
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the enthalpic preference of fragment binding†‡
György G.
Ferenczy
and
György M.
Keserű
*
Medicinal Chemistry Research Group, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar tudósok körútja 2, Budapest 1117, Hungary. E-mail: keseru.gyorgy@ttk.mta.hu
First published on 10th December 2015
Abstract
Detailed thermodynamic analysis of fragment binding revealed that unlike drug-like compounds, fragments bind with significant enthalpic preference. This observation is in line with the size dependency of binding enthalpy contributions and is also supported by a large body of experimental data from direct binding thermodynamic measurements. The enthalpy-driven binding of fragments represents a thermodynamic rationale for fragment-based drug discovery programs and suggests guidelines for fragment-based optimization programs.
Introduction
The thermodynamics of ligand–protein binding has recently gained increased attention due to the recognition that enthalpy and entropy changes accompanying binding can carry significant information on the quality of interactions. In the context of drug discovery, the thermodynamic quantities of binding have been suggested to be related to the drug-like properties of ligands and ultimately to the developability of drug candidates.1,2 The thermodynamic profile of ligand binding, i.e. the enthalpic and entropic contributions to the binding free energy, reflects the structural features and the interactions of the ligand and the protein. However, a detailed understanding of how structural changes affect thermodynamic profiles is still missing. It appears that the complex process of ligand binding which includes conformational changes, desolvation and formation of new interactions between the ligand and the protein gives various and, to a large extent, compensating contributions to both enthalpy and entropy. Nevertheless, it has been recognized that enthalpic contributions are often dominated by optimal polar interactions while entropic contributions come at a large extent from the desolvation that releases water molecules from the solvation shell and allows them to join bulk water. These trends manifest themselves in the ligand size dependence of thermodynamic profiles, that is, smaller ligands tend to bind with favourable enthalpy while favourable entropy changes become increasingly important in the binding of larger ligands.3 Since fragments usually bind in the high micromolar to the low millimolar range and endothermic binding for small molecule–protein interactions is relatively rare, (around 20% for all interactions in the SCORPIO and BINDING databases), the enthalpy gain of fragment binding is not unexpected. In the current contribution, however, we show that – in contrast to drug-like molecules – the contribution of favourable fragment binding enthalpy tends to dominate over entropic terms. We analyse this trend by investigating the theoretical background and experimental data of the binding of small compounds. We focus our analyses on fragment-sized compounds whose heavy atom number does not exceed 22. It is worth mentioning that fragments have emerged as a key tool in drug discovery as they provide highly suitable chemical starting points for medicinal chemistry optimizations. In the forthcoming analysis, we show that in contrast to other opinions which are more ambivalent about the enthalpic signature of fragments4,5 they do have some particular features that lead to specific thermodynamic profiles and this discriminates them from larger compounds.Theoretical considerations
Fragments used in drug discovery are small polar compounds. Owing to their small size, they typically exhibit low affinity towards protein targets. In order to detect their binding, a high screening concentration (typically >100 μM) is required and thus sufficient water solubility is needed. Thus, the fragments used in drug discovery must have a high polar surface area and a limited lipophilic character. Indeed, fragments have been shown to bind to protein hot spots through a limited number of optimal geometry H-bonds.6 It has to be noted that not all the H-bonds identified in ligand–protein complexes are expected to contribute to the binding free energy. Owing to the dependence of the H-bond energy on geometrical parameters, most notably on the donor–acceptor distance and the donor–hydrogen–acceptor angle,7 H-bonds with non-optimal geometrical parameters give little contribution to the binding free energy as they fail to overcome unfavourable desolvation effects. The estimated free energy gain for the formation of a single H-bond in water does not to exceed 6 kJ mol−1 in either peptide–peptide8,9 or small molecule interactions.10 Taking into account the fact that the average number of optimal geometry H-bonds in fragment–protein interactions was found to be 2, the free energy gain of H-bond formation would be 12 kJ mol−1 on average, based on these numbers. However, the above estimates for the free-energy of H-bonds are not directly related to fragment–protein binding whose thermodynamics may be different as protein hot spots are involved, as discussed below.Fragments bind to protein hot spots6,11,12 which are good binding sites in general.13 No universal characterization of hot spots is available but there are certain specific features attributed to them. It was shown14 that water molecules displaced by fragments in the hot spot have high unfavourable excess entropies as calculated by the WaterMap method.15 Various fragments binding to the same protein hot spot appear to displace the same set of water molecules without affecting distal water networks.16 This is in contrast to larger compounds which disturb the water network in the binding pocket to a larger extent and therefore need to expel water molecules at a larger free energy cost. Another feature of fragment–hot spot binding was pointed out by Vajda and co-workers showing that pose conservation upon fragment expansion is associated with binding to the hot spot.12 These observations make it reasonable to assume that the cited 6 kJ mol−1 estimation for the free energy gain of H-bonds represents a lower limit to that available for fragments at protein hot spots.
