DOI:
10.1039/C6MD00023A
(Research Article)
Med. Chem. Commun., 2016,
7, 1003-1006
Synthesis of radioiodinated probes to evaluate the biodistribution of a potent TRPC3 inhibitor†‡
Received
12th January 2016
, Accepted 12th March 2016
First published on 7th April 2016
Abstract
The transient receptor potential canonical 3 (TRPC3) channel is a member of the TRPC family that contributes to the entry of Ca2+ through the plasma membrane or modulates the driving force for Ca2+ entry channels. The pyrazole compound Pyr3 has recently been reported to be a selective TRPC3 inhibitor and has become an attractive research tool and therapeutic agent for the treatment of heart failure. However, the in vivo characteristics of Pyr3 have not been investigated. To monitor the fate of Pyr3 in vivo, we designed and synthesized a radioiodinated Pyr3 probe ([125I]I-Pyr3) by introducing radioiodine at the 2-position of the central phenyl ring of Pyr3. I-Pyr3 was shown to have direct TRPC3 inhibition activity similar to that of Pyr3 in TRPC3-overexpressing HEK293 cells. Using the tributyltin derivative as a radioiodination precursor, [125I]I-Pyr3 was successfully prepared with high radiochemical purity. Biodistribution studies of [125I]I-Pyr3 and [125I]I-Pyr8 (the esterolysis product of [125I]I-Pyr3) indicated high uptake of intact [125I]I-Pyr3 in the lung and rapid metabolism to [125I]I-Pyr8. These findings provide useful information about the in vivo kinetics of the selective TRPC inhibitor Pyr3.
1 Introduction
Changes in the intracellular concentration of the calcium ion ([Ca2+]i) modulate a wide range of cellular processes, including secretion, gene expression, proliferation, and cell death.1–4 Although [Ca2+]i is normally maintained in the 10–100 nM range, it is increased by the release of Ca2+ from intracellular stores and the endoplasmic reticulum (ER) on activation. In addition, [Ca2+]i is increased by the influx of Ca2+ across the plasma membrane in response to extracellular stimulation.5 Abnormal changes in [Ca2+]i influence intracellular Ca2+ homeostasis and can cause diseases.5
