
Mechanisms of resistance to membrane-disrupting antibiotics in Gram-positive and Gram-negative bacteria
Kfir B.
Steinbuch
and
Micha
Fridman
*
School of Chemistry, Beverly Raymond Sackler Faculty of Exact Sciences, Tel Aviv University, Tel Aviv, 6997801, Israel. E-mail: mfridman@post.tau.ac.il
First published on 19th November 2015
Abstract
Small-molecule-mediated disruption of bacterial membranes is an important component of the innate immune response in numerous organisms including humans. Although still under-represented in the clinically used repertoire of antibiotics, several antimicrobial agents that act by disrupting the structures and functions of bacterial membranes are used for treatment of topical and internal infections. Due to the relatively conserved structure compositions of bacterial membranes, antibiotics that disrupt bacterial membranes should be less likely to induce drug resistance than antibiotics that target other bacterial systems. However, drug resistance mechanisms that reduce the efficacy of membrane-disrupting antibiotics have evolved in a variety of bacterial pathogens. Similar to mechanisms that thwart antimicrobial agents that target intracellular bacterial elements, resistance to membrane-disrupting agents results from modifications to the target (in this case, lipids), the action of efflux pumps, expression of various drug deactivating agents, and proteolytic degradation. In this review, we describe recent progress in elucidating the various mechanisms of resistance to membrane-disrupting antibiotics.
 Kfir B. Steinbuch | Kfir Baruch Steinbuch received his M.Sc. degree under the supervision of Prof. Michael Gozin and his B.Sc. degree at Tel-Aviv University, both summa cum laude in 2014 and 2011, respectively. Currently, he is a Ph.D. student under the supervision of Dr. Micha Fridman at the Tel Aviv University School of Chemistry. His main interests are developing new membrane-targeting antibiotics and understanding the mechanism of action of membrane disruptors. |
 Micha Fridman | Micha Fridman received his PhD degree in Chemistry from the Technion: Israel Institute of Technology in 2005 under the supervision of Professor Timor Baasov. In 2005, he joined the group of Professor Daniel Kahne in the Harvard University Department of Chemistry & Chemical Biology as a post-doctoral fellow. Since October 2008, he has been a Senior Lecturer in the School of Chemistry of Tel-Aviv University, Israel. His research focuses on the exploration of novel approaches and concepts for the development of antimicrobial agents. |
The continuous rise in the number of infections caused by bacteria that are resistant to at least one of the clinically used antibiotics and the drop in the number of novel antibiotics in drug development pipelines have contributed to the revived interest in antimicrobial agents such as those that disrupt bacterial membranes. Three major benefits make bacterial membranes appealing targets for the development of antibiotics: first, targeting bacterial membranes is an effective strategy for combating infections caused by either slow-growing or dormant bacteria that are extremely challenging to treat using the current repertoire of antibiotics.1 Second, since bacterial membranes are composed of relatively conserved structures and since membrane-disrupting antibiotics are likely to exert a rapid bactericidal activity,2 it should be more difficult to select for resistance to these antibacterial agents than to agents that target intracellular systems. Hence, membrane targeting antibiotics are likely to maintain prolonged clinical efficacy. Third, the development of antibiotics that target bacterial membranes does not require cell permeability considerations that often pose a major obstacle to the development of antimicrobial agents.3 To be effective for internal use, however, membrane-disrupting antibiotics must selectively target bacterial, and not mammalian, cell membranes. Since membranes composed of lipid bilayers are essential to cells of all organisms, avoiding cytotoxicity to eukaryotic cells is a major challenge in designing membrane-disrupting antibiotics and is probably the reason that few membrane-disrupting antibiotics are currently in clinical use.
Although the concept of membrane disruption has relatively been poorly exploited as an antibiotic development strategy, it is a prevalent strategy for combating bacterial infections in nature. Cationic antimicrobial peptides (CAMPs) that are produced by the immune systems of mammals and by many other organisms act by permeabilizing bacterial membranes and are the best characterized family of membrane disruptors.4,5 These natural antimicrobial agents usually consist of 12 to 50 amino acids and, due to the presence of basic amino acids, have a net positive charge ranging between +2 and +7 under physiological conditions.5 CAMPs are amphiphilic; in most of these peptides, over 50% of their amino acids contain hydrophobic side chains. The amphiphilic nature of CAMPs is the key to the interactions with bacterial membranes.6 In mammals, these peptides are produced at sites of infection and usually possess broad-spectrum antimicrobial activity.6
Of the numerous CAMPs discovered to date, gramicidin S (Fig. 1) is an example of the very few that have been approved for clinical use. Isolated from Bacillus brevis, this cyclic peptide effectively combats Gram-positive and Gram-negative infections.7 A major obstacle that prevents the systemic clinical use of gramicidin S and other members of this family of antimicrobial agents is the fact that their therapeutic window is extremely narrow; these peptides cause red blood cell hemolysis at concentrations roughly equivalent to their minimal inhibitory concentrations (MICs) against many strains of pathogenic bacteria.
 | ||
| Fig. 1 Clinically used antimicrobial peptides and lipopeptides. (Left) Polymyxins B and E are mixtures of structurally related compounds that differ from one another in the chemical structure of their fatty acid. The best characterized of these are polymyxins B1 and B2 and polymyxins E1 and E2. (Top right) Gramicidin S. (Bottom right) Daptomycin. | ||
The cationic lipopeptide antibiotics are another example of molecules that evolved in nature and effectively disrupt the structure of bacterial membranes by highly selective binding to lipopolysaccharides (LPS). LPS is a major lipid component of the outer membrane of all Gram-negative bacteria. The lipopeptides polymyxin B and polymyxin E (also called colistin) are lipopeptide antibiotics isolated from the Gram-positive bacteria Bacillus polymyxa. Polymyxins consist of a hepta-peptide ring and a tripeptide side chain with a fatty acid tail (Fig. 1). They differ from one another only in one amino acid on the cyclic hepta-peptide.8–10 These antibiotics also share the same mechanism of action:11 polymyxins bind to the lipid A core of LPS at least three orders of magnitude more tightly than do the natural calcium and magnesium cations that hold the LPS molecules together in the membrane. By displacing the natural calcium and magnesium ions, these antibiotics disrupt the LPS assembly in the Gram-negative bacteria outer membrane.12 The short fatty acid side chain of lipopeptides interacts with the lipid region of the LPS and assists in anchoring these antibiotics to the outer membrane lipid bilayer. Their positively charged diaminobutyric acid side chains interact with the two phosphate groups of lipid A through ionic interactions.13,14
Another example of a clinically used membrane-disrupting antibiotic is the negatively charged antimicrobial lipopeptide daptomycin (Fig. 1). Daptomycin, produced by Streptomyces roseosporus, is a cyclic molecule with a decanoyl fatty acid side chain attached to an exocyclic N-terminal single tryptophan residue. It contains 13 amino acid residues, including several unique ones such as kynurenine, ornithine, and 3-methylglutamine acid, and it has a net negative charge under physiological conditions and is effective against Gram-negative bacteria.15,16 Although daptomycin's mechanism of action is not completely understood, it has been established that this anionic lipopeptide targets the membrane of the bacteria and that its activity is strictly dependent on the presence of physiological levels of Ca2+. The calcium ions are presumed to decrease or neutralize the overall negative charge of the lipopeptide to facilitate its insertion into the negatively charged membrane.15–18 Interestingly, although daptomycin is negatively charged and CAMPs are positively charged, certain mechanisms of resistance to CAMPs are also effective against daptomycin.
The increase in the number of bacterial pathogens that have acquired resistance to membrane-disrupting antibiotics urged us and other groups to pursue the development of non-peptide-derived cationic amphiphiles that cause membrane disruption and potentially evade resistance mechanisms.19–28 It was recently reported that a screen of “uncultivable” microorganisms for novel antimicrobials29–31 led to the identification of the CAMP teixobactin, which targets a membrane-bound precursor of the peptidoglycan.32 Teixobactin is highly effective against several Gram-positive pathogens and no resistance to this CAMP was detected so far, thus introducing novel cationic amphiphiles that will not be affected by some or all of the known resistance mechanisms.
In this review, we summarize recent progress in the elucidation and unravelling of resistance mechanisms that have evolved against membrane-disrupting antimicrobial agents. A better understanding of these mechanisms will hopefully guide the design of new membrane-disrupting antibacterial agents.
Glycosylation of membrane lipids by amino-sugars
Attachment of cationic sugar units to the lipid A core of LPS neutralizes the negative charge that results from the phosphate units of lipid A and thus lowers the affinity of the cationic polymyxins for LPS and prevents their antimicrobial action. The monosaccharide 4-amino-4-deoxy-L-arabinose (Ara4N, Scheme 1), which is positively charged at the C-4 amine under physiological conditions and is attached to the 4′-phosphate of lipid A, has been identified in lipopeptide-resistant Gram-negative bacterial strains including Salmonella typhimurium, Escherichia coli, and Pseudomonas aeruginosa. Minor species in which the Ara4N is attached at the 1-phosphate or on both phosphates of the lipid A have also been isolated.33,34 | ||
| Scheme 1 The biosynthesis pathway of Ara4N-lipid A. In the cytoplasm, UDP-glucose is oxidized by PmrE to UDP-glucuronic acid, which then undergoes oxidative decarboxylation by a two-step reaction catalyzed by PmrI. UDP-4-ketopentose undergoes transamination catalyzed by PmrH to form the UDP-β-4-amino-L-arabinose (green), which is formylated by PmrI. The UDP-β-4-formamido-L-arabinose is then transferred to an undecaprenyl phosphate unit on the inner leaflet of the inner membrane by PmrF. The resultant undecaprenyl phosphate-α-4-formamido-L-arabinose is then deformylated by PmrJ and is transferred through the inner membrane presumably by the flippase complex containing PmrL and PmrM. In the outer leaflet of the inner membrane, the Ara4N is transferred by PmrK (ArnT) to the lipid A core of LPS.44 | ||
One of the virulence strategies of Salmonella involves a two-component regulatory system termed PhoP–PhoQ. This regulatory system is activated following phagocytosis in response to the phagosome environment and induces expression of genes necessary for LPS modification and antimicrobial peptide resistance. PhoP–PhoQ also activates the transcription of the pmrCAB locus (pmr for polymyxin resistance), which encodes the PmrA–PmrB two-component regulatory system. This regulatory system is also induced in host cells (not necessarily by PhoP–PhoQ), activating genes necessary for resistance to polymyxins that modify the lipid A with Ara4N. Mutations in pmrA observed in S. typhimurium and in E. coli![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9,33,34 cause a constitutively active expression of pmrA, and thus, these species have high degrees of LPS modified with Ara4N, resulting in inherent resistance to polymyxin.
