
Structural hybridization of three aminoglycoside antibiotics yields a potent broad-spectrum bactericide that eludes bacterial resistance enzymes†
Juan Pablo
Maianti
and
Stephen
Hanessian
*
Department of Chemistry, Université de Montréal, CP 6128 Succ. Centre-Ville, Montréal, Québec H3C3J7, Canada. E-mail: stephen.hanessian@umontreal.ca
First published on 13th November 2015
Abstract
Vast numbers of prevalent aminoglycoside-modifying enzymes undermine the clinical use of aminoglycoside antibiotics. We present the design and synthesis of a potent broad-spectrum bactericidal aminoglycoside based on available X-ray co-crystal structures within the ribosomal binding-site. The resulting antibiotic displays broad protection of its functional groups from inactivation by clinically relevant resistance enzymes.
Aminoglycosides (AGs) are a class of broad-spectrum bactericidal antibiotics originally discovered among the arsenal of molecules evolved by soil microbes to wage warfare on each other.1 This class of natural and semi-synthetic antibiotics has been highly effective against Gram-positive and Gram-negative pathogens that are difficult to treat and cause life-threatening infections in humans, including the bacterial strains under the “E.S.K.A.P.E.” classification (Escherichia coli, Staphylococcus aureus, Klebsiella sp., Acinetobacter sp., Pseudomonas aeruginosa, and Enterobacter sp.).1,2 However, selective pressure from decades of clinical use of AGs has led to world-wide dissemination of more than 50 isoforms of aminoglycoside-modifying enzymes (AMEs) that render most AG antibiotics ineffective in clinical settings (Fig. 1A, Table 1).3–6
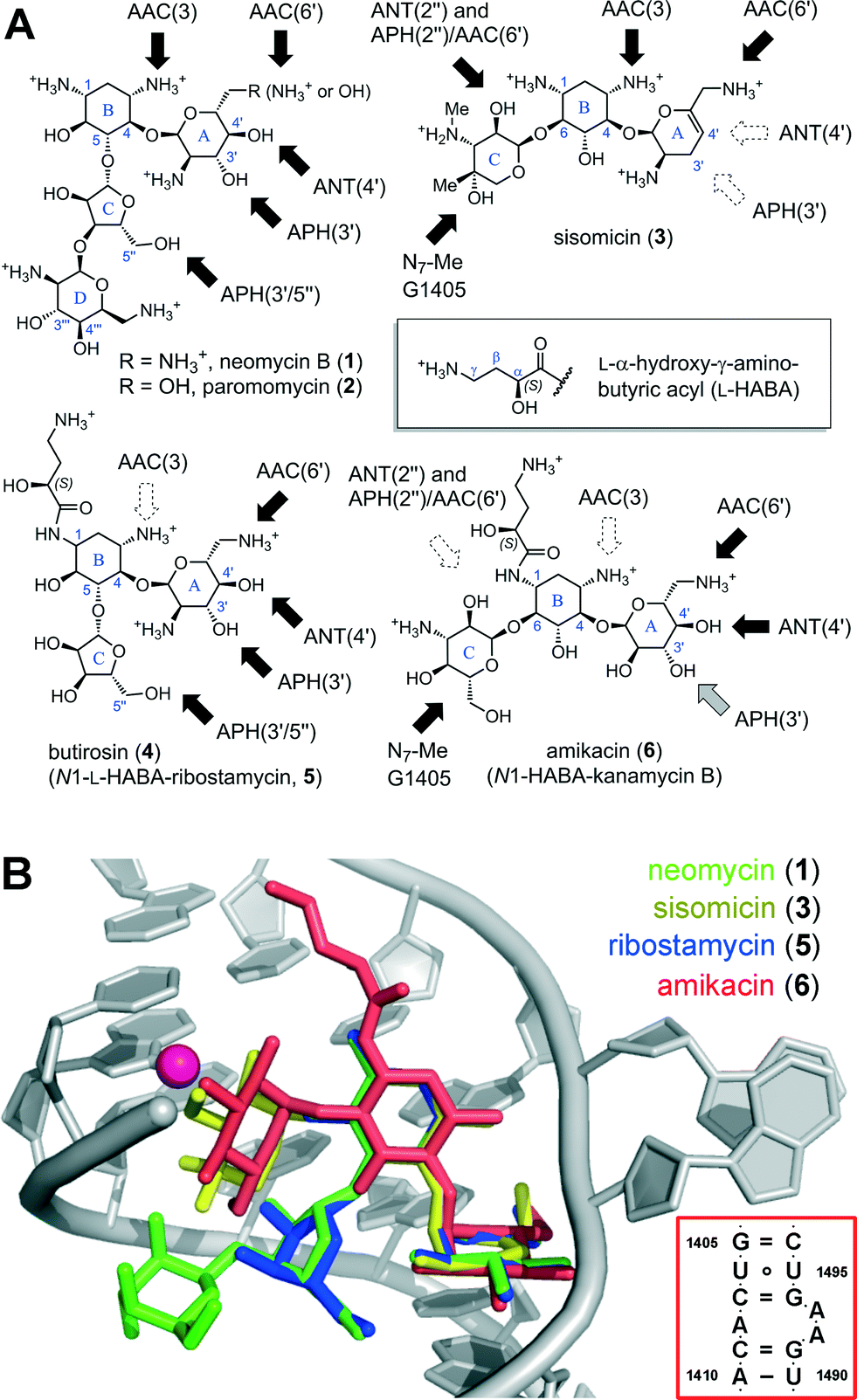 | ||
| Fig. 1 (A) Sub-classes of AGs based on the substitution pattern of ring B: the 4,5-disubstituted class includes neomycin (1), paromomycin (2), butirosin (4), and the simpler congener ribostamycin (5). The 4,6-disubstituted class includes natural products such as sisomicin (3), and semi-synthetic analogs such as amikacin (6), a kanamycin B analog acylated with the N1-L-HABA group originating from butirosin (4). Dark arrows indicate functional groups liable to AMEs. (B) Superimposed X-ray co-crystal structures of AGs bound in the A-site (red box): neomycin (1),10 sisomicin (3),11 ribostamycin (5),10 and amikacin (6).12 The location of the G1405-N7-Me product of RMT enzymes is highlighted as a magenta sphere. Rendered in PyMol.13 | ||
| Bacterium | Prevalent resistance mechanisms (ranked order) |
|---|---|
| Enterococci | AAC(6′)-Ie/APH(2′′)-Ia, APH(3′)-IIIa |
| S. aureus | AAC(6′)-Ie/APH(2′′)-Ia, ANT(4′)-Ia, APH(3′)-IIIa |
| K. pneumoniae | AAC(6′)-Ib, AAC(3)-I, ANT(2′′) |
| Acinetobacter | Efflux, APH(3′)-VI, AAC(6′)-I, AAC(3)-I/II, ANT(2′′)-I, RMT |
| P. aeruginosa | Efflux, AAC(6′)-II, ANT(2′′)-I, APH(3′)-II, AAC(3)-I |
| Enterobacter | AAC(6′)-Ie, AAC(3)-I, ANT(2′′), RMT |
The structural features common to all AGs include a positively charged pseudosaccharide scaffold (Fig. 1A), assembled on a 2-deoxystreptamine core (ring B) and two to four glycosidic linkages with neutral and basic sugar units, which are decorated with hydroxyl and ammonium groups. Owing to these unique and highly polar features, AGs belong to a distinct molecular-descriptor space compared to most drugs.14 Despite their structural diversity, the majority of AGs target overlapping binding sites within an evolutionarily conserved RNA helix deep in the core of the mRNA-tRNA decoding center of the bacterial 30S ribosomal subunit, called the A-site (Fig. 1B).7–10 When bound to the A-site AGs orchestrate a disruption of the protein synthesis machinery and coax the ribosome to accept mismatched tRNAs.7–10,15 The misfolded protein products resulting from the stalled and error-prone mRNA translation trigger a sequence of cellular stressors that lead to bacterial death.16 This broad-spectrum bactericidal effect is a key feature of AGs, which is distinctive from other classes of antibiotics that target the ribosome.15
AGs traverse the cell membrane exploiting an energy-dependent uptake mechanism conserved in all bacteria,22 and must build-up over a concentration threshold to engage in a self-reinforcing cycle of AG uptake.23 Resistant clinical isolates survive exposure to AGs by reducing the intracellular concentration of free AG under this critical threshold.23 Whereas β-lactam antibiotics are catalytically hydrolyzed by betalactamase enzymes, AGs are primarily rendered inactive by enzymatic modification with adducts that prevent binding to the A-site RNA helix (Fig. 1).3–6 AMEs are classified based on the targeted functional group and designated by a number indicating the position of attack on the sugar: N-acetyltransferases (AAC-#), O-phosphotransferases (APH-#), and O-adenyltransferases (ANT-#) (Fig. 2A–C).3–6 Secondary resistance mechanisms include AG-efflux pumps, mainly in P. aeruginosa and Acinetobacter strains.1,24 Moreover, there is growing concern over the number clinical isolates now carrying ribosomal RNA methyltransferase (RMT) enzymes targeting the A-site.25–27 Ribosomes modified with a N7-G1405 methyl adduct effectively repeal the entire sub-class of 4,6-disubstituted AGs including sisomicin (3), amikacin (5) and gentamicin (7) (Fig. 1B, Fig. S1†).1,25,26
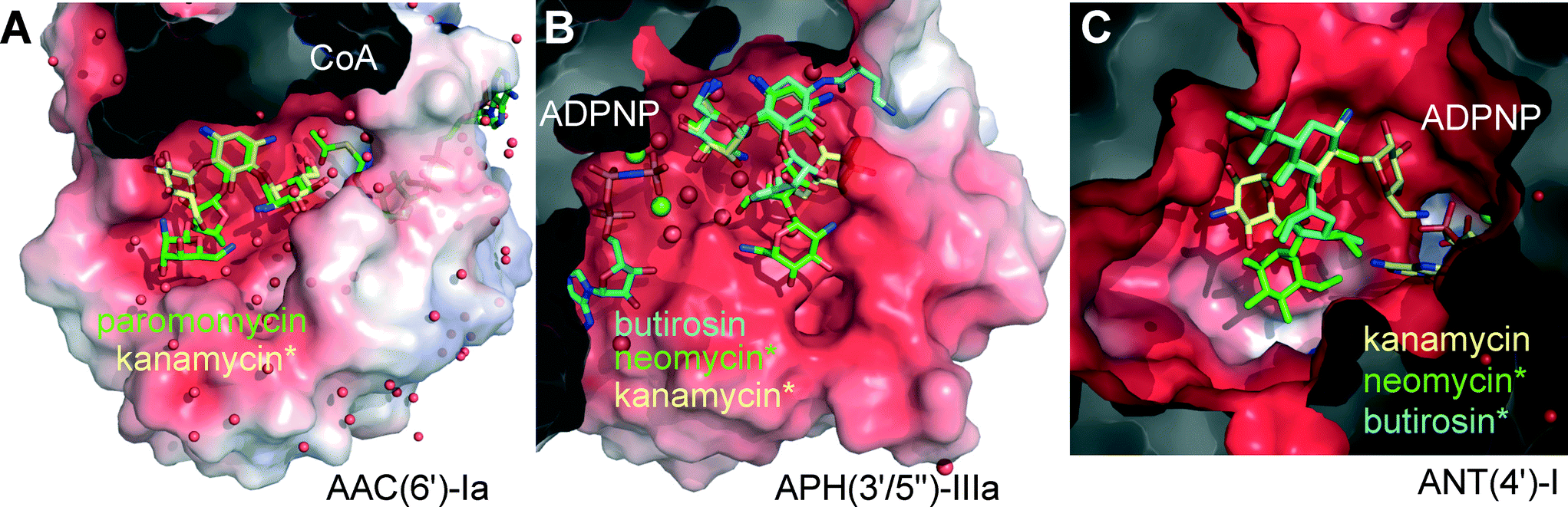 | ||
| Fig. 2 Active site pockets of representative AME isoforms co-crystallized with a cofactor and an AG substrate (coenzyme-A, CoA, or a non-hydrolysable ATP analog, ADPNP). Other substrates were superimposed (marked *). The protein structures are displayed as semi-transparent van der Waals surfaces colored with the calculated electrostatic potential (red = negative, blue = positive).17 AGs are displayed as sticks and color-coded uniformly: kanamycin is shown yellow, butirosin in cyan, and neomycin or paromomycin in green. Clipping planes were used to expose the active sites. Spheres represent water molecules (red) or magnesium ions (green). (A) AAC(6′)-Ia co-crystallized with paromomycin and Ac-CoA, overlaying kanamycin C in the same protein.18 PDB entries 2VQY and 1V0C.18 (B) APH(3′/5′′)-IIIa co-crystallized with butirosin and ADPNP, overlaying neomycin and kanamycin B in the same protein.19,20 PDB entries 3H8P,192B0Q and 1L8T.20 (C) ANT(4′)-I co-crystallized with kanamycin A and ADPNP,21 overlaying frameworks of butirosin and neomycin. PDB entry 1KNY.21 Rendered in PyMol.13 | ||
Surveillance of AME genes in AG resistant clinical isolates revealed that human pathogens express just one or two enzyme isoforms (Table 1, Fig. S2†).28–34 Expression of redundant AMEs may impose a metabolic burden by draining cellular cofactors leading to negative selection in the absence of antibiotic pressure.37 However, owing to the extended substrate preference of the AME active-site pockets, a few isoforms provide effective resistance against the majority of clinically deployed AGs (Fig. S1†).3–6 The development of small molecule inhibitors targeting a fraction of the >50 isoforms of AMEs could be helpful, but this approach cannot constitute a generalizable solution for all AME isoforms, which may also circumvent inhibition by rapid mutation.38 Most AMEs have evolved multi-AG substrate promiscuity by recognizing the common rings A and B, and harboring vast negatively charged active site surfaces (Fig. 2A–C). Nevertheless, the enzymes targeting hydroxyl groups, APHs and ANTs, are circumvented by AGs that lack these susceptible functionalities (Fig. 1A), such as the 3′- and 4′-OH groups missing in sisomicin (3) and gentamicin (7). Butirosin (4) is the only natural AG bearing a N1-(S)-hydroxy-aminobutyric acyl non-glycosidic appendage (“L-HABA”), which enhances its antibiotic potency compared to the unsubstituted trisaccharide congener, called ribostamycin (5).39 Many AME isoforms have not evolved a suitable pocket to accommodate the N1-L-HABA of butirosin (4) (Fig. 2a).40–43 Therefore, this side-chain has been widely exploited in semi-synthetic AG antibiotics, such as amikacin (6) (Fig. 1A).1,44,45
Despite the advancements over the past decades, the AGs of the 4,6-disubstituted 2-deoxystreptamine class remain liable to inactivation by a subset of AMEs prevalent in clinical isolates (Fig. 1A, Table 1). This class is also ineffective against A-sites modified by N7-G1405 rRNA methyltransferases (Fig. 1B).3–6
Although Nature's arsenal of diverse AG structures provides medicinal chemists with several examples that evolved to evade AMEs and A-site methylation, no natural structures are known that combine all the optimal features in one composite AG entity. Therefore, our group has envisioned the incorporation of AME-evasion features to the alternate 4,5-disubstituted AG series that includes neomycin (1) and paromomycin (2),40–43 which may render them inert to the AG resistance mechanisms. Herein, we report the systematic evaluation of synthetic AG analogs that sequentially incorporate segments of sisomicin (3), butirosin (4) and neomycin (1) in a single entity, culminating in one of the most potent AG antibiotics ever prepared, as evidenced by in vitro antibacterial assays against an extensive panel of highly relevant bacterial strains. The conceptual basis of this strategy of AG hybridization was inspired by pioneering structural endeavors by Westhof,10,12,46 Ramakrishnan7–9 and their respective groups, as well as our previous collaborative studies involving structure-based design in this series.40–42,47
Results and discussion
Synthetic strategy
In practice, the numerous hydroxyl and ammonium groups decorating AGs pose a daunting challenge for chemoselectivity. Paromomycin (2) is a versatile starting material to functionalize ring A using a 4′,6′-O,O-benzylidene protecting group (8) (Scheme 1).48 We recently reported a strategy to elaborate this intermediate into a 6′-aldehyde and the ensuing elimination of a 4′ leaving group,43 which provided the 4′,5′-unsaturated intermediate 11 in multi-gram scale (Scheme 1). Subsequently, we engaged the resulting allylic enal carbonate 11 in a palladium-catalyzed allylic deoxygenation reaction, yielding the 3′,4′-dideoxy intermediate 12 in 97% yield (gram scale and 70% overall yield from 2).43 The 6′-aldehyde functionality allowed access to N6′-substituted neomycin analogs by reductive amination (Scheme S1† and Fig. 3). Alternative elaboration of the 6′-OH and 6′-azido (13) intermediates delivered unsaturated 3′,4′-dideoxy paromomycin and neomycin analogs that we previously reported (Fig. 3, Table 2).43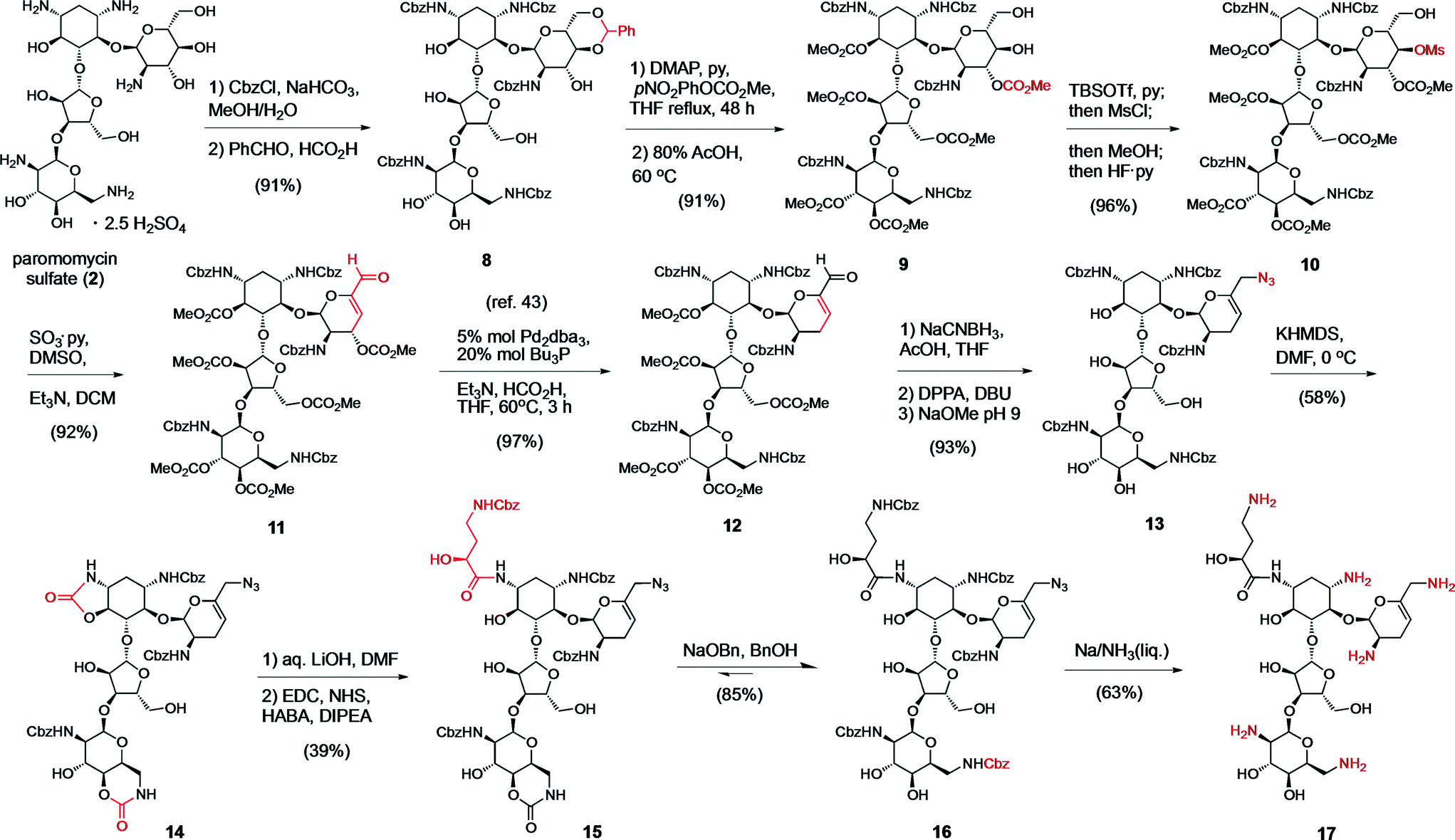 | ||
| Scheme 1 Synthesis of the sisomicin–butirosin–neomycin hybrid antibiotic 17. Functional group interconversions are highlighted in red. | ||
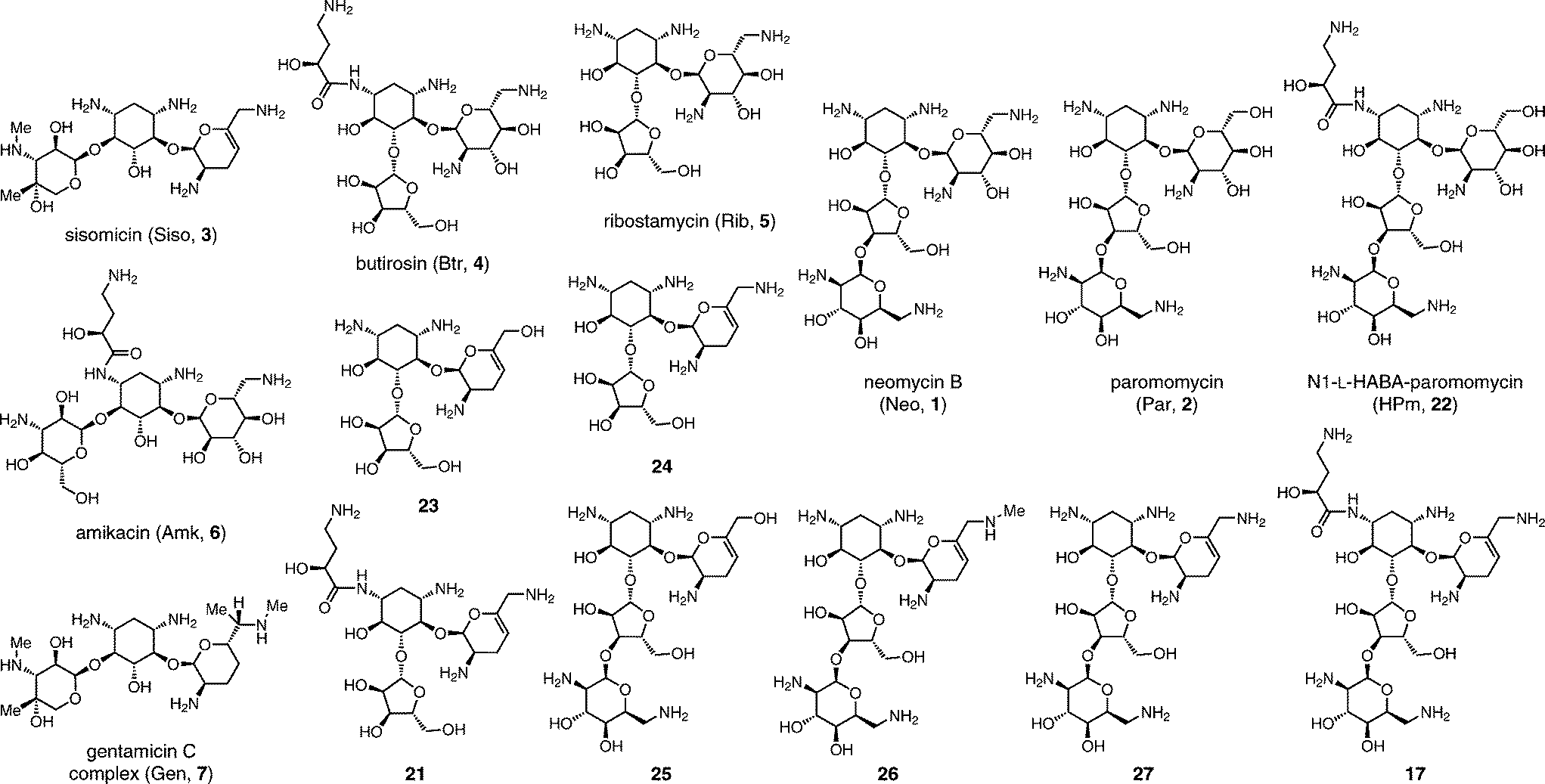 | ||
| Fig. 3 Structures of control AG antibiotics 1–7, new hybrid AGs antibiotics 17 and 21, and predecessor analogs 22–27. Reference for Table 2. | ||
| Entry | Bacterium/AME id. by PCR42 | Siso (3) | Amk (6) | Gen (7) | Btr (4) | Rib (5) | 23 | 24 | 21 | Neo (1) | Par (2) | HPm (22) | 25 | 26 | 27 | 17 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (a–d) strains were obtained from ATCC.a ATCC 25922.b ATCC 29213.c ATCC 10031.d ATCC 27853. Abbreviations: Neo, neomycin B (1); Par, paromomycin (2); HPm, N1-L-HABA-paromomycin (22); Siso, sisomicin (3); Amk, amikacin (5); Gen, gentamicin complex (7). Refer to structures in Fig. 3. | ||||||||||||||||
| 1 | E. coli | 0.5 | 2 | 0.5 | 2 | 8 | >64 | 64 | 8 | 2 | 4 | 2 | >64 | 8 | 2 | 0.5 |
| 2 | S. aureus | 0.5 | 2 | 0.5 | 8 | 16 | >64 | 16 | 8 | 0.5 | 2 | 2 | 8 | 1 | 1 | 0.5 |
| 3 | K. pnuemoniae | 0.25 | 0.5 | 0.25 | 1 | 2 | >64 | 16 | 4 | 0.5 | 2 | 1 | >64 | 2 | 0.5 | 0.5 |
| 4 | A. baumannii | 0.5 | 2 | 2 | 1 | 8 | >64 | >64 | 4 | 1 | 4 | 2 | >64 | 4 | 1 | 0.5 |
| 5 | P. aeruginosa APH(3′)-IIa | 0.5 | 2 | 0.5 | >32 | >64 | >64 | 16 | 4 | 32 | >64 | 8 | >64 | 2 | 0.5 | 0.25 |
| 6 | E. cloacae, APH(3′)-I, ANT(2′′)-I, AAC(6′) | >64 | 64 | 32 | 32 | >64 | >64 | >64 | >64 | >64 | >64 | 8 | >64 | >64 | >64 | 8 |
| 7 | E. coli APH(3′)-Ib | 0.25 | 0.5 | 0.25 | 1 | >64 | >64 | 64 | 4 | 64 | >64 | 2 | >64 | >64 | >64 | 0.25 |
| 8 | S. aureus APH(3′/5′′)-III | 0.25 | 8 | 0.5 | >32 | >64 | >64 | >64 | 16 | >64 | >64 | 32 | >64 | 16 | 16 | 1 |
| 9 | A. baumannii, AAC(3)-I, APH(3′)-VI, ANT(2′′)-I | >64 | >64 | >64 | >32 | >64 | >64 | >64 | 8 | >64 | >64 | 64 | >64 | >64 | >64 | 1 |
| 10 | S. aureus ANT(4′)-I | 1 | 64 | 0.5 | >32 | >64 | >64 | 16 | 8 | >64 | >64 | >64 | 16 | 2 | 1 | 1 |
| 11 | P. aeruginosa ANT(4′)-II | 1 | 32 | 2 | >32 | >64 | >64 | 64 | 8 | 8 | >64 | 4 | >64 | 4 | 1 | 0.5 |
| 12 | E. coli ANT(2′′)-I | 64 | 4 | 64 | 4 | 8 | >64 | >64 | 8 | 2 | 4 | 8 | >64 | 16 | 2 | 4 |
| 13 | P. aeruginosa AAC(6′)-II | 32 | 4 | 32 | >32 | >64 | >64 | >64 | 16 | 8 | >64 | 8 | >64 | >64 | >64 | 0.5 |
| 14 | S. aureus AAC(6′)/APH(2′′) | >64 | 64 | >64 | >32 | >64 | >64 | >64 | 16 | >64 | >64 | 32 | >64 | >64 | 64 | 2 |
| 15 | E. coli AAC(3)-IV | 32 | 2 | 16 | 2 | >64 | >64 | >64 | 8 | 2 | 8 | 4 | >64 | 8 | 8 | 1 |
| 16 | E. coli A-site methyltransferase ArmA | >64 | >64 | >64 | 2 | 8 | >64 | 64 | 8 | 1 | 4 | 4 | >64 | 16 | 2 | 1 |
For the final stage of this study, we exploited the absence of interfering functional groups on the ring-A of the advanced 6′-azido intermediate 13 to install the N1-L-HABA appendage (Scheme 1). We previously demonstrated that cyclic N1,O6-oxazolidinones are liable to mild alkaline hydrolysis in the presence of a 6-membered cyclic carbamate on ring D, which can be subsequently re-generated into a 6′′′N-carbobenzyloxy (Cbz) protecting group using sodium benzyloxide.42 Thus, exposing 13 to dry KHMDS forged two cyclic carbamates from the N1- and N6′′′-Cbz protecting groups (14) (Scheme 1). Selective cleavage of the N1,O6-oxazolidinone and acylation under standard amide coupling conditions afforded the N1-L-HABA-N6′′′,O4′′′-carbamate intermediate 15 in good yield. The ring D N6′′′-Cbz protecting group was then re-generated, affording intermediate 16 for global deprotection. Finally, Birch reduction cleanly removed the five Cbz protecting groups and reduced the 6′-azide in excellent yield (12% overall yield from 13), and outstanding purity (96%, LC trace), affording the novel sisomicin–butirosin–neomycin hybrid antibiotic 17 (Fig. 3, Table 2). Furthermore, using a similar N1,O6-oxazolidinone route we cleaved ring D from intermediate 13,43 and installed an N1-L-HABA substituent on the 3′,4′-deoxygenated trisaccharide 18 (Scheme 2) in order to boost the potency of analog 21, which represents a novel sisomicin–butirosin hybrid trisaccharide (Fig. 3, Table 2).
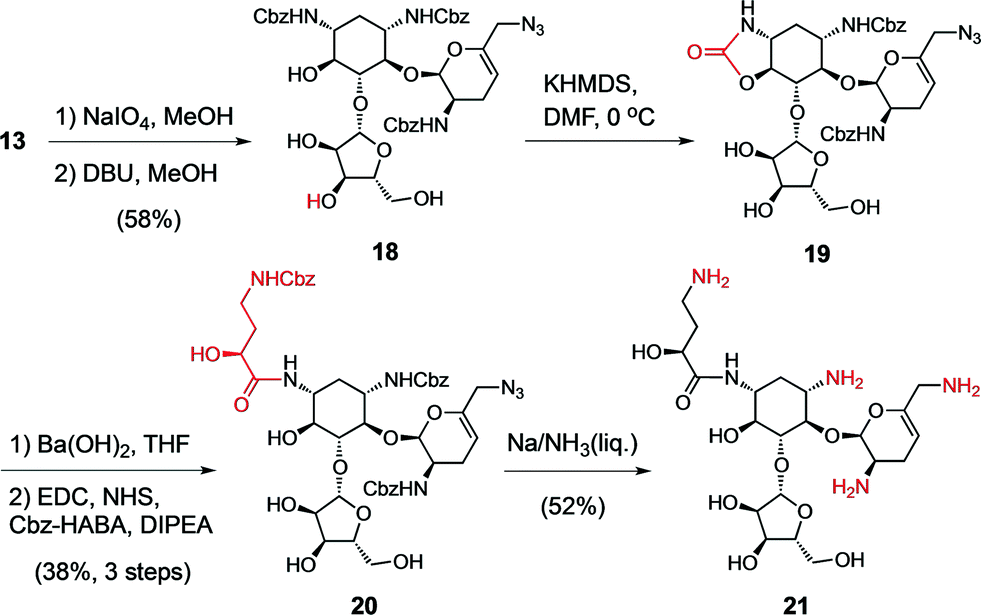 | ||
| Scheme 2 Synthesis of a sisomicin–butirosin hybrid antibiotic 21. | ||
Antibacterial potency and evasion of resistance mechanisms
The availability of seven hybrid analogs presented herein (Fig. 3) made possible a thorough examination of multiple structural modifications applied on a single AG series: the impact of ring-A dideoxygenation, the charge of the 6′-group, substitution of N1 and N6′, and the role of the distal L-idose ring D. We also compared the antibacterial potency of our novel analogs to clinically deployed antibiotics against a panel of 16 strains representative of susceptible and AG-resistant pathogens of the E.S.K.A.P.E. group (Table 2).Analysis of the minimum inhibitory concentration (MIC) results focusing on susceptible strains (Table 2, entries 1–5) highlights the fundamental antibiotic potency of each analog. A low MIC value is a result reflecting the AG transport and accumulation in the cell, the engagement of the ribosome target, and the downstream bactericidal action. The first unexpected observation in this series was the requirement for a 6′-ammonium group to achieve antibacterial activity in the 3′,4′-dideoxygenated analogs (Table 2, compare 1 to 2, 23 to 24, and 25 to 27).43 Structural analysis suggest that ribosomal binding would not be affected, therefore, the losses in potency suggest that the 6′-OH 3′,4′-dideoxygenated analogs 23 and 25 may fail to enter bacterial cells.43 On the other hand, we have shown that the sisomicin–paromomycin hybrid 6′-OH analog 25 displays antiprotozoal activity.11 The ring A 6′-OH versions of AGs, in particular paromomycin (2),49 display superior activity against eukaryotic ribosomes, owing to the A-site A1408G nucleotide variant (Fig. 1B, box).50,51
As expected, bacterial strains that rely on ANT(4′) enzymes to inactivate AGs were susceptible to the 3′,4′-dideoxygenated analogs (Table 2, entries 10 and 11). However, a subset of enzymes classified as APH(3′) isoforms are capable of binding alternate conformations to target either 3′- or 5′′-OH groups.52 Our data suggest that removal of the 3′-OH group is insufficient to prevent inactivation by strains expressing APH(3′)-IIIa and -Ia enzymes (Table 2, entries 7 and 8). Moreover, cleavage of ring D from 3′,4′-dideoxygenated analogs resulted in a considerable detriment to the potency to the trisaccharide series compared to the tetrasaccharide neomycin series (Table 2, compare 1 to 5, and 24 to 27). The ring D is an L-idose glycoside (Fig. 1A),53 which originates from additional biosynthetic elaboration of D-glucosamine,54 and suggests that eons of evolutionary pressures have optimized the neo/paromomycin series to bind the extended pocket of the A-site and exploit additional RNA contacts (Fig. 1B).10 The MIC data further suggest that 3′- and 4′-dideoxygenation are absolutely essential modifications for AME evasion, and when introduced in combination with a 6′-ammonium and ring D these features confer an enhancement in antibiotic potency (Table 2, Fig. 3).
These findings prompted us to further study the 6′-amino-3′,4′-dideoxygenated neomycin and butirosin series. The AG known as G52 is a naturally occurring N6′-methylated sisomicin congener (7).55 We introduced this modification in a G52-neomycin hybrid 26 (Scheme S1,†Fig. 3). However, no significant improvement against strains expressing AAC(6′) enzymes was observed (Table 2, entries 6, 13 and 14). We hypothesize that an extended library of C5′/N6′-modifications may be required for further optimization of this series, such as N6′-ethyl,55N6′-hydroxyethyl,45 or C5′-methylation.55
The most powerful feature that confers broad protection against AMEs, including N6′-acetylation, is the appendage of bulky N1-substituents, which are believed to be poorly accommodated by the active sites of most AME isoforms (Fig. 2A). Multiple side-chains have been reported: N1-L-HABA (e.g. butirosin (4), amikacin (6), and plazomycin),45 also the shorter variant N1-L-α-hydroxy-β-amino-propionic acyl (e.g. isepamicin), and N1-ethyl (e.g. netilmicin, a sisomicin analog).1 More recently, we reported an isosteric difluoro-substituted F2-HABA side-chain (N1-L-α-hydroxy-β,β-difluoro-γ-amino-butanoic acyl), as a general approach to reduce the cellular toxicity of AGs by partially neutralizing the neighboring γ-ammonium positive charge at physiological pH.56,57 We selected the 6′-amino 3′,4′-dideoxygenated tri- and tetra-saccharides to study the combined effect of N1-L-HABA and ring D on the antibiotic potency of the sisomicin-hybrid analogs 17 and 21 (Table 2, Fig. 3). Remarkably, the boost in antibiotic potency imparted by the N1-substituent combined with the neomycin ring D on analog 17 was of greater magnitude than that observed for the tetrasaccharide 21, or the comparable AG pairs butirosin (4) versus ribostamycin (5), or paromomycin (2) versus N1-HABA-paromomycin (22) (Fig. 3). The blending of numerous optimal features of the sisomicin–butirosin–neomycin hybrid structure 17 resulted in exceptional MIC values against Gram-negative and -positive E.S.K.A.P.E. bacterial strains, MIC range 0.25–0.5 μg mL−1 (Table 2).
The clinical AG antibiotics amikacin (4) and gentamycin (7) were ineffective against over half of the panel of E.S.K.A.P.E. strains expressing AMEs (MIC ranges 16 to >64 μg mL−1, Table 2, entries 6–16).3–6 This panel is representative of single-, double-, and multi-AME expression in clinical isolates, which impart pan-AG resistance in >10 to 60% of clinical isolates in hospitals throughout the world (Table 1, Fig. S1 and S2†). In particular, AGs of the 4,6-disubstituted class are ineffective against strains expressing the prevalent dual enzyme APH(2′′)/AAC(6′), as well as N7-G1405 A-site rRNA methyltransferases (e.g. ArmA).1,25,26 Analog 17 displays excellent MICs values for AME-expressing strains compared to neomycin B (1), sisomicin (3), amikacin (5), gentamicin (7), and other controls (entries 1 to 16, Table 2). The data further suggest that N1-L-HABA substitution imparted significant protection against strains expressing APH(3′), AAC(6′) or AAC(3) isoforms that display high to moderate resistance to the precursor analog 27. Moreover, the presence of ring D on 17, but not 21, appears to impart protection to the 5′′-OH of 17 from modification by the S. aureus APH(3′)-IIIa and E. coli APH(3′)-Ia enzymes, presumably by blocking alternate binding modes of the promiscuous AME active sites (Table 2, entries 7 and 8, compare 17 to 21 and 27). Taken together, the average MIC potency of analog 17 across the panel E.S.K.A.P.E. pathogens is less than 2 μg mL−1, which is excellent compared to the clinical AG antibiotics (Table 2). This comparison is favorable even taking into account the prevalent resistance mechanisms at the time the first generation of AGs were clinically deployed.44
Conclusions
The inactivation of AGs by widespread promiscuous AME isoforms and their synergistic combinations that endows bacterial pathogens with multi-AG resistance has posed a long-standing challenge. To reclaim their position in the arsenal of first-line antibiotic therapies the next generation of AG analogs must evade inactivation by three major types of AME mechanisms, and also engage the N7-G1405-methylated A-site variants without compromising the AG broad-spectrum bactericidal activity against clinical pathogens.This study presents the systematic evolution of AG hybrid structures blending the salient features of multiple natural and semi-synthetic AG antibiotics from the past five decades (Fig. 1A). Each of the hybrid analogs in this collection contributes to our understanding of different aspects of the AG structure-antibacterial potency relationships and the requirements for evasion of prevalent AME isoforms. We can discern the modular features that protect AGs from AMEs, some of which are essential regardless of their individual impact on potency, whereas other structural changes conveniently lead to both circumventing AMEs in addition to boosting antibacterial potency. Comparative analysis of the extended MIC panel revealed the individual and synergistic effects of the distal L-idose ring-D, the requisite 6′-ammonium group, the absence of susceptible 3′- and 4′-hydroxyls on ring A, and the appendage of the N1-HABA substituent. The deliberate blending of numerous optimal features into the sisomicin–butirosin–neomycin hybrid structure (17) provides us with a blueprint for the design of highly active next-generation AG antibiotics. Moreover, we anticipate that modulation of the pKa of the N1-L-HABA substituent,56 and subtle modification of the N6′-position,55 may enable further optimization of the therapeutic index for the pre-clinical examination of this remarkable AG antibiotic series.
Acknowledgements
We thank Rowena D. Matias, Lee Ann Feeney, and Eliana S. Armstrong at Achaogen Inc. San Francisco for the MIC measurements. This work was supported by the Achaogen Research Chair and the Natural Sciences and Engineering Research Council of Canada (NSERC). JPM received fellowship support from Fonds de Recherche en Santé du Québec (FRSQ).Notes and references
- D. P. Arya, Aminoglycoside antibiotics : from chemical biology to drug discovery, Wiley-Interscience, Hoboken, N.J., 2007 Search PubMed.
- H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Clin. Infect. Dis., 2009, 48, 1–12 CrossRef PubMed.
- K. J. Shaw, P. N. Rather, R. S. Hare and G. H. Miller, Microbiol. Mol. Biol. Rev., 1993, 57, 138–163 CAS.
- M. P. Mingeot-Leclercq, Y. Glupczynski and P. M. Tulkens, Antimicrob. Agents Chemother., 1999, 43, 727–737 CAS.
- E. Azucena and S. Mobashery, Drug Resist. Updates, 2001, 4, 106–117 CrossRef CAS PubMed.
- S. Magnet and J. S. Blanchard, Chem. Rev., 2005, 105, 477–498 CrossRef CAS PubMed.
- A. P. Carter, W. M. Clemons, D. E. Brodersen, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Nature, 2000, 407, 340–348 CrossRef CAS PubMed.
- B. T. Wimberly, D. E. Brodersen, W. M. Clemons, Jr., R. J. Morgan-Warren, A. P. Carter, C. Vonrhein, T. Hartsch and V. Ramakrishnan, Nature, 2000, 407, 327–339 CrossRef CAS PubMed.
- J. M. Ogle and V. Ramakrishnan, Annu. Rev. Biochem., 2005, 74, 129–177 CrossRef CAS PubMed.
- B. Francois, R. J. Russell, J. B. Murray, F. Aboul-ela, B. Masquida, Q. Vicens and E. Westhof, Nucleic Acids Res., 2005, 33, 5677–5690 CrossRef CAS PubMed.
- J. Kondo, M. Koganei, J. P. Maianti, V. L. Ly and S. Hanessian, ChemMedChem, 2013, 8, 733–739 CrossRef CAS PubMed.
- J. Kondo, B. Francois, R. J. Russell, J. B. Murray and E. Westhof, Biochimie, 2006, 88, 1027–1031 CrossRef CAS PubMed.
- W. L. DeLano, The PyMOL Molecular Graphics System, 2008 Search PubMed.
- R. O'Shea and H. E. Moser, J. Med. Chem., 2008, 51, 2871–2878 CrossRef PubMed.
- D. N. Wilson, Crit. Rev. Biochem. Mol. Biol., 2009, 44, 393–433 CrossRef CAS PubMed.
- M. A. Kohanski, D. J. Dwyer and J. J. Collins, Nat. Rev. Microbiol., 2010, 8, 423–435 CrossRef CAS PubMed.
- S. Jo, M. Vargyas, J. Vasko-Szedlar, B. Roux and W. Im, Nucleic Acids Res., 2008, 36, W270–275 CrossRef CAS PubMed.
- M. W. Vetting, C. H. Park, S. S. Hegde, G. A. Jacoby, D. C. Hooper and J. S. Blanchard, Biochemistry, 2008, 47, 9825–9835 CrossRef CAS PubMed.
- D. H. Fong and A. M. Berghuis, Antimicrob. Agents Chemother., 2009, 53, 3049–3055 CrossRef CAS PubMed.
- D. H. Fong and A. M. Berghuis, EMBO J., 2002, 21, 2323–2331 CrossRef CAS PubMed.
- J. Sakon, H. H. Liao, A. M. Kanikula, M. M. Benning, I. Rayment and H. M. Holden, Biochemistry, 1993, 32, 11977–11984 CrossRef CAS PubMed.
- H. W. Taber, J. P. Mueller, P. F. Miller and A. S. Arrow, Microbiol. Mol. Biol. Rev., 1987, 51, 439–457 CAS.
- B. D. Davis, Microbiol. Mol. Biol. Rev., 1987, 51, 341–350 CAS.
- I. T. Paulsen, M. H. Brown and R. A. Skurray, Microbiol. Rev., 1996, 60, 575–608 CAS.
- G. F. Liou, S. Yoshizawa, P. Courvalin and M. Galimand, J. Mol. Biol., 2006, 359, 358–364 CrossRef CAS PubMed.
- T. R. Fritsche, M. Castanheira, G. H. Miller, R. N. Jones and E. S. Armstrong, Antimicrob. Agents Chemother., 2008, 52, 1843–1845 CrossRef CAS PubMed.
- T. Tada, T. Miyoshi-Akiyama, R. K. Dahal, S. K. Mishra, H. Ohara, K. Shimada, T. Kirikae and B. M. Pokhrel, Int. J. Antimicrob. Agents, 2013, 42, 372–374 CrossRef CAS PubMed.
- F. J. Schmitz, A. C. Fluit, M. Gondolf, R. Beyrau, E. Lindenlauf, J. Verhoef, H. P. Heinz and M. E. Jones, J. Antimicrob. Chemother., 1999, 43, 253–259 CrossRef CAS PubMed.
- N. Kobayashi, M. M. Alam, Y. Nishimoto, S. Urasawa, N. Uehara and N. Watanabe, Epidemiol. Infect., 2001, 126, 197–204 CAS.
- K. J. Shaw, P. N. Rather, R. S. Hare and G. H. Miller, Microbiol. Mol. Biol. Rev., 1993, 57, 138–163 CAS.
- A. Endimiani, K. M. Hujer, A. M. Hujer, E. S. Armstrong, Y. Choudhary, J. B. Aggen and R. A. Bonomo, Antimicrob. Agents Chemother., 2009, 53, 4504–4507 CrossRef CAS PubMed.
- K. M. Hujer, A. M. Hujer, E. A. Hulten, S. Bajaksouzian, J. M. Adams, C. J. Donskey, D. J. Ecker, C. Massire, M. W. Eshoo, R. Sampath, J. M. Thomson, P. N. Rather, D. W. Craft, J. T. Fishbain, A. J. Ewell, M. R. Jacobs, D. L. Paterson and R. A. Bonomo, Antimicrob. Agents Chemother., 2006, 50, 4114–4123 CrossRef CAS PubMed.
- E. S. Armstrong, D. J. Biedenbach, R. N. Jones and G. H. Miller, Presented at The 19th European Congress of Clinical Microbiology and Infectious Diseases, Poster #643, Helsinki, Finland, 2009, http://www.achaogen.com/s/ECCMID_P-643.pdf (accessed Sept.2015) Search PubMed.
- K. Poole, Antimicrob. Agents Chemother., 2005, 49, 479–487 CrossRef CAS PubMed.
- G. H. Miller, F. J. Sabatelli, R. S. Hare, Y. Glupczynski, P. Mackey, D. Shlaes, K. Shimizu, K. J. Shaw, A. Bauernfeind, S. Schweighart, K. Shannon, J. Patzer, G. Molinari, G. C. Schito, R. GomezLus, S. GomezLus, H. Ferreira, J. C. Sousa, M. J. M. Vaz, E. Collatz, R. Bismuth, T. Lambert, P. Courvalin, C. Minozzi, K. Klugman, Y. Bilgeri, H. Giamarellou, G. Petrikkos, H. Akalin, D. Gur, M. Woloj, A. Rossi, J. Casellas, M. Tokumoto, E. Couto, C. Juliet, M. E. Pinto, R. Zemelman, W. Pedreira, M. Fernandez, I. Leal, M. Guzman, J. Murillo, P. Isturiz, A. Merentes, A. Bremner, B. Ho, K. Mayer, J. Ellal, W. Fu, D. Zhu, K. Dornbusch and E. Goransson, Clin. Infect. Dis., 1997, 24, S46–S62 CrossRef CAS PubMed.
- G. H. Miller, F. J. Sabatelli, L. Naples, R. S. Hare and K. J. Shaw, J. Chemother., 1995, 7, 17–30 Search PubMed.
- C. Kim, J. Y. Cha, H. Yan, S. B. Vakulenko and S. Mobashery, J. Biol. Chem., 2006, 281, 6964–6969 CrossRef CAS PubMed.
- J. L. Martinez and F. Baquero, Antimicrob. Agents Chemother., 2000, 44, 1771–1777 CrossRef CAS PubMed.
- J. D. Howells, L. E. Anderson, G. L. Coffey, G. D. Senos, M. A. Underhill, D. L. Vogler and J. Ehrlich, Antimicrob. Agents Chemother., 1972, 2, 79–83 CrossRef CAS PubMed.
- J. Kondo, K. Pachamuthu, B. Francois, J. Szychowski, S. Hanessian and E. Westhof, ChemMedChem, 2007, 2, 1631–1638 CrossRef CAS PubMed.
- S. Hanessian, K. Pachamuthu, J. Szychowski, A. Giguère, E. E. Swayze, M. T. Migawa, B. François, J. Kondo and E. Westhof, Bioorg. Med. Chem. Lett., 2010, 20, 7097 CrossRef CAS PubMed.
- S. Hanessian, A. Giguère, J. Grzyb, J. P. Maianti, O. M. Saavedra, J. B. Aggen, M. S. Linsell, A. A. Goldblum, D. J. Hildebrandt, T. R. Kane, P. Dozzo, M. J. Gliedt, R. D. Matias, L. A. Feeney and E. S. Armstrong, ACS Med. Chem. Lett., 2011, 2, 924–928 CrossRef CAS PubMed.
- S. Hanessian, J. P. Maianti, R. D. Matias, L. A. Feeney and E. S. Armstrong, Org. Lett., 2011, 13, 6476–6479 CrossRef CAS PubMed.
- K. E. Price, Antimicrob. Agents Chemother., 1986, 29, 543–548 CrossRef CAS PubMed.
- J. B. Aggen, E. S. Armstrong, A. A. Goldblum, P. Dozzo, M. S. Linsell, M. J. Gliedt, D. J. Hildebrandt, L. A. Feeney, A. Kubo, R. D. Matias, S. Lopez, M. Gomez, K. B. Wlasichuk, R. Diokno, G. H. Miller and H. E. Moser, Antimicrob. Agents Chemother., 2010, 54, 4636–4642 CrossRef CAS PubMed.
- Q. Vicens and E. Westhof, Structure, 2001, 9, 647–658 CrossRef CAS PubMed.
- S. Hanessian, O. M. Saavedra, M. A. Vilchis-Reyes, J. P. Maianti, H. Kanazawa, P. Dozzo, R. D. Matias, A. Serio and J. Kondo, Chem. Sci., 2014, 5, 4621–4632 RSC.
- S. Hanessian, T. Takamoto, R. Massé and G. Patil, Can. J. Chem., 1978, 56, 1482–1491 CrossRef CAS.
- R. N. Davidson, M. den Boer and K. Ritmeijer, Trans. R. Soc. Trop. Med. Hyg., 2009, 103, 653–660 CrossRef CAS PubMed.
- J. Kondo, Angew. Chem., Int. Ed., 2012, 51, 465–468 CrossRef CAS PubMed.
- I. Nudelman, A. Rebibo-Sabbah, M. Cherniavsky, V. Belakhov, M. Hainrichson, F. Chen, J. Schacht, D. S. Pilch, T. Ben-Yosef and T. Baasov, J. Med. Chem., 2009, 52, 2836–2845 CrossRef CAS PubMed.
- P. R. Thompson, D. W. Hughes and G. D. Wright, Biochemistry, 1996, 35, 8686–8695 CrossRef CAS PubMed.
- T. H. Haskell and S. Hanessian, J. Inorg. Chem., 1963, 28, 2598–2604 CAS.
- N. M. Llewellyn and J. B. Spencer, Nat. Prod. Rep., 2006, 23, 864–874 RSC.
- S. Hanessian, J. Szychowski and J. P. Maianti, Org. Lett., 2009, 11, 429–432 CrossRef CAS PubMed.
- J. P. Maianti, H. Kanazawa, P. Dozzo, R. D. Matias, L. A. Feeney, E. S. Armstrong, D. J. Hildebrandt, T. R. Kane, M. J. Gliedt, A. A. Goldblum, M. S. Linsell, J. B. Aggen, J. Kondo and S. Hanessian, ACS Chem. Biol., 2014, 9, 2067–2073 CrossRef CAS PubMed.
- M. Morgenthaler, E. Schweizer, A. Hoffmann-Roder, F. Benini, R. E. Martin, G. Jaeschke, B. Wagner, H. Fischer, S. Bendels, D. Zimmerli, J. Schneider, F. Diederich, M. Kansy and K. Muller, ChemMedChem, 2007, 2, 1100–1115 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, supplementary figures, and NMR spectra. See DOI: 10.1039/c5md00429b |
| This journal is © The Royal Society of Chemistry 2016 |