A clue to the enthalpic and entropic components of the free energy gain attributed to H-bonds is provided by an analysis of the polar surface area burial upon binding. Plots of enthalpy and entropy against buried polar surface area for an extensive data set show that 50–100 Å2 polar surface burial associated with the formation of two H-bonds corresponds to several tens of kJ mol−1 enthalpic gain and a smaller entropic penalty (Fig. 4c and d in ref. 17). It has to be noted that this high favourable enthalpy change is not proportional to the number of H-bonds; instead, it diminishes with increasing polar surface burial.17
Ligand–protein binding is accompanied by a rigid body entropy loss. Various estimates for its magnitude come from indirect experimental measurements and from computations (see e.g.ref 18). Specific analyses of fragment–protein binding suggest a 15–20 kJ mol−1 entropic loss.19,20 This value only slightly increases for larger ligands, and thus a rigid body entropy loss represents a relatively more important barrier to fragment binding owing to the limited number of interactions available to compensate for it.
The desolvation of the ligand and the protein typically contributes favourably to binding. The magnitude of this effect correlates with the apolar surface buried in the complex formation. An analysis of the buried apolar surface area and binding free energy data for a large number of protein–ligand complexes showed that the burial of approximately 20 Å2 apolar Connolly surface corresponds to 1 kJ mol−1 free energy gain.17 The enthalpy and entropy components of the binding free energy show weaker correlation with apolar surface area burial, but a trend of unfavourable enthalpy change and a twice as large favourable entropy change was identified.17 This is in line with the interpretation that these effects are dominated by the transition of water molecules from the solvation shell into the bulk water. The small size and polarity of the fragments represent limitations for the magnitude of their apolar surface area and for the corresponding thermodynamic effects upon binding. The apolar part of the Connolly surface of fragment-sized compounds in drug discovery is typically below 250 Å2 and in most cases it is much lower owing to the small size and the polar nature of the fragments.21 Taking into account the fact that the burial of the ligand surface is accompanied by the burial of the protein surface with a similar magnitude, the binding free energy gain corresponding to the burial of 250 Å2 of ligand apolar surface is 25 kJ mol−1. This is an upper limit, and the binding free energy gain due to apolar desolvation is significantly lower in most cases. This means that apolar desolvation alone typically does not give a high enough contribution to compensate for the rigid body entropy loss and to produce productive binding for the fragments.
The above analysis shows that fragment–protein binding has to be additionally driven by the enthalpy gain of few optimal geometry polar interactions, most notably by H-bonds that are able to overcome the rigid body entropy loss in spite of the typically less important entropy gain coming from desolvation. Consequently, theoretical considerations suggest that fragment binding should be enthalpic in nature.
Experimental fragment thermodynamics data
There are relatively few studies that are aimed at the experimental investigation of fragment binding thermodynamics. In a previous study, we analysed ITC data of fragment–protein complexes and found that the majority of the fragments bind enthalpically.3 The distribution of thermodynamic signatures showed that except for charged ions (7 out of 159) all other fragments bind enthalpically. In line with this observation, the large ITC fragment dataset of Astex measured under standardized conditions shows that 621 out of the 624 compounds bind with favourable enthalpy.22,23 This trend also is reflected the present dataset of 138 neutral fragments and druglike compounds24,26 on 17 targets where the pronounced tendency of fragments to bind with more favourable enthalpy and less favourable entropy with respect to drug-like compounds is apparent in the ΔH vs. −TΔS scatterplot (Fig. 1a). Furthermore, a statistical analysis of ΔH and −TΔS values showed a statistically significant difference between fragments and drug-like compounds (Mann–Whitney U-test, p < 0.005). On the other hand, charged compounds fail to show such a trend owing to the increased role of the desolvation penalty accompanying binding (Fig. 1b). Interestingly enough, for thrombin fragments the authors compared the thermodynamic profiles obtained by direct and displacement titration.25 This analysis revealed that only direct ITC data can be used to compare the thermodynamic profiles since these are not further affected by superimposed implications of the displacement ligand.16 The authors suggested that the surrounding water network has a significant impact on the thermodynamic profiles; however, we argue that these effects in fact do not override the enthalpic preference of the fragments as suggested by the size dependency of the binding enthalpy3,27 and the above analysis.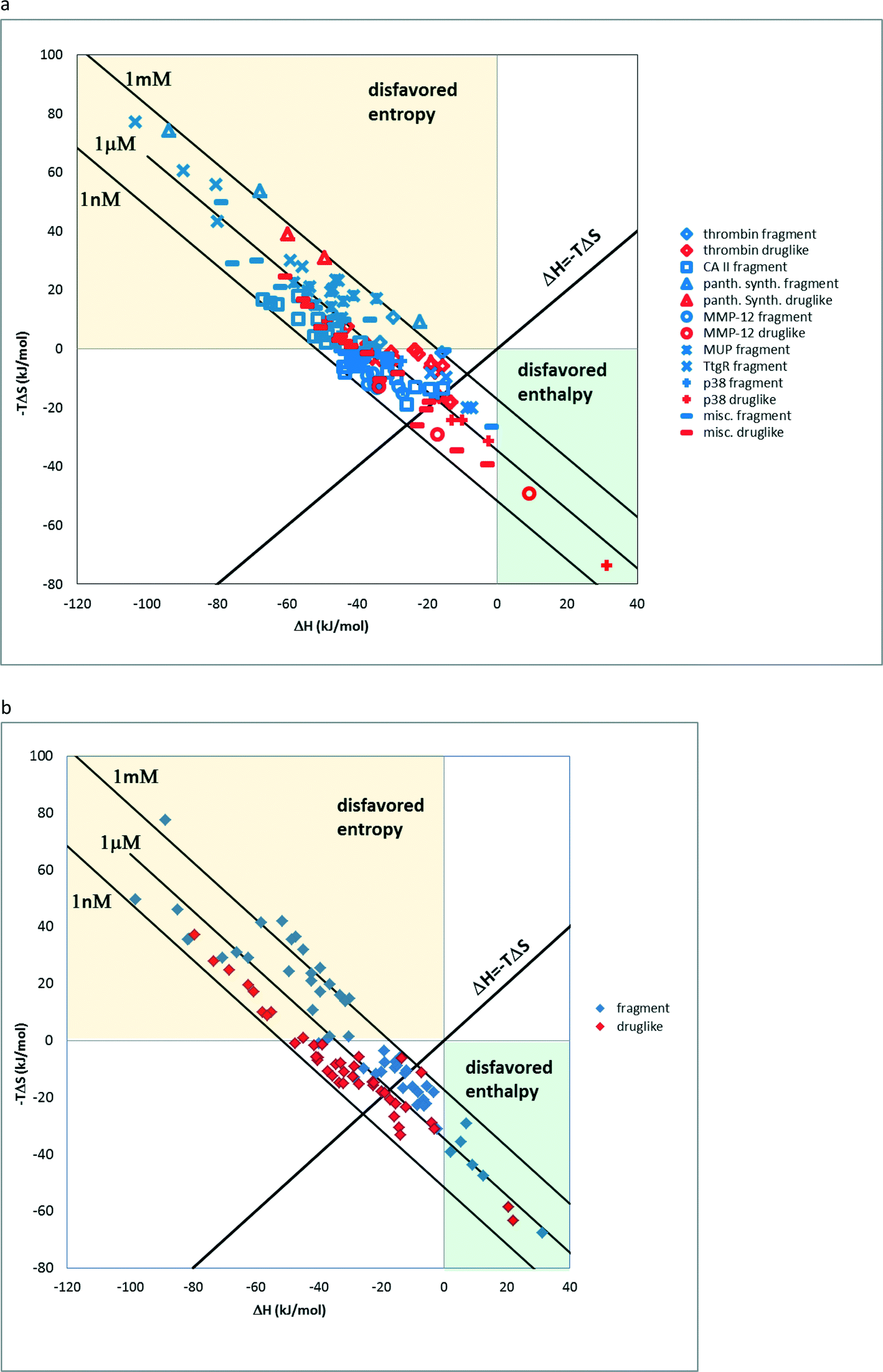 | ||
| Fig. 1 a) Enthalpic and entropic components of binding for complexes of neutral fragments and drug-like molecules. b) Enthalpic and entropic components of binding for complexes of charged fragments and drug-like molecules. | ||
Impact on fragment optimization
Investigating the enthalpic preference of fragment binding relative to optimized compounds, we performed a comparative structural and thermodynamic analysis of thrombin,25 carbonic anhydrase II (ref. 28) and MMP12 (ref. 29) ligands (Table 1). These examples were selected as both thermodynamic data and high resolution X-ray structures are available both for the fragments and for the optimized compounds. Owing to the selection of examples, our analysis focuses on true enzymatic hot spots and may not be valid for protein–protein interfaces. First we investigated the number of optimal H-bonds that contribute most significantly to the binding enthalpy gain upon binding. In the case of thrombin, we found that the benzamidine fragment forms four H-bonds within the S1 pocket of thrombin out of which two H-bonds fulfil the geometric criteria of optimal interactions.7 Although the corresponding optimized compound forms more H-bonds (6 optimized out of the 9 as reflected in the corresponding binding enthalpy contribution), the number of optimal H-bonds (2/4) does not change within the S1 pocket. FMAP calculations12 revealed that all the fragments bind to the hot spot of protein targets. In the case of thrombin, the S1 pocket was identified by FMAP as the hot spot. The benzamidine fragment displaced 5 water molecules from the S1 pocket and other thrombin fragments displaced the very same set of water molecules.16 The optimized ligand displaced basically the same set of water molecules from the S1 pocket; however, we found 3 more water molecules displaced from other thrombin pockets. This indicates that in the case of thrombin, the larger ligand disturbs the water network more significantly than the fragment. Apolar desolvation, as approximated by the change of the apolar contact surface area, was found to be also more significant for the optimized ligand relative to the fragment (217 Å2versus 172 Å2). Both the larger number of water molecules replaced and the larger extent of apolar desolvation contributed to the increased entropy gain observed for the optimized compound. Considering the larger improvement found in the binding enthalpy, this optimization is enthalpy driven which improved the potency with the simultaneous increase in MW but controlled the lipophilicity effectively.| Thrombin | CA-II | MMP12 | ||||
|---|---|---|---|---|---|---|
| Frag | Opt | Frag | Opt | Frag | Opt | |
| Ligand structure |
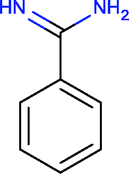
|
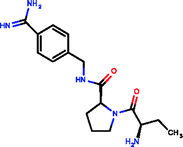
|

|
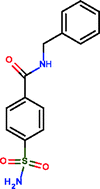
|
|
|
| H-bonds (opt/all) | 2/4 | 6/9 | 1/1 | 2/3 | 2/2 | 2/4 |
| ΔCSAapo (Å2) | 172 | 217 | 98 | 326 | 59 | 196 |
| # of H2O replaced | 4 | 8 | 5 | 5 | 2 | 5 |
| K d (μm) | 355 | 0.11 | 0.839 | 0.027 | 6180 | 0.0611 |
| ΔG (kcal mol−1) | −4.73 | −9.48 | −8.3 | −10.3 | −3.01 | −9.83 |
| ΔH (kcal mol−1) | −3.32 | −6.88 | −7.5 | −9.7 | −3.18 | −9.09 |
| −TΔS (kcal mol−1) | −1.41 | −2.60 | −0.8 | −0.6 | 0.17 | −0.74 |
| N heavy | 9 | 24 | 10 | 20 | 5 | 15 |
| MW | 120.2 | 331.4 | 157.2 | 290.3 | 75.1 | 230.2 |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
0.89 | −0.26 | 0.58 | 1.38 | −1.03 | −0.53 |
| Reference | 25 | 28 | 29 | |||
| PDB | 4UEH | 2ZGX | 4JSZ (H94C) | 1G4O (F131V) | 1Y93 | 3F1A |
| PDB (apo) | 2UUF | 1G3Z | 4IJO | |||
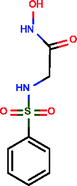
Benzenesulphonic amide, the carbonic anhydrase II fragment, forms only one H-bond within the binding cavity which is optimal. The most important interactions, however, are heteroatom–metal contacts formed between the Zn ion and sulphonic amide O and N atoms. The number of optimal H-bonds increased to 2 out of the total 3 for the optimized compound, which is in line with the enthalpy gain realized in the corresponding thermodynamic profile. The benzenesulphonic amide moiety binds to the hot spot of the protein in both cases and displaces the very same number of water molecules. The apolar contact surface area is much larger for the optimized compounds. However, only a small part of the contact surface is used upon binding since the phenyl group goes out from the binding pocket. Consequently, only a minor improvement in the binding entropy was detected. Comparing the thermodynamic profiles of the initial fragment and the optimized compound, this optimization is also enthalpy driven and improved the potency at the expense of some limited increase in both MW and lipophilicity.
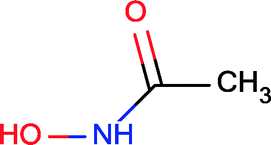
The starting fragment of the MMP12 optimization was hydroxamic acid that forms two H-bonds, both optimal, with the protein and also heteroatom–metal contacts with the active site Zn ion. The number of optimal H-bonds remained constant during the optimization; however, two new less than optimal H-bonds were detected. The fragment binds to the protein hot spot and displaces two water molecules. Although the position of the hydroxamic acid moiety does not change too much, the optimized compound displaces 3 more water molecules. The larger compound therefore disturbs the water network more significantly relative to the fragment. The apolar contact surface area is much increased and thus improved the contribution of the binding entropy to some extent. More importantly however, the introduced phenyl group forms van der Waals contacts with His218 that in addition to extra H-bonds make the binding of the optimized compound enthalpy dominant. Optimization of the initial fragment is again enthalpy driven which improves the potency with the simultaneous control of physicochemical parameters.
The above examples demonstrate that optimized fragment hits can be further developed with a more favourable binding enthalpy than the fragment screening hits they are derived from. In line with this finding, a thermodynamic analysis of 150 compounds from Astex drug discovery programs revealed that favorable binding enthalpy of fragment screening hits can be further improved by optimizing the compounds to leads (Fig. 2). However, lead optimization seems to be then associated with reduced favourable enthalpy, although with more favourable binding free energy. This is supported by the observation that late phase optimizations are usually entropy driven.30 In fact, Astex published a thermodynamic optimization scheme of HSP90 inhibitors indicating that both the fragment starting points and the optimized compounds are clearly enthalpic.31
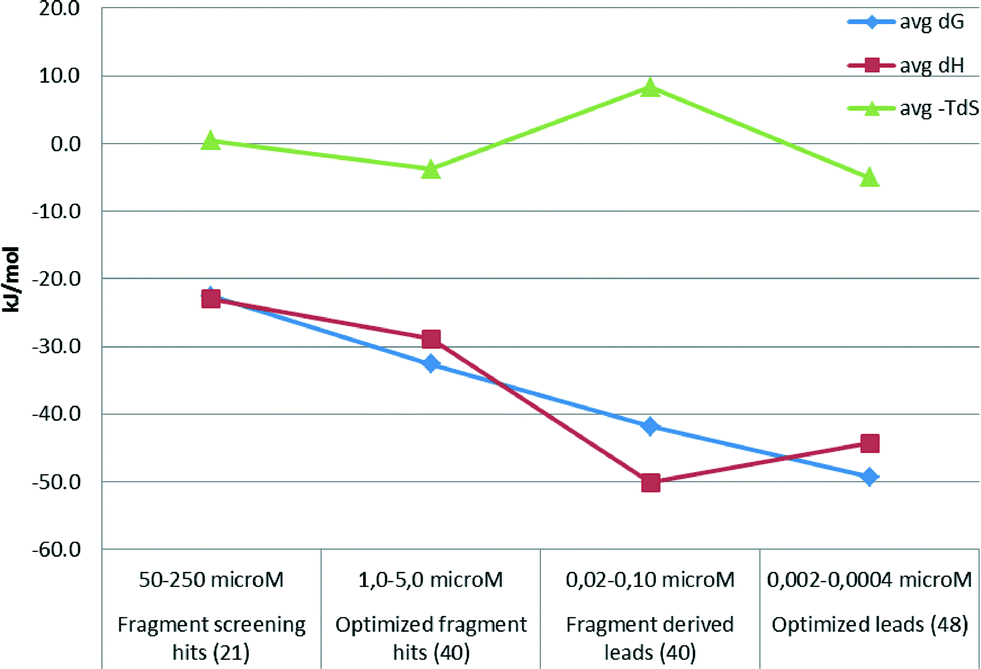 | ||
| Fig. 2 Fragment optimization data from Astex. | ||
These observations together with supporting theoretical considerations suggest that:
• the higher the enthalpy content of binding for a fragment hit, the higher the chance that it can be optimized to a clinical candidate with advantageous thermodynamic properties and associated beneficial physicochemical and pharmacokinetic profiles;
• early phase optimization offers more opportunity for increasing the enthalpic character of binding;
• although the measured binding enthalpy is multifactorial and the individual contributions of intermolecular interactions cannot be measured directly, the net enthalpy gain reflects the quality of interactions;32
• the optimization of fragment–protein interactions must be explored without looking for overall free energy improvement before lead optimization really starts.
Conclusions
In summary, theoretical considerations and detailed examples support the hypothesis that fragments form an optimal geometry via polar interactions with the protein hot spot without any important apolar desolvation contribution and without significantly disturbing the water network in the binding site. These findings together with the large amount of raw thermodynamic data all demonstrate that fragments bind enthalpically. Analyses of fragment optimizations support that appropriate screening hit selection and early phase optimization offer the best opportunity to maximize the favourable enthalpy content of binding.Acknowledgements
This work was supported by the National Brain Research Program KTIA-NAP-13-1-2013-0001.Notes and references
- J. E. Ladbury, G. Klebe and E. Freire, Nat. Rev. Drug Discovery, 2009, 9, 23 CrossRef PubMed.
- G. G. Ferenczy and G. M. Keserű, Drug Discovery Today, 2010, 15, 919 CrossRef CAS PubMed.
- G. G. Ferenczy and G. M. Keserű, J. Chem. Inf. Model., 2010, 50, 1536 CrossRef CAS PubMed.
- G. Klebe, Nat. Rev. Drug Discovery, 2015, 14, 95 CrossRef CAS PubMed.
- E. Ruhmann, M. Betz, A. Heine and G. Klebe, J. Med. Chem., 2015, 58, 6960 CrossRef PubMed.
- G. G. Ferenczy and G. M. Keserű, J. Chem. Inf. Model., 2012, 52, 1039 CrossRef CAS PubMed.
- A. V. Morozov, T. Kortemme, K. Tsemekhman and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6946 CrossRef CAS PubMed.
- A. R. Fersht, Trends Biochem. Sci., 1987, 12, 301 CrossRef CAS.
- S.-Y. Sheu, D.-Y. Yang, H. L. Selzle and E. W. Schlag, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12683 CrossRef CAS PubMed.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 CrossRef CAS PubMed.
- S. Barelier, J. Pons, O. Marcillat, J. M. Lancelin and I. Krimm, J. Med. Chem., 2010, 53, 2577 CrossRef CAS PubMed.
- D. Kozakov, D. R. Hall, S. Jehle, L. Luo, S. O. Ochiana, E. V. Jones, M. Pollastri, K. N. Allen, A. Whitty and S. Vajda, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E2585 CrossRef CAS PubMed.
- W. L. DeLano, Curr. Opin. Struct. Biol., 2002, 12, 14 CrossRef CAS PubMed.
- Y. Yang, F. C. Lightstone and S. E. Wong, Expert Opin. Drug Discovery, 2013, 1 Search PubMed.
- O. Ichiara, Y. Shimada and D. Yoshidome, ChemMedChem, 2014, 9, 2708 CrossRef PubMed.
- E. Rühmann, M. Betz, A. Heine and G. Klebe, J. Med. Chem., 2015, 58, 6960 CrossRef PubMed.
- T. S. Olsson, M. A. Williams, W. R. Pitt and J. E. Ladbury, J. Mol. Biol., 2008, 384, 1002 CrossRef CAS PubMed.
- S. J. Irudayam and R. H. Henchman, J. Phys. Chem. B, 2009, 113, 5871 CrossRef CAS PubMed.
- C. W. Murray and M. L. Verdonk, J. Comput.-Aided Mol. Des., 2002, 16, 741 CrossRef CAS PubMed.
- V. Borsi, V. Calderone, M. Fragai, C. Luchinat and N. Sart, J. Med. Chem., 2010, 53, 4285 CrossRef CAS PubMed.
- The apolar Connolly surfaces for the 91 fragments included in the analysis of thermodynamic profiles are available in the Supplementary Information.
- E. Chiarparin, N. Amin and G. Williams, The behaviour of physico-chemical properties during fragment progression. FRAGMENTS III - Third RSC-BMCS Fragment-based Drug Discovery meeting, March 7–8; Poster 31, GlaxoSmithKline, Stevenage, UK, 2011.
- F. E. Torres, M. I. Recht, J. E. Coyle, R. H. Bruce and G. Williams, Curr. Opin. Struct. Biol., 2007, 20, 598 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3 CrossRef CAS PubMed.
- E. Rühmann, M. Betz, M. Fricke, A. Heine, M. Schäfer and G. Klebe, Biochim. Biophys. Acta, 2015, 1850, 647 CrossRef PubMed.
- Compounds with either pKbasic higher than 7 or pKacidic lower than 7 were considered as charged. pK values were calculated with JChem for Excel, version 15.8, Chemaxon, Budapest, Hungary, 2014.
- C. H. Reynolds and M. K. Holloway, ACS Med. Chem. Lett., 2011, 2, 433 CrossRef CAS PubMed.
- A. D. Scott, C. Phillips, A. Alex, M. Flocco, A. Bent, A. Randall, R. O'Brien, L. Damian and L. H. Jones, ChemMedChem, 2009, 4, 1985 CrossRef CAS PubMed.
- I. Bertini, V. Calderone, M. Fragai, A. Giachetti, M. Loconte, C. Luchinat, M. Maletta, C. Nativi and K. J. Yeo, J. Am. Chem. Soc., 2007, 129, 2466 CrossRef CAS PubMed.
- M. M. Hann and G. M. Keserű, Nat. Rev. Drug Discovery, 2012, 11, 355 CrossRef CAS PubMed.
- G. Williams, The PyramidTM Approach to Fragment-Based Biophysical Screening, in Label-Free Technologies For Drug Discovery, ed. M. Cooper and L. M. Mayr, John Wiley & Sons, New York, USA, 2011, p. 241 Search PubMed.
- E. Freire, Drug Discovery Today, 2008, 13, 869 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interest. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5md00542f |
| This journal is © The Royal Society of Chemistry 2016 |