
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational identification and analysis of signaling subnetworks with distinct functional roles in the regulation of TNF production†‡
Maurizio
Tomaiuolo§
a,
Melissa
Kottke
b,
Ronald W.
Matheny
Jr.
b,
Jaques
Reifman
*a and
Alexander Y.
Mitrophanov
a
aDepartment of Defense Biotechnology High Performance Computing Software Applications Institute, Telemedicine and Advanced Technology Research Center, U.S. Army Medical Research and Materiel Command, ATTN: MCMR-TT, 504 Scott Street, Fort Detrick, MD, USA. E-mail: jaques.reifman.civ@mail.mil; Fax: +1 301 619 1983; Tel: +1 301 619 7915
bMilitary Performance Division, U.S. Army Research Institute of Environmental Medicine, 15 Kansas Street, Building 42, Natick, MA 01760, USA
First published on 4th January 2016
Abstract
Inflammation is a complex process driven by the coordinated action of a vast number of pro- and anti-inflammatory molecular mediators. While experimental studies have provided an abundance of information about the properties and mechanisms of action of individual mediators, essential system-level regulatory patterns that determine the time-course of inflammation are not sufficiently understood. In particular, it is not known how the contributions from distinct signaling pathways involved in cytokine regulation combine to shape the overall inflammatory response over different time scales. We investigated the kinetics of the intra- and extracellular signaling network controlling the production of the essential pro-inflammatory cytokine, tumor necrosis factor (TNF), and its anti-inflammatory counterpart, interleukin 10 (IL-10), in a macrophage culture. To tackle the intrinsic complexity of the network, we employed a computational modeling approach using the available literature data about specific molecular interactions. Our computational model successfully captured experimentally observed short- and long-term kinetics of key inflammatory mediators. Subsequent model analysis showed that distinct subnetworks regulate IL-10 production by impacting different temporal phases of the cAMP response element-binding protein (CREB) phosphorylation. Moreover, the model revealed that functionally similar inhibitory control circuits regulate the early and late activation phases of nuclear factor κB and CREB. Finally, we identified and investigated distinct signaling subnetworks that independently control the peak height and tail height of the TNF temporal trajectories. The knowledge of such subnetwork-specific regulatory effects may facilitate therapeutic interventions aimed at precise modulation of the inflammatory response.
Background
The inflammatory process is the initial and critical phase of the mammalian response to injury and infection, and is necessary for tissue repair.1 It involves the recruitment of several types of inflammatory cells and the production of both pro- and anti-inflammatory molecular mediators, many of which are categorized as cytokines.2 The coordinated balance in the dynamics of these mediators determines the inflammation status of the tissue after injury. Uncontrolled production of inflammatory mediators results in undesired and pathological outcomes, such as chronic inflammation and sepsis.3,4 Therefore, an understanding of inflammatory response regulation is key for the development of enhanced therapeutic anti-inflammatory strategies.A large number of molecular mediators are known to be involved in inflammation. Among them, tumor necrosis factor (TNF-α, or simply TNF) is known as the primary extracellular pro-inflammatory cytokine, whose expression plays a central role in the activation of inflammation.5 TNF is produced by different cell types, but its major source is macrophages. Exposure to bacterial lipopolysaccharide (LPS), which is the most frequently used inflammatory trigger for both in vitro and in vivo experimental studies, elicits robust production of TNF by macrophages. A lack of TNF expression leads to a disorganized inflammatory response, which could result in death,6 impaired bone repair,7 or increased susceptibility to infection,8 while excessive production of TNF may lead to tissue damage.9
The activity of anti-inflammatory mediators is necessary to prevent the damage inflicted by an otherwise unrestrained inflammatory response. In particular, interleukin 10 (IL-10) is an essential anti-inflammatory cytokine with important clinical applications.10,11 The signaling downstream of IL-10 inhibits pro-inflammatory cytokine production,12 thereby acting as a tissue protector. The LPS challenge in macrophages initiates a cascade of reactions that leads to the temporally regulated production of both TNF and IL-10. While TNF controls the initial phase of the inflammatory response, the expression of IL-10 serves as a negative feedback mechanism that downregulates TNF expression.
Research using both in vitro and in vivo systems has generated a wealth of mechanistic information regarding inflammatory signaling and its regulation by cytokines.2,13 This accumulated evidence renders inflammation as a process of immense complexity involving dozens (or perhaps hundreds) of functional elements engaged in intricate interactions. While it is known that the normal (i.e., physiological) resolution of inflammation results from the action of dedicated molecular mechanisms,14 the contributions of distinct mechanisms to different phases of inflammation resolution are unknown. Specifically, it is not known how the interactions of the stimulatory and inhibitory signaling circuits acting on different time scales shape the long-term dynamics (occurring over dozens of hours) of key pro-inflammatory mediators, such as TNF. Yet, it is the details of this long-term regulation that often define the difference between normal and pathological inflammation.15
Here, we attempted to address these challenges by using a research strategy that relied on computational modeling. Such an approach offers unique advantages for the integration of diverse data sets to develop a consistent representation of inflammatory signaling. Whereas computational models have been used to investigate different aspects of the inflammatory process, the majority of published studies focus on the short-term regulation of a key transcription factor16–20 or examine inflammation without detailed intracellular reactions.21–29 None, however, have examined the long-term regulation of inflammation by key intracellular signaling pathways.
Many mechanistic details of the signaling networks involved in the long-term inflammatory response are yet unresolved. We thus attempted to determine whether a self-consistent network of molecular interactions and the corresponding kinetic model could be constructed to reproduce a broad spectrum of experimental findings describing the short- and long-term inflammatory response. We performed an extensive literature analysis and used it as a basis to develop a mathematical model reflecting the biochemical reactions occurring in LPS-challenged macrophages and leading to the production of pro-inflammatory (i.e., TNF) and anti-inflammatory (i.e., IL-10) cytokines. We used the model to gain insights into the mechanistic regulation of the long-term inflammatory response. Analysis of our model's network topology revealed functional similarities between the transcriptional control of TNF and that of IL-10. These similarities lie in the two inhibitory mechanisms regulating the activity of nuclear factor κB (NF-κB) and cAMP response element-binding protein (CREB), which act as transcription factors regulating the production of TNF and IL-10, respectively. Furthermore, we found that the temporal regulation of IL-10 production can be naturally decomposed into two phases. The first phase is controlled by the mitogen-activated protein kinase kinase (MKK)-dependent signaling subnetwork, which is regulated only by intracellular mediators. In contrast, the second phase is controlled by the interferon-β (IFN-β)-dependent subnetwork, which is regulated by both intracellular and extracellular mediators. Finally, we found that the peak height and tail height of the TNF temporal trajectories are controlled by distinct signaling subnetworks. Specifically, the peak height is controlled by the direct negative feedback exerted by the protein IκBα on NF-κB, whereas the tail height is controlled by the negative feedback exerted by IL-10 on TNF transcription. This study suggests the possibility to independently regulate distinct quantitative features of inflammatory mediators' temporal trajectories, and highlights approaches for fine-tuning the inflammatory response.
Materials and methods
Computational model and simulations
We constructed a computational model that simulates the LPS-induced inflammatory response in a macrophage culture. Specifically, the model reflects key biochemical reactions that connect the extracellular concentrations of LPS and TNF with the nuclear localization of the transcription factors NF-κB and CREB, as well as with the subsequent synthesis of pro-inflammatory (i.e., TNF) and anti-inflammatory (i.e., IFN-β and IL-10) cytokines. The model is based on mass action kinetics and comprises 78 coupled ordinary differential equations (ODEs), each of which expresses the rate of change in the concentration of a biochemical species. The model's input is the extracellular LPS concentration and its output is the concentration time course (i.e., kinetic trajectory) for each biochemical species considered. The model contains 192 parameters (Table S1, ESI‡) representing the rates of different molecular and cellular processes, such as enzyme–substrate association/dissociation and cytokine production/degradation. Where possible, we included the reactions and equations from previously developed models.16,18,19,21 All computational analyses were performed in the software suite MATLAB R2014a (MathWorks, Natick, MA), and the ODEs were solved using the ODE15S solver with an absolute tolerance of 10−8 μM and a relative tolerance of 10−6.The expression of many inflammatory genes is activated in a specific temporal order, which previous studies (see, e.g., ref. 16) modeled using ad hoc time delays. Recently published experimental data demonstrated that the transcription of NF-κB-activated genes, such as nfkbia, nfkbie, tnf, and tnfaip3, is initiated simultaneously, whereas their expression timing differences are caused by splicing delays.30 To reflect this mechanism, we modeled the temporally ordered gene activation process by including model variables for immature mRNA (pre-mRNA), mRNA bound to the spliceosome complex (smRNA), and mature mRNA. We modeled gene transcription regulation using the equations defined as follows:
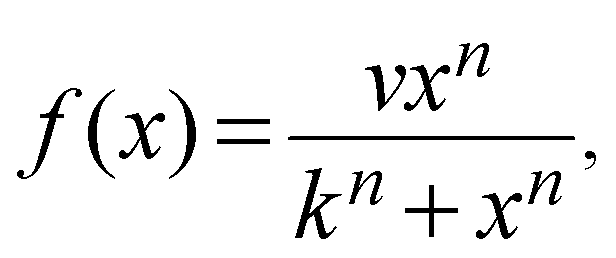 | (1) |
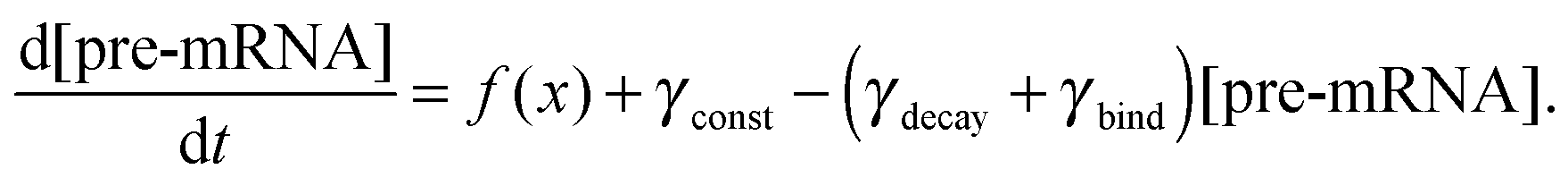 | (2) |
We modeled the transition from pre-mRNA to smRNA in the following way:
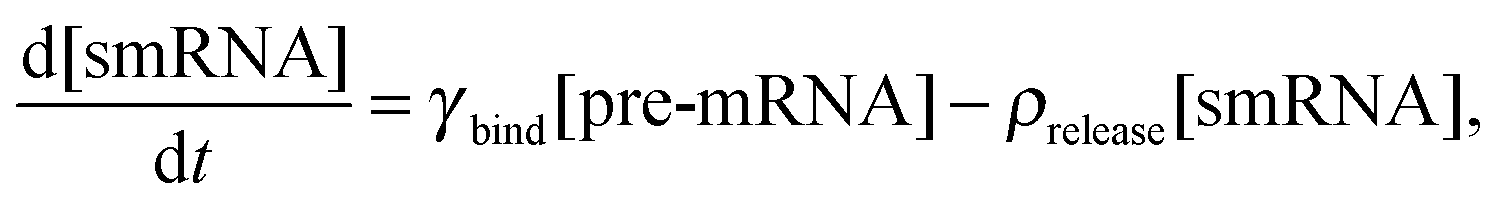 | (3) |
For all transcription factors in the model, we used eqn (1) with the same k and n values, which were selected so that the behavior of f(x) was nearly linear in the concentration range 0–100 nM. In contrast, the value of v was optimized for each individual transcription factor. We used the same approach (with the same set of k and n values) to model two additional processes. One of them was enzyme recruitment when the intermediate steps of this process were not included in the model, as in the case of the activation of myeloid differentiation primary response protein 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF)-dependent TNF receptor-associated factor 6 (TRAF6). The second process was the LPS-induced internalization of the TNF receptor.33
Our model reflected the IL-10-dependent inhibition of the TNF production. To achieve this, we modeled the signal transducer and activator of transcription 3 (STAT3)-dependent production of a repressor protein (REP) inhibiting NF-κB binding to the tnf promoter. While it is known that IL-10-mediated TNF inhibition is regulated by STAT3-dependent gene transcription, the exact regulatory mechanism or protein is not known.34,35 For this reason, the REP protein in our model represents a “placeholder” protein. We modeled the IL-10-mediated TNF inhibition using a linear function of the REP concentration:
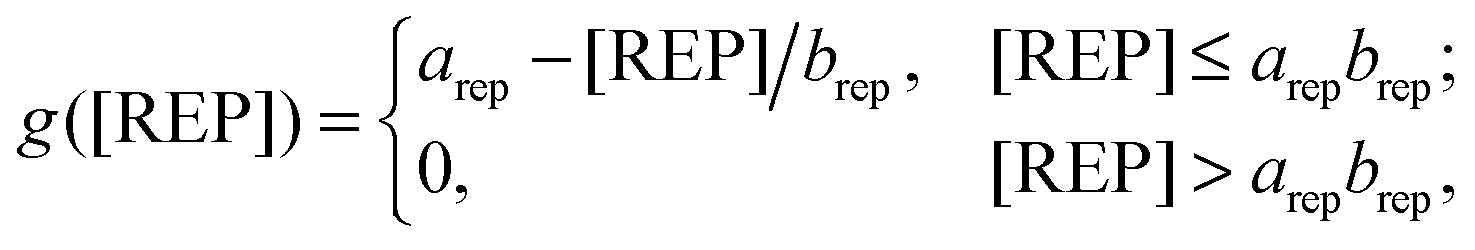 | (4) |
The initial concentrations for the biochemical species are defined in the model initial conditions. The initial conditions for model simulations were obtained as follows. We ran each simulation using a specific set of model parameters that corresponded to the wild type (WT) or to a specific gene knockout. For each specified parameter set, we first ran a simulation in the absence of any stimulatory challenge (i.e., no LPS) until it reached a steady state. In such a simulation, the initial concentrations of NF-κB, inactive IκB kinase (IKK), inactive transforming growth factor β activated kinase-1 (TAK1), and unbound toll-like receptor 4 (TLR4) were set to 0.125, 0.1, 0.1, and 0.1 μM, respectively [the concentrations for the first three species were taken from a previous study,16 and the unbound TLR4 concentration was assumed]. All other species in such a simulation were initialized at zero. From this simulation, we obtained the steady-state values of the species concentrations. We then used these steady-state values as initial conditions to simulate a challenge with 10 ng ml−1 of LPS.
We accounted for the dilution effect for the proteins being released into the extracellular compartment by multiplying the protein concentration by a constant. This multiplication effected the conversion of the intracellular to the extracellular concentrations, assuming spherical cells with a 10 μm radius and a volume of 4.18 × 10−9 ml.
Model parameter values and global optimization
Table S1 (ESI‡) gives the names, values, units, descriptions, and references for all the model parameters. The numerical values of the parameters were taken from available literature, assumed, or fitted (59, 54, and 79 parameters, respectively, of the total 192 parameters). We used assumed or fitted values for the parameters whose values could not be derived directly from available published data. We reflected the kinetics of some species by modeling the transition from an active (e.g., phosphorylated or bound to an activating ligand) to an inactive state, but we did not explicitly model their synthesis and degradation. Thus, we assumed that the total concentration of such species did not change over time; here, we refer to them as “conserved species”. As done in other studies,16,17,36 the total concentration of every conserved species was assumed to be 0.1 μM. We also assumed the values of the parameters representing the rates that were not critical for our model development. For example, the degradation of pre-mRNA was not considered, and thus the parameter value representing the rate of this process was set to zero. The binding of all the pre-mRNA species to the spliceosome complex is fast,30 thus, for all such binding rates, we assumed a single large value that was obtained after manual parameter tuning to match model output to available experimental data (Fig. S1, ESI‡). We tuned all the remaining parameters using the particle swarm constrained global optimization method,37 which is a population-based stochastic optimization technique, implemented in the MATLAB toolbox SBTOOLBOX2.38 We defined the lower and upper bounds for the parameters that needed tuning to be one order of magnitude below and above, respectively, a reference value characterizing a similar biological process for which the corresponding parameter value was known. For example, the measured value of the TNF mRNA degradation rate was 0.014 s−1.39 To calibrate the unknown value of the IFN-β mRNA degradation rate, we set the lower and upper bounds for this parameter to 0.001 and 0.1 s−1, respectively. The data used for the calibration procedure are referenced in the caption of Fig. 2 and in the “Model calibration” subsection of the Results section.Sensitivity analysis
Given the uncertainty in some of the model's parameter values, we tested the robustness of the model using single-parameter sensitivity analysis and global sensitivity analysis. To compute the single-parameter sensitivities, each kinetic trajectory of each modeled biochemical species was analyzed to extract four distinct quantitative features of response timing and intensity: the trajectory peak height, the peak time, the area under the curve, and the steady-state level. These features are informative characteristics of kinetic trajectories and are frequently used to analyze biological system behavior (see, e.g., ref. 40 and 41). Logarithmic local sensitivities were calculated according to the standard definition (see, e.g., ref. 23 and 31):sij = ∂![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logXi/∂logpj = (dXi/Xi)/(dpj/pj), logXi/∂logpj = (dXi/Xi)/(dpj/pj), | (5) |
000 simulations; for each simulation we generated a random set of parameters using the Latin hypercube sampling scheme, where the value of each parameter in the model was drawn from a uniform distribution with 50% and 200% of the default parameter value as lower and upper bounds, respectively. We evaluated the PRCCs at four time points (namely, at 1, 12, 24, and 48 hours) along the kinetic trajectory of each variable.
Results
Construction of the model network diagram
We used available literature data to construct a network of biochemical reactions with the goal of reproducing and predicting diverse experimental findings characterizing the LPS-induced macrophage signaling response (Fig. 1). The network comprised the biochemical reactions that facilitate the modulation of the pro- and anti-inflammatory cytokine production in response to an LPS challenge. To construct the network, we integrated and analyzed information from dozens of journal articles. In this process, whenever possible, we selected studies that examined the impact of specific signaling pathways on the inflammatory response in murine macrophages. As a result of this literature analysis, we proposed a comprehensive, non-redundant, and self-consistent picture of the short- and long-term regulation of pro- and anti-inflammatory cytokines, which is outlined below.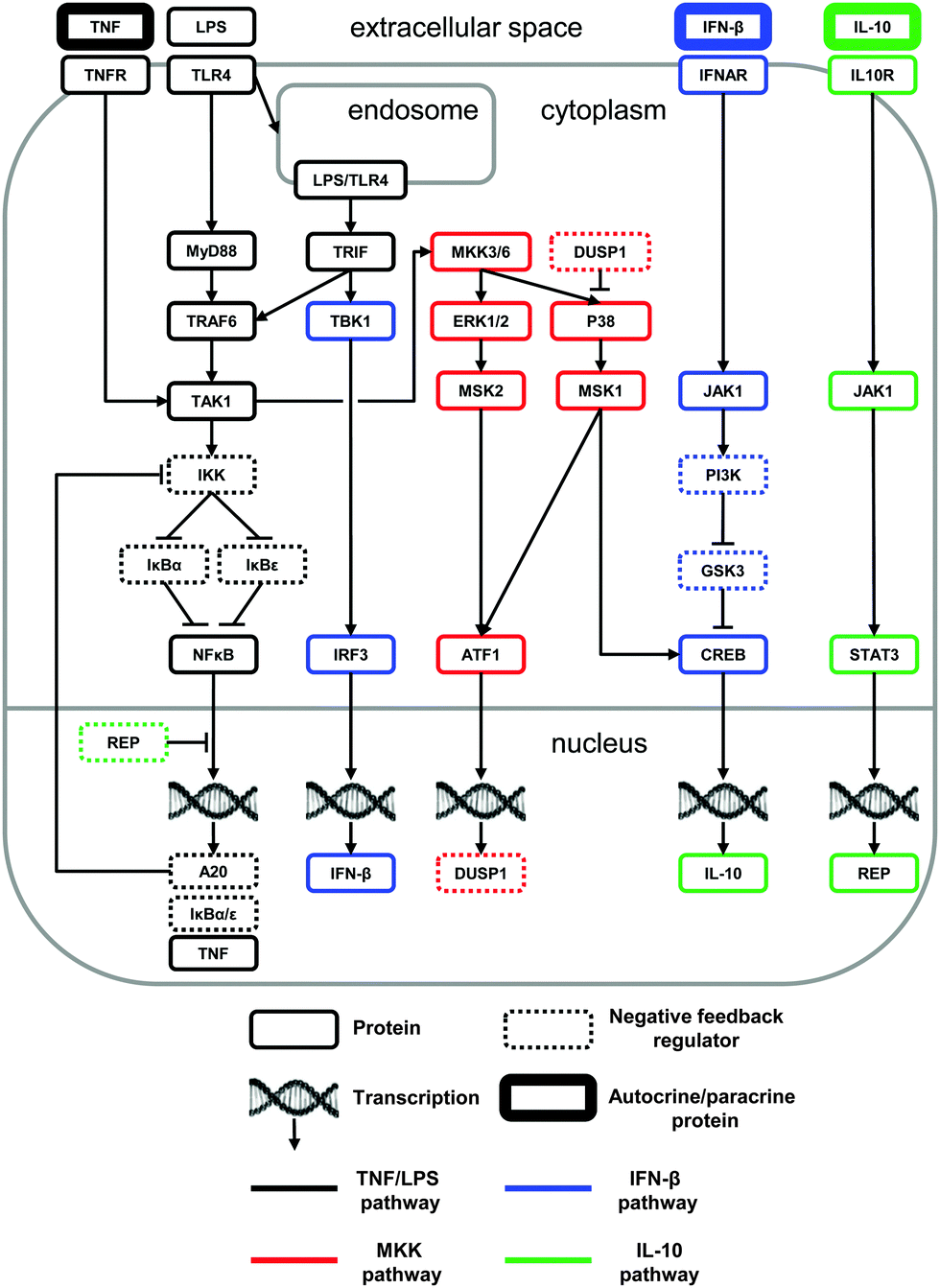 | ||
| Fig. 1 Color-coded network diagram of the signaling pathways represented in our computational model. Every protein reflected in the model is shown in the figure. The node coloring is provided to visually facilitate the network structure interpretation. The two boxes for JAK1 reflect the participation of JAK1 in distinct complexes with IFNAR and IL10R (i.e., with the cell-surface receptors for IFN-β and IL-10, respectively). See the construction of the model network diagram subsection of the Results section for a detailed description of the network components and interactions. | ||
As shown in Fig. 1, LPS activates two cytoplasmic adaptor proteins, MyD88 and TRIF.43 Activation of the MyD88-dependent pathway leads to the recruitment of TRAF6 and TAK1.44 The MyD88-independent pathway signals via TRIF after the internalization of the LPS:TLR4 complex.45 TAK1 activation results in IKK recruitment, thereby leading to the degradation of IκB proteins and freeing NF-κB to translocate to the nucleus and to initiate gene transcription.46 Although NF-κB regulates the transcription of many proteins, we restricted our attention to IκBα, IκBε, alpha-induced protein 3 (A20), and TNF. Both IκBα and IκBε act as direct negative feedback regulators of NF-κB via sequestration.36 Indirect negative feedback on NF-κB is mediated by A20 via IKK inhibition.47 We did not model other reported inhibitory mechanisms mediated by A20,48,49 because they would not affect the model's behavior.
