
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Droplet-based microfluidics in drug discovery, transcriptomics and high-throughput molecular genetics
Nachiket
Shembekar†
,
Chawaree
Chaipan†
,
Ramesh
Utharala†
and
Christoph A.
Merten
*
European Molecular Biology Laboratory (EMBL), Genome Biology Unit, Meyerhofstrasse 1, Heidelberg, Germany. E-mail: merten@embl.de
First published on 18th March 2016
Abstract
Droplet-based microfluidics enables assays to be carried out at very high throughput (up to thousands of samples per second) and enables researchers to work with very limited material, such as primary cells, patient's biopsies or expensive reagents. An additional strength of the technology is the possibility to perform large-scale genotypic or phenotypic screens at the single-cell level. Here we critically review the latest developments in antibody screening, drug discovery and highly multiplexed genomic applications such as targeted genetic workflows, single-cell RNAseq and single-cell ChIPseq. Starting with a comprehensive introduction for non-experts, we pinpoint current limitations, analyze how they might be overcome and give an outlook on exciting future applications.
Introduction
Droplet microfluidics holds great potential in drug discovery and genomics because of the conceptual advantages it offers, such as consuming just a few microliters of sample and requiring only small cell numbers – fewer than what can be obtained from a single human patient biopsy – for performing large-scale studies. Furthermore, droplet technology enables a high degree of automation and facilitates high throughput screens – several hundred thousand cell-based assays can be carried out in a single experiment. These advantages make droplet microfluidics a great tool for biomedical research and single-cell studies. In this review, we focus on published research in drug discovery and genomics using droplet microfluidics. We do not take into account other technologies such as valve-based compartmentalization or digital microfluidics and also excluded other applications of droplet technology. For readers interested in these or scientists who want to dive deeper into the physics and applications of microfluidics we refer to previously published reviews.1–8Microfluidic droplets containing just a few fLs to nLs can be generated at very high frequency (Hz–kHz).9 These droplets are usually generated by pressure-driven flow and are surrounded by an immiscible oil phase such that each droplet behaves as an individual micro-reactor. Usually, the minimum volume used for micro-titer plate assays is 1 μL. In that way, a single droplet accommodates 103–109 times less volume than conventional systems. Typically, aqueous samples containing biological material such as cells, DNA, bacteria, and molecules can be encapsulated into droplets.
Manufacturing chips for droplet microfluidics
Microfluidic chips are usually manufactured using soft lithography.10 This procedure requires specialized equipment and typically also a clean room facility. However, non-expert labs can get custom-made microfluidic devices manufactured by companies and service facilities such as Dolomite, Microfluidic Chip Shop or the Stanford Microfluidics Foundry.A microfluidic chip normally contains at least two layers: a substrate layer, which is usually made of glass or polydimethylsiloxane (PDMS), and another layer with the channel network. Using transparent materials such as glass and PDMS facilitates imaging and/or spectroscopic readouts, which is highly desirable for biological assays. Furthermore, PDMS is gas permeable, enabling the culture of live cells inside the chips. When using droplet-based microfluidics, the chip material also has to be highly hydrophobic to ensure efficient wetting of the channel walls by the carrier phase, while preventing surface interactions of the aqueous droplets. This can be achieved by coating the channel surface with hydrophobic chemicals such as silanes,11,12 Aquapel (PPG Industries),13 or Teflon.14 Begolo et al. have also shown that materials such as fluorinated polymers allow researchers to circumvent the need for surface treatment of channels.15
Droplet production
Droplets containing few fLs to nLs are produced when two immiscible liquids intersect each other. This process can be achieved either by active (on-demand droplet production using valves or electric fields) or by passive (continuous droplet production using pressure-driven flow and a special geometry of the channels) droplet production methods. The most commonly used methods are passive as this allows droplet production at very high rates (kHz). Because of the specific design of the microfluidic channels, an aqueous phase is sheared by an inert oil phase to produce uniform-sized drops surrounded by oil. The specific designs can be T-junction,16 flow-focusing,17,18 or co-flowing19 geometries as shown in Fig. 1.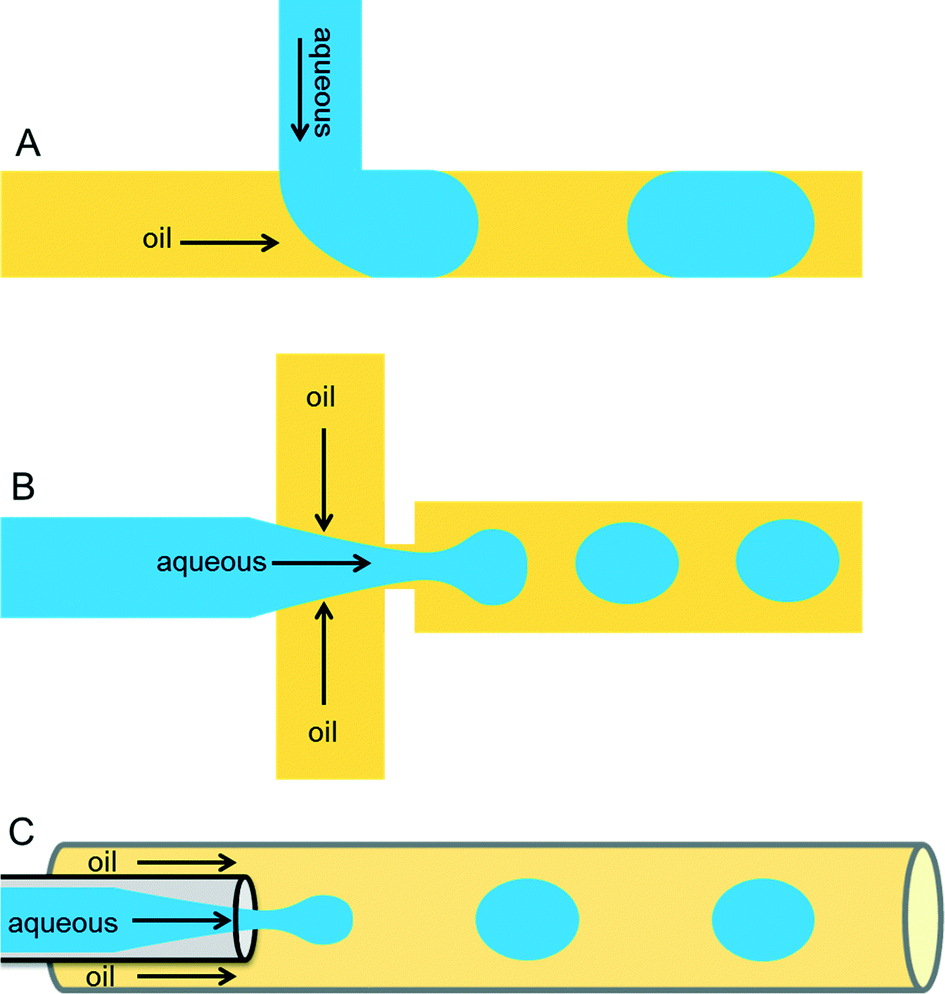 | ||
| Fig. 1 Different microfluidic drop maker geometries. (A) In the T-junction geometry, the perpendicular flow of the aqueous phase is sheared by oil and thereby generates droplets. (B) The flow-focusing geometry produces droplets by shearing the aqueous stream from two directions. (C) In the co-flow geometry, the aqueous phase is forced through a capillary, which is placed co-axially inside a bigger capillary, through which immiscible oil is pumped. | ||
Important requirements for a carrier phase are high affinity towards the channel surface and low viscosity to prevent problems with excessive back pressure. Numerous carrier phases were tested for various applications and the most commonly used carrier phases include hydrocarbon oils and fluorocarbon oils.16,20 Perfluorinated oils have specific advantages such as their immiscible nature with organic solvents, ∼20-times higher solubility of gases such as oxygen and carbon dioxide (compared to water),21 as well as physical and chemical stability. In order to stabilize the emulsion, one has to carefully select a suitable surfactant for the carrier phase according to parameters such as biocompatibility, (thermo-)stability of the resulting emulsion and potential micellar transport.22 Surfactants are surface active agents, which mainly act at the oil/water interface by reducing surface tension and thus increasing the stability of emulsions. In the absence of surfactants, a minimized surface area is energetically favorable and thus the droplets coalesce. This phenomenon can be nicely illustrated by a simple real-world example in cooking, where oil droplets in a pan come together and merge to reduce their surface area. Hydrocarbon oils such as mineral oil can be stabilized by commercially available surfactants, including Span 80 (sorbitone mono-oleate) and Abil EM (cetyle dimethicone copolyol), whereas in case of perfluorinated oils such as FC-40 and FC-70, use of surfactants such as polyethylene glycol (PEG), perfluoropolyethers (PFPEs), and dimorpholino phosphate have been demonstrated.13,16,20 Some companies such as BioRad, Sphere Fluidics, Dolomite and RainDance technologies offer different types of oil-surfactant systems commercially.
Laminar flow
One of the most important parameters describing the flow in microfluidic systems is the Reynold's number (Re), which is defined as the ratio of inertial to viscous forces, and is given by the equation below.where ρ is the density of the fluid (kg m−3), V is the mean velocity (m s−1), L is the characteristic length of the channel (m) and μ is the dynamic viscosity of the fluid (kg m−1 s−1). Re explains different flow regimes such as ‘laminar’ and ‘turbulent’ in microfluidic channels. Normally, Re for laminar flow systems is below 2000, and the streamlines are parallel. However, Re for turbulent flow systems is above 2000, and the streamlines are chaotic. Typically Re in a microfluidic channel is smaller than one and thus exclusively belongs to the ‘laminar’ flow category. As a direct consequence, two liquids co-flowing in a microfluidic device (e.g. a cell suspension and a lysis buffer) do not mix prior to droplet formation, but rapidly afterwards due to chaotic convection. This can be exploited to lyse cells directly upon encapsulation, while ensuring that no cellular contents are released before single cells have been compartmentalized.
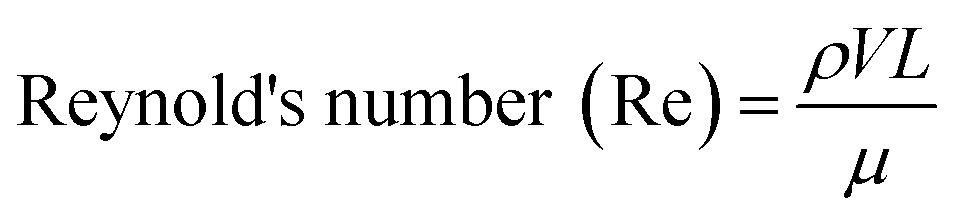
Cell encapsulation
Encapsulation of single cells into droplets is a powerful tool for single-cell studies.13,23 Usually, the cells are distributed randomly in an aqueous suspension and upon encapsulating them into droplets (Fig. 2A) random occupancies according to Poisson distribution are obtained. For single-cell studies, one can use a cell density of less than one cell per droplet volume on average (e.g. λ = 0.4, with λ = cell density divided by the droplet volume), resulting in about one quarter of total droplets hosting exactly one cell, while the remaining droplets contain either no cell (∼67%) or 2 or more cells (∼6%). Diluting the cell suspension further (e.g. λ = 0.1) enables researchers to largely avoid droplets with more than one cell (<0.5%), at the price of an increased fraction of empty droplets (>90%). Hence this is a tradeoff between accuracy (in terms of avoiding droplets with more than one cell) and throughput. In order to overcome Poisson statistics, Kemna et al. designed a microfluidic device to encapsulate single cells with high yield (77%) at high throughput (2700 cells s−1) using inertial ordering in a curved microchannel24 as shown in Fig. 2B. It should be noted that the high frequency of droplet generation also sets a lower limit for the required number of cells: according to our experience a starting amount of less than ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells is impracticable, since the system has to run for a couple of minutes before stable flow rates are obtained.
000 cells is impracticable, since the system has to run for a couple of minutes before stable flow rates are obtained.
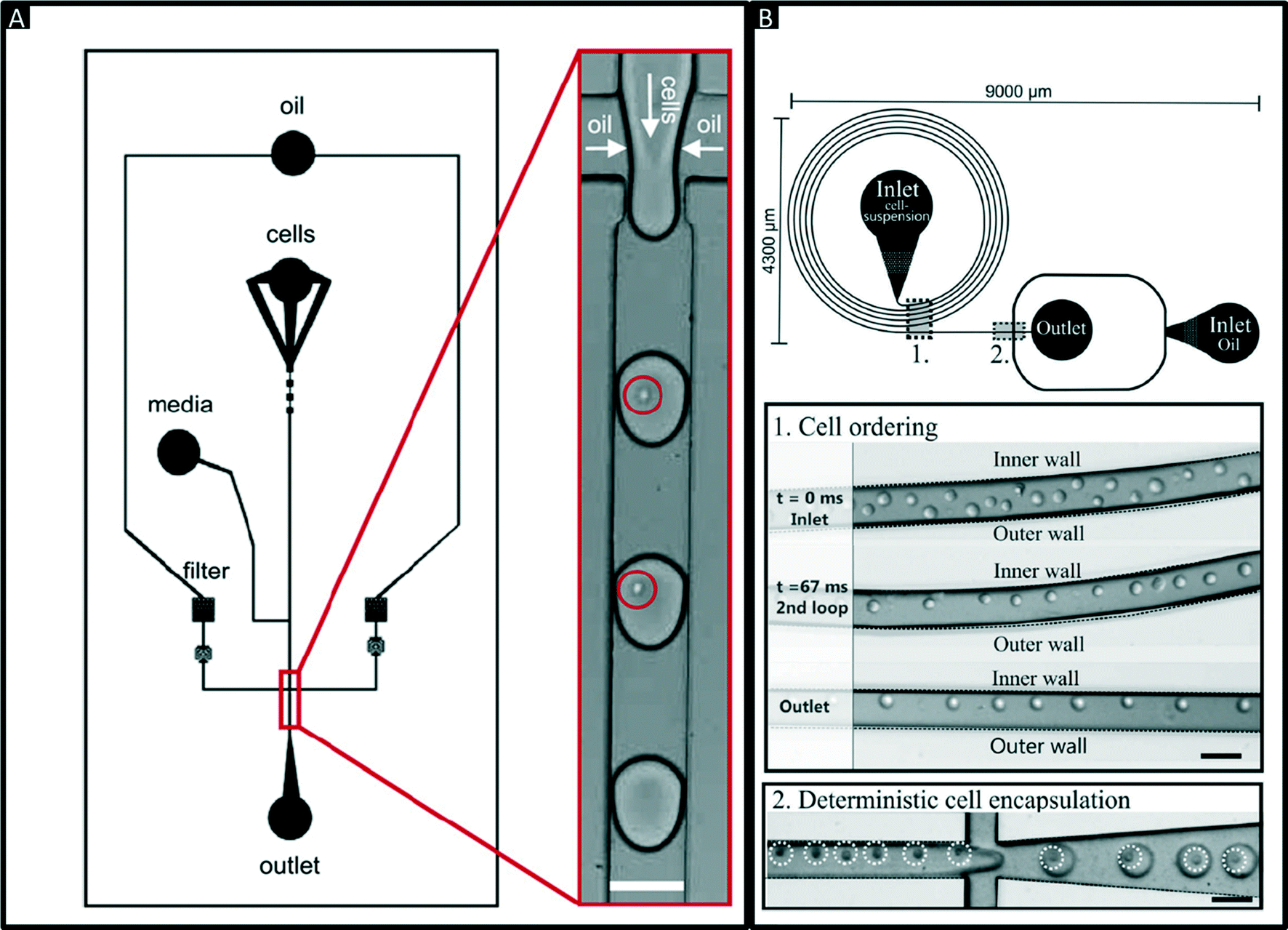 | ||
| Fig. 2 Encapsulation of single cells. (A) Design of a microfluidic chip for random encapsulation of cells and zoom-in showing individual human cells inside the droplets. Cells are highlighted by circles. White scale bar = 100 μm. Reproduced with permission from ref. 13 copyright 2008, Elsevier. (B) Design of a microfluidic chip enabling deterministic cell encapsulation. The cells flow through a long curved microchannel, where they align due to Dean force before reaching the drop maker. Cells are highlighted by dotted circles. Black scale bar = 100 μm. Reproduced with permission from ref. 24 copyright 2012, The Royal Society of Chemistry. | ||
Cells and their expressed products can easily be detected as the volume is minimal inside the droplet.25,26 Moreover, the cells can be lysed using lysis buffer or some mechanical (or) optical means to analyze the intracellular components such as DNA, RNA, and other biomolecules.27–29 In addition, mammalian cells can also be cultivated inside the droplets for several days. We performed quantitative studies for different incubation times of human cells (Jurkat and HEK293T) inside 660 pL droplets and observed more than 79% viability over the first 4 days.13 However, longer encapsulation times turned out to be problematic as the cells ultimately run out of nutrition and accumulate toxic metabolites. It is important to note that this cannot be overcome by simply increasing the droplet volume, which also results in decreased droplet stability and lowered throughput of all droplet manipulation steps. Therefore, proliferation assays using mammalian cells are difficult to implement, but apart from this the technology can be used for almost any study that does not require incubation times of more than a few days. This even includes studies requiring the encapsulation of two different cell types, such as monitoring the effect of a plasma cell-secreted antibody on a target cell.
Due to Poisson statistics, the co-encapsulation of different cell types can be problematic and the maximum probability for encapsulating exactly one cell of two different cell types into the same droplet is just 13.5%. Nonetheless, this number can be improved up to ∼29% using deterministic co-encapsulation procedures.30 Furthermore, we recently introduced an approach in which the two different cell types are stained with two different fluorescence dyes, before applying multicolor sorting. This allows to enrich the fraction of droplets with the correct occupancy up to 86.7% or even >97% when considering all droplets with at least one cell of each type.31
Droplet manipulations
There are several manipulations such as mixing,32,33 splitting,9,34 storage (incubation),35 droplet breakage (phase separation),13,36 re-injection,20 fusion,37–39 detection and sorting,40,41 which can be performed on the droplets based on the user requirements and design circuitry of microfluidic channels (Fig. 3A–J). | ||
| Fig. 3 Additional microfluidic modules. (A & B) Mixing in droplets. Mixing of different reagents inside droplets occurs due to chaotic advection and can be improved using serpentine channels. Reproduced with permission from ref. 33, copyright 2003, AIP Publishing LLC. (C) Droplet splitting. Droplets are flushed through consecutive branching channels where they are split according to the relative flow rates in each arm of the channel network. Reproduced with permission from ref. 34 copyright 2009, The Royal Society of Chemistry. (D) On-chip Incubation. Dynamic incubation of droplets flowing through a delay line. Reproduced with permission from ref. 25 copyright 2008, The Royal Society of Chemistry (E). Static incubation of droplets in an array of traps. The zoom in shows stained cells inside the droplets. Reproduced with permission from ref. 31 copyright 2015, The Royal Society of Chemistry. (F) Off-chip incubation. For longer incubation periods emulsions can be collected off-chip in conventional test tubes, syringes or capillaries. The zoom in shows individual droplets of the emulsion. (G) Re-injection of droplets. Re-injection of droplets into a microfluidic chip to allow for additional microfluidic operations after an incubation period. Reproduced with permission from ref. 20 copyright 2008, The Royal Society of Chemistry. (H) Pillar-induced passive droplet fusion enabling coalescence of surfactant stabilized big droplets with non-surfactant stabilized small droplets (e.g. to add a substrate after off-chip incubation),36 Copyright (2012) National Academy of Sciences, USA. (I) Active addition of reagents using a picoinjection system.43 In this approach the oil phase is supplemented with surfactants, so that in absence of an electric field no fusion occurs. Upon application of the electric field, the interphases merge and the pressurized dark liquid is injected through a narrow orifice into the incoming droplets. Copyright (2010) National Academy of Sciences, USA. (J) Sorting of droplets. Droplets passing a sorting junction with different fluidic resistances of the two channel arms. In absence of an electric field the droplets follow the main flow into the wider waste channel. Upon application of an AC pulse, the droplets are pulled into the narrower collection channel due to dielectrophoretic force. Reproduced with permission from ref. 54 copyright 2009, The Royal Society of Chemistry. | ||
Of major importance for almost any biological assay is an incubation step, which can be implemented by either on-chip or off-chip droplet storage. In both cases the droplets typically come into contact with each other and hence have to be stabilized with surfactants. On-chip incubation can be performed by flushing droplets through long delay lines or by trapping them in arrays (Fig. 3D and E).25,31,35,36 Off-chip incubation is done by collecting emulsions in a vial or in a syringe (Fig. 3F).28 This is advantageous if the samples must be incubated for longer periods of time (e.g. days, which cannot be achieved in delay lines) or if millions of droplets have to be stored (more than what can be accommodated on-chip). After off-chip incubation, re-injection is necessary for performing additional microfluidic manipulations (e.g. addition of further reagents, detecting and sorting droplets of interest). This can be performed by re-injecting stored droplets into another microfluidic chip (Fig. 3G).20,36
Additional reagents can be added to the droplets (e.g. to initiate a fluorescence readout) at a later time point using droplet fusion, being the equivalent to a pipetting step on the bench. Fusion is achieved by bringing the droplets into physical contact such that the interface is destabilized. The efficiency of this process is mainly dependent on surfactant concentration and viscosity of carrier and dispersed phase. Droplet fusion can be performed either by passive or by active merging methods. Passive fusion methods mainly rely on surfactant concentration (e.g. pairing droplets where at least one of the fusion partners is not surfactant stabilized) and channel geometry. Niu et al. described the use of pillars in a widening channel to achieve efficient 1:1 droplet fusion at a rate of ∼30 Hz.42 However, the above method required droplets that were produced on-chip and were not compatible with off-chip incubation steps. To overcome this limitation we developed a module (Fig. 3H) enabling the fusion of re-injected cell-laden droplets (stabilized by surfactant) with droplets produced in a pillar chamber (lacking surfactant).36 This approach allowed 1:1 droplet fusion at a frequency of 50 Hz with more than 99% efficiency. Using smaller droplets in a similar setup Mazutis et al. even achieved fusion rates in the kHz range.39 Active fusion methods include the application of electric fields37,38 and pico injection (Fig. 3I).43 In the latter method, the liquid to be added is pressurized and stored in a channel with a little orifice perpendicular to the channel with the incoming droplets. Due to the Laplace pressure (acting against the pressure of the reservoir) no liquid is ejected through the orifice and the liquid to be added forms a static meniscus. Even though the incoming droplets pass this meniscus, no fusion of the two aqueous surfactant-stabilized phases occurs unless an electric field is applied. Hence the fusion process can be well controlled electronically and performed at rates up to several kHz.
Detection of droplets is performed mainly by optical methods such as laser-induced fluorescence,44 IR spectroscopy,45 Raman spectroscopy46 and UV-visible absorption.47 Fluorescence spectroscopy is the most commonly used method as it can be carried out at high throughput (up to kHz rates) and due to the fact that many fluorescence assays for biological reactions are commercially available. Analytical methods such as liquid chromatography (LC),48,49 mass spectrometry50,51 and capillary electrophoresis (CE)52 have also been coupled with droplet microfluidic systems to characterize the droplet contents. However, these methods have limited throughput (<1 Hz) and furthermore show limited sensitivity due to the very small volumes. For example, mass spectrometry can be used to detect highly abundant metabolites or proteins, but it does not yet allow global single-cell proteomic analyses.
Similar to flow cytometry, droplets can be sorted based on their fluorescence. In contrast to FACS this not only enables researchers to sort cells or other solid particles, but rather entire assay vessels containing soluble molecules (e.g. secreted antibodies and assay reagents). This step can be performed by dielectrophoresis,53,54 valves,55 optics,56 acoustics57 or electric stream deflection.58,59 Dielectrophoresis (DEP) is the most commonly used technique for sorting droplets based on an optical readout (Fig. 3J). Baret et al. designed a fluorescence activated droplet sorting (FADS) device capable of sorting droplets at a rate of 2 kHz.54 Recently refined versions of this setup or the use of acoustic fields allowed increasing the throughput up to 30 kHz.57,60 After the sorting step, the droplet contents can be recovered, including the recovery of cells for recultivation in tissue culture flasks, by breaking the emulsion, resulting in separation of the aqueous and the oil phase. This can be achieved by adding surface-active chemicals such as 1H,1H,2H,2H-perfluoro-1-octanol (PFO),13 or by applying electric fields.61
Barcoding droplets
Indexing droplets is a crucial step to keep track of their identity – for example after off-chip incubation where droplets are stored in a random order. Optical methods have been designed for this purpose, such as the use of quantum dots and fluorescent dyes as barcodes (Fig. 4A).26,62 Mixtures of different fluorescence dyes at different concentrations can be read in real-time. However, their use is limited for encoding only a few hundred samples owing to the dynamic range of the optical set up – for instance detection of a “false” low fluorescence signal in one channel due to bleed-through of a high fluorescence signal in another channel. However, encoding larger sample numbers can be achieved with DNA oligonucleotides (Fig. 4B), whose barcoding capacity corresponds to 4n (with n being the number of nucleotides) and is hence almost unlimited. In 2010 we conceptualized and patented (PCT/EP20ll/004010) the use of oligonucleotide barcodes to label the droplet composition, with a special focus on combinatorial drug screening and the effect of compounds on gene expression. Using a similar approach, Weitz and co-workers published three seminal papers on the barcoding of single-cell transcriptomes and genomes in 2015,63–65 which are discussed in detail further below. The application part of this review also explains more comprehensively how to add different barcodes to the droplets, either directly upon generation or at a later time point by droplet fusion. One disadvantage of oligonucleotide barcodes is the fact that, in contrast to optical barcodes, they cannot be read in real time on-chip, but rather require downstream sequencing of the samples.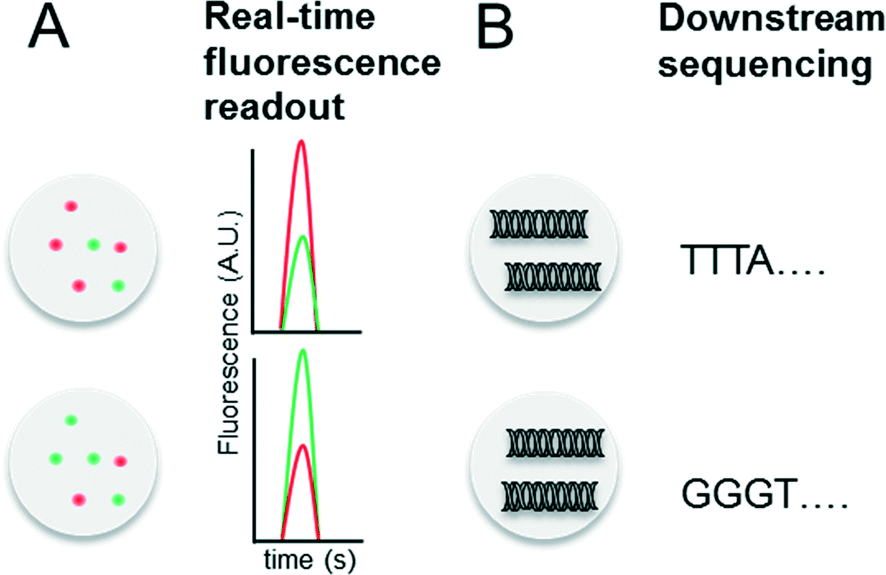 | ||
| Fig. 4 Barcoding of droplets. (A) Optical encoding using two different fluorescence dyes (red and green) at different concentrations. This allows to encode for a maximum of x samples, with x = number of dyes × number of dye concentrations. (B) Nucleotide sequence based barcoding. This allows to encode for a maximum of y samples, with y = 4(number of nucleotides). | ||
Conceptual limitations of compartmentalizing samples in droplets
While there are many advantages of droplet microfluidics, there are also a few fundamental limitations: first of all, exchanging media or implementing washing steps is barely possible. This not only restricts the survival times of encapsulated cells, but also makes multi-step assays challenging. Pioneering work in overcoming this problem by using magnetic beads that can be swapped between droplets of different composition has been published,66 but this technology is still in its infancy and has not yet been integrated into high-throughput cell-based screening platforms. Another significant problem is the undesired exchange of droplet contents via micellar transport: hydrophobic molecules with high log P values (a partition coefficient defined as the logarithm of the solute concentration in octanol divided by the solute concentration in water; log P = log([X in octanol]/[X in water]) tend to exchange over time between droplets, thus making the compartmentalization of particular chemicals very challenging.67 It would be interesting to see to what extent this affects the screening of highly diverse chemical libraries; however comprehensive data on this is still largely missing. Nonetheless, most biomolecules such as proteins and DNA are unaffected by micellar transport, for which reason applications such as antibody screening and single-cell genomics do not suffer from this limitation.Antibody screening and drug discovery
Antibodies are widely used in basic research and gained major importance in the treatment of various infectious diseases,68–70 several cancer malignancies71–73 and inflammations.74 In fact, the worldwide sales of monoclonal antibody products in 2013 reached almost 75 billion US$,75 demonstrating a clear need for efficient screening technology. Discovering new therapeutic antibodies has been conventionally achieved in microtiter plates using immortalized hybridoma cells (required for supplying sufficient numbers of cells for macroscopic screening methods) and robotic technology. However, during the generation of hybridoma cells, the very low fusion efficiency of the antibody producing B-cells and myeloma cells and a requirement to expand fused clones over several weeks, means that the output is rather low and only a small fraction of the immune repertoire (typically less than 1/104 for murine donors and less than 1/106 for human donors) can be screened.76 This can be drastically improved using droplet-based microfluidics, in which single cells can be assayed at very high throughput (up to several hundred thousand within a few hours). Due to the low volumes antibodies released from individual cells remain concentrated inside the droplets,77 thus bypassing clonal expansion, which is required for obtaining detectable concentrations of antibodies in microtiter plates.We exploited these conceptual advantages in a functional screen for antibodies inhibiting the hypertension and congestive heart failure drug target angiotensin-converting enzyme 1. Droplets were generated with a volume of 660 pL hosting single antibody producing hybridoma cells co-encapsulated with the enzymatic drug target (Fig. 5A). Incubation of the droplets for six hours resulted in an antibody concentration of ∼20 μg ml−1 within the droplets. The droplets were then fused 1:1 with smaller droplets (25 pL volume) hosting a fluorogenic substrate of the enzyme. Conversion of this substrate into a fluorescent product allowed the discrimination of cells releasing non-inhibitory antibodies (fluorescence positive droplets) from cells expressing potent inhibitors (fluorescence negative droplets). Starting with a 10000-fold excess of cells expressing non-related antibodies, fluorescence negative droplets were selectively sorted before the cells were recovered for further downstream analysis.36 Despite the fact that the system did not account for differences in the antibody expression levels of individual cells, 17 out of 18 positively sorted cells secreted antibodies inhibiting the drug target. More importantly, 300000 hybridoma clones could be screened in a single experiment.
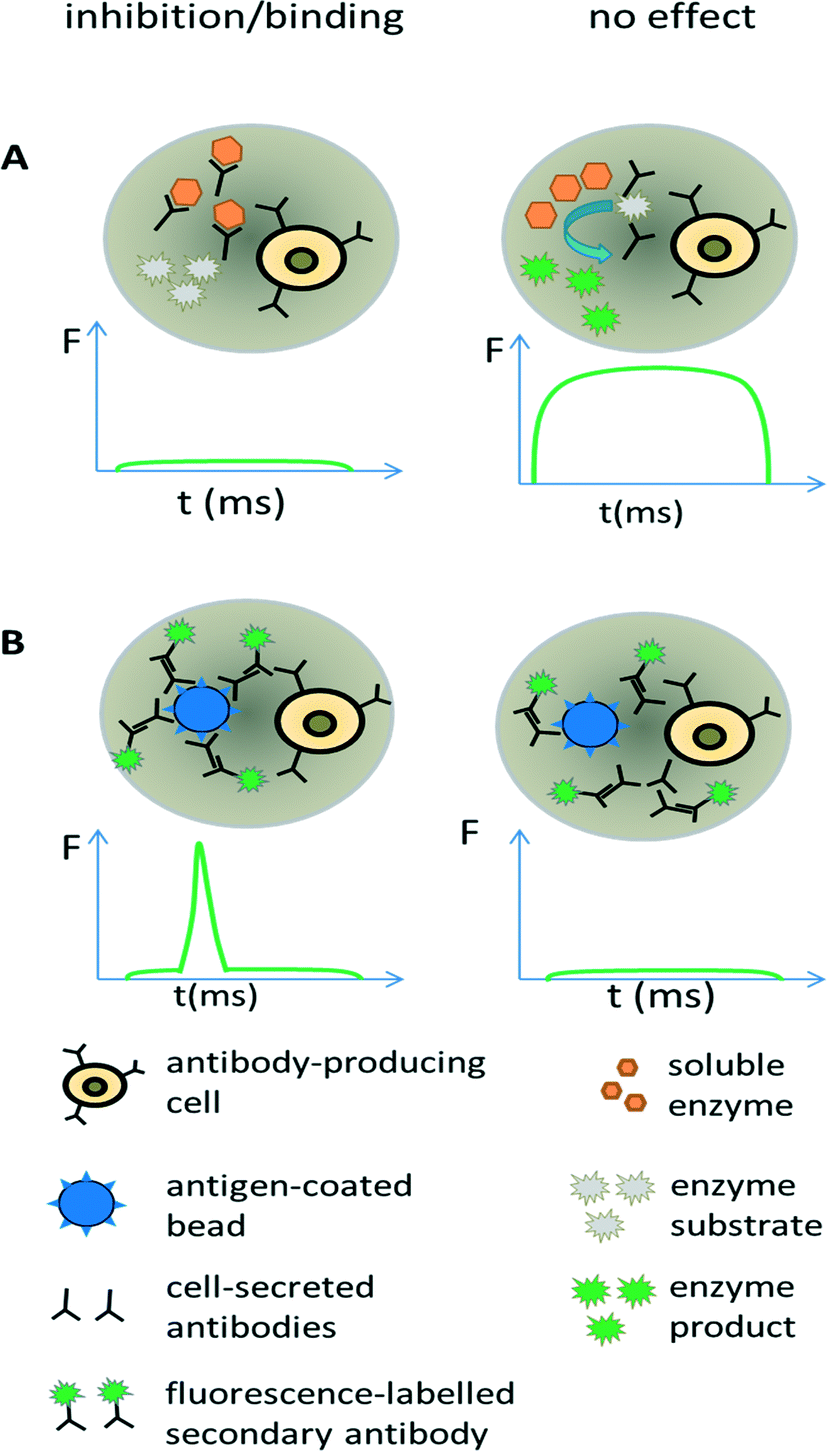 | ||
| Fig. 5 Screening for antibodies with desired properties. (A) Direct screening for inhibition of enzymatic drug targets. Antibody-releasing cells are encapsulated into droplets, together with the drug target of interest and a fluorogenic substrate. In case the enzymatic activity is inhibited by antibodies, no fluorescent product is generated and the droplet shows no or only a very low fluorescence signal (left). In contrast, the absence of inhibitory antibodies results in the conversion of the fluorogenic substrate into a fluorescent product and the entire droplets become strongly fluorescent (right). (B) Screening for antibody binding. Antibody-releasing cells are encapsulated into droplets, together with beads displaying the antigen of interest and a secondary fluorescently labelled antibody. Binding of the primary antibody to the beads leads to co-localization of the secondary fluorescently labelled antibodies, causing a narrow high intensity fluorescence peak (left). In case the primary antibody does not bind the antigen, also the secondary antibody and thus the fluorophores remain homogeneously distributed within the droplet, resulting in a broad but very weak fluorescence peak. Y axis = fluorescence (F); X axis = time (t). | ||
Screening for binding can also be performed in droplets, using an approach similar to conventional fluorometric microvolume assay technology (FMAT,78). This type of assay was first implemented in droplets by the Yarmush lab, focusing on the detection of cytokines secreted by single T-cells.79 It can also be exploited for antibody screening, by co-encapsulating antibody-secreting cells together with antigen-coated beads and secondary fluorescently labelled antibodies (Fig. 5B). In case the primary antibody binds to the bead, then the fluorophores of the secondary antibodies also get localized, resulting in a narrow high intensity fluorescence peak. However, in absence of the primary antibody (or in absence of target binding), the secondary antibodies remain homogeneously distributed, giving rise to a broad, low intensity fluorescence peak whenever the droplet passes the detector. Mazutis et al. used this setup to separate antibody expressing cells from a 10-fold excess of non-antibody expressing cells.77 However, they used neither higher dilution ratios nor demonstrated the selection of a binder from unrelated antibodies, which might indicate limited sensitivity of the system.
Extending the above approaches directly to primary B-cells will make it possible to circumvent the long procedure of generating hybridoma cells in the first place and facilitate the screening of a much larger fraction of the immune repertoire, including that of humans. This potential has also attracted the interest of several microfluidic companies offering services and/or instruments for large scale antibody screens. However, the proposed screening applications have never been published, which might indicate that the status of commercial technology is not as advanced as claimed on the non-peer reviewed company websites. It seems unlikely that ground breaking applications are routinely up and running without any paper preceding this (which, in an industrial context, would also be a free advertisement of the proprietary technology). Nonetheless it is just a question of time until we will see such applications, no matter if demonstrated by academic labs or by companies. Ultimately, this will for sure revolutionize antibody screening and furthermore enable completely novel approaches in immunology.
Small molecule screening
As illustrated above for antibody screens, the generation of genetically different droplets can be easily achieved by continuously encapsulating single cells of a cell library. In contrast, generating chemically distinct droplets is very difficult as it requires changing the composition of the aqueous phase. In the simplest case, experiments using different drugs can be done in parallel or sequentially (Fig. 6A). For example, a droplet drug library can be generated using multiple drop makers simultaneously and collecting the resulting emulsions in a single tube. However, this approach requires barcoding of the droplet composition. Brouzes et al. demonstrated such a work flow and generated a library of eight different drug samples, each one additionally labelled with a different concentration of Alexa Fluor 680 R-phycoerythrin (emitting in the red/magenta channel).26 Subsequent to library generation, the droplets were reinjected into a second microfluidic chip and fused with droplets hosting individual U937 cells, before incubating for 24 h to allow for the drug to take effect. Then, fluorescent live/dead stains (emitting in the green and orange channel) were added by droplet fusion, and the cytotoxicity of the drug samples was monitored by laser spectroscopy downstream of a delay line.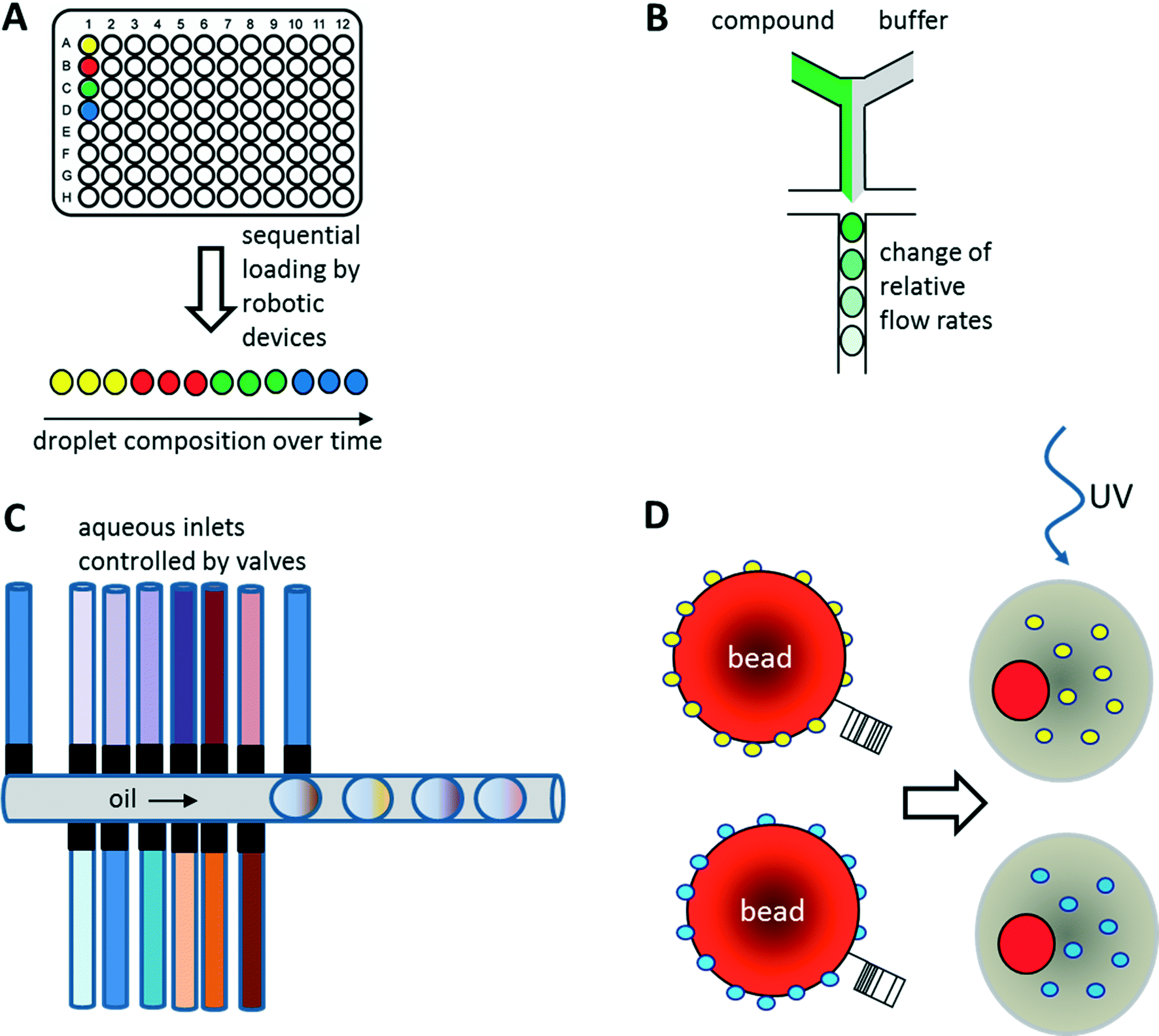 | ||
| Fig. 6 Strategies for the screening of small molecule libraries. (A) Generation of chemical diversity by sequential loading of samples from microtiter plates. (B) Generation of concentration gradients by changing the relative flow rates of compound and buffer. (C) Generation of combinatorial mixtures using multiple valve-controlled aqueous inlets. Opening several valves at the same time allows generating desired mixtures on-demand. Furthermore, unequal valve opening times can be exploited for changing the compound concentrations. (D) Encapsulation of beads displaying different compounds via photo cleavable linkers. Upon UV radiation the compounds can be released inside the droplets. Optionally the beads can also display unique barcodes to reveal the compound identity after screening. | ||
As well as generating droplets of different composition in parallel, they can also be generated sequentially and much effort has been put into automating this process using autosamplers or liquid handling robotic devices.80,81 The time for loading a single compound onto a microfluidic chip this way is typically in the range of seconds, as has been demonstrated using a variety of different approaches: initial studies by the Viovy lab used customized microtiter plates that were completely immersed in oil.82 The tip of an aspirator was then first immersed in the aqueous compound, before being lifted into the oil phase and starting a further cycle. As a consequence, droplets of different compositions were aspirated into the target tubing and spread out by oil. Similarly, Gielen et al. designed a co-axial aspirator in which the outer capillary pumped oil into the wells, while the inner one aspirated the samples.80 This resulted in the direct generation of droplet trains. In an alternative approach, we used an autosampler to aspirate samples and fluorescent barcodes from wells and then co-injected oil to generate droplets inside a target tubing.81 While all these approaches enable the automated feeding of different compounds into the microfluidic system, the number of chemically distinct samples that can be screened in a single experiment still remains quite limited and typically does not exceed a few hundred at maximum. To exploit the power of droplet-based microfluidics further, several other strategies have been developed: i) increasing the chemical diversity by generating concentration gradients for high resolution dose–response studies ii) generating systematic combinatorial mixtures on chip iii) using drugs immobilized on beads.83
Generation of concentration gradients for dose–response studies
Concentration gradients of compounds can be easily generated on-chip by continuously changing the relative flow rates of samples and buffer (Fig. 6B). This can even be done with more than one compound at a time, resulting in multidimensional concentration gradients. Doing so for antibiotics, Cao et al. generated 1150 distinct mixtures of ampicillin, caffeine and captopril and tested their effect on the growth of E. coli.84 These dose response studies not only enabled the identification of optimal antibiotic concentrations and mixtures, but also revealed antagonistic effects between the compounds.Another way of screening drugs in a dose-dependent manner in droplet-based microfluidics is to exploit Taylor–Aris dispersion for generating concentration gradients. Miller et al. directly injected aqueous compounds spread out with buffer into the microfluidic chip using an autosampler.85 During flow of the samples, the compounds dispersed into the buffer and hence arrived at the microfluidic droplet maker at different concentrations. This enabled the screening of a total of 704 drugs for the inhibition of protein tyrosine phosphatase 1B (PTP1B) in a high resolution dose response study with a concentration range of more than three orders of magnitude. As a direct result of this work, sodium cefsulodine could be identified as a new inhibitor of PTP1B.
Combinatorial screening in microfluidic droplets
Combinatorial drug cocktails have shown improved efficacy in the treatment of several types of cancer or infectious diseases.86 In contrast to screening for entirely new drugs, this approach also ensures well documented and certified safety, toxicity and pharmaceutical profiles.87,88 Furthermore, systematic mixing of compounds also holds great potential in combinatorial (medicinal) chemistry. Therefore, many corresponding screens have been carried out previously using microtiter plate-based robotic systems.89–92 Similar approaches have also been implemented in microfluidic systems making use of either droplet fusion or valves controlling the addition of different drugs during droplet generation. For example, Theberge et al. synthesized a library of 7 × 3 potential enzyme inhibitors on-chip by fusing droplets containing amine reagents A1–3 with droplets containing aldehyde reagents B1–7. However, droplets containing reagent A1–3 or B1–7 had to be generated individually and mixed to generate a library prior to the fusion process, which is very time consuming for larger screens. Furthermore, the approach was lacking any barcodes in the droplets, and thus did not allow the assignment of a specific effect to a particular compound. Instead, using this approach, it is only possible to test if a given library contains any members showing an inhibitory potential.93As an alternative to exploiting droplet fusion, combinatorial samples can also be generated by mixing different compounds on-chip prior to the generation of droplets (Fig. 6C). This can be done at relatively high rates (∼1 s−1) using microfluidic valves. The Wang lab used such an approach to screen 5 Matrix Metalloproteinase (MMP) substrates against 5 different MMPs at 5 different concentrations (resulting in 650 samples when including controls) to infer substrate specificity.94 All reagents on the microfluidic device were constantly kept under pressure, such that a drop was released whenever a valve was opened. The relative volumes of different reagents within the mixture and hence their concentration could furthermore be controlled by the valve opening time.94
Beads
Instead of loading drugs onto microfluidic chips in solution, they can also be co-encapsulated into droplets on beads (Fig. 6D). This can be done at high frequency (similar to the encapsulation of cells) and enables the screening of diverse one-bead-one-compound (OBOC) libraries, synthesized in a combinatorial split and pool approach. We have used such an approach to set up a screening system for enzymatic inhibitors.95 However, instead of using a conventional droplet maker, we seeded the beads into nanoliter wells (with a diameter slightly larger than a single bead) together with all components of a fluorescence assay for horseradish peroxidase activity. In a second step, the nanowells were covered with oil, resulting in the generation of arrayed droplets, each one hosting a single bead. Subsequently the samples were irradiated with UV light to cleave the photolabile linker and trigger compound release. Then the samples were incubated and a microscopic readout for enzyme inhibition was performed. Hits were manually recovered from the nanowell plates and applied to the deconvolution of the gas chromatography barcodes, indicating the compound identity on each bead. Performing a model screen of 35000 samples including the positive control 4-aminobenzoic acid exclusively resulted in the selection of true hits, thus demonstrating the power of the approach.
Integrating a microfluidic drop maker into such a system can help to improve throughput and facilitate further droplet manipulation steps. However, sedimentation of the beads typically hinders their use in microfluidic devices. To circumvent this limitation, Paegel and co-workers developed the “suspension hopper” which consists of a pipette tip inserted into a microfluidic chip.83 This approach enables the loading of beads by gravity in a very controlled way: the beads sediment to the bottom of the chip where buffer from a perpendicular channel transports them to the drop maker. There, the beads are co-encapsulated together with assay compounds at high throughput. Paegel and co-workers successfully used this system to assay 112000 samples for inhibition of HIV protease. Even though they did not demonstrate the sorting of positive samples or the use of barcodes for hit identification, it is easy to imagine how powerful such an integrated platform would be. Taken together, it seems very likely that the coming years will show many more examples in which small molecule compounds rather than genetically encoded factors are screened in high throughput fashion using droplet-based microfluidics.
Droplet based microfluidics in genomics
Single-cell heterogeneity is attracting increasing attention, largely fueled by the possibilities of novel microfluidic systems. As a direct result, it is becoming more and more obvious that differences on the single-cell level have crucial significance in development, differentiation, signaling and disease. For example, genomic heterogeneity is of major importance in cancer therapy, since it can cause the selection of resistant cells. Droplet based microfluidic technology that can allow for compartmentalization not only of single cells but also single genomes is thus an ideal technique to study single-cell genomics at high throughput. Since the beginning, when on-chip polymerase chain reaction (PCR) and reverse transcription (RT) – PCR was performed in droplets,96–98 significant progress has been made with respect to studying genomics using droplet based microfluidics.Single-cell DNA and genomic studies
The cellular genome can be altered in comparison to the parental cell due to various environmental cues, errors in replication or exposure to various substances. Similarly, varying expression patterns of the cell during differentiation are well known. One of the major challenges of detecting such differences in single-cell genomic studies is the identification of rare mutations in large cell populations. This can be achieved by precise and sensitive quantification of mutated DNA using digital techniques such as Digital PCR (dPCR). Digital PCR is a sensitive and specific technique for quantification of a target DNA molecule, as well as identification of rare variants by making use of large limiting dilution of the starting sample to achieve less than one template per aliquot.99 dPCRs have been done previously in microtiter plates, on beads in picotiter plates, in water-in-oil emulsions, or even by using valve-based microfluidic platforms.100–102 However, these platforms can analyze no more than a few hundred reactions, whereas droplet-based microfluidics can enable the screening of millions of such reactions.Pekin et al. developed a picoliter droplet based microfluidic platform enabling emulsion based digital PCR to identify and sensitively detect mutations in the KRAS oncogene within a large excess of wildtype genomic DNA.103 Using a dual probe TaqMan assay, resulting in green droplets upon amplification of mutant DNA and red droplet upon amplification of wildtype DNA, precise mutant alleles could be detected in several cancer cell lines. The authors were able to sensitively detect a single mutated KRAS gene over a background of 200000 unmutated KRAS genes. Furthermore they demonstrated the screening for 6 common mutations in a single experiment by encapsulating six different probes in picoliter droplets which were then fused with the droplets containing DNA templates and PCR reagents. The clinical utility of this technique was also shown by detection of KRAS mutations in circulating DNA from the plasma of colorectal cancer patients: in 14 out of 19 clinically confirmed KRAS mutation positive cases, the genotype could be determined correctly from blood plasma, rather than requiring invasive biopsies as in conventional diagnosis.104 Shuga et al. carried out a nested digital PCR in droplets to quantitatively measure a rare carcinogenic translocation, t(14;18), in healthy subjects, which is a predictive biomarker for follicular lymphoma.105 The key feature of the study was a pre-amplification step to increase sensitivity. From the clot genomic DNA an amplicon was produced which was then used as a template in a second, nested-PCR. To quantify the nested-PCR amplified products, a primer attached to beads and a second FAM-labelled primer were used, resulting in fluorescent labelling and capture of the amplified products on the beads. The beads were then analyzed using flow-cytometry, enabling the detection and sequencing of a single t(14;18) copy in 9 μg (∼3 × 106 copies) of clot genomic DNA (gDNA).105 Similarly, a duplex droplet digital PCR has also been used to detect pathogenic E. coli and Listeria monocytogenes from a drinking water supply, using two color fluorescent probes.106 Digital PCR can also be carried out in agarose beads rather than using aqueous droplets.107 In this method, uniform-sized agarose solution-in-oil droplets are formed, which essentially serve as miniaturized PCR-reactors. After the PCR, the droplets are cooled down, resulting in solidified beads in which the amplicons are trapped. Thus, the agarose beads serve as a capturing matrix to retain the monoclonality of the amplicons, even when the oil phase is removed. The agarose beads can then be processed for genotyping or sequencing.107 Using agarose droplet microfluidics, Geng et al. developed a short tandem repeat (STR) genotyping method to identify a target sequence from a mixture of cells and DNA.108 Interestingly, the cells were lysed inside the agarose droplets by allowing the diffusion of lysis buffer into it, followed by diffusion of PCR mix into the droplets. The authors could decipher the STR profile of GM09947 cells in the presence of contaminating GM09948 genomic DNA molecules at high concentrations.
A study by Hatch et al. showed an alternative method for analyzing droplet PCR amplification using fluorescence imaging.109 The technique involved an integrated microfluidic platform for droplet generation, on-chip thermocycling for dPCR and fluorescence imaging of up to 1 million droplets in an array enabling quantitative analysis of the PCR products.109 However, all these sequence analysis studies require prior knowledge of sequences or mutations associated with the particular disease or genomic target.
To overcome this limitation, complementary developments in DNA sequencing and mapping have been established. For example, Abate et al., have put forward a proof of concept for a droplet-based microfluidic platform for analyzing DNA sequences in a high throughput manner using a FRET based ligation assay.110 In order to analyze a 64 bp region of a lacZ operon, the authors employed two probes, one labelled with quencher and another with FAM, that bind adjacent to one another on the target molecule. In order to decipher the identity of probes, droplets were labelled with different dyes and at various concentrations. The probes were then co-encapsulated into the droplets which were then picoinjected with templates and analyzed by fluorescence spectroscopy. The results showed that the perfect complementarity between some probes and a template could be identified using this technique and mismatch of even a single base could be discriminated. By increasing the number as well as the length of probes and considering all possible sequence combinations, this assay can be extrapolated to detect arbitrary target sequences.110 However, one likely limitation for upscaling is the cross talk between different fluorescence channels, which probably restricts the number of distinguishable fluorescence signals (for barcoding different ligation probes) to no more than a few hundred. In 2015, Eastburn et al., developed a droplet based sequence enrichment and analysis method called, MESA (Microfluidic droplet Enrichment for Sequence Analysis),111 to identify a particular genomic region of interest from 50–200 kb size targets. The method is an alternative to hybridization based sequence capture-enrichment methods and does not rely on PCR amplification of a whole target to be sequenced. Instead, MESA makes use of selective amplification of small (∼100 bp) regions within the 50–200 kb targets, using TaqMan probes. The TaqMan amplified target sequences were then separated by sorting of fluorescent droplets. Subsequently the resultant TaqMan amplicons were enzymatically cleaved so that only the enriched full-length target sequences could be applied to next generation sequencing (NGS). As key advantages, this method requires only minimal knowledge of the target sequence and circumvents PCR based artifacts or bias.111 Such studies can be pursued for discovering novel mutations associated with a particular disease using multiple probes and subsequent sequence combinations. With the advances in the field of droplet microfluidics allowing PCR amplification of templates in droplets and sequencing of the templates; researchers have also made some progress with respect to whole genome amplification/sequencing of single cells. In 2008, Kumaresan et al., developed a high throughput single-cell/single genome amplification technique using nanoliter droplets.112 Single cells or DNA template molecules were encapsulated into droplets along with PCR mix, dye labelled forward primers and microbeads displaying reverse primers. The emulsion PCR produced attomole quantities of long amplicons (>600 bp) in individual droplets, which is sufficient for Sanger sequencing. One of the significant highlights of the study is the use of a microfabricated droplet generator that produced nanoliter droplets which could hold larger amounts of nucleotides, PCR mix and also allow for longer (>500 bp) amplicons. In contrast, previous studies that performed the PCR in pico to femtoliter droplets,96,102 did not allow for uniform amplification of sufficient amounts of templates that could be further used for next generation sequencing. Fu et al. have recently shown emulsion-based whole genome amplification using multiple displacement amplification (MDA) chemistry.113,114 In this study, single-cell genomic DNA fragments were distributed into a large number (105) of picoliter droplets and were amplified to the saturation level using the MDA method. MDA is a non-PCR based DNA amplification technique that can efficiently amplify even minute amounts of DNA samples, such as from single cells, using random hexamers and high fidelity enzyme which can work at constant temperature. Since MDA is prone to external DNA contamination, dilution of the templates in emulsion significantly reduced the rate of false-positives. Fu et al., have also been able to show that using this method the amplification bias was reduced while retaining the sensitivity and it was possible to simultaneously detect small copy number variations (CNVs) and high confidence single-nucleotide variations (SNV).114 Sidore et al. have reported the use of MDA for the amplification of femtogram amounts of starting material from E. coli single cell genomes.115 Some of the shortcomings of the MDA method such as amplification of contaminated DNA or non-uniform amplification of templates can normally cause errors and/or lead to decreased sequencing coverage. To overcome this, the authors have described a digital droplet MDA method involving the compartmentalization of single molecules for amplification, which ultimately resulted in significantly better sequencing data. The authors also compared the efficiency of bulk emulsion MDA, polydisperse droplet MDA and digital droplet MDA, with the latter showing the most reliable results.115
Characterization of single-cell genomes has also been interesting for the discovery and analysis of novel microbial species which cannot be cultivated in vitro. As a proof of concept, Lim et al., have used PCR activated cell sorting (PACS116) for identification and isolation of rare microbial species from a heterogeneous population.117 In this work, small genomic regions of hundreds of bases in length served as sequence biomarkers and were used to amplify specific genomic regions using TaqMan PCR. The specific genomic amplification resulted in bright fluorescence droplets which could then be sorted and processed for further analysis (Fig. 7). The TaqMan PCR could potentially even facilitate simultaneous interrogation of several genomic regions in the same microbe by mutiplexing.117
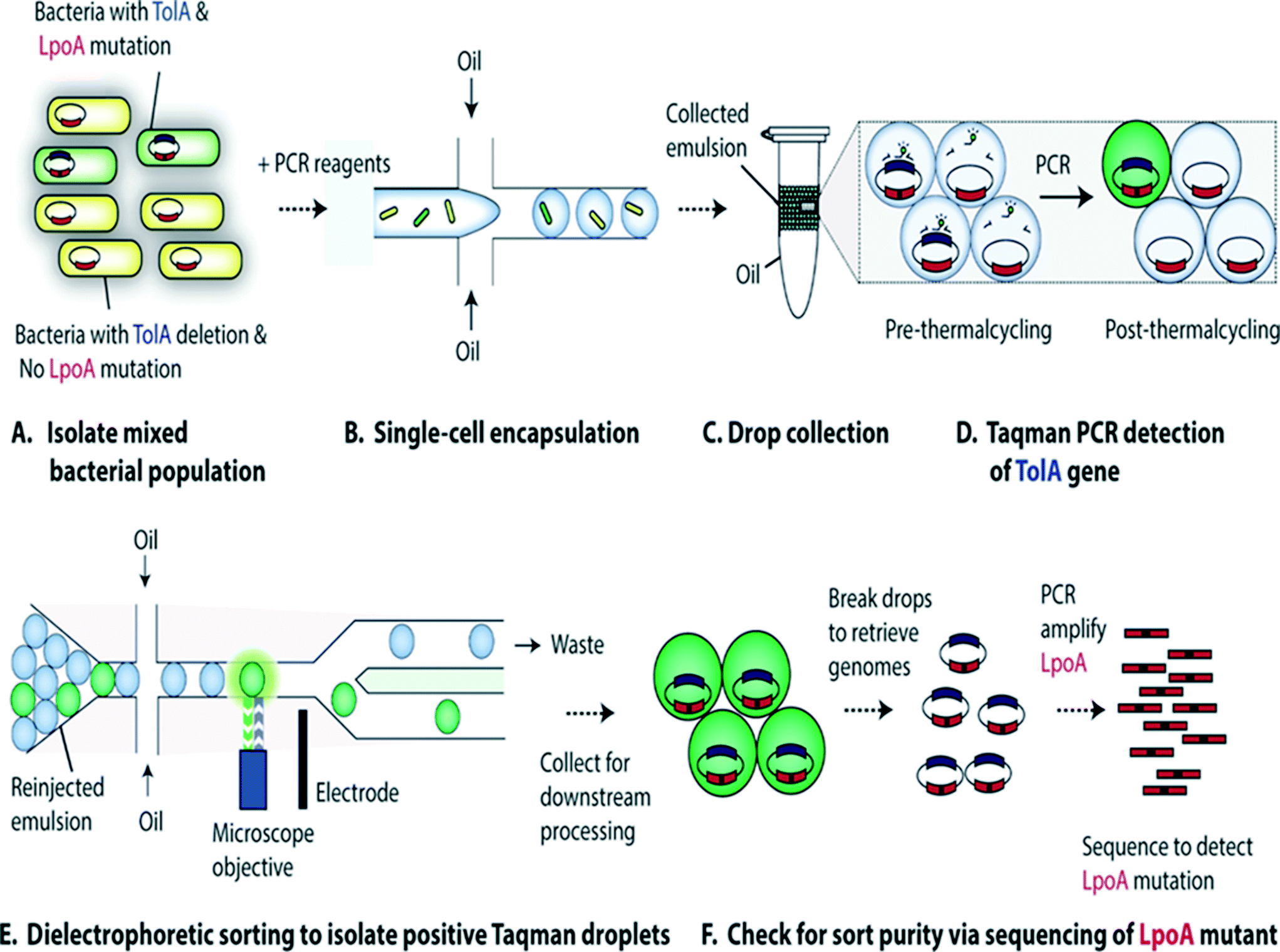 | ||
| Fig. 7 PCR Activated Cell Sorting (PACS) methodology. (A) A mixture of 2 bacterial strains (e.g. containing wild type TolA with LpoA mutation and a ΔTolA spike-in variant) are encapsulated into droplets, together with PCR reagents and TaqMan probes (B). The emulsion is then collected and thermocycled (C) & (D). The specific amplification of the targeted TolA region produces TaqMan fluorescence, enabling the sorting of positive droplets (E) from which the genomic content can be recovered for validation and to determine the efficiency of this genotype based sorting procedure (F). Figure reproduced from ref. 117 by Lim et al. published in Plos One. | ||
In another step forward, Rotem et al. recently performed droplet based single-cell Chromatin Immunoprecipitation (ChIP)-Seq, a chromatin profiling technique on the single-cell level that could identify subpopulations of the cells based on the chromatin state.65 In this procedure, cells were lysed and chromatin was fragmented inside the droplets. In order to annotate the chromatin from single cells, these droplets were allowed to fuse with another set of droplets containing a barcode library of oligonucleotide adapters (a unique barcode for each droplet and hence each individual cell). These barcode adapters were ligated to both ends of nucleosomal DNA fragments and contained a unique ‘barcode’ sequence, an Illumina-compatible adaptor and a restriction site for removing concatemers. Contents of the droplets were then pooled together and immunoprecipitated with an antibody, followed by PCR amplification and sequencing of the enriched DNA (Fig. 8). In this manner, the authors screened thousands of individual cells and were able to segregate embryonic stem cell subpopulations by their chromatin signatures of pluripotency- or differentiation-associated transcription factors or by targeted epigenetic repressors. The data indicated that a single-cell chromatin profile showed only about 1000 unique peaks but with very high specificity and correlating with previously known positive sites. Although the cell to cell variations could be detected, the authors admit that there is still scope for improvements in the efficiency of ligation, amplification and barcoding using alternative strategies.65 This could lead to higher data output per cell as well as extensive coverage for all the chromatin fragments that were generated. Nevertheless, the system already enabled the identification of a “cell type” from a given population with nearly 100% accuracy.65
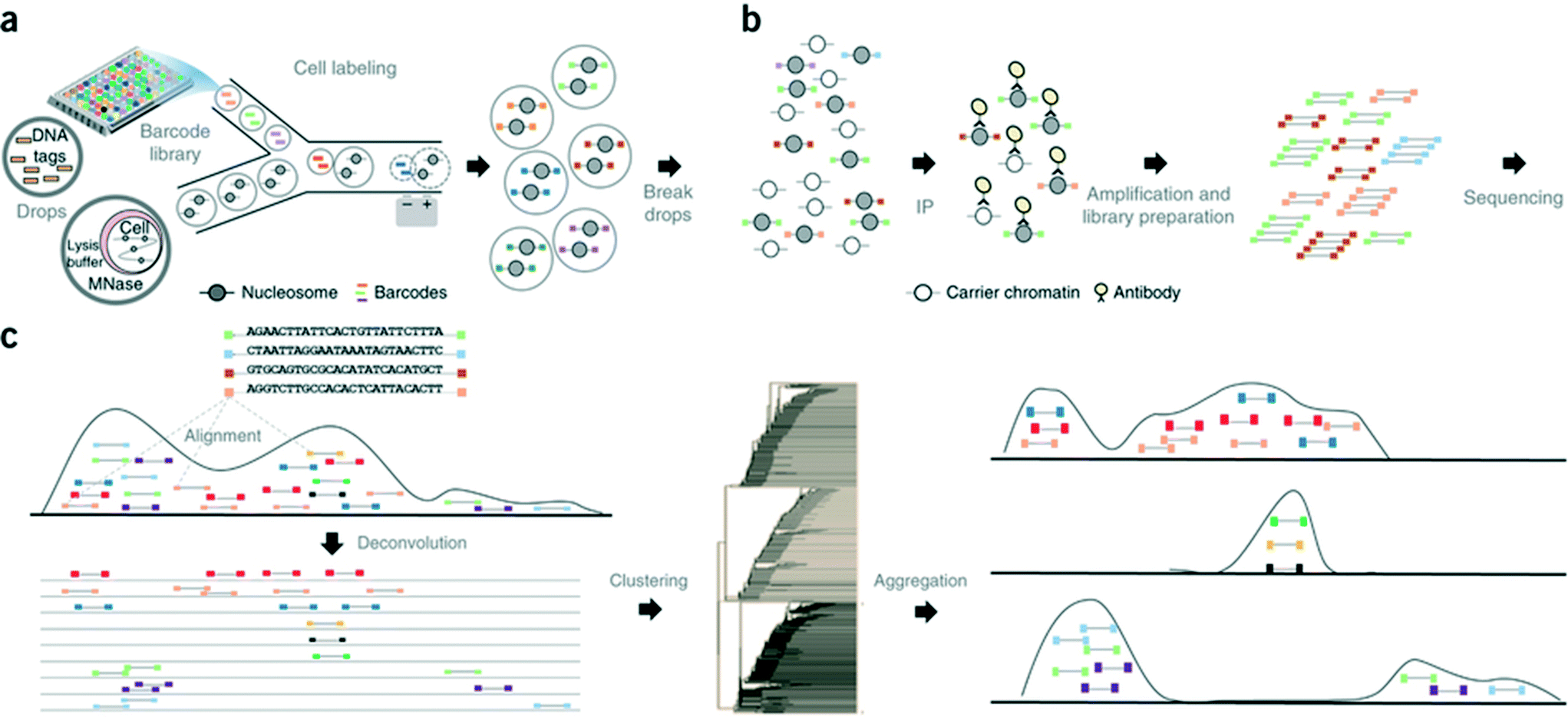 | ||
| Fig. 8 Workflow of the Drop-ChIP method. (a) A DNA barcode library is emulsified from plates into the droplets. Separately, cells are lysed inside further droplets and chromatin is fragmented using micrococcal nuclease (MNase). The barcode droplets and the cell contents bearing droplets are then fused in a microfluidic device, with ligation buffer being simultaneously added through a third inlet (not shown). This 3 point merging step triggers the labelling of fragmented chromatin with unique barcodes. (b) The contents of all the drops are then pooled together and immunoprecipitated along with carrier DNA. (c) Subsequently the enriched fragments are sequenced enabling the assignment of chromatin profiles to individual cells (based on the barcodes). Reproduced with permission from ref. 65 copyright 2015, Nature publishing group. | ||
Single-cell RNA and transcriptomic studies
The ability to perform RT-PCR in droplet-based microfluidic systems opened tremendous avenues for exploring gene expression profiles and transcriptomic studies on single cells at high throughput scale. For example, Eastburn et al. developed a droplet based high throughput microfluidic platform for single-cell TaqMan RT-PCR of up to 50000 cells in parallel.118 One of the highlights of the technique is the dilution of the cell lysate by splitting of the drops before picoinjection of the RT-PCR reagents, which avoids the lysate mediated inhibition of RT-PCR. The study described the specific identification of PTPRC (protein tyrosine phosphatase, receptor type, C) transcript expressing cells, from a mixture of dually stained cells, PTPRC expressing calcein green stained Raji cells and PTPRC-negative calcein violet labelled PC3 cells. The cell staining allowed for analyzing the specificity of the TaqMan reaction, elimination of false positive amplifications and identification of the TaqMan signal arising from free transcripts or lysate. Importantly, about 50000 RT-PCR reactions were performed within 2 h and with as little as 400 μL reaction mix.
As a further breakthrough, novel approaches for analyzing single cells transcripts by NGS rather than qPCR were introduced. In contrast to previously existing non-droplet microfluidic systems such as the Fluidigm C1 platform, these droplet-based approaches enable the processing of thousands of individual cells in parallel. The first published system allowing to process large cell numbers was termed CytoSeq. While Cytoseq did not make use of microfluidic droplets119 and was based on miniaturized assay wells, the assay volumes and many of the assay principles are similar to very recent droplet approaches and it seems worth to briefly discuss CytoSeq here. In this method, single cells were seeded into microwells (with 100000 wells per plate; each having a diameter of ∼30 μm) with a volume of ∼20 pL. Each well also received a bead barcoded with oligonucleotides that contained a universal PCR priming site, a combinatorial cell label, a unique molecular identifier (to circumvent mRNA amplification biases), and an mRNA capture sequence. After cell lysis, mRNAs hybridized to beads, which were pooled for reverse transcription, amplification and sequencing119 (Fig. 9). Using this technique, the authors could segregate a human PBMC population of about 750 cells into major cell types. Additionally, close to 5000 T cells were analyzed based on a subset of 93 genes for their activation profiles to in vitro stimuli. About 7000 T cells were also screened for identifying novel antigen specific T cells after exposure to cytomegalovirus (CMV) peptide. After these successful demonstration experiments the technology has been made available commercially by Cellular Research, Inc. This might be of particular interest for non-microfluidics labs, especially as the approach does not involve complex microfluidic handling procedures.
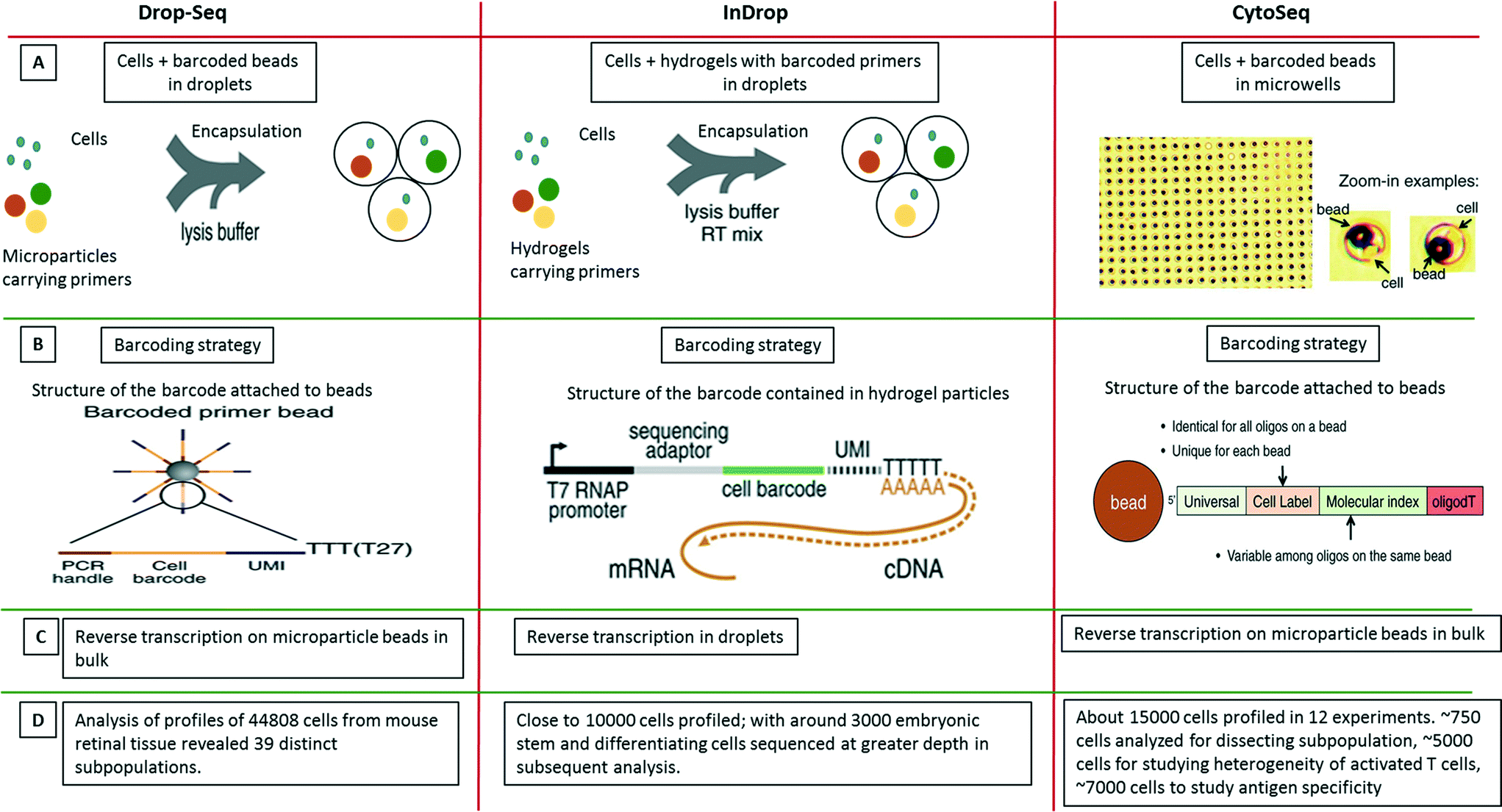 | ||
| Fig. 9 Single-cell transcriptomics using Drop-seq, inDrop and Cyto-seq methods. The figure represents a schematic summary and comparison of 3 recently published techniques for studying single-cell transcriptomics: Drop-seq,63 inDrop64 and Cyto-seq.119 (A) The Drop-seq method involves co-encapsulation of cells along with beads coated with primers into droplets. The inDrop method involves co-encapsulation of cells along with a reverse transcription (RT) mix and hydrogel particles carrying primers that can be released upon UV excitation. The Cyto-seq method involves the seeding of single cells and beads in microwells with picoliter volumes. (B) The barcoding strategy is fairly similar amongst all the methods, with initial universal sequence for PCR/sequencing, followed by a cell barcode, then a unique molecular identifier (UMI) sequence and a poly-dT sequence to capture mRNAs. (C) There are few differences in RT reaction of the captured mRNAs. In Drop-seq method, the RT reaction is carried out on the captured mRNAs on the beads after breaking the droplets. In InDrop method, the RT reaction is carried out inside the droplets. In Cyto-seq, the RT reaction is carried out in bulk after pooling all the beads and thereby the captured mRNAs with them. (D) Published throughput of the different methods (in terms of single cells that have been analyzed). Reproduced with permission from ref. 119 copyright 2015, The American Association for the Advancement of Science and ref. 63 and 64 copyright 2015, Elsevier. | ||
Only months after the CytoSeq paper, two further single-cell transcriptomics approaches based on droplet technology were published back to back: inDrop and Drop-seq, both enabling global RNAseq of thousands of individual cells.63,64 The inDrop is a single-cell transcriptomic profiling method that includes unique labeling of mRNAs from single cells by co-encapsulating the cells with a barcode library consisting of photo-releasable primers on hydrogel microspheres, lysis buffer and RT-PCR mix. As for the CytoSeq method, the immobilized barcodes were generated in a solid phase split and pool synthesis.64 The mRNAs released from each cell were captured and uniquely barcoded during cDNA synthesis inside the droplets. After barcoding, the droplets were broken, all the material was combined and the cDNA library was sequenced using established methods (CEL-seq) (Fig. 9). Interestingly, due to the close packing and regular release of deformable hydrogels, ∼100% droplet occupancy in terms of gel beads could be achieved, while only ∼10% of the droplets hosted a cell. Each microsphere contained more than 109 covalently coupled primers, encoding one of 147456 barcodes and thus a total of over 10000 barcoded cells and controls could be profiled. Analysis of mRNA from thousands of mouse embryonic stem cells and differentiating cells provided a map of gene regulatory elements and corresponding population profiles.
In the simultaneously developed Drop-seq methodology, a single-cell suspension from a tissue was co-encapsulated with microparticle beads containing the DNA-barcodes (Fig. 9). The unique barcoding strategy that retained the origin of the transcript with its cell, consisted of four parts of a barcode: i) a constant sequence that is identical on all beads and is used for downstream PCR and sequencing, ii) a cell barcode that is unique to a particular bead and essential for the co-encapsulated cell to be identified, iii) a unique molecular identifier (UMI) which is different on each primer to differentiate between PCR duplicates and iv) an oligo-dT sequence for capturing polyadenylated mRNAs and priming RT reaction (Fig. 9). After the cells were lysed and mRNAs were captured on microparticles inside the droplets; the emulsion was broken. The reverse transcription was carried out on beads following which sequencing and analysis was performed. Both techniques, inDrop and Drop-seq highlight the problem of contaminations in single-cell studies and thus have used species mixing studies to evaluate it. Altogether, transcriptomic analysis of 44808 cells from retinal tissue identified 39 distinct cell populations using Drop-seq.63
Taken together, inDrop and Drop-seq have provided an impressive demonstration of the throughput that can be achieved by droplet microfluidics. Nonetheless there are still a few limitations. Firstly, the low capture efficiency of mRNAs (∼7%), could be problematic for transcripts with extremely low abundance. Secondly, though the barcoding allows for complete transcript profiling of a single-cell, the randomness in barcoding does not account for cell size, shape, or lineage.64 In other words, it does not allow coupling of genotype with phenotype. We are confident that means to overcome these limitations, as well as the development of other exciting applications will be addressed in future work. Furthermore, the techniques can be extrapolated to DNA studies as well and with coupling to inhibitors, mutations or pathogens, multi-dimensional data could be obtained.
DeKosky et al. have described another application of droplet-based mRNA capture for the sequencing of paired antibody variable heavy and light chain (VH–VL) genes.120 The VH–VL chains of an antibody are encoded by separate mRNA transcripts, thus preventing the sequencing of samples containing more than a single B-cell (for which the information about the correct chain pairing gets lost). To overcome this limitation, the authors encapsulated single B cells into droplets, together with lysis buffer and individual poly(dT) magnetic beads for mRNA capture. After annealing of mRNA from single cells, the beads were recovered and re-emulsified into droplets with RT-PCR mix to amplify and fuse all individual VH–VL genes. This approach allowed the processing of up to 6 million human B-cells on a single day at an efficiency of >97% in terms of correct VH–VL chain pairing.120
In parallel to studying cellular mRNA molecules in droplets, the Weitz lab also applied droplet microfluidics to analyze single viral RNA templates and characterized mutations/recombinations associated with it.121 The mutations including recombinations that happen in viral genomes are an important aspect of host-pathogen interaction, since such rare recombinant viruses often cause epidemics and pandemics. Tao et al. developed a droplet based microfluidic platform for specific identification and sequencing of rare recombinant Noroviruses emerging from co-infection of 2 strains of Norovirus (MNV-1 and WU20) in macrophage cells. The viral template population was encapsulated in droplets along with RT-PCR mix and TaqMan fluorescent probes. Only recombinant templates were amplified since the two primers used were each specific for the two virus strains. This amplification step was coupled to an increase in fluorescence using the DNA-intercalating dye Eva green, which allowed the sorting of positive droplets and subsequent Sanger sequencing. Due to the analysis of individual RNA molecules, the technique allowed for bias and artifact free amplification of rare recombinant templates in the large background of the genome. Additionally, it also minimized the generation of artificial chimera.121 The advantage of droplet microfluidics over the current genotyping methods in this case is that along with identification of recombination, one can also elucidate the frequency of recombination events. The study was further supported by in vivo mouse model studies, where coinfection of noroviruses in an animal was carried out. The resulting viral templates were processed similarly in a droplet microfluidic platform for detection of recombinant viral templates. The analysis revealed recombination events among the recovered viruses that were mapped to multiple viral loci and occurred at a frequency as low as 1/300000. Thus the study provides an insight into the importance of such single viral genome microfluidic studies that could otherwise be limited by common cell culture/in vitro assays as well as error prone molecular biology techniques.122
In summary, the last decade brought significant developments in the field of single-cell or single molecule genomics studies using droplet-based microfluidics. From the ability to perform PCRs in droplets, to analyze the mutations, and map the sequences, through chromatin profiling at single-cell level to analyze whole transcriptome of individual cells with precise barcoding strategies, droplet microfluidics is undoubtedly widening the scope for single-cell genomic studies. While this technology unquestionably provides the throughput required for large scale single-cell genomics studies, at the moment these studies are still at the level of proof-of-concept based research. It will be exciting to see how these techniques will spread and how they will be applied in other research fields, for example in developmental biology or biomedical science (e.g. for analyzing tumour heterogeneity). Furthermore it will be interesting to see to what extent different methods can be combined to obtain highly multiplexed data.
Outlook
Here, we have presented an overview of the latest developments, potentialities and promises of droplet-based microfluidics in drug discovery and molecular genetics applications. While the number of publications in this field seems to grow exponentially, for now most of the studies show proof-of-concepts instead of addressing important biomedical questions. With due appreciation for all the technological innovations so far, now is the time to apply the technology successfully with the goal of delivering biological breakthroughs such as the discovery of new drugs and drug targets or detailed knowledge on cellular pathways and mechanisms. Progress in the following areas seems to be of major importance for achieving this:In conclusion, significant future discoveries and breakthroughs are not necessarily dependent on the development of further microfluidic modules, but rather on combining knowledge from different disciplines. This is already the goal of particular conferences and meetings (such as the EMBL Microfluidics Conference series and the Single Cell Genomics (SCG) meetings), but requires further interdisciplinary initiatives from academia and industry. We feel confident that such initiatives will grow over the next few years.
Acknowledgements
The authors would like to thank Adam Gristwood for proof-reading the manuscript.References
- M. Junkin and S. Tay, Lab Chip, 2014, 14, 1246–1260 RSC.
- S. Vyawahare, A. D. Griffiths and C. A. Merten, Chem. Biol., 2010, 17, 1052–1065 CrossRef CAS PubMed.
- K. Choi, A. H. C. Ng, R. Fobel and A. R. Wheeler, Annu. Rev. Anal. Chem., 2012, 5, 413–440 CrossRef CAS PubMed.
- I. E. Araci and P. Brisk, Curr. Opin. Biotechnol., 2014, 25, 60–68 CrossRef CAS PubMed.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem., Int. Ed., 2010, 49, 5846–5868 CrossRef CAS PubMed.
- P. Day, A. Manz and Y. Zhang, Microdroplet Technology Principles and Emerging Applications in Biology and Chemistry, Springer, New York, 2012 Search PubMed.
- T. M. Tran, F. Lan, C. S. Thompson and A. R. Abate, J. Phys. D: Appl. Phys., 2013, 46, 114004 CrossRef.
- M. T. Guo, A. Rotem, J. A. Heyman and D. A. Weitz, Lab Chip, 2012, 12, 2146–2215 RSC.
- D. R. Link, S. L. Anna, D. A. Weitz and H. A. Stone, Phys. Rev. Lett., 2004, 92, 054503 CrossRef CAS PubMed.
- D. C. Duffy, J. C. McDonald, O. J. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- N. R. Beer, E. K. Wheeler, L. Lee-Houghton, N. Watkins, S. Nasarabadi, N. Hebert, P. Leung, D. W. Arnold, C. G. Bailey and B. W. Colston, Anal. Chem., 2008, 80, 1854–1858 CrossRef CAS PubMed.
- T. Kawakatsu, G. Tragardh, C. Tragardh, M. Nakajima, N. Oda and T. Yonemoto, Colloids Surf., A, 2001, 179, 29–37 CrossRef CAS.
- J. Clausell-Tormos, D. Lieber, J. C. Baret, A. El-Harrak, O. J. Miller, L. Frenz, J. Blouwolff, K. J. Humphry, S. Koster, H. Duan, C. Holtze, D. A. Weitz, A. D. Griffiths and C. A. Merten, ACS Chem. Biol., 2008, 15, 427–437 CrossRef CAS PubMed.
- S. Kuhn, R. L. Hartman, M. Sultana, K. D. Nagy, S. Marre and K. F. Jensen, Langmuir, 2011, 27, 6519–6527 CrossRef CAS PubMed.
- S. Begolo, G. Colas, J. L. Viovy and L. Malaquin, Lab Chip, 2011, 11, 508–512 RSC.
- T. Thorsen, R. W. Roberts, F. H. Arnold and S. R. Quake, Phys. Rev. Lett., 2001, 86, 4163–4166 CrossRef CAS PubMed.
- S. L. Anna, N. Bontoux and H. A. Stone, Appl. Phys. Lett., 2003, 82, 364–366 CrossRef CAS.
- L. Yobas, S. Martens, W. L. Ong and N. Ranganathan, Lab Chip, 2006, 6, 1073–1079 RSC.
- C. Cramer, P. Fischer and E. J. Windhab, Chem. Eng. Sci., 2004, 59, 3045–3058 CrossRef CAS.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. B. Hutchison, F. E. Angile, C. H. J. Schmitz, S. Koster, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano and D. A. Weitz, Lab Chip, 2008, 8, 1632–1639 RSC.
- K. C. Lowe, J. Fluorine Chem., 2002, 118, 19–26 CrossRef CAS.
- J. C. Baret, Lab Chip, 2012, 12, 422–433 RSC.
- A. Huebner, M. Srisa-Art, D. Holt, C. Abell, F. Hollfelder, A. J. deMello and J. B. Edel, Chem. Commun., 2007, 1218–1220 RSC.
- E. W. Kemna, R. M. Schoeman, F. Wolbers, I. Vermes, D. A. Weitz and A. van den Berg, Lab Chip, 2012, 12, 2881–2887 RSC.
- S. Koster, F. E. Angile, H. Duan, J. J. Agresti, A. Wintner, C. Schmitz, A. C. Rowat, C. A. Merten, D. Pisignano, A. D. Griffiths and D. A. Weitz, Lab Chip, 2008, 8, 1110–1115 RSC.
- E. Brouzes, M. Medkova, N. Savenelli, D. Marran, M. Twardowski, J. B. Hutchison, J. M. Rothberg, D. R. Link, N. Perrimon and M. L. Samuels, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14195–14200 CrossRef CAS PubMed.
- A. C. Hatch, J. S. Fisher, A. R. Tovar, A. T. Hsieh, R. Lin, S. L. Pentoney, D. L. Yang and A. P. Lee, Lab Chip, 2011, 11, 3838–3845 RSC.
- L. Mazutis, A. F. Araghi, O. J. Miller, J. C. Baret, L. Frenz, A. Janoshazi, V. Taly, B. J. Miller, J. B. Hutchison, D. Link, A. D. Griffiths and M. Ryckelynck, Anal. Chem., 2009, 81, 4813–4821 CrossRef CAS PubMed.
- J. U. Shim, L. F. Olguin, G. Whyte, D. Scott, A. Babtie, C. Abell, W. T. Huck and F. Hollfelder, J. Am. Chem. Soc., 2009, 131, 15251–15256 CrossRef CAS PubMed.
- T. P. Lagus and J. F. Edd, RSC Adv., 2013, 3, 20512–20522 RSC.
- H. Hu, D. Eustace and C. A. Merten, Lab Chip, 2015, 15, 3989–3993 RSC.
- M. R. Bringer, C. J. Gerdts, H. Song, J. D. Tice and R. F. Ismagilov, Philos. Trans. R. Soc., A, 2004, 362, 1087–1104 CrossRef CAS PubMed.
- H. Song, M. R. Bringer, J. D. Tice, C. J. Gerdts and R. F. Ismagilov, Appl. Phys. Lett., 2003, 83, 4664–4666 CrossRef CAS PubMed.
- A. T. H. Hsieh, N. Hori, R. Massoudi, P. J. H. Pan, H. Sasaki, Y. A. Lin and A. P. Lee, Lab Chip, 2009, 9, 2638–2643 RSC.
- L. Frenz, K. Blank, E. Brouzes and A. D. Griffiths, Lab Chip, 2009, 9, 1344–1348 RSC.
- B. El Debs, R. Utharala, I. V. Balyasnikova, A. D. Griffiths and C. A. Merten, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11570–11575 CrossRef CAS PubMed.
- L. Frenz, A. El Harrak, M. Pauly, S. Begin-Colin, A. D. Griffiths and J. C. Baret, Angew. Chem., Int. Ed., 2008, 47, 6817–6820 CrossRef CAS PubMed.
- D. R. Link, E. Grasland-Mongrain, A. Duri, F. Sarrazin, Z. D. Cheng, G. Cristobal, M. Marquez and D. A. Weitz, Angew. Chem., Int. Ed., 2006, 45, 2556–2560 CrossRef CAS PubMed.
- L. Mazutis, J. C. Baret and A. D. Griffiths, Lab Chip, 2009, 9, 2665–2672 RSC.
- M. Chabert and J. L. Viovy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3191–3196 CrossRef CAS PubMed.
- Y. C. Tan, J. S. Fisher, A. I. Lee, V. Cristini and A. P. Lee, Lab Chip, 2004, 4, 292–298 RSC.
- X. Niu, S. Gulati, J. B. Edel and A. J. deMello, Lab Chip, 2008, 8, 1837 RSC.
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163–19166 CrossRef CAS PubMed.
- M. Srisa-Art, A. J. deMello and J. B. Edel, Anal. Chem., 2007, 79, 6682–6689 CrossRef CAS PubMed.
- K. L. A. Chan, X. Niu, A. J. deMello and S. G. Kazarian, Anal. Chem., 2011, 83, 3606–3609 CrossRef CAS PubMed.
- M. P. Cecchini, J. Hong, C. Lim, J. Choo, T. Albrecht, A. J. Demello and J. B. Edel, Anal. Chem., 2011, 83, 3076–3081 CrossRef CAS PubMed.
- N. Li, Y. A. Gao, L. Q. Zheng, J. Zhang, L. Yu and X. W. Li, Langmuir, 2007, 23, 1091–1097 CrossRef CAS PubMed.
- J. S. Edgar, G. Milne, Y. Zhao, C. P. Pabbati, D. S. Lim and D. T. Chiu, Angew. Chem., Int. Ed., 2009, 48, 2719–2722 CrossRef CAS PubMed.
- X. Z. Niu, B. Zhang, R. T. Marszalek, O. Ces, J. B. Edel, D. R. Klug and A. J. deMello, Chem. Commun., 2009, 6159–6161 RSC.
- L. M. Fidalgo, G. Whyte, B. T. Ruotolo, J. L. Benesch, F. Stengel, C. Abell, C. V. Robinson and W. T. Huck, Angew. Chem., Int. Ed., 2009, 48, 3665–3668 CrossRef CAS PubMed.
- T. Hatakeyama, D. L. Chen and R. F. Ismagilov, J. Am. Chem. Soc., 2006, 128, 2518–2519 CrossRef CAS PubMed.
- G. T. Roman, M. Wang, K. N. Shultz, C. Jennings and R. T. Kennedy, Anal. Chem., 2008, 80, 8231–8238 CrossRef CAS PubMed.
- X. Y. Hu, P. H. Bessette, J. R. Qian, C. D. Meinhart, P. S. Daugherty and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15757–15761 CrossRef CAS PubMed.
- J. C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El-Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison, J. J. Agresti, D. R. Link, D. A. Weitz and A. D. Griffiths, Lab Chip, 2009, 9, 1850–1858 RSC.
- A. Y. Fu, C. Spence, A. Scherer, F. H. Arnold and S. R. Quake, Nat. Biotechnol., 1999, 17, 1109–1111 CrossRef CAS PubMed.
- Z. C. Landry, S. J. Giovanonni, S. R. Quake and P. C. Blainey, Methods Enzymol., 2013, 531, 61–90 CAS.
- L. Schmid, D. A. Weitz and T. Franke, Lab Chip, 2014, 14, 3710–3718 RSC.
- C. H. Chen, S. H. Cho, H. I. Chiang, F. Tsai, K. Zhang and Y. H. Lo, Anal. Chem., 2011, 83, 7269–7275 CrossRef CAS PubMed.
- P. S. Dittrich and P. Schwille, Anal. Chem., 2003, 75, 5767–5774 CrossRef CAS PubMed.
- A. Sciambi and A. R. Abate, Lab Chip, 2015, 15, 47–51 RSC.
- A. R. Thiam, N. Bremond and J. Bibette, Phys. Rev. Lett., 2009, 102, 188304 CrossRef PubMed.
- Y. Zhao, H. C. Shum, H. Chen, L. L. Adams, Z. Gu and D. A. Weitz, J. Am. Chem. Soc., 2011, 133, 8790–8793 CrossRef CAS PubMed.
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef CAS PubMed.
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS PubMed.
- A. Rotem, O. Ram, N. Shoresh, R. A. Sperling, A. Goren, D. A. Weitz and B. E. Bernstein, Nat. Biotechnol., 2015, 33, 1165–1172 CrossRef CAS PubMed.
- A. Ali-Cherif, S. Begolo, S. Descroix, J. L. Viovy and L. Malaquin, Angew. Chem., Int. Ed., 2012, 51, 10765–10769 CrossRef CAS PubMed.
- P. Gruner, B. Riechers, B. Semin, J. Lim, A. Johnston, K. Short and J. C. Baret, Nat. Commun., 2016, 7, 10392 CrossRef CAS PubMed.
- A. Casadevall, E. Dadachova and L. A. Pirofski, Nat. Rev. Microbiol., 2004, 2, 695–703 CrossRef CAS PubMed.
- S. Kotsovilis and E. Andreakos, Methods in molecular biology, 2014, vol. 1060, pp. 37–59 Search PubMed.
- X. Qiu, G. Wong, J. Audet, A. Bello, L. Fernando, J. B. Alimonti, H. Fausther-Bovendo, H. Wei, J. Aviles, E. Hiatt, A. Johnson, J. Morton, K. Swope, O. Bohorov, N. Bohorova, C. Goodman, D. Kim, M. H. Pauly, J. Velasco, J. Pettitt, G. G. Olinger, K. Whaley, B. Xu, J. E. Strong, L. Zeitlin and G. P. Kobinger, Nature, 2014, 514, 47–53 CAS.
- P. Carter, Nat. Rev. Cancer, 2001, 1, 118–129 CrossRef CAS PubMed.
- R. T. Strait, M. T. Posgai, A. Mahler, N. Barasa, C. O. Jacob, J. Kohl, M. Ehlers, K. Stringer, S. K. Shanmukappa, D. Witte, M. M. Hossain, M. Khodoun, A. B. Herr and F. D. Finkelman, Nature, 2015, 517, 501–504 CrossRef CAS PubMed.
- M. X. Sliwkowski and I. Mellman, Science, 2013, 341, 1192–1198 CrossRef CAS PubMed.
- A. C. Chan and P. J. Carter, Nat. Rev. Immunol., 2010, 10, 301–316 CrossRef CAS PubMed.
- D. M. Ecker, S. D. Jones and H. L. Levine, mAbs, 2015, 7, 9–14 CrossRef PubMed.
- A. P. Kodituwakku, C. Jessup, H. Zola and D. M. Roberton, Immunol. Cell Biol., 2003, 81, 1e63–170 CrossRef PubMed.
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. D. Griffiths and J. A. Heyman, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS PubMed.
- S. Miraglia, E. E. Swartzman, J. Mellentin-Michelotti, L. Evangelista, C. Smith, I. Gunawan, K. Lohman, E. M. Goldberg, B. Manian and P. M. Yuan, J. Biomol. Screening, 1999, 4, 193–204 CrossRef CAS PubMed.
- T. Konry, M. Dominguez-Villar, C. Baecher-Allan, D. A. Hafler and M. L. Yarmush, Biosens. Bioelectron., 2011, 26, 2707–2710 CrossRef CAS PubMed.
- F. Gielen, T. Buryska, L. Van Vliet, M. Butz, J. Damborsky, Z. Prokop and F. Hollfelder, Anal. Chem., 2015, 87, 624–632 CrossRef CAS PubMed.
- J. Clausell-Tormos, A. D. Griffiths and C. A. Merten, Lab Chip, 2010, 10, 1302–1307 RSC.
- M. Chabert, K. D. Dorfman, P. de Cremoux, J. Roeraade and J. L. Viovy, Anal. Chem., 2006, 78, 7722–7728 CrossRef CAS PubMed.
- A. K. Price, A. B. MacConnell and B. M. Paegel, Anal. Chem., 2014, 86, 5039–5044 CrossRef CAS PubMed.
- J. Cao, J. Goldhan, K. Martin and J. M. Köhler, Investigation of mixture toxicity of widely used drugs caffeine and ampicillin in the presence of an ACE inhibitor on bacterial growth using droplet-based microfluidic technique, Green Process. Synth., 2013, 2(6), 591–601 CAS.
- O. J. Miller, A. El Harrak, T. Mangeat, J. C. Baret, L. Frenz, B. El Debs, E. Mayot, M. L. Samuels, E. K. Rooney, P. Dieu, M. Galvan, D. R. Link and A. D. Griffiths, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 378–383 CrossRef CAS PubMed.
- A. A. Borisy, P. J. Elliott, N. W. Hurst, M. S. Lee, J. Lehar, E. R. Price, G. Serbedzija, G. R. Zimmermann, M. A. Foley, B. R. Stockwell and C. T. Keith, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7977–7982 CrossRef CAS PubMed.
- T. T. Ashburn and K. B. Thor, Nat. Rev. Drug Discovery, 2004, 3, 673–683 CrossRef CAS PubMed.
- C. R. Chong and D. J. Sullivan, Jr., Nature, 2007, 448, 645–646 CrossRef CAS PubMed.
- R. E. White, Annu. Rev. Pharmacol. Toxicol., 2000, 40, 133–157 CrossRef CAS PubMed.
- L. A. Mathews Griner, R. Guha, P. Shinn, R. M. Young, J. M. Keller, D. Liu, I. S. Goldlust, A. Yasgar, C. McKnight, M. B. Boxer, D. Y. Duveau, J. K. Jiang, S. Michael, T. Mierzwa, W. Huang, M. J. Walsh, B. T. Mott, P. Patel, W. Leister, D. J. Maloney, C. A. Leclair, G. Rai, A. Jadhav, B. D. Peyser, C. P. Austin, S. E. Martin, A. Simeonov, M. Ferrer, L. M. Staudt and C. J. Thomas, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2349–2354 CrossRef CAS PubMed.
- T. Kodadek, Chem. Commun., 2011, 47, 9757–9763 RSC.
- J. P. Kennedy, L. Williams, T. M. Bridges, R. N. Daniels, D. Weaver and C. W. Lindsley, J. Comb. Chem., 2008, 10, 345–354 CrossRef CAS PubMed.
- A. B. Theberge, E. Mayot, A. El Harrak, F. Kleinschmidt, W. T. Huck and A. D. Griffiths, Lab Chip, 2012, 12, 1320–1326 RSC.
- T. D. Rane, H. C. Zec and T. H. Wang, Anal. Chem., 2015, 87, 1950–1956 CrossRef CAS PubMed.
- G. Upert, C. A. Merten and H. Wennemers, Chem. Commun., 2010, 46, 2209–2211 RSC.
- N. R. Beer, B. Hindson, E. K. Wheeler, S. B. Hall, K. A. Rose, I. M. Kennedy and B. W. Colston, Anal. Chem., 2007, 79, 8471–8475 CrossRef CAS PubMed.
- N. R. Beer, E. Wheeler, L. Lee-Houghton, N. Watkins, S. Nasarabadi, N. Hebert, P. Leung, D. W. Arnold, C. G. Bailey and B. W. Colston, Anal. Chem., 2008, 80, 1854–1858 CrossRef CAS PubMed.
- P. Mary, L. Dauphinot, N. Bois, M. C. Potier, V. Studer and P. Tabeling, Biomicrofluidics, 2011, 5, 24109 CrossRef PubMed.
- E. Day, P. Dear and F. McCaughan, Methods, 2013, 59, 101–107 CrossRef CAS PubMed.
- J. H. Leamon, W. L. Lee, K. R. Tartaro, J. R. Lanza, G. J. Sarkis, A. D. deWinter, J. Berka, M. Weiner, J. M. Rothberg and K. L. Lohman, Electrophoresis, 2003, 24, 3769–3777 CrossRef CAS PubMed.
- D. Dressman, H. Yan, G. Traverso, K. W. Kinzler and B. Vogelstein, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8817–8822 CrossRef CAS PubMed.
- E. A. Ottesen, J. Hong, S. R. Quake and J. R. Leadbetter, Science, 2006, 314, 1464–1467 CrossRef CAS PubMed.
- D. Pekin, Y. Skhiri, J. C. Baret, D. Le Corre, L. Mazutis, C. B. Salem, F. Millot, A. El Harrak, J. B. Hutchison, J. W. Larson, D. R. Link, P. Laurent-Puig, A. D. Griffiths and V. Taly, Lab Chip, 2011, 11, 2156–2166 RSC.
- V. Taly, D. Pekin, L. Benhaim, S. K. Kotsopoulos, D. Le Corre, X. Li, I. Atochin, D. R. Link, A. D. Griffiths, K. Pallier, H. Blons, O. Bouché, B. Landi, J. B. Hutchison and P. Laurent-Puig, Clin. Chem., 2013, 59, 1722–1731 CAS.
- J. Shuga, Y. Zeng, R. Novak, Q. Lan, X. Tang, N. Rothman, R. Vermeulen, L. Li, A. Hubbard, L. Zhang, R. A. Mathies and M. T. Smith, Nucleic Acids Res., 2013, 41, e159 CrossRef PubMed.
- X. Bian, F. Jing, G. Li, X. Fan, C. Jia, H. Zhou, Q. Jin and J. Zhao, Biosens. Bioelectron., 2015, 74, 770–777 CrossRef CAS PubMed.
- X. Leng, W. Zhang, C. Wang, L. Cui and C. J. Yang, Lab Chip, 2010, 10, 2841–2843 RSC.
- T. Geng, R. Novak and R. A. Mathies, Anal. Chem., 2014, 86, 703–712 CrossRef CAS PubMed.
- A. C. Hatch, J. Fisher, A. R. Tovar, A. T. Hsieh, R. Lin, S. L. Pentoney, D. L. Yang and A. P. Lee, Lab Chip, 2011, 11, 3838–3845 RSC.
- A. R. Abate, T. Hung, R. A. Sperling, P. Mary, A. Rotem, J. J. Agresti, M. A. Weiner and D. A. Weitz, Lab Chip, 2013, 13, 4864–4869 RSC.
- D. J. Eastburn, Y. Huang, M. Pellegrino, A. Sciambi, L. J. Ptáček and A. R. Abate, Nucleic Acids Res., 2015, 43, e86 CrossRef PubMed.
- P. Kumaresan, C. J. Yang, S. A. Cronier, R. G. Blazej and R. A. Mathies, Anal. Chem., 2008, 80, 3522–3529 CrossRef CAS PubMed.
- R. S. Lasken, Curr. Opin. Microbiol., 2007, 10, 510–516 CrossRef CAS PubMed.
- Y. Fu, C. Li, S. Lu, W. Zhou, F. Tang, X. S. Xie and Y. Huang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11923–11928 CrossRef CAS PubMed.
- A. M. Sidore, F. Lan, S. W. Lim and A. R. Abate, Nucleic Acids Res., 2015 DOI:10.1093/nar/gkv1493.
- D. J. Eastburn, A. Sciambi and A. R. Abate, Nucleic Acids Res., 2014, 42, e128 CrossRef PubMed.
- S. W. Lim, T. Tran and A. R. Abate, PLoS One, 2015, 10, e0113549 Search PubMed.
- D. J. Eastburn, A. Sciambi and A. R. Abate, Anal. Chem., 2013, 85, 8016–8021 CrossRef CAS PubMed.
- H. C. Fan, G. Fu and S. P. Fodor, Science, 2015, 347, 1258367 CrossRef PubMed.
- B. J. DeKosky, T. Kojima, A. Rodin, W. Charab, G. C. Ippolito, A. D. Ellington and G. Georgiou, Nat. Med., 2015, 21, 86–91 CrossRef CAS PubMed.
- Y. Tao, A. Rotem, H. Zhang, S. K. Cockrell, S. Koehler, C. B. Chang, L. W. Ung, P. Cantalupo, Y. Ren, J. S. Lin, A. B. Feldman, C. E. Wobus, J. M. Pipas and D. Weitz, ChemBioChem, 2015, 16, 2167–2171 CrossRef CAS PubMed.
- H. Zhang, S. Cockrell, A. O. Kolawole, A. Rotem, A. W. Serohijos, C. B. Chang, Y. Tao, T. S. Mehoke, Y. Han, J. S. Lin, N. S. Giacobbi, A. B. Feldman, E. Shakhnovich, D. A. Weitz, C. E. Wobus and J. M. Pipas, J. Virol., 2015, 89, 7722–7734 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |