studies included DFT and TD-DFT calculations, carried out using Gaussian 09,30 and ab initio coupled-cluster methods involving the Laplace transformed density-fitted local CC2 (LT-DF-LCC2)31,32 using Molpro.33The molecular structures of 1, 3, and 6, obtained from single-crystal X-ray diffraction measurements, were used as the geometric starting point for the calculations, which were then optimized at the PBE1PBE/6-31G(p) level of theory.34–37 The initial geometries for 2, 4, 5, and 7 were estimated from their closest-matching crystal structures, modified to match the chemical structure, and then geometry optimized. Solvent effects for methanol were included via the polarizable continuum model (PCM).38,39 Initial calculations showed that the presence of the perchlorate counterion neither affected the geometry, nor the energies of the calculated structures (see ESI† S.5) and it was therefore excluded from calculations. In contrast, the sodium counterion of 5 was included (see ESI† S.5). Positive vibrational frequencies were calculated for 1–4, which suggests that reliable ground-state structures were encountered.
Single-point DFT energy calculations were performed on the optimized geometries in order to determine HOMO energies and spatial distributions of molecular orbitals. These calculations were performed at the PBE1PBE/6-311+G(2d,p) level of theory40 and solvent effects for methanol were considered using the PCM model. Orbitals were processed using cubgen30 and visualized in Avogadro (isovalues = 0.0007).41 In order to establish an appropriate level of theory, benchmark calculations were carried out for a range of functionals with respect to experimental values. Lowest vertical excitation energies (EV) were calculated using LT-DF-LCC2 with extended domains, including solvent effects (for benchmark values and full details, see ESI† S.5).
Theoretical studies of the dye⋯TiO2 interface were conducted on a (TiO2)9 cluster. The starting model for the (TiO2)9 cluster was a previously determined structure42 that was also geometrically optimized (for details, see ESI† S.5). This cluster represents the semiconductor surface structure at a level of approximation that has already been shown to be sufficient in order to model DSC working electrodes.42 More generally, its size represents a good compromise between an accurate description of the TiO2 surface and computational cost, which increases with cluster size.43–45 The geometry of the (TiO2)9 cluster was initially optimized at the B3LYP level of theory, using the 6-31G* basis set and the PCM model. Each selected dye was then attached to the TiO2 surface, initially in a bidentate chelating (bc) and bidentate bridging (bb) adsorption mode configuration, since they represent the strongest adsorption modes on TiO2.24 However, upon geometry optimization, the structure of the dye⋯TiO2 system can converge to different anchoring modes. This optimization process involved single-point energy DFT and TD-DFT calculations at the same level of theory.
3. Results and discussion
3.1 Assessing the innate molecular suitability of 1–7 for DSC dye applications
The molecular attributes of 1–7 were first investigated in order to determine the extent by which they are innately suited for DSC dye applications. In particular, a DSC dye should exhibit intramolecular charge transfer, whose associated optical absorption properties feature LUMO energy levels that are higher than the energy of the TiO2 conduction band edge (ECB), and HOMO energy levels that are lower than the electrolyte redox energy (Eredox).33.1.1.1 Molecular structures of 1–7. The molecular structures of a dye can be used to determine the nature of the intramolecular charge transfer (ICT) that governs their photophysical properties.46 Accordingly, these structures were determined as a pre-requisite to assessing the ICT characteristics of 1–7.
It was possible to solicit the molecular structures of 1, 3, and 6via single-crystal X-ray diffraction experiments, since these dyes formed good quality crystals for viable crystal structure determination. Fig. 5 depicts their structures while salient bond geometries are summarized in Table 1. Further crystallographic details are provided in the ESI† S.1.
 | ||
| Fig. 5 Asymmetric units of the crystal structures of 1, 3 and 6 (CCDC deposition numbers 1498038–1498040, respectively). Atomic displacement ellipsoids are drawn at the 50% probability level. The perchlorate counter-ion and trapped solvent molecules have been omitted for clarity. | ||
| Dye | C–NXRD [Å] | C–NDFT [Å] | TAXRD [°] | TADFT [°] | RMSD [Å] |
|---|---|---|---|---|---|
| a Since 2 features a lactone configuration in its solution-state modeling but a cationic structure in the dye⋯TiO2 system (see §3.2.2), two values are given for the DFT-generated TA of 2 according to: lactone {cationic}. | |||||
| 1 | 1.348(5)–1.331(5) | 1.339–1.339 | 77.8(4) | 88.9 | 0.199 |
| 2 a | — | 1.372–1.372 {1.351–1.352} | — | 90.0 {78.7} | — |
| 3 | 1.347(3)–1.348(3) | 1.345–1.345 | 83.6(2) | 89.4 | 0.262 |
| 4 | — | 1.351–1.352 | — | 73.9 | — |
| 5 | — | 1.352–1.353 | — | 69.2 | — |
| 6 | 1.347(4)–1.359(4) | 1.350–1.355 | 84.9(3) | 87.3 | 0.127 |
| 7 | — | 1.352–1.353 | — | 89.6 | — |
Fig. 5 shows that the xanthene ring and the carboxyphenyl moiety are highly planar in themselves, but they adopt an almost perpendicular arrangement with respect to each other, with torsion angles (TAs) ranging from 77.8(4)–84.9(3)° across the three dyes (Table 1). As a consequence, π-orbitals cannot overlap at the bridge between the xanthene and carboxyphenyl moieties. This bridge therefore separates two distinct regions of π-conjugated electron density that manifest in the overall molecular architecture. Indeed, this is further evidenced by the bridging carbon–carbon bond lengths being longer than that expected for a π-conjugated C⋯C bond (cf. bond lengths, C13–C14 = 1.494(5) Å for 1; C19–C20 = 1.511(3) Å for 3, C25–C26 = 1.496(4) Å for 6, relative to a regular C⋯C bond length (1.397 Å (ref. 47)). Accordingly, the intramolecular charge transfer (ICT) in these dyes is bifurcated.
If the ICT bifurcation observed in these crystal structures persists through to the solution state, the viability of rhodamine dyes for DSC application is placed very much in question. This is because a DSC dye molecule needs to contain an ICT path that connects its electron donor to its anchoring group – the substituent that enables dye adsorption onto the TiO2 surface to afford a dye⋯TiO2 interface (the DSC working electrode). The NR2 groups on the xanthene ring have the electron donating capacity for these rhodamine dyes, while the carboxylic acid on their carboxyphenyl moiety is needed to act as the anchoring group.3,24 Since the crystal structures of 1, 3 and 6 show that the ICT between these ring systems is distinct, there is a complete disconnection in ICT between the electron donor and anchoring group in these rhodamine dyes in their solid-state form.
That said, DSC dyes adsorb onto the TiO2 surface while in their solution state, during the device fabrication process. Since the bridging carbon–carbon bond between these ring systems bears somewhat single-bond characteristics, it is possible that it may possess sufficient rotational freedom in solution that ICT can traverse these ring systems. In order to explore this possibility, DFT studies were employed to calculate the lowest-energy structures of 1–7 in solution. Indeed, the structures of all seven subject dyes could be considered in this fashion, since computation can cover for the shortfall in being unable to characterize 2, 4, 5, and 7 experimentally as single crystals for these dyes could not be obtained. Furthermore, the DFT calculations afforded quantum-energy information to complement each electronic structure, which will prove useful in later discussion.
It transpires that the computed molecular structures of 1, 3 and 6 are very similar to those of their experimentally determined crystal structures, as evidenced by their modest root-mean-square deviations (RMSDs) of 0.1–0.3 Å (see Table 1). The calculated dye structures bear large torsion angles within a few degrees of 90° in all cases except for 4 and 5; in these two cases, the xanthene rings appear less planar which explains this ostensible discrepancy. The DFT-optimized geometries of the electron donor groups for 1–7, as judged via the C–N bond lengths (1.339–1.372 Å), are also similar to those in the experimentally determined structures of 1, 3, and 6. All of these comparisons suggest that packing forces in the crystal structures of 1, 3 and 6 have minimal impact on their molecular form; and that, assuming that these crystal structures are representative of all dyes, their molecular structures in the solid state and in solution behave similarly.
This assumption may not entirely carry for 2 since its DFT structure optimization converged to its lactone form whereby the carboxylic acid deprotonates and forms a ring in conjunction with part of the xanthene ring. These anomalous computational results for 2 were actually corroborated via1H and 13C{1H} NMR spectroscopic analysis which indicated that the lactone structure of 2 forms in methanol (a hydrogen-bond donating solvent) but the cation forms when 2 resides in dimethylsulfoxide solvent (a non-hydrogen-bond donating solvent). See the ESI† S.2 for further details. Nonetheless, if 2 forms a lactone structure in any type of solution, it will lose its anchoring group via that ring formation process, thereby negating its viability for DSC functionality; while in its cationic form, its structure will be similar to those of the other subject dyes, i.e. bear an interrupted ICT path between the ring systems in the molecular architecture. Moreover, as will be evidenced later (§3.2.2), DFT modeling shows that 2 binds to TiO2 in its cationic form, in the same way as all of the subject dyes that can adsorb to TiO2; it is the structural form of 2 within this dye⋯TiO2 interface that matters, since this interface defines the operational mechanism of the DSC working electrode.
In summary, these experimental and computational structural analyses both indicate that the ICT path is indeed interrupted between the two ring systems in these rhodamine dyes. Given these findings and the need for a DSC dye to possess some form of D–π–A molecular architecture, one might intuitively think that rhodamine dyes cannot be viable for DSC applications. Yet it was noted earlier that there are previous reports of rhodamine dyes being successfully employed in DSC devices.19–23 How can this ostensible discrepancy be resolved? Is there perhaps another way that ICT could traverse the two ring systems, or might a more complicated structural influence on DSC functionality be unaccounted for at this stage of structural analysis?