The transient receptor potential canonical (TRPC) channel is a subfamily of TRP channels and contributes to changes in [Ca2+]i by serving as a Ca2+ entry pathway in the plasma membrane or by modulating the driving force for Ca2+ entry channels.6 It has been reported that TPRC channels are closely related to heart diseases, including cardiac hypertrophy and fibrosis, and cancer.7–10 TRPC channels can be divided into seven isoforms (TRPC1–TRPC7), and TRPC1 and TRPC3–7 are broadly expressed in mammals.11,12 TRPC channels are activated in response to membrane receptors linked to phospholipase C (PLC), including PLCβ and PLCγ. TRPC3, TRPC6, and TRPC7 are directly activated by PLC-derived diacylglycerol (DAG).6
To elucidate the functions of a TRPC channel, several small molecules, such as SKF 96365, LOE908, and 2-APB, have been evaluated as TRP inhibitors.13,14 However, the roles played by the TRPC channels have not been adequately characterized because these inhibitors exhibit low selectivity toward TRPC channels. BTP2 has been reported to be a TRPC channel inhibitor that does not influence mitochondrial Ca2+ signaling, ER Ca2+ release, or K+ channels in T-cells. However, BTP2 lacks TRPC subtype selectivity.15–17 The pyrazole compound Pyr3 has been recently reported to be a selective TRPC3 inhibitor that directly inhibits the TRPC3 channel by binding to its extracellular side.18 Pyr3 regulated the TRPC3-mediated Ca2+ influx in B lymphocytes and eliminated the B cell receptor-induced Ca2+ oscillation. Pyr3 also inhibited pressure overload-induced cardiac hypertrophy in mice with transverse aortic constriction.18 Furthermore, in an in vitro model of stent-induced arterial injury, Pyr3 prevented arterial injury by suppressing agonist-induced Ca2+ entry into vascular smooth muscles.19
