
One-dimensional assembly of functional proteins: toward the design of an artificial cellulosome†
Y.
Mori‡
a,
H.
Nakazawa
b,
G. A. L.
Gonçalves
a,
T.
Tanaka
c,
M.
Umetsu
b and
N.
Kamiya
*ad
aDepartment of Applied Chemistry, Graduate School of Engineering, Kyushu University, 744 Motooka, Fukuoka 819-0395, Japan. E-mail: nori_kamiya@mail.cstm.kyushu-u.ac.jp; Fax: +81 (0)92 802 2810; Tel: +81 (0)92 802 2807
bDepartment of Biomolecular Engineering, Graduate School of Engineering, Tohoku University, Aoba 6-6-11, Aramaki, Aoba-ku, Sendai, 980-8579, Japan
cDepartment of Chemical Science and Engineering, Graduate School of Engineering, Kobe University, 1-1 Rokkodaicho, Nada, Kobe 657-8501, Japan
dDivision of Biotechnology, Center for Future Chemistry, Kyushu University, Japan
First published on 15th March 2016
Abstract
In biological systems, proteins can form well-organized, higher-order structures with unique functions that would be difficult to achieve with a single protein. These proteinaceous supramolecular structures form by self-assembly, and the spatial arrangement of the protein building blocks in them is very important. In the present study, an artificial system was developed using recombinant proteins as building blocks, which were assembled in a one-dimensional manner. The assembly of these building blocks was based on the avidin–biotin interaction. A tetrameric biotin ligand unit was designed so that the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 stoichiometry of the avidin–biotin interaction was altered to a 1:2 directional interaction between the streptavidin and tetrabiotinylated protein units. In a proof-of-concept study, site-specifically tetrabiotin-labeled endoglucanase and cellulose-binding module units were prepared, and then these components were self-assembled by mixing with streptavidin to mimic a natural cellulosome. The formation of one-dimensional assemblies of the protein units depended on the stoichiometry of the avidin–biotin interaction sites in the system. Interestingly, the saccharification efficiency improved when the component ratio of protein units in the assemblies was changed. The presence of the optimal ratio of the building blocks implies the modularity of the present protein assembly system.
:4 stoichiometry of the avidin–biotin interaction was altered to a 1:2 directional interaction between the streptavidin and tetrabiotinylated protein units. In a proof-of-concept study, site-specifically tetrabiotin-labeled endoglucanase and cellulose-binding module units were prepared, and then these components were self-assembled by mixing with streptavidin to mimic a natural cellulosome. The formation of one-dimensional assemblies of the protein units depended on the stoichiometry of the avidin–biotin interaction sites in the system. Interestingly, the saccharification efficiency improved when the component ratio of protein units in the assemblies was changed. The presence of the optimal ratio of the building blocks implies the modularity of the present protein assembly system.
Introduction
Proteins are essential in the control of many biological functions. When proteins are used practically as nanomaterials, a bottom-up approach based on self-assembly is useful.1 In nature, homo- and heteromeric supramolecular complexes of proteins can form via self-assembly. These supramolecular protein assemblies are one-, two- or three-dimensional structures as required for their function, such as actin, tubulin, S-layer proteins or capsids.2 In some cases in nature, non-covalent interactions and post-translational covalent modifications are used to facilitate the growth and stabilization of proteinaceous structures. For example, pili, which are extracellular structures involved in adhesion and bacterial conjugation, form one-dimensional polymeric structures by self-assembly of monomeric protein units and subsequent covalent attachment at specific sites. In some cases, the functions of individual protein building blocks can be enhanced by a protein assembly. For example, antibodies can cluster together in homomultimers to greatly increase the binding affinity to target molecules. In addition, some cellulolytic bacteria and fungi produce multi-enzyme complexes called cellulosomes, which show improved efficiency for biomass hydrolysis.3–5Many studies have investigated strategies for combining functional proteins via self-assembly to produce novel artificial supramolecular complexes. The two main methods that have been used for protein assembly are a template-based method where the target proteins are assembled onto a template and a ligand-based method where the target proteins are labeled with ligands and assembled using the binding affinity between receptor/ligand pairs (affinity pairs). For the template-based method, DNA,6 RNA,7 protein scaffolds,8–18 organic and non-organic materials19 have been used as templates. For the ligand-based method, the avidin–biotin system has been very important for supramolecular protein assembly in nanotechnology and biotechnology.20–22 (Strept)avidin is a tetrameric protein that forms a non-covalent, stable bond with the small organic ligand molecule, biotin. Upon biotin labeling to a protein of interest, the biotinylated target proteins can be assembled through the avidin–biotin interaction.23–27 The use of this non-covalent interaction in protein assembly is attractive because dynamic assembly can be realized.28–30 However, precise control of the formation, including the number of protein units, topology, solubility and expected functionality of the artificial supramolecular complex, is challenging.
In an initial investigation with the biotinylated dimeric enzyme alkaline phosphatase from bacteria, we formed protein supramolecular complexes using the avidin–biotin interaction.31 This research suggested that the formation of a one-dimensional protein assembly would be possible with the introduction of a bis-biotinylated ligand on the long-axis edge of the enzyme. The investigation with the dimeric protein also suggested that controlled assembly of functional proteins as building blocks could be achieved by altering the avidin–biotin interaction topology. In the present study, we performed a proof-of-concept study to create a one-dimensional protein nanostructure composed of monomeric functional proteins using a multivalent ligand and its affinity pair (Fig. 1). The proposed concept was validated by fabrication of an artificial cellulosome as a model molecular system.