3.1.1.2 Quantifying the ICT characteristics of 1–7. Given this conundrum of demonstrated photovoltaic performance of DSCs that use rhodamine dyes in other studies, despite the evidence of a disconnected ICT pathway in their core molecular structure, it seemed prudent to perform a more quantitative study of the π-electron density within their ring systems. A simple comparison of the arene bond lengths in the carboxylphenyl moiety, from the available crystal structures, shows that they are all similar (1.369(6)–1.402(5) Å for 1; 1.378(4)–1.408(3) Å for 3; 1.378(4)–1.406(4) Å for 6), suggesting that this ring is essentially delocalized. The extent of delocalization in the xanthene rings is less clear, judging simply from a raw comparison of the associated bond lengths; rather, the π-structure in these rings appears to be somewhere in between delocalized and canonical in form.
Indeed, various possible resonance structures are conceivable for 1–7, and Fig. 6 presents several of the more likely representations, as judged by chemical intuition and the nature of the molecular structures observed. From a theoretical perspective, it is possible to quantify the ICT characteristics by resonance theory, which describes the overall architecture of a molecule in terms of a collection of its possible resonance structures.48 These resonance structures can be used in conjunction with the bond geometry in 1–7 to quantify the nature of ICT taking place within these molecules, via the harmonic oscillator stabilization energy (HOSE) model.49,50 This is a measure of the energy needed to transform the idealized bond geometry of a particular resonance structure to the geometry of a real molecule.51 The HOSE for each resonance structure, j, is defined by:
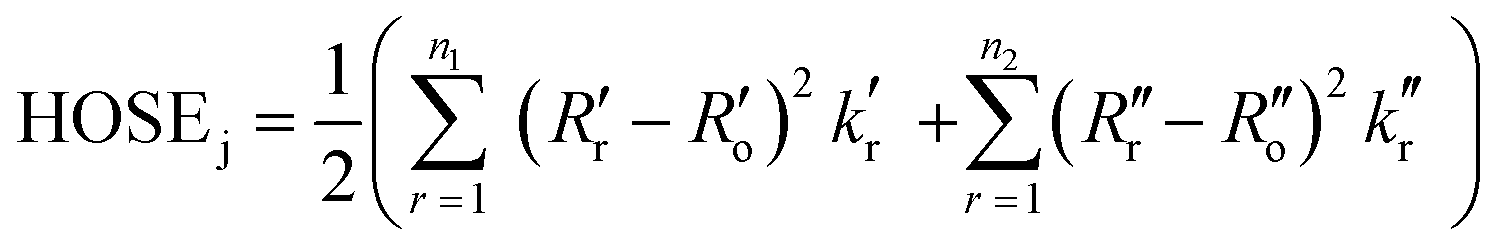 | (1) |
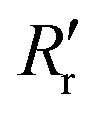 and
and 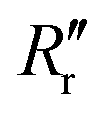 are the experimentally observed single and double bond values, respectively;
are the experimentally observed single and double bond values, respectively; 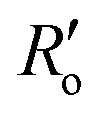 (1.467 Å) and
(1.467 Å) and 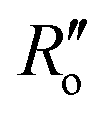 (1.349 Å) are reference values for single and double bonds, respectively.47 The HOSE model can thus be used to calculate the relative contributions, Ci, of each canonical resonance structure to that of the real molecule, according to:
(1.349 Å) are reference values for single and double bonds, respectively.47 The HOSE model can thus be used to calculate the relative contributions, Ci, of each canonical resonance structure to that of the real molecule, according to: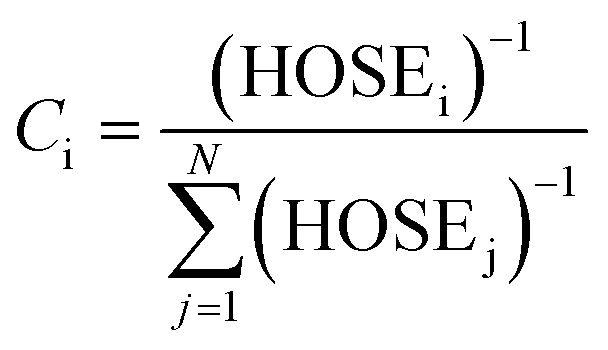 | (2) |
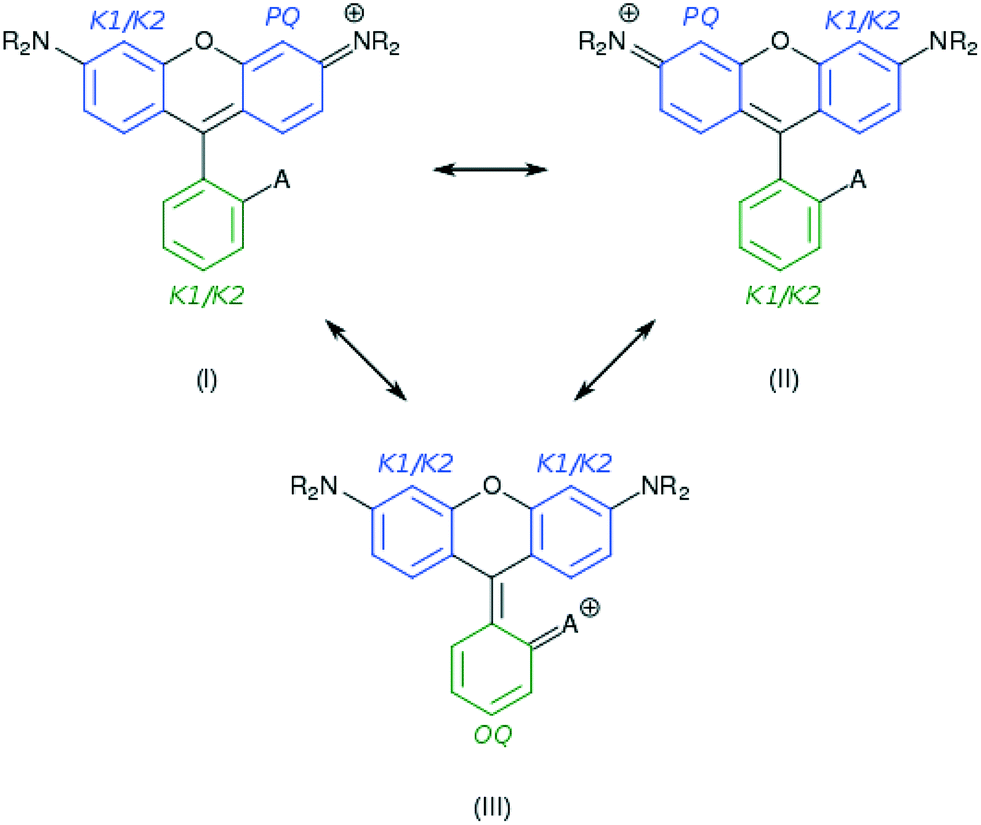 | ||
| Fig. 6 A selection of representative resonance structures for 1–7; the conjugated rings of the xanthene fragment are shown in blue, while the carboxylphenyl moiety is displayed in green. Kekulé variations, K1 and K2, as well as para- and ortho-quinoidal options, pQ and oQ, are annotated. A resembles the anchoring group of the dye. | ||
As Ci is inversely proportional to the HOSE value, an energetically more stable structure affords a larger contribution, Ci. In order to establish the dominant resonance structure, low-energy resonance states have to be included in the model. For 1–7, the possible contributions to the ICT include: two Kekulé (CK1 and CK2) and one para-quinoidal (CPQ) resonance state on the left and right arene rings of the xanthene moiety, and two Kekulé and one ortho-quinoidal (COQ) resonance on the adjoining substituted phenyl ring (see Fig. 6). Compound 5 was excluded from the calculation of COQ, since the model takes only carbon atoms into account.
For the calculations using the experimental crystal structure data for 1, 3, and 6, the HOSE results suggested dominant contributions (40–60%) from the CPQ states. Moreover, an analysis of the correlation between the CPQ contributions of these crystal structures and their experimental EV values revealed a linear relationship (see ESI† S.1). For the corresponding theoretically-calculated structures, the dominant contributions from the CPQ states were even higher (55–60%). The predominance of CPQ in both rings prevents the determination of one dominant resonance structure. However, the presence of two electron donors on the xanthene moiety (the –NR2 substituents) suggests a mixture of the two symmetric resonant structures, with a para-quinoidal character on the left or the right ring, and a Kekulé character on the alternate ring. For the substituted phenyl moiety, a relatively low difference in resonance energies (∼3–8 kJ mol−1) between contributions was observed, and a dominant resonance structure could thus not be identified. These dominating resonance structures (I) and (II) corroborate the single-bonded character of the carbon–carbon bond that bridges the xanthenes and substituted phenyl rings.
Optical absorption spectra of 1–7 (0.01 mM) were measured in methanol (Fig. 7; Table 2). All spectra exhibited one dominant absorption band (∼500–570 nm), accompanied by a shoulder at shorter wavelengths. Differences between the maximum absorption wavelengths of individual dyes can be attributed to ICT perturbations that result from various chemical substitutions to the molecular core fragment that is highlighted as blue in Fig. 2. The optical properties are found to change upon the addition of alkyl substituents on the terminal nitrogen atoms, whereby each additional ethyl group induces a bathochromic shift of ∼25 nm. In addition, substituting the carboxylate with an ester or sulfonate anchoring group appears to induce a bathochromic shift of ∼10 nm. In contrast, the additional hydrocarbon substituents on the xanthene moiety in 2, 3, and 6, as well as the counter ions in 1 and 3–7, did not affect the optical properties. Molar extinction coefficients were obtained from linear regression curves including five measurements of the optical absorption per concentration. Within the tested range of concentrations and solvents, aggregation of the dyes was not observed (ESI† S.3).