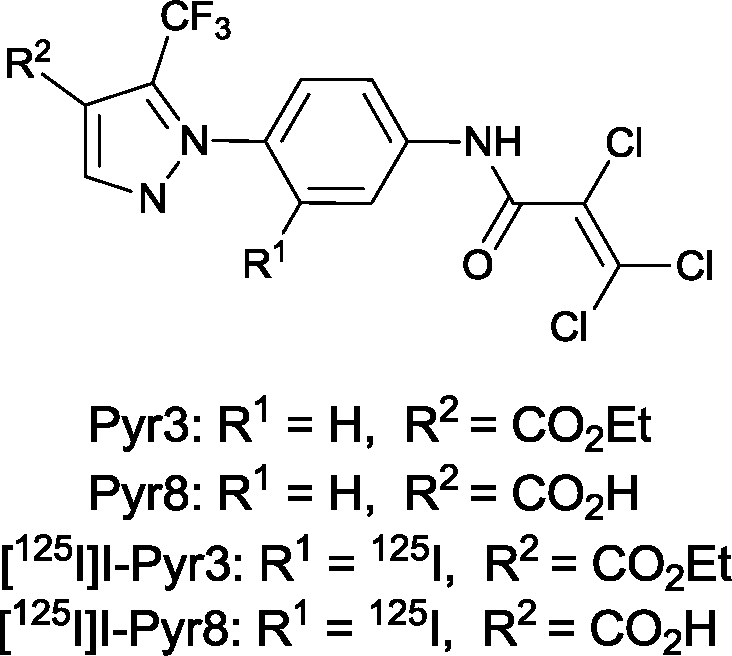
These findings suggest that Pyr3 could be used as a research tool and a therapeutic agent for the treatment of heart failure. Nevertheless, little is known about the in vivo characteristics of Pyr3, including its distribution, metabolism, and elimination. In addition, because the ester moiety of Pyr3 is easily hydrolyzed, Pyr3 is converted to Pyr8, the carboxylic acid derivative, which shows little TRPC3 inhibition. Radiolabeled probes are widely used to study the in vivo characteristics of drugs. Iodine radioisotopes (123I, 125I, and 131I) have higher specific activities than β-emitters, such as 3H or 14C, and are commonly used in biological assay and in vivo imaging. 125I has a relatively long half-life and emits low energy photons, thus the pyrazole compounds were labeled with 125I. In this study, we synthesized the radiolabeled probes ([125I]I-Pyr3 and [125I]I-Pyr8) (Fig. 1) and used them to monitor the fate of Pyr3 in vivo.
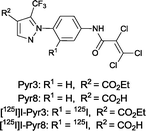 |
| | Fig. 1 Structure of TRPC3-targeted compounds. | |
2 Results and discussion
2.1 Chemistry
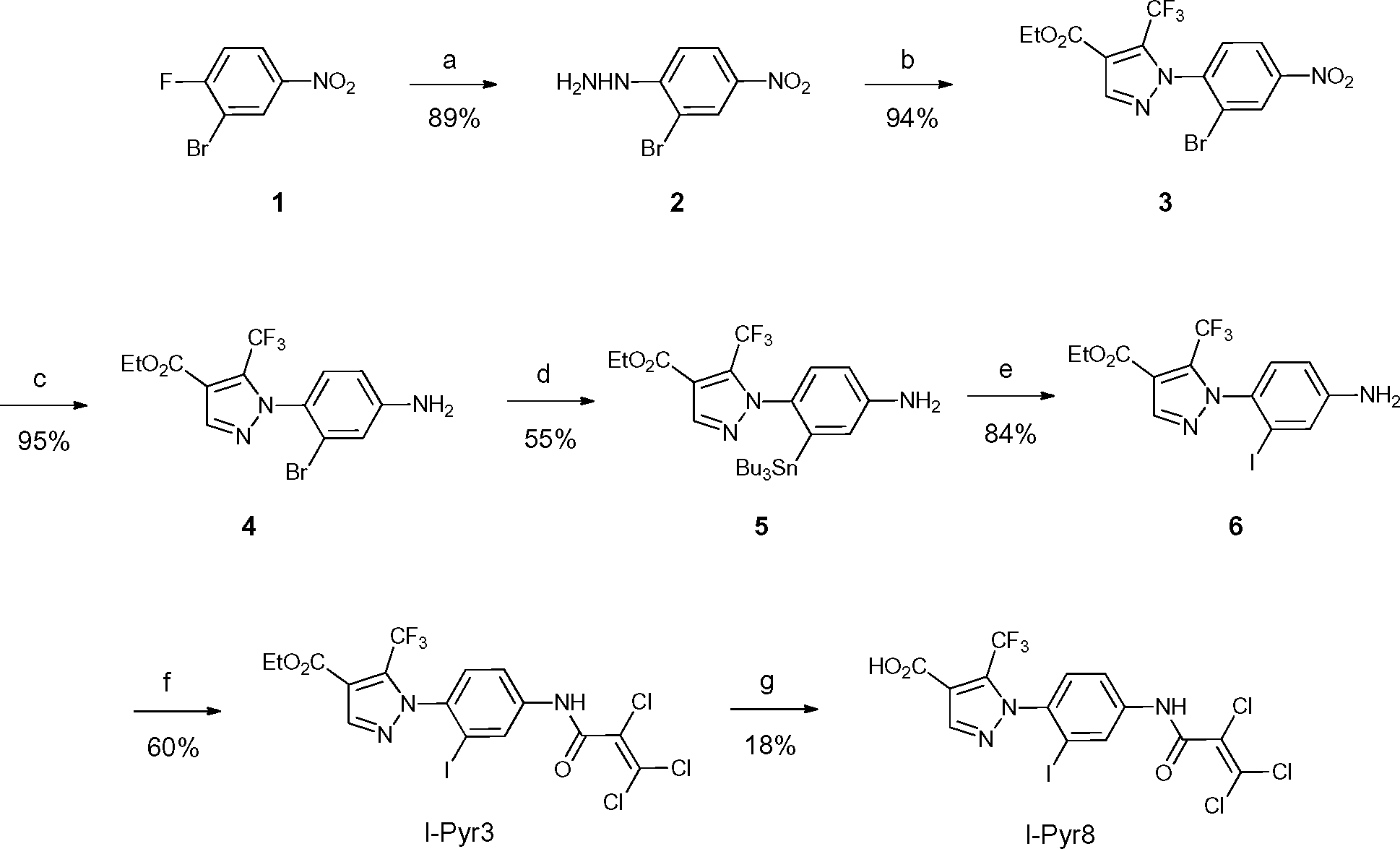
Pyr3 comprises two structural motifs: an ethyl-3-trifluoromethylpyrazole-4-carboxylate group and a trichloroacrylic amide group. The trichloroacrylic amide group of Pyr3 is critical for the observed TRPC3-selective inhibition.18 The effect of introducing a functional group to the central phenyl ring of Pyr3 on TRPC3 channel inhibition or selectivity has not been reported. We designed ethyl 1-(2-[125I]iodo-4-(2,3,3-trichloroacrylamido)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate, [125I]I-Pyr3, to