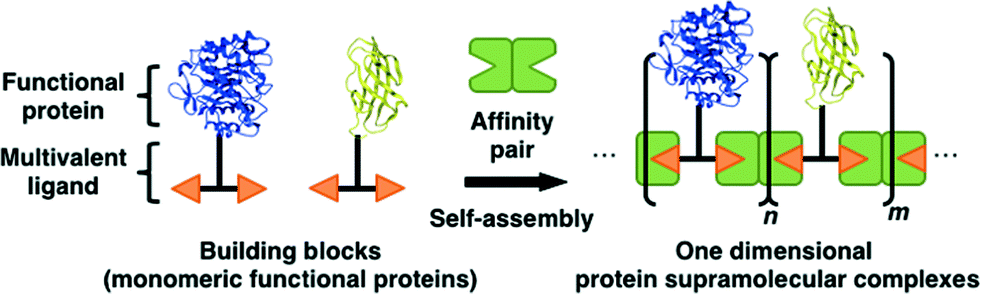 | ||
| Fig. 1 Concept for the design of a one-dimensional protein supramolecular assembly using a multivalent ligand and its affinity pair. | ||
Results and discussion
Design concept for one-dimensional protein assembly
For protein assembly via the avidin–biotin interaction, the design of the biotin ligands was key to the control of the structures of the protein supramolecular complexes. The avidin-like protein streptavidin (SA) forms a tetramer and has four biotin-binding sites. In its quaternary structure, the cis-related dimer has two binding sites facing in the same direction. The tetramer is assembled with two cis-related dimers facing in opposite directions32 (Fig. S1†). Ward et al. designed a bis-biotin ligand to bind two biotin-binding sites of the cis-related dimer in SA,33 which could control the structure of avidin–biotin complexes.34In this study, a one-dimensional structure was fabricated from monomeric functional proteins via the avidin–biotin interaction. Enzymatic biotinylation was conducted with microbial transglutaminase (MTG), which can catalyze an acyl-transfer reaction between the side chain of Gln (Q) and Lys (K) in specific peptide sequences. Given that a Lys-containing peptide tag (MRHKGS; K-tag) which could be recognized by MTG was fused to the monomeric proteins, we designed a novel tetra-biotinylation substrate, bis(bis(biotin-GGG)-K)-KGLQG (Q-biotin4) (Fig. 2). The tri-peptide Leu-Gln-Gly (LQG) was selected as the Q-donor sequence for the biotinylation of the K-tagged proteins. For the combined bis-biotin ligands and SAs, the length between biotin moieties in the ligand does not promote intramolecular interactions (e.g. binding to cis-related dimer in SA) but rather intermolecular interactions (e.g. binding to two SAs). Based on the distance between the cis-related biotin-binding sites (20 Å), the length between the biotin moieties in the bis-biotin moiety of Q-biotin4 was set to avoid branching of the complexes and to promote the formation of the one-dimensional structure. The length between the two bis-biotin moieties of Q-biotin4 was shorter than the distance between trans-related biotin-binding sites (35 Å) to prevent occupation of the four biotin-binding sites in SA with one tetra-biotin ligand and to promote extension of the avidin–biotin complexes.
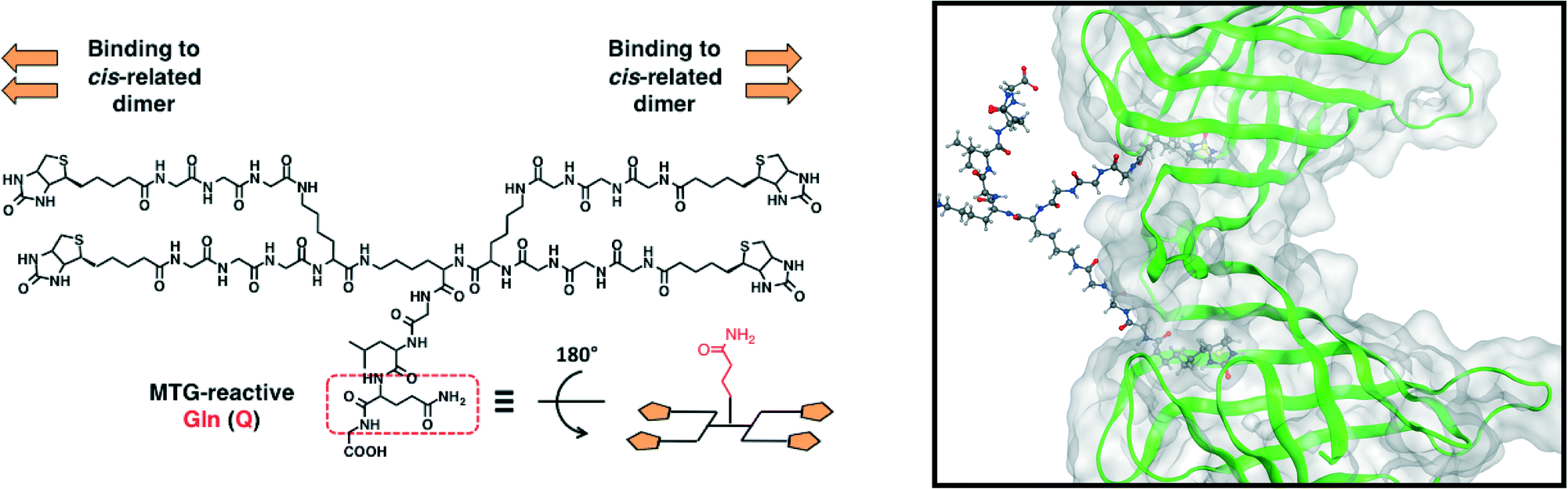 | ||
| Fig. 2 Molecular design of a novel tetra-biotinylation substrate (left), bis(bis(biotinGGG)-K)-KGLQG (Q-biotin4). Bis-biotin moieties can bind to cis-related biotin-binding sites in SA (right box, only the cis-related dimer is shown. Structural data from PDB: 1STP). | ||
Q-biotin4 was synthesized by solid-phase peptide synthesis and its function as a MTG substrate was confirmed by conjugation with dansylcadaverine, which is a MTG-reactive primary amine substrate. Q-biotin4 was recognized by MTG (Fig. S2†) and was employed for further study as a MTG-reactive tetrabiotinylation substrate.
Design of an artificial cellulosome system
In the 1980s, Bayer et al. discovered the first multi-enzyme complexes called as cellulosomes at the surface of the anaerobic thermophilic bacterium Clostridium thermocellum.35 After the discovery of cellulosomes, many studies have been conducted to explore the molecular mechanisms of the formation of multi-dimensional structures and the sophisticated biological functions of cellulosomes.3–5,36,37 In a natural cellulosome system, the one-dimensional core structure of the cellulosome is formed through cohesin–dockerin interactions, which are non-covalent and high-affinity protein–protein interactions between several cohesin domains on the scaffold protein and a dockerin domain fused to the biomass-degrading enzymes. The cellulosome could bind to the biomass surface and degrade the lignocellulosic biomass efficiently on the cell surface of the host bacteria because of many factors such as spatial enzyme proximity, synergistic action among biocatalytic components, and the rapid uptake of produced soluble oligosaccharides to prevent substrate inhibition. Since the design of artificial cellulosomes was proposed in 1994,38 to improve the degradation efficiency of cellulosic biomass, designer and artificial cellulosomes have been fabricated using the cohesin–dockerin interaction,12–18 other non-covalent affinity systems39–42 or covalent conjugation.43–45 In our previous study, an artificial cellulosome was fabricated in the form of several cellulases covalently conjugated onto DNA scaffolds and the improved saccharification efficiency was observed.43 In terms of the heteroprotein assembly, however, quantitative crosslinking of different types of enzymes on the same DNA scaffold was difficult (unpublished data). The use of non-covalent affinity pairs as a driving force is thus advantageous to the assembly and diversification of the protein components. As depicted in the cellulosome system, the different types of protein units fused with the affinity domain can be assembled with the counterpart affinity pair spatially arranged on the scaffolds.In the present study, we designed a new artificial cellulosome system using the avidin–biotin interaction (Fig. 3). For this system, we selected endoglucanase Cel5A from Thermobifida fusca (EG) as a model biomass-degrading enzyme. EG can randomly hydrolyze the β-1,4-glycosidic bonds of internal cellulose chains in the amorphous region, and has a catalytic domain (CD) and a cellulose-binding module (CBM). Cel5A has been used in previous reports on designer cellulosomes as the cellulase components.12–14 While endoglucanase Cel5A belongs to the GH5 family and this enzyme family is commonly found in a natural cellulosomal system,4 EG from T. fusca is a free cellulase (non-cellulosomal enzyme).46 Thus, we aimed at the improvement of the saccharification efficiency by assembling EG into the artificial cellulosomal system. A K-tag was fused to the C-terminus of CD (CD-CK) and EG (EG-CK) for quantitative and site-specific tetrabiotinylation with Q-biotin4 (Step 1, Fig. 3). By mixing with SA, tetrabiotinylated CD-CK (b4-CD) and EG-CK (b4-EG) were then complexed into a one-dimensional assembly with a topology similar to that of a natural cellulosome (Step 2, Fig. 3). Finally, the catalytic function of the one-dimensional avidin–biotin complex was investigated in the enzymatic saccharification of a cellulose substrate.
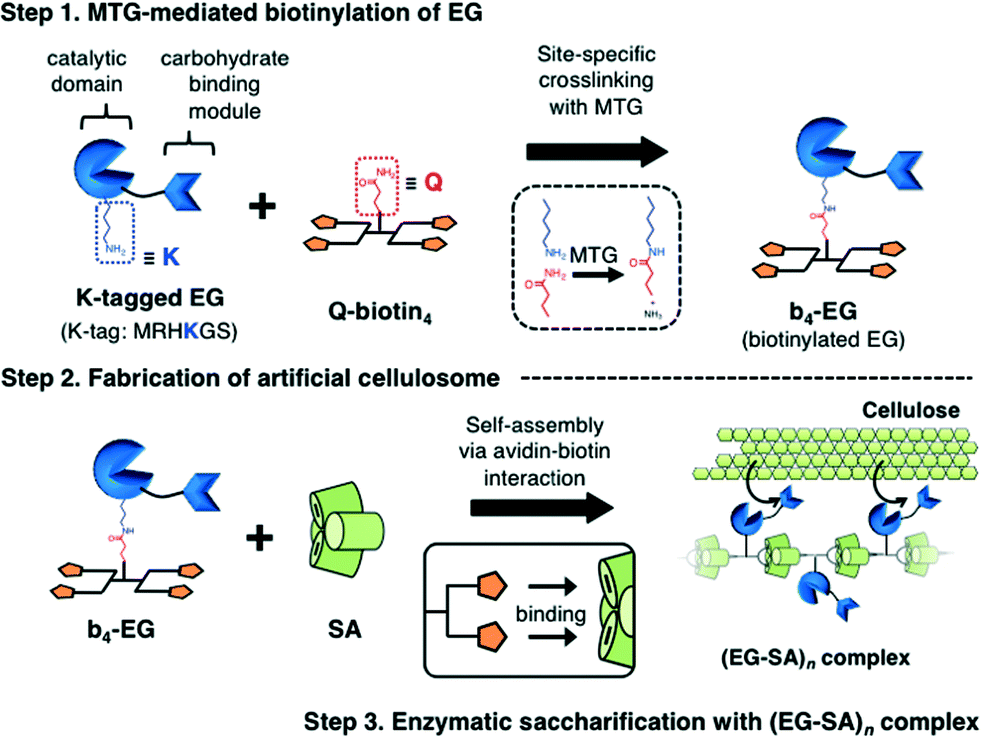 | ||
| Fig. 3 Schematic illustration of MTG-mediated fabrication of the artificial cellulosome (EG–SA)n complexes. | ||
Formation of (CD–SA)n assembly and its characterization
After synthesis of Q-biotin4, MTG-mediated biotin labeling of CD-CK was conducted. The degree of biotinylation (i.e. the number of labeled biotins per protein) was calculated to be 4.12 ± 0.06 for b4-CD. MTG cannot recognize the intrinsic Q and K residues of wild-type EG,43 and almost quantitative and peptide-tag-specific biotin labeling was achieved. The catalytic activity of the biotinylated enzymes was measured with a cellulose substrate (Avicel) by quantifying the reducing ends. The activity before biotinylation was defined as 1.00, and the relative activity for b4-CD was 0.87 ± 0.04. These results confirmed that MTG-mediated quantitative and site-specific biotinylation occurred with only a slight loss of the enzymatic activity.Then, the b4-CD and SA assembly (labeled (CD–SA)n) produced using the avidin–biotin interaction was characterized. Rapid and spontaneous formation of the (CD–SA)n complex was observed when b4-CD and SA were simply mixed together. Size-exclusion chromatography (SEC) was used to study the formation of (CD–SA)n complexes with different molar ratios of the biotin-binding sites of SA (mSA) to the modified biotin groups of CD (Fig. 4A). The results showed that the b4-CD peak, which occurred at an elution volume of 15.6 mL, gradually decreased as the SA content increased, and new broad peaks appeared. With [mSA]/[biotin] ratios of 1:8 and 1:4, the apparent molecular weights of the (CD–SA)n complexes at an elution volume of 13.1 mL were calculated using a standard curve (Fig. S3†) at approximately 145 kDa. This value corresponds closely to the theoretical molecular weight of a CD–SA–CD complex (152.6 kDa). With a [mSA]/[biotin] ratio of 1:2, larger (CD–SA)n complexes formed and some of the complexes were excluded in the void volume of the column (M.W. >1.3 MDa). Then, with equal concentrations of biotin and the biotin-binding site (i.e. [mSA]/[biotin] ratio of 1:1) in the sample, the b4-CD peak almost disappeared and most of the complexes were excluded in the void volume. The molecular weight of this complex was estimated to be larger than 1.3 MDa, which showed that supramolecular (CD–SA)n complexes (n >13) were fabricated.
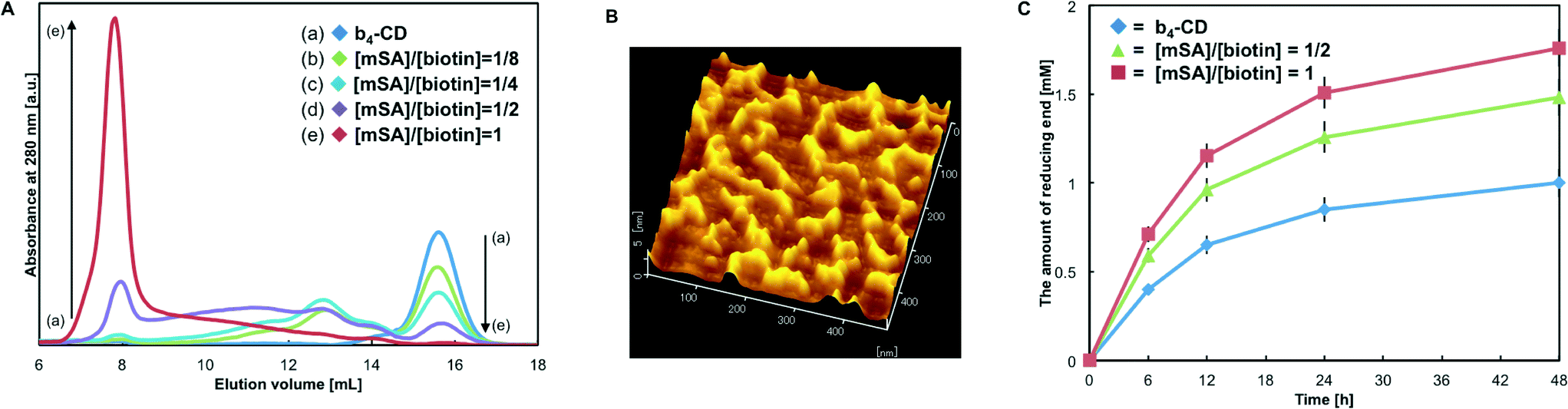 | ||
| Fig. 4 (A) Formation of artificial cellulosome (CD–SA)n complexes at different molar ratios of SA to b4-CD. The complexes were characterized by size-exclusion chromatography. The blue line is for tetra-biotinylated CDs, while the green, cyan, purple and red lines are for complexes with SA biotin-binding site-to-CD biotin group ratios of 1:8, 1:4, 1:2 and 1:1, respectively. (B) AFM image of the (CD–SA)n complex. The approximate sizes of the building blocks were as follows (width × depth × height): CD (5 nm × 5 nm × 5 nm) and SA (5 nm × 5 nm × 5 nm). (C) Hydrolysis reaction of the cellulose substrate (Avicel) with the (CD–SA)n complex. The amount of reducing ends produced from 0.5% (mass fraction) Avicel, 100 nM CD and 100 nM β-glucosidase in 50 mM phosphate buffer (pH 7.0) at 50 °C for 48 h. The blue line is for tetra-biotinylated CDs, while the green and red lines are for complexes with SA biotin-binding site-to-CD biotin group ratios of 1:2 and 1:1, respectively. | ||
For direct observation of the protein assemblies, the samples produced with a [mSA]/[biotin] ratio of 1:1 were analyzed by AFM. The AFM images showed fibrous one-dimensional protein complexes (Fig. 4B). The complexes were about 4–5 nm high and some of the protein units formed the circular structure (20–50 nm), but the predominant form was the linear structure (50–200 nm). The heights were consistent with the heights of CD (ca. 5 nm) and SA (ca. 5 nm). Based on the characterization, we found that the tetra-biotinylation substrate (Q-biotin4), which was designed based on the quaternary structure of SA, allowed for complex formation between the resultant tetrabiotinylated monomeric proteins and SA and the subsequent formation of one-dimensional assemblies.
To investigate how the molecular structure affected the function of the complex, enzymatic degradation of cellulose with the (CD–SA)n complex was conducted. Complexes prepared with [mSA]/[biotin] ratios of 1:2 and 1:1 were added to a reaction solution, and under these conditions, half and all of the CD complexes self-assembled, respectively (Fig. 4C). Compared with free b4-CDs, the (CD–SA)n complexes promoted the hydrolysis of Avicel more efficiently. Increasing the SA content enhanced the saccharification reaction, and the maximum enhancement was almost 1.7-fold. Compared with that of free b4-CDs, the saccharification efficiency was better even with assemblies that contained only one type of enzyme because of the spatial proximity of CDs and the possible increase in binding affinity to the cellulose surface.13,40 These results imply that prolonged retention on the surface of the cellulose substrate may provide assembled enzymes with more opportunities to encounter the hydrolyzed ends of the cellulose chains.
Enzymatic saccharification by (EG)n(CBM)m assemblies
One unique feature of protein self-assembling systems is that different types of protein units can be used as building blocks and assembled into one complex.12,15,18,40 In this study, tetrabiotinylated heteroprotein units can form proteinaceous supramolecular complexes through the avidin–biotin interaction. Although the order of protein units in the one-dimensional structure cannot be controlled, incorporation of different molecular units to alter the function of the system would be of great interest. In fact, the natural cellulosome system accepts different types of enzymes through cohesin–dockerin interactions to optimize the hydrolysis activity depending on the kind of biomass.47 Generally, the adsorption process via the CBM greatly affects the activity of biomass-degrading enzymes.12,48–50 Thus, we explored the formation of assemblies composed of two protein units, b4-EG and b4-CBM. The original CBM of EG is classified as CBM2 and preferably binds to the crystalline region of cellulose.12,49 In contrast, the CBM from Bacillus halodurans belongs to CBM46 and has the substrate-binding cleft to bind the extended internal regions of beta-1,4-glycan chains.51 Given that EG hydrolyzes the cellulose chains in the amorphous region, we envisioned the enhancement of the saccharification efficiency through the combination of EG and CBM46. An EG-CK and K-tagged CBM46 (CBM-CK) was biotinylated with MTG in the same manner as CD-CK. The degrees of tetra-biotinylation were 4.26 ± 0.07 for b4-EG and 3.98 ± 0.07 for b4-CBM, and the relative activity for b4-EG was 0.94 ± 0.06. These confirmed that the quantitative site-specific labeling without impairing the activity of the enzyme was achieved.Next, b4-EG and b4-CBM were assembled simultaneously by adding SA to a solution of the tetrabiotinylated protein units. With a [mSA]/[biotin] ratio of 1:1 and a constant concentration of b4-EG, protein complexes were prepared with 1, 4 and 9 equiv. of CBM to EG. SEC analyses clearly showed that the avidin–biotin interaction resulted in the assembly of the b4-EG and b4-CBM units into complexes, and the complexes were excluded in the void volume (M.W. >1.3 MDa) under all the conditions (Fig. 5A). A small quantity of free b4-CBM was observed when the concentrations of b4-CBM and SA were increased. However, even at the highest concentrations of the components ([EG]:[CBM]:[SA] = 1:9:10), the free form content corresponded to approximately 2.7% of the total b4-CBM applied to the system. These results suggest that heteroprotein assemblies, such as the (EG)n(CBM)m–(SA)n+m complex, formed.
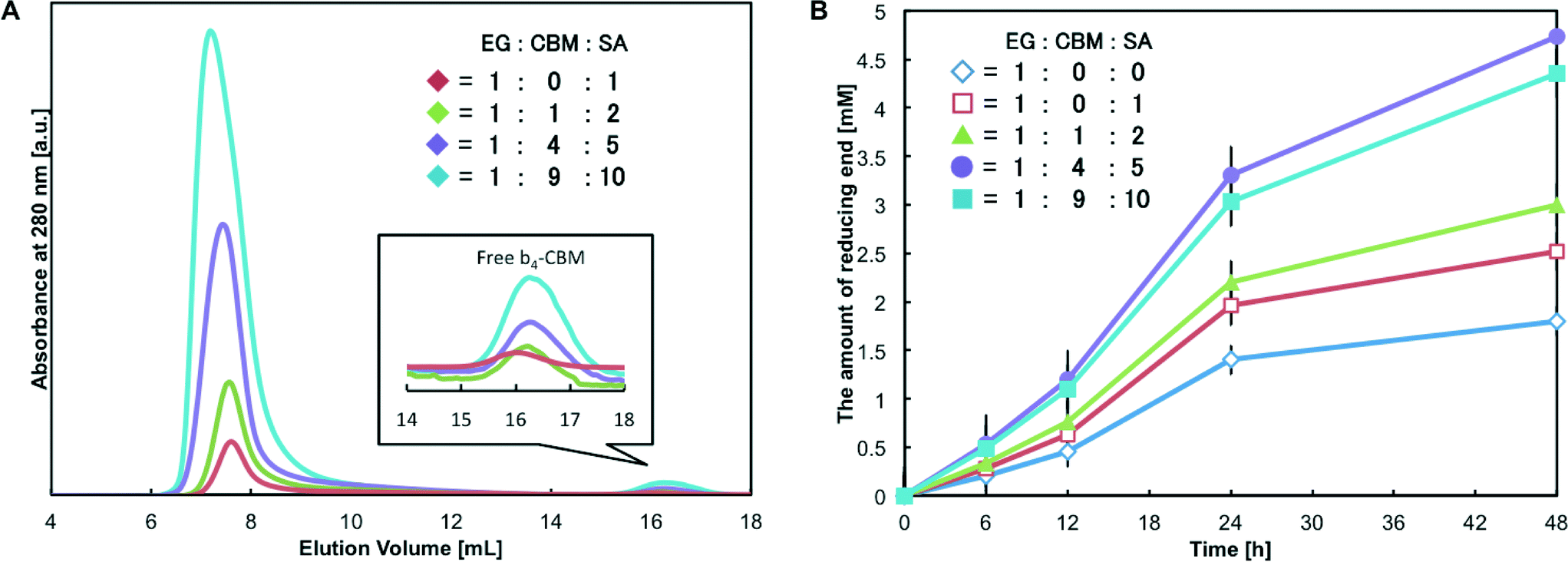 | ||
| Fig. 5 (A) Formation of artificial cellulosome (EG)n(CBM)m–(SA)n+m complexes at different molar ratios of b4-EG:b4-CBM: SA tetramer. The complexes were characterized by size-exclusion chromatography. The red, green, purple and cyan lines are for b4-EG:b4-CBM:SA tetramer ratios of 1:0:1, 1:1:2, 1:4:5 and 1:9:10, respectively. The ratio of the biotin-binding sites of SA to the total biotin groups in the reaction system was 1. (B) Hydrolysis reaction of the cellulose substrate (Avicel) with the (EG)n(CBM)m–(SA)n+m complex. The amount of reducing ends produced from 0.5% (mass fraction) Avicel, 100 nM EG and 100 nM β-glucosidase in 50 mM phosphate buffer (pH 7.0) at 50 °C for 48 h. The blue line is for b4-EGs, while the red, green, purple and cyan lines are for b4-EG:b4-CBM:SA tetramer ratios of 1:0:1, 1:1:2, 1:4:5 and 1:9:10, respectively. The ratio of biotin-binding sites in SA to total biotin groups in the reaction system was 1:1. | ||
Finally, a saccharification reaction with the (EG)n(CBM)m–(SA)n+m complex was conducted (Fig. 5B). As was observed with the (CD–SA)n complex, the (EG–SA)n complex slightly improved the saccharification efficiency (1.4-fold) because of the increasing binding affinity, compared with free b4-EGs. Since the cellulosic substrate Avicel consisted of both crystalline and amorphous regions, the incorporation of CBM46 into the heteroprotein complexes would promote the binding to the amorphous region. We thus expected the improvement of hydrolysis activity in comparison with complexes composed solely of EG. Under the condition of [b4-EG]/[b4-CBM] = 1, (EG)1(CBM)1 complexes, of which the ratio of CBM2 to CBM46 in heteroprotein assemblies was 1:1 in the reaction system, showed a slight increase in hydrolytic activity. Under the condition of [b4-EG]/[b4-CBM] = 1/4, the resultant (EG)1(CBM)4 complexes achieved further enhancement of the saccharification reaction, and a 2.6-fold enhancement was achieved, compared with free b4-EGs. In these heteroprotein assemblies, it is likely that the CBM46-driven binding affinity to the amorphous region was superior to the CBM2-driven binding affinity to the crystalline region. In a previous report on the clustering endoglucanase and CBM on nanoparticles, amorphous-cellulose-binding CBM was more effective for the improvement of endoglucanase activity than crystalline-cellulose-binding CBM.40 The results suggested that the incorporation of a suitable CBM unit into the complexes could improve the access of the EG units to the amorphous region.
In contrast, further increase in the CBM content (under the condition of [b4-EG]/[b4-CBM] = 1/9, the ratio of CBM2 to CBM46 in the heteroprotein assemblies was 1:9 in the system) decreased the saccharification efficiency. With respect to the formation of protein complexes via non-covalent interaction, an increase in the concentration of protein units leads to the growth of the protein complexes in general.30 Thus, (EG)1(CBM)9 complexes formed the larger assemblies and had more CBMs than the (EG)1(CBM)4 complexes. The incorporation of more CBMs into one complex could promote adsorption, while desorption might be hindered by multi-point affinity among CBMs and cellulose. Judging from the results obtained, (EG)1(CBM)4 complexes exhibited sufficient binding affinity, whereas the lower activity of (EG)1(CBM)9 complexes compared to that of (EG)1(CBM)4 complexes could be caused by the delay of the desorption process from the cellulose surface. Another possibility is that the large number of CBMs in the system might occupy the substrate sites on the cellulose surface and block the binding of EGs. Overall, the results obtained confirmed the formation of hetero-functional proteinaceous supramolecular complexes and indicated that functional control could be achieved by simply changing the ratio of the building blocks.
Conclusions
In this study, artificial one-dimensional supramolecular complexes containing monomeric functional proteins were designed and assembled using avidin–biotin interactions. In a proof-of-concept study, the designed tetrabiotin ligand, which was based on the quaternary structure of SA, was used to produce an artificial cellulosome. The possibility of catalytic functional control of the enzyme assemblies, depending on the loading ratio of building blocks, was demonstrated. It is feasible that this concept could be expanded to other types of supramolecular complexes, and could be useful in a wide range of biotechnological applications. This study also showed the potential of designed small molecules as ligands that could be incorporated into specific sites in the protein of interest. This offers a powerful approach for the design of novel functional protein assemblies.Experimental
Materials
Avicel® PH-101 and 3,5-dinitrosalicylic acid (DNS) were obtained from Sigma-Aldrich (St. Louis, MO). Sodium chloride, magnesium chloride hexahydrate, acetic acid, acetonitrile and Quick-CBB were purchased from Wako Pure Chemical Industries (Osaka, Japan). N,N′-Dimethylformamide (DMF) and imidazole were purchased from Kishida Chemical (Osaka, Japan). The In-Fusion HD Cloning kit, EX Taq® and PrimeSTAR HS vector were obtained from Takara Bio (Shiga, Japan). KOD FX® DNA polymerases were from Toyobo (Osaka, Japan). dNTPs were purchased from Roche Applied Science (Tokyo, Japan). The pET22b(+) plasmid vector was purchased from Novagen (Madison, WI). A Vivaspin ultrafiltration spin column was purchased from Sartorius (Göttingen, Germany). HisTrap™ HP columns, PD-10 columns and full-range rainbow molecular weight markers were purchased from GE Healthcare Bio-Sciences (Uppsala, Sweden). Sodium dodecyl sulfate and tris(hydroxymethyl)aminomethane were from Nacalai Tesque (Kyoto, Japan). PCR primers were synthesized at GeneNet (Fukuoka, Japan). Microbial transglutaminase (MTG) was supplied by Ajinomoto Co., Ltd. (Tokyo, Japan). A BCA protein assay kit was purchased from Thermo Fisher Scientific (Waltham, MA). PBS buffer for the preparation of solutions was prepared using 137 mM NaCl, 2.68 mM KCl and 25 mM phosphate buffer (pH 7.4).Expression and purification of recombinant proteins
The recombinant Ktag-CD, Ktag-EG and BGL were prepared as ΔCBM-EG(Tfu0901)-CK, EG(Tfu0901)-CK and BGL(Tfu0937) as previously described.21A gene fragment encoding the CBM in CelB from Bacillus halodurans C-125 was selected from the Carbohydrate-Active enZYmes database, and the synthesized gene was purchased from Takara Bio and Thermo Fisher Scientific. The synthesized gene was cloned into the NdeI and XhoI sites of a modified pET22 vector with the DNA fragment encoding the IgA hinge linker (SPSTPPTPSPSTPP)–biotin acceptor peptide (AviTag: GGLNDIFEAQKIEWH) and polyhistidine tag (HHHHHH) at the C terminus. To replace the AviTag fragment with the HAtag (YPYDVPDYA)–linker(GGGS)–MTG-reactive K-tag (MRHKGS) fragment, each CBM expression vector, except for AviTag, was prepared using inverse PCR. A DNA fragment encoding the HAtag–linker–MTG-reactive K-tag fragment with 15 base flanking regions homologous to vector ends was purchased from Fasmac Co., Ltd. (Kanagawa, Japan). To generate each CBM-CK expression vector, the vector fragment and the HAtag–linker–MTG-reactive K-tag fragment were fused using the In-Fusion™ system. Protein expression and purification were performed as described previously.22
Synthesis of a new biotinylation substrate of MTG
Bis(bis(biotin-Gly-Gly-Gly)-Lys)-Lys-Gly-Leu-Gln-Gly (Q-biotin4) was manually synthesized on an Fmoc-Gly-Alko resin (0.1 mmol) through an Fmoc strategy using an empty PD-10 column. Fmoc amino acids were used in the synthesis. Coupling reactions employed 3 equiv. (relative to the resin) of Fmoc amino acids activated in situ with 3 equiv. of O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate, 3 equiv. of 1-hydroxybenzotriazole and 6 equiv. of N,N-diisopropylethylamine in N,N-dimethylformamide (DMF) for 60–180 min. Removal of the Fmoc-protecting group was achieved by treatment with piperidine/DMF (20%, v/v) for 15 min. After completion of the synthesis of the peptidyl moiety on the resin, biotin was conjugated to the amino group of the N-terminal glycine. The coupling reactions were conducted using 12 equiv. of biotin, 3 equiv. of O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate, 3 equiv. of 1-hydroxybenzotriazole and 6 equiv. of N,N-diisopropylethylamine in DMF. The reaction mixture was incubated at room temperature overnight. The deprotection and cleavage from the resin were accomplished by the addition of trifluoroacetic acid–triisopropylsilane–H2O (95:2.5:2.5, v/v/v) for 1 h. The biotinylation substrates in the cleavage mixture were precipitated with water, collected by filtration and dried under vacuum. Reversed-phase HPLC purification was employed with the absorbance monitored at 230 nm, and the collected fractions were lyophilized. The column was an Inertsil ODS-3 (GL Sciences, Inc., 4.6 × 250 mm). The mobile phase was CH3CN–H2O with the following gradient: 0 min, 10:90 (v/v); 30 min, 30:70 (v/v); 40 min, 80:20 (v/v). Synthesis of the biotinylation substrates was confirmed by reversed-phase HPLC and MALDI-TOF MS. The purified biotinylation substrate was eluted at 22.1 min. MALDI-TOF MS: m/z calculated for C97H155N31O29S4Na ([M + Na]+) 2370.6, found 2370.6.
MTG-mediated biotinylation of recombinant proteins
Q-biotin4 (200 μM) and MTG (0.5 U mL−1) were added to an aqueous solution of CD-CK, EG-CK or CBM-CK (10 μM in 10 mM phosphate buffer, pH 5.8) at 25 °C. After 30 min, enzymatic site-specific biotinylated proteins (b4-CD, b4-EG and b4-CBM) were obtained using spin-filtering with Vivaspin 10000 molecular weight cutoff columns to stop the reaction. The buffer exchange and spin-down steps were repeated until the theoretical concentration of Q-biotin4 in the reaction solution reached <0.1% of the concentration of the target protein.
The degree of biotinylation (i.e. the number of labeled biotins per protein) was calculated using the 2-hydroxyazobenzene-4′-carboxylic acid (HABA)–avidin method.52 For this method, the HABA reagent was prepared by dissolving 14.0 mg of HABA in 500 μL of DMSO. The HABA–avidin reagent was then prepared by adding 200 μL of the HABA reagent and 19.8 mL of deionized water to 10 mg of avidin. Then, 900 μL of the HABA–avidin reagent was pipetted into a 1 mL cuvette, and the optical density (OD) was measured at 500 nm (AbsA). Next, 100 μL of the biotinylated sample was mixed with the HABA–avidin reagent in the cuvette. After 30 min, the OD at 500 nm was measured again (AbsB). The AbsA and AbsB values were used in eqn (1) to calculate the degree of biotinylation.
| [Biotin](mol L−1) = 10−3 × (0.9 × AbsA − AbsB)/34 | (1) |
Enzymatic activity measurements of biotinylated EGs
To measure the enzymatic activity, 1 mL of a 0.5% (mass fraction) solution of the cellulose substrate Avicel was mixed with a 100 nM solution of b4-CD or b4-EG and a 100 nM solution of BGL. All solutions were prepared in 50 mM phosphate buffer (pH 7.0). The reaction was carried out at 50 °C with stirring. The relative activities were measured using the DNS method.53 The DNS assay quantified the number of reducing ends of cellulose after the enzymatic reaction. To perform the assay, 100 μL of the DNS reagent (1.3 M DNS, 1.0 M potassium sodium tartrate and 0.4 M NaOH) was added to 100 μL of the reaction mixture after the enzymatic reaction was completed. This mixture was incubated at 95 °C for 5 min to label the reducing ends of the hydrolyzed cellulose. As a control, a DNS assay of the cellulose substrate without the conjugates was also conducted in the same manner. The OD was measured at 540 nm, and the activity was determined from a glucose standard curve.Self-assembly of biotinylated proteins
A 200 μM SA solution was prepared in PBS. To prepare the (CD–SA)n complexes, 0, 0.125, 0.25, 0.5 or 1 equiv. (0, 1, 2, 4 or 8 μM) of the SA solution was gradually added to 500 μL of a 2 μM b4-CD solution in PBS. To prepare the (EG–SA)n complexes, 1 equiv. (8 μM) of the SA solution was gradually added to 500 μL of a 2 μM b4-EG solution in PBS. For the (EG)n(CBM)m–(SA)n+m complexes, the solutions containing 2 μM b4-EG and 0, 2, 8 or 18 μM b4-CBM were prepared. Then, the solutions were mixed with 0, 16, 40 or 80 μM SA. In each of the assembled samples, the ratios of b4-EG to b4-CBM to SA tetramer were 1:0:1, 1:1:2, 1:4:5 and 1:9:10. In all solutions, the ratio of the biotin-binding sites of SA to the total biotin groups was 1. All solutions were stirred vigorously and then incubated at 4 °C for 30 min. After this, each sample was applied to a size-exclusion chromatography (SEC) column using a 0.5 mL sample loop. SEC was carried out at a flow rate of 0.5 mL min−1, and the samples were observed using an atomic force microscope (AFM, AFM5100N, Hitachi High-Tech Science Co., Ltd., Tokyo, Japan). To prepare the samples for AFM measurement, they were first dried under a stream of N2 gas and then washed with 100 μL of water to remove any salts. Then, the samples were transferred onto the stage of the AFM and measured in dynamic force mode with an SI-DF20 tip. All AFM measurements were performed at ambient temperature in air.
Enzymatic saccharification of cellulose with (CD–SA)n and (EG)n(CBM)m–(SA)n+m complexes
An incubated solution of the (CD–SA)n or (EG)n(CBM)m–(SA)n+m complex was added to a reaction mixture containing 0.5% (mass fraction) Avicel and 100 nM BGL in 50 mM PBS (pH 7.0). The final concentrations of CD and EG in the reaction solution were 100 nM. The enzymatic saccharification reaction was conducted in the same manner as the DNS assay.Acknowledgements
We are grateful to Ajinomoto Co., Inc. for providing MTG. This research was supported by the Advanced Low Carbon Technology Research and Development Program (ALCA) from the Japan Science and Technology Agency (JST). Y. M. was supported by a Research Fellowship from the Japan Society for the Promotion of Science (JSPS) for Young Scientists.References
- T. O. Yeates and J. E. Padilla, Curr. Opin. Struct. Biol., 2002, 12, 464 CrossRef CAS PubMed.
- B. J. G. E. Pieters, M. B. Eldijk, R. J. M. Nolte and J. Mecinović, Chem. Soc. Rev., 2016, 45, 24 RSC.
- C. M. Fontes and H. J. Gilbert, Annu. Rev. Biochem., 2010, 79, 655 CrossRef CAS PubMed.
- R. H. Doi and A. Kosugi, Nat. Rev. Microbiol., 2004, 2, 541 CrossRef CAS PubMed.
- E. A. Bayer, R. Lamed, B. A. White and H. J. Flint, Chem. Rec., 2008, 8, 364 CrossRef CAS PubMed.
- C. Teller and I. Willner, Trends Biotechnol., 2010, 28, 619 CrossRef CAS PubMed.
- C. J. Delebecque, A. B. Lindner, P. A. Silver and F. A. Aldaye, Science, 2011, 333, 470 CrossRef CAS PubMed.
- I. J. Minten, L. J. Hendriks, R. J. Nolte and J. J. Cornelissen, J. Am. Chem. Soc., 2009, 131, 17771 CrossRef CAS PubMed.
- C. You, S. Myung and Y. H. Zhang, Angew. Chem., Int. Ed., 2012, 51, 8787 CrossRef CAS PubMed.
- D. Men, Z. P. Zhang, Y. C. Guo, D. H. Zhu, L. J. Bi, J. Y. Deng, Z. Q. Cui, H. P. Wei and X. E. Zhang, Biosens. Bioelectron., 2010, 26, 1137 CrossRef CAS PubMed.
- J. E. Dueber, G. C. Wu, G. R. Malmirchegini, T. S. Moon, C. J. Petzold, A. V. Ullal, K. L. Prather and J. D. Keasling, Nat. Biotechnol., 2009, 27, 753 CrossRef CAS PubMed.
- J. Caspi, Y. Barak, R. Haimovitz, D. Irwin, R. Lamed, D. B. Wilson and E. A. Bayer, Appl. Environ. Microbiol., 2009, 75, 7335 CrossRef CAS PubMed.
- A. Heyman, Y. Barak, J. Caspi, D. B. Wilson, A. Altman, E. A. Bayer and O. Shoseyov, J. Biotechnol., 2007, 131, 433 CrossRef CAS PubMed.
- S. Moraïs, A. Heyman, Y. Barak, J. Caspi, D. B. Wilson, R. Lamed, O. Shoseyov and E. A. Bayer, J. Biotechnol., 2010, 147, 205 CrossRef PubMed.
- H. P. Fierobe, A. Mechaly, C. Tardif, A. Belaich, R. Lamed, Y. Shoham, J. P. Belaich and E. A. Bayer, J. Biolumin. Chemilumin., 2001, 276, 21257 CAS.
- J. Krauss, V. V. Zverlov and W. H. Schwarz, Appl. Environ. Microbiol., 2012, 78, 4301 CrossRef CAS PubMed.
- S. Mitsuzawa, H. Kagawa, Y. Li, S. L. Chan, C. D. Paavola and J. D. Trent, J. Biotechnol., 2009, 143, 139 CrossRef CAS PubMed.
- S. Moraïs, Y. Barak, J. Caspi, Y. Hadar, R. Lamed, Y. Shoham, D. B. Wilson and E. A. Bayer, mBio, 2010, 1, e00285 CrossRef PubMed.
- J. M. Hannink, J. J. Cornelissen, J. A. Farrera, P. Foubert, F. C. De Schryver, N. A. Sommerdijk and R. J. Nolte, Angew. Chem., Int. Ed., 2001, 40, 4732 CrossRef CAS.
- T. Matsumoto, T. Tanaka and A. Kondo, Langmuir, 2012, 28, 3553 CrossRef CAS PubMed.
- S. Noda, T. Matsumoto, T. Tanaka and A. Kondo, Microb. Cell Fact., 2015, 14, 5 CrossRef PubMed.
- M. Fairhead, G. Veggiani, M. Lever, J. Yan, D. Mesner, C. V. Robinson, O. Dushek, P. A. van der Merwe and M. Howarth, J. Am. Chem. Soc., 2014, 136, 12355 CrossRef CAS PubMed.
- M. Wilchek and E. A. Bayer, Anal. Biochem., 1988, 171, 1 CrossRef CAS PubMed.
- N. M. Green, Methods Enzymol., 1990, 184, 51 CAS.
- E. P. Diamandis and T. K. Christopoulos, Clin. Chem., 1991, 37, 625 CAS.
- M. Wilchek, E. A. Bayer and O. Livnah, Immunol. Lett., 2006, 103, 27 CrossRef CAS PubMed.
- O. H. Laitinen, H. R. Nordlund, V. P. Hytönen and M. S. Kulomaa, Trends Biotechnol., 2007, 25, 269 CrossRef CAS PubMed.
- M. M. C. Bastings, T. F. A. de Greef, J. L. J. van Dongen, M. Merkx and E. W. Meijer, Chem. Sci., 2010, 1, 79 RSC.
- Q. Li, C. R. So, A. Fegan, V. Cody, M. Sarikaya, D. A. Vallera and C. R. Wagner, J. Am. Chem. Soc., 2010, 132, 17247 CrossRef CAS PubMed.
- T. F. Chou, C. So, B. R. White, J. C. T. Carlson, M. Sarikaya and C. R. Wagner, ACS Nano, 2008, 2, 2519 CrossRef CAS PubMed.
- Y. Mori, R. Wakabayashi, M. Goto and N. Kamiya, Org. Biomol. Chem., 2013, 11, 914 CAS.
- M. Fairhead, D. Krndija, E. D. Lowe and M. Howarth, J. Mol. Biol., 2014, 426, 199 CrossRef CAS PubMed.
- S. Burazerovic, J. Gradinaru, J. Pierron and T. R. Ward, Angew. Chem., Int. Ed., 2007, 46, 5510 CrossRef CAS PubMed.
- K. Oohora, S. Burazerovic, A. Onoda, Y. M. Wilson, T. R. Ward and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 3818 CrossRef CAS PubMed.
- R. Lamed, E. Setter and E. A. Bayer, J. Bacteriol., 1983, 156, 828 CAS.
- O. Grépinet and P. Béguin, Nucleic Acids Res., 1986, 14, 1791 CrossRef.
- K. Tokatlidis, S. Salamitou, P. Béguin, P. Dhurjati and J. P. Aubert, FEBS Lett., 1991, 291, 185 CrossRef CAS PubMed.
- E. A. Bayer, E. Morag and R. Lamed, Trends Biotechnol., 1994, 12, 379 CrossRef CAS PubMed.
- S.-L. Tsai, M. Park and W. Chen, Biotechnol. J., 2013, 8, 257 CrossRef CAS PubMed.
- D. M. Kim, H. Nakazawa, M. Umetsu, T. Matsuyama, N. Ishida, A. Ikeuchi, H. Takahashi, R. Asano and I. Kumagai, Catal. Sci. Technol., 2012, 2, 499 CAS.
- H. Nakazawa, D.-M. Kim, T. Matsuyama, N. Ishida, A. Ikeuchi, Y. Ishigaki, I. Kumagai and M. Umetsu, ACS Catal., 2013, 3, 1342 CrossRef CAS.
- Q. Sun, B. Madan, S. L. Tsai, M. P. DeLisa and W. Chen, Chem. Commun., 2014, 50, 1423 RSC.
- Y. Mori, S. Ozasa, M. Kitaoka, S. Noda, T. Tanaka, H. Ichinose and N. Kamiya, Chem. Commun., 2013, 49, 6971 RSC.
- G. A. L. Gonçalves, Y. Mori and N. Kamiya, Sustainable Chem. Processes, 2014, 2, 1 CrossRef.
- C. Blanchette, C. Lacayo, N. O. Fischer, M. Hwang and M. P. Thelen, PLoS One, 2012, 7, e42116 CAS.
- A. Lykidis, K. Mavromatis, N. Ivanova, I. Anderson, M. Land, G. DiBartolo, M. Martinez, A. Lapidus, S. Lucas, A. Copeland, P. Richardson, D. B. Wilson and N. Kyrpides, J. Bacteriol., 2007, 189, 2477 CrossRef CAS PubMed.
- B. Raman, C. Pan, G. B. Hurst, M. Rodriguez Jr., C. K. McKeown, P. K. Lankford, N. F. Samatova and J. R. Mielenz, PLoS One, 2009, 4, e5271 Search PubMed.
- D. N. Bolam, A. Ciruela, S. McQueen-Mason, P. Simpson, M. P. Williamson, J. E. Rixon, A. Boraston, G. P. Hazlewood and H. J. Gilbert, Biochem. J., 1998, 331, 775 CrossRef CAS PubMed.
- A. B. Boraston, D. N. Bolam, H. J. Gilbert and G. J. Davies, Biochem. J., 2004, 382, 769 CrossRef CAS PubMed.
- K. L. Strobel, K. A. Pfeiffer, H. W. Blanch and D. S. Clark, J. Biol. Chem., 2015, 290, 22818 CrossRef CAS PubMed.
- H. Zhang, G. Zhang, C. Yao, M. Junaid, Z. Lu, H. Zhang and Y. Ma, PLoS One, 2015, 10, e0142107 Search PubMed.
- N. M. Green, Methods Enzymol., 1970, 18, 418 CrossRef.
- G. L. Miller, Anal. Chem., 1959, 31, 426 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5me00011d |
| ‡ Current address: Biomass Engineering Program, RIKEN, 1-7-22, Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan. |
| This journal is © The Royal Society of Chemistry 2016 |