Two additional pathways are activated by LPS signaling, resulting in the production of anti-inflammatory cytokines. The first pathway (which we term the “MKK-dependent pathway”) leads to the sequential activation of TAK1, dual specificity mitogen-activated protein kinase kinase (MKK) 3 and 6, extracellular-signal-regulated kinase (ERK) 1 and 2, mitogen-activated protein kinase (P38), and mitogen- and stress-activated protein kinase (MSK) 1 and 2 (reviewed in ref. 50). For simplicity, in our model we treated MKK3 and MKK6 as a single species (labeled MKK3/6), and we did the same with ERK1 and ERK2 (labeled ERK1/2). MSK1 and MSK2 phosphorylate two transcription factors.51 The first is cyclic AMP-dependent transcription factor 1 (ATF1), which mediates the transcription of dual-specificity phosphatase 1 (DUSP1), a negative regulator of P38 activity.52 The second is CREB, which mediates the IL-10 gene (i.e., il10) transcription.
The second pathway (which we term the “IFN-β-dependent pathway”) activated by LPS is TRIF-dependent and involves the recruitment of TANK-binding kinase 1 (TBK1), leading to the phosphorylation of interferon regulatory factor 3 (IRF3) and the expression of IFN-β.53 IFN-β signals via the Janus kinase 1 (JAK1), which is bound to the IFN-β receptor (IFNAR),54 phosphatidylinositol-3-kinase (PI3K), and glycogen synthase kinase 3 (GSK3).55 PI3K inactivates the constitutively active GSK3, which inhibits CREB phosphorylation. For modeling purposes, we considered a lack of GSK3-induced CREB inhibition as a CREB activation process.
Thus, we modeled two pathways that control the LPS-induced IL-10 production: the MKK-dependent and the IFN-β-dependent pathways. Immediately downstream of these pathways, IL-10 signals via its receptor (IL10R) and its receptor-associated JAK1, which, in combination with its associated tyrosine kinase-2, facilitates the phosphorylation of STAT3.56 This, in turn, leads to TNF downregulation, although the precise mechanism of this inhibition is unknown. In our model, STAT3 leads to the transcription of the protein REP (see the Materials and methods section) that acts as a repressor of the NF-κB-mediated TNF production.
Model calibration
We calibrated our model by adjusting (i.e., fitting) its parameters using multiple in vitro data sets (from different research groups) that were available for key model components. Whenever possible, we used experimental data from LPS-challenged murine macrophages, such as bone marrow derived macrophages, alveolar macrophages, or the RAW264.7 macrophage-derived cell line. We favored published studies where the data were recorded over a period of several hours to days.LPS-induced TNF production and secretion occurs within hours, a process followed by the production and secretion of IL-10. The result is an increase and a subsequent decrease in the TNF level. This level is modulated by IκBα/ε-mediated NF-κB inhibition in the short-term,57 and by IL-10-mediated inhibition in the long-term,58 which is further investigated in the following subsections. These dynamics were captured, both quantitatively and qualitatively, by our model (Fig. 2A and B). The initial delay in the simulated trajectory of IL-10 compared to experimental data (Fig. 2B) could have resulted from missing biochemical components in the model network diagram, from an oversimplification of the IL-10 production mechanism, or from the constraints of the multiple-parameter, multiple-data-set fitting procedure. The simulated trajectory of secreted IFN-β (Fig. 2C) was in a satisfactory agreement with the experimental IFN-β data.59
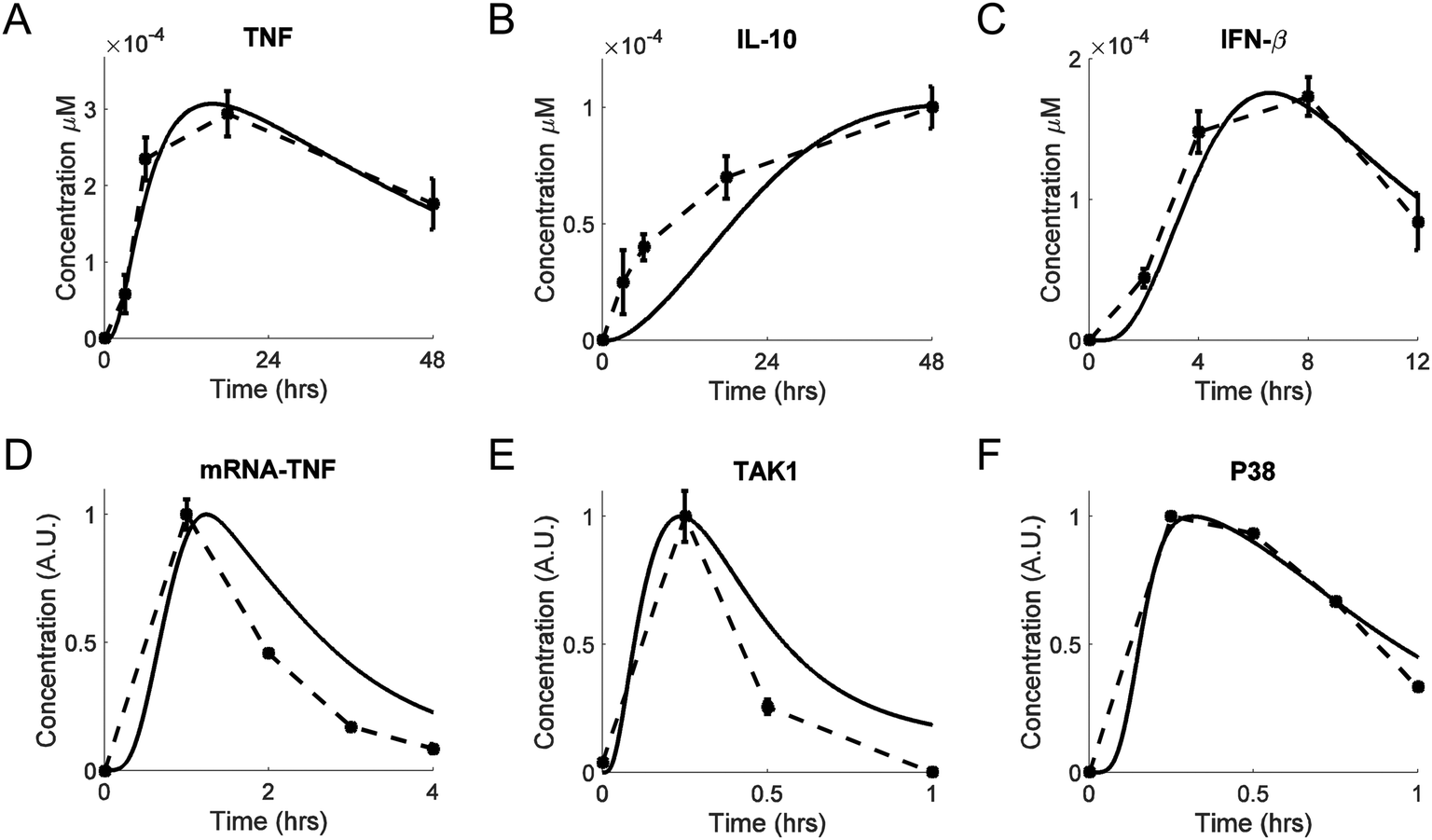 | ||
| Fig. 2 Model calibration. Shown are experimental traces (dashed lines), with error bars where available, and model fits (solid lines). (A and B) Extracellular TNF and IL-10 concentrations, respectively, measured from bone marrow-derived macrophages challenged with LPS (100 ng ml−1) for 48 h.58 (C) Extracellular IFN-β concentrations, measured from RAW264.7 cells challenged with LPS (10 ng ml−1) for 12 h.59 (D) TNF mRNA concentrations measured from RAW264.7 cells challenged with LPS (100 ng ml−1) for 4 h.83 (E) Phos-TAK1/total TAK1 concentrations measured from bone marrow-derived macrophages challenged with LPS (100 ng ml−1) for 1 h.60 (F) Phos-P38/total P38 concentrations measured from RAW 264.7 cells challenged with LPS (250 ng ml−1) for 1 h.84 In panels D, E, and F, the concentrations of the selected species are shown in normalized arbitrary units obtained by first reducing all values of a particular species by subtracting the minimum of that species, and then dividing all values of the species by the maximum value of that species. | ||
Our model correctly reproduced the short-term dynamics of the LPS-induced production of TNF mRNA (Fig. 2D) and TAK1 (Fig. 2E). The phosphorylation and dephosphorylation of TAK1 have been demonstrated experimentally,60 but they have not been modeled computationally.21 TAK1 can be dephosphorylated by several phosphatases.61–63 For modeling simplicity, we modeled TAK1 inactivation as was previously done for IKK.19 The transient activation of P38 was also matched reasonably well by our simulated P38 trajectory (Fig. 2F).
We used available data on the dynamics of unspliced (pre-mRNA) and spliced (mRNA) transcripts30 to computationally reproduce the timing differences in protein expression (Fig. S1, ESI‡). Thus, our model involves a causal mechanism to explain observed kinetic differences, instead of using artificially introduced delays lacking explanatory power.16 Moreover, our model accurately reproduced the temporal trajectories of the intermediate species following TAK1 activation. These signaling events were modeled using the equations from previously published studies.16,19,21 As expected, the model-simulated dynamics of IκBα, IκBε, A20, NF-κB, and IKK were quantitatively and qualitatively similar to the published results (results not shown).
Model validation
We performed model validation to ensure that our model can facilitate predictive analyses. As validation data sets, we selected four experimental studies in which different components of the signaling network leading to IL-10 production were blocked. LPS activates two biochemical pathways leading to the production of IL-10, namely, the MKK-dependent pathway and IFN-β-dependent pathway. The first pathway we analyzed was the sequential activation of MKK, P38, and MSK1 occurring after TAK1 activation (the MKK-dependent pathway, shown in red in Fig. 1). In this pathway, MSK1 phosphorylates CREB, thereby enabling it to translocate into the nucleus and initiate IL-10 transcription. The importance of this pathway has been established experimentally by blocking the IFN-β contribution to CREB phosphorylation and measuring the IL-10 and IL-10 mRNA production either one time after 24 h53 or at several time points.64 Our model correctly reproduced the results from both of those studies (Fig. 3A and B, respectively).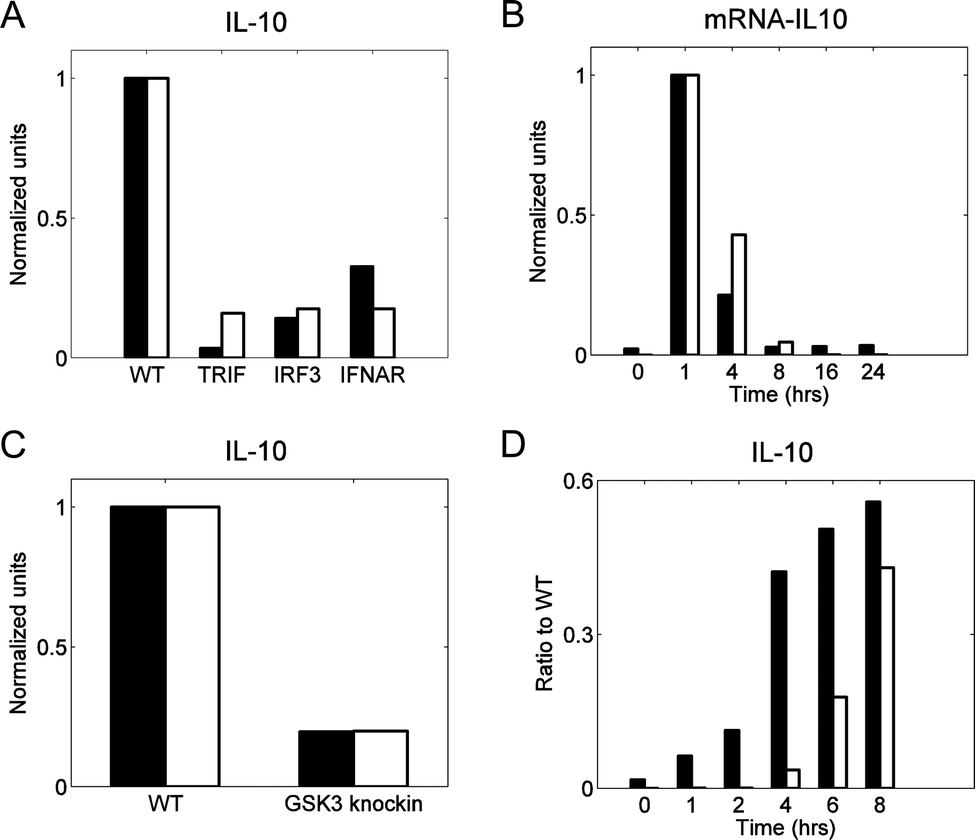 | ||
| Fig. 3 Model validation: IFN-β-dependent and MKK-dependent pathway contributions to IL-10 production. In all panels black bars represent published data, whereas white bars are the results of simulations using the calibrated model. We normalized both experimental and simulated values for comparison purposes. Normalization was obtained by first reducing all values of a species by subtracting the minimum of that species, and then dividing all values of the species by the maximum value of that species. In all studies, “WT” (wild type) represents the control case, which was simulated using the default values of the model parameters. (A) IL-10 measured from bone marrow-derived macrophages challenged with LPS (100 ng ml−1) for 24 h.53 The labels “TRIF,” “IRF3,” and “IFNAR” refer to specific knockouts of proteins upstream of IFN-β synthesis (i.e., TRIF and IRF3) or of the IFN-β receptor (i.e., IFNAR). All the knockout conditions abrogate the IFN-β-dependent pathway contribution to IL-10 production. The knockout conditions were simulated by setting to zero the model parameters representing total concentrations of TRIF, IRF3, or IFNAR. (B) IL-10 mRNA measured from bone marrow-derived macrophages challenged with LPS (100 ng ml−1) for up to 24 h.64 Both the experimental data and the simulations show the relative amount of the IL-10 mRNA for the IFN-β receptor knockout with respect to that for the control case at different time points. The receptor knockout condition was simulated by setting to zero the model parameter representing the total IFNAR concentration. (C) IL-10 measured from dendritic cells challenged with IFN-β (1000 IU ml−1) for 24 h.55 “GSK3 knockin” represents a genetic modification that prevents the phosphorylation (and, therefore, deactivation) of the constitutively active inhibitory kinase GSK3. This knockin condition was simulated by a 90% reduction in the parameter reflecting the inhibitory activity of PI3K towards GSK3. (D) IL-10 measured from bone marrow-derived macrophages challenged with LPS (100 ng ml−1) for 8 h.51 Both the experimental data and the simulations show the ratio of the IL-10 concentration for the MSK double knockout to that for the control case at each measured time point. The double knockout condition was simulated by setting to zero the concentration of MSK1/2. | ||
The second pathway we considered was the sequential activation of TRIF, TBK, and IRF3 occurring after LPS-induced activation (the IFN-β-dependent pathway, shown in blue in Fig. 1). In this pathway, phosphorylated IRF3 initiates the transcription of IFN-β, which, once secreted, binds to its receptor and activates a JAK1-dependent signaling pathway, resulting in CREB phosphorylation. The contribution of this pathway to IL-10 production has been tested by challenging dendritic cells with IFN-β while blocking the GSK3 kinase downstream of IFN-β,55 which resulted in an attenuated production of IL-10. To reproduce this result, we assumed an incomplete blockage of GSK3 activity (90%), and under this assumption, the model predictions were in a good agreement with the experimental data (Fig. 3C). In a different study, the contribution of the MKK pathway was removed by blocking the kinases MSK, and the contribution of the LPS-induced, IFN-β-dependent IL-10 production was measured.51 This resulted in delayed IL-10 synthesis, which occurred because the synthesis and secretion of IFN-β needed to occur first. The delayed synthesis of IL-10 was captured by our model with some quantitative discrepancies (Fig. 3D). This disagreement between the model and the data could have resulted from network structure simplifications in the model, from the imperfections of our model calibration strategy, or from inter-laboratory and inter-assay variability in the data sets used to calibrate and validate the model. Overall, however, the model satisfactorily reproduced the experimental behavior from a variety of genetic and pharmacologic perturbations (Fig. 3).
We tested the robustness of the model to parameter variations using local and global sensitivity analysis (see the Materials and methods section). A small value of the sensitivities, usually less than 3, indicates that the corresponding model variable is robust to a small perturbation in a parameter value and, therefore, the uncertainty in that parameter's value is unlikely to strongly affect the results.23 We used two different perturbation magnitudes (namely, 1% and 50% of the default parameter value) and computed the local sensitivities for the quantitative features calculated for the species' temporal trajectories. We found that our model was robust to local perturbations for all the four features considered, i.e., the trajectory peak height, the peak time, the area under the curve, and the steady-state level (Fig. S2 and S3, ESI‡). Indeed, only 0.13% and 12% (for the 1% and 50% perturbation magnitudes, respectively) of the 59904 computed sensitivity values exceeded a threshold value of 3. We also evaluated how the entire simulated trajectory of selected species (i.e., TNF, NF-κB, IFN-β, and IL-10) was modified after perturbing the values of each parameter in the model by ±50%. These selected species demonstrated a reasonable sensitivity to perturbations (Fig. S4, ESI‡). Taken together, these results demonstrate that the uncertainty in the parameter values is expected to only moderately affect the conclusions derived from our modeling results.
The results of the global sensitivity analysis evaluated at 1, 12, 24, and 48 h (Fig. S5–S8, ESI‡) can be briefly summarized as follows. First, the majority of the parameters had little or no effect on the model behavior. Indeed, 13662, 13700, 13701, and 13723 PRCCs (out of a total of 14976, evaluated at 1, 12, 24, and 48 h, respectively) did not exceed 0.5. Second, only a few parameters (controlling gene transcription rates) affected the kinetic trajectories of ≥20 biochemical species (out of a total of 78), which was reflected by the corresponding parameter-species PRCC exceeding 0.5. Specifically, this condition was satisfied for 3, 3, 3, and 5 parameters (out of a total of 192) evaluated at 1, 12, 24, and 48 h, respectively. Third, some parameters affected the kinetics of biochemical species (PRCC > 0.5) only at early, but not at late, time points, or vice versa (563 of the combined 59904 PRCCs evaluated for all 4 considered times). To sum up, both local and global sensitivity analyses showed that our model is generally robust to parameter perturbations.
Distinct pathways control different temporal phases of CREB phosphorylation
The transcription factor CREB can be phosphorylated by distinct kinases that are under the control of different signaling pathways (Fig. 4A). Here, we examined how the topology of those pathways shapes the kinetics of LPS-induced il10 gene transcription via CREB phosphorylation.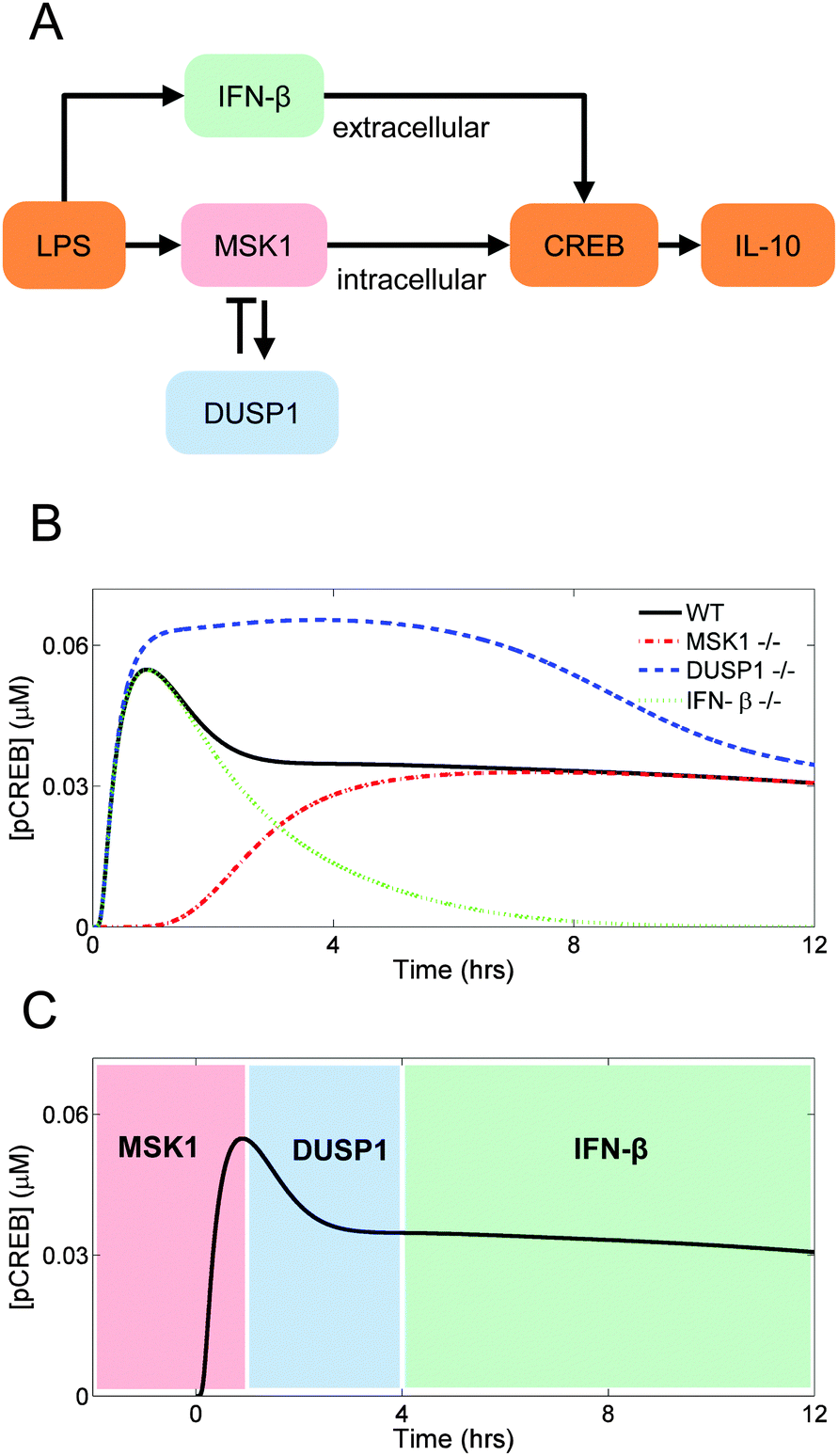 | ||
| Fig. 4 Control of CREB activation. Plotted curves represent nuclear pCREB (phosphorylated CREB). (A) Diagram illustrating the MSK1-driven (intracellular) and the IFN-β-driven (extracellular) CREB activation pathways. (B) Absence of MSK1 delays CREB phosphorylation (red dash-dotted line), absence of DUSP1 enhances CREB phosphorylation (blue dashed line), and absence of IFN-β fails to sustain the late phase of CREB phosphorylation (green dotted line). See the Results section for details about the simulation protocols. (C) Simulated trajectory of LPS-induced CREB phosphorylation illustrating the distinct phases of CREB activation under the control of MSK1, DUSP1, and IFN-β. | ||
First, we eliminated the contribution of the MSK1 pathway to CREB phosphorylation, which was accomplished by setting the MSK1 initial concentration to zero. As a result, the activation of CREB was delayed. This delay occurred because CREB phosphorylation became entirely dependent on the IFN-β-activated pathway, which needed IFN-β synthesis and secretion to occur first. This result implies that the initial rise in CREB phosphorylation is controlled primarily by MSK1 (Fig. 4B, red dash-dotted line).