introduce radioiodine at the 2-position of the central phenyl ring of Pyr3. Nonradioactive I-Pyr3 was synthesized via the route outlined in Scheme 1. Reaction of 2-bromo-1-fluoro-4-nitrobenzene with hydrazine hydrate, followed by treatment with ethyl 2-(ethoxymethylene)-4,4,4-trifluoro-3-oxobutyrate, afforded the pyrazole derivative 3 in 94% yield. Treatment of 3 with zinc dust and aqueous NH4Cl afforded the amine 4 in 95% yield. A bromo to tributyltin exchange reaction using Pd(PPh3)4 and a subsequent iododestannylation reaction yielded the iodine derivative 6. I-Pyr3 was prepared from 6 in 60% yield by treatment with 2,3,3-trichloroacryloyl chloride. Alkaline hydrolysis of the ester moiety of I-Pyr3 afforded the carboxylic acid derivative, I-Pyr8, in 18% yield.
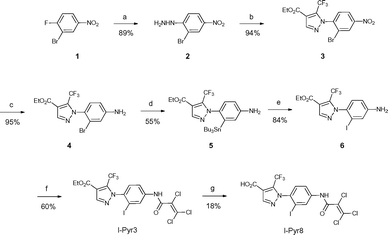 |
| | Scheme 1 Syntheses of I-Pyr3 and I-Pyr8. Reagents and conditions: (a) NH2NH2·H2O, EtOH, reflux, 2 h, (b) ethyl 2-(ethoxymethylene)-4,4,4-trifluoro-3-oxobutyrate, H2SO4, EtOH, reflux, 4 h, (c) zinc dust, aqueous NH4Cl solution, EtOAc, 0 °C, 4 h, (d) Pd(PPh3)4, SnBu3, toluene, 60 °C, 15 h, (e) I2, CH2Cl2, rt, 2.5 h, (f) 2,3,3-trichloroacryloyl chloride, CH2Cl2, N,N-diisopropylethylamine, 0 °C, 1 h, and (g) 1 M KOH (aq.), EtOH, H2O, rt, 8 h. | |
2.2 Measurement of intracellular Ca2+ concentration
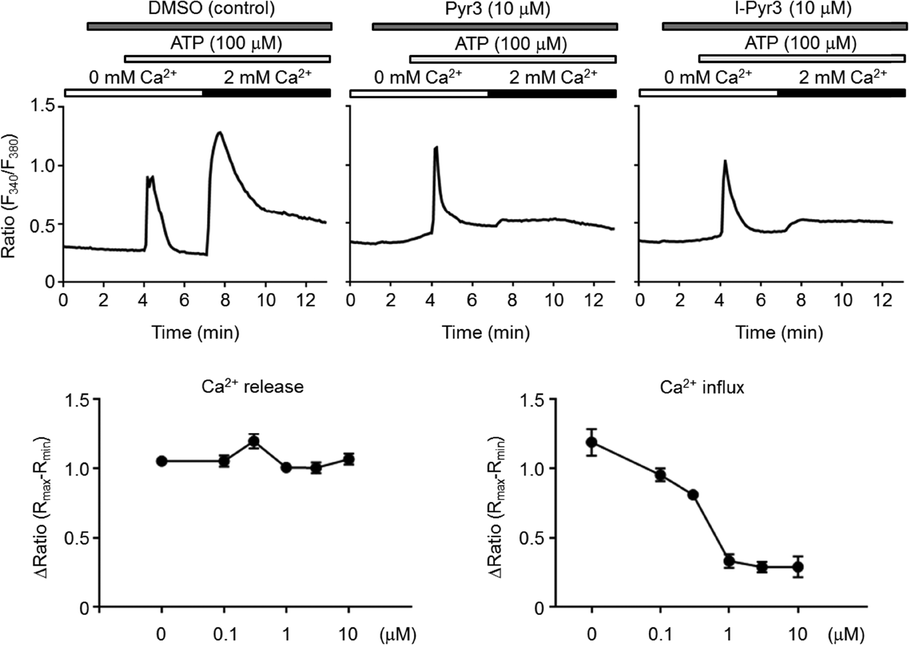
The ability of Pyr derivatives to inhibit TRPC3-mediated Ca2+ influx in vitro was evaluated using TRPC3-overexpressing HEK293 cells (Fig. 2). I-Pyr3 showed potent inhibition of the TRPC3 channel-mediated Ca2+ influx that was only slightly poorer than that of the mother compound (Pyr3).18 This result suggested that the introduction of iodine at the 2-position of the central phenyl ring of Pyr3 did not affect its ability to inhibit the TRPC3-mediated Ca2+ influx and that 125I-labeled I-Pyr3 could be a promising probe to monitor the fate of Pyr3 in plasma.