9,33,34 cause a constitutively active expression of pmrA, and thus, these species have high degrees of LPS modified with Ara4N, resulting in inherent resistance to polymyxin.
Recently, a PmrAB system has been identified in P. aeruginosa that mediates the modification of Ara4N on lipid A.35 Additionally, strains of P. aeruginosa isolated from infants with cystic fibrosis, who develop chronic airway P. aeruginosa infections, have Ara4N modified lipid A, implying early adaptation to selective pressure from CAMPs.36 Furthermore, as inhaled polymyxin E (colistin) is commonly used to treat airway infections in cystic fibrosis patients, colistin-resistant strains of P. aeruginosa have been isolated from patients receiving this treatment, and these strains have alterations in the lipid A structure, among them, the addition of Ara4N.35,37
PmrA–PmrB regulates the transcription of the pmrE gene (also termed ugd) and the pmrHFIJKLM operon (also called pbgPE or arnBCADTEF). All of the product proteins of these loci are involved in the biochemical pathway necessary for Ara4N biosynthesis, its attachment to LPS and in polymyxin resistance (Scheme 1).34,38–44 The biochemical pathway commences from UDP-α-D-glucose. PmrE (Ugd), a UDP-glucose dehydrogenase, oxidizes UDP-α-D-glucose to yield UDP-α-D-glucuronic acid. The C-terminal domain of PmrI (ArnA) then catalyzes an oxidative decarboxylation reaction of the UDP-α-D-glucuronic acid, first to form the intermediate UDP-α-4-keto-D-glucuronic acid (oxidation step) and then to form the novel UDP-α-4-ketopentose (decarboxylation step).43 The latter undergoes a transamination reaction catalyzed by the aminotransferase PmrH (ArnB), which uses glutamic acid as the amine donor to form UDP-β-4-amino-L-arabinose and releases α-ketoglutarate.39–42,44 The N-terminal domain of PmrI (ArnA) catalyzes N-formylation of the aminated arabinose using N-10-formyltetrahydrofolate (N-10-THF) as a donor to form UDP-β-4-formamido-L-arabinose,41 which is then transferred to undecaprenyl phosphate (also known as bactoprenol phosphate) by PmrF (ArnC), an undecaprenyl transferase that replaces the UDP group with undecaprenyl. The resulting product is then rapidly deformylated by PmrJ (ArnD) and transferred to the 4′-phosphate of lipid A by the lipid A transferase PmrK (ArnT) releasing the undecaprenyl phosphate.41,42,44 Two lines of evidence suggest that the transfer of the Ara4N from undecaprenyl phosphate-α-L-Ara4N to the lipid A occurs on the periplasmic surface of the inner membrane. First, many enzymatic systems that utilize undecaprenyl phosphate derivatives function in the periplasm. The second reason relates to the substrate specificity of the 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) transferase (KdtA), which requires the presence of an unsubstituted 4′-monophosphate moiety for catalysis. Premature transfer of Ara4N to the 4′-phosphate on the cytoplasmic surface of the inner membrane would prevent Kdo addition, which occurs in the periplasm.34,40–44 Recently, the products of pmrL and pmrM (recently renamed arnE and arnF) have been suggested to be subunits of a putative undecaprenyl phospho-L-4-aminoarabinose flippase that transfer the undecaprenyl phospho-L-4-aminoarabinose from the cytoplasmic surface of the inner membrane to the periplasmic surface of the inner membrane.44
Modification of lipid A through glycosylation with galactosamine is also associated with resistance to lipopeptide antibiotics. For example, colistin-resistant strains of Acinetobacter baumannii have a D-galactosamine unit attached to the 1-phosphate group of lipid A (Fig. 2A).45 This modification is similar to the Ara4N modification observed in S. typhimurium and E. coli as both are amino-sugars carrying a positively charged group under physiological conditions attached to the lipid A phosphate. This modification was also observed in Francisella tularensis (Fig. 2B), and several studies have suggested a homologous biosynthesis pathway.46–49 It is important to note that unlike E. coli and S. typhimurium, in which only some of LPS molecules carry modified lipid A, in F. tularensis subsp. novicida almost all of the lipid A are found as “free” lipid A rather than in LPS, and most of the “free” lipid A is modified with a galactosamine residue on the 1-phosphate.50 The galactosamine addition was first described in F. tularensis subsp. holarctica, and the same report suggested that this modification might confer resistance to CAMPs.46 It was presumed that a homologue of the enzyme ArnT, which transfers the Ara4N to the lipid A in S. typhimurium and E. coli, transfers the galactosamine to the lipid A in F. tularensis.46,47 Indeed, it was found in F. tularensis subsp. novicida that deletion of the single arnT orthologue present in its genome (later termed flmK) causes the synthesis of lipid A molecules lacking the galactosamine moiety.47
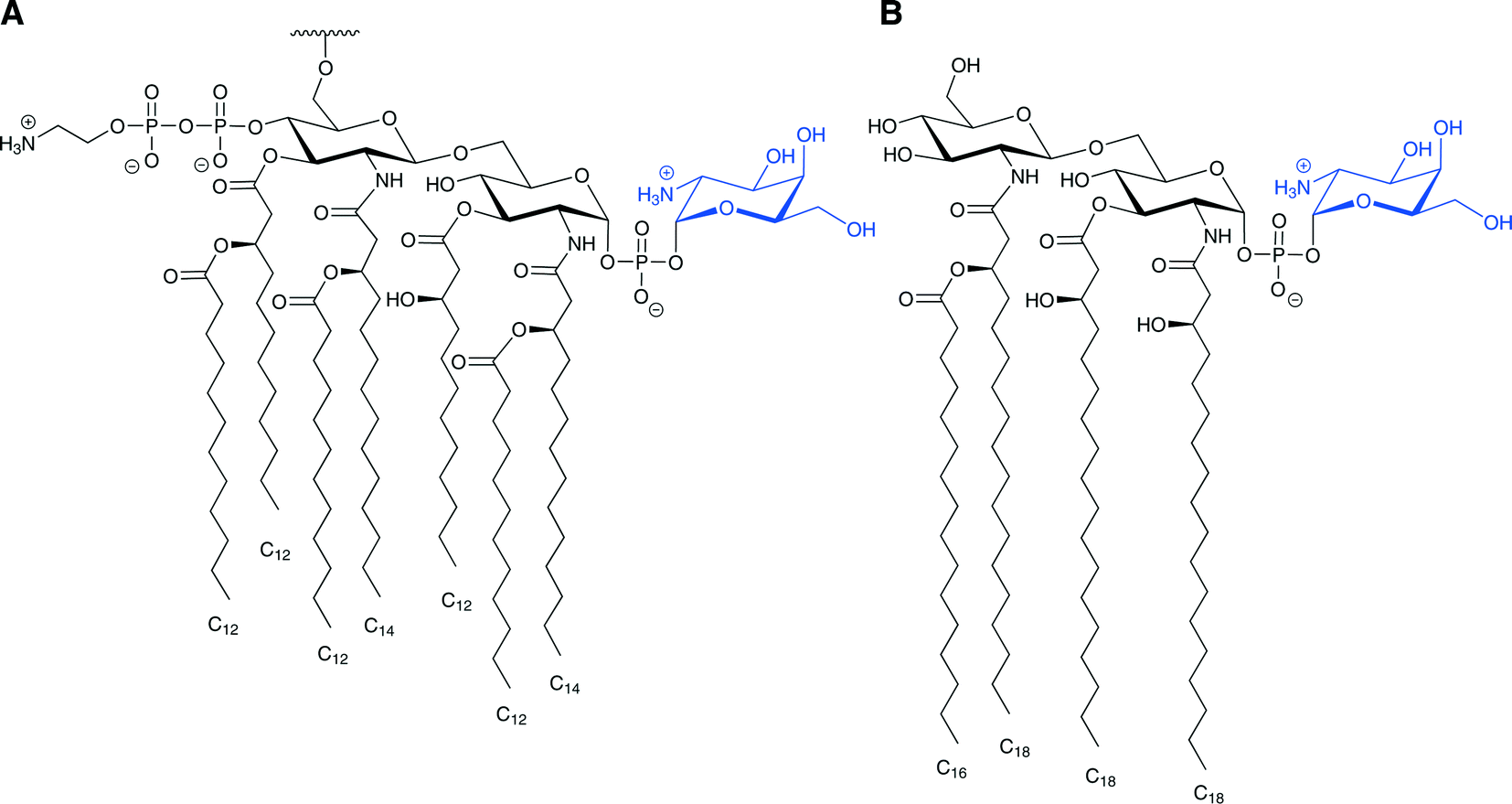 | ||
| Fig. 2 Proposed structures of lipid A modified with galactosamine (blue). A. The proposed structure of lipid A isolated from colistin-resistant strains of A. baumannii. This lipid A was also found to be hepta-acylated and modified with phosphoethanolamine as well. B. The lipid A structure of F. tularensis subsp. novicida. | ||
Undecaprenyl phosphate-β-D-galactosamine was subsequently isolated from F. tularensis subsp. novicida; this enzyme is the homologue of undecaprenyl phosphate-α-L-Ara4N from the Ara4N-modified LPS synthetic pathway. This finding suggests that the addition of galactosamine to the lipid A occurs on the periplasmic surface of the inner membrane.48 It was also demonstrated that FlmK, the orthologue of ArnT, transfers the galactosamine unit from the undecaprenyl phosphate-β-D-galactosamine to lipid A.48 In this biosynthetic pathway, FlmF2 (the homologue of ArnC) selectively catalyzes the condensation of undecaprenyl phosphate and UDP-N-acetylgalactosamine to generate undecaprenyl phosphate-N-acetylgalactosamine, which is de-acetylated by a deacetylase present in membranes of F. tularensis subsp. novicida. The reaction resembles the de-formylation in the Ara4N-LPS biosynthesis.49 It is likely that this modification confers at least some resistance to antimicrobial peptides, since it also decreases the net negative charge of the membrane, but this has not yet been demonstrated.
Incorporation of amino acids into bacterial membrane lipids
Most major human bacterial pathogens achieve CAMP resistance by expression of the multiple peptide resistance factor (MprF).51 MprFs are integral membrane proteins consisting of two domains that are responsible for two specific functions. First, these enzymes catalyze incorporation of lysine or alanine onto the anionic phospholipid phosphatidyl glycerol (PG), one of the most common bacterial phospholipids. This converts the charge on this lipid to positive in the case of lysine or neutralizes it in the case of alanine (Fig. 3). Second, a flippase domain translocates the lysine-attached PG (Lys-PG) or alanine-attached PG (Ala-PG) to the outer leaflet of the membrane.52,53 Incorporation of these positively charged or neutral lipids leads to a reduction in the overall negative charge of the membrane reducing the affinity of the membrane for positively charged membrane-disrupting antibiotics. | ||
| Fig. 3 Chemical structures of phosphatidyl glycerol (PG) and the aminoacylated phosphatidyl glycerol (aa-PG). (Left) Unmodified PG, mono anionic. (Middle) L-Lys-PG, positively charged. (Right) L-Ala-PG, neutral. Both modified PGs are the products of a reaction catalyzed by MprF. | ||
The substrates for the acylation reaction catalyzed by MprF is either lysyl- or alanyl-tRNA and the negatively charged phospholipid (Fig. 4).52,53 The aminoacyl tRNA used by the MprF varies with bacteria strain.51Staphylococcus aureus MprF only synthesizes Lys-PG, and P. aeruginosa MprFs only synthesize Ala-PG,53,54 whereas Enterococcus faecalis has both of these lipids.55 In Enterococcus faecium, Ala-PG, Lys-PG, and slight amounts of arginyl PG are produced, implying that this MprF has a rather relaxed specificity for the donor substrate.56 The phospholipid substrate also varies. In most strains, the aminoacylation occurs on PG, but in Listeria monocytogenes, MprF activity results in cardiolipin linked to Lys as well as Lys-PG.57 The molecular basis for the substrate specificities of MprF proteins is still not fully understood. Recently, a point mutation in S. aureus MprF that led to a gain of function has been found to grant resistance to daptomycin.18,58–62 It is still unclear whether the mutation leads to increased Lys-PG synthesis, causes Lys-PG to flip to the outer leaflet of the membrane, or results in another change in the membrane properties.51
 | ||
| Fig. 4 Incorporation of amino acid lysine or alanine onto PG catalyzed by the transmembrane, two domain, protein MprF. The synthase domain of MprF catalyzes the reaction between the lysyl- or alanyl-tRNA and PG (magenta), neutralizing the negative charge of PG. The flippase domain translocates the synthesized lipid to the outer leaflet of the membrane, reducing the overall negative charge of the membrane and thus decreasing the affinity to positively charged membrane disruptors. | ||
A reduction in affinity of the membrane for positively charged membrane disruptors, similar to the effect of alteration of negatively charged bacterial membrane lipids, results from D-alanylation of teichoic acids found in the cell walls of many Gram-positive strains (Fig. 5). This D-alanylation is typically mediated by the proteins encoded by the dlt operon.63,64 Over-expression of this operon induced by the regulatory gene system aps (also called graRS) is also associated with the resistance found in S. aureus to the antimicrobial anionic amphiphile daptomycin.65,66D-Alanylation of teichoic acids neutralizes and reduces the overall negative charge of the cell wall and thus lowers the affinity for positively charged membrane disruptors (Fig. 6).
 | ||
| Fig. 5 Structures of teichoic acids. These acids are composed of a long linear chain of monomers attached through anionic phosphodiester bonds. (Top) The prevalent lipoteichoic acid presented is composed of 1,3-glycerol-phosphate (Gro-P) monomers that are attached to the membrane by a glycolipid anchor. (Bottom) In wall teichoic acid, the poly-(alditol P) units are either 1,5-D-ribitol-phosphate (Rbo-P) or Gro-P and are linked via a sugar to the peptidoglycan. In both WTA and LTA structures, X indicates sites at which D-alanylation may occur.67 High levels of D-alanylations are correlated with CAMP resistance. The other variations shown are not related to CAMP resistance. | ||
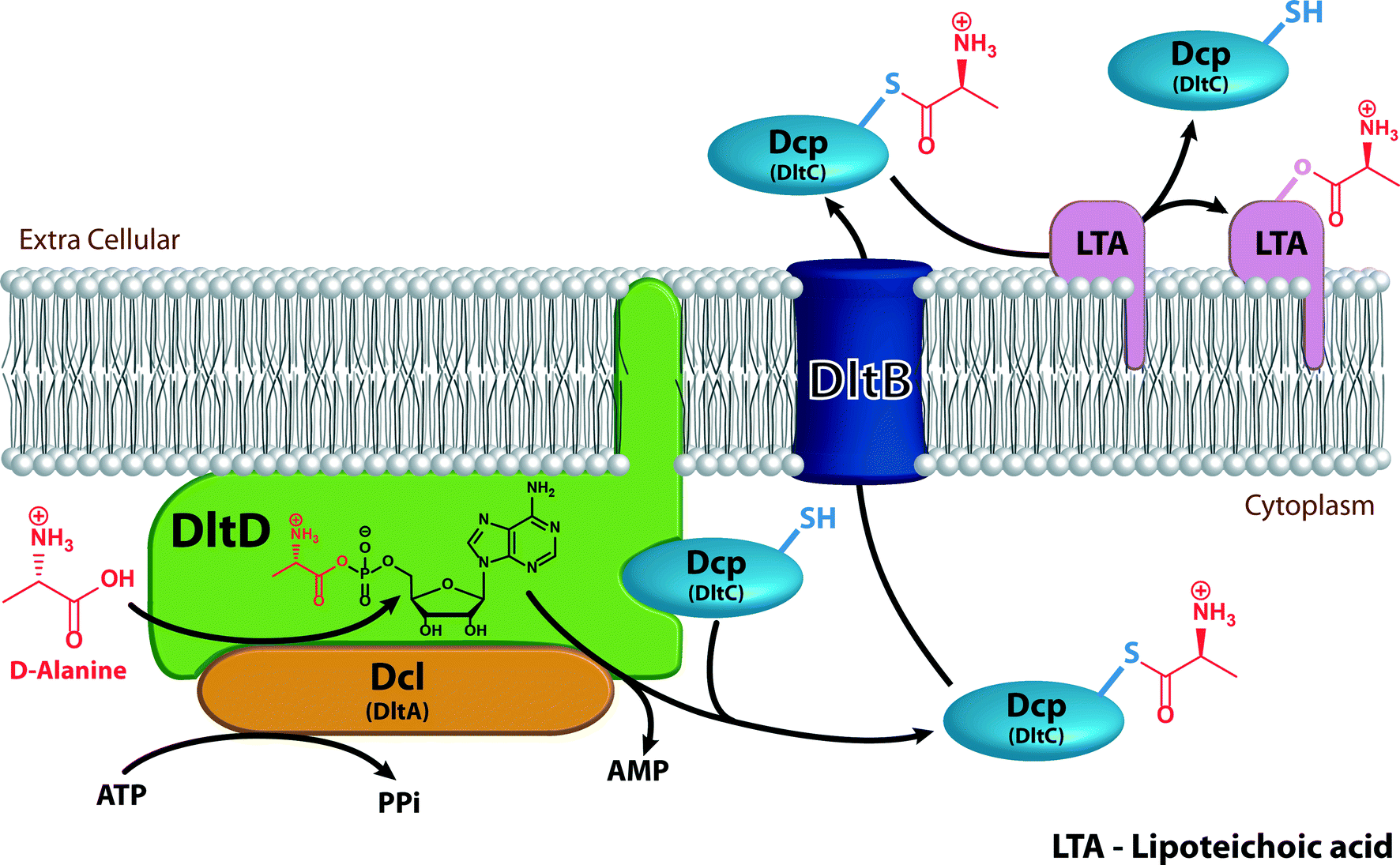 | ||
| Fig. 6 Mechanism of the D-alanylation of lipoteichoic acids. Dcl (orange) activates D-alanine (red) using ATP and transfers it to Dcp (light blue) in a reaction that occurs on the membrane-anchored DltD (green). The D-alanyl-Dcp is then transferred through the transmembrane transfer protein DltB (dark blue) to the extracellular environment where it delivers the D-alanine to a membrane-bound lipoteichoic acid (magenta). | ||
The dlt operon encodes four proteins: Dcl (encoded by dltA), a D-alanine-D-alanyl carrier protein ligase; DltB (encoded by dltb), a D-alanyl transfer protein; Dcp (encoded by dltC), a D-alanyl carrier protein; and DltD (encoded by dltD), a membrane protein that ensures the ligation of D-alanine to Dcp. All four proteins are required for successful addition of D-alanine to cell wall components (Fig. 6).64,67 Dcl activates D-alanine and transfers it to Dcp in an ATP-consuming reaction that yields AMP and pyrophosphate (PPi).68 The reaction between Dcl and Dcp occurs on DltD, a membrane protein anchored to the inner leaflet of the membrane by a hydrophobic tail. DltD selects the correct carrier protein, Dcp, for ligation with D-alanine and hydrolyses mischarged D-alanyl-acyl carrier proteins. Dcl can transfer D-alanine to acyl carrier proteins other than Dcp that are unable to transfer the D-alanyl to lipoteichoic acid (LTA), hence the importance of DltD.69D-Alanyl-Dcp is then transferred out of the membrane by the transmembrane transfer protein DltB. In the periplasm, Dcp transfers the D-alanyl to the membrane-associated LTA (Fig. 5). An enzyme that catalyzes the transfer of D-alanyl from Dcp to LTA has not been identified. The transfer does require that the acceptor LTA is bound to the membrane. D-Alanyl wall teichoic acids (WTAs) are formed by trans-acylation between D-alanyl-LTA and a WTA (Fig. 5).67
It was recently revealed that Vibrio cholerae O1 El Tor, a Gram-negative bacterium that is resistant to the antimicrobial peptide polymyxin B, modifies the lipid A anchor of its LPS with glycine and diglycine residues on the 3′-linked secondary acyl chain of the glucosamine disaccharide (Scheme 2).70,71 This acyl chain is a unique hydroxylauryl chain that possesses a hydroxyl group on its 3′ carbon and is found specifically in Vibrio cholerae O1 El Tor.72 Biosynthesis of the glycine-modified lipid A involves enzymes similar to those in Gram-positive bacteria that modify teichoic acids. Three enzymes are involved in glycine incorporation: AlmG, a membrane-associated protein that contains a domain similar to that of late acetyl transferases; AlmE, which contains an amino acid adenylation domain and shares homology with S. aureus Dcl; and AlmF, which has structural similarity to Dcp. The model suggests that apo-AlmF undergoes post-translational modification on a conserved serine residue by a 4′-phosphopantetheine (Ppant) transferase to form the holo-AlmF. The reaction occurs between the conserved serine residue and coenzyme A and releases adenosine 3′,5′ bisphosphate. The holo-AlmF then activates a glycine on AlmE; this results in formation of a glycine thioester on holo-AlmF and release of AMP and PPi. AlmG transfers the glycine from AlmF-S-Gly to the secondary hydroxylauryl acyl chain of hexa-acylated lipid A. The reaction occurs in the inner membrane. The glycine-modified lipid A units are then transported to the bacterial surface (Scheme 2). The diglycine modification is suggested to occur by two glycylation reactions catalyzed by AlmG.70,71
 | ||
| Scheme 2 Proposed model for the glycine modification of lipid A from V. cholerae O1 El Tor. Ppant (green) is transferred by Ppant transferase (green) from coenzyme A to a conserved serine residue of apo-AlmF (orange) forming the holo-AlmF. AlmE (red) activates glycine (blue) using ATP and transfers it to the holo-AlmF (orange). The membrane-associated AlmG (dark blue) then transfers the glycine from the holo-AlmF to the 3′ alcohol of the secondary acyl chain of lipid A. The modified lipid A is transferred (in several steps) to the outer leaflet of the outer membrane where it decreases the overall negative charge of the membrane and promotes resistance to polymyxins.71 | ||
Incorporation of phosphoethanolamine residues
Another lipid A modification, found in Gram-negative bacteria, that is related to the resistance toward polymyxins and CAMPs is the addition of phosphoethanolamine (pEtN) to the 1-phosphate of lipid A (Fig. 7A). This modification introduces a positive charge at the lipid A reducing the overall negative charge of the membrane and the affinity for cationic membrane disruptors. Additionally, the positively charged amine group can reduce the electrostatic repulsion between neighboring lipid A molecules and thus can lower the dependency of membrane stability on divalent cations such as Mg2+ and Ca2+, which are known to be released from the membrane in the presence of membrane disruptors.73 Finally, the positive charge masks the phosphate groups on lipid A further lowering the affinity for membrane disruptors.74 | ||
| Fig. 7 Phosphoethanolamine modification. A. (Left) Phosphoethanolamine (red) attached to the 1-phosphate of lipid A. This modification usually occurs coincidentally with the Ara4N modification on the 4′-phosphate since enzymes involved in biosynthesis of both modifications are under the same regulatory system. (Middle) Phosphoethanolamine (red) modification on the distal Kdo unit; attachment is mediated by EptB. (Right) Phosphoethanolamine modification on the 4′-phosphate observed in colistin-resistant A. baumannii. B. The proposed reaction for the attachment of phosphoethanolamine to the 1-phosphate of lipid A (red) by EptA (red) and to the distal Kdo (blue) by EptB (blue). The phosphoethanolamine donor is phosphatidylethanolamine. Diacylglycerol is the proposed by-product. | ||
The enzyme that attaches the phosphoethanolamine to the 1-phosphate of the lipid A is called PmrC or EptA. This enzyme is regulated by the PmrAB two-component regulatory system. The PmrAB two-component system consists of the PmrA response regulator and the PmrB sensor, which is activated by high concentrations of Fe3+. In addition, this regulatory system can also be activated by low Mg2+ through the PhoP–PhoQ two-component system.75 The phosphoethanolamine donor is phosphatidylethanolamine, an abundant lipid of the inner membrane, and the reaction occurs in the periplasmic surface of the inner membrane releasing diacylglycerol (Fig. 7B).42,76 This modification usually occurs coincidentally with the addition of the Ara4N at the 4′-phosphate of the lipid A since expression of the proteins responsible for these processes is regulated by the same system. Uncommon species are present in which the locations of the Ara4N and pEtN groups are reversed or in which both phosphates are modified with the same substituent.34 Furthermore, when the function of a gene necessary for the synthesis of one of these modifications is disrupted, the amount of the other modification is increased relative to levels in the wild-type strain.76
It was recently shown that the lipid A of colistin-resistant A. baumannii is modified with phosphoethanolamine at the 4′-phosphate group and a galactosamine (discussed in the glycosylation section above) at the 1-phosphate group (Fig. 7A).45A. baumannii is a nosocomial opportunistic pathogen that can cause severe infections including hospital-acquired pneumonia, wound infections, and sepsis. Multidrug-resistant (MDR) strains are prevalent, and colistin serves as a last resort treatment for some A. baumannii infections. Hence, resistance to the action of this lipopeptide is particularly alarming.45
Addition of phosphoethanolamine can also take place on a Kdo sugar unit of LPS as is observed in certain strains of E. coli and S. typhimurium (Fig. 7A).42,77 The incorporation of this modification is catalyzed by the enzyme EptB, which is homologous to EptA, and yet this phosphoethanolamine transferase is not regulated by PmrA. Instead, EptB is induced by Ca2+ and is under the control of the transcription factor σE.77,78 EptB transfers the phosphoethanolamine to the outer Kdo residue. The phosphoethanolamine donor is again phosphatidylethanolamine, and diacylglycerol is released as a by-product (Fig. 7B). It has been suggested that this modification functions in the maintenance of the outer membrane stability.78 The phosphoethanolamine modification on the distal Kdo unit might also promote resistance to membrane disruptors; however, there is no experimental evidence for this.
Efflux pumps
ATP binding cassette transporter (ABC transporter) superfamily
Various types of efflux pumps capable of expelling small molecules, including many drugs and antibiotics that act on intracellular targets, may also promote resistance toward cell membrane disruptors. The ABC transporters couple the hydrolysis of ATP to substrate transport across the cell membrane. ABC transporters belong to one of the largest superfamilies of proteins that import or export (efflux pumps) a broad range of substrates such as amino acids, ions, sugars, lipids, and drugs across membranes.79–82 Bacterial genomes encode for both export and import classes of ABC transporters, whereas eukaryotes have only exporters.79 In humans, ABC transporters have an important role in resistance to antitumor drugs.80 Some species of bacteria express many different ABC transporters; for instance, E. coli has 69 different ABC transporters that account for almost 5% of its genomic coding capacity.81 ABC transporters most frequently result in resistance to antibiotics that target elements inside the cell, but some transporters are reported to promote resistance to cell-envelope-targeting antibiotics.VraFG is an ABC transporter that has been shown to induce resistance to CAMPs in S. aureus and Staphylococcus epidermidis (Fig. 8A).65,83 The VraFG transporter system is one of the three main CAMP resistance mechanisms in Gram-positive bacteria, although there are interspecies differences in the induction and the regulations of these mechanisms. In S. epidermidis, expression of the VraFG homologues is up-regulated by the aps system (along with the dlt operon and the mprF system) in the presence of the human CAMP hBD3.83 In S. aureus, only the vraFG gene system is up-regulated and only at high concentrations of hBD3. In the presence of a different CAMP, indolicidin, the three resistance mechanisms are all up-regulated in S. aureus, with the vraFG gene system the most up-regulated of the three.65 It is important to note that VraFG only confers resistance to CAMPs, not to uncharged or negatively charged antimicrobial peptides.65,83
 | ||
| Fig. 8 Schematic representation of transporters. A. ABC exporters (purple), such as the one found in S. aureus, utilize ATP to drive export of positively charged membrane disruptors (blue). B. ABC importers (purple), such as the one found in Salmonella, work in conjunction with solute binding proteins that bind membrane disruptors in the periplasm and transfer them through an ABC importer, which also utilizes ATP for energy, into the cytoplasm, where they are degraded by peptidases. C. RND transporters (yellow), such as the one found in Yersinia, utilize proton pumps to provide the energy necessary to extrude membrane disruptors from the membrane. | ||
ABC transporters can also induce drug resistance by importing the membrane-targeting antimicrobial peptides into the cell where they are degraded by proteases.83 This mechanism was demonstrated in S. typhimurium toward the CAMPs melittin and protamine (Fig. 8B). An operon of five genes encodes the ABC transporter and periplasmic binding proteins. The co-expression of the periplasmic binding proteins suggests that this system is likely to mediate resistance to antimicrobial peptides by transporting them into the cytoplasm, and away from their putative membrane targets, where they could either be degraded by peptidases or initiate a regulatory cascade resulting in the activation of the resistance determinants.84
Resistance-nodulation-cell division (RND) efflux pump superfamily
RND transporters, like transporters from other superfamilies, are secondary transporters that utilize electrochemical potential differences created by pumping hydrogen or sodium ions from or to the outside of the cell as an energy source. In contrast, ABC transporters are primary transporters that utilize ATP as the energy source for the transfer.85–88 The RosA/RosB system includes an RND-type efflux pump coupled to a potassium antiporter. The system is regulated by temperature and responds to the presence of CAMPs. This system in Yersinia strains confers resistance to polymyxin B (Fig. 8C). RosA serves as the efflux pump protein and RosB serves as the potassium antiporter that removes potassium ions from the cell and imports hydrogen ions. RosA uses the proton-motive force created by RosB as an energy supply in order to force polymyxin B outside the cytoplasmic membrane. The ros locus is up-regulated by the presence of polymyxin B. Both RosA and RosB are essential for the resistance to polymyxin B.89The MtrCDE (for multiple transferable resistance) efflux pump of the RND family is expressed in Neisseria gonorrhoeae, a Gram-negative bacteria responsible for a sexually transmitted infection, and promotes resistance toward the CAMPs protegrin-1 (PG-1) and LL-37. Protegrins are found in porcine leukocytes; these cationic, cysteine-rich peptides assume β-sheet structures in solution. LL-37 is an α-helical human antimicrobial peptide expressed constitutively by granulocytes and testis that is induced by keratinocytes. Loss of the gene encoding the Mtr efflux pump results in increased susceptibility of gonococci to these CAMPs.90 MtrCDE also promotes resistance to polymyxin B in Neisseria meningitidis, the pathogen that causes meningitis and other forms of meningococcal disease. Mutants with increased sensitivity to polymyxin B were identified from a library of mariner transposon mutants generated in a meningococcal strain N. meningitidis serogroup B. More than half of the initial polymyxin B-sensitive mutants had insertions within the mtrCDE operon. Additionally, mutations within the mtrD also result in susceptibility to the two structurally unrelated CAMPs human LL-37 and PG-1.91
AcrAB, another RND-type efflux pump, is found in Klebsiella pneumoniae. This Gram-negative pathogen causes respiratory infections characterized by high rates of mortality and morbidity. Expression of AcrAB is associated with resistance to polymyxin B and several human CAMPs. Notably, although RND efflux pumps are capable of recognizing structurally diverse α helical, β sheet, or cyclic peptides, it has been reported that resistance to polymyxin B, LL-37, and various other antimicrobial peptides is not mediated by the AcrAB of E. coli or the MexAB of P. aeruginosa.92
Finally, it is well established that efflux pumps play a role in the resistance to CAMPs; however, the mechanism by which the efflux pumps transport substrates from within the bacterial membrane is under-explored.93–95 Since the transporters promote resistance to membrane disruptors and since they are known to have other roles in bacterial cell maintenance, these systems could be targets for the development of new antibiotics or of new additives for administration with an existing drug to prevent transporter-induced resistance.
Proteolytic degradation
Proteolytic degradation of CAMPs results in resistance in both Gram-positive and Gram-negative bacteria. There are two general types of proteases in bacteria that interfere with CAMPs, secreted and membrane bound. The secreted proteases degrade CAMPs even before they reach the bacterial membrane, and membrane-bound proteases degrade CAMPs on the cell surface.The Omptin family proteases are membrane-bound proteases found in Gram-negative bacteria including Shigella flexneri, Yersinia pestis, E. coli, and Salmonella enterica. Despite high sequence identity and a conserved β-barrel fold in the outer membrane, individual Omptins exhibit different substrate specificities. Only OmpT of E. coli and PgtE of S. enterica degrade CAMPs. The differences in Omptin family protease substrate specificities are suggested to be due to minor sequence variations in the surface-exposed regions of these proteins.96 OmpT and PgtE, as well other members of this protease family, contain aspartate in their catalytic sites, which is negatively charged under physiological conditions and, hence, bind positively charged substrates. The cleavage of peptide bonds by OmpT and PgtE is also dependent on the presence of bound LPS, which is speculated to induce adoption of the correct active-site geometry. OmpT from E. coli hydrolyses protamine, a DNA-binding CAMP from salmon sperm.97 PgtE expressed by S. typhimurium cleaves the human α-helical CAMP LL-37 and C18G, a synthetic 18-residue α-CAMP.98 CroP is another member of the Omptin family that was recently identified in Citrobacter rodentium.99 This protein is responsible for the resistance of C. rodentium to α-helical CAMPs. CroP is so effective that the peptides do not reach the periplasm to induce the PhoP–PhoQ system. In a strain that expresses a mutant CroP, the PhoP–PhoQ was activated but the strain was more susceptible to antimicrobial peptides than the wild-type strain.
P. aeruginosa expresses a secreted zinc-dependent metalloproteinase called elastase. This protease is associated with the degradation of LL-37.100,101 The metalloproteinases of E. faecalis (called gelatinase) and of Proteus mirabilis degrade the human CAMP LL-37. The cysteine protease of Streptococcus pyogenes also degrades LL-37. It is important to mention that LL-37 does effectively kill each of these strains of bacteria when the peptide is present at concentrations between 2 and 10 μg ml−1.100 Isolated aureolysin, the metalloproteinase of S. aureus, cleaves LL-37; an S. aureus mutant that secretes high levels of aureolysin is less susceptible to LL-37 than the wild-type strain, implying that this protease confers resistance to CAMPs.102Bacillus anthracis secretes a metalloprotease that cleaves LL-37; however, susceptibility levels vary among species of Bacillus and even between different strains of B. anthracis.103
The clinically used anionic lipopeptide antibiotic daptomycin that is synthesized by the actinomycete Streptomyces roseosporus undergoes proteolytic degradation in the presence of proteins isolated from the environmental bacteria actinomycetes. Several sites in daptomycin are prone to hydrolysis, and the development of future generations of this drug may be guided by analysis of its degradation pattern.104 Not all of the CAMPs are prone to protease degradation. The lipopeptide polymyxin B is resistant to proteolytic cleavage due to its cyclic structure, which protects it from degradation by exopeptidases and other peptidases.99,105
Systems that prevent CAMPs from reaching the cell membrane
An evasive strategy used by several bacterial species prevents membrane disruptors from reaching their target, thus promoting resistance and virulence. In this mode of action, the bacteria release factors that interact directly or indirectly with the membrane disruptors. E. coli and V. cholerae secrete vesicles in response to stress signals such as the presence of membrane disruptors. In both of these species and in other Gram-negative bacteria, vesicle release occurs during normal growth. Vesicles are composed of the outer membrane and periplasm contents only. In E. coli, an independent stress response system is activated by a variety of conditions, among them the presence of membrane disruptors, that increases the production of these vesicles. Increased vesiculation correlates with enhanced resistance to polymyxin B, and these vesicles may both remove toxic components from the membrane and also act as decoys that bind to the membrane disruptors.106 A hyper-vesiculating mutant of E. coli is more resistant to both polymyxins B and E than is the wild-type strain (Fig. 9A).107 | ||
| Fig. 9 Schematic representation of agents that block CAMP activity secreted by different bacterial species. A. Vesicle formation can either remove segments of the membrane disturbed by CAMPs or serve as decoys that sequester CAMPs present in the environment. B. A plasmid released upon lysis of bacterial cells is associated with resistance in Shigella species toward LL-37. The plasmids penetrate the host cells and inhibit production of LL-37. C. Proteases secreted by bacteria that are propagating in connective tissues or wounds, such as P. aeruginosa, cleave host decorin releasing negatively charged dermatan sulfate units, which bind the CAMPs through electrostatic interactions and prevent them from reaching the bacterial cell membrane. | ||
Growth of V. cholerae O1 El Tor strain A1552 in sub-lethal concentrations of polymyxin B induces production of larger vesicles with modified protein content than does growth in the absence of the membrane disruptor; in the absence and presence of the CAMP, the same number of vesicles are produced. These vesicles contain an extracellular biofilm associated protein (Bap1) that is bound to the vesicle through the porins of OmpT. When wild-type V. cholerae O1 El Tor strain A1552 was incubated with isolated polymyxin-enhanced modified vesicles, it was more resistant to the CAMP LL-37 than when the modified vesicles were not present. Bap1 binds the LL-37 but does not degrade it, although its presence is necessary for the resistance. It appears that growth in sub-lethal concentrations of polymyxin B enhances resistance to LL-37 as the polymyxin-modified vesicles bind to LL-37 and prevent it from reaching the membrane.108
Shigella species dysenteriae type I and flexneri down-regulate transcription of the host antimicrobial peptides LL-37 and β defensin both in cell lines and in vivo as shown in gut biopsies of patients infected with Shigella. A DNA plasmid carried by the bacteria appears to interfere with the signaling pathway involved in the expression of LL-37. It is hypothesized that the plasmid is released upon lysis of first intruders (caused by antimicrobial compounds including LL-37), and then the plasmid is responsible for interference with inhibitory signals from epithelial cells, thereby down-regulating the expression of the antimicrobial peptides (Fig. 9B).109
P. aeruginosa, E. faecalis, and S. pyogenes all propagate and multiply in connective tissues and are wound-infection promoters. Proteoglycans, among them decorin, are abundant in this type of environment. The proteases of these species, in addition to targeting CAMPs directly, also cleave decorin, releasing negatively charged dermatan sulfate. Dermatan sulfates bind to the highly cationic antimicrobial α-defensins neutralizing their bactericidal activity (Fig. 9C).110
Systems that sense cationic membrane disruptors
Since constant expression of genes responsible for the resistance to CAMPs would consume a great amount of energy and is not necessary under many conditions, regulatory systems have evolved to ensure that the bacteria express these genes only in the presence of CAMPs.65 Both Gram-positive and Gram-negative bacteria have such regulatory systems. These systems regulate expression of numerous genes that encode proteins that are responsible for various modifications on the cell envelope and that promote resistance to CAMPs by other mechanisms. The Gram-positive bacteria CAMP-induced regulatory system is called either GraRS (glycopeptide resistance associated111) or Aps (for antimicrobial peptide sensor65). The Gram-negative CAMP-induced regulatory system is PhoP–PhoQ. The Pho designation typically denotes loci involved in phosphate metabolism.83The Aps regulatory system is highly conserved in staphylococci. It was first characterized in S. epidermidis83 and was later identified and studied in strains of S. aureus.65,112 It is composed of three components, ApsS, ApsR, and ApsX; however, it resembles a classical two-component histidine kinase response regulator.83 ApsS is the membrane-inserted sensor histidine kinase, and ApsR is the transcriptional response regulator that becomes activated upon phosphorylation by ApsS. The function of the third protein, ApsX, is unknown. ApsS has a single nine-amino-acid extracellular loop with a high density of negative charges through which it interacts with various CAMPs (Fig. 10A).65 The amino acid composition varies among Staphylococcus strains, and therefore, the CAMPs sensed by the system are strain dependent.51,65 The system up-regulates expression of genes involved in CAMP resistance including the dltABCD and the MprF systems that neutralize negatively charged lipids by the addition of positively charged groups and the ABC transporters such as VraFG. Interestingly, the Aps system also up-regulates genes involved in lysine biosynthesis. This finding supports the hypothesis that these regulatory systems give an evolutionary advantage: there is a need for high levels of lysine in the presence of CAMPs for its incorporation on phosphatidyl glycerol, a reaction that is catalyzed by MprF; under normal conditions, this overexpression would be an energetic burden.
 | ||
| Fig. 10 Schematic representation of the Aps and PhoP–PhoQ two-component systems of Gram-positive and Gram-negative bacteria, respectively. A. ApsS (green) contains a single nine-amino-acid extracellular loop with a high density of negative charges. ApsR (orange) is the cytoplasmic transcription regulator. When CAMPs are present (magenta), they bind the negatively charged extracellular loop, enhancing auto-phosphorylation and phosphorylation of ApsR, which in turn up-regulates resistance-related gene expression. B. PhoQ (blue) is inserted through the membrane as a dimer; each monomer has an extracellular loop containing acidic residues that are held next to the membrane by divalent cations such as Mg2+ and Ca2+ (pink). PhoP (brown) is the cytoplasmic transcription regulator. CAMPs, when present, release the divalent cations bridging between PhoQ and the membrane resulting in a conformational change that induces autophosphorylation and phosphorylation of PhoP, which in turn up-regulates gene expression related to CAMP resistance. | ||
The PhoP–PhoQ system is a two-component regulatory system found in Gram-negative species including P. aeruginosa, E. coli K12, S. flexneri, Y. pestis, and S. typhimurium.113–115 Many of the features of the well-characterized Salmonella PhoP–PhoQ system apply to the PhoP–PhoQ homologues in other Gram-negative species.113 In this two-component system, PhoQ is the membrane-bound kinase that senses environmental signals, and PhoP is the cytoplasmic transcriptional response regulator (Fig. 10B). This system up- and down-regulates expression of more than 40 different genes, termed PhoP-activated (pag) and PhoP-repressed (prg) genes, respectively.114,116 PhoP–PhoQ is activated by low concentrations of Mg2+ and repressed by high concentrations of Mg2+. The PhoQ protein of Salmonella has a periplasmic domain containing several acidic residues that could be involved in sensing divalent cations. Furthermore, in vivo experiments have established that PhoQ has binding sites for Mg2+ and Ca2+ and that the PhoP–PhoQ system responds to the periplasmic concentrations of these ions.113 Various publications have shown that growth of Salmonella and other Gram-negative species under low concentrations of Mg2+ induces resistance to CAMPs including polymyxin.115–117 Other studies have shown that modifications in lipid A associated with resistance to CAMPs and polymyxin are induced by Mg2+-deficient conditions such as those inside phagosomes. Since microorganisms such as Salmonella, Shigella, and Yersinia are capable of surviving and proliferating inside host phagsomes,113 sensing this environment and activating the resistance mechanisms accordingly would contribute to bacterial cell survival. Until recently, it was unclear why low concentrations of Mg2+ correlate with the presence of CAMPs and why microorganisms that do not have an intracellular life cycle would have this system. Recent studies of the PhoQ structure suggest that the Mg2+ ions bound to the acidic residues of PhoQ in its un-activated form bridge between the acidic residues and the negatively charged lipids of the membrane. When CAMPs replace some of these ions, it interferes with both the PhoQ protein, by binding to its acidic domains, and the membrane beneath it, causing structural changes that together initiate signal transduction (Fig. 10B).118–121
Amongst the genes regulated by PhoP–PhoQ, there are genes responsible for another two-component system, PmrAB. This system is responsible for the additions of Ara4N and phosphoethanolamine to LPS. In some bacteria, such as in E. coli, the PmrAB system is not regulated by PhoP–PhoQ, but rather through sensing of Fe3+, which at high concentrations binds to the sensor PmrB in the periplasm, or by acidic conditions sensed through an unknown mechanism.
Conclusions
Both Gram-positive and Gram-negative bacterial species have evolved various mechanisms that limit the efficacy of several families of membrane disruptors including the clinically used lipopeptides polymyxins and daptomycin. Although the basic structure and composition of bacterial membranes are relatively conserved, several chemical modifications on bacterial membrane lipids and cell wall components result in resistance to membrane-disrupting antibiotics. Moreover, bacteria have evolved a diverse array of resistance pathways involving efflux pumps, proteolytic degradation, and secretion of factors that capture membrane-disrupting antibiotics to prevent damage to the bacterial cell membrane. The multiple proteins that are involved in conferring resistance to membrane-disrupting antibiotics should be considered as drug targets as the inhibition of activities of these proteins will maintain efficacy of important last-resort antimicrobials such as polymyxins. Finally, understanding the resistance to membrane-disrupting antibiotics at the molecular level offers opportunities to develop next generation antibiotics capable of evading these mechanisms. Membrane-disrupting agents offer a solution to treatment of infections caused by pathogens that grow slowly and those that are resistant to first-line antibiotics; therefore, the fact that resistance has developed to certain membrane-disrupting agents should not quell enthusiasm for this type of antibiotic.Acknowledgements
We would like to thank Ms. Tesh (Natalya) Weber for the graphic design of figures and schemes. The study of membrane-disrupting antibiotics in the Fridman research group is supported by the Israel Science Foundation (Grant 6/14).References
- J. G. Hurdle, A. J. O'Neill, I. Chopra and R. E. Lee, Nat. Rev. Microbiol., 2011, 9, 62–75 CrossRef CAS.
- R. Daugelavicius, E. Bakiene and D. H. Bamford, Antimicrob. Agents Chemother., 2000, 44, 2969–2978 CrossRef CAS PubMed.
- H. Nikaido, Science, 1994, 264, 382–388 CAS.
- H. Jenssen, P. Hamill and R. E. W. Hancock, Clin. Microbiol. Rev., 2006, 19, 491–511 CrossRef CAS PubMed.
- R. E. Hancock and G. Diamond, Trends Microbiol., 2000, 8, 402–410 CrossRef CAS.
- L. M. Yin, M. A. Edwards, J. Li, C. M. Yip and C. M. Deber, J. Biol. Chem., 2012, 287, 7738–7745 CrossRef CAS.
- L. H. Kondejewski, S. W. Farmer, D. S. Wishart, R. E. Hancock and R. S. Hodges, Int. J. Pept. Protein Res., 1996, 47, 460–466 CrossRef CAS.
- D. R. Storm, K. S. Rosenthal and P. E. Swanson, Annu. Rev. Biochem., 1977, 46, 723–763 CrossRef CAS PubMed.
- A. P. Zavascki, L. Z. Goldani, J. Li and R. L. Nation, J. Antimicrob. Chemother., 2007, 60, 1206–1215 CrossRef CAS PubMed.
- H. Tsubery, I. Ofek, S. Cohen and M. Fridkin, J. Med. Chem., 2000, 43, 3085–3092 CrossRef CAS PubMed.
- M. E. Falagas and S. K. Kasiakou, Clin. Infect. Dis., 2005, 40, 1333–1341 CrossRef CAS.
- R. E. Hancock, Lancet, 1997, 349, 418–422 CrossRef CAS.
- H. Tsubery, I. Ofek, S. Cohen, M. Eisenstein and M. Fridkin, Mol. Pharmacol., 2002, 62, 1036–1042 CrossRef CAS.
- P. Pristovšek and J. Kidrič, J. Med. Chem., 1999, 42, 4604–4613 CrossRef.
- R. M. Humphries, S. Pollett and G. Sakoulas, Clin. Microbiol. Rev., 2013, 26, 759–780 CrossRef CAS.
- L. Jeu and H. B. Fung, Clin. Ther., 2004, 26, 1728–1757 CrossRef CAS PubMed.
- S. K. Straus and R. E. W. Hancock, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1215–1223 CrossRef CAS PubMed.
- A. S. Bayer, T. Schneider and H.-G. Sahl, Ann. N. Y. Acad. Sci., 2013, 1277, 139–158 CrossRef CAS PubMed.
- I. M. Herzog and M. Fridman, MedChemComm, 2014, 5, 1014–1026 RSC.
- R. F. Epand, J. E. Pollard, J. O. Wright, P. B. Savage and R. M. Epand, Antimicrob. Agents Chemother., 2010, 54, 3708–3713 CrossRef CAS.
- N. Srinivas, P. Jetter, B. J. Ueberbacher, M. Werneburg, K. Zerbe, J. Steinmann, B. Van der Meijden, F. Bernardini, A. Lederer, R. L. A. Dias, P. E. Misson, H. Henze, J. Zumbrunn, F. O. Gombert, D. Obrecht, P. Hunziker, S. Schauer, U. Ziegler, A. Käch, L. Eberl, K. Riedel, S. J. DeMarco and J. A. Robinson, Science, 2010, 327, 1010–1013 CrossRef CAS.
- S. K. Vooturi, C. M. Cheung, M. J. Rybak and S. M. Firestine, J. Med. Chem., 2009, 52, 5020–5031 CrossRef CAS PubMed.
- Y. Eun, M. H. Foss, D. Kiekebusch, D. A. Pauw, W. M. Westler, M. Thanbichler and D. B. Weibel, J. Am. Chem. Soc., 2012, 134, 11322–11325 CrossRef CAS PubMed.
- H. Zou, J.-J. Koh, J. Li, S. Qiu, T. T. Aung, H. Lim, R. Lakshminarayanan, X. Dai, C. Tang, F. H. Lim, L. Zhou, A. L. Tan, C. S. Verma, D. Tan, H. S. O. Chan, P. Saraswathi, D. Cao, S. Liu and R. W. Beuerman, J. Med. Chem., 2013, 56, 2359–2373 CrossRef CAS PubMed.
- J. J. Zhang, F. I. Chiang, L. Wu, P. G. Czyryca, D. Li and C. W. T. Chang, J. Med. Chem., 2008, 51, 7563–7573 CrossRef CAS PubMed.
- S. Bera, G. G. Zhanel and F. Schweizer, J. Med. Chem., 2010, 53, 3626–3631 CrossRef CAS PubMed.
- L. Zimmermann, A. Bussière, M. Ouberai, I. Baussanne, C. Jolivalt, M. P. Mingeot-Leclercq and J. L. Décout, J. Med. Chem., 2013, 56, 7691–7705 CrossRef CAS PubMed.
- Y. Berkov-Zrihen, I. M. Herzog, R. I. Benhamou, M. Feldman, K. B. Steinbuch, P. Shaul, S. Lerer, A. Eldar and M. Fridman, Chem. – Eur. J., 2015, 21, 4340–4349 CrossRef CAS.
- T. Kaeberlein, K. Lewis and S. S. Epstein, Science, 2002, 296, 1127–1129 CrossRef CAS.
- A. D'Onofrio, J. M. Crawford, E. J. Stewart, K. Witt, E. Gavrish, S. Epstein, J. Clardy and K. Lewis, Chem. Biol., 2010, 17, 254–264 CrossRef PubMed.
- D. Nichols, N. Cahoon, E. M. Trakhtenberg, L. Pham, A. Mehta, A. Belanger, T. Kanigan, K. Lewis and S. S. Epstein, Appl. Environ. Microbiol., 2010, 76, 2445–2450 CrossRef CAS.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- Z. Zhou, A. A. Ribeiro, S. Lin, R. J. Cotter, S. I. Miller and C. R. H. Raetz, J. Biol. Chem., 2001, 276, 43111–43121 CrossRef CAS PubMed.
- M. S. Trent, A. A. Ribeiro, S. Lin, R. J. Cotter and C. R. H. Raetz, J. Biol. Chem., 2001, 276, 43122–43131 CrossRef CAS PubMed.
- S. M. Moskowitz, R. K. Ernst and S. I. Miller, J. Bacteriol., 2004, 186, 575–579 CrossRef CAS.
- R. K. Ernst, E. C. Yi, L. Guo, K. B. Lim, J. L. Burns, M. Hackett and S. I. Miller, Science, 1999, 286, 1561–1565 CrossRef CAS PubMed.
- M. Denton, K. Kerr, L. Mooney, V. Keer, A. Rajgopal, K. Brownlee, P. Arundel and S. Conway, Pediatr. Pulmonol., 2002, 34, 257–261 CrossRef CAS PubMed.
- J. S. Gunn, K. B. Lim, J. Krueger, K. Kim, L. Guo, M. Hackett and S. I. Miller, Mol. Microbiol., 1998, 27, 1171–1182 CrossRef CAS PubMed.
- J. S. Gunn, J. Endotoxin Res., 2001, 7, 57–62 CrossRef CAS PubMed.
- Z. Zhou, L. Shanhua, R. J. Cotter and C. R. H. Raetz, Biochemistry, 1999, 274, 18503–18514 CAS.
- S. D. Breazeale, A. A. Ribeiro, A. L. McClerren and C. R. H. Raetz, J. Biol. Chem., 2005, 280, 14154–14167 CrossRef CAS PubMed.
- C. R. H. Raetz, C. M. Reynolds, M. S. Trent and R. E. Bishop, Annu. Rev. Biochem., 2007, 76, 295–329 CrossRef CAS PubMed.
- S. D. Breazeale, A. A. Ribeiro and C. R. H. Raetz, J. Biol. Chem., 2002, 277, 2886–2896 CrossRef CAS PubMed.
- A. Yan, Z. Guan and C. R. H. Raetz, J. Biol. Chem., 2007, 282, 36077–36089 CrossRef CAS PubMed.
- M. R. Pelletier, L. G. Casella, J. W. Jones, M. D. Adams, D. V. Zurawski, K. R. O. Hazlett, Y. Doi and R. K. Ernst, Antimicrob. Agents Chemother., 2013, 57, 4831–4840 CrossRef CAS.
- N. J. Phillips, B. Schilling, M. K. McLendon, M. A. Apicella and B. W. Gibson, Infect. Immun., 2004, 72, 5340–5348 CrossRef CAS.
- X. Wang, A. A. Ribeiro, Z. Guan, S. C. McGrath, R. J. Cotter and C. R. H. Raetz, Biochemistry, 2006, 45, 14427–14440 CrossRef CAS.
- X. Wang, A. A. Ribeiro, Z. Guan and C. R. H. Raetz, Biochemistry, 2009, 48, 1162–1172 CrossRef CAS PubMed.
- F. Song, Z. Guan and C. R. H. Raetz, Biochemistry, 2009, 48, 1173–1182 CrossRef CAS.
- C. R. H. Raetz, Z. Guan, B. O. Ingram, D. A. Six, F. Song, X. Wang and J. Zhao, J. Lipid Res., 2009, 50(Suppl.), S103–S108 Search PubMed.
- C. M. Ernst and A. Peschel, Mol. Microbiol., 2011, 80, 290–299 CrossRef CAS PubMed.
- C. M. Ernst, P. Staubitz, N. N. Mishra, S. J. Yang, G. Hornig, H. Kalbacher, A. S. Bayer, D. Kraus and A. Peschel, PLoS Pathog., 2009, 5, e1000660 Search PubMed.
- S. Klein, C. Lorenzo, S. Hoffmann, J. M. Walther, S. Storbeck, T. Piekarski, B. J. Tindall, V. Wray, M. Nimtz and J. Moser, Mol. Microbiol., 2009, 71, 551–565 CrossRef CAS PubMed.
- P. Staubitz, H. Neumann, T. Schneider, I. Wiedemann and A. Peschel, FEMS Microbiol. Lett., 2004, 231, 67–71 CrossRef CAS.
- J. M. dos Santos Mota, J. A. den Kamp, H. M. Verheij and L. L. van Deenen, J. Bacteriol., 1970, 104, 611–619 CAS.
- H. Roy and M. Ibba, J. Biol. Chem., 2009, 284, 29677–29683 CrossRef CAS PubMed.
- K. Thedieck, T. Hain, W. Mohamed, B. J. Tindall, M. Nimtz, T. Chakraborty, J. Wehland and L. Jänsch, Mol. Microbiol., 2006, 62, 1325–1339 CrossRef CAS PubMed.
- L. Friedman, J. D. Alder and J. A. Silverman, Antimicrob. Agents Chemother., 2006, 50, 2137–2145 CrossRef CAS PubMed.
- D. Patel, M. Husain, C. Vidaillac, M. E. Steed, M. J. Rybak, S. M. Seo and G. W. Kaatz, Int. J. Antimicrob. Agents, 2011, 38, 442–446 CrossRef CAS PubMed.
- C. I. Montera, F. Stock and P. R. Murray, Antimicrob. Agents Chemother., 2008, 52, 1167–1170 CrossRef.
- H. W. Boucher and G. Sakoulas, Clin. Infect. Dis., 2007, 45, 601–608 CrossRef CAS PubMed.
- C. M. Ernst, S. Kuhn, C. J. Slavetinsky, B. Krismer, S. Heilbronner, C. Gekeler, D. Kraus, S. Wagner and A. Peschel, mBio, 2015, 6, e02340 CrossRef CAS PubMed.
- F. C. Neuhaus, M. P. Heaton, D. V. Debabov and Q. Zhang, Microb. Drug Resist., 1996, 2, 77–84 CrossRef CAS PubMed.
- M. Kovács, A. Halfmann, I. Fedtke, M. Heintz, A. Peschel, W. Vollmer, R. Hakenbeck and R. Brückner, J. Bacteriol., 2006, 188, 5797–5805 CrossRef.
- M. Li, D. J. Cha, Y. Lai, A. E. Villaruz, D. E. Sturdevant and M. Otto, Mol. Microbiol., 2007, 66, 1136–1147 CrossRef CAS.
- S.-J. Yang, B. N. Kreiswirth, G. Sakoulas, M. R. Yeaman, Y. Q. Xiong, A. Sawa and A. S. Bayer, J. Infect. Dis., 2009, 200, 1916–1920 CrossRef CAS PubMed.
- F. C. Neuhaus and J. Baddiley, Microbiol. Mol. Biol. Rev., 2003, 67, 686–723 CrossRef CAS PubMed.
- M. P. Heaton and F. C. Neuhaus, J. Bacteriol., 1994, 176, 681–690 CAS.
- D. V. Debabov, M. Y. Kiriukhin, C. Francis and F. C. Neuhaus, J. Bacteriol., 2000, 182, 2855–2864 CrossRef CAS PubMed.
- J. V. Hankins, J. A. Madsen, D. K. Giles, J. S. Brodbelt and M. S. Trent, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8722–8727 CrossRef CAS PubMed.
- J. C. Henderson, C. D. Fage, J. R. Cannon, J. S. Brodbelt, A. T. Keatinge-Clay and M. S. Trent, ACS Chem. Biol., 2014, 9, 2382–2392 CrossRef CAS PubMed.
- J. V. Hankins, J. A. Madsen, D. K. Giles, B. M. Childers, K. E. Klose, J. S. Brodbelt and M. S. Trent, Mol. Microbiol., 2011, 81, 1313–1329 CrossRef CAS.
- S. R. Murray, R. K. Ernst, D. Bermudes, S. I. Miller and K. B. Low, J. Bacteriol., 2007, 189, 5161–5169 CrossRef CAS.
- A. X. Tran, M. E. Lester, C. M. Stead, C. R. H. Raetz, D. J. Maskell, S. C. McGrath, R. J. Cotter and M. S. Trent, J. Biol. Chem., 2005, 280, 28186–28194 CrossRef CAS.
- J. V. Farizano, M. D. L. M. Pescaretti, F. E. López, F. F. Hsu and M. A. Delgado, J. Biol. Chem., 2012, 287, 38778–38789 CrossRef CAS PubMed.
- H. Lee, F. F. Hsu, J. Turk and E. A. Groisman, J. Bacteriol., 2004, 186, 4124–4133 CrossRef CAS PubMed.
- M. I. Kanipes, S. Lin, R. J. Cotter and C. R. H. Raetz, J. Biol. Chem., 2001, 276, 1156–1163 CrossRef CAS PubMed.
- C. M. Reynolds, S. R. Kalb, R. J. Cotter and C. R. H. Raetz, J. Biol. Chem., 2005, 280, 21202–21211 CrossRef CAS PubMed.
- G. Chang, FEBS Lett., 2003, 555, 102–105 CrossRef CAS PubMed.
- W. T. Bellamy, Annu. Rev. Pharmacol. Toxicol., 1996, 36, 161–183 CrossRef CAS PubMed.
- K. J. Linton and K. J. Linton, Physiology, 2007, 22, 122–130 CrossRef CAS.
- A. L. Davidson, E. Dassa, C. Orelle and J. Chen, Microbiol. Mol. Biol. Rev., 2008, 72, 317–364 CrossRef CAS PubMed.
- M. Li, Y. Lai, A. E. Villaruz, D. J. Cha, D. E. Sturdevant and M. Otto, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9469–9474 CrossRef CAS PubMed.
- C. Parra-Lopez, M. T. Baer and E. A. Groisman, EMBO J., 1993, 12, 4053–4062 CAS.
- M. H. Saier, Microbiol. Rev., 1994, 58, 71–93 CAS.
- T. T. Tseng, K. S. Gratwick, J. Kollman, D. Park, D. H. Nies, A. Goffeau and M. H. Saier, J. Mol. Microbiol. Biotechnol., 1999, 1, 107–125 CAS.
- J. M. Blair and L. J. Piddock, Curr. Opin. Microbiol., 2009, 12, 512–519 CrossRef CAS.
- D. Du, Z. Wang, N. R. James, J. E. Voss, E. Klimont, T. Ohene-Agyei, H. Venter, W. Chiu and B. F. Luisi, Nature, 2014, 509, 512–515 CrossRef CAS PubMed.
- J. A. Bengoechea and M. Skurnik, Mol. Microbiol., 2000, 37, 67–80 CrossRef CAS PubMed.
- W. M. Shafer, X. Qu, A. J. Waring and R. I. Lehrer, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1829–1833 CrossRef CAS.
- Y. Tzeng, K. D. Ambrose, S. Zughaier, X. Zhou, Y. K. Miller, W. M. Shafer and D. S. Stephens, J. Bacteriol., 2005, 187, 5387–5396 CrossRef CAS PubMed.
- S. Rieg, A. Huth, H. Kalbacher and W. V. Kern, Int. J. Antimicrob. Agents, 2009, 33, 174–176 CrossRef CAS PubMed.
- H. Nikaido, J. Bacteriol., 1996, 178, 5853–5859 CAS.
- X. Q. Yao, N. Kimura, S. Murakami and S. Takada, J. Am. Chem. Soc., 2013, 135, 7474–7485 CrossRef CAS.
- J. Dreier and P. Ruggerone, Front. Microbiol., 2015, 6, 660 Search PubMed.
- M. Kukkonen and T. K. Korhonen, Int. J. Med. Microbiol., 2004, 294, 7–14 CrossRef CAS PubMed.
- S. Stumpe, R. Schmid, D. L. Stephens, G. Georgiou and E. P. Bakker, J. Bacteriol., 1998, 180, 4002–4006 CAS.
- T. Guina, E. C. Yi, H. Wang, M. Hackett and S. I. Miller, J. Bacteriol., 2000, 182, 4077–4086 CrossRef CAS.
- V. Le Sage, L. Zhu, C. Lepage, A. Portt, C. Viau, F. Daigle, S. Gruenheid and H. Le Moual, Mol. Microbiol., 2009, 74, 98–111 CrossRef CAS.
- A. Schmidtchen, I. M. Frick, E. Andersson, H. Tapper and L. Björck, Mol. Microbiol., 2002, 46, 157–168 CrossRef CAS PubMed.
- Z. Kuang, Y. Hao, B. E. Walling, J. L. Jeffries, D. E. Ohman and G. W. Lau, PLoS One, 2011, 6, e27091 CAS.
- M. Sieprawska-lupa, P. Mydel, K. Wójcik, M. Puklo, B. Lupa, P. Suder, J. Silberring, M. Reed, J. Pohl, W. Shafer, F. Mcaleese, T. Foster, J. Potempa, K. Krawczyk, K. Wo and J. Travis, Antimicrob. Agents Chemother., 2004, 48, 4673–4679 CrossRef CAS.
- J. E. Thwaite, S. Hibbs, R. W. Titball and T. P. Atkins, Antimicrob. Agents Chemother., 2006, 50, 2316–2322 CrossRef CAS.
- V. M. D'Costa, T. A. Mukhtar, T. Patel, K. Koteva, N. Waglechner, D. W. Hughes, G. D. Wright and G. De Pascale, Antimicrob. Agents Chemother., 2012, 56, 757–764 CrossRef.
- J. L. Thomassin, J. R. Brannon, B. F. Gibbs, S. Gruenheid and H. Le Moual, Infect. Immun., 2012, 80, 483–492 CrossRef CAS.
- A. J. McBroom and M. J. Kuehn, Mol. Microbiol., 2007, 63, 545–558 CrossRef CAS PubMed.
- A. J. Manning and M. J. Kuehn, BMC Microbiol., 2011, 11, 258 CrossRef CAS PubMed.
- M. Duperthuy, A. E. Sjöström, D. Sabharwal, F. Damghani, B. E. Uhlin and S. N. Wai, PLoS Pathog., 2013, 9, e1003620 Search PubMed.
- D. Islam, L. Bandholtz, J. Nilsson, H. Wigzell, B. Christensson, B. Agerberth and G. Gudmundsson, Nat. Med., 2001, 7, 180–185 CrossRef CAS PubMed.
- A. Schmidtchen, I. M. Frick and L. Björck, Mol. Microbiol., 2001, 39, 708–713 CrossRef CAS PubMed.
- M. Falord, G. Karimova, A. Hiron and T. Msadeka, Antimicrob. Agents Chemother., 2012, 56, 1047–1058 CrossRef CAS.
- H. Roy, IUBMB Life, 2009, 61, 940–953 CrossRef CAS.
- E. A. Groisman, J. Bacteriol., 2001, 183, 1835–1842 CrossRef CAS PubMed.
- L. Guo, K. B. Lim, J. S. Gunn, B. Bainbridge, R. P. Darveau, M. Hackett and S. I. Miller, Science, 1997, 276, 250–253 CrossRef CAS.
- L. Guo, K. B. Lim, C. M. Poduje, M. Daniel, J. S. Gunn, M. Hackett and S. I. Miller, Cell, 1998, 95, 189–198 CrossRef CAS.
- J. S. Gunn and S. I. Miller, J. Bacteriol., 1996, 178, 6857–6864 CAS.
- M. E. Falagas, P. I. Rafailidis and D. K. Matthaiou, Drug Resist. Updates, 2010, 13, 132–138 CrossRef CAS PubMed.
- M. W. Bader, S. Sanowar, M. E. Daley, A. R. Schneider, U. Cho, W. Xu, R. E. Klevit, H. Le Moual and S. I. Miller, Cell, 2005, 122, 461–472 CrossRef CAS PubMed.
- K. S. Molnar, M. Bonomi, R. Pellarin, G. D. Clinthorne, G. Gonzalez, S. D. Goldberg, M. Goulian, A. Sali and W. F. DeGrado, Structure, 2014, 22, 1239–1251 CrossRef CAS PubMed.
- K. G. Hicks, S. P. Delbecq, E. Sancho-Vaello, M.-P. Blanc, K. K. Dove, L. R. Prost, M. E. Daley, K. Zeth, R. E. Klevit and S. I. Miller, eLife, 2015, 4, 1–23 Search PubMed.
- T. Lemmin, C. S. Soto, G. Clinthorne, W. F. DeGrado and M. Dal Peraro, PLoS Comput. Biol., 2013, 9, e1002878 CAS.
| This journal is © The Royal Society of Chemistry 2016 |