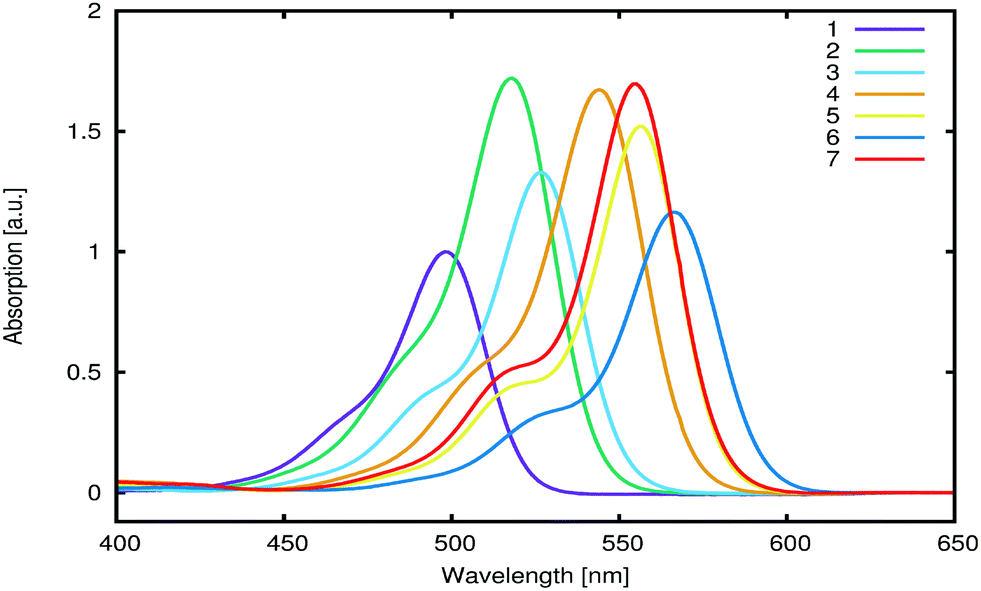 | ||
| Fig. 7 UV/vis absorption spectra of 1–7 (0.01 mM in methanol). Spectra for 2–7 are normalized with respect to 1. | ||
| Dye | E exptalV [eV] | E calcV [eV] | λ exptal,absmax [nm] | λ exptal,emimax [nm] | λ exptal,emiexcit [nm] | ε × 104 [L mol−1 cm−1] |
|---|---|---|---|---|---|---|
| a Calculated using LT-DF-LCC2. b Estimated from the dye⋯TiO2 system. | ||||||
| 1 | 2.49 ± 0.1 | 2.67 | 498 | 529 | 400 | 7.6 ± 0.1 |
| 2 | 2.39 ± 0.1 | 4.58(2.43b) | 518 | 551 | 430 | 12.2 ± 0.7 |
| 3 | 2.35 ± 0.1 | 2.53 | 527 | 558 | 430 | 11.2 ± 0.2 |
| 4 | 2.28 ± 0.1 | 2.30 | 544 | 582 | 440 | 9.9 ± 0.6 |
| 5 | 2.23 ± 0.1 | 2.29 | 556 | 584 | 450 | 11.8 ± 0.5 |
| 6 | 2.19 ± 0.1 | 2.22 | 566 | 600 | 460 | 8.4 ± 0.7 |
| 7 | 2.23 ± 0.1 | 2.34 | 555 | 587 | 450 | 12.9 ± 0.6 |
Optical emission spectra for 1–7 were measured (Table 2), in order to estimate the lowest vertical excitation energy (EV) from the intersection between the optical absorption and emission curves.
3.1.3.1 ICT characteristics during photoexcitation. In order to obtain complementary quantum energy information about the ICT characteristics of 1–7, single-point DFT calculations were undertaken. The topologies of their HOMOs and LUMOs provide useful insight on the charge dynamics within the molecule upon photo-excitation. In particular, the topologies of their squared differences, LUMO2–HOMO2 (Fig. 8) revealed shifts in electron density upon excitation of an electron from the HOMO to the LUMO. These difference plots appear different for 2 owing to its solution-state DFT calculations converging anomalously to a lactone-based molecular configuration (see §3.1.1.1). All other dyes evidence only a slight depletion of electron density for the terminal nitrogen atoms in the HOMO-to-LUMO transition, indicating that ICT propagates from these electron donors by only a small amount during photoexcitation. Meanwhile, photoexcitation is accompanied by increased electron density that lies primarily in the xanthene ring, which impinges slightly on the inter-ring bridge and its immediate arene bond neighbors in the adjoining phenyl ring. However, this increase in electron density does not extend to the anchoring group of this phenyl ring in any of the seven subject dyes. The overall magnitude of ICT during photoexcitation can be quantified by measuring the orbital overlap between HOMO and LUMO orbitals across the dye molecule (Λ).52 For 1–7, Λ values between 65–67% were observed, which indicates a decent level of overlap (Table 3). The major contribution to this orbital overlap stems from the xanthene ring (see Fig. 8); there are minimal electron density differences in the region of the electron donors and anchoring groups in all cases. This would seem to suggest that ICT cannot traverse the dye molecule from the donor moieties to the anchoring group, in order to stimulate successful electron injection into the conduction band of the TiO2 to which the dye will be adsorbed in the working electrode of a DSC. Since the function of a dye as a DSC sensitizer needs this D–π–A type of ICT pathway, these results are internally consistent with the rest of this study, but diverge from the DSC observation reported by other studies on rhodamine dyes.19–23
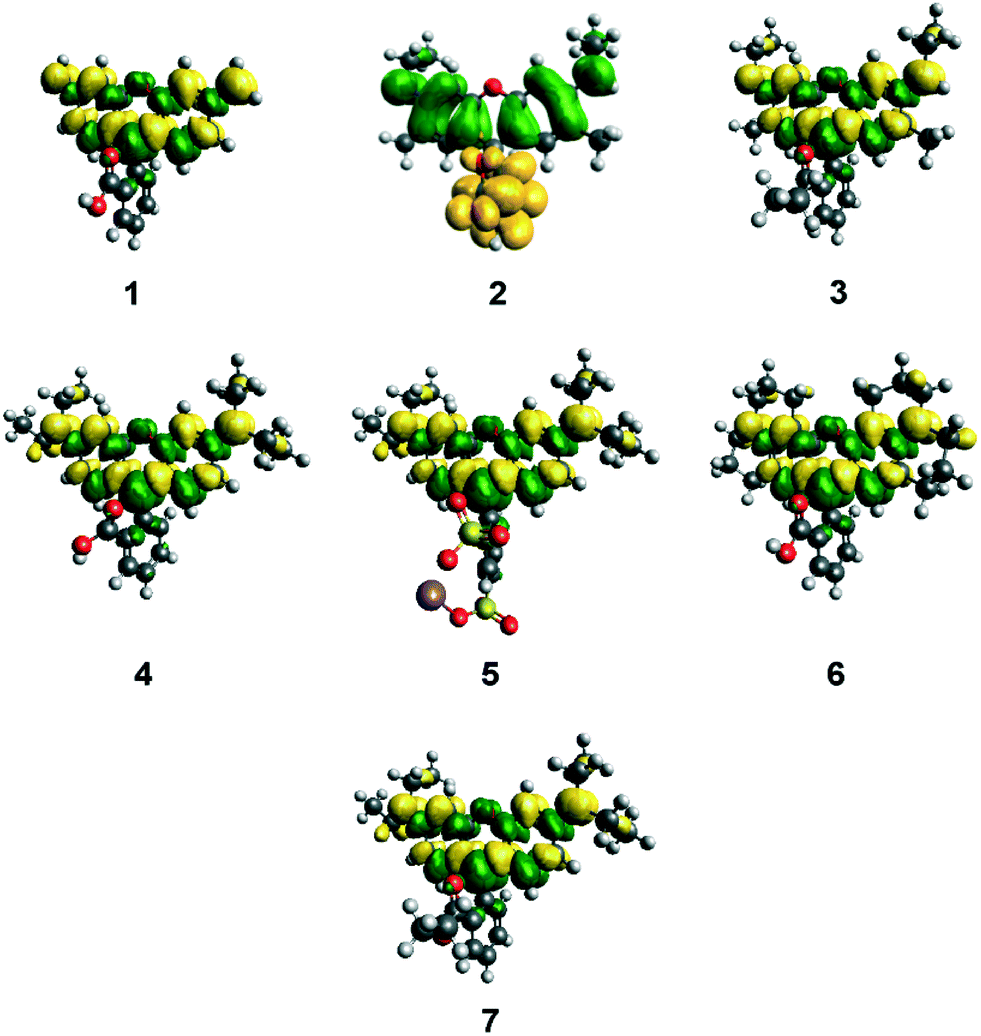 | ||
| Fig. 8 LUMO2–HOMO2 orbital plots for 1–7 (isovalue = 0.0007). Green areas represent regions of increased electron density, while yellow areas represent regions of decreased electron density. 2 differs from the rest of the dyes, because of its convergence to the lactone form in the solution-state calculations. | ||
| Dye | HOMOexptal [eV] | HOMOcalc [eV] | Λ [%] |
|---|---|---|---|
| a Since 2 features a lactone configuration in its solution-state modeling but a cationic structure in the dye⋯TiO2 system (see §3.2.2), two values are given for the DFT calculated HOMO and Λ value for 2 according to: lactone (cationic). | |||
| 1 | −5.81 ± 0.1 | −6.26 | 67 |
| 2 | — | −5.89 (−5.91a) | 67 (67a) |
| 3 | 5.62 ± 0.1 | −5.95 | 67 |
| 4 | 5.52 ± 0.1 | −5.92 | 66 |
| 5 | — | −5.85 | 65 |
| 6 | 5.41 ± 0.01 | −5.52 | 67 |
| 7 | 5.78 ± 0.01 | −5.90 | 66 |
3.1.3.2 Assessing energy levels of 1–7 relative to those of other DSC device components. Despite the lack of sufficient evidence for a rational mechanism by which ICT can traverse between the two ring systems in rhodamine dyes, the HOMO and LUMO energies of 1–7 can nonetheless be checked for their suitable alignment against the corresponding energy levels of the other DSC device components. As stated earlier, the LUMO energy levels of a DSC dye must be higher than the energy of the TiO2 conduction band edge (ECB) in order to provide a driving force for electron injection. Meanwhile, the HOMO energy levels of a DSC dye must be lower than the electrolyte redox energy (Eredox) in order to afford a sufficient driving force for dye regeneration.3 A combined set of experiments and calculations were employed to obtain this information.
Low-lying EV values for 1–7 were estimated from their maximum peak absorption (λpeakabs) bands in the experimental UV/vis spectra. Complementary calculated values employed the LT-DF-LCC2 model (see ESI† S.5). Experimental HOMO (HOMOexptal) energies for 1, 3, 4, 6, and 7 were measured by cyclic voltammetry (voltammograms in ESI† S.4). HOMO energies for 1, 3, and 4 were determined using the “onset” method, while those for 6 and 7 were determined by the “peak” method (see ESI† S.4). For 2 and 5, HOMOexptal values could not be obtained, as the voltammograms did not show oxidation or reduction peaks. Theoretical HOMO energies (HOMOcalc) and spatial distributions of molecular orbitals were obtained from single-point DFT energy calculations at the PBE1PBE/6-311+G(2d,p) level of theory,40 whereby solvent effects for methanol were included using the PCM model. A summary of these values together with molecular orbital overlap (Λ) values, is presented in Fig. 9 and Table 3.
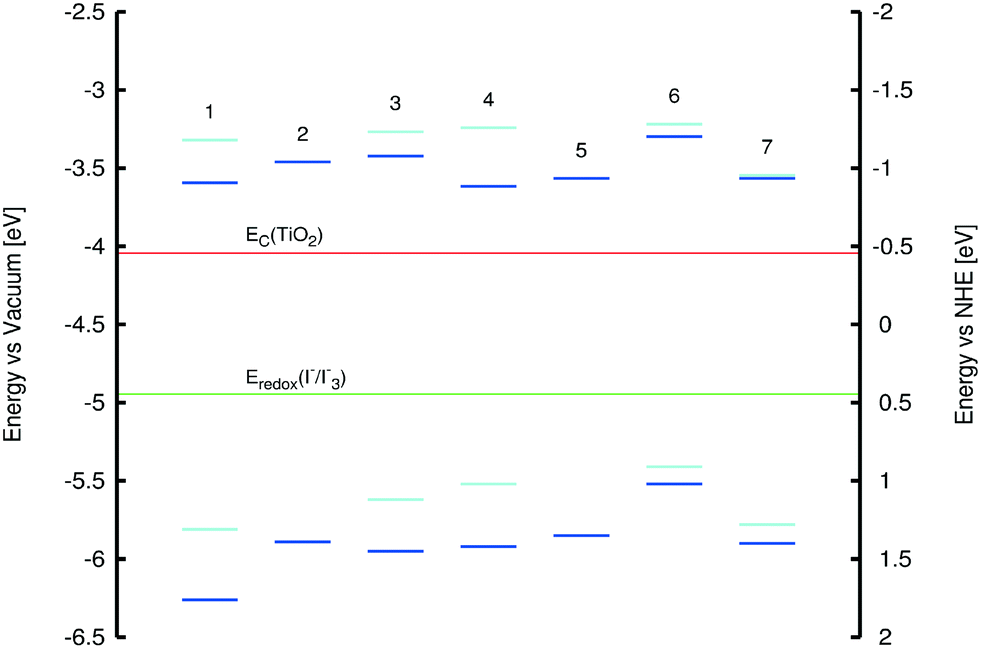 | ||
| Fig. 9 HOMO and LUMO energies of 1–7; color code: dark blue = calculated energies, light blue = experimental values from cyclic voltammetry. | ||
All HOMO energies were found to lie below that of the I−/I3− electrolyte redox potential (−4.94 eV), thus permitting dye-regeneration after electron injection. The LUMO energies were estimated from EV and HOMO energy values; these exceeded that of the TiO2 conduction band edge (−4.04 eV), thus confirming that electron injection from the dye to TiO2 upon photoexcitation is permitted on energy grounds (Fig. 9). All experiments and calculations described in this section included the provision for solvent effects which was deemed mandatory (for computational details, see ESI† S.5).
3.2 Studying the dye⋯TiO2 interface
Since the solution-phase dye is used to sensitize the TiO2 semiconductor in the DSC fabrication stage, it was natural to first establish the compatibility of the solution-phase energy levels of 1–7 with those of the other DSC device material components, for viable dye function; section §3.1.3.2 described the studies carried out to confirm this hypothesis. Nonetheless, the dyes adsorb onto a TiO2 surface during the fabrication process, to form a dye⋯TiO2 composite that represents the DSC working electrode. The true potential of these prospective sensitizers can therefore be judged more accurately via examination of the way in which the complex dye⋯TiO2 interfacial characteristics impinge upon the optical and structural behavior of the DSC. The extension of this work to these interfacial studies is especially pertinent in light of the solution-based dye results evidencing a lack of ICT pathway suitable for functionalizing the DSC working electrode, since this remains a conundrum if rhodamines are in fact viable as DSC dyes as other reports suggest.To this end, UV/vis absorption spectroscopy experiments on thin films of 1–7 adsorbed onto TiO2, which resemble DSC working electrodes, were performed in order to appreciate how the optical properties are affected by the complex nature of adsorption onto the TiO2 surface; possible complex factors of the dyes include dye aggregation, anchoring mode variation, or dye⋯dye interactions. Meanwhile, a computational approach was employed to unravel the nature and extent by which the formation of this interface affects the dye structure and its respective energy levels.
3.2.1.1 Anchoring group suitability. Fig. 10 shows the UV/vis absorption spectra of 1–7 adsorbed onto TiO2. While 1–6 successfully adsorbed onto the TiO2 surface, 7 did not adsorb onto TiO2, presumably due to its lack of a carboxylic acid group; instead, its substituted phenyl moiety features an ester group, which is not a suitable anchor.24 The only potentially viable anchoring groups in 7 are the amine substituents which can, in principle, adsorb onto TiO2via Lewis acid/basic interactions.24 However, in practice, the doubly-substituted alkyl chains on these amine groups sterically encumber the nitrogen atoms such that this negates potential bonding interactions with a TiO2 surface; i.e.7 is not tenable as a DSC dye. These steric effects are evident by comparing the results from 3 and 7. Dye 3 contains the same ester group as 7, and yet it adsorbs onto TiO2, presumably via its less encumbered Lewis basic amine groups. That said, while such Lewis acid/base interactions with TiO2 seem to be facilitated for 3, the use of the amines as anchoring modes, as well as their pre-existing electron donor role, would lead to an inappropriate molecular architecture for DSC dye function; as such, 3 adsorbed onto TiO2 will not favor dye-to-TiO2 charge injection, and so 3 was dismissed as a viable DSC dye. The only other subject dye to not contain a terminal carboxylate group is 5. However, a SO3− substituent lies in its place on the phenyl ring which also bears a second SO3− substituent. While employing sulfonate ions, rather than carboxylic acids, as anchoring groups generally appears to afford dyes with lower DSC device efficiencies,241 and 5 displayed one of the most substantial optical absorption intensities of all subject dyes. One might speculate that the duplicative existence of the sulfonate groups in 5 may have a bearing on this measure. As a result, 5 therefore remains an interesting DSC dye candidate. Dyes 1, 2 and 4 also showed strong optical absorption, as one might expect given that they bear a classical carboxylic acid group as an anchoring substituent.
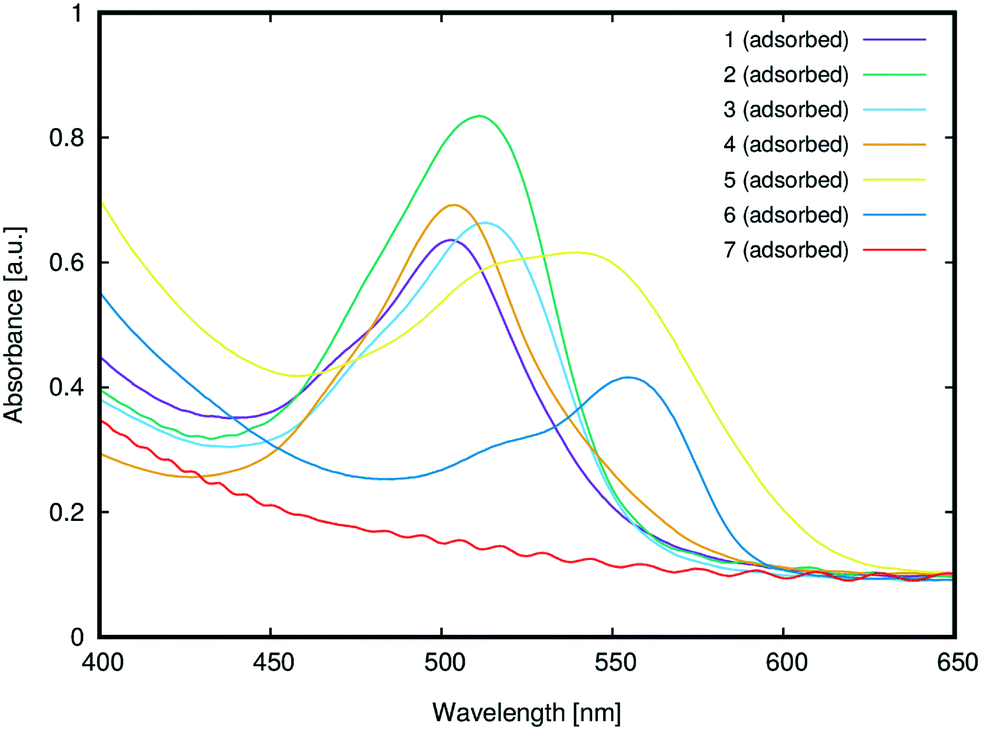 | ||
| Fig. 10 UV/vis absorption spectra of TiO2 films sensitized with 1–7. | ||
3.2.1.2 Dye aggregation effects. A comparison of the λpeakmax of the adsorbed dyes, relative to those in solution (cf.Fig. 5), offers the chance to glean insights into possible dye aggregation processes that may occur upon sensitization. To this end, a bathochromic shift of λpeakmax upon adsorption indicates J-aggregation of the adsorbed dye, while a hypsochromic shift of λpeakmax upon adsorption indicates H-aggregation.53 For example, bathochromic shifts of ∼6 nm were observed for 1 and 5, indicating the formation of J-aggregates upon adsorption. Weak hypsochromic shifts were observed upon the sensitization of 2 (6 nm) and 3 (15 nm), while stronger hypsochromic shifts were observed for 4 (40 nm) and 6 (27 nm). Based on the observed batho- and hypsochromic shifts and their respective extents, 1–6 can be divided into three categories: J-aggregates (1 and 5), weak H-aggregates (2 and 3) and strong H-aggregates (4 and 6). The type of aggregation determines the nature of the dynamics of dye adsorption on TiO2; it can be therefore used to predict successful DSC dye candidates.
Previous reports on the nature of dye aggregation in rhodamines54 suggested that H-aggregation is undesirable in DSC applications for several reasons: dimers that form are unable to attach onto TiO2 and the dimerization can also cause their excited states to quench. This would suggest that dyes 2, 3, 4 and 6 are compromised for DSC application by dye aggregation, especially 4 and 6 where the H-aggregation appears stronger. In contrast, the J-aggregation in 1 and 5 seems to indicate that they may be suitable for DSC applications. The fact that this J-aggregation is weak bolsters their potential viability, since weak J-aggregates are preferred in DSCs on account of the potentially harmful effects that intermolecular interactions can cause upon dye photoexcitation, which may diminish the electron injection.3 Moreover, according to molecular exciton theory,55 rhodamine J-aggregates will form dimers that are oriented in a head-to-tail fashion, thereby maintaining exposure of their dye anchoring groups to TiO2; as such, electron injection involving each dye component of the dimers is not necessarily compromised. Rather, the presence of some J-aggregate dimers may in fact increase JSC since the dyes stand to pack together more efficiently on the TiO2 surface, offering higher dye loading abilities; dimerization also frequently results in interfacial dye⋯dye interactions (e.g. π⋯π intermolecular stacking) which may enhance ICT.22
Bearing in mind the aforementioned complex dye⋯TiO2 adsorption factors, 1 and 5 were short-listed for further consideration as possible DSC dye candidates (cf.Fig. 3 and 4).
The calculations yielded the bidentate chelating mode as the most favorable dye anchoring configuration for 2, 4 and 6. In case of 1, calculations converged to the monodentate anchoring configuration, irrespective of the two starting models for adsorption; this result was assumed to reflect a limitation of using a (TiO2)9 cluster, which might be too small to rule out alternative sensitization methods on account of edge effects. Nonetheless, this behavior is hereby only reported for this dye, and it consistently yields the same result for both converged structures, confirming the validity of the calculation.
However, the salient results of this study relate to the main differences between anchoring modes, which lies in the adsorption energy (Eads) that can be quantified according to:
| Eads = Edye + ETiO2 − Edye/TiO2, | (3) |
Frontier molecular orbital overlap indexes, Λ, for 1, 2, 4, and 6 were 66% on average, with variations of 0.8%, similar to the results on the isolated dyes in gas or solution phase discussed in §3.1.3.1. No significant changes in HOMO energies and EV values for each dye, between TiO2-adsorbed and non-adsorbed dyes, were observed. Furthermore, the main orbitals involved in the lowest-lying excitations (HOMO-2/HOMO-1/HOMO → LUMO) orbitals did not show a clear transfer of charge density from the dyes to the (TiO2)9 cluster, which is required for successful charge injection (ESI† S.5). This is in line with the findings for the gas-phase calculations for these dyes.
3.3 Co-sensitization of short-listed DSC dye candidates
Previous reports demonstrating that rhodamine dyes successfully function in DSC devices22,23 employed the rhodamine as a co-sensitizer to another dye. It therefore seems to make sense to co-sensitize the subject dyes in order to gain better insight into those reports as well as our own. Moreover, as was discussed earlier, co-sensitization is the natural way forward to test the DSC prospects of these organic dyes, whereby dyes with complementary optical absorption features are paired together to maximize their extent of panchromatic optical absorption.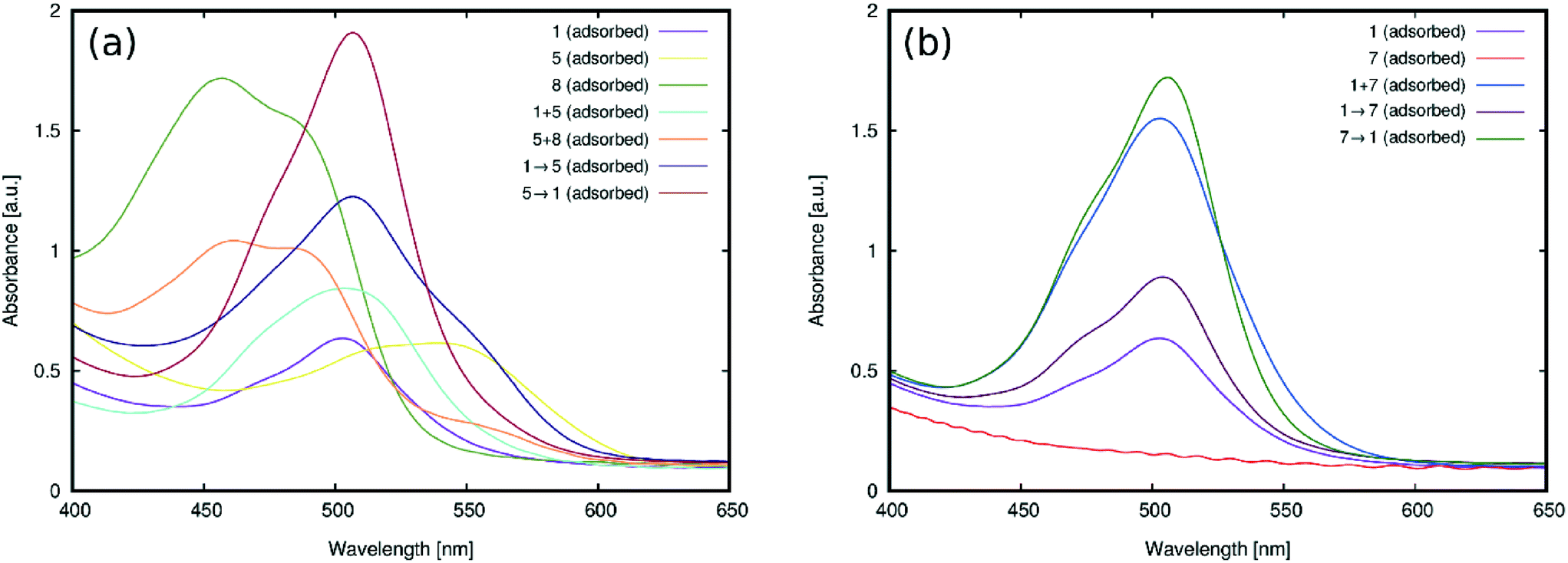 | ||
| Fig. 11 UV/vis absorption spectra of TiO2 films co-sensitized via (a) the cocktail approach for (5 and 8), and cocktail and sequentially sensitized approaches for (1 and 5), compared with the singly-sensitized contributions to the optical absorption for each pair of dyes; (b) cocktail and sequentially sensitized approaches for (1 and 7) compared with the optical absorption for each singly-sensitized dye. Plus (+) symbols between dye labels indicate the use of the cocktail method, while the sequential approach is demarcated by an arrow whereby the dye label before the arrow is the dye sensitized first. | ||
It was also considered helpful to compare the co-sensitization results of 1 and 5 against those of 1 with a non-adsorbing dye from the same chemical family, in order to gain insight into any non-adsorption based competition between the two dyes, e.g. resulting from dye⋯dye interactions. Pairing dyes 1 and 7 for co-sensitization tests seemed to be the natural choice in this regard, and because they offer complementary optical absorption spectra.
Co-sensitization of 5 and 8 was also carried out in order to analyze the effects of pairing a rhodamine dye (5) with a non-rhodamine dye (8). The fluorescein dye 8 seemed to be a good candidate for co-sensitization with 5 since it belongs to a different chemical family, and yet presents similar steric and electronic properties to 5, and offers complementary optical absorption characteristics.
Each of these three pairs of dyes were co-sensitized onto TiO2 thin films twice: using one or the other of the two most common fabrication methods: the cocktail and sequential approach. The results were then compared to each other and against those films that were singly sensitized with dyes (§3.2.1).
The UV/vis absorption spectra for the films of (1 and 5), (5 and 8), and (1 and 7) on TiO2 are shown in Fig. 11. The films fabricated by the cocktail approach are herein denoted by a plus sign (+) between the labels of the dye pairs to symbolize that they are simply mixed together. 1 + 5 and 5 + 8 produced spectra with absorption profiles that resemble the successful merger of the dye pairs which bear an overall better panchromatic range than the analogous results from TiO2 films where 1, 5 or 8 had been singly sensitized (Fig. 11(a)). The pairing of the two subject dyes that show the most potential for co-sensitization with complementary absorption profiles therefore seems justified. Likewise, the test in co-sensitizing a rhodamine dye with a non-rhodamine dye bears out good chemical compatibility.
Co-sensitization of 1 + 7 using the cocktail approach produced more curious results: substantial improvements in optical absorption intensity, with respect to the films that had been singly sensitized with 1 (Fig. 11(b)). As was found in §3.2.1, 7 does not adsorb onto TiO2 owing to its lack of a viable anchoring group. So its role in improving the optical absorption intensity of 1via co-sensitization must be non-adsorption specific in its origin. While the current findings do not reveal its precise role, one possible option for this auxiliary effect of 7 on 1, as a result of cocktail-fabricated co-sensitization, is that 1 and 7 may combine to form aggregates in acetone which, in turn, could ameliorate the adsorption of 1. This process may be driven by the smaller affinity of 1 to remain in solution, which renders the 1 + 7 dimer a better adsorber. This hypothesis is supported by the emergence of a slight bathochromic shift (by 3 nm) in the peak optical absorption wavelength for the spectrum of the 1 + 7 co-sensitized film, compared to that of singly-sensitized 1. The resulting peak wavelength for 1+7 corresponds closely to that of 7 in solution, suggesting the presence of 7 in the co-sensitized film.
 | ||
| Fig. 12 UV/vis absorption spectra of TiO2 films co-sensitized with 5 and 8 using the sequential approach. The spectra for films singly sensitized with 5 or 8 are added for comparison. Table on top-right: dye pairings employed in sequential co-sensitization with different solvents. The dye on each row is the first dye in the sequence to be sensitized, while the one on each column is the second in the co-sensitization sequence. The two digits in the non-empty cells indicate a successfully tested pairing, with the first letter representing the solvent used for the first dye in the sequence, while the second letter represents the solvent used for the second dye in the sequence of co-sensitization. In this context, A corresponds to acetone, E corresponds to ethanol. | ||
More details of UV/vis absorption experiments on TiO2 films co-sensitized by 5 and 8 using the sequential approach are shown in Fig. 12. Different orders of dye sensitization and solvents pairings were tested. The best dye adsorption was observed for 8 → 5, when ethanol and acetone were used for 8 and 5, respectively. This choice of solvent and sensitization order is the only option providing the overall optimal adsorption of 5, compared to other co-adsorbers, while maintaining high adsorption of its co-adsorber, 8. Fig. 12 also shows the detrimental effects of less optimal co-sensitization orders and solvents on the absorption intensity of the spectra. Indeed, 5 and 8 differ in their preferred solvents (8: ethanol; 5: acetone), as was similarly noted when applying the cocktail approach to co-sensitization (§3.3.2); as Fig. 12 demonstrates, desorption of 5 was evidenced whenever ethanol was used in the second sequential step of co-sensitization.
3.4 DSC device fabrication and testing
Experimental results in §3.3.2 and §3.3.3 showed that 1 and 5 successfully adsorb onto TiO2. By extension, one might therefore assume that all rhodamine dyes that contain a carboxylic acid group can do the same, even though 1 and 5 may exhibit more intense optical absorption. Nonetheless, this expression of optical properties in the DSC working electrode still needs to be translated into the full DSC device.These poor JSC results can be understood by the manifold evidence, which has unfolded during the course of this study, that the subject dyes lack a suitable D–π–A type ICT pathway for DSC applications. In the absence of such ICT characteristics, electron injection from the dye into the conduction band of TiO2 cannot occur, and yet it is needed in order to initiate the electrical current in the DSC working electrode. It is significant that the structure–function relationships which appear to be at the heart of the ICT issue all concern a fundamental property of all rhodamine dyes: the lack of π-conjugation between the xanthenes and substituted phenyl rings to link the electron donor to the anchoring group to afford the basic D–π–A molecular architecture needed for DSC applications.
This would seem to suggest a more general notion that, despite their good optical properties, no rhodamine dye should be able to produce photovoltaic output in a DSC device. The structure–function relationships revealed from this study of seven rhodamine dyes are internally consistent with each other and concur with our photovoltaic DSC device performance test results. But how can we explain our findings in terms of the previously reported successful DSC studies using rhodamine dyes?
The other DSC performance focused report23 explores the use of Rhodamine 575 (known as 2 in this study) as a co-sensitizer with the deprotonated form of the standard industrial standard dye, N3, to enhance the photovoltaic performance conversion efficiency (PCE) of N3-based DSCs. Their salient finding was that TiO2 co-sensitized with 2 and N3 augments the PCE of their devices from 2.3% (singly-sensitized N3) to 4.7% (2 and N3 co-sensitized) while DSCs sensitized with only 2 produce a PCE of 0.6%.
All other reports on rhodamine dyes for DSC applications19–21 are focused on developing DSC fabrication technology rather than DSC performance. Moreover, all of their subject rhodamine dyes are one or more of the same dyes that are contained in the two reports focused on DSC device performance.22,23 So it would appear that the two reports, discussed above, can substitute for the associated DSC performance considerations for the rhodamine dyes in these other studies.
The results from Bhattacharya and co-workers22 can be rationalized in a manner consistent with our findings herein. They similarly report essentially negligible photovoltaic performance for DSCs containing singly-sensitized 3 and 4. The hydrogen-bonding mechanism that they propose to explain their positive co-sensitization results also stands to reason given our studies evidence that dye aggregation effects can occur with 3 and 4 (both causing hypochromic shifts in the UV/vis absorption spectra; see §3.2.1). Moreover, the presence of dye aggregation effects does not mandate that all dyes in the aggregate must be involved in dye-to-TiO2 electron injection in order to have an auxiliary role in boosting the DSC performance of one of the dyes involved. Indeed, Bhattacharya and co-workers nicely exemplified this via their UV/vis absorption and fluorescence spectroscopy results which reveal that 3 and 4 do adsorb onto TiO2, but behave as ground-state species given their invariance in fluorescence lifetime upon co-sensitization with their subject porphryin. So it is entirely possible that 3 and 4 could facilitate the DSC performance of the porphyrin dye in the study by Bhattacharya and co-workers,22 without themselves acting directly as a DSC dye.
The results from Saxena and co-workers23 are more difficult to reconcile with our findings. Indeed, they would appear to contradict our results directly when considered at face value. However, DSC results are notoriously hard to compare, owing to consistency issues associated with various control parameters in the DSC device fabrication process.3 It therefore seemed prudent to attempt to reproduce their N3 + 2 co-sensitization experiments with our laboratory set-up for DSC fabrication and testing, in order to diagnose the apparent discrepancy. That way, we can generate internally consistent results on N3 + 2 co-sensitization to compare with our photovoltaic studies on all of our subject dyes, as well as produce an independent set of data to understand better this co-sensitization process. For the purposes of providing more general reference data on the role of rhodamines in their co-sensitization with N3, the co-sensitization of N3 with 1 was also explored during the course of these tests. 1 was the target dye for this reference data collection since its core molecular fragment is the least chemically substituted of all subject dyes while it demonstrates strong gains in optical absorption intensity upon adsorption to TiO2 in both singly- and co-sensitized forms (§3.2.1 and §3.3, respectively).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile/tert-butanol solution of N3 for 24 h; Saxena et al. used an ethanol solution of N3 and performed this sensitization step under three different dye soaking times: 14 h, 20 h and 40 h. Their tabulated values for the DSC device performance pertain to the 40 h soaking time. These values are reproduced in Table 4 together with the results from our study. For the purposes of ensuring internal consistency throughout our study, the soaking times and solvents used in our co-sensitization work with N3 were constrained to be the same as those used in all of our singly sensitized DSC device studies.
:1 acetonitrile/tert-butanol solution of N3 for 24 h; Saxena et al. used an ethanol solution of N3 and performed this sensitization step under three different dye soaking times: 14 h, 20 h and 40 h. Their tabulated values for the DSC device performance pertain to the 40 h soaking time. These values are reproduced in Table 4 together with the results from our study. For the purposes of ensuring internal consistency throughout our study, the soaking times and solvents used in our co-sensitization work with N3 were constrained to be the same as those used in all of our singly sensitized DSC device studies.
| Dyes | J SC [mA cm−2] | V OC [mV] | FF [%] |
η
dye:ηN3 [%] |
|---|---|---|---|---|
| N3 (this work) | 9.5 ± 0.5 | 629 ± 14 | 62 ± 8 | 100.0 |
| N3 (Saxena et al.) | 6.97 | 641 | 53 | 100 |
| 2 → N3 (this work) | 6.7 ± 1.1 | 595 ± 15 | 69 ± 2 | 71.0 |
| 2 → N3 (Saxena et al.) | 11 | 695 | 62 | 200 |
| 1 → N3 (this work) | 4.9 ± 2.0 | 560 ± 19 | 66 ± 3 | 47.4 |
Fig. 13 presents the UV/vis absorption spectra of our TiO2 films co-sensitized with 1 or 2; those for singly sensitized 1, 2 and N3 are also presented for the ease of comparison. These optical results show that co-sensitizing 1 or 2 with N3 enhances the overall UV/vis absorption intensity in both cases. The corresponding spectral profiles for 1 and 2 appear to be largely unaffected by the introduction of N3.