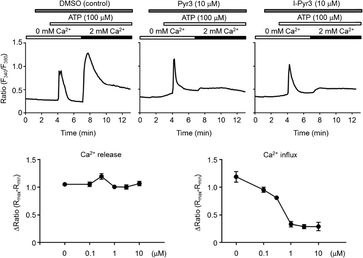 |
| | Fig. 2 Concentration-dependent inhibition of TRPC3-mediated Ca2+ influx by I-Pyr3. Upper panels: average changes in intracellular Ca2+ concentration ([Ca2+]i) induced by endogenous purinergic receptor stimulation in TRPC3-overexpressing HEK293 cells (n = 45–92 cells) in the presence or absence of extracellular Ca2+ (2 mM). The cells were treated with Pyr3 2 min prior to ATP stimulation. Lower panels: concentration-dependent inhibition of TRPC3-mediated Ca2+ influx by I-Pyr3. | |
2.3 Radiosynthesis
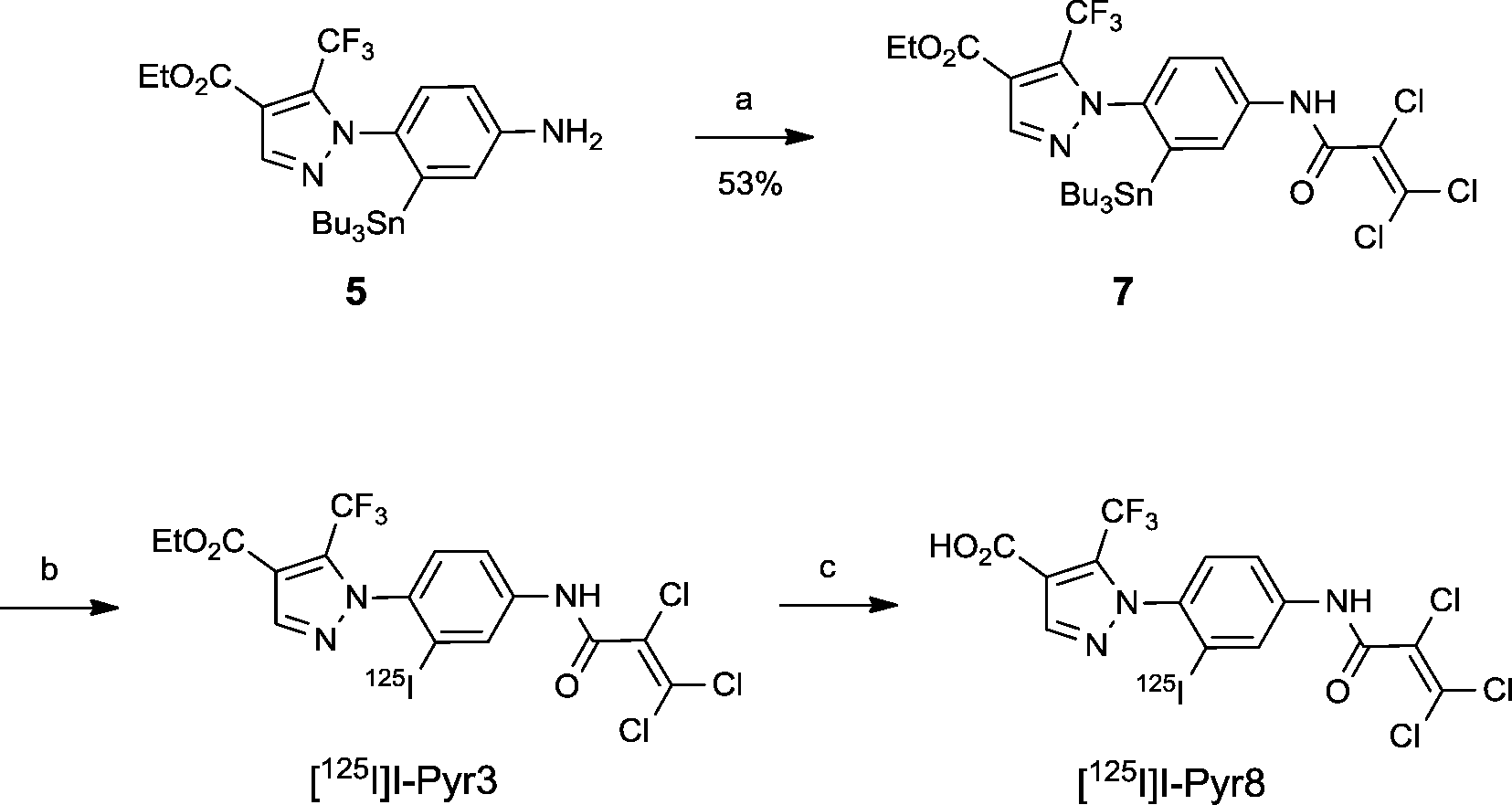
The radioiodinated analog [125I]I-Pyr3 was obtained via the iododestannylation of tributyltin precursor 7, which was obtained from the reaction of amine 5 with 2,3,3-trichloroacryloyl chloride. However, its radiochemical yield was only 15% when N-chlorosuccinimide was used as an oxidant. After the optimization of reaction conditions, the radiochemical yield of [125I]I-Pyr3 improved to 39% when chloramine-T was used as an oxidant (Scheme 2). The carboxylate analog [125I]I-Pyr8 was also obtained in high yield (84%) via the alkaline hydrolysis of the ester moiety of [125I]I-Pyr3. The radiochemical purities and specific activities of these [125I]I-labeled compounds were greater than 98% and greater than 26.3 GBq μmol−1, respectively.
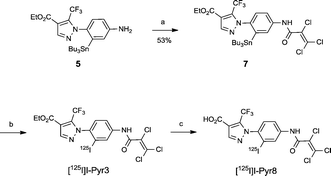 |
| | Scheme 2 Syntheses of [125I]I-Pyr3 and [125I]I-Pyr8. Reagents and conditions: (a) 2,3,3-trichloroacryloyl chloride, CH2Cl2, DIEPA, 0 °C, 1 h, (b) chloramine-T, AcOH, MeOH, [125I]NaI, 35 °C, 2 h, and (c) 1 M KOH (aq.), EtOH, H2O, rt, 8 h. | |
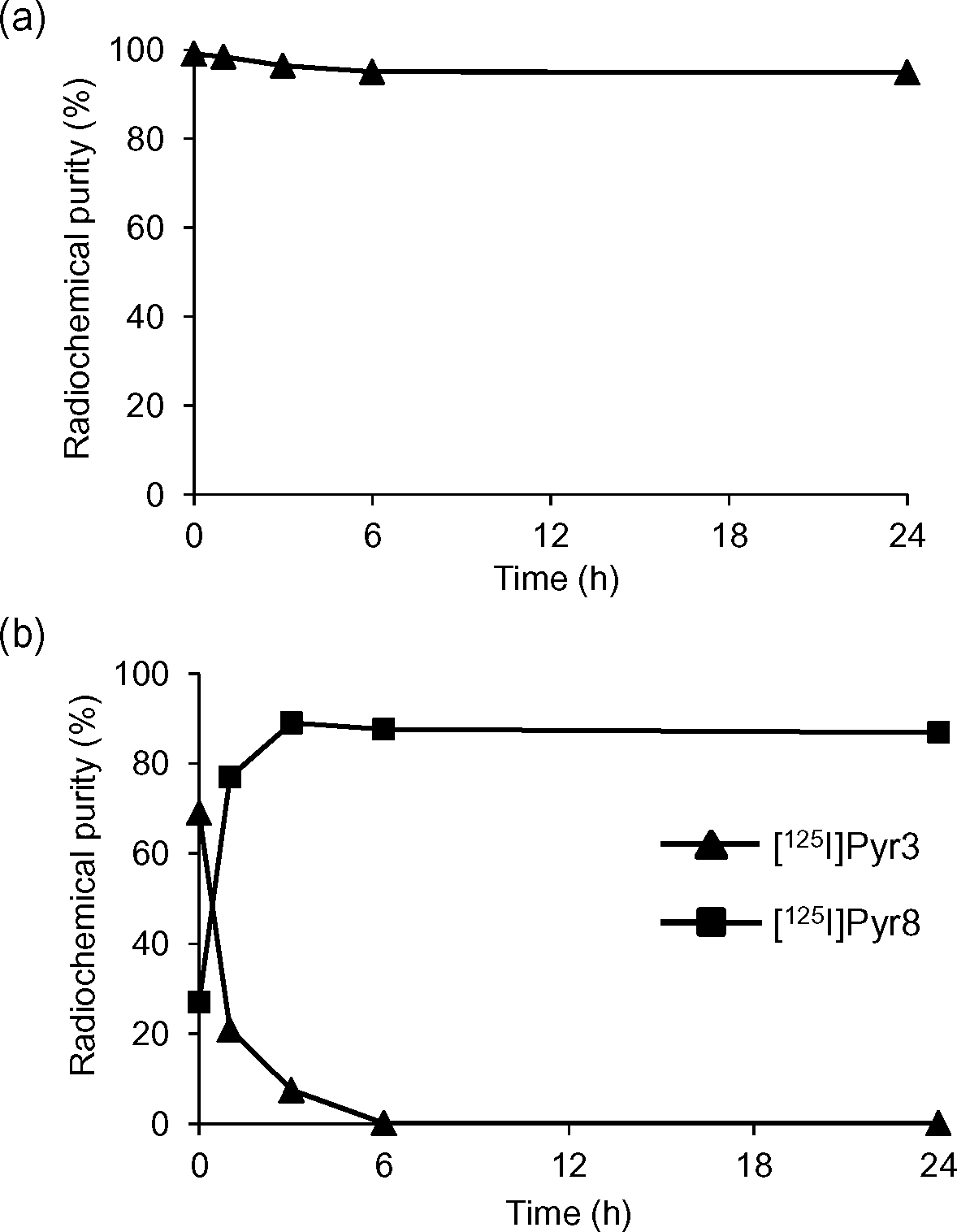
2.4 Stability studies
The in vitro stability of [125I]I-Pyr3 in 20 mM phosphate buffer (PB; pH 7.4) or mouse plasma at 37 °C was evaluated using radio-TLC analysis (Fig. 3). The radioactivity of [125I]I-Pyr3 remained >95% intact for 24 h in PB, while the radioactivity of [125I]I-Pyr3 rapidly decreased and completely disappeared after 6 h in mouse plasma. The metabolite of [125I]I-Pyr3 was identified as [125I]I-Pyr8, indicating the rapid hydrolysis of the ester group of [125I]I-Pyr3.
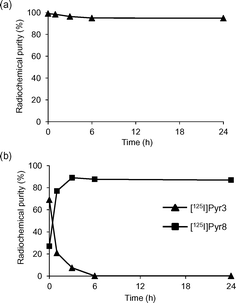 |
| | Fig. 3 Stability studies of (a) [125I]I-Pyr3 in 20 mM phosphate buffer (pH 7.4) and (b) [125I]I-Pyr3 in mouse plasma. | |
2.5
In vivo biodistribution in normal mice
To characterize the in vivo kinetics of Pyr3, a biodistribution study of [125I]I-Pyr3 was carried out in normal ddY mice (Table 1). The radioactivity of [125I]I-Pyr3 cleared rapidly from the blood pool and showed considerable accumulation in the lung, small intestine, and liver 10 min after injection. In addition, accumulation of radioactivity was broadly observed in the stomach, heart, kidney, and muscle. The thyroid accumulation was low, indicating high stability against in vivo deiodination. Because [125I]I-Pyr3 was hydrolysed to [125I]I-Pyr8, the distribution of radioactivity after administration of [125I]I-Pyr3 might reflect the distribution of [125I]I-Pyr8. Therefore, we performed a biodistribution study of [125I]I-Pyr8 (Table 2). Similar to [125I]I-Pyr3, [125I]I-Pyr8 showed rapid blood clearance and considerable accumulation of radioactivity in the small intestine and liver. However, the lung radioactivity after administration of [125I]I-Pyr8 was dramatically lower compared to that after [125I]I-Pyr3 administration. These results indicated that [125I]I-Pyr3 is broadly distributed, especially to the lung, as an intact form at early times post-injection, and that this is followed by rapid metabolism to [125I]I-Pyr8.
