
Drug-target networks in aminoglycoside resistance: hierarchy of priority in structural drug design†‡
Valjean R.
Bacot-Davis
 ac,
Angelia V.
Bassenden
ac and
Albert M.
Berghuis
*abcd
ac,
Angelia V.
Bassenden
ac and
Albert M.
Berghuis
*abcd
aDepartment of Biochemistry, McGill University, Montréal, Québec, Canada. E-mail: albert.berghuis@mcgill.ca; Tel: 514 398 8795
bDepartment of Microbiology & Immunology, McGill University, Montréal, Québec, Canada
cGroupes de recherche GRASP et PROTEO, Montréal, Québec, Canada
dBellini Pavilion, 3649 Promenade Sir William Osler, Room 466, Montréal, Quebec H3G 0B1, Canada
First published on 10th December 2015
Abstract
Antibiotic resistance is a multifactorial problem that demands multifaceted strategies to address. Here we present a drug-target network analysis of the clinically most prominent mechanism of resistance to aminoglycoside antibiotics, i.e. enzyme mediated modification of the antibiotics. This drug-target network displays prominent resistance preferences for 4,6-disubstituted aminoglycosides such as tobramycin and gentamicin, reflective of their extensive clinical usage. Further analysis also highlights aminoglycosides that remain more resilient to modifications by various bacterial resistance enzymes. This aminoglycoside resistance drug-target network conveys a compelling case for prioritization of next-generation aminoglycosides development exploiting 4,5-disubstituted and non-deoxystreptamine aminoglycoside scaffolds to surmount rising drug-resistance, in conjunction with advancing inhibitor/adjuvant leads effective against multiple aminoglycoside modifying enzyme.
Introduction
Devastating diseases caused by primary and opportunistic microbial infections have plagued modern humans since their evolution 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years ago.1,2 Prehistoric human fossils and ancient written records detail the ravages of leprosy, pneumonia, meningitis, rabies, and many other microbial plagues, some of which persist as problems today.3–6 Infections caused by bacterial transmission remain a leading cause of human mortality despite centuries of medical progression.7 Foremost among these advancements was the clinical introduction of antimicrobial molecules, known as antibiotics, which allowed the successful treatment of potentially fatal bacterial infections.8,9 Decades after the unparalleled success of early antibiotic regimens, selective pressures have led to an increasing prevalence of microbial populations capable of surviving exposure to formerly toxic antibiotics.8 Now, it is estimated that by 2050, emerging antibiotic resistance will cost $100 trillion in lost GDP revenue, and 10 million global deaths every year.10–13
000 years ago.1,2 Prehistoric human fossils and ancient written records detail the ravages of leprosy, pneumonia, meningitis, rabies, and many other microbial plagues, some of which persist as problems today.3–6 Infections caused by bacterial transmission remain a leading cause of human mortality despite centuries of medical progression.7 Foremost among these advancements was the clinical introduction of antimicrobial molecules, known as antibiotics, which allowed the successful treatment of potentially fatal bacterial infections.8,9 Decades after the unparalleled success of early antibiotic regimens, selective pressures have led to an increasing prevalence of microbial populations capable of surviving exposure to formerly toxic antibiotics.8 Now, it is estimated that by 2050, emerging antibiotic resistance will cost $100 trillion in lost GDP revenue, and 10 million global deaths every year.10–13
Antibiotics
Currently, there are eighteen main classes of antibiotics, of which many have predominant roles in human therapy, e.g.: β-lactams, macrolides, quinolones, sulfonamides, tetracyclines, and aminoglycosides.14 Antibiotics, such as β-lactams, generally affect Gram-positive bacteria, while other antibiotics such as tetracyclines, macrolides, and aminoglycosides, largely affect both Gram-negative and Gram-positive bacteria.15 Of these, bactericidal aminoglycoside antibiotics are particularly attractive for the treatment of serious bacterial infections, unlike other bacteriostatic antibiotics that target protein synthesis, such as macrolides, lincosamides, and tetracyclines.15 In addition to spectral potential and bactericidal effectiveness, aminoglycosides are conducive to next-generation, analogue development, thereby extending their clinical lifetime for patient treatment. A profitable number of derivative antibiotics have already been constructed using the aminoglycoside core.14 Here, we will focus on the valuable potential remaining in studying the structural mechanisms of resistance in order to tailor intractable analogue developments for the aminoglycoside class of antibiotics as well as adjuvant/inhibitors against key resistance mechanisms.Aminoglycosides
Aminoglycosides interfere with bacterial protein synthesis by binding the 30S ribosomal subunit to obstruct tRNA translocation from the A-site and P-site. This interaction thereby counteracts bacterial translation and propagation.16,17 Chemically, the majority of aminoglycosides are characterized by a deoxystreptamine ring that is substituted at two positions.18–20 Specifically, the deoxystreptamine core can be 4,5-disubstituted, or 4-6-disubstituted. 4,5-Disubstituted aminoglycosides include neomycin, lividomycin, ribostamycin, paromomycin, and butirosin, while 4,6-disubstituted aminoglycosides encompass kanamycin, amikacin, isepamicin, tobramycin, gentamicin, dibekacin, netilmicin, and sisomicin. In addition to these two main classes of aminoglycoside antibiotics there is a third small but diverse group of non-deoxystreptamine aminoglycosides, which possess a slightly different pseudo-oligosacharride core. This group contains the founding member of the aminoglycoside class of antibiotics, streptomycin, and two additional drugs: hygromycin, and spectinomycin (Fig. 1). Note that unlike all other aminoglycosides the non-deoxystreptamine compounds hygromycin and spectinomycin do not have the same mode of action, nor is spectinomycin considered a bactericidal antibiotic.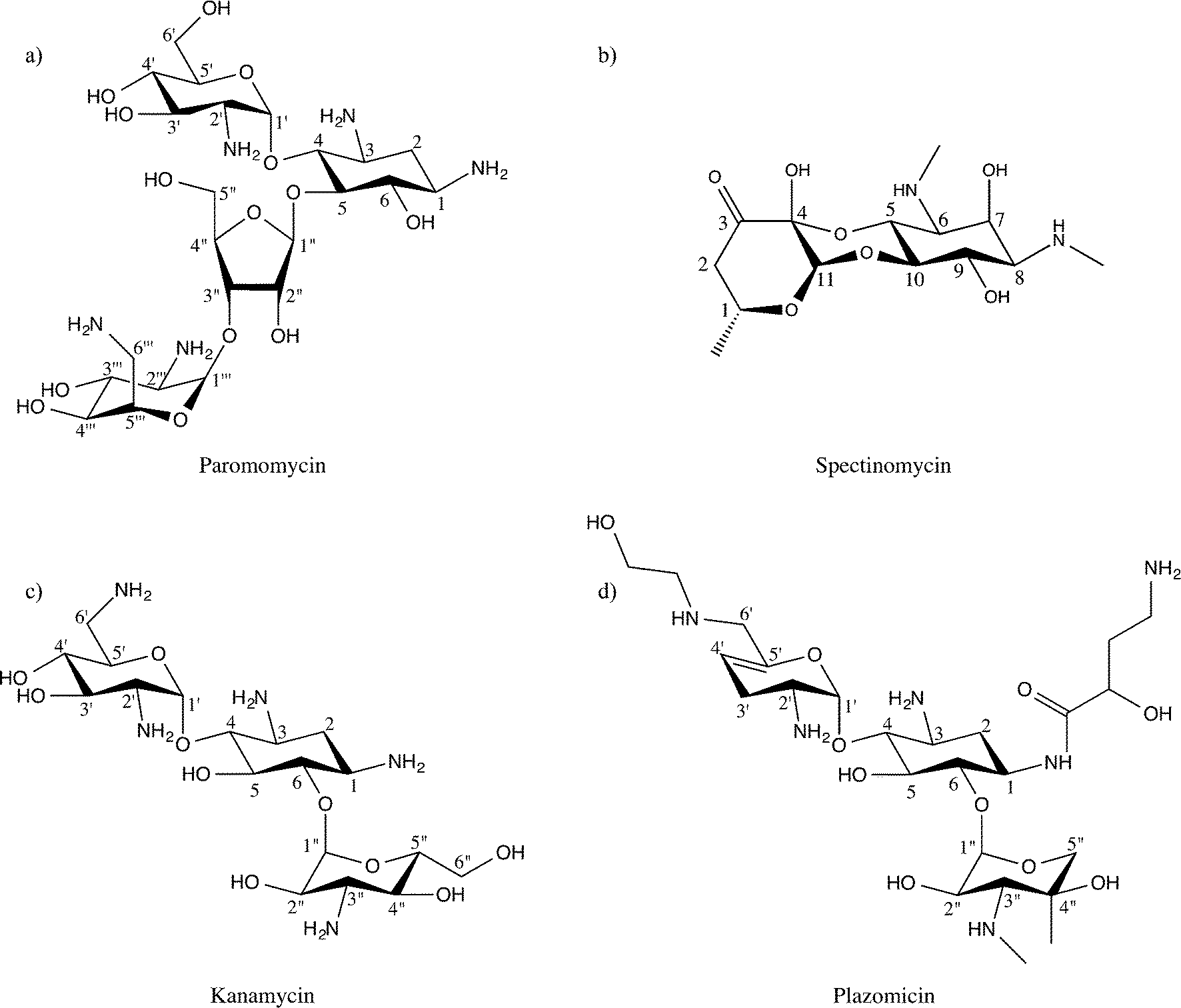 | ||
| Fig. 1 Chemical structures of representative aminoglycoside antibiotic classes. a) 4,5-Disubstituted aminoglycoside paromomycin. b) Non-deoxystreptamine aminoglycoside spectinomycin. c) 4,6-Disubstituted aminoglycoside kanamycin. d) Next-generation 4,6-disubstituted aminoglycoside plazomicin, currently in phase III trials. | ||
4,5-Disubstituted aminoglycosides remain very influential drugs to human health. Neomycin has common antiseptic topical uses, and is the active ingredient in commercially available Neosporin. In addition, lividomycin is utilized as a second-line antibiotic to treat Mycobacterium tuberculosis, Pseudomoas aeruginosa, and urinary tract infections.21–23 Likewise, ribostamycin was declared a critical antibiotic in human health by the World Health Organization, and is commonly employed against pelvic inflammatory diseases that can go on to cause life-long infertility,24,25 while paromomycin is effective against gastrointestinal and liver infections.
