
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A quinazoline-based HDAC inhibitor affects gene expression pathways involved in cholesterol biosynthesis and mevalonate in prostate cancer cells†
Z.
Lin
a,
K. S.
Bishop
*a,
H.
Sutherland
a,
G.
Marlow
b,
P.
Murray
a,
W. A.
Denny
a and
L. R.
Ferguson
ab
aAuckland Cancer Society Research Centre, University of Auckland, New Zealand. E-mail: k.bishop@auckland.ac.nz
bDiscipline of Nutrition and Dietetics, University of Auckland, New Zealand
First published on 6th January 2016
Abstract
Chronic inflammation can lead to the development of cancers and resolution of inflammation is an ongoing challenge. Inflammation can result from dysregulation of the epigenome and a number of compounds that modify the epigenome are in clinical use. In this study the anti-inflammatory and anti-cancer effects of a quinazoline epigenetic-modulator compound were determined in prostate cancer cell lines using a non-hypothesis driven transcriptomics strategy utilising the Affymetrix PrimeView® Human Gene Expression microarray. GATHER and IPA software were used to analyse the data and to provide information on significantly modified biological processes, pathways and networks. A number of genes were differentially expressed in both PC3 and DU145 prostate cancer cell lines. The top canonical pathways that frequently arose across both cell lines at a number of time points included cholesterol biosynthesis and metabolism, and the mevalonate pathway. Targeting of sterol and mevalonate pathways may be a powerful anticancer approach.
1. Introduction
Although inflammation is a necessary immune response, chronic inflammation can lead to the development of cancers as well as other chronic diseases, and there are ongoing efforts to either block pro-inflammatory mediators or to stimulate resolution of inflammation.1 Deregulation of the epigenome can result in a range of chronic diseases associated with inflammation, and a number of compounds that modify the epigenome are in clinical use today.2Chromatin remodelling plays a key role in gene expression, and epigenetic modifications may be as important as mutations, insertions and deletions in tumour development and progression.3 Unlike genetic mutations, epigenetic changes that have resulted in gene activation or silencing can sometimes be reversed by small molecules that modify the epigenome. One such group of small molecules are the histone deacetylase (HDAC) inhibitors. HDACs influence the expression of a number of key enzymes involved in pathways associated with apoptosis, cell cycle, tumour cell proliferation and inflammation, amongst others4 and tumour progression is associated with an increase in HDAC activity.5 However, although HDAC inhibitors have an impact on tumour and T cell lymphomas rather than non-malignant cells, their mechanism of action remains unclear.4
HDACs and histone acetyltransferases (HATs) act in opposition to modify chromatin and thus control gene expression.6 HDACs can repress transcription by bringing about chromatin condensation in response to the removal of acetyl groups from histone tails.6 Not only have HDACs been found to be aberrantly recruited to “inappropriate” loci, but abnormal expression of HDACs 1, 2, 3 and 6 have been reported in numerous types of cancer e.g. gastric, breast, prostate, colorectal and cervical.6 SN30028 is an HDAC inhibitor that was identified from an in-house compound library.7 SN30028 (Fig. 1) is regarded as a quinazoline drug and was selected from the aforementioned compound library based on its anti-inflammatory activity and the strength of HDAC inhibition.7,8
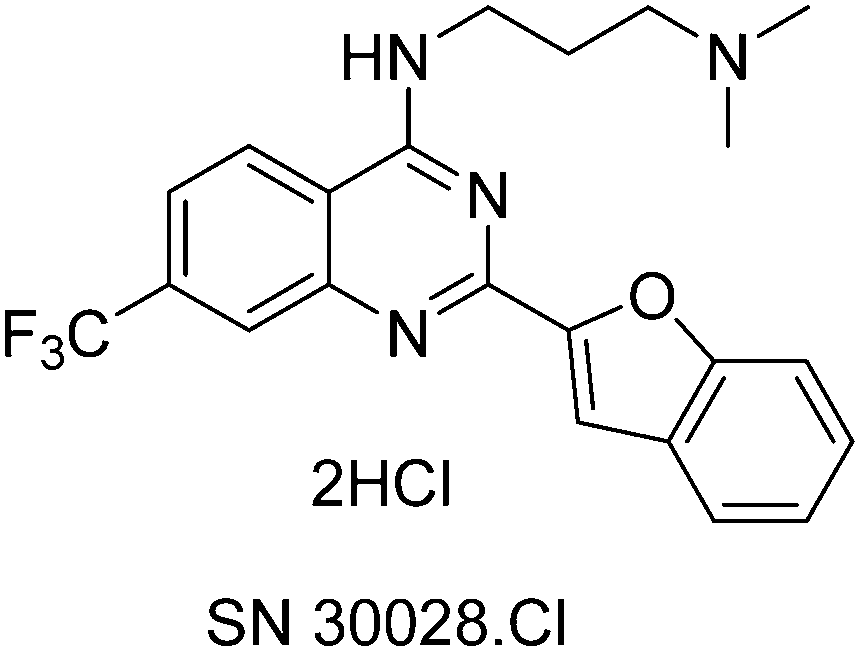 | ||
| Fig. 1 Chemical structure of SN30028. | ||
SN30028 decreased the activity of HDAC 1, 3 and 6 by 23%, 76% and 48% respectively.9 HDACs 1 and 3, HDAC class I compounds, are restricted to the nucleus and are believed to play a key role in cell survival and proliferation.10 Loss of HDAC 1 activity results in an overall reduction of deacetylase activity, reduced proliferation rates and increased levels of the cyclin-dependent kinase inhibitors p21 and p27.11 HDAC 3 is important as it mediates gene expression of tumour necrosis factor (TNF) as well as the expression of other genes.12
HDAC 6 belongs to the HDAC Class 2b group of compounds and is unique as it has two catalytic domains and a zinc finger.6 HDAC 6 is of interest as it helps to protect against cellular stress by the regulation of heat shock protein 90 and alpha tubulin and down-regulation can bring about apoptosis and inhibition of metastasis.13,14
Determining the effect of a particular compound on cancer cell lines can be challenging as effects on any one gene can be small, and these effects can also be broad. For this reason the anti-inflammatory and anti-cancer effects of a quinazoline epigenetic-modulator compound were determined, in prostate cancer cell lines, using a transcriptomics approach.
One advantage of transcriptomics is that experimental design is non-hypothesis driven, and provides sufficient sensitivity and breadth to examine the expression of thousands of genes simultaneously.15 The Affymetrix PrimeView GeneChip Human Microarray was used as it is a “perfect-match-only” (probe to transcript) array16 and therefore the false signal changes referred to by Li et al.17 are less likely to arise.
In addition biological processes are likely to be represented by complex networks consisting of multiple signalling modules rather than a series of linear pathways18 and thus a transcriptomics approach, followed by network analysis, was deemed preferable. The aim of this study was to determine the effect of SN30028 on differential gene expression in prostate cancer cell lines with a particular focus on inflammation and epigenetic modulation. Compounds, that restore histone acetylation, have potential as anti-inflammatory and anti-cancer drugs,19 and we show evidence that the known7 quinazoline-based HDAC inhibitor SN30028 (Fig. 1) influenced cholesterol biosynthesis and mevalonate pathways.
2. Methods
2.1 Cell culture
The prostate cancer cell lines PC3, DU145 and LNCaP were obtained from the American Type Culture Collection (VA, USA) and maintained in Alpha 5 minimum essential media (Gibco, Invitrogen Corporation, New York, USA) with 10% fetal calf serum (Morgate Biotech, Hamilton, New Zealand) and 100 IU penicillin/streptomycin (Sigma Chemical Company, St Louis, MO, USA). Cells were cultured in a water jacketed incubator (Thermo Fisher Scientific, MA, USA) at 37 °C with 5% CO2, and passaged twice weekly. The 50% inhibitory concentrations (IC50s) for SN30028 and a number of other compounds were established and the anti-proliferative activity was determined relative to the reference drugs suberoylanilide hydroxamic acid (SAHA) (Sigma Chemical Company, St Louis, MO, USA) and 5′-aza-2-deoxycytidine (Sigma-Aldrich, St Louis, MO, USA). Dimethyl sulphoxide (DMSO) (Merck, Darmstadt, Germany) was used as a solvent for the compounds tested and hence was also used as a control. The IC50s were performed on PC3, DU145 and LNCaP using the sulforhodamine B colorimetric assay.20 Three independent experiments were performed in quadruplicate.2.2 HDAC activity
HDAC activity was assessed from total protein extracted from DU145, PC3 and LNCaP cells following treatment for 24 hours with a number of compounds (TSA, SAHA, SN30028, SN30029, SN30140, SN29887, SN29984, SN30711, 5-aza-2-deoxycitidine (5AZA)) (Sigma Aldrich, St Louis, USA) at their respective IC50 concentrations. Total protein was extracted from each treated cell line grown in P100 plates. The cells were washed with PBS, lysed (lysis buffer containing 1% Triton X-100 (Sigma Aldrich, St Louis, USA)), frozen, then thawed and removed to a micro-centrifuge tube, whereupon the cells were centrifuged and the supernatant collected. Protein concentration was assessed using the bicinchoninic acid (Sigma Aldrich, St Louis, USA) protein assay (as outlined in Lin et al.).7Using the manufacturer's protocol the HDAC Fluorometric Activity Assay (BIOMOL International – Cayman Chemical, Ann Arbor, USA) was used to measure the effect of compounds on HDAC activity from the extracted protein. Three independent experiments were performed in duplicate.
2.3 Transcriptomics
Two prostate cancer cell lines, namely DU145 and PC3 cells, were treated with SN30028 and harvested at 4, 24 and 96 hours after dosing (Fig. 2). In addition, cell lines were incubated with an equivalent amount of water for each of the three time points. These served as controls. DU145 and PC3 were chosen ahead of LNCaP as the aforementioned were shown to have greater sensitivity to HDAC inhibition than LNCaP9 (see Section 3.1) A QIAshredder (Qiagen, MD, USA) was used to disrupt the cells and an RNeasy Mini kit (Qiagen, MD, USA) was used to isolate the RNA in triplicate, following the manufacturer's recommended protocol. The quantity and quality of RNA was determined using the NanoDrop ND 1000 spectrophotometer (NanoDrop Technologies, Wilmington, USA) and the Experion RNA StdSens Analysis Kit (Biorad, Hercules, USA) respectively. Approximately 500 ng of RNA with a RIN value of >8 were required for the PrimeView® Human Gene Expression arrays (Affymetrix, CA, USA). The RNA was reverse transcribed, biotin labelled, fragmented, hybridised to the gene chip and scanned21 by the Centre for Genomics and Proteomics (New Zealand Genomics Ltd, University of Auckland, New Zealand).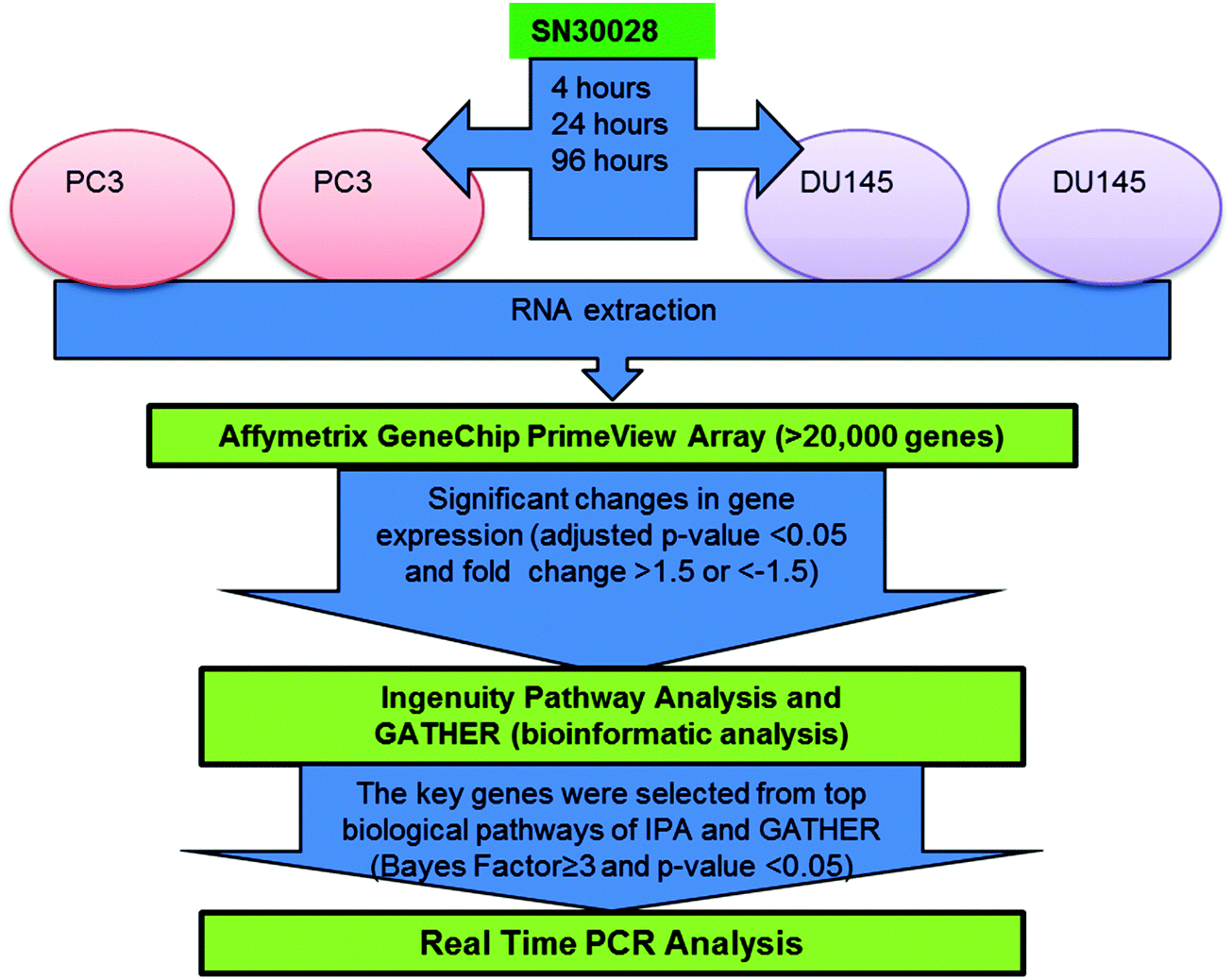 | ||
| Fig. 2 Workflow for microarray analysis of differential gene expression generated from prostate cancer cells treated with SN30028 (ref. 9 adapted from ref. 23). | ||
The PrimeView® Human Gene Expression array uses 530![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 probes covering 36000 transcripts and variants located in more than 20000 genes.22 Transcripts were measured independently by using multiple probes. The level of gene expression was associated with the probe/s targeting that specific gene and following adjustment for the solvent/media controls, differential gene expression was calculated. The workflow for the analysis of the gene expression array data is outlined in Fig. 2.
000 probes covering 36000 transcripts and variants located in more than 20000 genes.22 Transcripts were measured independently by using multiple probes. The level of gene expression was associated with the probe/s targeting that specific gene and following adjustment for the solvent/media controls, differential gene expression was calculated. The workflow for the analysis of the gene expression array data is outlined in Fig. 2.
Confirmation of the microarray results was carried out using quantitative reverse transcription polymerase chain reaction (qRT-PCR) and 27 statistically significant differentially expressed genes were selected for validation. The 27 TaqMan probe sets were obtained from ThermoFisher (Pleasanton, USA). The aforementioned RNA (Section 2.2) was isolated from DU145 and PC3 following treatment for 4 and 24 h, converted to cDNA using a Quantitect Reverse Transcription Kit (Qiagen, Victoria, Australia) and PCR was performed (on the three biological repeats as well as non-template controls) on an Applied Biosystems 7900 thermocycler (Waltham, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), hypoxanthine phosphor-ribosyltransferase 1 (HPRT1), and β-actin (ACTB) were tested for assessment as normalisation genes for RNA expression. A standard curve was calculated from technical triplicates using SDS2.3 and RQ Manager 2.2 software (Applied Biosystems, USA). The relative expression of each of the genes was calculated as fold change using a delta delta cycle threshold (Ct) method.24 Thereafter fold changes from the Affymetrix and qRT-PCR experiments were compared.
2.4 Data analysis
To identify the differentially expressed genes, the data from the microarray experiments were subjected to Robust Multiarray Analysis25 to assess for quantile normalisation and then filtered based on the following parameters: (adjusted p value <0.05; fold change >1.5). The “Limma” package within the statistical language R26 was used to analyse the CEL files generated by the Affymetrix GeneChip Instrument System (Affymetrix, MD, USA). Gene expression values were based on the absorbance values of the probes targeting each specific gene. Changes in average gene expression in response to treatment were calculated as fold changes. p-Values were calculated so as to determine whether the changes in gene expression were significant (Fig. 2). A multiple testing correction was applied using Benjamini–Hochberg's false-discovery rate27 set at 5%. Functional, pathway and network analyses were performed by using GATHER (Gene Annotation Tool to Help Explain Relationships28) and IPA (Ingenuity Pathway Analysis29) software. Networks were ranked by a score that was assigned based on the probability that a collection of nodes within that network can occur by chance. A number of networks scored highly, but only the highest scoring network per cell line – treatment time combination were presented here so as to avoid biasing results through “fishing” (Fig. S1–S6, ESI†). The network score indicates the likelihood of Focus Genes in a network being found together, and it is clear from the high scores that one should be confident that these associations were not generated by chance.30 IPA constructs networks using a stepwise process. Networks are constructed in such a way that both interconnectivity between Focus Genes and the number of Focus Genes in the network are optimised (using a network size of 35 or 70 genes/proteins, as specified in the supplementary figure legends).3. Results
The four day IC50 concentrations were established for PC3, DU145 and LNCaP cell lines and the results are listed in Table 1.| Compounds (μM) | PC3 | DU145 | LNCaP |
|---|---|---|---|
| IC50 | IC50 | IC50 | |
| SAHA = suberoylanilide hydroxamic acid. | |||
| SN30028 | 4.58 | 7.30 | 0.82 |
| SN30029 | 2.87 | 3.82 | 0.84 |
| SN30140 | 2.31 | 3.9 | 0.71 |
| SN29887 | 3.61 | 12.7 | 6.92 |
| SN29984 | 7.13 | 15.66 | 6.38 |
| SN26855 | 1.65 | 0.49 | 1.11 |
| SAHA | 0.88 | 0.92 | 0.58 |
| 5′-Aza-2-deoxycytidine | 0.25 | 0.31 | 0.53 |
With the exception of SN26855, DU145 cells showed the greatest tolerance to the novel compounds tested. In general, LNCaP cells appeared to be the most sensitive to the novel compounds.
3.1 HDAC inhibition
Following treatment of cells for 24 h, total protein was extracted. The effects of compounds of interest on HDAC activity are shown in Fig. 3–5. The HDAC activity of total protein extracted from the cell lines, varied from one cell line to another and varied amongst the novel compounds. HDAC activity of DU145 cells was markedly reduced following treatment with all the novel compounds, particularly SN30028 (Fig. 4), whilst SN30140 had the largest reduction in HDAC activity in PC3 cells (Fig. 3). The novel compounds did not appear to reduce HDAC activity in LNCaP cells (Fig. 5).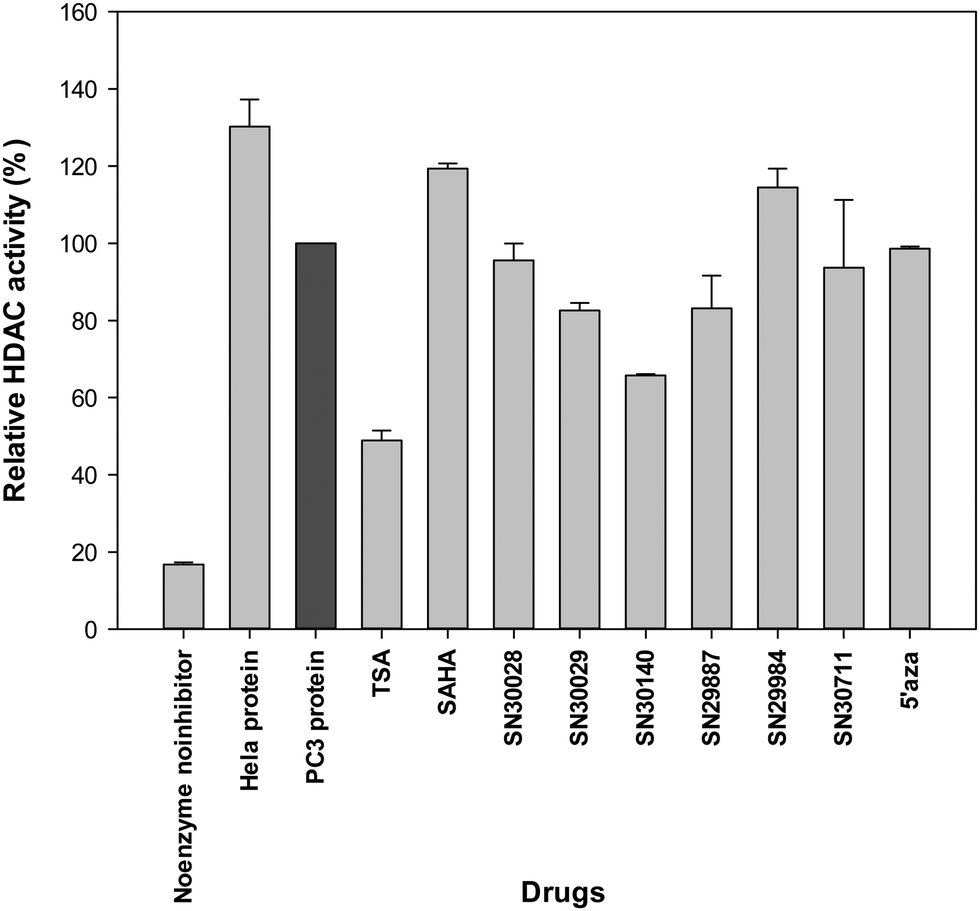 | ||
| Fig. 3 Effect of selected compounds on HDAC activity in PC3 cells. | ||
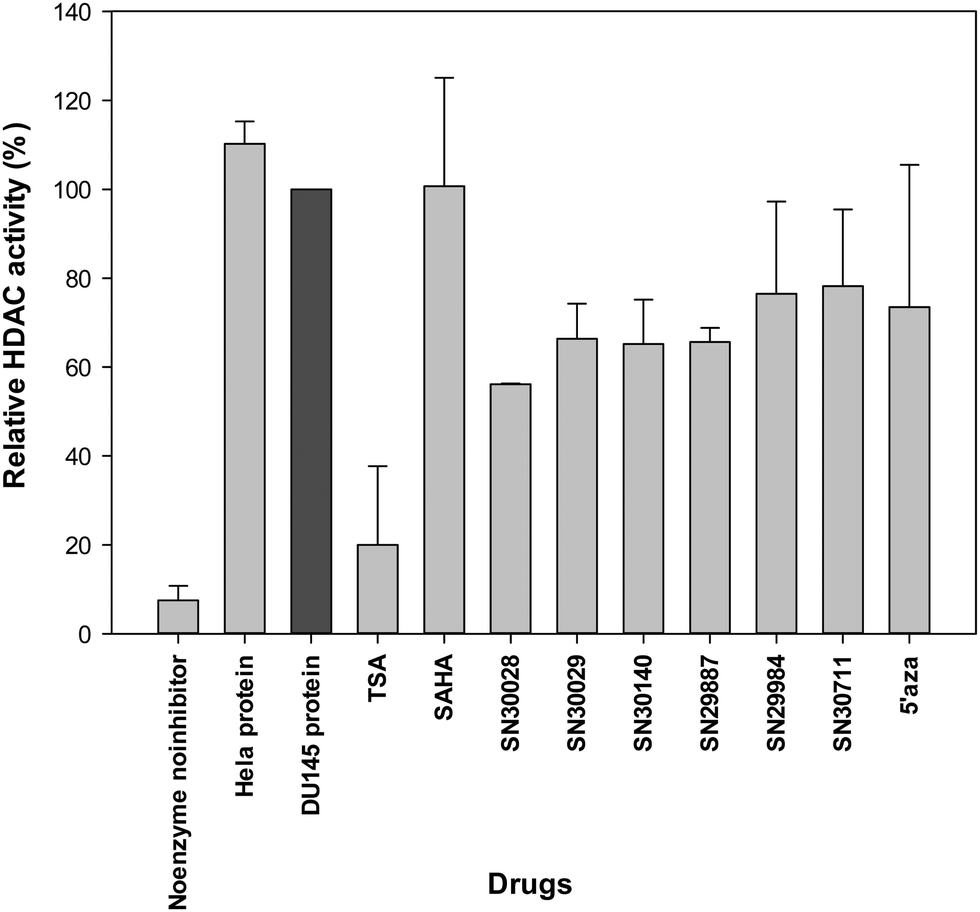 | ||
| Fig. 4 Effect of selected compounds on HDAC activity in total protein extracted from DU145 cells. | ||
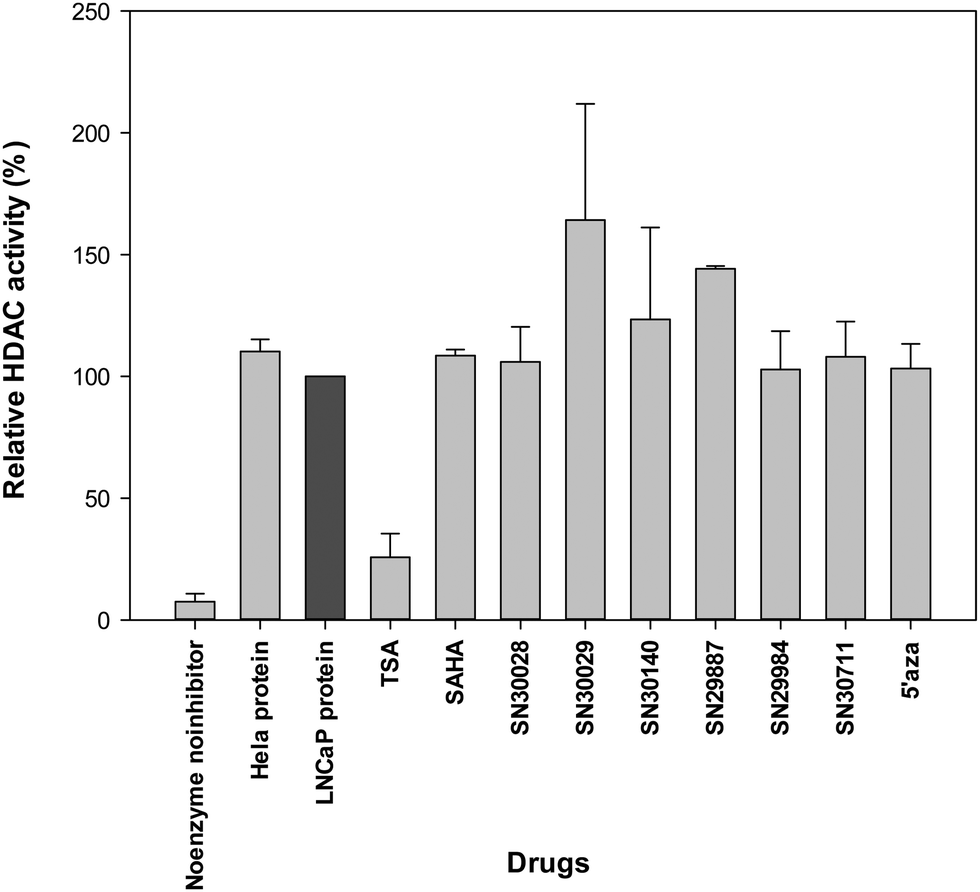 | ||
| Fig. 5 Effect of selected compounds on HDAC activity in total protein extracted from LNCaP cells. | ||
3.2 Differential gene expression (Affymetrix PrimeView™ Microarrays)
Following the analysis of the Affymetrix PrimeView™ microarray data, it was found that a number of genes in PC3 and DU145 were differentially expressed (p < 0.05; fold change >1.5) following treatment with SN30028. In total 59, 7 and 64 genes were up-regulated and 38, 6 and 34 genes were down-regulated in PC3 at 4, 24 and 96 hours respectively. In DU145 49, 108 and 796 genes were up-regulated and 3, 4 and 399 genes were down-regulated at 4, 24 and 96 hours respectively. A list of ten of the most differentially expressed genes for each cell line–time point combination, with statistically significant p-values and a fold change of >1.5 are shown in Table 2 (PC3) and Table 3 (DU145). The genes listed in the table were selected based on the absolute fold change and presented from largest to smallest, regardless of the direction of change.| Cell line time | Gene symbol | Gene name | Fold change | p-Value (≤) |
|---|---|---|---|---|
| PC3 4 hours | MMP3 | Matrix metallopeptidase 3 (stromelysin 1, progelatinase) | 2.25 | 2.00 × 10−5 |
| HEXA | Hexosaminidase A (alpha polypeptide) | −2.17 | 1.07 × 10−2 | |
| CSF2 | Colony stimulating factor 2 (granulocyte-macrophage) | 2.16 | 3.00 × 10−4 | |
| CYP1B1 | Cytochrome P450, family 1, subfamily B, polypeptide 1 | −2.03 | 9.20 × 10−4 | |
| PHC1 | Polyhomeotic homolog 1 (Drosophila) | −1.96 | 1.50 × 10−4 | |
| PNRC1 | Proline-rich nuclear receptor coactivator 1 | 1.91 | 3.30 × 10−4 | |
| VNN1 | Vanin 1 | 1.87 | 7.00 × 10−5 | |
| RAB5C | RAB5C, member RAS oncogene family | 1.86 | 1.06 × 10−3 | |
| SNORA28 | Small nucleolar RNA, H/ACA box 28 | −1.73 | 6.40 × 10−4 | |
| SPEN | Spen homolog, transcriptional regulator (Drosophila) | −1.73 | 1.85 × 10−2 | |
| PC3 24 hours | PTGS2 | Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) | −2.47 | 4.34 × 10−2 |
| EPGN | Epithelial mitogen homolog (mouse) | −2.26 | 4.16 × 10−2 | |
| CYP1B1 | Cytochrome P450, family 1, subfamily B, polypeptide 1 | −2.07 | 1.25 × 10−2 | |
| RGS4 | Regulator of G-protein signalling 4 | 1.98 | 4.34 × 10−2 | |
| AREG | Amphiregulin | −1.87 | 3.80 × 10−2 | |
| GULP1 | GULP, engulfment adaptor PTB domain containing 1 | 1.83 | 4.10 × 10−2 | |
| SC4MOL | Sterol-C4-methyl oxidase-like | 1.76 | 1.12 × 10−2 | |
| SGK2 | Serum/glucocorticoid regulated kinase 2 | −1.76 | 1.85 × 10−2 | |
| RNF144A | Ring finger protein 144A | 1.75 | 1.25 × 10−2 | |
| KRT17 | Keratin 17 | −1.71 | 4.16 × 10−2 | |
| PC3 96 hours | SCNN1G | Sodium channel, non-voltage-gated 1, gamma | 2.92 | 1.72 × 10−2 |
| DDIT3 | DNA-damage-inducible transcript 3 | −2.77 | 4.57 × 10−2 | |
| CXCR7 | Chemokine (C–X–C motif) receptor 7 | 2.49 | 3.12 × 10−2 | |
| CHRNA1 | Cholinergic receptor, nicotinic, alpha 1 (muscle) | 2.38 | 2.13 × 10−2 | |
| HERPUD1 | Homocysteine-inducible, endoplasmic reticulum stress-inducible, ubiquitin-like domain member 1 | −2.11 | 3.67 × 10−2 | |
| STC1 | Stanniocalcin 1 | 2.10 | 4.22 × 10−2 | |
| TMSB15A | Thymosin beta 15a | 2.04 | 2.40 × 10−2 | |
| HSPA5 | Heat shock 70 kDa protein 5 (glucose-regulated protein, 78 kDa) | −2.04 | 2.86 × 10−2 | |
| SESN2 | Sestrin 2 | −1.98 | 3.12 × 10−2 | |
| AREG | Amphiregulin | −1.95 | 3.12 × 10−2 | |
| Cell line time | Gene symbol | Gene name | Fold change | p-Value (≤) |
|---|---|---|---|---|
| DU145 4 hours | INSIG1 | Insulin induced gene 1 | 2.74 | 2.03 × 10−2 |
| ULBP1 | UL16 binding protein 1 | 2.24 | 2.02 × 10−3 | |
| HMGCS1 | 3-Hydroxy-3-methylglutaryl-CoA synthase 1 (soluble) | 2.17 | 3.71 × 10−2 | |
| DDIT4 | DNA-damage-inducible transcript 4 | 2.07 | 2.03 × 10−2 | |
| BDP1 | B double prime 1, subunit of RNA polymerase III transcription initiation factor IIIB | 2.06 | 3.00 × 10−2 | |
| SC4MOL | Sterol-C4-methyl oxidase-like | 2.01 | 2.03 × 10−2 | |
| LPIN1 | Lipin 1 | 1.95 | 5.55 × 10−3 | |
| EGFR | Epidermal growth factor receptor | 1.94 | 3.53 × 10−2 | |
| CEP350 | Centrosomal protein 350 kDa | 1.91 | 2.29 × 10−2 | |
| MLL3 | Myeloid/lymphoid or mixed-lineage leukemia 3 | 1.89 | 1.24 × 10−2 | |
| DU145 24 hours | IFIT2 | Interferon-induced protein with tetratricopeptide repeats 2 | 3.09 | 2.23 × 10−3 |
| HMGCS1 | 3-Hydroxy-3-methylglutaryl-CoA synthase 1 (soluble) | 3.02 | 9.05 × 10−3 | |
| SC4MOL | Sterol-C4-methyl oxidase-like | 3.01 | 4.72 × 10−4 | |
| IFIT3 | Interferon-induced protein with tetratricopeptide repeats 3 | 2.98 | 1.34 × 10−2 | |
| INSIG1 | Insulin induced gene 1 | 2.89 | 2.14 × 10−3 | |
| IFIT1 | Interferon-induced protein with tetratricopeptide repeats 1 | 2.82 | 4.72 × 10−4 | |
| AKR1B10 | Aldo-keto reductase family 1, member B10 (aldose reductase) | 2.58 | 2.21 × 10−3 | |
| IFI44 | interferon-induced protein 44 | 2.52 | 1.71 × 10−2 | |
| OASL | 2′-5′-Oligoadenylate synthetase-like | 2.44 | 4.72 × 10−4 | |
| DDX60 | DEAD (Asp–Glu–Ala–Asp) box polypeptide 60 | 2.41 | 5.30 × 10−3 | |
| DU145 96 hours | MMP1 | Matrix metallopeptidase 1 (interstitial collagenase) | 7.00 | 8.28 × 10−6 |
| S100A9 | S100 calcium binding protein A9 | −5.47 | 1.96 × 10−4 | |
| IGFBP3 | Insulin-like growth factor binding protein 3 | −5.41 | 2.92 × 10−5 | |
| C3 | Complement component 3 | −4.79 | 7.94 × 10−5 | |
| SLPI | Secretory leukocyte peptidase inhibitor | −4.51 | 3.21 × 10−4 | |
| GKN2 | Gastrokine 2 | 4.50 | 9.97 × 10−6 | |
| AKR1C1 | Aldo-keto reductase family 1, member C1 (dihydrodiol dehydrogenase 1; 20-alpha (3-alpha)-hydroxysteroid dehydrogenase) | −4.47 | 1.04 × 10−3 | |
| CDH1 | Cadherin 1, type 1, E-cadherin (epithelial) | −4.39 | 1.69 × 10−4 | |
| CYP4F11 | Cytochrome P450, family 4, subfamily F, polypeptide 11 | −4.08 | 2.99 × 10−4 | |
| AKR1C1///AKR1C2 | Aldo-keto reductase family 1, member C1 (dihydrodiol dehydrogenase 1; 20-alpha (3-alpha)-hydroxysteroid dehydrogenase)///aldo-keto reductase family 1, member C2 (dihydrodiol dehydrogenase 2; bile acid binding | −4.06 | 1.12 × 10−3 | |
| AKR1C2 | Protein; 3-alpha hydroxysteroid dehydrogenas | |||
3.3 Validation of the differential gene expression data generated by Affymetrix PrimeView™ microarray
To corroborate the differential gene expression data generated by the Affymetrix assay a validation was undertaken by comparing the aforementioned data with that obtained from a select number of genes tested using qRT-PCR. qRT-PCR is widely used for accurate expression profiling of key genes31 and is a suitable technique to validate differential gene expression data generated from microarray experiments. Genes were selected based on a high level of differential gene expression in one or more cell line – time point combinations, an important role in a biological pathway/network relevant to cancer or inflammation, or effects on epigenetics.Gene expression was measured using qRT-PCR in the following genes: AREG, ARID5B, CDKN2B, CYP1B1, CYP51A1, DHCR7, DUSP10, EGFR, EPGN, IFIT2, HMGCR, HMGCS1, GULP1, IDI1, INSIG1, KRT17, LDLR, MMP1, MMP3, NR4A3, PTGS2, RSG4, SCD, SGK2, SQLE, TM7SF2 and TP53INP1 (Table S1, ESI†). The aforementioned genes were selected based on the level of differential gene expression and relevance to cancer/epigenetic mechanisms. Although the magnitude of change varied between the two methods with qRT-PCR generating the higher value in general, the direction of change remained consistent, with the exception of the gene ARID5B. The gene expression level of ARID5B was down-regulated according to the results obtained from the Affymetrix array (fold change of −1.82), and up-regulated according to the results generated by qRT-PCR (fold change of 1.43).
GAPDH and HPRT1 were used as normalisation genes as they showed consistent results in both PC3 and DU145 cell lines across the Ct range of the 27 genes tested. Following normalisation, the fold changes in expression of the selected genes were compared between those generated from the Affymetrix microarray and those generated from qRT-PCR experiments (Table S1, ESI†).
3.4 GATHER
GATHER software was used to analyse the relationships between individual or groups of genes. Only those genes that were differentially expressed with a p-value ≤0.05 and a minimum fold change of 1.5 were uploaded to GATHER. SN30028 treatment modulated biological processes and the involved genes are summarised in Table S2 (ESI†). Those biological processes with Bayes factors ≥3 and p-values ≤ 0.05 were nominated.32 The Gene Ontology (GO) database was used to obtain definitions of biological processes.33The biological processes with the highest Bayes factors following treatment of PC3 cells with SN30028 at 4, 24 and 96 hours were: the transforming growth factor beta receptor signalling process (3.53), cyclooxygenase process (4.72), and response to nutrients process (5.69), respectively. In DU145 cells the sterol biosynthesis and sterol metabolism processes had the highest Bayes factors at 4 (29.66 and 26.51 respectively) and 24 hours (51.26 and 49.75 respectively), and cell proliferation and cell cycle were the most affected biological processes at 96 hour (25.21 and 21.49 respectively) following SN30028 treatment.
3.5 Ingenuity pathways analysis
IPA software (IPA®, QIAGEN Redwood City, USA) was used to transform a list of genes into a set of relevant networks29 and this software is widely used for biological pathway, function and disease, and molecular network analysis.34,35 All results from each time-point-cell line combination were submitted to IPA with the filters and settings as follows: adjusted p-value of 0.05 and fold-change of 1.5. The most significantly affected canonical pathways and associated network functions in response to treatment by SN30028 at three time points in PC3 and DU145 cell lines generated through IPA are shown in Table 4. Using Fischer's exact test, the results were ranked. The ratios listed in the table represent the number of genes in the dataset that were in the canonical pathway stated, divided by the total number of genes in that particular pathway.| PC3 | Top canonical pathways | p-Value | Differentially expressed genesa | Ratio |
|---|---|---|---|---|
| Ratio = differentially expressed genes/total number of genes in that pathway.a = up to five genes with the smallest p-values were selected. | ||||
| 4 hours | Glucocorticoid receptor signalling | 2.76 × 10−4 | BCL2, CSF2, NCOR2, MMP1, PTGS2 | 7/272 |
| Docosahexaenoic acid (DHA) signalling | 7.88 × 10−4 | BCL2, BIK, FOXO1 | 3/39 | |
| Chondroitin sulfate degradation | 1.61 × 10−3 | HEXA, MGEA5 | 2/13 | |
| Dermatan sulfate degradation | 1.88 × 10−3 | HEXA, MGEA5 | 2/14 | |
| PI3K/AKT signalling | 2.46 × 10−3 | BCL2, FOXO1, GDF15, PTGS2 | 4/121 | |
| Associated network functions | Score | |||
| Developmental disorder, cell death and survival, organismal injury and abnormalities. | 60 | |||
| Cellular growth and proliferation, cell death and survival, cancer. | 56 | |||
| Cellular development, cellular growth and proliferation, haematological system development and function. | 37 | |||
| Post-translational modification, cancer, gastrointestinal disease. | 2 | |||
| Cell morphology, cellular function and maintenance, DNA replication, recombination, and repair. | 2 | |||
| PC3 | Top canonical pathways | p-Value | Differentially expressed genesa | Ratio |
|---|---|---|---|---|
| 24 hours | Role of IL-17A in arthritis | 5.43 × 10−4 | MMP1, PTGS2 | 2/54 |
| Zymosterol biosynthesis | 3.89 × 10−3 | MSMO1 | 1/6 | |
| Airway pathology in chronic obstructive pulmonary disease | 5.18 × 10−3 | MMP1 | 1/8 | |
| Prostanoid biosynthesis | 5.83 × 10−3 | PTGS2 | 1/9 | |
| Cholesterol biosynthesis I | 8.41 × 10−3 | MSMO1 | 1/13 | |
| Associated network functions | Score | |||
| Cancer, dermatological diseases and conditions, tissue morphology. | 38 | |||
| PC3 | Top canonical pathways | p-Value | Differentially expressed genesa | Ratio |
|---|---|---|---|---|
| 96 hours | Unfolded protein response | 1.52 × 10−4 | DDIT3, DNAJB9, HSPA5, INSIG1 | 4/53 |
| Superpathway of cholesterol biosynthesis | 3.41 × 10−4 | ACAT2, HMGCS1, SQLE | 3/27 | |
| Ketogenesis | 1.12 × 10−3 | ACAT2, HMGCS1 | 2/10 | |
| Mevalonate pathway I | 1.63 × 10−3 | ACAT2, HMGCS1 | 2/12 | |
| Superpathway of geranylgeranyl-diphosphate biosynthesis I | 2.93 × 10−3 | ACAT2, HMGCS1 | 2/16 | |
| Associated network functions | Score | |||
| Cancer, organismal injury and abnormalities, neurological disease. | 46 | |||
| Cardiovascular disease, hereditary disorder, metabolic disease. | 30 | |||
| Lipid metabolism, molecular transport, small molecule biochemistry. | 30 | |||
| Cell-to-cell signalling and interaction, cellular assembly and organization, cellular function and maintenance. | 30 | |||
| Cancer, endocrine system disorders, organismal injury and abnormalities. | 28 | |||
| DU145 | Top canonical pathways | p-Value | Differentially expressed genesa | Ratio |
|---|---|---|---|---|
| 4 hours | Superpathway of cholesterol biosynthesis | 4.03 × 10−15 | HMGCR, MSMO1, MVK, SC5D, SQLE | 8/27 |
| Cholesterol biosynthesis I | 1.66 × 10−10 | DHCR7, MSMO1, NSDHL, SC5D, SQLE | 5/13 | |
| Cholesterol biosynthesis II | 1.66 × 10−10 | DHCR7, MSMO1, NSDHL, SC5D, SQLE | 5/13 | |
| Cholesterol biosynthesis III | 1.66 × 10−10 | DHCR7, MSMO1, NSDHL, SC5D, SQLE | 5/13 | |
| Mevalonate pathway I | 4.28 × 10−6 | HMGCR, HMGCS1, MVK | 3/12 | |
| Associated network functions | Score | |||
| Cancer, cell morphology, cellular function and maintenance. | 43 | |||
| Cardiovascular disease, metabolic disease, lipid metabolism. | 35 | |||
| Drug metabolism, small molecule biochemistry, cellular assembly and organization. | 35 | |||
| DU145 | Top canonical pathways | p-Value | Differentially expressed genesa | Ratio |
|---|---|---|---|---|
| 24 hours | Superpathway of cholesterol biosynthesis | 3.25 × 10−32 | HMGCS1, MSMO1, SC5D, HSD17B7, SQLE | 17/27 |
| Cholesterol biosynthesis I | 1.53 × 10−23 | MSMO1, SC5D, HSD17B7, SQLE, TM7SF2 | 11/13 | |
| Cholesterol biosynthesis II | 1.53 × 10−10 | MSMO1, SC5D, HSD17B7, SQLE, TM7SF2 | 11/13 | |
| Cholesterol biosynthesis III | 1.53 × 10−10 | MSMO1, SC5D, HSD17B7, SQLE, TM7SF2 | 11/13 | |
| Zymosterol biosynthesis | 4.07 × 10−6 | CYP51A1, HSD17B7, MSMO1, NSDHL, TM7SF2 | 5/6 | |
| Associated network functions | Score | |||
| Lipid metabolism, small molecule biochemistry, vitamin and mineral metabolism. | 66 | |||
| Carbohydrate metabolism, lipid metabolism, small molecule biochemistry. | 63 | |||
| Lipid metabolism, small molecule biochemistry, vitamin and mineral metabolism. | 60 | |||
| Antimicrobial response, inflammatory response, infectious disease. | 23 | |||
| DU145 | Top canonical pathways | p-Value | Differentially expressed genesa | Ratio |
|---|---|---|---|---|
| 96 hours | Molecular mechanisms of cancer | 7.24 × 10−7 | RHOU, CDH1, SHC1, BBC3, PLCB4 | 46/359 |
| Cell cycle control of chromosomal replication I | 1.75 × 10−6 | ORC6, CDC6, CDT1, CDC45, MCM2 | 10/27 | |
| Estrogen-mediated S-phase entry | 5.18 × 10−6 | CCNE2, CDKN1B, E2F5, CCND1, CDK1 | 9/24 | |
| Glioblastoma multiforme signaling I | 1.49 × 10−5 | RHOU, SHC1, PLCB4, PLCB1, PTEN | 23/145 | |
| Glioma signaling | 3.45 × 10−5 | SHC1, PTEN, IGF1R, PRKCA, PA2G4 | 17/94 | |
| Associated network functions | Score | |||
| Skeletal and muscular system development and function, protein synthesis, cellular compromise. | 84 | |||
| Cellular development, cellular growth and proliferation, digestive system development and function. | 77 | |||
| Post-translational modification, cell cycle, hair and skin development and function. | 73 | |||
| Cell cycle, cellular assembly and organization, DNA replication, recombination, and repair. | 68 | |||
| DNA replication, recombination, and repair, cell cycle, connective tissue disorders. | 67 | |||
Obtaining a list of genes and related pathways is informative, but it is the identification of the connections between the pathways that is important. The IPA networks are assembled based on connectivity between genes. Several networks were generated by IPA software from each microarray experiment using the IPA knowledge base, but only the strongest network for each cell line – time point combination are shown here (Fig. S1–S6, ESI†). The scores of the IPA network indicate how relevant the network is to the genes in the uploaded dataset (Score = −log10(p-value)). It is evident from Table 4 that cancer-related pathways are highly affected by SN30028 treatment in both cell lines. The parameters were set at either 70 or 35 molecules per network and a direct interaction between the molecules. Only relevant genes with an adjusted p-value <0.05 and fold change >1.5 or <−1.5 were selected for network analysis.
IPA generated results regarding the most prominent diseases and functions associated with the network depicted in Fig. S1–S6 (ESI†). These diseases and functions included developmental disorders, cancer, inflammatory disorders, cell death and survival, cellular growth and proliferation, cellular function and maintenance and DNA replication amongst others. Using the Fisher's exact test IPA calculates a network score which is the log10 of the p-value of the network.29 The network scores ranged from 38 (Fig. S2, ESI†) to 84 (Fig. S6, ESI†) and between 13% (Fig. S2, ESI†) and 100% (Fig. S6, ESI†) of the genes in each network were differentially expressed.
4. Discussion
Regulation of gene expression is encoded by the genome and by the epigenome. The focus of this discussion is on the modulation of gene expression and the molecular interaction of differentially expressed genes, represented in the form of networks, generated using transcriptomics and analysed using IPA. The most common pathways, as shown in Table 4, will be examined. Similarly, central nodes in networks will be discussed if they are evident in more than one network.Novel compounds were used to treat prostate cancer cell lines, inhibition of cell proliferation was observed and HDAC activity was assessed to determine which compound-cell line combination to use in the transcriptomics experiments. DU145 was found to be more tolerant to the novel compounds than PC3 and LNCaP, and LNCaP was found to be the most sensitive to the novel compounds with respect to cell proliferation. In contrast, DU145 had the greatest response to the novel compounds with respect to HDAC inhibition, whereas LNCaP had the least or no inhibitory response to these compounds. In addition, there was a far greater response observed in DU145 than in PC3 when considering the number of genes differentially expressed, as well as the size of the response.
There are a number of phenotypic and genotypic differences amongst PC3, DU145 and LNCaP. LNCaP is androgen sensitive, whilst PC3 and DU145 are androgen independent. HDAC inhibitors interfere with androgen receptor activity36 and therefore it is likely that the cell lines would respond differently. Seeing that PC3 and DU145 are androgen independent, it was not surprising that a change in the AR gene was not observed.
In addition to androgen sensitivity, the three cell lines of interest also differ with regards to TNFβ. When treated with TNFβ, cell proliferation was initially inhibited, whilst no effect was observed in LNCaP cells.37 TGFβ induces epigenetic changes to modulate cell proliferation, differentiation and migration, and TGFβ may initiate cellular changes that facilitate its role as both a tumour suppressor during the early stages of tumour development, and as a tumour promoter in metastatic or later stage disease.5 HDAC inhibitors may inhibit the activation of TGFβ in epithelial cells5 by blocking TGFβ mediated epithelial-mesenchymal transition (EMT), which is essential for cell growth and invasion.38 In our study we found that TGFβ was down regulated in response to SN30028, with fold changes between 1.074 and 1.707 (adjusted p values were significant for DU145 96 h treatment only).
4.1 Biological processes and pathways
Based on the transcriptomics results and analysis using GATHER and IPA, a number of cancer related “biological processes,” and “network associated functions” were affected by SN300028 treatment of the prostate cancer cell lines, DU145 and PC3 at all three time points tested (Table 4). Using GATHER, the most commonly modulated biological processes in DU145 (with high Bayes factor values) were in relation to sterol and cholesterol metabolism and biosynthesis (Table S2, ESI†) at the 4 and 24 h time points, whilst at 96 h the more directly cancer related biological processes of cell proliferation, cell cycle and DNA replication were evident. This is consistent with data generated using IPA. The top canonical pathways generated using IPA included those related to cholesterol biosynthesis (PC3 – 24 and 96 h, and DU145 – 4 and 24 h) and the mevalonate pathway (PC3 – 96 h and DU145 – 4 h).Other studies have been carried out using gene expression arrays, or targeted gene expression to assess levels of gene expression in normal versus adenocarcinoma or precursor adenocarcinoma tissues as well as in cell lines in response to HDAC inhibitors.39–43 Variations were observed in different cancer cell lines in response to HDAC inhibitors, with, in some cases, non-overlapping cellular targets.43 Using different cell lines and a different HDAC inhibitor, it is not surprising that different pathways were modulated, although some overlap was observed with respect to modification of gene expression.
Cholesterol metabolism plays an important role in providing cells with compounds for growth and sterol biosynthesis is an essential metabolic component of cancers.44 In addition, overexpression of cholesterol biosynthesis pathways has been previously detected in refractory breast cancers45 and this is consistent with the data observed from the metastatic prostate cancer cell lines we tested. More specifically, cytochrome P450 1B1 (CYP1B1) is important for the synthesis of cholesterol steroids and lipids, and is well known for its role in drug metabolism.46CYP1B1 activity is inhibited by a number of anti-cancer agents and is commonly over-expressed in a variety of tumours.47 In our study the expression of CYP1B1 was down regulated in PC3 cells treated for 4 and 24 h, and it is suggested that the inhibition of CYP1B1 is brought about through the inhibition of HDAC6 activity.48 In contrast, inconsistencies have arisen, for example the HDAC inhibitors SAHA and TSA induced CYP1B1 expression in the human breast cancer MCF-7.49
The mevalonate pathway has a broad influence and is associated with the cholesterol related biosynthesis pathways; is important in cellular metabolism; plays a role in the maintenance of cell membranes; is involved in steroid biosynthesis and can be disrupted by medication prescribed for bone-density disorders and high cholesterol levels.50,51 In addition, the mevalonate pathway is an important target for anti-cancer therapy and inhibitors of this pathway target malignant cell growth50,51 and are believed to act through the modification of methylation status of CpG sites in gene promoter regions involved in apoptosis and/or cell proliferation.52 In addition, DU145 cells, after 96 hours of treatment, showed the top canonical pathway affected was “molecular mechanisms of cancer” with 46 of the 359 genes involved were differentially expressed.
All cell line – treatment time combinations showed cancer to be one of the top five diseases and disorders, with the exception of DU145 at 24 h with a closely related disease/disorder, namely inflammatory response listed in the top five. The number of cancer related genes that were differentially expressed for each cell line – treatment time combination ranged from 11 to 445 genes with p values ranging from 8.00 × 10−2 to 2.15 × 10−17. It is clear that numerous cancer related genes were differentially expressed in prostate cancer cell lines in response to treatment and therefore the compound, SN30028 is of interest. Similarly, Chang et al. found that a large number of genes were differentially expressed in response to the HDAC inhibitor, TSA, in non-small cell lung cancer.43 However, in the study by Chang et al.,43 the fold-changes observed were much higher than those reported here.
“Associated network functions” were also listed as an output from IPA for each cell line – treatment time combination. Cell cycle, cellular growth and proliferation, DNA replication and repair, inflammatory response, and cancer all ranked highly in one or more of the cell line – treatment time combinations and thus it is evident that SN30028 has an impact on cancer related mechanisms.
4.2 Networks
The networks shown in Fig. S1 and S2 (ESI†) demonstrate that prostaglandin H synthase (PTGS2), otherwise known as COX2, connects with many genes in networks generated from PC3 treated for 4 and 24 hours and was one of the top ten differentially expressed genes following PC3 treatment for 4 h (Table 2). The down regulation of COX2 was corroborated in qRT-PCR experiments (Table S1, ESI†). COX2 is down-regulated by treatment with SN30028 for 4 and 24 hours (PC3) by a fold change of −1.71 and −2.47 respectively. COX2 plays a key role in prostanoid production by catalysing the conversion of arachidonic acid to prostaglandin G2 and H2 resulting in an inflammatory response.53,54 Numerous studies have been conducted to investigate COX2 inhibition and the reduction in inflammatory response55 and its role in oncogenesis.56 Non-steroidal anti-inflammatory drugs are a class of drugs that inhibit COX2 enzyme activity and thereby reduce the inflammatory response.57,58 This supports the notion that SN30028 has anti-inflammatory effects.Matrix metalloproteinase 3 (MMP3) was initially up-regulated in PC3 cells treated with SN30028 (Table 2 and Fig. S1, ESI†), but this response was not maintained. In addition, although MMP3 acts on a number of different genes, the expression of these genes was not modified when MMP3 was up-regulated (Fig. S1, ESI†). Although MMP3 is over-expressed in most human cancers, and is known to induce initial cancer cell-growth and differentiation, rather than act at a later stage in cancer progression,59,60 it is also known to have many opposing functions59 and to date MMP inhibitors have not been successful in the clinic.60
Differential expression of numerous other central node or core genes is evident, but none of the central node genes arise in more than two top ranked networks representing each cell line – treatment time combination. Some of the differentially expressed central node genes include FOXO1 and 3, thought to be involved in triggering apoptosis or cell survival and are induced by oxidative stress;61amphiregulin (AREG) was down-regulated in PC3 (Fig. S2 and S3, ESI†) supporting the idea that SN30028 reduces inflammation and inhibits tumour development.62 AREG is a ligand of epidermal growth factor receptor (EGFR)62 and the fact that EGFR is up-regulated in DU145 (Fig. S4, ESI†) could either be a chance occurrence as this finding was only attained in DU145 treated for 4 h and therefore is an initial response that is not sustained, or it could be that SN30028 works through different mechanisms in the two cell lines. Up-regulation of EGFR is associated with prostate cancer progression and EGFR dysfunction induces cell survival, proliferation, invasion and metastasis and therefore was not an anticipated response. The AR gene is known to interact with EGFR and we wouldn't expect to see a change in expression as DU145 and PC3 are androgen insensitive. However, although these cell lines are regarded as androgen non-responsive, some authors have reported low level expression of AR mRNA, and treatment with interferon (IFN) resulted in up-regulation of AR protein levels.63 This may be due to AR phosphorylation. In the experiments reported herein a change in AR expression in response to treatment with SN30028 was not noted.
Similar to the unexpected up-regulation of EGFR, the ETS-related gene (ERG) was up-regulated (at low intensity) after 96 h (Fig. S6, ESI†) and is an unexpected response. ERG is a proto-oncogene, regulates cell proliferation, differentiation, angiogenesis, inflammation, apoptosis and can result in gene fusion products associated with prostate and other cancers.64ERG, when over-expressed, is able to regulate oncogenic pathways involving cMyc, AR and EZH2,64 none of which were up-regulated in this study. ERG acts on neuronally expressed developmentally down-regulated 4 (NEDD4),64 which is a central node gene that was down-regulated in DU145 cells (Fig. S6, ESI†). NEDD4 is an oncoprotein that promotes degradation through the ubiquitination of its substrates and it is also thought to promote colon and lung carcinogenesis, be over-expressed in prostate, breast and bladder cancers, and promotes growth of colon cancer cells independently of PTEN and PI3K/AKT signalling.65 Despite the fact that ERG is over-expressed in DU145 after 96 h of treatment and acts on NEDD4 (amongst other genes), SN30028 appears to inhibit NEDD4 which is desirable, although further work is required for the elucidation of the mechanisms involved in treating cancers in this way.
5. Conclusions
In conclusion SN30028, an HDAC inhibitor was synthesised and used in the treatment of prostate cancer cell lines for 4, 24 and 96 h, and assessed for differential gene expression. Using IPA software the data were analysed and pathways involved in cholesterol biosynthesis and mevalonate were affected most commonly in the cell line DU145. In addition to pathway and network analysis the most frequently affected diseases and disorders were assessed. Those related to inflammatory response and/or cancers were one of the top five diseases and disorders listed for each cell line-time point tested. SN30028 showed it could be a potential therapeutic agent in treating prostate cancer by targeting the sterol and mevalonate pathways, however further work is required to confirm these results.Funding
This work was funded by the Auckland Cancer Society Research Centre.Acknowledgements
Liam Williams is acknowledged for performing the microarray assays and Daniel Hurley carried out the statistical analysis and normalisation of the Affymetric data (New Zealand Genomics Limited, Auckland, New Zealand). Matthew Barnett (AgResearch Ltd, Palmerston North, New Zealand) provided guidance on the use of IPA software.References
- C. N. Serhan, FASEB J., 2011, 25, 1441–1448 CrossRef CAS PubMed.
- C. H. Arrowsmith, C. Bountra, P. V. Fish, K. Lee and M. Schapira, Nat. Rev. Drug Discovery, 2012, 11, 384–400 CrossRef CAS PubMed.
- N. Dali-Youcef, S. Froelich, F.-M. Moussallieh, S. Chibbaro, G. Noël, I. J. Namer, S. Heikkinen and J. Auwerx, Sci. Rep., 2015, 5, 9087 CrossRef CAS PubMed.
- O. Khan and N. B. La Thangue, Immunol. Cell Biol., 2012, 90, 85–94 CrossRef CAS PubMed.
- M. Pickup, S. Novitskiy and H. L. Moses, Nat. Rev. Cancer, 2013, 13, 788–799 CrossRef CAS PubMed.
- J. E. Bolden, M. J. Peart and R. W. Johnstone, Nat. Rev. Drug Discovery, 2006, 5, 769–784 CrossRef CAS PubMed.
- Z. Lin, P. M. Murray, Y. Ding, W. A. Denny and L. R. Ferguson, Mutat. Res., 2010, 7, 81–88 CrossRef PubMed.
- Z. W. Lin, Assay and characteristics of histone deacetylase inhibitors, Masters thesis, University of Auckland, 2009 Search PubMed.
- Z. W. Lin, Modulation of epigenetic regulation as a novel mechanism in anti-inflammatory and anti-cancer drugs, PhD thesis, University of Auckland, 2015 Search PubMed.
- A. J. de Ruijter, A. H. van Gennip, H. N. Caron, S. Kemp and A. B. van Kuilenburg, Biochem. J., 2003, 370, 737–749 CrossRef CAS PubMed.
- G. Lagger, D. O'Carroll, M. Rembold, H. Khier, J. Tischler, G. Weitzer, B. Schuettengruber, C. Hauser, R. Brunmeir, T. Jenuwein and C. Seiser, EMBO J., 2002, 21, 2672–2681 CrossRef CAS PubMed.
- U. Mahlknecht, J. Will, A. Varin, D. Hoelzer and G. Herbein, J. Immunol., 2004, 173, 3979–3990 CrossRef CAS.
- C. Boyault, Y. Zhang, S. Fritah, C. Caron, B. Gilquin, S. H. Kwon, C. Garrido, T. P. Yao, C. Vourc'h, P. Matthias and S. Khochbin, Genes Dev., 2007, 21, 2172–2181 CrossRef CAS PubMed.
- G. I. Aldana-Masangkay and K. M. Sakamoto, J. Biomed. Biotechnol., 2011, 2011, 10 Search PubMed.
- G. Marlow, S. Ellett, I. R. Ferguson, S. Zhu, N. Karunasinghe, A. C. Jesuthasan, D. Y. Han, A. G. Fraser and L. R. Ferguson, Hum. Genomics, 2013, 7, 24 CrossRef PubMed.
- Affymetrix, GeneChip® PrimeView™ Human Gene Expression Array Datasheet, http://media.affymetrix.com/support/technical/datasheets/primeview_array_cartridge_datasheet.pdf, (accessed 4 Aug 2015).
- Q. Li, N. J. Birkbak, B. Gyorffy, Z. Szallasi and A. C. Eklund, BMC Bioinf., 2011, 12, 474 CrossRef PubMed.
- J. T. Chang, C. Carvalho, S. Mori, A. H. Bild, M. L. Gatza, Q. Wang, J. E. Lucas, A. Potti, P. G. Febbo, M. West and J. R. Nevins, Mol. Cell, 2009, 34, 104–114 CrossRef CAS PubMed.
- A. G. Wilson, J. Periodontol., 2008, 79, 1514–1519 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- F. J. Staal, M. van der Burg, L. F. Wessels, B. H. Barendregt, M. R. Baert, C. M. van den Burg, C. van Huffel, A. W. Langerak, V. H. van der Velden, M. J. Reinders and J. J. van Dongen, Leukemia, 2003, 17, 1324–1332 CrossRef PubMed.
- H. Auer, D. L. Newsom and K. Kornacker, Methods Mol. Biol., 2009, 509, 35–46 CAS.
- C. H.-J. Kao, K. S. Bishop, D. Y. Han, P. M. Murray, M. P. Glucina, G. J. Marlow and L. R. Ferguson, Funct. Foods Health Dis., 2014, 4, 182–207 Search PubMed.
- J. S. Yuan, A. Reed, F. Chen and C. N. Stewart Jr., BMC Bioinf., 2006, 7, 85 CrossRef PubMed.
- R. A. Irizarry, B. Hobbs, F. Collin, Y. D. Beazer-Barclay, K. J. Antonellis, U. Scherf and T. P. Speed, Biostatistics, 2003, 4, 249–264 CrossRef PubMed.
- R. F. f. S. Computing, R: A language and environment for statistical computing, http://www.R-project.org).
- Y. Benjamini and Y. Hochberg, J. R. Stat. Soc. Series B Stat. Methodol., 1995, 57, 289–300 Search PubMed.
- J. Chang and J. Nevins, Bioinformatics, 2006, 22, 2926–2933 CrossRef CAS PubMed.
- IPA, White Paper: IPA Network Generation Algorithm, 2005.
- F. Long, H. Liu, C. Hahn, P. Sumazin, M. Zhang and A. Zilberstein, In Silico Biol., 2004, 4, 0033 Search PubMed.
- J. Vandesompele, K. De Preter, F. Pattyn, B. Poppe, N. Van Roy, A. De Paepe and F. Speleman, Genome Biol., 2002, 3, 0034 CrossRef.
- R. E. Kass and A. E. Raftery, J. Am. Stat. Assoc., 1995, 90, 773–795 CrossRef.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Nat. Genet., 2000, 25, 25–29 CrossRef CAS PubMed.
- H. Lv, L. Liu, Y. Zhang, T. Song, J. Lu and X. Chen, Mol. BioSyst., 2010, 6, 2056–2067 RSC.
- G. Marlow, S. Ellett, I. R. Ferguson, S. Zhu, N. Karunasinghe, A. C. Jesuthasan, D. Y. Han, A. G. Fraser and L. R. Ferguson, Hum. Genomics, 2013, 7, 24 CrossRef PubMed.
- D. S. Welsbie, J. Xu, Y. Chen, L. Borsu, H. I. Scher, N. Rosen and C. L. Sawyers, Cancer Res., 2009, 69, 958–966 CrossRef CAS PubMed.
- G. Wilding, G. Zugmeier, C. Knabbe, K. Flanders and E. Gelmann, Mol. Cell. Endocrinol., 1989, 62, 79–87 CrossRef CAS PubMed.
- E. Smolle, V. Taucher, E. Petru and J. Haybaeck, Anticancer Res., 2014, 34, 1519–1530 CAS.
- N. Sato, H. Matsubayashi, T. Abe, N. Fukushima and M. Goggins, Clin. Cancer Res., 2005, 11, 4681–4688 CrossRef CAS PubMed.
- E. Capobianco, A. Mora, D. La Sala, A. Roberti, N. Zaki, E. Badidi, M. Taranta and C. Cinti, PLoS One, 2014, 9, e95596 Search PubMed.
- F. Wisnieski, D. Calcagno, M. Leal, E. Chen, C. Gigek, L. Santos, T. Pontes, L. Rasmussen, S. Payão, P. Assumpão, L. Loureno, S. Demachki, R. Artigiani, R. Burbano and M. Cardoso Smith, Tumor Biol., 2014, 35, 6373–6381 CrossRef CAS PubMed.
- N. Mahmud, B. Petro, S. Baluchamy, X. Li, S. Taioli, D. Lavelle, J. G. Quigley, M. Suphangul and H. Araki, Biol. Blood Marrow Transplant., 2014, 20, 480–489 CrossRef CAS PubMed.
- J. Chang, D. S. Varghese, M. C. Gillam, M. Peyton, B. Modi, R. L. Schiltz, L. Girard and E. D. Martinez, Br. J. Cancer, 2012, 106, 116–125 CrossRef CAS PubMed.
- A. Gorin, L. Gabitova and I. Astsaturov, Curr. Opin. Pharmacol., 2012, 12, 710–716 CrossRef CAS PubMed.
- S. P. Pitroda, N. N. Khodarev, M. A. Beckett, D. W. Kufe and R. R. Weichselbaum, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5837–5841 CrossRef CAS PubMed.
- P. B. Danielson, Curr. Drug Metab., 2002, 3, 561–597 CrossRef CAS PubMed.
- H. Piotrowska, M. Kucinska and M. Murias, Mol. Cell. Biochem., 2013, 383, 95–102 CrossRef CAS PubMed.
- V. D. Kekatpure, A. J. Dannenberg and K. Subbaramaiah, J. Biol. Chem., 2009, 284, 7436–7445 CrossRef CAS PubMed.
- L. A. Hooven, B. Mahadevan, C. Keshava, C. Johns, C. Pereira, D. Desai, S. Amin, A. Weston and W. M. Baird, Bioorg. Med. Chem. Lett., 2005, 15, 1283–1287 CrossRef CAS PubMed.
- A. Ilyas, Z. Hashim, N. Naeem, K. Haneef and S. Zarina, Int. J. Proteomics, 2014, 532953 Search PubMed.
- K. M. Swanson and R. J. Hohl, Curr. Cancer Drug Targets, 2006, 6, 15–37 CrossRef CAS PubMed.
- R. Thaler, S. Spitzer, H. Karlic, C. Berger, K. Klaushofer and F. Varga, Biochem. Pharmacol., 2013, 85, 173–185 CrossRef PubMed.
- E. Ricciotti and G. A. FitzGerald, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 986–1000 CrossRef CAS PubMed.
- W. L. Smith, D. L. DeWitt and R. M. Garavito, Annu. Rev. Biochem., 2000, 69, 145–182 CrossRef CAS PubMed.
- H. Yokouchi, K. Kanazawa, T. Ishida, S. Oizumi, N. Shinagawa, N. Sukoh, M. Harada, S. Ogura, M. Munakata, H. Dosaka-Akita, H. Isobe and M. Nishimura, Mol. Clin. Oncol., 2014, 2, 744–750 Search PubMed.
- I. Stasinopoulos, T. Shah, M. F. Penet, B. Krishnamachary and Z. M. Bhujwalla, Front. Pharmacol., 2013, 4, 34 Search PubMed.
- C. Luong, A. Miller, J. Barnett, J. Chow, C. Ramesha and M. F. Browner, Nat. Struct. Biol., 1996, 3, 927–933 CrossRef CAS PubMed.
- M. Annamanedi and A. M. Kalle, PLoS One, 2014, 9, e99285 Search PubMed.
- M. D. Sternlicht, A. Lochter, C. J. Sympson, B. Huey, J. P. Rougier, J. W. Gray, D. Pinkel, M. J. Bissell and Z. Werb, Cell, 1999, 98, 137–146 CrossRef CAS PubMed.
- J. Cathcart, A. Pulkoski-Gross and J. Cao, Genes Dis., 2015, 1, 26–34 CrossRef PubMed.
- R. C. Poulsen, A. J. Carr and P. A. Hulley, Biochem. Biophys. Res. Commun., 2015, 462, 78–84 CrossRef CAS PubMed.
- C. Berasain and M. A. Avila, Semin. Cell Dev. Biol., 2014, 28, 31–41 CrossRef CAS PubMed.
- F. Alimirah, J. Chen, Z. Basrawala, H. Xin and D. Choubey, Fed. Eur. Biochem. Soc., Lett., 2006, 580, 2294–2300 CrossRef CAS PubMed.
- L. Wu, J. C. Zhao, J. Kim, H.-J. Jin, C.-Y. Wang and J. Yu, Cancer Res., 2013, 73, 6068–6079 CrossRef CAS PubMed.
- X. Ye, L. Wang, B. Shang, Z. Wang and W. Wei, Curr. Cancer Drug Targets, 2014, 14, 549–556 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00554j |
| This journal is © The Royal Society of Chemistry 2016 |