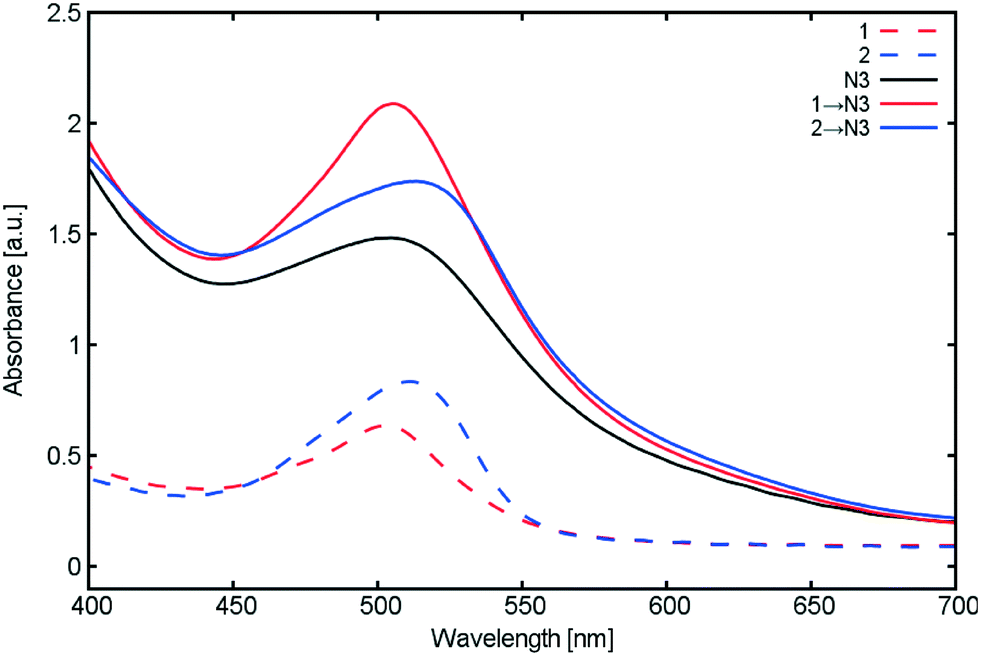 | ||
| Fig. 13 UV/vis absorption spectra of TiO2 films sensitized with 1, 2 and N3, together with those for the singly-sensitized dyes for the purposes of comparison. | ||
Similarly, Saxena and co-workers23 similarly report an increase in UV/vis absorption between singly-sensitized N3 and 2 → N3 co-sensitized TiO2 films. The authors state that the shape of the UV/vis absorption spectrum upon introduction of N3 is only affected by intensity. In the absence of any spectral shifts that would evidence solvatochromism, they consequently rule out the possibility of dye aggregation occurring as a result of co-sensitization process, which is consistent with our results shown in Fig. 13.
The performance characteristics of the co-sensitized DSC devices that incorporate N3 (Table 4) were then analyzed. To this end, the two DSC device fabrication approaches from each investigation were first benchmarked against each other, by comparing results on N3 singly-sensitized DSC devices that act as reference data. The photovoltaic output for the singly-sensitized N3 devices in our work and that of Saxena et al.23 are comparable in terms of open-circuit voltage (VOC) and fill factor (FF). In stark contrast, the short-circuit current densities (JSC) differ significantly between the two investigations, with a higher value presenting in our work. This indicates that we obtained higher dye coverage in this work, according to the following argument: a larger JSC signifies a more extensive dye-to-TiO2 charge injection process as it is this which initiates electrical current; in turn, better electron injection requires greater levels of dye adsorption onto the TiO2 surface pending that the dyes adsorbed bear an efficient injection mechanism (which is proven for N3).
It is well known that JSC measurements tend to bear the greatest variability in DSC testing, out of the three salient photovoltaic performance characteristics shown in Table 4. As a consequence, it is usually the primary source of variation in the corresponding PCE calculation. Since the variation in VOC and FF between independent device tests on DSCs tends to be so much lower than that of JSC, it has been found that the reliability of statements on PCE results can be much improved, and are therefore far more informative, if they are reported as a ratio, rather than as an absolute PCE value.57 This ratio is a measure of the PCE for a DSC containing the subject dye being assessed, against that of a DSC which incorporates an industrial standard dye (e.g.N3) where this device has been fabricated and tested under the same conditions. The ratio, ηdye:ηN3 [%], rather than absolute PCE values, are therefore reported in Table 4. Nonetheless, for this particular comparison, it is helpful to additionally provide the absolute PCE values obtained for DSCs containing the reference dye, N3, for the purposes of judging the respective extents of dye loading in each study; accordingly, ηN3 obtained by Saxena et al.23 was 2.37%, while this report issued a ηN3 of 3.78%.
With these reference data understood, the DSC device performance characteristics of N3 co-sensitized with 2 from both studies were compared (Table 4). This is where discrepancies deepen. Whilst the FF results are comparable, VOC and JSC values between the two studies not only differ substantially in magnitude but also in the sense by which they change relative to their cognate singly-sensitized N3 findings, with JSC showing the biggest contributions to the change. The same trend is echoed by the ηdye:ηN3 percentages, whereby the co-sensitization of N3 with 2 in our study is found to actually diminish the PCE, affording 71% of the original value of a DSC containing N3; in stark contrast, Saxena and co-workers23 observed a 200% increase in PCE. For the purposes of making a more general comparison about the role of rhodamines in this process, N3 was also co-sensitized with 1 in our study; as Table 4 shows, this rendered an even more significant drop in DSC device performance, to less than 50% of that of the singly-sensitized N3 reference cell.
Comparing our results with those of Saxena and co-workers,23 one marked difference is that our DSC fabrication process afforded significantly higher dye coverage, as judged by the difference in the reference DSC device performance data on the N3 singly-sensitized DSCs. This would tend to suggest that the origins of this ostensible discrepancy between these two sets of independent rhodamine and N3 co-sensitized photovoltaic data could have something to do with dye concentration in some fashion. Dye coverage is a parameter that is highly controlled by the solvents used in the co-sensitization process. The findings of our study (§3.3.2 and §3.3.3) concluded that ethanol was a strongly dye-interacting solvent which would inhibit co-sensitization of the dyes on TiO2, and it was ethanol that was used in the report by Saxena et al.23 for all of their fabricated DSCs. This could indicate that different chemical processes are at play during the sequential co-sensitization process of 2 and N3 dyes between these two studies. Our results, on the contrary, might imply a higher level of sensitization of 2, which subsequently lowers the number of TiO2 adsorption sites available for N3, thereby lowering the overall power conversion efficiency. Given our findings on solvent effects on dye adsorption to TiO2 (§3.3.2 and §3.3.3), a different mechanism of sensitization might be reversing this effect in the study by Saxena et al.,23 that could be opening more adsorption sites on TiO2 and allowing higher sensitization of N3, justifying the impressive improvements from the reference cells, without 2 necessarily playing an active role in the co-DSC functions. This is in line with the improvements in both JSC and VOC reported by Saxena et al.,23 which contrasted with our results. Furthermore, it reconciles our finding that rhodamines are not exploiting the ICT characteristics to improve performance of co-DSCs, as was presumed for 2 in the report by Saxena and co-workers;23 rather, it seems that solvent influences may be behind the ostensible discrepancies in DSC photovoltaic outputs between the two independent studies.
Another hypothesis that can explain these discrepancies is the presence of unmeasurable aggregation of different types which is reducing the performance in JSC reported in our results. Given that dye aggregation is a dye concentration-dependent process, and that Bhattacharya and co-workers22 found that dye aggregation played a significant role in their findings, it seems natural to also consider dye aggregation as a possible source of these origins.
Interestingly, Bhattacharya et al.22 showed that rhodamine dye aggregation had a facilitatory effect on the porphyrin dye performance, in stark contrast to the deleterious effect that they display on N3. This makes sense when one considers that a porphyrin molecule is essentially two-dimensional in form, and so it can interact directly with rhodamine dye molecules, as Bhattacharya et al.22 observed, presumably via π⋯π stacking between the flat π-conjugated portions of the respective dyes, which are substantial. In contrast, N3 is a three-dimensional and bulky dye molecule, and so it cannot interact easily with rhodamine dyes; indeed, no interactions between N3 and rhodamine dyes were observed; rather, the bathochromic effects observed in this study evidence that the rhodamine dyes aggregate with themselves.
Additional mitigating factors that affect the disparity between the PCEs obtained from this study and those of Saxena and co-workers23 could be found due to the variation in the fabrication procedures. Although the fabrication process control parameters reported for each investigation seem similar, DSC device performance results are notoriously hard to compare across independent data sets. Nonetheless, the use of the PCE ratio method57 in comparing PCE values from each study should make our assessment reliable.
4. Conclusions
This systematic molecular design study has explored the prospects of seven well-known rhodamine dyes (1–7) for DSC applications. A concerted experimental and computational approach was applied to investigate the structural attributes and optical properties of 1–7 in relation to how well they are aligned to the photovoltaic functionality that is required for them to act as a DSC dye. All seven dyes were initially studied in the solution state, and those bearing appropriate anchoring groups were then investigated within the molecular architecture of the DSC working electrode: a dye⋯TiO2 interface. The collective results indicated that the subject dyes could adsorb onto a TiO2 surface, wheresoever the subject dyes possessed suitable anchoring groups; but the core molecular framework that defines these dyes as rhodamines does not bear a suitable ICT pathway to enable electron injection from the dye to the TiO2 surface once adsorbed. Accordingly, rhodamine dyes are not able to produce photovoltaic output in their own right. DSC performance tests on devices singly-sensitized with these subject rhodamine dyes, corroborated this notion.That said, the literature evidences successful DSC device outputs when rhodamines are co-sensitized with other types of dyes.19–23 So co-sensitization tests were carried out on selective pairings of subject dyes (1 and 5; 1 and 7) which, when combined, afforded panchromatic absorption owing to their complementary optical absorption signatures. 5 was also co-sensitized with a fluorescein chromophore (8) that belongs to a chemically related family of dyes, in order to explore chemical compatibility. DSC working electrodes that incorporated these pairs of dyes were fabricated using two co-sensitization methods: the cocktail and sequential approaches. It transpired that the sequential approach produced superior results.
DSC device tests on 1–6 yielded negligible photovoltaic output. On the one hand, our photovoltaic performance tests could be understood in terms of the structure–function relationships that have unfolded during the course of this study, especially regarding the ability of rhodamine dyes to adsorb onto TiO2 but not possess a suitable ICT pathway to effect dye-to-TiO2 electron injection which is needed to initiate the electrical circuit in DSCs. These findings appeared to be consistent with a DSC device report in the literature that employs rhodamines as co-sensitizers.22 On the other hand, our results initially seemed to be in discord with one report in the literature23 that demonstrates successful photovoltaic output of DSCs containing rhodamine dyes which are co-sensitized with another type of dye. These literature results were therefore further inspected, which included the internal reproduction of some co-sensitization experiments on 1 and 2 with the industrial standard reference dye, N3. A collective comparison of our results with those from the literature22,23 revealed a consistent and general finding, i.e. rhodamine dyes cannot actually function as DSC dyes owing to their lack of suitable ICT pathway to create electron injection to enable DSC function (see §3.1.1.2); yet, their demonstrated successful use as DSC co-sensitizers22,23 suggest that alternative chemical processes ensuing during dye adsorption onto TiO2 can contribute towards improving the co-DSC performance, even though they are not actively participating in the light absorption mechanism (see §3.4.4). This can be related to dye aggregation or to solvent influences that could increase the available adsorption sites for the co-sensitized dye.
In broader terms, the chemical processes ensuing at the dye⋯TiO2 interface during adsorption needs to be understood a lot better at the molecular level, especially dye aggregation and the choice of solvent for co-sensitization; yet, it remains complex to determine by experiment or to model, given that intermolecular interactions are so difficult to predict even in regular crystal structures,59 let alone in solution mixtures. Nonetheless, this work has shown that our combined efforts on experimental and computational work has provided a consistent rationale behind the role of rhodamine dyes in DSC research, and has shed more light on their true role as co-sensitizers. With further work that continues to employ a concerted experimental and computational approach, we hope to unravel further insights into the dye-adsorption process of other families of dyes.
Acknowledgements
G. P. thanks the EPSRC for a DTA Studentship (Reference: EP/K503009/1). J. M. C. is grateful to the 1851 Royal Commission for the 2014 Design Fellowship, and Argonne National Laboratory where work done was supported by DOE Office of Science, Office of Basic Energy Sciences, under Contract No. DEAC02-06CH11357. The Bragg Institute at ANSTO is gratefully acknowledged for funding (for P. G. W.). All authors thank the EPSRC UK National Service for Computational Chemistry Software (NSCCS) and acknowledge contributions from its staff in supporting this work.References and notes
- S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181 CrossRef.
- D. J. Hofmann, J. H. Butler and P. P. Tans, Atmos. Environ., 2009, 43, 2084 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- Z. Xie, et al. , Chem. Commun., 2014, 50, 608 RSC.
- C. Bechinger, S. Ferrere, A. Zaban, J. Sprague and B. A. Gregg, Nature, 1996, 383, 608 CrossRef CAS.
- K.-S. Ahn, S. J. Yoo, M.-S. Kang, J.-W. Lee and Y.-E. Sung, J. Power Sources, 2007, 168, 533 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- A. Yella, et al. , Science, 2011, 334, 629 CrossRef CAS PubMed.
- K. Kakiage, et al. , Chem. Commun., 2015, 51, 15894 RSC.
- M. Kimura, H. Nomoto, N. Masaki and S. Mori, Angew. Chem., 2012, 124, 4447 CrossRef.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed.
- M. Cheng, X. Yang, J. Li, F. Zhang and L. Sun, ChemSusChem, 2012, 6, 70 CrossRef PubMed.
- G. D. Sharma, M. S. Roy and S. P. Singh, J. Mater. Chem., 2012, 22, 18788 RSC.
- S. L. Bayliss, J. M. Cole, P. G. Waddell, S. McKechnie and X. Liu, J. Phys. Chem. C, 2014, 118, 14082 CAS.
- F. A. Y. N. Schröder, J. M. Cole, P. G. Waddell and S. McKechnie, Adv. Energy Mater., 2015, 5, 1401728 CrossRef.
- G. Pepe, J. M. Cole, P. G. Waddell and S. McKechnie, Mol. Syst. Des. Eng., 2016, 1, 86 Search PubMed.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465 RSC.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410 RSC.
- H. K. Singh, et al. , Vacuum, 2013, 87, 21 CrossRef CAS.
- P. K. Baviskar, et al. , J. Alloys Compd., 2012, 510, 33 CrossRef CAS.
- X. Zhang, et al. , Nanoscale, 2012, 4, 1707 RSC.
- S. Bhattacharya, et al. , J. Nanosci. Nanotechnol., 2011, 11, 7735 CrossRef CAS PubMed.
- V. Saxena, et al. , Appl. Phys. Lett., 2012, 100, 133303 CrossRef.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427 CAS.
- Materials Genome Initiative for Global Competitiveness, U.S. Government White Paper; National Science and Technology Council, Executive Office of the President of the United States, 2011 Search PubMed.
- Crystal Clear-SM Expert, 2.0 Software Package, 2009 Search PubMed.
- A. Messerschmidt, M. Schneider and R. Huber, J. Appl. Crystallogr., 1990, 23, 436 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112 CrossRef PubMed.
- J. C. Williams, Treatise Mater. Sci. Technol., 1976, 9, 173 CAS.
- M. J. Frisch, et al., Gaussian 09 Revision D.01, Gaussian Inc. Wallingford CT, 2009 Search PubMed.
- D. Kats, T. Korona and M. Schutz, J. Chem. Phys., 2006, 125, 104106 CrossRef PubMed.
- D. Kats and M. Schutz, J. Chem. Phys., 2009, 131, 124117 CrossRef PubMed.
- H.-J. Werner, et al., MOLPRO, version 2012.1, a package of ab initio programs ( 2012) Search PubMed.
- B. Hammer, L. Hansen and J. Norskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- D. M. Chipman, J. Chem. Phys., 2000, 112, 5558 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- M. D. Hanwell, et al. , J. Cheminf., 2012, 4, 17 CAS.
- R. S. de Armas, M. A. S. Miguel, J. Oviedo and J. F. Sanz, Phys. Chem. Chem. Phys., 2012, 14, 225 RSC.
- R. S. de Armas, et al. , J. Chem. Theory Comput., 2010, 6, 2856 CrossRef PubMed.
- R. S. de Armas, M. A. San-Miguel, J. Oviedo, A. Marquez and J. F. Sanz, Phys. Chem. Chem. Phys., 2011, 13, 1506 RSC.
- R. S. de Armas, J. Oviedo, M. A. S. Miguel and J. F. Sanz, J. Phys. Chem. C, 2011, 115, 11293 Search PubMed.
- X. Liu, J. M. Cole and K. S. Low, J. Phys. Chem. C, 2013, 117, 14723 CAS.
- International Union of Crystallography, International tables for X-ray crystallography, Kynock Press, 1959, vol. 2 Search PubMed.
- W. P. Oziminski and T. M. Krygowski, J. Phys. Org. Chem., 2010, 23, 551 CrossRef CAS.
- F. H. Allen, et al. , J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- T. M. Krygowski, R. Anulewicz and J. Kruszewski, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 732 CrossRef.
- J. Karolak-Wojciechowska, Acta Crystallogr., Sect. B: Struct. Sci., 1987, 43, 574 CrossRef.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- T. Horiuchi, H. Miura and S. Uchida, Chem. Commun., 2003, 3036 RSC.
- D. Setiawan, A. Kazaryan, M. A. Martoprawiro and M. Filatov, Phys. Chem. Chem. Phys., 2010, 12, 11238 RSC.
- M. Kasha, Radiat. Res., 1963, 20, 55 CrossRef CAS PubMed.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2014, 6, 15760 CAS.
- J. M. Cole, K. S. Low, H. Ozoe, P. Stahi, C. Kitamura, P. Rudolf and T. Kawase, Phys. Chem. Chem. Phys., 2014, 16, 26684 RSC.
- S. Wenger, et al. , J. Am. Chem. Soc., 2010, 132, 5164 CrossRef CAS PubMed.
- E. Gibney, Nature, 2015, 527, 20 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1498038–1498040. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6me00076b |
| ‡ Current address: School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. |
| This journal is © The Royal Society of Chemistry 2016 |