4,6-Disubstituted aminoglycosides also play pivotal roles in treating human disease. Tobramycin, amikacin, and gentamicin are all used in inhalable forms to clear chronic infections associated with cystic fibrosis.26,27 Furthermore, isepamicin and dibekacin, in combination with penicillin derivatives, treat potentially deadly, methicillin-resistant Staphylococcus aureus (MRSA) and P. aeruginosa skin, lung, and soft-tissue infections.28,29 Netilmicin is only used for gentamicin resistant infections because it must be injected,30 while sisomicin is largely employed to clear systemic urinary tract infections.31 Equally important, non-deoxystreptamine aminoglycosides, like streptomycin, are used in combination to clear serious M. tuberculosis infections, especially tuberculosis un-responsive to other antibiotics (drug-resistant TB).32 Also, spectinomycin is given to patients with Neisseria gonorrhoeae (gonorrhea), and particular those with penicillin allergies.
Aminoglycoside resistance
There are five major mechanisms microbes utilize to impart aminoglycoside resistance: (a) revision of the 30S ribosomal subunit target by mutation, (b) methylation of aminoglycoside binding targets, (c) decrease in intracellular aminoglycoside concentrations by active efflux pumps, (d) reduction of effective aminoglycoside concentration by decreased inner membrane transport or changes of outer membrane permeability, and (e) substrate deactivation by aminoglycoside modifying enzymes (AMEs).33–36 30S ribosomal mutations can occur in the A-site, which alters physiochemical base-stacking interactions to affect shape, dynamics, and electrostatic properties. The resulting changes disrupt aminoglycoside–RNA binding interactions necessary for tRNA recognition.37M. tuberculosis is a significant human health example of 30S ribosome mutational accumulations that evade aminoglycoside utility. M. tuberculosis directly mutates its 16S rRNA, as well as the ribosomal S12 protein, to achieve resistance. Interestingly, some variants can then go on to become aminoglycoside dependent, with the antibiotic actually lowering rates of translation error instead of the intended, opposite effect.38Ribosomal RNA modification by methyltransferases to circumvent aminoglycoside recognition also presents a dire dilemma.39,40 Most notably, New Delhi metallo-β-lactamase 1 (NDM1) drug-resistant microorganisms are a cause for alarm, as these bacteria are often resistant to several different classes of antibiotics.41–43 With respect to aminoglycoside susceptibility, the NDM1 gene has been observed to be associated with the gene that encodes RmtF 16S rRNA methyltransferase.42 This implies that bacteria possessing such a resistance plasmid, as has been observed in Klebsiella pneumoniae, are completely unresponsive to many clinically used β-lactam and all 4,6-disubstituted aminoglycoside antibiotics.41,44
Furthermore, efflux pumps play a significant role in aminoglycoside resistance amongst Salmonella and Escherichia coli clinical isolates.45,46E. coli such as pathogenic E. coli O157:H7,47 and opportunistic pathogen Acinetobacter baumannii, are just two examples of Gram-negative bacteria with a high rate of aminoglycoside resistance due to encoding aminoglycoside efflux pumps.46,48 In E. coli, AcrD is one such transporter, whose presence decreases bacterial aminoglycoside concentration to ineffective levels by an impressive factor of two to eight.46
Notwithstanding, anaerobic bacteria alter outer membrane permeability as well as inner membrane transport in response to environmental cues, and are inherently more difficult to target with aminoglycoside antibiotics.33E. coli and Clostridium perfringens are both major causes of food poisoning. When grown anaerobically, E. coli and C. perfringens become resistant to gentamicin and other aminoglycoside antibiotics due to decreased membrane potential. A quinone oxidation–reduction cycle is required for aminoglycoside trafficking, rendering the aminoglycoside class ineffectual for the majority of anaerobes as well as facultative anaerobes under such low oxygen conditions.49 However, the principal aminoglycoside resistance mechanism remains direct enzyme catalysis as carried out by aminoglycoside modifying enzymes (AMEs).36,50 For this reason, AMEs are of great medical and pharmacological interest, and will be discussed in further detail below.
Aminoglycoside modifying enzymes
Enzymatic aminoglycoside inactivation is carried out by acetylation, phosphorylation, or nucleotidylation. A class of enzymes expressed by pathogenic bacteria has the ability to carry out each mechanism: N-acetyltransferases (AACs), O-phosphotransferases (APHs), and O-nucleotidyltransferases (ANTs).51 Aminoglycoside N-acetyltransferases (AACs) are a major source of resistance in Gram-negative bacteria such as Enterobacteriacae, but are also found in Gram-positive organisms, such as Staphylococci, Enterococci, and Streptococci.52–54 AACs utilize acetylCoA as a donor to facilitate N-acetylation of aminoglycoside antibiotics. Aminoglycosides have many amine functional groups, and AACs have been found that modify the deoxystreptamine ring and 6-aminohexose ring through amino group acetylation.55–58Aminoglycoside O-phosphotransferases (APHs) are primarily found in Gram-positive bacteria such as S. aureus, complications of which can cause serious skin infection and toxic shock syndrome.59–61 Interestingly, APH presence is known to confer a high level of aminoglycoside resistance.27,62 APHs bind ATP or GTP as the phosphate donor. The APHs then transfer the γ-phosphate from the nucleotide triphosphate to a hydroxyl group of the aminoglycoside, succeeded by release of modified substrate and ADP or GDP.27,62,63
Aminoglycoside O-nucleotidyltransferases (ANTs) are a major source of aminoglycoside resistance found amongst Gram-negative clinical organisms, such as Enterobacteriaceae and Pseudomonas, which are prevalent pathogens in food poisoning and cystic fibrosis.52,64,65 The ANT class catalyzes a nucleotidyltransfer reaction, where ATP binds to the enzyme followed by the aminoglycoside substrate. ANTs nucleotidylate the aminoglycoside by direct nucleophilic attack of the aminoglycoside hydroxyl on the ATP molecule α-phosphate.66 This reaction covalently links AMP to the now modified-aminoglycoside, releasing pyrophosphate, and the modified aminoglycoside no longer efficiently impedes bacterial translation. Acetylation, phosphorylation, or nucleotidylation all decrease modified-aminoglycoside recognition of the 30S ribosome by 10–15 fold, such that bacterial translational error never approaches toxic levels.52,67–70
Aminoglycoside drug-target network
The number of enzymes identified that are able to confer resistance to aminoglycoside antibiotics through acetylation, phosphorylation, or nucleotidylation have been steadily growing. Currently, nearly a hundred different bacterial enzymes have been described, most identified in clinical isolates. Anticipating the deluge, Shaw in 1993, systematized the nomenclature of these enzymes, in which the enzyme name reflects its modification activity, the site of modification on the antibiotic, and the antibiotic substrate spectrum.51 If multiple enzymes were found with the identical activity, subscripts were additionally used as unique identifier. Nonetheless, the plethora of enzymes and the diversity of aminoglycosides they provide resistance to have become sufficiently complex to present and describe aminoglycoside resistance in the format of a drug-target network.Previously, drug-target networks have been taken advantage of to present a novel perspective that enables the identification of additional protein targets, new drug ligands, and guide experimental drug synthesis.71 Characterization of these networks has been applied to treating endocrine, hematological, cardiovascular, and psychiatric gene diseases.72 As such, understanding enzymes and the drugs they target in association with vast protein networks aids prospective and increasingly rational drug/inhibitor discovery and design.72
Recasting the description of enzyme mediated aminoglycoside resistance as a drug-target network highlights the complexity facing those that pursue avenues to combat antibiotic resistance. As shown in Fig. 2, aminoglycoside antibiotics are able to interact with the ribosome, thereby precipitating a cascade of events that ultimately results in a bactericidal outcome, or alternatively can interact with a large number of different resistance enzymes that will detoxify the drug. Simplistically, this illustration emphasizes that overcoming resistance implies blocking or avoiding interactions with a large number of AMEs. To illustrate the scope, half of the aminoglycoside antibiotics can be rendered useless by more than 40 different enzymes (Table 1). Most impressively, kanamycin can be rendered ineffective by no less than 64 different AMEs, and the clinically used tobramycin and gentamicin antibiotics are susceptible to detoxification by more than 50 AMEs. While it is understood that the prevalence of different AMEs varies, and some of these are only associated with a select subset of bacterial pathogens, nonetheless, the numbers indicate that the chance of encountering AME mediated resistance for kanamycin is higher than, for example, isepamicin or paromomycin.
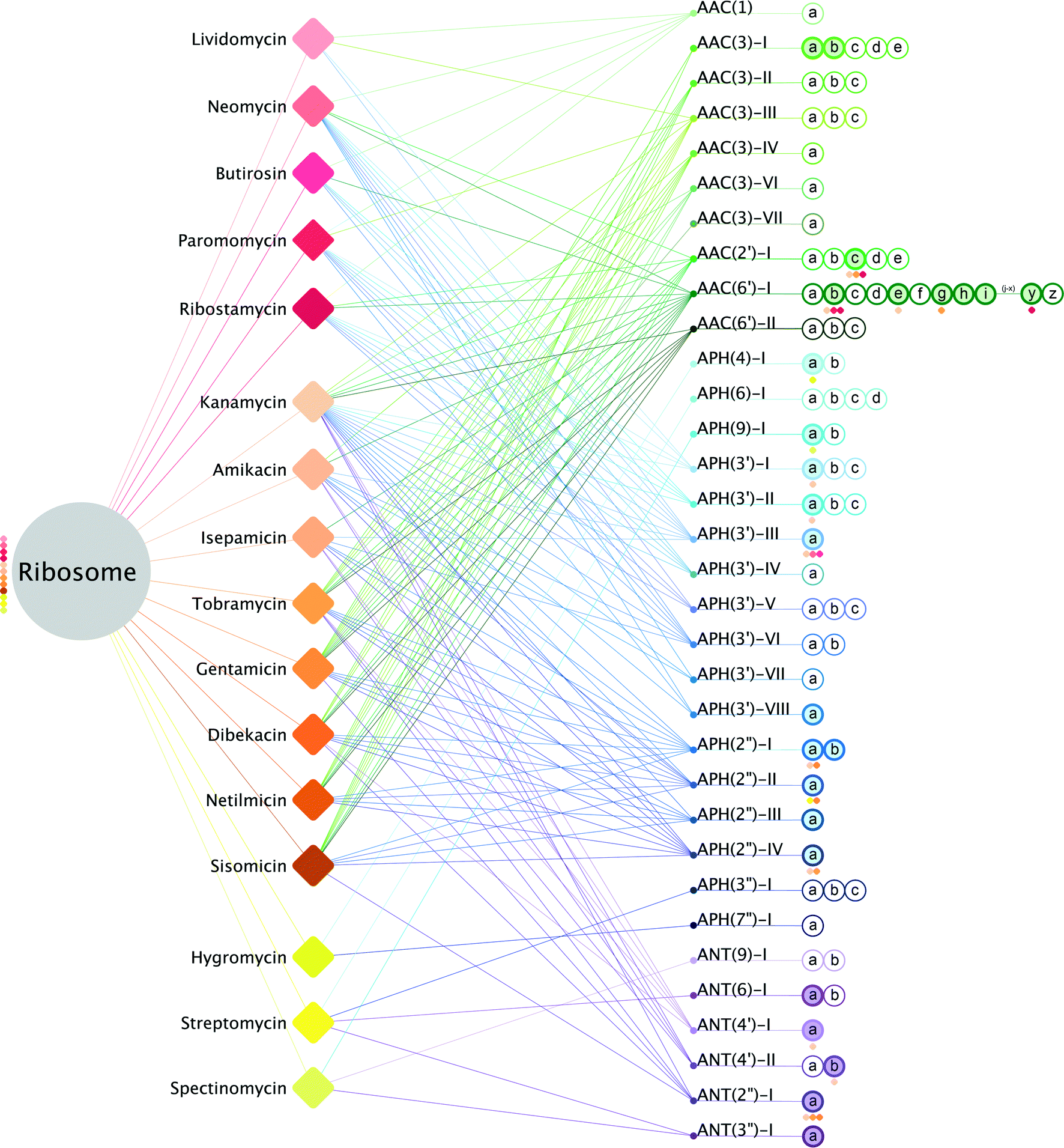 | ||
| Fig. 2 Aminoglycoside drug-target network. Drug-target network of known aminoglycosides with modifying enzymes. The bacterial ribosome is the primary node (grey). Aminoglycosides are represented as diamonds and are separated into their three classes: 4,5-disubstituted aminoglycosides (red), 4,6-disubstituted aminoglycosides (orange) and non-deoxystreptamine aminoglycosides (yellow). Aminoglycoside interactions with the ribosome are depicted as lines colored according to their corresponding diamond. AME nodes are colored by enzyme class: AAC's (green), APH's (blue) and ANT's (purple). Lines colored according to their corresponding modifying enzyme represent interactions of AMEs with their respective aminoglycosides. Enzymes are listed in numerical order and according to their gene nomenclature (ex: a, b, c) as hollow nodes. Solid colored nodes represent enzymes with a currently known structure; structures with bound aminoglycosides have diamonds beneath with corresponding aminoglycoside color.36,51,102,103 | ||
| Antibiotic | Position of modification by: | Num. AACs | Num. APHs | Num. ANTs | Total AMEs | ||
|---|---|---|---|---|---|---|---|
| AACs | APHs | ANTs | |||||
| Lividomycin | 1, 3 | 3′ | — | 4 | 4 | 0 | 8 |
| Neomycin | 1, 2′, 6′ | 3′ | — | 32 | 15 | 0 | 47 |
| Butirosin | 1, 6′ | 3′ | — | 27 | 15 | 0 | 42 |
| Paromomycin | 1, 3 | 3′ | — | 15 | 15 | 0 | 30 |
| Ribostamycin | 1, 2′, 6′ | 3′ | — | 32 | 13 | 0 | 45 |
| Kanamycin | 3, 2′, 6′ | 3′, 2′′ | 4′, 2′′ | 40 | 20 | 4 | 64 |
| Amikacin | 3, 6′ | 3′, 2′′ | 4′ | 27 | 8 | 3 | 38 |
| Isepamicin | 6′ | 3′, 2′′ | 4′ | 26 | 7 | 3 | 36 |
| Tobramycin | 3, 2′, 6′ | 2′′ | 4′, 2′′ | 48 | 5 | 4 | 57 |
| Gentamicin | 3, 2′, 6′ | 2′′ | 2′′ | 48 | 5 | 1 | 54 |
| Dibekacin | 3, 2′, 6′ | 3′, 2′′ | 4′, 2′′ | 41 | 5 | 2 | 48 |
| Netilmicin | 3, 2′, 6′ | 2′′ | — | 42 | 5 | 0 | 47 |
| Sisomicin | 3, 2′, 6′ | 2′′ | 2′′ | 48 | 5 | 1 | 54 |
| Hygromycin | — | 4, 7′′ | — | 0 | 3 | 0 | 3 |
| Streptomycin | — | 6, 3′′ | 6, 3′′ | 0 | 7 | 3 | 10 |
| Spectinomycin | — | 9 | 9, 3′′ | 0 | 2 | 3 | 5 |
While the aminoglycoside drug-target network emphasizes and highlights the complexity of AME mediated resistance, it also can enable the prioritization of avenues for addressing this phenomenon. Specifically, if additional data is incorporated into the network diagram, it could suggest a hierarchy of strategies to pursue. Most obviously, integrating prevalence of distinct resistance enzymes encountered within a specific clinical setting could readily suggest optimal aminoglycoside antibiotics for treatment. For example, AAC(6')-Ie and APH(2′′)-Ia are always found together as a single bi-functional polypeptide; furthermore, in clinical isolates this bi-functional AME is occasionally found together with APH(3')-IIIa.73–75 When these three AMEs co-exist, treatment by aminoglycoside antibiotics is essentially made impossible, as all 4,5- and 4,6-disubstituted aminoglycosides are substrates for at least one of these three enzymes. But, if APH(3')-IIIa is not found in those clinical isolates, then treatment with the 4,5-disubstituted aminoglycosides paromomycin or lividomycin is viable. Unfortunately, within clinical settings, such detailed data on AME prevalence is nearly always incomplete and highly fluid as observed resistance mechanisms closely track antibiotic usage.76,77 To advance new chemotherapeutic strategies for addressing AME mediated antibiotic resistance, we have instead linked the available structural data for aminoglycoside drug-target interactions to the network.
Since the first crystal structure of an AME by Holden and co-workers, more than 20 years ago,78 our knowledge on these enzymes has increased, and currently we have structural data for 21 different enzymes. Furthermore, for nearly all of these enzyme structures, the data also includes detailed information on drug target interactions, and in several cases for multiple drug interactions (Fig. 2 and ESI‡Table 1). Unfortunately, the available structural data do not provide a comprehensive coverage of the entire network, and significant and substantial gaps remain to be filled. Nonetheless, sufficient structural information is now accessible to begin exploiting these for advancing new chemotherapeutic avenues. Specifically, we explore two separate, but not mutually exclusive, avenues for combating emerging aminoglycoside antibiotic resistance: (a) adjuvants that block aminoglycoside interactions with unwanted targets, including AME inhibitors, or (b) novel analogues in the form of next-generation aminoglycosides.
Structural drug design – I: AME inhibitors
The aminoglycoside drug-target network would suggest that to block aminoglycoside resistance by inhibiting AMEs, nearly a hundred different inhibitors would be required to ensure complete coverage. This is therapeutically not a viable strategy. Fortunately, that may not be required. First, the choice of aminoglycoside with which an inhibitor is co-administrated as an adjuvant may substantially reduce the AMEs to be targeted. Secondly, it is to be expected that several of the AMEs to be inhibited will share considerable structural similarity, such that a single inhibitor may target multiple AMEs. For example, while paromomycin can be rendered ineffective by 15 different APHs (Table 1), one can reason that, given the extensive structural similarity observed between several of the different APH(3') enzymes, a single inhibitor might be able to accomplish this task. Thirdly, not all of the AMEs that can potentially confer resistance will likely be present in a specific clinical setting, further reducing the number of adjuvants that may need to be co-administered. Guided by these considerations, efforts have been pursued to arrive at pan-AAC, APH and ANT inhibitors.Applying structure-based inhibitor/adjuvant design to AMEs has already proven useful in the past. For example, AACs share structural and functional homology with the GCN5-related histone acetyltransferase (HAT) superfamily, despite poor sequence identity.79 Compellingly, histone acetyltransferase inhibitors have gone into clinical trial that treat cancers, such as neuroblastomas.80,81 Therefore, these inhibitors present a promising avenue for adjuvant development to combat antibiotic resistance conferred by the AACs, though ensuring absence of cross-reactivity with host enzymes is a concern. Even more striking, APHs share homology with eukaryotic protein kinases, many of whom have large libraries of well-characterized, anti-cancer inhibitors. Interestingly, some of these protein kinase inhibitors have already demonstrated the ability to impede APH(3′)-III activity.82–84 The knowledge could help tailor inhibitors against the multiple APHs. Moreover, the ANT class also belongs to the much larger DNA polymerase β – like nucleotidyltransferase superfamily (polβ superfamily), which includes enzymes involved in DNA replication and repair.85 Previously reported DNA polymerase β inhibitors are another promising avenue that could also help guide ANT adjuvant and inhibitor designs.86,87
Structural drug design – II: next-generation aminoglycosides
Exploiting the available structural data on AMEs for next-generation aminoglycoside development is particularly attractive given that, thanks to the Nobel Prize awarded work by Steitz and colleagues, detailed information is also available for how aminoglycoside antibiotics interact with their intended target, the bacterial ribosome.88,89It must be acknowledged that even prior to having acquired any structural insights into drug-target interactions, drug developers actually exploited inferred binding topologies for aminoglycosides and AMEs to arrive at next-generation antibiotics. Guided by observed AME interactions with butirosin, a next-generation derivative of kanamycin, amikacin, was developed that incorporated the (S)-4-amino-2-hydroxybutyrate group on the N1-position. This single modification substantially altered the susceptibility of the antibiotic to numerous AMEs. While 64 different enzymes can deactivate kanamycin, the additional N1-substitution limits this to 38 enzymes for amikacin, i.e. a reduction of nearly 40%.
With the three-dimensional structural data that has become available, we can now dissect and analyze the structural basis for the significantly decreased susceptibility to AME mediated resistance for amikacin vs. kanamycin (see Fig. 3). Examination of the structural data for kanamycin in complex with its various partners reveals that the actual conformation of the antibiotic does not significantly change.60,90–93 The rational given for this observation has been that the biosynthesis of aminoglycosides has evolved to result in bioactive compounds that in their lowest energy conformation are potent binders to the ribosome. For AMEs to effectively compete with the ribosome they have also evolved to bind to this identical lowest energy conformation.60 Also, the hydrogen bonding interactions the drug makes with different targets shows extensive similarities, which is not surprising, as similar to the ribosome, AMEs aim to satisfy all hydrogen bond opportunities presented by the antibiotic so as to enhance binding affinity. However, what is significantly different is the van der Waals interactions made, i.e. the walls of the aminoglycoside-binding pocket in the various targets are located in different places. This observation implies that bulky substitutions, such as the (S)-4-amino-2-hydroxybutyrate group modification on the N1-position, will differentially impact different target interactions. Hence, 40% of the enzymes that can deactivate kanamycin are unable to provide resistance to amikacin.