Second, we simulated the absence of DUSP1 by setting to zero the dusp1 gene transcription rate. Our simulation showed that the inhibition exerted by DUSP1 on the P38 kinase activity limited the short-term CREB phosphorylation. Without DUSP1, P38 remained active for a longer time period, and thus CREB phosphorylation continued to occur over a longer period of time compared to the control case (Fig. 4B, blue dashed line).
Third, we simulated the absence of IFN-β by setting to zero the ifnb1 gene transcription rate. CREB phosphorylation showed a fast increase followed by a decrease, which were MSK1- and DUSP1-dependent, respectively. Thus, the long-term dynamics of CREB phosphorylation are controlled by IFN-β, and the contribution of the IFN-β-dependent pathway controls the sustained activation of CREB (Fig. 4B, green dotted line).
In sum, the simulated trajectory of LPS-induced CREB phosphorylation appeared to be shaped by the interactions of distinct biological mechanisms acting on different temporal phases of CREB phosphorylation and, consequently, of il10 gene transcription (Fig. 4C).
NF-κB and CREB are modulated by functionally similar, but structurally different, inhibition mechanisms
Temporal regulation via negative feedback has been demonstrated experimentally for LPS-induced NF-κB translocation to the nucleus. After an LPS challenge, NF-κB is activated and controls the transcription of several genes, including two genes, nfkbia and tnfaip3, that encode the proteins IκBα and A20, respectively, which mediate NF-κB inhibition (Fig. 5A). Specifically, IκBα controls the short-term inhibition, while A20 controls the long-term inhibition of NF-κB.16 Interestingly, our model predicted a functionally similar, but structurally different, negative regulation of CREB phosphorylation (Fig. 5A and B), as described below.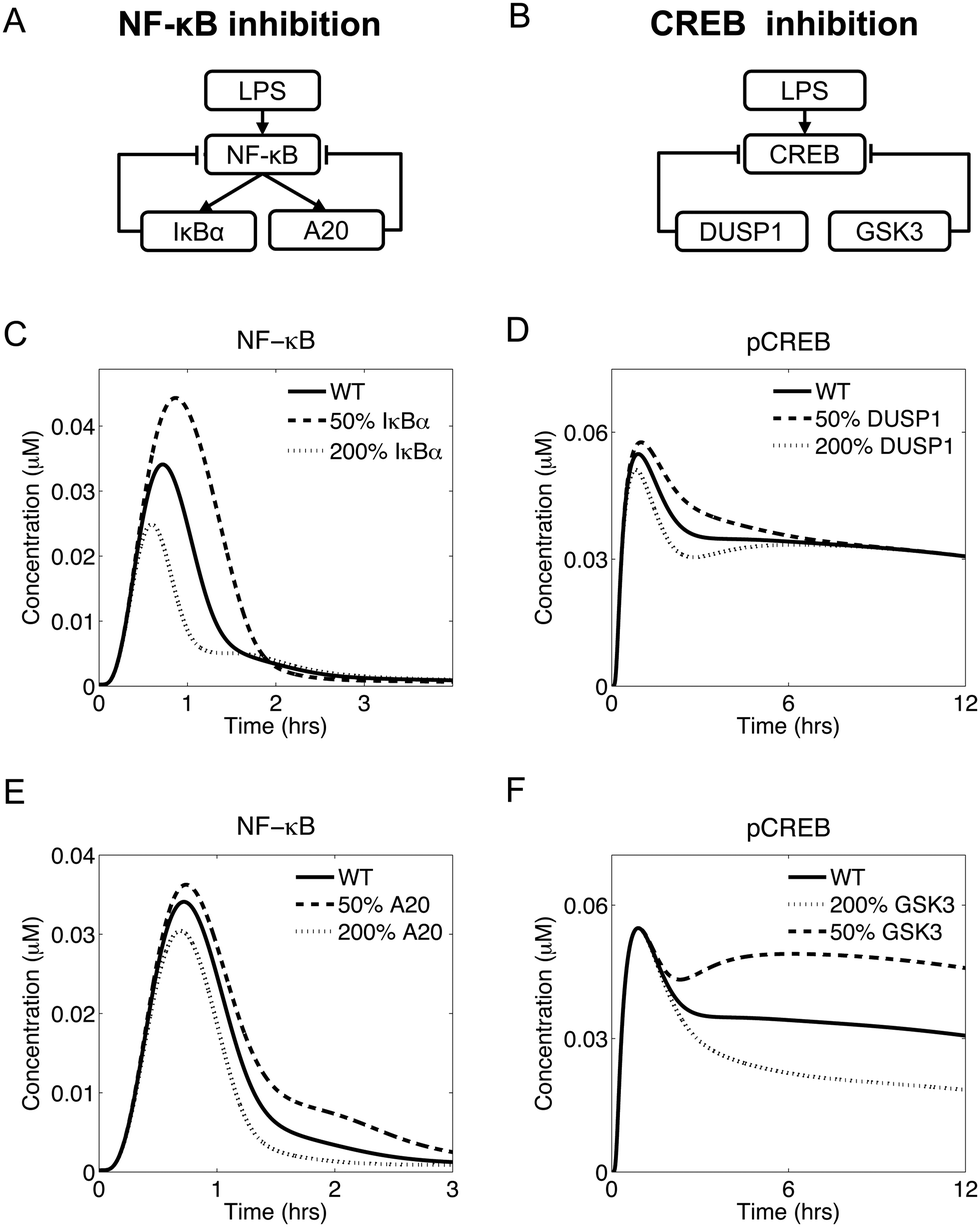 | ||
| Fig. 5 Functionally similar inhibitory control of nuclear NF-κB and pCREB. (A and B) Diagrams illustrating the direct and indirect inhibitory mechanisms regulating NF-κB and CREB activity, respectively. (C) IκBα expression modulates the early activation of NF-κB. All simulated NF-κB trajectories returned to baseline within 2 h, but displayed different levels of activation during the first hour. (D) DUSP1 expression modulated early CREB activation. DUSP1 underexpression (overexpression) resulted in enhanced (reduced) early CREB phosphorylation. (E) A20 expression controlled the late phase of NF-κB activation. When A20 was underexpressed, NF-κB took longer to return to baseline. (F) GSK3 overexpression (underexpression) led to reduced (enhanced) CREB activation. | ||
In our simulations, upregulation of IκBα (by changing the NF-κB-induced transcription rate of the nfkbia gene) led to diminished short-term NF-κB translocation to the nucleus. Conversely, IκBα downregulation led to increased short-term NF-κB activity (Fig. 5C). In contrast, the modulation of A20 expression (by changing the NF-κB-induced transcription rate of the tnfaip3 gene) regulated the long-term NF-κB activation after the initial peak (Fig. 5E). Thus, IκBα and A20 represented two negative feedback mechanisms controlling distinct phases of the NF-κB dynamics.
Modeling revealed that CREB is under the control of a functionally similar inhibitory mechanism. In our model, overexpression of DUSP1 (by changing the dusp1 gene transcription rate) led to a weaker early phase of CREB activation, while DUSP1 downregulation led to a stronger initial CREB activation (Fig. 5D). Similarly, overexpression of GSK3 (by changing the total GSK3 concentration) led to reduced long-term CREB activation, while GSK3 downregulation resulted in a more pronounced long-term CREB activation (Fig. 5F). These data suggest that DUSP1 was responsible for the initial phase of CREB activation, while GSK3 was responsible for the late phase. Thus, the roles of DUSP1 and GSK3 with respect to CREB activation were functionally similar to those of IκBα and A20, respectively, in regard to NF-κB activation.
In summary, our modeling suggests that the LPS-induced inflammatory response presented some functional similarities between the regulation of NF-κB and CREB. Indeed, both transcription factors were regulated by inhibitory mechanisms controlling different temporal phases of their activation.
Distinct signaling subnetworks regulate the peak height and tail height of the TNF temporal trajectory
We used our model network topology (Fig. 1) to examine the regulation of TNF synthesis. In our model, TNF synthesis was under the direct control of NF-κB and under the indirect control of IL-10. We investigated the differences between the direct and the indirect inhibition of TNF synthesis by examining the effects of changes in the IκBα and IL-10 levels on the kinetic trajectory of TNF. We found that the peak height and tail height of the TNF trajectory could be fine-tuned independently.First, we simulated a range of values (50–200% of the default value) for the NF-κB-induced transcription rate of the nfkbia gene and the transcription rate of the il10 gene (encoding IκBα and IL-10, respectively), to determine their effects on secreted TNF. The modulation of IκBα resulted in variations of the TNF peak height, but the peak time and the TNF tail height remained unaltered (Fig. 6A). When we followed the same protocol for a virtual IL-10 titration, the TNF peak height did not change, but the TNF tail height was altered (Fig. 6B).
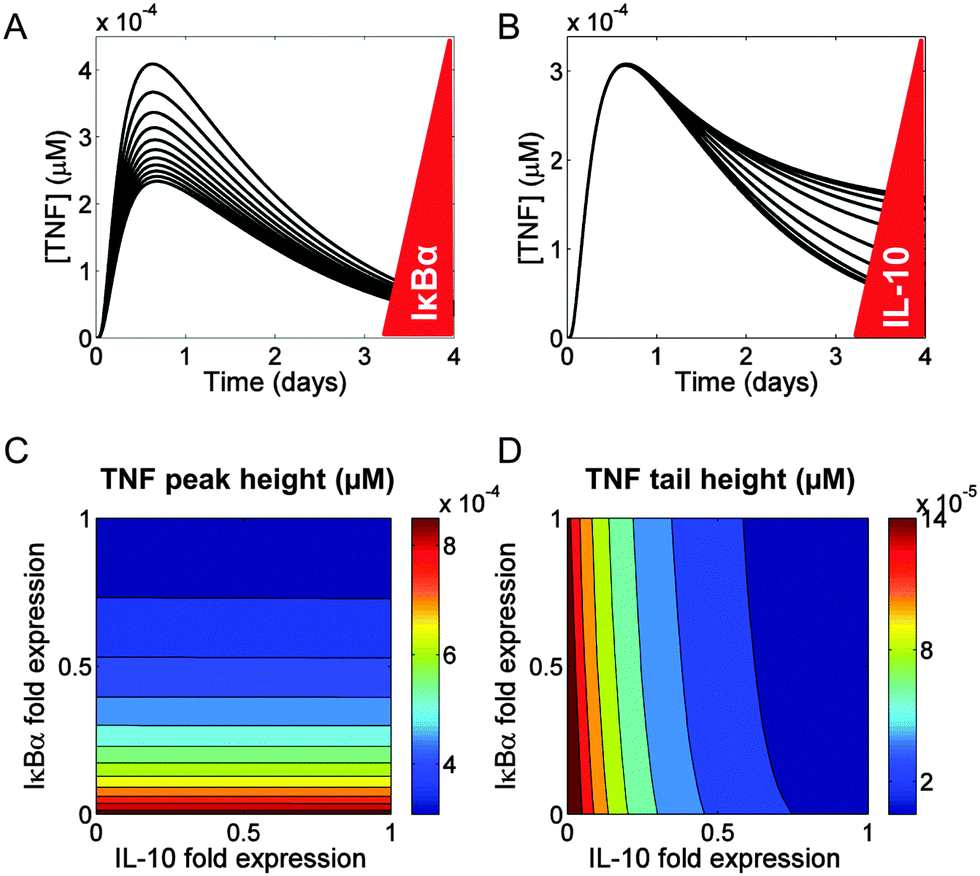 | ||
| Fig. 6 Regulation of the extracellular TNF trajectory. (A) IκBα expression modulation impacted the peak height of the TNF trajectory, but not the location of its peak. The red triangle to the right of the plot illustrates the direction of IκBα expression level decrease (i.e., upward in the plot). The bottom trajectory corresponds to IκBα expressed to 200% of the default value, while the top trajectory shows IκBα expressed to 50% of the default value. (B) Modulation of IL-10 expression affected the tail height of the TNF trajectory. The red triangle to the right of the plot illustrates the direction of IL-10 expression level decrease (i.e., upward in the plot). The bottom trajectory corresponds to IL-10 expressed to 200% of the default value, while the top trajectory shows IL-10 expressed to 50% of the default value. (C and D) Simultaneous modulation of IκBα and IL-10 expression. In both panels, we calculated fold expression by dividing the value of the transcription rate parameter for IκBα and IL-10 by their default values. The peak height of the TNF trajectory in (C) was only affected by IκBα and not by IL-10 expression levels. All the variation occurred along the vertical axis (reflecting IκBα expression). Conversely the tail height of the TNF trajectory in (D) was only affected by IL-10 and not by IκBα expression levels. | ||
To investigate the combined effects of IκBα and IL-10 on the TNF temporal trajectory, we modulated their expression at the same time. We selected two quantitative features from the TNF trajectory, the peak height and the tail height. The expression of IκBα, but not IL-10, determined the peak height in our simulations (Fig. 6C). Conversely, the expression of IL-10, but not IκBα, determined the tail height (Fig. 6D). We also found that the timing of the TNF peak was not modified by the expression changes of either IκBα or IL-10 (Fig. S9, ESI‡).
Finally, we examined the effects of specific knockouts on the TNF kinetic trajectory (Fig. 7). Changes in IκBα expression, effected by setting to zero the NF-κB-induced transcription rate of the nfkbia gene, led to early changes in the TNF trajectory (Fig. 7, dashed black line). Changes to the intracellular MKK-dependent pathway (Fig. 7, dashed red line), effected by setting to zero the rate of MSK1 binding with p38, or changes to the extracellular IFN-β-dependent pathway (Fig. 7, dashed blue line) pathway, effected by setting to zero the transcription rate of the ifnb1 gene encoding IFN-β, modified later phases of the TNF trajectory. In summary, using our model, we showed that distinct parts of the inflammatory signaling network controlled different phases and different features of TNF production.
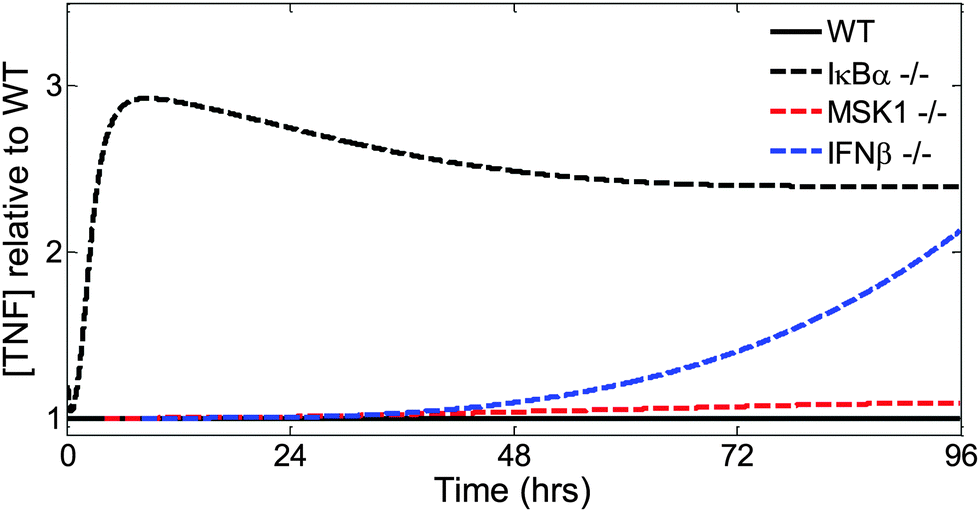 | ||
| Fig. 7 Contributions of distinct signaling subnetworks to extracellular TNF production dynamics. IκBα knockout leads to early changes in the TNF temporal trajectory. MSK1 knockout leads to weak late changes in the TNF temporal trajectory, detectable about 24 h after an LPS challenge. IFN-β knockout leads to more robust late changes in the TNF temporal trajectory. | ||
Discussion and conclusions
Robust regulation of the inflammatory response is critical for adequate resolution of inflammation65 and effective tissue repair after injury.66 Here, we applied a computational modeling approach to propose a self-consistent set of biochemical reactions reflecting the intra- and extracellular inflammatory signaling network in macrophages (Fig. 1). Our computational model was calibrated (Fig. 2) and validated (Fig. 3) using published experimental data. Using the model, we investigated the contributions of distinct signaling subnetworks to the kinetics of the inflammatory response. Model analysis demonstrated that distinct temporal phases of the phosphorylation kinetics of CREB, the essential activator of il10 transcription, were controlled by MSK1 and DUSP1 in a differential manner (Fig. 4). Moreover, our model elucidated a functional similarity between the negative regulation of NF-κB by IκBα and A20, and the negative regulation of CREB by DUSP1 and GSK3. Finally, our simulations allowed us to associate the regulation of early and late phases of TNF production with specific subnetworks of the considered signaling network (Fig. 7).Of all the numerous components and interactions involved in the regulation of inflammation, this study focused primarily on the interplay between the production and activity of TNF and IL-10. This choice of research objective naturally followed from the global regulatory logic of inflammatory signaling. Indeed, LPS, which represents pathogen-associated molecular patterns (PAMPs) that are a hallmark of pathogen-induced inflammation,67 activates the TLR4 receptor, which necessarily leads to the activation of NF-κB (Fig. 1). While other PAMP types, as well as damage-associated molecular patterns that accompany tissue injury,68 may engage other TLRs instead of (or in addition to) TLR4, the resulting signaling responses ultimately converge to NF-κB.69 NF-κB activation inevitably results in TNF induction.30 Therefore, the pro-inflammatory activity of the latter is a primary feature of the general pro-inflammatory response. IL-10 production is activated as a result of TNF activity, and also by LPS via a TNF-independent pathway50 (Fig. 1). Moreover, IL-10 is known as the main anti-inflammatory cytokine capable of counteracting the pro-inflammatory effects of TNF.70 This tight and antagonistic functional connection requires that TNF regulation be analyzed in conjunction with that of IL-10. While other prominent cytokines, such as IL-1β and IL-6, can modulate the NF-κB/TNF/IL-10 axis, they can neither override nor replace its functional impact.
Our choice to analyze a simpler network was motivated by the reductionist paradigm prevalent in molecular biology. Indeed, the signaling network centered on the NF-κB/TNF/IL-10 axis (Fig. 1) is simpler, and more amenable to analysis, than the overall network of interactions among all known cytokines. However, the complexity of this simpler network can still be prohibitive if we desire to understand how the synergy between network elements gives rise to the normal and pathological inflammation time courses. Our network overview (see the first subsection of the Results section) can serve as an illustration of the difficulties encountered by qualitative intuition attempting to predict inflammatory network kinetics. We addressed this complexity challenge by using a computational modeling approach to relate network structure with its function on different time scales. While some of our model's components were derived from earlier published studies,16–19,21 development of a mechanistically accurate kinetic model for the entire NF-κB/TNF/IL-10 axis has not been previously undertaken.
The detailed representation of signaling mechanisms in our model allowed us to tease out the contributions of specific network segments to short- and long-term dynamics of TNF production. We were particularly interested in understanding the determinants of long-term dynamics, because of their association with chronic inflammatory conditions underlying many pathologies.65,71 It can generally be expected that different pathways may exert different influence at distinct time scales, because of the delays associated with the pathways' length or the nature of their constituent biochemical reactions. Thus, it could perhaps be anticipated that IL-10 is involved at a later phase of TNF production. Our modeling analysis, however, offered deeper insights by demonstrating how the IL-10-dependent modulation of TNF can be determined by the specific network component (e.g., MSK1 or IFN-β, respectively) being modulated (Fig. 4B, C and 7). While these conclusions require direct experimental testing, partial independent validation of one of our modeling predictions comes from a study in airway smooth muscle cells.72 There, inhibition of the MSK-DUSP1 axis led to an increase in the level of phosphorylated CREB, which is consistent with our results (Fig. 4B). These modeling-based insights suggest that the coexistence of multiple IL-10 regulation pathways is evolutionarily justified by the necessity to independently fine-tune distinct quantitative characteristics of TNF and IL-10 production.
Our modeling elucidated both differences and similarities between the effects of different signaling pathway components on the specific quantitative features of the temporal trajectories for the regulated network elements. A functional similarity was detected in the regulation of NF-κB and CREB by negative regulators. Indeed, both IκBα and DUSP1 regulate the trajectory peak, but not the post-peak “tail”, of their respective targets (i.e., NF-κB and CREB), whereas A20 and GSK3 exert a comparatively weaker regulation of the peak but can also modulate the tail (Fig. 5). This similarity was not expected given that both IκBα and A20 are activated by NF-κB and therefore are involved in negative feedback loops, whereas DUSP1 and GSK3 are not activated by their target CREB.73 Extensive research into the roles of negative feedback in biological regulation has focused on the properties that distinguish feedback from simple (i.e., one-directional) negative regulation, suggesting that feedback itself typically plays a defining role in feedback-regulated regulatory circuits (see, e.g., ref. 31 and 74). Our results, however, suggest that feedback per se may not always be the main determinant of a circuit's function, and other factors may define the distinct roles of multiple regulators acting on the same target.
The differential regulation of the TNF trajectory peak height and tail height by two distinct signaling proteins (i.e., IκBα and IL-10, respectively) (Fig. 6A and B) was consistent with the notion that early and late TNF kinetics are controlled by these respective regulators. Simultaneous variation in the IκBα and IL-10 levels resulted in ∼2.5-fold changes in the TNF peak height (Fig. 6C) and in ∼10-fold change in the tail height (Fig. 6D). The same IκBα and IL-10 variation, however, resulted in only ∼1 hour change in the TNF peak time (Fig. S9, ESI‡), which suggests that the TNF peak height and tail height are more “tunable” than the TNF peak timing. Interestingly, this result is consistent with the properties of bacterial signal transduction circuits, for which the response intensity was predicted to be much more sensitive to circuit parameter variations than response time.32 These patterns support the notion that the increased controllability of response intensity compared with that of response timing may be a frequent feature of biological control circuits.
Taken together, our results underscore the possibility of differential regulation of the quantitative features and temporal phases of the inflammatory response. This possibility may be essential for the control of inflammation in pathological situations characterized by abnormally high cytokine levels (i.e., a “cytokine storm”, such as in sepsis)75 or by delayed inflammation resolution (such as in chronic inflammation).65,71 Moreover, such differential regulation may not only impact the time course of inflammation per se, but could also define the subsequent phases of wound healing, i.e., the tissue proliferation and remodeling phases.66,76 Targeted inflammation modulation may be critical for situations in which injury-induced inflammation results in delayed wound healing and hypertrophic scarring, such as severe combat injuries in military settings. While the kinetics of inflammatory signaling in vivo may differ from those in a macrophage culture, the defining role of the NF-κB/TNF/IL-10 axis is expected to be similar. Therefore, we anticipate that our findings can inform hypothesis generation aimed at understanding the regulation of inflammation kinetics and its contribution to wound healing as in vivo phenomena.
The limitations of our approach arise from the necessary model simplifications and assumptions. First, our model simulates the response of an “average” macrophage, rather than individual macrophages, to an LPS challenge. Although heterogeneity in the response of individual cells has been documented for the NF-κB signaling pathway,18 the collective response of a cell population can be approximated by employing an “average” cell model. Second, the inflammatory response is defined by the interactions between chemical mediators and the different cell types participating in the response. Our model only captures the kinetics of three extracellular mediators (i.e., TNF, IL-10, and IFN-β) produced by only one cell type (the macrophage) in response to an LPS challenge. Yet, the chemical mediators that we modeled are regarded as key players in inflammation,50,77–79 and macrophages strongly impact the kinetic trajectories of other cells participating in the response.80 Furthermore, the LPS challenge is a standard approach to experimentally study the inflammatory response both in vitro and in vivo. Thus, our modeling results reflect a simplified version of what drives inflammation. Third, our modeling results depend on the network topology and the parameter values. While the network topology represented by our model was a direct result of our analysis of the published data, the available data appear insufficient for completely adequate parameterization of all reactions in the model. This topic has been intensively studied81 and is the reason why model validation is a crucial step to assess the model's predictive power. A lack of an experimental phase aimed to validate our modeling predictions with newly generated data is another limitation of the present study. Whereas the use of experimental data from future studies may allow us to improve our model's accuracy, the model's current version provides a comprehensive representation of the known short- and long-term regulation of pro- and anti-inflammatory cytokines.
Temporal regulation of cytokine production requires further research. It is known that the short-term (hours) TNF secretion is as important for inflammation resolution as its long-term (days) inhibition.15 Research suggests that simple upregulation of anti-inflammatory mediators does not always promote the timely resolution of the inflammatory process.82 Thus, an ability to independently modulate specific phases of the trajectory of an inflammatory mediator would enable one to fine-tune the inflammatory response. Our study may provide a mechanistic framework to explain the nature of effective inflammatory regulation and to design strategies for therapeutic control of distinct temporal phases of inflammation.
Acknowledgements
The authors are grateful to Dr Sridevi Nagaraja for her assistance with global sensitivity analysis and to three anonymous reviewers for their comments on an earlier version of the paper. This work was supported by the Military Operational Medicine Research Program of the U.S. Army Medical Research and Materiel Command, Fort Detrick, MD.References
- R. Medzhitov and C. Janeway, Jr., N. Engl. J. Med., 2000, 343, 338–344 CrossRef CAS PubMed.
- C. A. Dinarello, Eur. J. Immunol., 2007, 37(suppl 1), S34–S45 CrossRef CAS PubMed.
- D. Rittirsch, M. A. Flierl and P. A. Ward, Nat. Rev. Immunol., 2008, 8, 776–787 CrossRef CAS PubMed.
- M. Murakami and T. Hirano, Front. Immunol., 2012, 3, 323 Search PubMed.
- B. B. Aggarwal, S. C. Gupta and J. H. Kim, Blood, 2012, 119, 651–665 CrossRef CAS PubMed.
- M. W. Marino, A. Dunn, D. Grail, M. Inglese, Y. Noguchi, E. Richards, A. Jungbluth, H. Wada, M. Moore, B. Williamson, S. Basu and L. J. Old, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8093–8098 CrossRef CAS.
- W. Lehmann, C. M. Edgar, K. Wang, T. J. Cho, G. L. Barnes, S. Kakar, D. T. Graves, J. M. Rueger, L. C. Gerstenfeld and T. A. Einhorn, Bone, 2005, 36, 300–310 CrossRef CAS PubMed.
- A. Zganiacz, M. Santosuosso, J. Wang, T. Yang, L. Chen, M. Anzulovic, S. Alexander, B. Gicquel, Y. Wan, J. Bramson, M. Inman and Z. Xing, J. Clin. Invest., 2004, 113, 401–413 CrossRef CAS PubMed.
- K. J. Tracey, B. Beutler, S. F. Lowry, J. Merryweather, S. Wolpe, I. W. Milsark, R. J. Hariri, T. J. Fahey, 3rd, A. Zentella and J. D. Albert, et al. , Science, 1986, 234, 470–474 CAS.
- A. O'Garra, F. J. Barrat, A. G. Castro, A. Vicari and C. Hawrylowicz, Immunol. Rev., 2008, 223, 114–131 CrossRef PubMed.
- W. Ouyang, S. Rutz, N. K. Crellin, P. A. Valdez and S. G. Hymowitz, Annu. Rev. Immunol., 2011, 29, 71–109 CrossRef CAS PubMed.
- R. de Waal Malefyt, J. Abrams, B. Bennett, C. G. Figdor and J. E. de Vries, J. Exp. Med., 1991, 174, 1209–1220 CrossRef CAS PubMed.
- S. I. Grivennikov, F. R. Greten and M. Karin, Cell, 2010, 140, 883–899 CrossRef CAS PubMed.
- C. N. Serhan and J. Savill, Nat. Immunol., 2005, 6, 1191–1197 CrossRef CAS PubMed.
- C. N. Serhan, S. D. Brain, C. D. Buckley, D. W. Gilroy, C. Haslett, L. A. O'Neill, M. Perretti, A. G. Rossi and J. L. Wallace, FASEB J., 2007, 21, 325–332 CrossRef CAS PubMed.
- S. L. Werner, J. D. Kearns, V. Zadorozhnaya, C. Lynch, E. O'Dea, M. P. Boldin, A. Ma, D. Baltimore and A. Hoffmann, Genes Dev., 2008, 22, 2093–2101 CrossRef CAS PubMed.
- S. L. Werner, D. Barken and A. Hoffmann, Science, 2005, 309, 1857–1861 CrossRef CAS PubMed.
- S. Tay, J. J. Hughey, T. K. Lee, T. Lipniacki, S. R. Quake and M. W. Covert, Nature, 2010, 466, 267–271 CrossRef CAS PubMed.
- L. Ashall, C. A. Horton, D. E. Nelson, P. Paszek, C. V. Harper, K. Sillitoe, S. Ryan, D. G. Spiller, J. F. Unitt, D. S. Broomhead, D. B. Kell, D. A. Rand, V. See and M. R. White, Science, 2009, 324, 242–246 CrossRef CAS PubMed.
- T. K. Lee, E. M. Denny, J. C. Sanghvi, J. E. Gaston, N. D. Maynard, J. J. Hughey and M. W. Covert, Sci. Signaling, 2009, 2, ra65 Search PubMed.
- M. Fallahi-Sichani, D. E. Kirschner and J. J. Linderman, Front. Physiol., 2012, 3, 170 CrossRef PubMed.
- N. A. Cilfone, C. R. Perry, D. E. Kirschner and J. J. Linderman, PLoS One, 2013, 8, e68680 CAS.
- S. Nagaraja, A. Wallqvist, J. Reifman and A. Y. Mitrophanov, J. Immunol., 2014, 192, 1824–1834 CrossRef CAS PubMed.
- M. Fallahi-Sichani, M. El-Kebir, S. Marino, D. E. Kirschner and J. J. Linderman, J. Immunol., 2011, 186, 3472–3483 CrossRef CAS PubMed.
- M. Rodriguez-Fernandez, B. Grosman, T. M. Yuraszeck, B. G. Helwig, L. R. Leon and F. J. Doyle, 3rd, PLoS One, 2013, 8, e73393 CAS.
- S. Mathew, J. Bartels, I. Banerjee and Y. Vodovotz, J. Theor. Biol., 2014, 358, 132–148 CrossRef CAS PubMed.
- P. T. Foteinou, S. E. Calvano, S. F. Lowry and I. P. Androulakis, PLoS One, 2009, 4, e4706 Search PubMed.
- X. Dong, P. T. Foteinou, S. E. Calvano, S. F. Lowry and I. P. Androulakis, PLoS One, 2010, 5, e9249 Search PubMed.
- Y. Vodovotz, G. Constantine, J. Rubin, M. Csete, E. O. Voit and G. An, Math. Biosci., 2009, 217, 1–10 CrossRef PubMed.
- S. Hao and D. Baltimore, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11934–11939 CrossRef CAS PubMed.
- A. Y. Mitrophanov, G. Churchward and M. Borodovsky, J. Theor. Biol., 2007, 246, 113–128 CrossRef CAS PubMed.
- A. Y. Mitrophanov and E. A. Groisman, J. Mol. Biol., 2010, 396, 1398–1409 CrossRef CAS PubMed.
- A. H. Ding, E. Sanchez, S. Srimal and C. F. Nathan, J. Biol. Chem., 1989, 264, 3924–3929 CAS.
- A. P. Hutchins, D. Diez and D. Miranda-Saavedra, Briefings Funct. Genomics, 2013, 12, 489–498 CrossRef CAS PubMed.
- K. C. El Kasmi, J. Holst, M. Coffre, L. Mielke, A. de Pauw, N. Lhocine, A. M. Smith, R. Rutschman, D. Kaushal, Y. Shen, T. Suda, R. P. Donnelly, M. G. Myers, Jr., W. Alexander, D. A. Vignali, S. S. Watowich, M. Ernst, D. J. Hilton and P. J. Murray, J. Immunol., 2006, 177, 7880–7888 CrossRef CAS.
- A. Hoffmann, A. Levchenko, M. L. Scott and D. Baltimore, Science, 2002, 298, 1241–1245 CrossRef CAS PubMed.
- A. I. Vaz and L. N. Vicente, J. Glob. Optim., 2007, 39, 197–219 CrossRef.
- H. Schmidt and M. Jirstrand, Bioinformatics, 2006, 22, 514–515 CrossRef CAS PubMed.
- J. R. Pfeiffer, B. L. McAvoy, R. E. Fecteau, K. M. Deleault and S. A. Brooks, Mol. Cell. Biol., 2011, 31, 277–286 CrossRef CAS PubMed.
- M. Tomaiuolo, R. Bertram, A. E. Gonzalez-Iglesias and J. Tabak, J. Neuroendocrinol., 2010, 22, 1279–1289 CrossRef CAS PubMed.
- A. Y. Mitrophanov, F. R. Rosendaal and J. Reifman, Transfusion, 2012, 52, 2475–2486 CrossRef PubMed.
- S. Marino, I. B. Hogue, C. J. Ray and D. E. Kirschner, J. Theor. Biol., 2008, 254, 178–196 CrossRef PubMed.
- S. Akira and K. Takeda, Nat. Rev. Immunol., 2004, 4, 499–511 CrossRef CAS PubMed.
- K. Takeda, T. Kaisho and S. Akira, Annu. Rev. Immunol., 2003, 21, 335–376 CrossRef CAS PubMed.
- J. C. Kagan, T. Su, T. Horng, A. Chow, S. Akira and R. Medzhitov, Nat. Immunol., 2008, 9, 361–368 CrossRef CAS PubMed.
- S. Ghosh, M. J. May and E. B. Kopp, Annu. Rev. Immunol., 1998, 16, 225–260 CrossRef CAS PubMed.
- B. Skaug, J. Chen, F. Du, J. He, A. Ma and Z. J. Chen, Mol. Cell, 2011, 44, 559–571 CrossRef CAS PubMed.
- K. Newton, M. L. Matsumoto, I. E. Wertz, D. S. Kirkpatrick, J. R. Lill, J. Tan, D. Dugger, N. Gordon, S. S. Sidhu, F. A. Fellouse, L. Komuves, D. M. French, R. E. Ferrando, C. Lam, D. Compaan, C. Yu, I. Bosanac, S. G. Hymowitz, R. F. Kelley and V. M. Dixit, Cell, 2008, 134, 668–678 CrossRef CAS PubMed.
- N. Shembade, A. Ma and E. W. Harhaj, Science, 2010, 327, 1135–1139 CrossRef CAS PubMed.
- M. Saraiva and A. O'Garra, Nat. Rev. Immunol., 2010, 10, 170–181 CrossRef CAS PubMed.
- O. Ananieva, J. Darragh, C. Johansen, J. M. Carr, J. McIlrath, J. M. Park, A. Wingate, C. E. Monk, R. Toth, S. G. Santos, L. Iversen and J. S. Arthur, Nat. Immunol., 2008, 9, 1028–1036 CrossRef CAS PubMed.
- Q. Zhao, E. G. Shepherd, M. E. Manson, L. D. Nelin, A. Sorokin and Y. Liu, J. Biol. Chem., 2005, 280, 8101–8108 CrossRef CAS PubMed.
- E. Y. Chang, B. Guo, S. E. Doyle and G. Cheng, J. Immunol., 2007, 178, 6705–6709 CrossRef CAS.
- T. Decker, M. Muller and S. Stockinger, Nat. Rev. Immunol., 2005, 5, 675–687 CrossRef CAS PubMed.
- H. Wang, J. Brown, C. A. Garcia, Y. Tang, M. R. Benakanakere, T. Greenway, P. Alard, D. F. Kinane and M. Martin, J. Immunol., 2011, 186, 675–684 CrossRef CAS PubMed.
- X. Hu, J. Chen, L. Wang and L. B. Ivashkiv, J. Leukocyte Biol., 2007, 82, 237–243 CrossRef CAS PubMed.
- S. L. Werner, J. D. Kearns, V. Zadorozhnaya, C. Lynch, E. O'Dea, M. P. Boldin, A. Ma, D. Baltimore and A. Hoffmann, Genes Dev., 2008, 22, 2093–2101 CrossRef CAS PubMed.
- J. M. Yuk, D. M. Shin, H. M. Lee, J. J. Kim, S. W. Kim, H. S. Jin, C. S. Yang, K. A. Park, D. Chanda, D. K. Kim, S. M. Huang, S. K. Lee, C. H. Lee, J. M. Kim, C. H. Song, S. Y. Lee, G. M. Hur, D. D. Moore, H. S. Choi and E. K. Jo, Nat. Immunol., 2011, 12, 742–751 CrossRef CAS PubMed.
- H. Qin, C. A. Wilson, S. J. Lee, X. Zhao and E. N. Benveniste, Blood, 2005, 106, 3114–3122 CrossRef CAS PubMed.
- C. Tiruppathi, D. Soni, D. M. Wang, J. Xue, V. Singh, P. B. Thippegowda, B. P. Cheppudira, R. K. Mishra, A. Debroy, Z. Qian, K. Bachmaier, Y. Y. Zhao, J. W. Christman, S. M. Vogel, A. Ma and A. B. Malik, Nat. Immunol., 2014, 15, 239–247 CrossRef CAS PubMed.
- S. I. Kim, J. H. Kwak, L. Wang and M. E. Choi, J. Biol. Chem., 2008, 283, 10753–10763 CrossRef CAS PubMed.
- H. Zheng, Q. Li, R. Chen, J. Zhang, Y. Ran, X. He, S. Li and H. B. Shu, J. Biol. Chem., 2013, 288, 819–825 CrossRef CAS PubMed.
- T. Kajino, H. Ren, S. Iemura, T. Natsume, B. Stefansson, D. L. Brautigan, K. Matsumoto and J. Ninomiya-Tsuji, J. Biol. Chem., 2006, 281, 39891–39896 CrossRef CAS PubMed.
- M. J. Pattison, K. F. Mackenzie and J. S. Arthur, J. Immunol., 2012, 189, 2784–2792 CrossRef CAS PubMed.
- C. Nathan and A. Ding, Cell, 2010, 140, 871–882 CrossRef CAS PubMed.
- S. A. Eming, T. Krieg and J. M. Davidson, J. Invest. Dermatol., 2007, 127, 514–525 CrossRef CAS PubMed.
- H. Kumar, T. Kawai and S. Akira, Int. Rev. Immunol., 2011, 30, 16–34 CrossRef CAS PubMed.
- M. T. Lotze, H. J. Zeh, A. Rubartelli, L. J. Sparvero, A. A. Amoscato, N. R. Washburn, M. E. Devera, X. Liang, M. Tor and T. Billiar, Immunol. Rev., 2007, 220, 60–81 CrossRef CAS PubMed.
- K. Takeda and S. Akira, Semin. Immunol., 2004, 16, 3–9 CrossRef CAS PubMed.
- J. G. Bode, C. Ehlting and D. Haussinger, Cell. Signalling, 2012, 24, 1185–1194 CrossRef CAS PubMed.
- P. Hunter, EMBO Rep., 2012, 13, 968–970 CrossRef CAS PubMed.
- W. Che, M. Manetsch, T. Quante, M. M. Rahman, B. S. Patel, Q. Ge and A. J. Ammit, Biochim. Biophys. Acta, 2012, 1823, 1658–1665 CrossRef CAS PubMed.
- A. Y. Wen, K. M. Sakamoto and L. S. Miller, J. Immunol., 2010, 185, 6413–6419 CrossRef CAS PubMed.
- O. Brandman and T. Meyer, Science, 2008, 322, 390–395 CrossRef CAS PubMed.
- J. R. Tisoncik, M. J. Korth, C. P. Simmons, J. Farrar, T. R. Martin and M. G. Katze, Microbiol. Mol. Biol. Rev., 2012, 76, 16–32 CrossRef CAS PubMed.
- M. L. Novak and T. J. Koh, Am. J. Pathol., 2013, 183, 1352–1363 CrossRef CAS PubMed.
- H. Wajant, K. Pfizenmaier and P. Scheurich, Cell Death Differ., 2003, 10, 45–65 CrossRef CAS PubMed.
- L. B. Ivashkiv and L. T. Donlin, Nat. Rev. Immunol., 2014, 14, 36–49 CrossRef CAS PubMed.
- J. J. O'Shea and P. J. Murray, Immunity, 2008, 28, 477–487 CrossRef PubMed.
- P. J. Murray and T. A. Wynn, Nat. Rev. Immunol., 2011, 11, 723–737 CrossRef CAS PubMed.
- P. Meyer, T. Cokelaer, D. Chandran, K. H. Kim, P. R. Loh, G. Tucker, M. Lipson, B. Berger, C. Kreutz, A. Raue, B. Steiert, J. Timmer, E. Bilal, Dream Six, Seven Parameter Estimation Consortium, H. M. Sauro, G. Stolovitzky and J. Saez-Rodriguez, BMC Syst. Biol., 2014, 8, 13 CrossRef PubMed.
- F. R. Greten, M. C. Arkan, J. Bollrath, L. C. Hsu, J. Goode, C. Miething, S. I. Goktuna, M. Neuenhahn, J. Fierer, S. Paxian, N. Van Rooijen, Y. Xu, T. O'Cain, B. B. Jaffee, D. H. Busch, J. Duyster, R. M. Schmid, L. Eckmann and M. Karin, Cell, 2007, 130, 918–931 CrossRef CAS PubMed.
- R. M. Rowlett, C. A. Chrestensen, M. Nyce, M. G. Harp, J. W. Pelo, F. Cominelli, P. B. Ernst, T. T. Pizarro, T. W. Sturgill and M. T. Worthington, Am. J. Physiol.: Gastrointest. Liver Physiol., 2008, 294, G452–G459 CrossRef CAS PubMed.
- X. Xia, J. Cui, H. Y. Wang, L. Zhu, S. Matsueda, Q. Wang, X. Yang, J. Hong, Z. Songyang, Z. J. Chen and R. F. Wang, Immunity, 2011, 34, 843–853 CrossRef CAS PubMed.
Footnotes |
| † The opinions and assertions contained herein are private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Army or of the U.S. Department of Defense. This article has been approved for public release with unlimited distribution. |
| ‡ Electronic supplementary information (ESI) available: Tables S1–S3, Fig. S1–S9, MATLAB code description, and MATLAB code. See DOI: 10.1039/c5mb00456j |
| § Current address: Department of Medicine, University of Pennsylvania, Philadelphia, PA, USA. |
| This journal is © The Royal Society of Chemistry 2016 |