
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective reaction monitoring utilizing two-dimensional heart-cut liquid chromatography on an integrated microfluidic chip†
Carsten
Lotter
,
Elisabeth
Poehler
,
Josef J.
Heiland
,
Laura
Mauritz
and
Detlev
Belder
 *
*
Institute of Analytical Chemistry, Leipzig University, Linnéstraße 3, 04103 Leipzig, Germany. E-mail: belder@uni-leipzig.de; Tel: +49 341 97 36091
First published on 8th November 2016
Abstract
Chip-integrated, two-dimensional high performance liquid chromatography is introduced to monitor enantioselective continuous micro-flow synthesis. The herein described development of the first two-dimensional HPLC-chip was realized by the integration of two different columns packed with reversed-phase and chiral stationary phase material on a microfluidic glass chip, coupled to mass spectrometry. Directed steering of the micro-flows at the joining transfer cross enabled a heart-cut operation mode to transfer the chiral compound of interest from the first to the second chromatographic dimension. This allows for an interference-free determination of the enantiomeric excess by seamless hyphenation to electrospray mass spectrometry. The application for rapid reaction optimization at micro-flow conditions is exemplarily shown for the asymmetric organocatalytic continuous micro-flow synthesis of warfarin.
Integrated microfluidic systems combining different functionalities such as reaction compartments and analytical tools for real time information are regarded as an enabling technology for the future of chemical synthesis.1–4 With such devices the development of optimal conditions and the identification of new chemical routes can be performed at significantly higher speed and lower resource consumption compared to conventional technologies. For such automated chemical devices continuous flow chemistry is very appealing.5–18 Due to its modular nature it can be combined with downstream analytical tools19–22 to realize self-optimizing and self-controlling machines. In this context, integrated lab-on-a-chip devices are very attractive as they allow a seamless integration of various analytical and synthetic functionalities.23–32
While such miniaturized chemical flow processes can be monitored spectroscopically,20,26 the often mandatory separation of compounds from crude reaction mixtures in real time poses a challenge. Such separation techniques are especially mandatory when compounds cannot be distinguished by spectroscopic means alone, as in the important case of asymmetric syntheses.33–35
For this purpose chip-based flow reactors have been combined with electrophoresis,36,37 which has however a rather limited application range in organic syntheses. In order to study a broad range of organic reactions, the use of high performance liquid chromatography (HPLC), preferably with mass spectrometric detection (MS), is much more appealing. To date the highly demanded combination of continuous flow chemistry and HPLC is mostly performed off-chip with common chromatographic instrumentation.29,30 The rather slow development in the area of chemical chip laboratories with integrated HPLC functionality can be explained with technical constraints. The integration of chip-based liquid chromatography is by far more technically demanding than chip-electrophoresis.
The realization of on-chip liquid chromatography poses different challenges depending on the applied column technology.38 Open tubular liquid chromatography (OTLC) utilizing open channels as in chip electrophoresis was applied in the very first chip-LC device.39 While OTLC is technically rather straightforward the integration of columns filled with stationary phases is more complex and can cause high back pressures which complicates world-to-chip interfacing.40,41 While monoliths and pillar arrays can be generated on-chip, the introduction and immobilization of packed particulate columns is more challenging. Although technically most demanding, slurry packed chip columns, using common particulate HPLC-phases, are very attractive as it facilities method transfer from classical HPLC. Furthermore, with the commercial availability of a countless selection of various phase materials, with different selectivity and proven performance, it opens up an unmatched application range to chip-HPLC, including chiral and multidimensional separation as disclosed herein.
Besides the very broad application range and the straightforward coupling to mass spectrometry, another very appealing feature of HPLC is the use of multidimensional chromatography by column coupling. Two-dimensional HPLC is especially useful for the reliable quantitative determination of enantiomers in complex mixtures.42–44 While one dimensional chip-HPLC is the current state of the art,38,40,45–50 there is very little to no progress in the development of respective chip-integrated multidimensional methods.51
Herein, we present an approach to realize what is to the best of our knowledge the first 2D-LC chip. This is achieved by sequential coupling of a reversed-phase and a chiral separation column on a single device. The essential sample transfer from one column to the other in a heart-cut mode is realized by directed flow steering at the microfluidic transfer cross, interconnecting the columns. This process builds up on a low pressure sample injection strategy described earlier, where a channel cross is used for sample injection and flow splitting.52 In the current work the fluid flows at the cross are steered via a 6-port valve and 2 HPLC pumps, as schematically shown in Fig. 1. In elution mode (A) the cross is flushed with fresh solvent which deflects the eluate from the first dimension to the waste and supplies fresh eluent to the second dimension. To transfer an analyte band from the first to the second dimension, the 6-port valve is switched to position B. This closes off the open channel of the cross, allowing a direct transfer of the first dimensional eluate onto the column head of the second dimension. By switching back the 6-port valve, the sample transfer is stopped and the elution in the second dimension begins. This process can be monitored by video microscopy as shown in Fig. 1, with respective fluorescence microscopy video images for the transfer of 7-amino-4-methylcoumarin from the first to the second column.
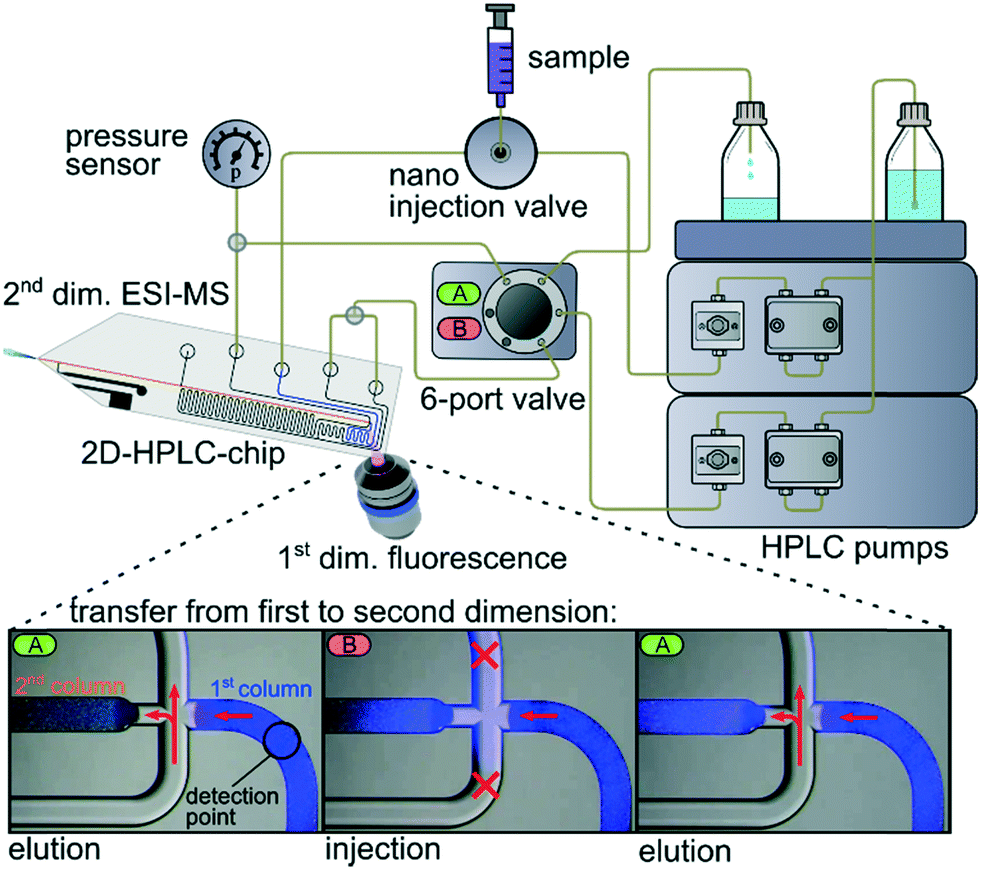 | ||
| Fig. 1 Schematic drawing of the instrumental setup and operation principal for heart-cut 2D chip LC/MS. Connection lines in the top-part represent PEEK capillaries attached via clamps to the chip. Bottom: Fluorescence microscopic images of the on-chip cross, depicting the actual transfer of 7-amino-4-methylcoumarin from the first to the second column. The fluid flow situations are illustrated with red symbols: closed line and stopped flow (cross); flow direction (arrows). | ||
The setup further includes a 5 nL injection valve for sample injection onto the first column and a miniaturized epifluorescence microscope tracing analytes at the end of the first column. This fluorescence signal can be used to trigger the valve for sample transfer. This triggering should also be feasible with other techniques like Raman signals, changes in refractive index or absorbance and of course by simple time control as applied below. The end of the second column transfers seamlessly into the integrated monolithic electrospray emitter for mass spectrometry coupling. As particulate columns, a reversed-phase material (ProntoSIL C18 SH, 5 μm) was used in the first dimension and a chiral stationary phase (CSP, CHIRALPAK IB 5 μm) in the second dimension. The columns were generated via a slurry packing technique and retained within the chip by monolithic frits, as disclosed in more detail in the ESI.53†
After developing and realizing the whole setup, including the 2D-HPLC-MS glass chip as shown in the photographic image in Fig. 2a, the proper 2D-separation functionality was examined. In these initial studies we utilized fluorescent model compounds to monitor and document the microfluidic processes via epi-fluorescence microscopy. For this purpose a mixture of 2 μg mL−1 7-amino-4-methylcoumarin, 10 μg mL−1 (R)-Pirkle's alcohol, 20 μg mL−1 (S)-Pirkle's alcohol and 20 μg mL−1 anthracene was dissolved in MeOH/H2O (50/50 vol%) and separated on the first chip column in reversed phase mode. As shown in a representative chromatogram in Fig. 2b, obtained by monitoring the fluorescence intensity at the column end, nearly baseline separation is achieved on the 30 mm short RP column. The fluorescence signal now triggers the transfer of the second elution compound, which is a enantiomeric mixture of Pirkle's alcohol, onto the chiral stationary phase column. A representative chromatogram, recorded via epi-fluorescence microscopy at the end of the second column is shown in Fig. 2b revealing that a baseline separation of the transferred enantiomers can be achieved on the only 35 mm long CSP column.
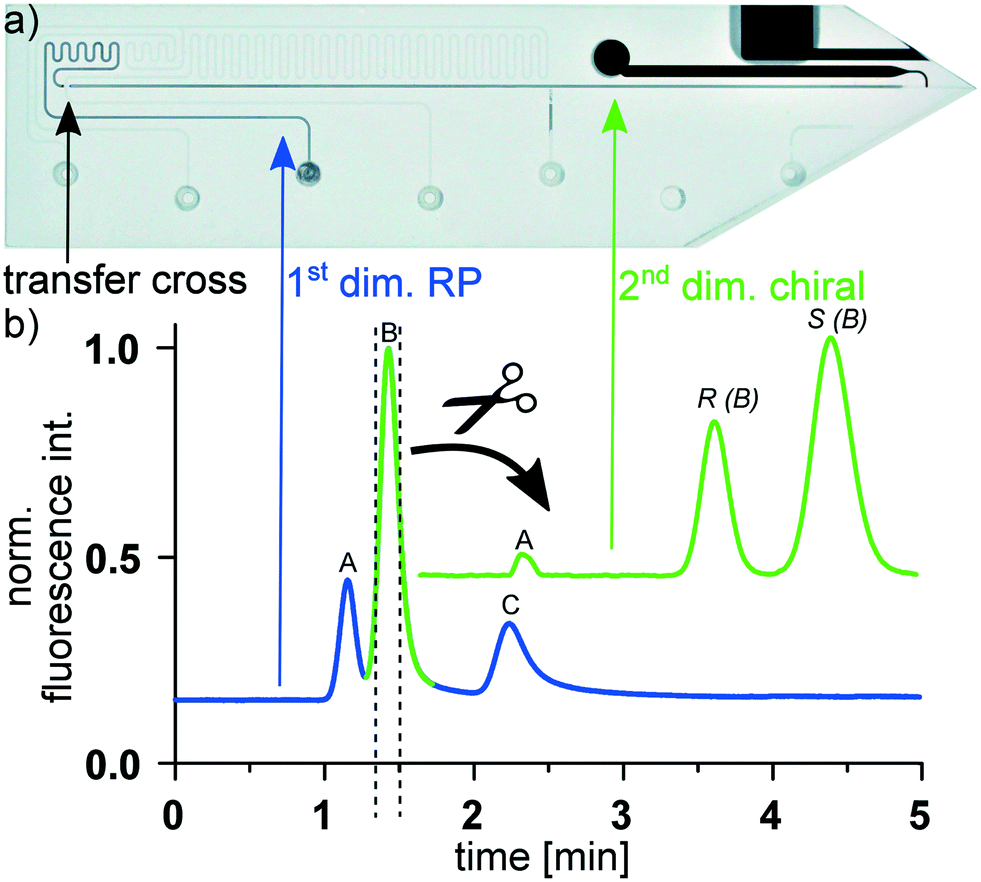 | ||
| Fig. 2 a) Photograph of the utilized glass chip. Dimensions: 10 × 45 × 2.2 mm. b) Two-dimensional heart-cut separation of the compounds: (A) 7-amino-4-methylcoumarin, (B) Pirkle's alcohol and (C) anthracene. Blue line: Reversed phase separation in first dimension. Green line: Chiral separation of transferred Pirkle's alcohol in second dimension. On-chip columns: ProntoSIL C18, 5 μm, 30 mm; CHIRALPAK IB, 5 μm, 35 mm. Elution: MeOH/H2O (70/30 vol%) with 0.1% formic acid; 1.5 μl min−1 in first dimension, 60 μl min−1 at chip cross for second dimension. Epi-fluorescence detection: λEX = 360–370 nm; λEM ≥ 390 nm. | ||
After successfully demonstrating the functionality of the 2D-HPLC-chip with model compounds, we further developed the device to monitor an organocatalytic asymmetric continuous flow reaction downstream of a chip reactor. For this purpose we coupled a flow reactor chip via the 5 nL injection valve to the 2D-LC chip and utilized mass spectrometry for detection after the second dimension as depicted schematically in Fig. 3.48
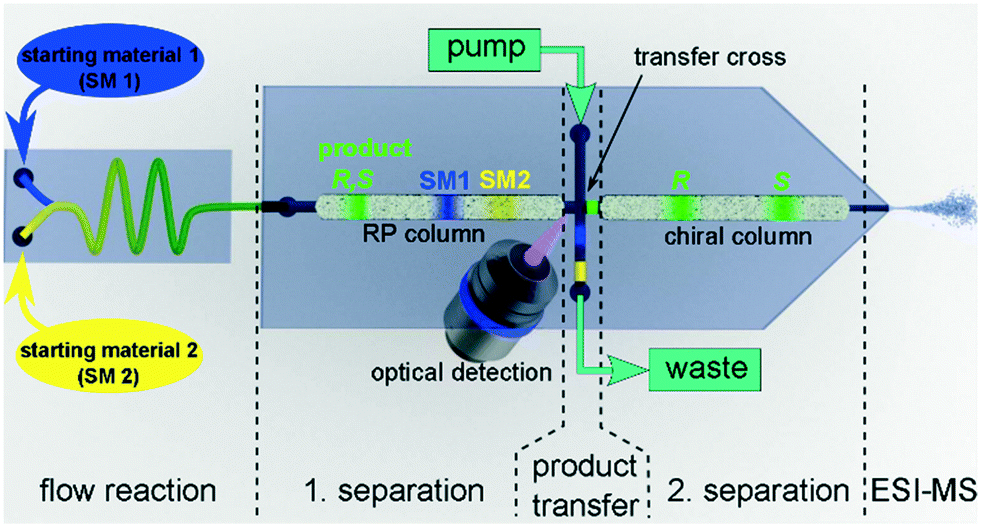 | ||
| Fig. 3 Illustration of the developed two-dimensional hearth-cut chip-HPLC-MS system for the online reaction screening application. | ||
As model reaction we chose the organocatalyzed asymmetric continuous flow synthesis of warfarin (3) from the starting materials 4-hydroxycoumarin (1) and benzalacetone (2), as shown in Scheme 1.54–58 The utilized catalyst (1S,2S)-1,2-diphenylethylenediamine (4a) is reported to steer the reaction enantioselectively with (S)-warfarin as main product, while ethylenediamine (4b) is a common achiral catalyst.56,58
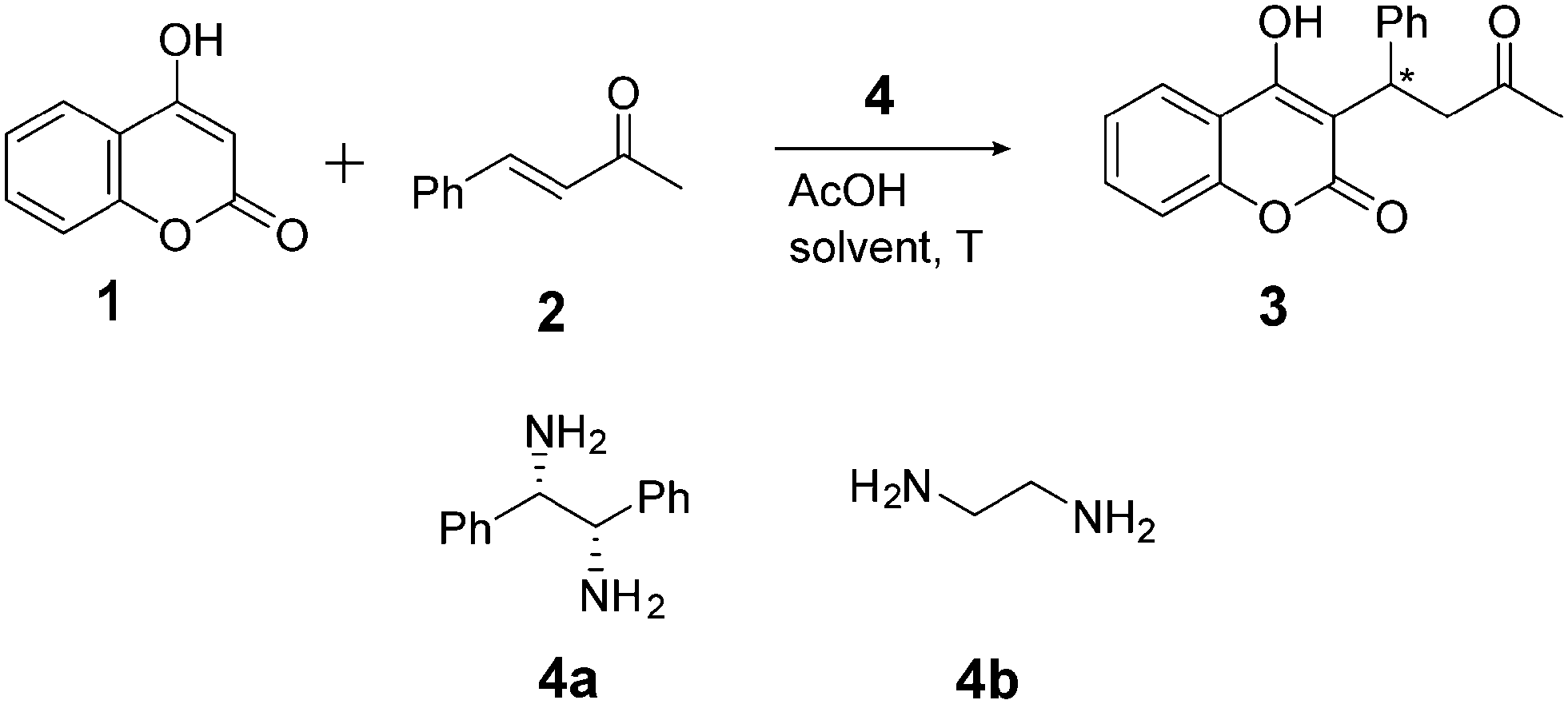 | ||
| Scheme 1 Chiral warfarin synthesis. | ||
Prior to the reaction monitoring experiments, the device was tested by analyzing a sample mixture consisting of the respective pure compounds as standards. The result is shown in Fig. 4, revealing a successful separation of the product from the starting materials in the first dimension, which was monitored by epi-fluorescence detection. The transfer of the product to the second dimension results in a baseline separation of the enantiomers in less than 4 min. This second dimension was monitored by mass spectrometry. For this purpose the 2D-HPLC chip with the monolithically integrated emitter was placed in front of a mass spectrometer.48 A representative base peak chromatogram of this second dimension is shown in Fig. 4b.
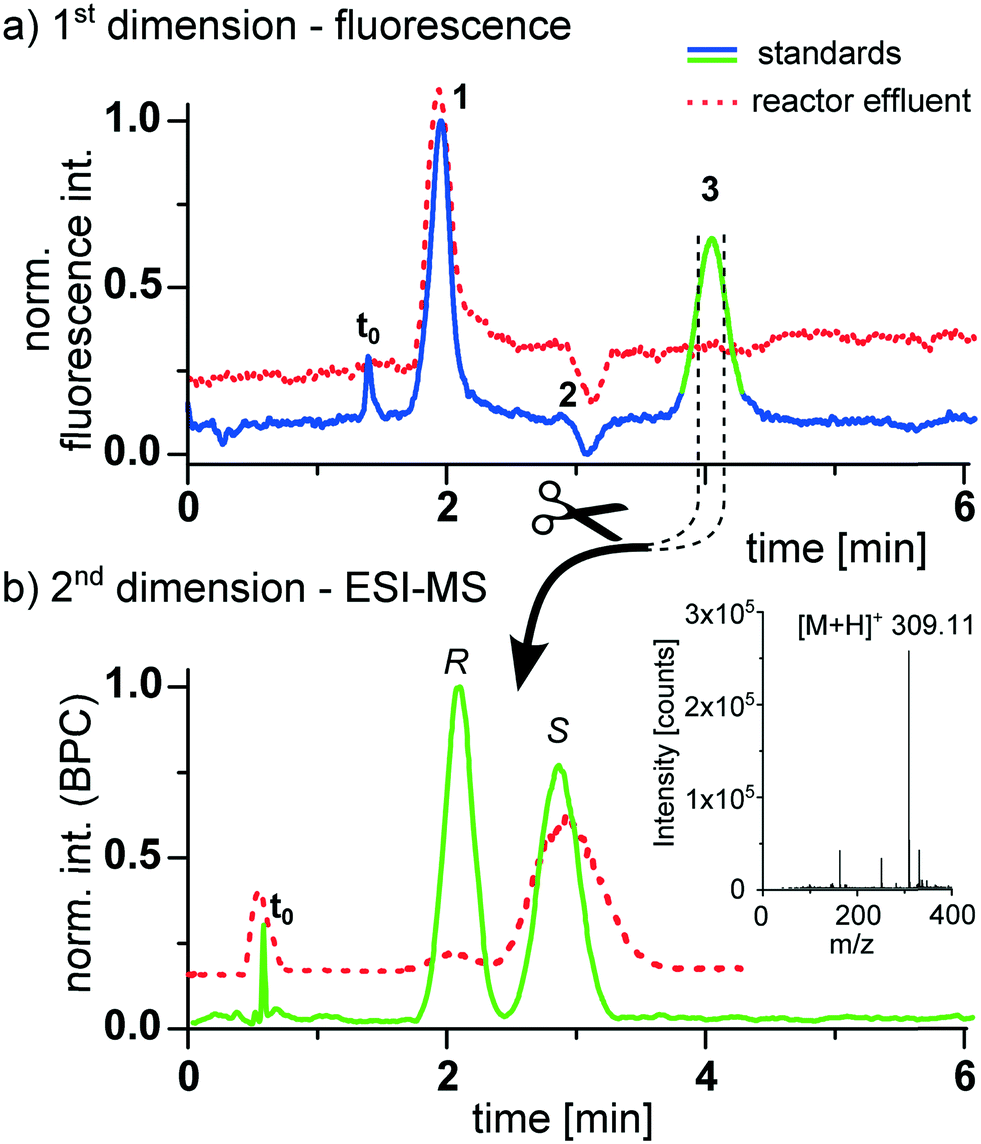 | ||
| Fig. 4 Two-dimensional heart-cut chip-HPLC-MS analysis of the asymmetric continuous flow warfarin synthesis. a) First-dimensional reversed phase on-chip separation of the crude reactor effluent with fluorescence detection. Analytes: 1 4-hydroxycoumarin, 2 benzalacetone, 3 warfarin b) mass spectrometric base peak chromatogram (BPC) of the chiral warfarin separation in the second dimension. On-chip columns: ProntoSIL C18, 5 μm, 30 mm; CHIRALPAK IB, 5 μm, 35 mm. Elution: 1.5 μl min−1 ACN/H2O (50/50 vol%) in first dimension; 20 μl min−1 MeOH/H2O (70/30 vol%) with 0.1% formic acid at chip cross for second dimension. Sample: reactor effluent at a flow rate of 200 nL min−1 in ACN/H2O 95 vol% (dotted line), mixed standard with 0.06 M of each compound in ACN/H2O 95 vol% (solid line). Peak identification of enantiomers was achieved via spiking with pure (S)-enantiomer. | ||
After we could successfully prove the performance of the device with standards we applied it to monitor the enantioselective continuous flow synthesis of warfarin. While the micro-reactor could in principal be integrated on the same device, we attached in this proof of concept study a second reactor chip to the 2D-HPLC device as schematically shown in Fig. 3 and described in detail in the ESI.† The continuous flow synthesis was performed by pumping the reagents 0.2 M 4-hydroxycoumarin (1) as well as a mixture of 0.22 M benzalacetone (2) with 0.02 M of the catalyst (4a) through the chip. As solvent a mixture of ACN/H2O (95/5 vol%) containing 1 M acetic acid was used. A small portion of the reaction mixture from the continuous flow reactor (5 nL) was then transferred by an injection valve onto the 2D-HPLC-MS-chip and analyzed as described above. Representative chromatograms obtained at a reactor flow rate of 200 nL min−1 with a corresponding residence time of 57 min are presented as dotted lines in Fig. 4. The data presented in Fig. 4 illustrates that the two-dimensional separation can also be steered reliably if the product concentration is too low to be detected via epi-fluorescence detection in the first dimension. In this case the heart-cut experiment is triggered by the known and highly reproducible elution time of the product. The high sensitivity of the mass spectrometer allows a reliable detection of the compounds eluting from the chiral column, with a determined ee-value of 83%. The slightly broader chromatographic signals of the effluent compared to the standards can be explained by degradation of the second column over time (several weeks of excessive measurements) due to imperfect packing. As in classical 2D-HPLC-MS, this two-dimensional separation can improve quantitation in electrospray LC mass spectrometry by cutting off potential coeluting compounds, eliminating troublesome matrix effects.44,59
After this successful proof of concept, we further investigated the potential of the approach for rapidly studying the influence of reaction parameters in continuous flow. The seamless hyphenation of the chip reactor and the 2D-LC-MS-chip allows a rapid screening of the reaction conditions in a semi-automated manner. At first, we varied the solvent composition and residence times by adjusting the flow rates in the micro-flow reactor. A set of 69 experiments were conducted for 4 different solvents at various flow rates, resulting in reaction times between 5 and 57 min. The results of the solvent screening experiments are summarized in Table 1 and are presented in detail in the ESI.† As evident from the data in Table 1, a solvent mixture of acetonitrile with 5 vol% water resulted in the highest ee-values.
| Catalysta | Solvent | Reaction time [min] | ee [%]b |
|---|---|---|---|
| a All reactions were performed at 23 °C. b Average and standard deviation of three successive chip-2D-HPLC-MS measurements. | |||
| 4a | ACN/H2O (95 vol%) | 57 | 82.9 ± 1.2 |
| 4a | MeOH | 57 | 73.3 ± 1.7 |
| 4a | THF | 57 | 71.1 ± 1.2 |
| 4a | DMF | 57 | 57.7 ± 0.8 |
| 4b | ACN/H2O (95 vol%) | 11 | Racemic |
Besides the solvent composition and residence time, we also studied the effect of the temperature on the ee-value by placing the micro-flow reactor in a thermostat. The results are shown in Fig. S4 in the ESI.† As expected, an increase in reaction temperature reduces the ee-values drastically, while the overall turnover increases. This trend and the achievable ee-values are in good agreement with literature data.54,56,58
In conclusion, we were able to develop the first example of a two-dimensional heart-cut HPLC-chip. It was coupled to mass spectrometry and exemplarily applied for the reliable determination of the ee-values following asymmetric reactions in continuous micro-flow systems. This new addition to the chemist's toolbox opens up new possibilities for studying and developing chemical transformations at the micro scale. The system is well suited for the investigation of extremely short reaction times and is a good starting point towards the development of comprehensive microfluidic 2D-LC by the adaption of multi-heart-cutting and peak parking methods. Furthermore, it can be an important step in the development of enabling technology for the future chemistry, such as automated micro-synthesis platforms.
Acknowledgements
We would like to thank the Deutsche Forschungsgemeinschaft for funding. The author Josef J. Heiland would like to thank the Studienstiftung des deutschen Volkes for the financial support in form of a PhD-scholarship. The authors would like to thank Chiral Technologies Europe (esp. Dr. Pilar Franco) for providing chiral stationary phase materials and columns.References
- V. Sans and L. Cronin, Chem. Soc. Rev., 2016, 45, 2032–2043 RSC.
- R. J. Ingham, C. Battilocchio, D. E. Fitzpatrick, E. Sliwinski, J. M. Hawkins and S. V. Ley, Angew. Chem., Int. Ed., 2015, 54, 144–148 CrossRef CAS PubMed.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, ACS Cent. Sci., 2016, 2, 131–138 CrossRef CAS PubMed.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed.
- C. Wiles and P. Watts, Green Chem., 2014, 16, 55–62 RSC.
- I. R. Baxendale, J. Chem. Technol. Biotechnol., 2013, 88, 519–552 CrossRef CAS.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456 RSC.
- V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed.
- S. V. Ley, Catal. Sci. Technol., 2016, 6, 4676–4677 CAS.
- J. Hartwig, J. B. Metternich, N. Nikbin, A. Kirschning and S. V. Ley, Org. Biomol. Chem., 2014, 12, 3611–3615 CAS.
- J. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- K. Gilmore and P. H. Seeberger, Chem. Rec., 2014, 14, 410–418 CrossRef CAS PubMed.
- K. S. Elvira, X. C. I. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed.
- D. Ghislieri, K. Gilmore and P. H. Seeberger, Angew. Chem., Int. Ed., 2015, 54, 678–682 CAS.
- T. Rodrigues, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2014, 53, 5750–5758 CrossRef CAS PubMed.
- D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed.
- J. Hereijgers, T. Breugelmans and W. De Malsche, J. Chem. Technol. Biotechnol., 2015, 90, 2122–2131 CrossRef CAS.
- M. Trojanowicz, Talanta, 2016, 146, 621–640 CrossRef CAS PubMed.
- J. Yue, J. C. Schouten and T. A. Nijhuis, Ind. Eng. Chem. Res., 2012, 51, 14583–14609 CrossRef CAS.
- X. Feng, B. F. Liu, J. Li and X. Liu, Mass Spectrom. Rev., 2015, 34, 535–557 CrossRef CAS PubMed.
- X. Wang, L. Yi, N. Mukhitov, A. M. Schrell, R. Dhumpa and M. G. Roper, J. Chromatogr. A, 2015, 1382, 98–116 CrossRef CAS PubMed.
- D. E. W. Patabadige, S. Jia, J. Sibbitts, J. Sadeghi, K. Sellens and C. T. Culbertson, Anal. Chem., 2016, 88, 320–338 CrossRef PubMed.
- C. T. Culbertson, T. G. Mickleburgh, S. A. Stewart-James, K. A. Sellens and M. Pressnall, Anal. Chem., 2014, 86, 95–118 CrossRef CAS PubMed.
- S. Stockinger, J. Troendlin, F. Rominger and O. Trapp, Adv. Synth. Catal., 2015, 357, 3513–3520 CrossRef CAS.
- V. Sans, L. Porwol, V. Dragone and L. Cronin, Chem. Sci., 2015, 6, 1258–1264 RSC.
- K. F. Jensen, B. J. Reizman and S. G. Newman, Lab Chip, 2014, 14, 3206–3212 RSC.
- A. G. O'Brien, Z. Horváth, F. Lévesque, J. W. Lee, A. Seidel-Morgenstern and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 7028–7030 CrossRef PubMed.
- A. Odedra and P. H. Seeberger, Angew. Chem., Int. Ed., 2009, 48, 2699–2702 CrossRef CAS PubMed.
- J. P. McMullen, M. T. Stone, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 7076–7080 CrossRef CAS PubMed.
- H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., 2007, 119, 5806–5810 CrossRef.
- O. Trapp, S. K. Weber, S. Bauch and W. Hofstadt, Angew. Chem., Int. Ed., 2007, 46, 7307–7310 CrossRef CAS PubMed.
- I. Atodiresei, C. Vila and M. Rueping, ACS Catal., 2015, 5, 1972–1985 CrossRef CAS.
- F. G. Finelli, L. S. M. Miranda and R. O. M. A. de Souza, Chem. Commun., 2015, 51, 3708–3722 RSC.
- X. Y. Mak, P. Laurino and P. H. Seeberger, Beilstein J. Org. Chem., 2009, 5, 19 Search PubMed.
- K. M. Krone, R. Warias, C. Ritter, A. Li, C. G. Acevedo-Rocha, M. T. Reetz and D. Belder, J. Am. Chem. Soc., 2016, 138, 2102–2105 CrossRef CAS PubMed.
- S. Fritzsche, S. Ohla, P. Glaser, D. S. Giera, M. Sickert, C. Schneider and D. Belder, Angew. Chem., Int. Ed., 2011, 50, 9467–9470 CrossRef CAS PubMed.
- J. P. Grinias and R. T. Kennedy, TrAC, Trends Anal. Chem., 2016, 81, 110–117 CrossRef CAS PubMed.
- A. Manz, Y. Miyahara, J. Miura, Y. Watanabe, H. Miyagi and K. Sato, Sens. Actuators, B, 1990, 1, 249–255 CrossRef CAS.
- C. Lotter, J. J. Heiland, V. Stein, M. Klimkait, M. Queisser and D. Belder, Anal. Chem., 2016, 88, 7481–7486 CrossRef CAS PubMed.
- Y. Temiz, R. D. Lovchik, G. V. Kaigala and E. Delamarche, Microelectron. Eng., 2015, 132, 156–175 CrossRef CAS.
- X. Shi, S. Wang, Q. Yang, X. Lu and G. Xu, Anal. Methods, 2014, 6, 7112 RSC.
- H. Malerod, E. Lundanes and T. Greibrokk, Anal. Methods, 2010, 2, 110–122 RSC.
- M. E. León-González, N. Rosales-Conrado, L. V. Pérez-Arribas and V. Guillén-Casla, Biomed. Chromatogr., 2014, 28, 59–83 CrossRef PubMed.
- J. Šesták, D. Moravcová and V. Kahle, J. Chromatogr. A, 2015, 1421, 2–17 CrossRef PubMed.
- J. P. Kutter, J. Chromatogr. A, 2012, 1221, 72–82 CrossRef CAS PubMed.
- G. Desmet and S. Eeltink, Anal. Chem., 2013, 85, 543–556 CrossRef CAS PubMed.
- C. Lotter, J. J. Heiland, S. Thurmann, L. Mauritz and D. Belder, Anal. Chem., 2016, 88, 2856–2863 CrossRef CAS PubMed.
- S. Thurmann, C. Lotter, J. J. Heiland, B. Chankvetadze and D. Belder, Anal. Chem., 2015, 87, 5568–5576 CrossRef CAS PubMed.
- J. Xie, Y. Miao, J. Shih, Y.-C. Tai and T. D. Lee, Anal. Chem., 2005, 77, 6947–6953 CrossRef CAS PubMed.
- E. Davydova, S. Wouters, S. Deridder, G. Desmet, S. Eeltink and P. J. Schoenmakers, J. Chromatogr. A, 2016, 1434, 127–135 CrossRef CAS PubMed.
- S. Thurmann, A. Dittmar and D. Belder, J. Chromatogr. A, 2014, 1340, 59–67 CrossRef CAS PubMed.
- S. Thurmann, L. Mauritz, C. Heck and D. Belder, J. Chromatogr. A, 2014, 1370, 33–39 CrossRef CAS PubMed.
- R. Porta, M. Benaglia, F. Coccia, S. Rossi and A. Puglisi, Symmetry, 2015, 7, 1395–1409 CrossRef.
- N. Halland, T. Hansen and K. A. Jørgensen, Angew. Chem., 2003, 115, 5105–5107 CrossRef.
- H. Kim, C. Yen, P. Preston and J. Chin, Org. Lett., 2006, 8, 5239–5242 CrossRef CAS PubMed.
- J.-W. Xie, L. Yue, W. Chen, W. Du, J. Zhu, J.-G. Deng and Y.-C. Chen, Org. Lett., 2007, 9, 413–415 CrossRef CAS PubMed.
- T. C. Wong, C. M. Sultana and D. A. Vosburg, J. Chem. Educ., 2010, 87, 194–195 CrossRef CAS.
- P. J. Taylor, Clin. Biochem., 2005, 38, 328–334 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed technical and optical setup, reaction time dependent solvent screenings, experimental details. See DOI: 10.1039/c6lc01138a |
| This journal is © The Royal Society of Chemistry 2016 |