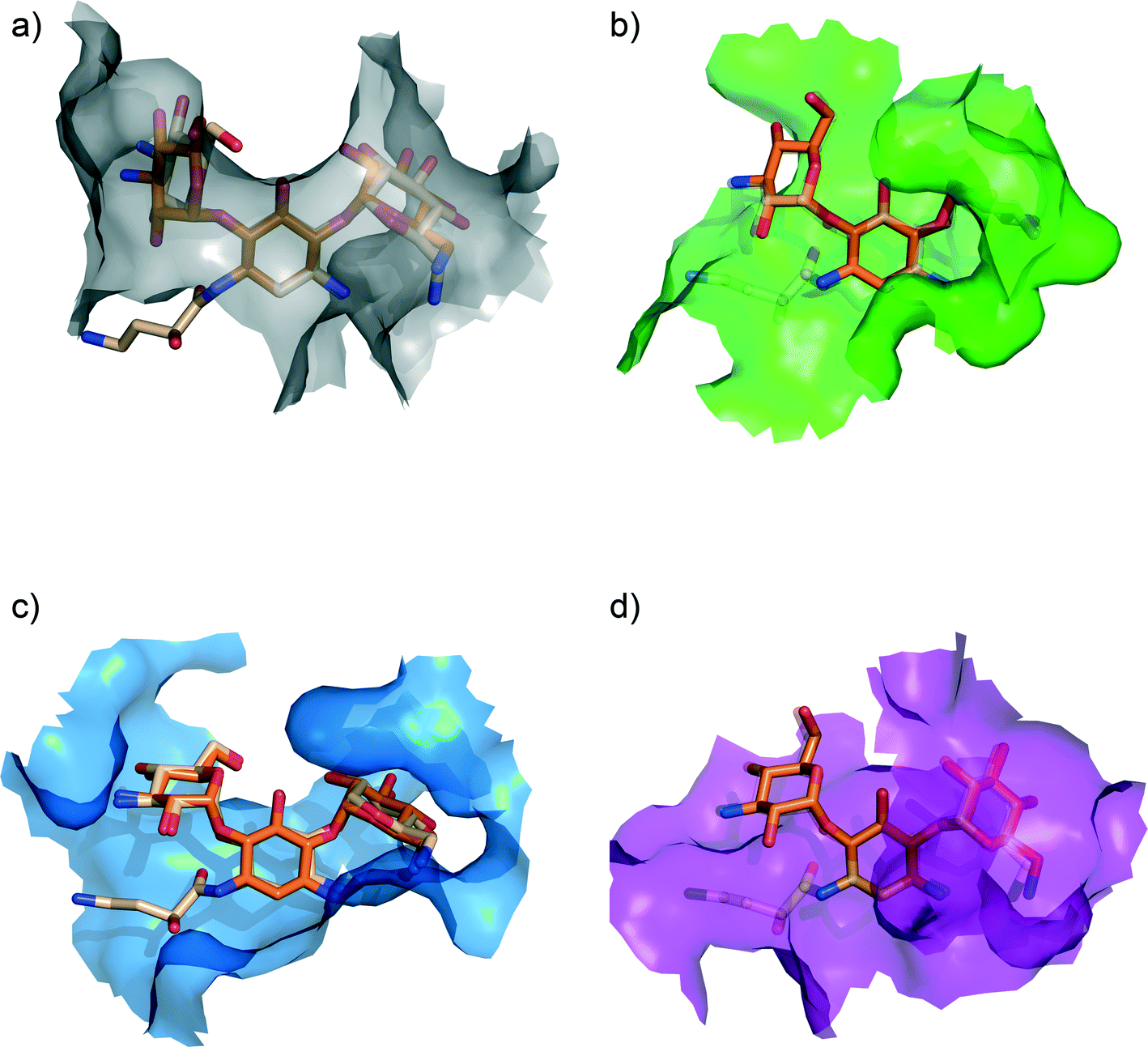 | ||
| Fig. 3 Ribosome bound kanamycin and amikacin versus AMEs. van der Waals surface of the (a) bacterial ribosome A-site (grey) [PDB ID: 2ESI], (b) AAC(2′)-Ic (green) [PDB ID: 1M4I], (c) APH(3′)-IIIa (blue) [PDB ID: 1L8T], and (d) ANT(2′′)-Ia (purple) [PDB ID: 4WQL] bound to kanamycin (orange). Amikacin (light orange) bound to the bacterial ribosome A-site [PDB ID: 4P20] and modeled into the active sites of aminoglycoside modifying enzymes (by adding the (S)-4-amino-2-hydroxybutyrate group on the N1-position of kanamycin).104 Amikacin is depicted as semi-transparent in the active sites of AAC(2′)-Ic and ANT(2′′)-Ia as binding results in steric clashes, rationalizing its inability to interact with those particular enzymes. | ||
Building on this identical paradigm, the latest aminoglycoside that is presently entering phase III clinical trials, plazomicin, is derived from the sisomicin, with an identical modification at the N1 position as amikacin, and a second modification on the N6′ position (see Fig. 1). This new antibiotic has already proven effective against methicillin-resistant S. aureus and other multi-drug resistant, pathogenic, Gram-negative bacteria.94–98 It is predicted that AME mediated resistance for plazomicin will be limited to select enzymes capable of modifying amino moieties at the 2′ position.99 However, when this antibiotic becomes widely available, it is perhaps inevitable that additional enzymes will make their way from the “resistome” to pathogenic bacteria.100,101 Nonetheless, the strategy is effective and allows the much needed expansion of the available armament of antibiotics. Moreover, amikacin and plazomicin, in the context of the aminoglycoside drug-target network and the available structural data, suggests new avenues for next-generation aminoglycoside development. For example, based on the current predominance of 4,6-disubstituted aminoglycoside AME interactions, 4,5-disubstituted aminoglycosides should be considered an attractive scaffold for future antibiotic development (see Table 1). The suitable extensions on this scaffold can be informed by current structural information, though an expansion of this knowledge is needed. Also, the non-deoxystreptamine aminoglycoside appear to represent an underexplored area.
Conclusions
The aminoglycoside resistance drug-target network illustrates the complexity of AME mediated resistance. It also provides a platform to aid in prioritizing different strategies to overcome and combat resistance to these antibiotics. Specifically incorporating the expanding insights on the structural details of aminoglycoside-target interactions will facilitate the development of AME inhibitors that could be exploited as adjuvants, and the design of next-generation aminoglycoside antibiotics. It is extremely encouraging that much progress has been made in the development of next-generation aminoglycoside antibiotics with reduced affinity for AMEs. Exploiting structural details on AME-aminoglycoside interactions provides opportunities to further expand the available armament of aminoglycoside antibiotics with dramatically reduced susceptibility to clinically relevant resistance mechanisms.Abbreviations
| AAC | Aminoglycoside N-acetyltransferase |
| AMEs | Aminoglycoside modifying enzymes |
| ANT | Aminoglycoside O-nucleotidyltransferase |
| APH | Aminoglycoside O-phosphotransferase |
Acknowledgements
We would like to thank past and present members of the Berghuis lab for discussions and helpful suggestions. Some of the research presented was made possible by a grant from the Canadian Institutes of Health Research (CIHR MOP-114889) awarded to A. M. B. V. R. B.-D. and A. V. B acknowledge fellowship and scholarship support from the CIHR Strategic Initiative in Chemical Biology, and PROTEO. A. M. B. holds a Canada Research Chair in Structural Biology.References
- D. R. Riley, K. B. Sieber, K. M. Robinson, J. R. White, A. Ganesan and S. Nourbakhsh, et al., Bacteria-human somatic cell lateral gene transfer is enriched in cancer samples, PLoS Comput. Biol., 2013, 9(6), e1003107 CAS.
- J. T. Stock, Are human still evolving? Technological advances and unique biological characteristics allow us to adapt to environmental stress. Has this stopped genetic evolution?, EMBO Rep. Georg von Holtzbrinck Publishing Group, 2008, vol. 9(Suppl 1), pp. S52–S54 Search PubMed.
- E. V. Hulse, Leprosy and ancient Egypt, Lancet, 1972, 2(7785), 1024–1025 CrossRef CAS.
- M. Schultz, Paleohistopathology of bone: a new approach to the study of ancient diseases, Am. J. Phys. Anthropol., 2001,(Suppl 33), 106–147 CrossRef CAS PubMed.
- S. J. Olshansky, B. Cernes, R. G. Rogers and L. Smith, Infectious disases--new and ancient threats to world health, Popul Bull., 1997, 52(2), 1–52 CAS.
- G. M. Baer, Rabies--an historical perspective, Infect. Agents Dis., 1994, 3(4), 168–180 CAS.
- Boundless. History of Bacterial Diseases [Internet]. Boundless Biology. 2015 [cited 2015 Aug 30]. Available from: https://www.boundless.com/biology/textbooks/boundless-biology-textbook/prokaryotes-bacteria-and-archaea-22/bacterial-diseases-in-humans-143/history-of-bacterial-diseases-566-11779/.
- B. Spellberg, R. Guidos, D. Gilbert, J. Bradley, H. W. Boucher and W. Michael Scheld, et al., The epidemic of antibiotic-resistance infections: a call to action for the medical community from the infectious diseases society of america, Clin. Infect. Dis., 2008, 46(2), 155–164 CrossRef PubMed.
- M. Bonafede, LB Rice LB. Emerging antibiotic resistance, J. Lab. Clin. Med., 1997, 130(6), 558–566 CrossRef CAS PubMed.
- A. Demirjian, G. V. Sanchez, J. A. Finkelstein, S. M. Ling, L. A. Pollack and L. A. Hicks, et al., CDC grand rounds: getting smart about antibiotics, MMWR Morb. Mortal. Wkly. Rep., 2015, 64(32), 871–873 CrossRef PubMed.
- J. O'Neill, Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. [Internet]. London, UK; 2014. Available from: http://amr-review.org/sites/default/files/AMR Review Paper - Tackling a crisis for the health and wealth of nations_1.pdf.
- H. W. Boucher, G. H. Talbot, D. K. J. Benjamin, J. Bradley, R. J. Guidos and R. N. Jones, et al., 10 × '20 Progress--development of new drugs active against Gram-negative bacilli: an update from the Infectious Diseases Society of America, Clin. Infect. Dis., 2013, 56(12), 1685–1694 CrossRef PubMed.
- Organization WH, Antimicrobial resistance: global report on surveillance [Internet]. 2014. Available from: http://www.who.int/drugresistance/documents/surveillancereport/en.
- A. R. M. Coates, G. Halls and Y. Hu, Novel classes of antibiotics or more of the same?, Br. J. Pharmacol., 2011, 162(1), 184–194 CrossRef PubMed.
- R. W. Finberg, R. C. Moellering, F. P. Tally, W. A. Craig, G. A. Pankey and E. P. Dellinger, et al., The importance of bactericidal drugs: future directions in infectious disease, Clin. Infect. Dis., 2004, 39(9), 1314–1320 CrossRef CAS PubMed.
- J. Davies, L. Gorini and B. D. Davis, Misreading of RNA codewords induced by aminoglycoside antibiotics, Mol. Pharmacol., 1965, 1, 93–106 CAS.
- M. A. Borovinskaya, R. D. Pai, W. Zhang, B. S. Schuwirth, J. M. Holton and G. Hirokawa, et al., Structural basis for aminoglycoside inhibition of bacterial ribosome recycling, Nat. Struct. Mol. Biol., 2007, 14(8), 727–732 CAS.
- D. S. Pilch, M. Kaul, C. M. Barbieri and J. E. Kerrigan, Thermodynamics of aminoglycoside r-RNA recognition, Biopolymers, 2003, 70(1), 58–79 CrossRef CAS PubMed.
- M. Chittapragada, S. Roberts and Y. W. Ham, Aminoglycosides: molecular insights on the recognition of RNA and aminoglycoside mimics, Perspect. Med. Chem., 2009, 3, 21–37 CAS.
- M.-P. Mingeot-Leclercq, Y. Glupczynski and P. M. Tulkens, Aminoglycosides: activity and resistance, Antimicrob. Agents Chemother., 1999, 43(4), 727–737 CAS.
- M. Tsukamura, A preliminary trial of lividomycin in patients with pulmonary tuberculosis, Tubercle, 1972, 53(1), 43–46 CrossRef CAS PubMed.
- F. Kobayashi, M. Yamaguchi and S. Mitsuhashi, Activity of Lividomycin Against Pseudomonas aeruginosa: Its Inactivation by Phosphorylation Induced by Resistant Strains, Antimicrob. Agents Chemother., 1972, 1, 17–21 CrossRef CAS PubMed.
- A. Leroy, G. Humbert, G. Oksenhendler and J. P. Fillastre, Comparative pharmacokinetic of lividomycin, amikacin and sisomicin in normal subjects and in uraemic patients, J. Antimicrob. Chemother., 1976, 2, 373 CrossRef CAS PubMed.
- Organization WH, Critically important antimicrobials for human medicine, (3rd revision), 2011 Search PubMed.
- N. P. Kurumbang, K. Liou and J. K. Shong, Biosynthesis of Ribostamycin Derivatives by Reconstitution and Heterologous Expression of Required Gene Sets, Appl. Biochem. Biotechnol., 2011, 163, 373–382 CrossRef CAS PubMed.
- A. Prayle and A. R. Smyth, Aminoglycoside use in cystic fibrosis: therapeutic strategies and toxicity, Curr. Opin. Pulm. Med., 2010, 16, 604–610 CrossRef CAS PubMed.
- K. Shi, S. J. Caldwell, D. H. Fong and A. M. Berghuis, Prospects for circumventing aminoglycoside kinase mediated antibiotic resistance, Front. Cell. Infect. Microbiol., 2013, 3, 22, DOI:10.3389/fcimb.2013.00022.
- K. Deguchi, M. Kogushi, Y. Suzuki, S. Tanaka, S. Fukayama and R. Ishihara, et al., Antibacterial activities of combination uses of isepamicin and beta-lactams in vitro against clinically isolated strains. Part 1. Activities against Staphylococcus aureus, Jpn. J. Antibiot., 1996, 49(2), 194–202 CrossRef CAS PubMed.
- S. Aonuma, F. Ariji, K. Oizumi and K. Konno, Electron microscopy of Pseudomoas aeruginosa treated with sulbenicillin and dibekacin, Tohoku J. Exp. Med., 1987, 152(2), 119–128 CrossRef CAS PubMed.
- A. Santé, française de sécurité sanitaire des produits de. Update on good use of injectable aminoglycosides, gentamycin, tobramycin, netilmicin, amikacin. Pharmacological properties, indications, dosage, and mode of administration, treatment monitoring, Med. Mal. Infect, 2012, 42(7), 301–308 CrossRef PubMed.
- T. A. Rodriquez, Sisomicin sulfate: its efficacy in the treatment of urologic infections, Arch. Esp. Urol., 1979, 32(4), 387–396 Search PubMed.
- A. Zumla, P. Nahid and S. T. Cole, Advances in the development of new tuberculosis drugs and treatment regimens, Nat. Rev. Drug Discovery, 2013, 12, 388–404 CrossRef CAS PubMed.
- S. Shakil, R. Khan, R. Zarrilli and A. U. Khan, Aminoglycosides versus bacteria--a description of the action, resistance mechanism, and nosocomial battleground, J. Biomed. Sci., 2008, 15(1), 5–14 CrossRef CAS PubMed.
- S. Jana and J. K. Deb, Molecular understanding of aminoglycoside action and resistance, Appl. Microbiol. Biotechnol., 2006, 70(2), 140–150 CrossRef CAS PubMed.
- H. I. Zgurskaya, J. W. Weeks, A. T. Ntreh, L. M. Nickels and D. Wolloscheck, Mechanism of coupling drug transport reactions located in two different membranes, Front Microbiol., 2015, 6, 100 Search PubMed.
- M. S. Ramirez and M. E. Tolmasky, Aminoglycoside modifying enzymes, Drug Resist. Updates, 2010, 13, 151–171 CrossRef CAS PubMed.
- J. Romanowska, J. A. McCammon and J. Rylska, Understanding the origins of bacterial resistance to aminoglycosides through molecular dynamics mutational study of the ribosomal A-site, PLoS Comput. Biol., 2011, 7(7), e1002099 CAS.
- D. L. Mayers, Antimicrobial Drug Resistance: Mechanisms of Drug Resistance, 1st ed. ed. D. L. Mayers, S. A. Lerner, M. Ouellette and J. D. Sobel, Humana Press, Springer Science+Business Media, LLC, New York, NY, 2009, pp. 89–91 Search PubMed.
- G. F. Liou, S. Yoshizama, P. Courvalin and M. Galimand, Aminoglycoside resistance by ArmA-mediated ribosomal 16S methylation in human bacterial pathogens, J. Mol. Biol., 2006, 359(2), 358–364 CrossRef CAS PubMed.
- J. A. Dunkle, K. Vinal, P. M. Desai, N. Zelinskaya, M. Savic and D. M. West, et al., Molecular recognition and modification of the 30S ribosome by the aminoglycoside-resistance methyltransferase NpmA, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(17), 6275–6280 CrossRef CAS PubMed.
- L. Hidalgo, K. L. Hopkins, B. Gutierrez, C. M. Ovejero, S. Shukla and S. Douthwaite, et al., Association of the novel aminoglycoside resistance determinant RmtF with NDM carbapenemase in Enterobacteriaceae isolated in India and the UK, J. Antimicrob. Chemother., 2013, 68(7), 1543–1550 CrossRef CAS PubMed.
- C. S. Lee, S. Vasoo, F. Hu, R. Patel and Y. Doi, Klebsiella pneumoniae ST147 Coproducing NDM-7 Carbapenemase and RmtF 16S rRNA Methyltransferase in Minnesota, J. Clin. Microbiol., 2014, 52(11), 4109–4110 CrossRef CAS PubMed.
- R. J. Fair and Y. Tor, Antibiotics and Bacterial Resistance in the 21st Century, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed.
- M. Galimand, P. Courvalin and T. Lambert, RmtF, a new member of the aminoglycoside resistance 16S rRNA N7 G1405 methyltransferase family, Antimicrob. Agents Chemother., 2012, 56(7), 3960–3962 CrossRef CAS PubMed.
- Y. Samadi, I. Pakzad, A. Monadi Sefidan, H. Hosainzadegan and A. Tanomand, Study of aminoglycoside resistance genes in enterococcus and salmonella strains isolated from ilam and milad hospitals, iran. Jundishapur, J. Microbiol, 2015, 8(4), e18102 Search PubMed.
- E. Y. Rosenberg, D. Ma and H. Nikaido, AcrD of Escherichia coli is an aminoglycoside efflux pump, J. Bacteriol., 2000, 182(6), 1754–1756 CrossRef CAS PubMed.
- Secret Agents: The Menace of Emerging Infections. Revised ed. Books P, ed. M. Drexler, New York, NY, 2009 Search PubMed.
- S. Magnet, P. Courvalin and T. Lambert, Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454, Antimicrob. Agents Chemother., 2001, 45(12), 3375–3380 CrossRef CAS PubMed.
- L. E. Bryan and S. Kwan, Mechanisms of aminoglycoside resistance of anaerobic bacteria and facultative bacteria grown anaerobically, J. Antimicrob. Chemother., 1981, 8(Suppl 500), 1–8 CrossRef CAS PubMed.
- F. Vaziri, S. N. Peerayeh, Q. B. Nejad and A. Farhadian, The prevalence of aminoglycoside-modifying enzyme genes (aac (6′)-I, aac (6′)-II, ant (2′′)-I, aph (3′)-VI) in Pseudomonas aeruginosa, Clinics, 2011, 66(9), 1519–1522 Search PubMed.
- K. J. Shaw, P. N. Rather, R. S. Hare and G. H. Miller, Molecular genetic of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes, Microbiol. Rev., 1993, 57, 138–163 CAS.
- G. H. Miller, F. J. Sabatelli, R. S. Hare, Y. Glupczynski, P. Mackey and D. Shlaes, The most frequent aminoglycoside resistance mechanisms-changes with time and geographic area: a reflection of aminoglycoside usage patterns?, Clin. Infect. Dis., 1997, 24(Suppl 1), S46–62 CrossRef CAS PubMed.
- E. Wolf, A. Vassilev, Y. Makino, A. Sali, Y. Nakatani and S. K. Burley, Crystal structuer of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase, Cell, 1998, 94, 439–449 CrossRef CAS PubMed.
- J. J. Ferretti, K. S. Gilmore and P. Courvalin, Nucleotide sequence analysis of the gene specifying the bifunctional 6′-aminoglycoside acetyltransferase 2′′-aminoglycoside phosphotransferase enzyme in Streptococcus faecalis and identification and cloning of gene regions specifying the two activities, J. Bacteriol., 1986, 167, 631–638 CAS.
- R. Benveniste and J. Davies, Mechanism of antibiotic resistance in bacteria, Annu. Rev. Biochem., 1973, 42, 471–506 CrossRef CAS PubMed.
- T. J. Foster, Plasmid-determined resistance to antimicrobial drugs and toxic metal ions in bacteria, Microbiol. Rev., 1983, 47, 361–409 CAS.
- A. M. Lovering, L. O. White and D. S. Reevew, AAC(1): a new aminoglycoside-acetylating enzyme modifying the C1 aminogroup of apramycin, J. Antimicrob. Chemother., 1987, 20, 803–813 CrossRef CAS PubMed.
- S. Mitsuhashi, L. Rosival and V. Krcmery, Drug inactivating enzymes and antibiotic resistance, Springer-Verlag KG, Berlin, 1975, pp. 115–119 Search PubMed.
- G. S. Gray and W. M. Fitch, Evolution of antibiotic resistance genes: the DNA sequence of a kanamycin resistance gene from Staphylococcus aureus, Mol. Biol. Evol., 1983, 1(1), 57–66 CAS.
- D. H. Fong and A. M. Berghuis, Substrate promiscuiting of an aminoglycoside antibiotic resistance enzyme via target mimicry, EMBO J., 2002, 21, 2323–2331 CrossRef CAS PubMed.
- P. R. Thompson, D. D. Boehr, A. M. Berghuis and G. D. Wright, Mechanism of Aminoglycoside Antibiotic Kinase APH(3′)-IIIa: Role of the Nucleotide Positioning Loop, Biochemistry, 2002, 41(22), 7001–7007 CrossRef CAS PubMed.
- K. Shi and A. M. Berghuis, Structural basis for dual nucleotide selectivity of aminoglycoside 2′′-phosphotransferase IVa provides insight on determinants of nucleotide specificity of aminoglycoside kinases, J. Biol. Chem., 2012, 287(16), 13094–13102 CrossRef CAS PubMed.
- A. van Broekhoven, F. Shapiro and J. Anné, Novel frontiers in the production of compounds for biomedical use, ed. M. Hofman and J. Anné, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1st edn, 2001, pp. 87–96 Search PubMed.
- R. G. Wax, K. Lewis, A. A. Salyers and H. Taber, Bacterial resistance to antimicrobials, ed. R. G. Wax, K. Lewis, A. A. Salyers and H. Taber, CRC Press, Boca Raton, 1st edn, 2001, pp. 106–114 Search PubMed.
- J. C. Davies, Pseudomonas aeruginosa in cystic fibrosis: pathogenesis and persistence, Paediatr. Respir. Rev., 2002, 3(2), 128–134 CrossRef PubMed.
- J. E. Van Pelt, R. Iyengar and P. A. Frey, Gentamicin nucleotidyltransferase. Stereochemical inversion at phosphorus in enzymatic 2′-deoxyadenylyl transfer to tobramycin, J. Biol. Chem., 1986, 261, 15995–15999 CAS.
- S. Salian, T. Matt, R. Akbergenov, S. Harish, M. Meyer and S. Duscha, et al., Structure-Activity relationships among the kanamycin aminoglycosides: role of ring I hydroxyl and amino groups, Antimicrob. Agents Chemother., 2012, 56(12), 6104–6108 CrossRef CAS PubMed.
- M. D. Disney, Studying modification of aminoglycoside antibiotics by resistance-causing enzymes vis microarray, Methods Mol. Biol., 2012, 808, 303–320 CAS.
- G. A. McKay, P. R. Thompson and G. D. Wright, Broad spectrum aminoglycoside phosphotransferase type III from Enterococcus: overexpression, purification, and substrate specificity, Biochemistry, 1994, 33, 6936–6944 CrossRef CAS PubMed.
- P. R. Thompson, D. W. Hughes and G. D. Wright, Regiospecificity of aminoglycoside phosphotransferase from Enterococci and Staphylococci (APH(3′)-IIIa), Biochemistry, 1996, 35, 8686–8695 CrossRef CAS PubMed.
- A. D. W. Boran and R. Iyengar, Systems approaches to polypharmacology and drug discovery, Curr. Opin. Drug Discovery Dev., 2010, 13, 297–309 CAS.
- M. A. Yildirim, K. I. Goh, M. E. Cusick, A. L. Barabási and M. Vidal, Drug-target network, Nat. Biotechnol., 2007, 25(10), 1119–1126 CrossRef CAS PubMed.
- S. J. Caldwell and A. M. Berghuis, Small-angle X-ray scattering analysis of the bifunctional antibiotic resistance enzyme aminoglycoside (6′) acetyltransferase-ie/aminoglycoside (2′′) phosphotransferase-ia reveals a rigid solution structure, Antimicrob. Agents Chemother., 2012, 56(4), 1899–1906 CrossRef CAS PubMed.
- W. Li, J. Li, Q. Wei, Q. Hu, X. Lin and M. Chen, et al., Characterization of aminoglycoside resistance and virulence genes among Enterococcus spp. isolated from a hospital in China, Int. J. Environ. Res. Public Health, 2015, 12(3), 3014–3025 CrossRef CAS PubMed.
- E. Padmasini, R. Padmaraj and S. S. Ramesh, High level aminoglycoside resistance and distribution of aminoglycoside resistant genes among clinical isolates of Enterococcus species in Chennai, India, Sci. World J., 2014, 2014, 329157 Search PubMed.
- J. M. Munita, A. S. Bayer and C. A. Arias, Evolving Resistance Among Gram-positive Pathogens, Clin. Infect. Dis., 2015, 61(Suppl 2), S48–57 CrossRef PubMed.
- P. M. Mira, J. C. Meza, A. Nandipati and M. Barlow, Adaptive landscapes of resistance genes change as antibiotic concentrations change, Mol. Biol. Evol., 2015, 32(10), 2707–2715 CrossRef PubMed.
- J. Sakon, H. H. Liao, A. M. Kanikula, M. M. Benning, I. Rayment and H. M. Holden, Molecular structure of kanamycin nucleotidyltransferase determined to 3.0 A resolution, Biochemistry, 1993, 32, 11977–11984 CrossRef CAS PubMed.
- L. E. Wybenga-Groot, K. Draker, G. D. Wright and A. M. Berghuis, Crystal structure of an aminoglycoside 6′-N-acetyltransferase: defining the GCN5-related N-acetyltransferase superfamily fold, Structure, 1999, 7, 497–507 CrossRef CAS PubMed.
- J. M. Gajer, S. D. Furdas, A. Gründer, M. Gothwal, U. Heinicke and K. Keller, et al., Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo, Oncogenesis, 2015, 4, e137 CrossRef CAS PubMed.
- F. Manzo, F. P. Tambaro, A. Mai and L. Altucci, Histone acetyltransferase inhibitors and preclinical studies, Expert Opin. Ther. Pat., 2009, 19(6), 761–774 CrossRef CAS PubMed.
- W.-C. Hon, G. A. McKay, P. R. Thompson, R. M. Sweet, D. S. C. Yang and G. D. Wright, et al., Structure of an Enzyme required for aminoglycoside antibiotic resistance reveals homology to eukaryotic protein kinases, Cell, 1997, 89, 887–895 CrossRef CAS PubMed.
- D. M. Daigle, G. A. McKay and G. D. Wright, Inhibition of aminoglycoside antibiotic resistance enzymes by protein kinase inhibitors, J. Mol. Biol., 1997, 272, 24755–24758 CAS.
- H. Hidaka, M. Inagaki, S. Kawamoto and Y. Sasaki, Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C, Biochemistry, 1984, 23, 5036–5041 CrossRef CAS PubMed.
- L. Aravind and E. V. Koonin, DNA polymerase beta-like nucleotidyltransferase superfamily: Identification of three new families, classification and evolutionary history, Nucleic Acids Res., 1999, 27(7), 1609–1618 CrossRef CAS PubMed.
- Z. Gao, D. J. Maloney, L. M. Dedkova and S. M. Hecht, Inhibitors of DNA polymerase beta: activity and mechanism, Bioorg. Med. Chem., 2008, 16(8), 4331–4340 CrossRef CAS PubMed.
- K. H. Barakat, M. M. Gajewski and J. A. Tuszynski, DNA polymerase beta (pol β) inhibitors: a comprehensive overview, Drug Discovery Today, 2012, 17, 913–920 CrossRef CAS PubMed.
- J. L. Hansen, P. B. Moore and T. A. Steitz, Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit, J. Mol. Biol., 2003, 330(5), 1061–1075 CrossRef CAS PubMed.
- M. J. Cabanas, D. Vazquez and J. Modolell, Inhibition of ribosomal translocation by aminoglycoside antibiotics, Biochem. Biophys. Res. Commun., 1978, 83, 991–997 CrossRef CAS PubMed.
- B. Francois, R. J. M. Russel, J. B. Murray, F. Aboul-ela, B. Masquida and Q. Vicens, et al., Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding, Nucleic Acids Res., 2005, 33, 5677–5690 CrossRef CAS PubMed.
- M. W. Vetting, S. S. Hedge, F. Javid-Majd, J. S. Blanchard and S. L. Roderick, Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates, Nat. Struct. Biol., 2002, 9, 653–658 CrossRef CAS PubMed.
- G. Cox, P. J. Stogios, A. Savchenko and G. D. Wright, Structural and Molecular Basis for Resistance to Aminoglycoside Antibiotics by the Adenylyltransferase ANT(2′′)-Ia, mBio, 2015, 6(1), e02180 CrossRef CAS PubMed.
- J. Kondo, B. Francois, R. J. Russell, J. B. Murray and E. Westof, Crystal structure of the bacterial ribosomal decoding site complexed with amikacin containing the gamma-amino-alpha-hydroxybutyryl (haba) group, Biochimie, 2006, 88, 1027–1031 CrossRef CAS PubMed.
- G. G. Zhanel, C. D. Lawson, S. Zelenitsky, B. Findlay, F. Schweizer and H. Adam, et al., Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin, Expert Rev. Anti-Infect. Ther., 2012, 10(4), 459–473 CrossRef CAS PubMed.
- S. C. Olsen and S. A. Carlson, In vitro bactericidal activity of aminoglycosides, including the next-generation drug plazomicin, against Brucella spp, Int. J. Antimicrob. Agents, 2015, 45(1), 76–78 CrossRef CAS PubMed.
- M. Bassetti and E. Righi, Development of novel antibacterial drugs to combat multiple resistant organisms, Langenbecks Arch. Surg., 2015, 400(2), 153–165 CrossRef PubMed.
- M. Bassetti and E. Righi, New antibiotics and antimicrobial combination therapy for the treatment of Gram-negative bacterial infections, Curr. Opin. Crit. Care, 2015, 21(5), 402–411 CrossRef PubMed.
- E. Bettiol and S. Harbarth, Development of new antibiotics: taking off finally?, Swiss Med. Wkly., 2015, 145, w14167 Search PubMed.
- E. S. Armstrong and G. H. Miller, Combating evolution with intelligent design: the neoglycoside ACHN-490, Curr. Opin. Microbiol., 2010, 13(5), 565–573 CrossRef CAS PubMed.
- G. D. Wrights, The antibiotic resistome: the nexus of chemical and genetic diversity, Nat. Rev. Microbiol., 2007, 5, 175–186 CrossRef PubMed.
- S. M. Hatosy and A. C. Martiny, The ocean as a global reservoir of antibiotic resistance genes, Appl. Environ. Microbiol., 2015, 81(21), 7593–7599 CrossRef PubMed.
- B. Becker and M. A. Cooper, Aminoglycoside antibiotics in the 21st century, ACS Chem. Biol., 2013, 8(1), 105–115 CrossRef CAS PubMed.
- G. D. Wright and P. Ladak, Overexpression and characterization of the chromosomal aminoglycoside 6′-N-acetyltransferase from Enterococcus faecium, Antimicrob. Agents Chemother., 1997, 41(5), 956–960 CAS.
- D. H. Fong and A. M. Berghuis, Structural basis of APH(3′)-IIIa-mediated resistance to N1-substituted aminoglycoside antibiotics, Antimicrob. Agents Chemother., 2009, 53, 3049–3055 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interest. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5md00384a |
| This journal is © The Royal Society of Chemistry 2016 |