Table 1 Biodistribution in normal mice after iv injection of [125I]I-Pyr3a
| Organ |
10 min |
30 min |
1 h |
3 h |
24 h |
|
Each value represents the mean ± s.d. (n = 5). Expressed as % injection dose per gram of organ.
Expressed as % injection dose.
|
| Blood |
1.74 ± 0.31 |
0.84 ± 0.48 |
0.73 ± 0.10 |
0.40 ± 0.10 |
0.26 ± 0.09 |
| Spleen |
1.52 ± 0.21 |
1.02 ± 0.17 |
0.93 ± 0.08 |
0.98 ± 0.14 |
0.98 ± 0.19 |
| Pancreas |
1.20 ± 0.12 |
0.98 ± 0.14 |
1.10 ± 0.16 |
0.88 ± 0.21 |
0.62 ± 0.08 |
| Stomach |
3.14 ± 2.97 |
3.42 ± 2.64 |
3.27 ± 2.96 |
1.47 ± 1.04 |
0.07 ± 0.02 |
| Intestine |
18.20 ± 2.25 |
21.09 ± 2.28 |
26.51 ± 3.75 |
26.91 ± 3.72 |
0.05 ± 0.01 |
| Kidney |
3.10 ± 0.45 |
1.23 ± 0.40 |
0.93 ± 0.17 |
0.60 ± 0.09 |
0.27 ± 0.03 |
| Liver |
14.59 ± 2.91 |
6.16 ± 2.57 |
7.80 ± 2.64 |
4.29 ± 1.51 |
0.21 ± 0.03 |
| Heart |
2.68 ± 0.67 |
1.14 ± 0.15 |
0.98 ± 0.03 |
0.82 ± 0.05 |
0.62 ± 0.06 |
| Lung |
20.58 ± 2.64 |
5.65 ± 1.12 |
4.68 ± 0.78 |
1.61 ± 0.23 |
0.66 ± 0.10 |
| Muscle |
2.24 ± 0.46 |
2.66 ± 0.78 |
1.92 ± 0.24 |
2.02 ± 0.81 |
1.29 ± 0.34 |
| Brain |
1.11 ± 0.12 |
0.94 ± 0.09 |
1.04 ± 0.25 |
0.57 ± 0.11 |
0.23 ± 0.04 |
| Thyroidb |
0.13 ± 0.03 |
0.14 ± 0.03 |
0.16 ± 0.05 |
0.19 ± 0.08 |
0.29 ± 0.11 |
| Urineb |
|
|
|
|
3.77 ± 1.45 |
| Fecesb |
|
|
|
|
73.81 ± 10.52 |
Table 2 Biodistribution in normal mice after iv injection of [125I]I-Pyr8a
| Organ |
10 min |
30 min |
1 h |
3 h |
24 h |
|
Each value represents the mean ± s.d. (n = 5). Expressed as % injection dose per gram of organ.
Expressed as % injection dose.
|
| Blood |
0.64 ± 0.04 |
0.46 ± 0.14 |
0.36 ± 0.04 |
0.31 ± 0.04 |
0.28 ± 0.05 |
| Spleen |
1.40 ± 0.71 |
0.97 ± 0.18 |
0.92 ± 0.23 |
0.89 ± 0.11 |
0.69 ± 0.17 |
| Pancreas |
1.75 ± 1.70 |
0.64 ± 0.13 |
0.54 ± 0.20 |
0.88 ± 0.30 |
0.60 ± 0.16 |
| Stomach |
1.59 ± 0.46 |
0.47 ± 0.25 |
0.91 ± 0.13 |
1.15 ± 0.41 |
0.12 ± 0.04 |
| Intestine |
18.06 ± 7.65 |
22.83 ± 6.08 |
27.53 ± 3.09 |
21.44 ± 1.48 |
0.10 ± 0.04 |
| Kidney |
2.34 ± 0.68 |
1.03 ± 0.43 |
0.83 ± 0.17 |
0.85 ± 0.58 |
0.23 ± 0.03 |
| Liver |
12.22 ± 2.60 |
9.37 ± 1.87 |
8.94 ± 6.27 |
8.08 ± 2.75 |
0.12 ± 0.02 |
| Heart |
1.00 ± 0.36 |
0.84 ± 0.32 |
0.75 ± 0.07 |
0.64 ± 0.04 |
0.63 ± 0.16 |
| Lung |
1.31 ± 1.02 |
0.76 ± 0.40 |
0.59 ± 0.03 |
0.57 ± 0.10 |
0.45 ± 0.07 |
| Muscle |
2.34 ± 0.78 |
1.90 ± 0.85 |
1.93 ± 0.83 |
1.78 ± 0.99 |
1.03 ± 0.35 |
| Brain |
0.31 ± 0.07 |
0.36 ± 0.06 |
0.55 ± 0.49 |
0.31 ± 0.04 |
0.23 ± 0.05 |
| Thyroidb |
0.10 ± 0.02 |
0.10 ± 0.01 |
0.10 ± 0.01 |
0.13 ± 0.02 |
0.13 ± 0.03 |
| Urineb |
|
|
|
|
2.50 ± 0.65 |
| Fecesb |
|
|
|
|
77.08 ± 8.38 |
3 Conclusions
We successfully designed and synthesized [125I]I-Pyr3 and [125I]I-Pyr8 and used these compounds for the in vivo characterization of the selective TRPC3 inhibitor Pyr3. The ability of I-Pyr3 to inhibit TRPC3-mediated Ca2+ influx was comparable to Pyr3. Biodistribution studies of [125I]I-Pyr3 and [125I]I-Pyr8 in normal mice showed that [125I]I-Pyr3 is highly distributed to the lung as an intact form at early times post-injection and that this is followed by rapid metabolism to [125I]I-Pyr8. These findings provide useful information about the in vivo kinetics of the selective TRPC inhibitor Pyr3.
References
- S. N. Yang and P. O. Berggren, Endocr. Rev., 2006, 27, 621 CrossRef CAS PubMed.
- S. W. Flavell and M. E. Greenberg, Annu. Rev. Neurosci., 2008, 31, 563 CrossRef CAS PubMed.
- J. M. Schmitt, E. Abell, A. Wagner and M. A. Davare, Mol. Cell. Biochem., 2010, 335, 155 CrossRef CAS PubMed.
- S. P. Yu, L. M. Canzoniero and D. W. Choi, Curr. Opin. Cell Biol., 2001, 13, 405 CrossRef CAS PubMed.
- D. E. Clapham, Cell, 2007, 131, 1047 CrossRef CAS PubMed.
- S. F. Pedersen, G. Owsianik and B. Nilius, Cell Calcium, 2005, 38, 233 CrossRef CAS PubMed.
- M. M. El-Mas, H. M. El-Gowelli and M. C. Michel, Naunyn-Schmiedeberg's Arch. Pharmacol., 2013, 386, 929 CrossRef CAS PubMed.
- Z. Yue, Y. Zhang, J. Xie, J. Jiang and L. Yue, Curr. Top. Med. Chem., 2013, 13, 270 CrossRef CAS PubMed.
- E. Aydar, S. Yeo, M. Djamgoz and C. Palmer, Cancer Cell Int., 2009, 9, 23 CrossRef PubMed.
- S. L. Yang, Q. Cao, K. C. Zhou, Y. J. Feng and Y. Z. Wang, Oncogene, 2009, 28, 1320 CrossRef CAS PubMed.
- A. Riccio, A. D. Medhurst, C. Mattei, R. E. Kelsell, A. R. Calver, A. D. Randall, C. D. Benham and M. N. Pangalos, Mol. Brain Res., 2002, 109, 95 CrossRef CAS PubMed.
- Y. Jang, Y. Lee, S. M. Kim, Y. D. Yang, J. Jung and U. Oh, Arch. Pharmacal Res., 2012, 35, 1823 CrossRef CAS PubMed.
- K. Treiber, A. Singer, B. Henke and W. E. Müller, Br. J. Pharmacol., 2005, 145, 75 CrossRef CAS PubMed.
- J. P. Lievremont, G. S. Bird and J. W. Putney Jr., Mol. Pharmacol., 2005, 68, 758 CAS.
- L. P. He, T. Hewavitharana, J. Soboloff, M. A. Spassova and D. L. Gill, J. Biol. Chem., 2005, 280, 10997 CrossRef CAS PubMed.
- J. Ishikawa, K. Ohga, T. Yoshino, R. Takezawa, A. Ichikawa, H. Kubota and T. Yamada, J. Immunol., 2003, 170, 4441 CrossRef CAS.
- C. Zitt, B. Strauss, E. C. Schwarz, N. Spaeth, G. Rast, A. Hatzelmann and M. Hoth, J. Biol. Chem., 2004, 279, 12427 CrossRef CAS PubMed.
- S. Kiyonaka, K. Kato, M. Nishida, K. Mio, T. Numaga, Y. Sawaguchi, T. Yoshida, M. Wakamori, E. Mori, T. Numata, M. Ishii, H. Takemoto, A. Ojida, K. Watanabe, A. Uemura, H. Kurose, T. Morii, T. Kobayashi, Y. Sato, C. Sato, I. Hamachi and Y. Mori, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5400 CrossRef CAS PubMed.
- S. Koenig, M. Schernthaner, H. Maechler, C. O. Kappe, T. N. Glasnov, G. Hoefler, M. Braune, E. Wittchow and K. Groschner, J. Pharmacol. Exp. Ther., 2013, 344, 33 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00023a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence