
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn automated microreactor for semi-continuous biosensor measurements†
Nina
Buffi‡
a,
Siham
Beggah‡
b,
Frederic
Truffer
c,
Martial
Geiser
c,
Harald
van Lintel
a,
Philippe
Renaud
a and
Jan Roelof
van der Meer
*b
aLaboratory for Microsystems Engineering, Ecole Polytechnique de Lausanne, Station 17, CH-1015 Lausanne, Switzerland
bDepartment of Fundamental Microbiology, University of Lausanne, 1015 Lausanne, Switzerland. E-mail: janroelof.vandermeer@unil.ch
cInstitute of Systems Engineering, University of Applied Sciences and Arts of Western Switzerland, 1950 Sion, Switzerland
First published on 8th March 2016
Abstract
Living bacteria or yeast cells are frequently used as bioreporters for the detection of specific chemical analytes or conditions of sample toxicity. In particular, bacteria or yeast equipped with synthetic gene circuitry that allows the production of a reliable non-cognate signal (e.g., fluorescent protein or bioluminescence) in response to a defined target make robust and flexible analytical platforms. We report here how bacterial cells expressing a fluorescence reporter (“bactosensors”), which are mostly used for batch sample analysis, can be deployed for automated semi-continuous target analysis in a single concise biochip. Escherichia coli-based bactosensor cells were continuously grown in a 13 or 50 nanoliter-volume reactor on a two-layered polydimethylsiloxane-on-glass microfluidic chip. Physiologically active cells were directed from the nl-reactor to a dedicated sample exposure area, where they were concentrated and reacted in 40 minutes with the target chemical by localized emission of the fluorescent reporter signal. We demonstrate the functioning of the bactosensor-chip by the automated detection of 50 μgarsenite-As l−1 in water on consecutive days and after a one-week constant operation. Best induction of the bactosensors of 6–9-fold to 50 μg l−1 was found at an apparent dilution rate of 0.12 h−1 in the 50 nl microreactor. The bactosensor chip principle could be widely applicable to construct automated monitoring devices for a variety of targets in different environments.
Introduction
Whole cell living bacterial bioreporters1 (or “bactosensors”)2 present excellent and robust tools for rapid and accurate sensing of select target chemicals in e.g., water,3 food stuff,4 medical specimens,2 gut,5 industrial processes6 or soils.7 The bactosensor platform allows easy and flexible engineering of a variety of chemical target recognition specificities8 while maintaining a single output format, such as fluorescence or bioluminescence.9 A large variety of bactosensors has been constructed in the laboratory targeting different analytes or analyte groups with the help of reporter gene circuits embedded in the cell.1 Bactosensor assays are typically conducted by suspending living sensor cells in aqueous solution with the sample extract of interest and reading out the reporter signal (e.g., bioluminescence, fluorescence or other) after a predefined incubation period.1,8 Alternatively, paper-based in vitro assays which include the reporter gene circuit have also been proposed without the need of live bactosensor cells.10Because of their robustness and flexibility, bactosensors would be ideal for constant monitoring of specific analytes, but they have so far only been deployed in manual and single-use assays. Although permitting accurate analyte quantification, the manual and single-use suspension assay has major drawbacks. Primarily, the physiological state of the sensor cells is poorly controlled in suspension assays. A fast reaction with the target analyte is optimally achieved with exponentially growing sensor cells, but this requires preculturing and dilution procedures, which are cumbersome in matching routine sampling and analyses with regard to time. Alternatively, sensor cells can be cultured and stored to maintain optimal activity, but typical storage procedures of sensor cells by freeze-drying or freezing at −80 °C lead to loss of immediate activation potential and require much manual handling. Further alternative storage options that include cell encapsulation11 or formation of spores12 also need longer cellular reactivation times.
As an alternative strategy we considered continuous cultivation of sensor cells in a miniaturized reactor, which would constantly produce actively growing cells optimally suited for analyte detection and synthesis of the reporter signal. Taking advantage of their small size, bactosensor cells are ideal to be embedded into microengineered platforms that would constitute the complete biosensor unit. Various miniaturized systems for cell culturing have been described that allow continuous growth conditions on chip for multiple days,13–18 and also pre-loaded immobilized cells-on-chip cartridges have been deployed for biosensor measurements.19 However, none of these studies investigated the possibility of using the on-chip cultivated cells for downstream bioassays.
The major question and goal of the underlying work was thus the design and fabrication of an autonomously operating biosensor system, which would permit retrieval on demand of a pool of active reporter cells to perform measurements on external samples. As a proof of concept we used an Escherichia coli reporter strain for the detection of arsenic in water,20 which is a major contaminant of drinking water around the world.21,22 Detection of arsenic requires specialized instruments, which are mostly unavailable in developing countries or remote areas. Simpler technologies for analysis of arsenic in water samples could therefore be useful alternatives. In order to find a suitable solution for an automated and unsupervised biosensor, we designed a miniaturized microfluidic growth reactor for E. coli bactosensor cells. The main idea is that the constant growth in the reactor will maintain cells in a physiologically active state needed for reporter measurements, and constant cell division will produce the quantity of cells needed for incubation with a sample at any desired moment. The microfluidic chip we designed contained, therefore, in addition to the growth reactor, a specific measurement zone into which actively dividing cells can be transferred, and where the cells can be exposed to an aqueous sample with the analyte. After analysis the cells are removed from the chip, readying the measurement zone for the next batch of bactosensor cells. We extensively calibrated the reporter cell growth conditions in the two variants of the microchip reactor with different volumes and demonstrated that over the course of at least a week, growth can be maintained and cells react immediately to the presence of arsenic (here 50 μg l−1 of arsenite), allowing multiple consecutive measurements. Our results thus pave the way for the development of automated monitoring systems incorporating bactosensors.
Materials and methods
Biochip design and fabrication
The bactosensor biochips were fabricated by multilayer soft lithography.23 Biochips are composed of a two-layered polydimethylsiloxane (PDMS) block bonded on a transparent glass slide with a thickness of 1 mm (Fig. 1a). The bottom (flow) layer contains the designed chamber, channels and filter zones, while the top (control) layer contains a set of channels that can be filled with water and pressure-controlled, acting as valves to close the channels in the flow layer below (Fig. 1c and h).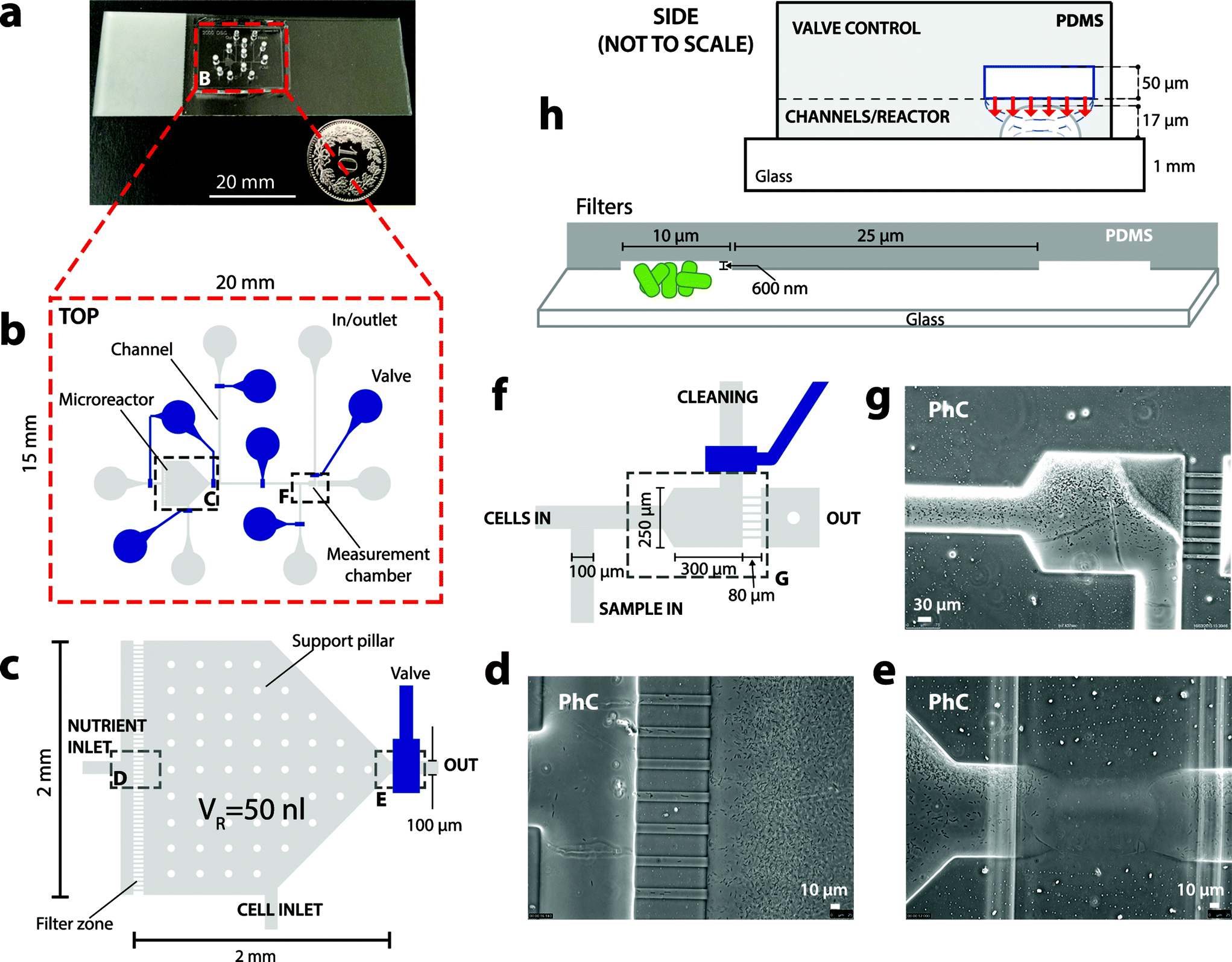 | ||
| Fig. 1 Outline of the microfluidic biochip for bactosensor growth and assays. (a) The biochip is composed of a PDMS block bonded on a glass slide. (b) Top view of the design of the nl-reactor growth and measurement zones with inlets (grey) and the valves for fluid control (blue). (c) Enlargement of the nl-reactor area. Photographic details of the nutrient inlet and microfilter zone (d), and valves (e). (f) Design detail of the microfilter measurement zone. (g) Photographic detail of cells accumulated in the measurement zone. (h) Schematic side-views of the two layered PDMS biochip: a thin bottom layer (30 μm) containing the flow channels (17 μm height), and a thick top layer (ca. 5 mm) containing the control channels. Temporary overpressure (1 bar) in the upper control channels deflects the thin membrane between the two layers and closes the underlying flow channel. Microfilters (10 × 80 × 0.6 μm) block most E. coli bactosensor cells but let the liquid sample pass, allowing exposure of the cells to the target analyte. PhC: phase-constrast. | ||
The microreactor for cell culturing was designed as a pentagonal chamber of 0.8 or 2.9 mm2 with a height of 17 μm, resulting in volumes of 13 or 50 nl (Fig. 1c). Shallow flow filters (600 nm height × 10 μm width × 80 μm length) were installed between the nutrient inlet and the microreactor to prevent backflow of cells into the nutrient solution (Fig. 1d). Further backflow was prevented by valves surrounding the microreactor. The measurement zone also has a similar filter with 6 lines, which prevent reporter cells but not liquid from passing through (Fig. 1f and g). All other flow channels on the microfluidics chip have a height of 17 μm and a width of 100 μm.
Both PDMS layers were fabricated separately using a negative-imprint silicon mold, then superimposed and irreversibly bonded (Fig. S1†). To fabricate single depth molds, silicon wafers were structured by dry etching (Bosch process, Alcatel 601E) using a photolithographically structured AZ1512 resist (Clariant) as a mask. Molds with the shallow filters (600 nm depth) and flow channels (17 μm depth) were produced in two steps: first, dry etching with AZ1512 resist and masking as before, to fabricate the filters. Next, to fabricate the negative of the channels, a layer of AZ9260 resist (Clariant) was deposited (EVG150) on the wafer, exposed to UV (MA6), developed and baked to obtain rounded structures thanks to the resist reflow. This last step is crucial because only rounded channels can be completely closed by the deflection of the membrane in the upper flow control layer.23
The control PDMS valve layer (Fig. S1c†) was prepared from a liquid PDMS mixture containing 60 g of Sylgard 184 silicone elastomer base (Dow Corning) and 12 g of Sylgard 184 silicone elastomer curing agent (ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After degassing, the first mix was poured on the control layer mold (thickness ca. 5 mm). The (thinner) PDMS layer containing reactor, filters and channels (Fig. S1b†) was prepared from a liquid PDMS mixture containing 10 g of base and 0.5 g of curing agent (ratio 20:1), which was spin-coated on the respective mold (thickness ca. 30 μm). Both PDMS layers were cured separately at 80 °C for 13 min. The control PDMS layer was cut into small blocks (1.5 × 2 cm), which were peeled off from the mold and surface activated by oxygen plasma treatment (30 s at 29 W and 300–500 mTorr). The bottom thin layer was also surface activated and the control layer blocks were aligned and superimposed on top, after which the double-layered structure was cured at 80 °C for 16 h (Fig. S1d†). This final curing step binds the two PDMS layers together to withstand a pressure of 2.5 bar. After curing, the PDMS blocks were cut out from the lower (thin) layer, peeled off from the mold, after which flow channel inlets were punched with a 1.5 mm diameter Harris Uni-Core puncher (TED PELLA, Inc). The block and a standard glass slide (1 mm thickness, RS France, Milian, Fig. S1e†) were again activated by oxygen plasma treatment (0.1 min, 100 W and 0.6 mbar, FEMTO plasma cleaner from Diener Electronic), then placed together to be bonded.
:1). After degassing, the first mix was poured on the control layer mold (thickness ca. 5 mm). The (thinner) PDMS layer containing reactor, filters and channels (Fig. S1b†) was prepared from a liquid PDMS mixture containing 10 g of base and 0.5 g of curing agent (ratio 20:1), which was spin-coated on the respective mold (thickness ca. 30 μm). Both PDMS layers were cured separately at 80 °C for 13 min. The control PDMS layer was cut into small blocks (1.5 × 2 cm), which were peeled off from the mold and surface activated by oxygen plasma treatment (30 s at 29 W and 300–500 mTorr). The bottom thin layer was also surface activated and the control layer blocks were aligned and superimposed on top, after which the double-layered structure was cured at 80 °C for 16 h (Fig. S1d†). This final curing step binds the two PDMS layers together to withstand a pressure of 2.5 bar. After curing, the PDMS blocks were cut out from the lower (thin) layer, peeled off from the mold, after which flow channel inlets were punched with a 1.5 mm diameter Harris Uni-Core puncher (TED PELLA, Inc). The block and a standard glass slide (1 mm thickness, RS France, Milian, Fig. S1e†) were again activated by oxygen plasma treatment (0.1 min, 100 W and 0.6 mbar, FEMTO plasma cleaner from Diener Electronic), then placed together to be bonded.
Microreactor setup
Before starting cell culturing, the PDMS block on glass was again treated with oxygen plasma to make the surfaces hydrophilic, after which the channels in the bottom (flow) layer were filled with a sterile nutrient solution (see below) at a pressure of 0.3 bar with all the valves open (Fig. S2a†). A sterile nutrient solution, (arsenic-containing) sample and cleaning solution were prepared in 15 ml glass vials with PTFE/silicone stoppers (Supelco), with the gas phase connected to an air pressure bottle (1.3 bar) and the solution connected to the respective biochip inlet (Fig. S2a†). Gas and liquid connections were made by using Teflon tubing (1.06 mm øin and 1.68 mm øout) connected to the respective biochip inlet by custom-made stainless steel capillary metal adapters (length ∼5 cm, bend to 90° angle, øin 0.25 mm, øout 1.59 mm, MetrOhm, Fig. S2c†).Subsequently all valve channels in the upper layer were filled with water, connected by custom-made stainless steel capillary adapters to Teflon tubing, which were connected to 1/13′′ gauge needles to a set of eight Solenoid valves (24V DC 3-Way, Pneumadyne, Inc.), joined to an air pressure bottle. Valves were operated at an overpressure of 1 bar with respect to the lower flow channels (i.e., 1.3 bar), by which the 13 μm thin PDMS membrane separation between the upper and lower channel deflects and closes the channel below (Fig. 1h). Time intervals and the sequence of valve opening and closing was controlled by a custom-written LabVIEW program (National Instruments). The biochip was operated at controlled room temperature (21 °C).
Microreactor principle of operation
The microreactor principle of operation is illustrated in Fig. S3† and can be summarized as follows. Reporter cells are inoculated by air pressure (0.3 bar) for a few seconds through the cell inlet, which is then permanently closed (Fig. S3b†). Cells are then given fresh nutrients by opening the nutrient in- and outflow valves simultaneously (Fig. S3c†). A sterile external nutrient reservoir (ca. 10 mL in a glass vial) is hereto connected to the nutrient inlet. The frequency of opening in- and outflow valves surrounding the microreactor while maintaining constant pressure-controlled flow determines the growth rate of the cells (Fig. S3b†). In order to make a measurement, part of the cells from the chemostat is released during 30 min and accumulated into the measurement zone (Fig. S3d†). Cells in the measurement zone are exposed to the sample (Fig. S3e†). During the measurement period (180 min) the middle valve is closed and culturing in the reactor continues as before. After measurement, the cage and channels are cleaned by backflow from the second external reservoir, to remove accumulating cells (Fig. S3f†). The valve controlling the cleaning inlet is opened and the flow of the sterile cleaning solution removes the cells in the measurement zone to the waste outlet (Fig. S3f†). In the first design (13 nl reactor, Fig. S4a†), the cells were removed from the measurement zone by increasing the pressure by 0.7 bar, as a result of which the (flexible) filter channels deflect and let the cells pass.Bioreporter strain
The arsenic bioreporter E. coli DH5α strain 1598 (pPR-ArsR-ABS)20 was used for the present work, which carries a plasmid with a transcriptional fusion between the promoter Pars and the gene encoding the enhanced green fluorescent protein (egfp). It also encodes the regulatory protein ArsR, which controls the level of GFP expression in response to arsenite (AsIII as in AsO33− or AsO2−).24The mechanism of response to arsenite is the following: in the absence of arsenite in the cell, ArsR binds the operator sites within the Pars promoter and directly upstream of egfp (Fig. 2a), preventing transcription to occur, and resulting in basal expression of arsR and the downstream egfp gene. In contrast, when arsenite enters the cell, it binds with the ArsR repressor, which undergoes a conformational change and dissociates from its operator, leading to a higher expression of arsR and egfp. The GFP levels attained in the cells over time are linearly proportional to the external arsenite concentration in the range of approximately 1–100 μgAsIII l−1.20,24
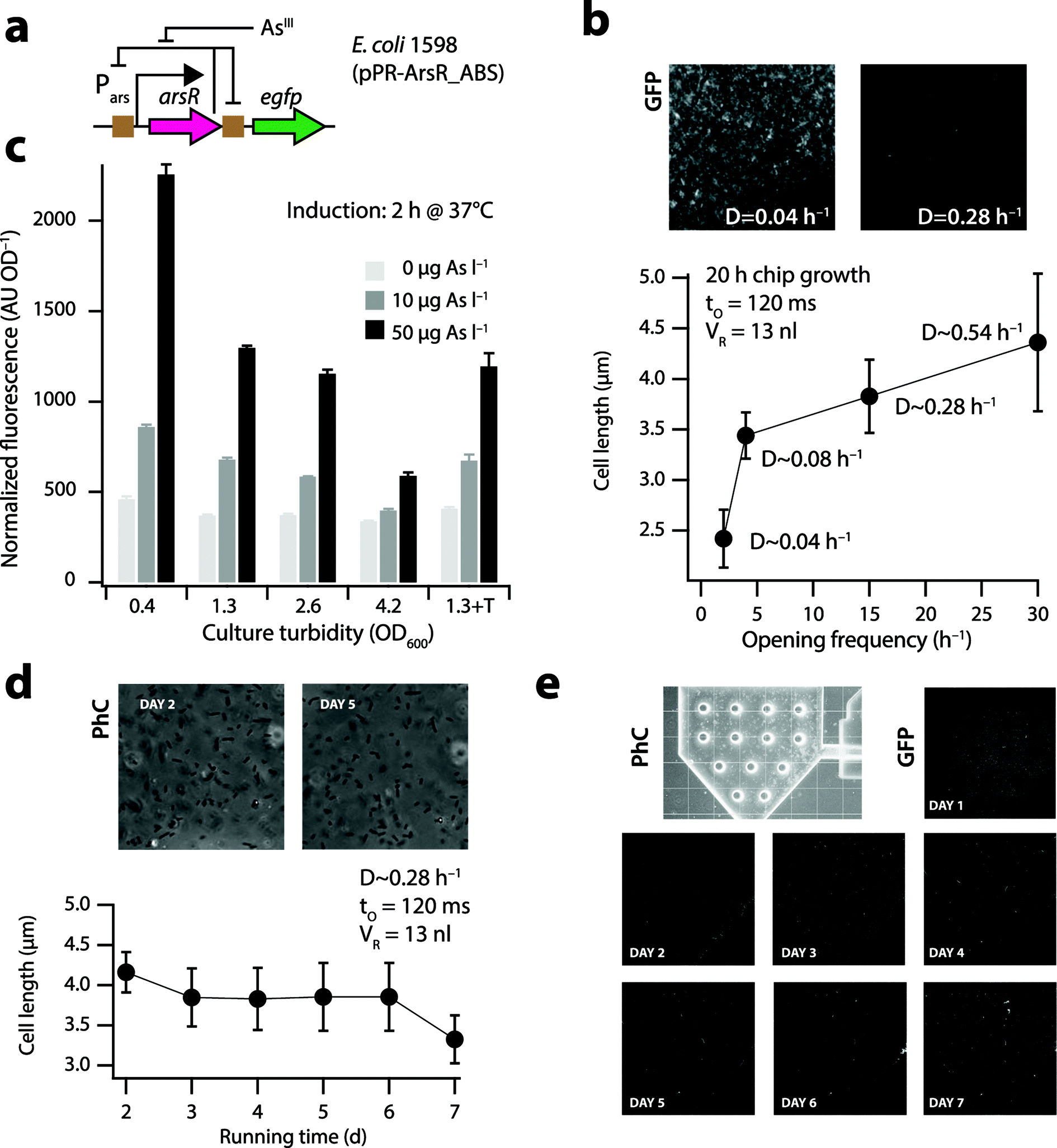 | ||
| Fig. 2 Physiology of E. coli cells in the nl-reactor. (a) Bactosensor cells of E. coli strain 1598 (ref. 20) carry a plasmid-borne genetic circuit in which ArsR controls the expression of itself and of an egfp reporter gene (brown squares indicate ArsR binding sites [ABS]). Arsenite (AsIII) derepresses ArsR, leading to reporter gene expression. (b) Mean cell lengths of E. coli strain 1598 in the 13 nl reactor under different dilution rates, measured at 400× magnification on phase-contrast images of cells taken 20 h after inoculation and continuous growth on the biochip. Inset shows GFP background signals in the nl-reactor at two dilution rates. (c) Mean GFP induction from triplicate assays (normalized to culture turbidity) 2 h after exposure to arsenite by E. coli 1598 at different physiological states in batch culture. Culture turbidity of 4.2 corresponds to stationary phase cells. 1.3 + T, OD600 of 1.3 in the presence of 0.1% Triton X-100. (d) Mean cell length and (e) GFP background (scaled to same maximal intensity) of E. coli 1598 in the 13 nl-reactor during a 7 day continuous culturing period. | ||
In order to qualitatively monitor the physiological state of E. coli DH5α in the microreactor, we created an E. coli strain carrying a plasmid with the rrnB1 ribosomal promoter fused to an unstable GFP.25,26 This strain (E. coli 4224) expresses the GFP protein primarily during exponential growth but less so when the cells are entering or in a stationary phase, in which case the degradation rate of the fluorescent protein (half-life of ∼110 min as a result of the ASV-degradation tag)25 will surpass its de novo synthesis and fluorescence of the cells will remain very low.
Growth media and culture conditions
Starting from a single colony, E. coli DH5α strain 1598 (pPR-arsR-ABS) or 4224 (rrnB1p-gfpASV) was grown in a Luria-Bertani (LB) medium in the presence of 50 μg ml−1 kanamycin at 37 °C for 18 h and with 180 rpm agitation of the culture flask. The bacterial culture was then 50-fold diluted into the fresh LB medium plus kanamycin and incubated for a further 2 h at 37 °C and with 180 rpm agitation. At the culture turbidity between 0.2 and 0.5 at 600 nm (representative for exponential phase), cells from 20 ml of the culture were harvested by centrifugation at 4000 × g for 7 min at room temperature. The cell pellet was resuspended into the MOPS medium (MOPS medium contains 10% [v/v] of MOPS buffer, 2 mM MgCl2, 0.1 mM CaCl2, 2 gglucose l−1, pH 7.0) to obtain a final concentration of cells in the suspension of about 5 × 109 ml−1. The MOPS buffer itself was prepared by dissolving, per liter: 5 g of NaCl, 10 g of NH4Cl, 98.4 g of 3-([N-morpholino]propanesulfonic acid, sodium salt), 0.59 g of Na2HPO4·2H2O and 0.45 g of KH2PO4. An aliquot of 8 μl of cell suspension was then pipetted into the dedicated inoculation inlet of the microreactor and driven into the growing chamber by opening the inlet valve (Fig. 1c, Fig. S3b†).Nutrient solution for biochip cell culturing (also used as cleaning solution) consisted of 50% MOPS medium and 50% LB (v/v) plus 50 μg ml−1 of kanamycin. The carbon content of this medium equals 0.4 gC l−1 from glucose and approximately 2.95 gC l−1 from LB.27 0.1% Triton X-100 was added to the nutrient and cleaning solution in order to prevent cells from clogging at the polymer and glass surfaces.
To test the effect of Triton X-100 addition on the inducibility of the bactosensor cells with arsenite, the cells were cultured in growth media with or without different Triton X-100 concentrations (w/v, 0.05, 0.1 and 0.2%) to a culture turbidity of 1.3, washed and resuspended in MOPS medium.
Cells used for batch induction of reporter formation outside the biochip were precultured as described above to exponential phase (or to the required culture turbidity), resuspended (from 20 ml culture) in 20 ml of MOPS medium and immediately used for assays.
Induction of the bactosensor cells to arsenite
Arsenite solutions equivalent to 10 and 50 μgAsIII l−1 were prepared by dilution from a 50 mM stock solution of sodium arsenite (NaAsO2, Merck) in MOPS medium. Cells in the biochip measurement zone were exposed to the arsenite solution under constant flow (0.4 bar, Fig. S3†) for up to 180 min. In the case of batch assays outside the biochip the cell suspension in MOPS was mixed in a ratio of 1:1 (v/v) with the arsenite solution (10 or 50 μgAsIII l−1) or with pure water as negative control, incubated for 3 h at 30 °C at 500 rpm, and measured every 1 h for GFP fluorescence (480 nm excitation and 520 nm collection) and culture turbidity (at 600 nm) in a 96-well plate reader (FLUOstar Omega, BMG LABTECH). The GFP fluorescence values were normalized for culture turbidity. Cell counting and single cell GFP fluorescence measurements were carried out per flow cytometry as described elsewhere.24 In short, the culture samples were diluted to about 106 cells per ml, of which 100 μl was aspired at 3 μl s−1 on a Becton Dickinson LSR-Fortessa flow cytometer (BD Biosciences, Erembodegem, Belgium). Cells were identified on the basis of their GFP-fluorescence (‘FITC’ channel: emission wavelength of 530 nm, 30 nm bandwidth).
Quantification of cell length and GFP fluorescence in the biochip measurement zone
Reporter cells in the microreactor chamber and in the measurement cage were imaged using a Leica DFC320 cooled black and white charge-coupled device camera (Leica Microsystem CMS GmbH, Wetzlar, Germany) mounted on a Leica DMI6000B inverted epifluorescence microscope. Cells in the chamber and measurement zone were imaged every 20–30 min during incubation at a 100-, 200- or 400-fold magnification (Leica AF6000 program). GFP fluorescence was measured at an exposition time of 66 ms (or 33 ms in the case of signal saturation), using a BP470/40 filter (Leica). In order to estimate the biomass in the microreactor chambers, cells were manually counted in 25 × 25 μm2 areas (at 17 μm depth) on phase-contrast images at a 400-fold magnification. The cell count average was then multiplied over the total chamber volume (13 or 50 nl), assuming homogenous distribution. Cell lengths in the reactor chamber were measured manually on phase-contrast images at a 400-fold magnification, taking into consideration only the individual cells well-aligned to the coverslip surface and excluding occasional aberrantly long cells. Images were recorded as 16-bit TIFF files and the fluorescence signal intensity per unit surface of the measurement cage occupied by cells was determined using ImageJ.28 GFP fluorescence at each induction time point was measured and averaged across ten regions covering the zone of cells in the measurement cage. For display, GFP images were opened and “auto toned” in Adobe Photoshop (CC2014), and levelled to the same minimum and maximum grey values, in order to have all images displayed at the same intensity, cropped to the final size at 300 dpi and saved as 8-bit TIFF-files.Results
Operation of a semi-continuous growth chip for bactosensor cells
We designed and fabricated double-layered polydimethylsiloxane (PDMS) microfluidic biochips (Fig. 1a, Fig. S1†) with a nl-reactor (13 or 50 nl) and integrated valves (Fig. 1b–g), operated by air-pressurized water-filled channels in the upper PDMS layer to control the substrate, sample and cell flow in the bottom layer (Fig. 1h, Fig. S2†). As a case in point we used E. coli 1598 carrying a plasmid with a dedicated genetic circuit (pPR-ArsR-ABS, Fig. 2a),20 as the bactosensor for the measurement of arsenic, a recurrent contaminant of potable water sources across the globe.21E. coli cells were inoculated at a concentration of 5 × 109 cells ml−1 from the inlet into the reactor by air-pressure (0.3 bar and few s, Fig. S3b†) until approximately one-fifth of the reactor visibly contained cells. The inlet valve was closed after inoculation and continuous culturing was started (Fig. S3†). Continuous culturing was achieved by feeding sterile nutrient solutions at a constant flow pressure (0.5 bar) to the cells in the microreactor by simultaneous opening of the nutrient in- and outlet valves during short intervals (120 ms) (Fig. 1b and c, Fig. S3†). As a result, a fixed volume of cell suspension is pushed out from the microreactor to the waste when the same fixed volume of nutrient solution is pushed in. The frequency of opening of the nutrient valves can be varied in order to regulate the growth rate of the cells in the microreactor. The growth phase of E. coli in the reactor as a function of nutrient inlet opening frequency was estimated by measuring the mean cell length after 20 h under a given flow regime (Fig. 2b). Flow pressure and inlet opening rates were converted into an apparent reactor dilution rate (D, h−1). First the flux (F, l h−1) was calculated from the valve opening frequency (e.g., 15 h−1) and the opening time of the nutrient in- and outlet valves (e.g., 120 ms), and by using a calculated flow resistance of 1.44 × 1016 Pa s m−3. D is then derived from F by dividing by the reactor volume (VR, here 13 or 50 nl).Bactosensor physiology under continuous culturing conditions
Cell length measurements in the 13 nl reactor (Fig. S4a†) indicated that cells at high dilution rates (e.g., 0.28 h−1, 4.4 ± 0.7 μm) are longer than those at low dilution rates of 0.04 h−1 (2.4 ± 0.3 μm, Fig. 2b). This is similar to the cell length trends reported in the literature for chemostat-grown E. coli cells at different dilution rates.29 Importantly, cells at low dilution rates displayed a much higher GFP background in the reactor compared to cells at higher dilution rates (Fig. 2b, inset), which will lead to poor induction and low signal-to-noise ratios upon exposure to arsenic. Indeed, arsenite solutions of 10 and 50 μgAsIII l−1 induced the GFP formation 1.5-5 fold in exponentially growing batch cultures (Fig. 2c, culture turbidities of 0.4, 1.3 and 2.6), but only 1.2–2 fold in the stationary phase (Fig. 2c, OD600 = 4.2). For optimal measurement with bactosensor cells, their growth rates in the microreactor should thus be sufficiently high. The microreactor chambers in the steady-state contained on average 9.0 × 104 (13 nl reactor) and 3.5 × 105 cells (50 nl reactor), estimated from cell counts on microscope images. This is close to biomass calculations expected from the carbon content of the nutrient solution (3.35 gC l−1), namely 6.5 × 104 cells in the 13 nl and 2.5 × 105 cells in the 50 nl reactor, assuming 0.2 pg of C (dry weight) per E. coli cell and a yield of 0.3 g g−1.30E. coli cells started to grow in biofilms on the glass and PDMS surfaces after an operation period of 3 days, which caused preferential flow of nutrients and local occurrence of slow-growing or stationary phase cells. This could be effectively counteracted by adding 0.1% Triton X-100 to the culture medium, which did not influence the inducibility of the bactosensor cells by arsenite (Fig. 2c, S5†). Video-imaging showed rapidly swimming cells near the nutrient inlet (Video S1†) and approximately similar amounts of cells released during every opening cycle over the course of the whole experiment. The mean length of the cells at a dilution rate of 0.28 h−1 remained constant over time and close to 3.8 ± 0.4 μm (Fig. 2d). GFP background expression of the bactosensor cells in the 13 nl-reactor growing in media containing 0.1% Triton X-100 remained more or less equally low over the course of 7 days of constant operation, although occasional regions of a higher cell density as well as individual bright cells with aberrant long shapes were observed (Fig. 2e).In order to better examine the physiology of E. coli cells in the reactor, we repeated continuous growth with an E. coli strain (4224) in which the expression of an unstable GFP (GFP-ASV, half life 2 h)25 is coupled to the rrnB1 ribosomal promoter (Fig. S6a†). When cells are actively growing, the rrnB1 promoter is turned on26 and the GFP fluorescence increases (Fig. S6b†). In contrast, the promoter is poorly expressed in cells at stationary phase and GFP fluorescence remains low as a result of degradation rates surpassing new synthesis (Fig. S6b†). Freshly inoculated cells showed homogenous GFP fluorescence across the reactor after 1 day of incubation (Fig. S6c,† day 1), which decreased slightly and became a bit patchier over time under the continuous growth regime (Fig. S6c,† days 2–4), suggesting that locally regions of a high cell density may form. Average GFP signals from E. coli 4224 cells in the 13 nl reactor decreased after initial batch growth (t = 20 h) during continuous culturing (D ∼ 0.28 h−1), suggesting the removal of a part of the biomass until the new steady-state (t = 40 h, Fig. S6d†). Apart from a few patches where cell clusters formed (visible as bright spots in Fig. S6c†), the average GFP signal from E. coli 4224 remained more or less constant over the next four days (Fig. S6d†). Arrest of in- and outflow during 2 h did not dramatically influence cellular GFP fluorescence values (Fig. S6d,† light brown-shaded zones). Collectively, these results showed that E. coli cells on average grow exponentially at the set dilution rates in the nl-reactor. Even with Triton X-100 in the nutrient medium, some patchy growth of the bactosensor cells occurred over time in the reactor, likely as a result of preferential flow paths and the absence of turbulent mixing. However, this did not have a major influence on the overall inducibility of the cells after exposure to arsenite (see below).
Consecutive bactosensor measurements from continuously growing cells in microreactors
Two cultivation and induction regimes were tested with the E. coli 1598 arsenic bactosensor cells. In the first chip design (13 nl reactor, Fig. S4a,†D ∼ 0.28 h−1) the constantly growing cells in the nl-reactor are removed at a rate of ∼2 × 103 cells per opening cycle, which are directed to a waste outlet on the bactosensor-chip (Fig. S4a†). When making a measurement, the cells are redirected to a dedicated filter zone (Fig. S4b†), where they are retained by liquid outlets as shallow as 600 nm (Fig. 1f–h). Cells were collected in the filter zone for 30 min and then exposed to a sample from the sample inlet (Fig. 1f, Fig. S4a†), upon which accumulation of the reporter signal over time was measured. This has the additional advantage that the absolute reporter signal is enhanced by the higher quantity of cells compared to measuring with lower suspended cell density in solution. After measurement, cells were removed from the filter zone by increasing the air pressure, which slightly deforms the PDMS and increases the height of the pores through which the cells disappear (Fig. S4b†). By subsequently decreasing the pressure, the measurement zone was readied for collecting the next batch of cells. At a 0.28 h−1 dilution rate (in the 13 nl reactor) the background GFP signal of the E. coli cells after 3 days of continuous operation was low enough (Fig. 2b, inset) to enable repetitive induction assays with an arsenite solution of 50 μgAsIII l−1 in the measurement cage for three subsequent days (Fig. 3b). In a second independent experiment, the E. coli bactosensor cells were cultured for 6 days on the chip (VR = 13 nl, D ∼ 0.28 h−1), before being directed to the filter zone and exposed to 50 μgAsIII l−1. Cells in both independent experiments displayed slightly different backgrounds but were induced 3–5 fold in the presence of 50 μgAsIII l−1 after 180 min (Fig. 3b).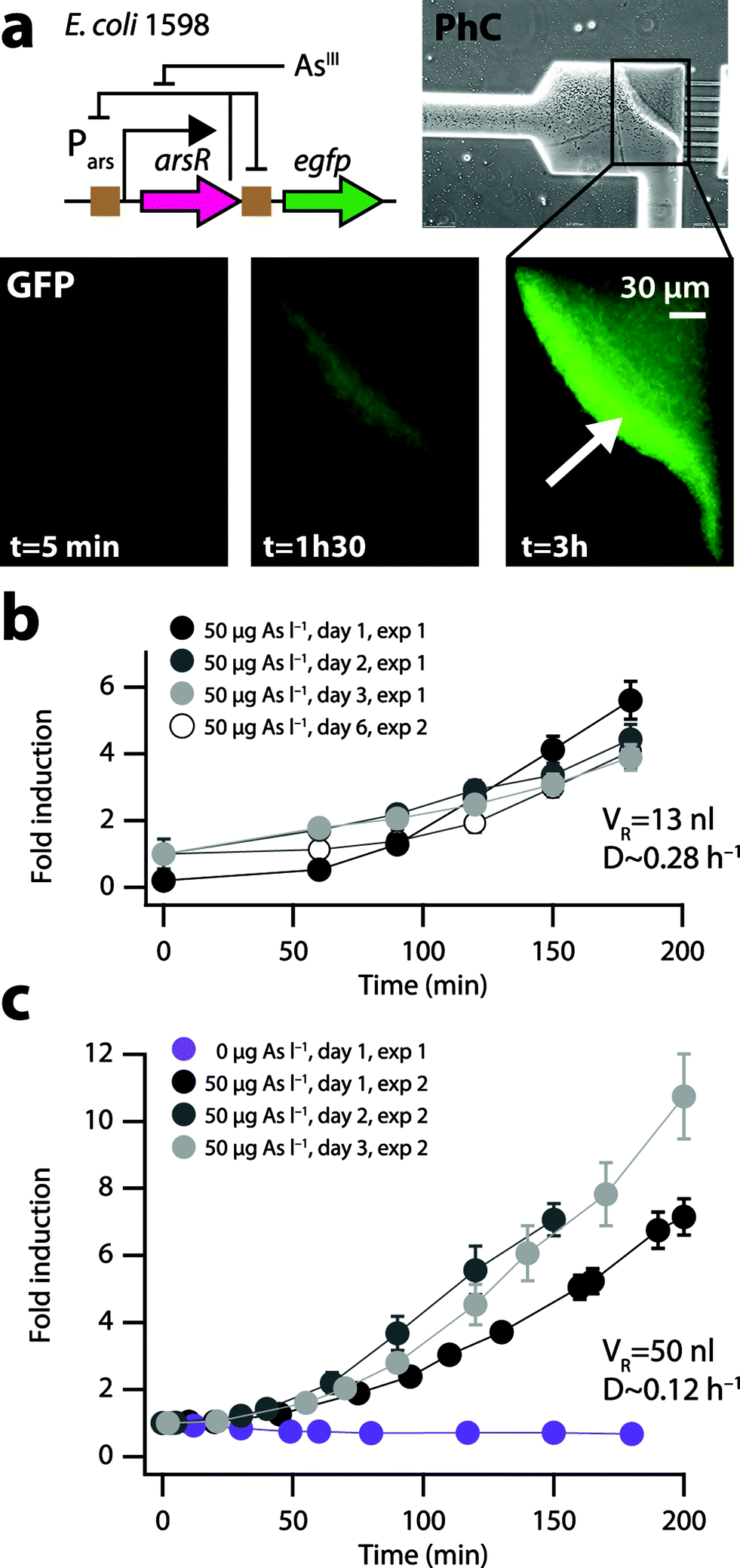 | ||
| Fig. 3 Bactosensor cell induction to arsenite on the biochip. (a) Fluorescence images (GFP, scaled to same maximal intensity, corresponding to experiment in panel c) at selected time points of E. coli 1598 arsenite bactosensor cells in the microfilter measurement zone (PhC, phase contrast inset). (b) Fold change of the mean GFP fluorescence in the measurement zone over time to 50 μgarsenite-AsIII l−1 on three consecutive days (exp 1) or after six days biochip growth (exp 2) in the 13 nl reactor at a dilution rate of 0.28 h−1. Values are normalized to a mean GFP fluorescence at t = 0 over all four series. (c) Same as (b) for cells growing in the 50 nl reactor at a dilution rate of 0.12 h−1. | ||
Because the efficacy of cleaning the filter zone from cells by pressure-induced deformation of the PDMS was not entirely satisfactory, a new valve was incorporated in a second design chip to allow backflushing (Fig. 1f). Additionally, the volume of the reactor was increased from 13 to 50 nl to avoid emptying too many cells from the reactor for each individual measurement. This permitted more reproducible functionality of the biochip and improved inducibility of the bactosensor cells. For example, the bactosensor cells induced the GFP fluorescence on three consecutive days between 6 and 9-fold after 180 min of exposure to an arsenite solution of 50 μgAsIII l−1 at a dilution rate of 0.12 h−1 in the 50 nl reactor compared to a sample without arsenite (Fig. 3c). The variation of the mean GFP fluorescence after 150 min induction on different consecutive days was comparable between cells cultured in either of the two microreactors (13 nl, 10.7% variation; 50 nl reactor: 11.1% variation). We observed that the bactosensor cells were not induced homogenously in the measurement filter zone (Fig. 3a, arrow), suggesting that an arsenite gradient forms over time across the dense cell layer, by which cells closest to the sample flow become induced the highest. The shortest exposure time reproducibly leading to a GFP signal development with 50 μgarsenite-AsIII l−1 significantly different from a control without arsenite was 40 min (P < 0.01).
Discussion
We have shown that it is possible to create an automated biosensor system allowing multiple consecutive measurements of a specific analyte in water samples with constantly growing E. coli bactosensor cells on a single nl-reactor microfluidic biochip. Dividing cells maintain good physiological properties in the microreactor for at least one week to enable immediate reaction to the analyte. For every measurement, a quantity of bactosensor cells is removed from the microreactor and directed by the on-chip flow control to a specific measurement cage, where they are exposed to the aqueous sample. With a biomass geometry in the measurement cage as in Fig. 3a and assuming three cell layers, some 3 × 104 cells are used for a measurement, which represents about one-tenth of the total number of cells in the 50 nl and one-third for the 13 nl microreactor. In the case of the 50 nl microreactor, the cells continue to divide in the microreactor chamber during the exposure of the retrieved and concentrated cells to the sample in the measurement cage (Fig. S3e†). At a dilution rate of 0.12 h−1, carbon inflow concentration of 3.35 gC l−1 and maximum cell growth rate of 0.28 h−1, it will thus take less than 1 h to replenish the bactosensor biomass used for a single measurement. In principle, therefore, one could make new measurements with the system every 3–4 h (30 min to fill the measurement cage, 180 min exposure time, 30 min to clean the cage). The measurement cage contains a set of filter pores so shallow that the bactosensor cells are mostly retained but liquid will pass. This has the advantage that the cells can first be accumulated in the cage, forming a concentrated cell “zone” (Fig. 3a), and subsequently can be percolated with the aqueous sample, allowing intensive contact. This is probably the reason as to why we observed the appearance of a fluorescence gradient when the bactosensor cell layer is exposed to a sample containing 50 μgarsenite-AsIII l−1 (Fig. 3a). The cells closest to the sample source take up the arsenic, depleting somewhat its concentration for the adjacent cells. After a measurement, the bactosensor cells can be removed from the measurement cage, either by augmenting the flow pressure that slightly increases the pore size of the filters, or (more effectively in our hands) by temporary backflow (Fig. 1h). Although we deployed an epifluorescence microscope here to image the fluorescent reporter signal from the bactosensors, the accumulating bactosensor cells in the measurement cage produce a concentrated spot of GFP fluorescence, which can be detected using low-cost photodiodes, as previously demonstrated.31A variety of studies have reported the construction and maintenance of continuous growth conditions for bacterial or yeast cells on microfluidic chips.13–18 Since reactor miniaturization leads to potentially very different and irregular growth conditions compared to larger (ml to l volumes) well-mixed controlled reactors,32 wide emphasis has been placed on studying single cell physiology in microreactors. Most results so far demonstrated clearly that continuous and relatively homogenous growth of bacteria and yeast can be achieved in a variety of both microreactor formats and sizes (nl to μl volumes).32 Depending on the specific aims, for example, single cell physiology analysis, reactors can be configured very differently,13,16 and also active17 as well as passive diffusion substrate flow regimes14 have been deployed successfully. For the purpose of this study we chose a growth volume (13–50 nl) that would be suitable to hold sufficient cells from which a few ten thousand could be removed for downstream biosensor analysis, without disturbing the growth in the microreactor too much. This was most easily achieved by adopting and miniaturizing a chemostat principle with a single nutrient in- and outflow, removing the excess biomass growth with every opening cycle. We acknowledge that we cannot formally claim that chemostat conditions are achieved, since we could not properly quantify the biomass in the reactor over time but could only show constant mean cell length (Fig. 2d) and demonstrate overall growth using unstable fluorescent reporters (Fig. S6†). It is also very likely that substrate gradients are formed across the microreactor in the absence of mixing. Reporter fluorescence data and video cell imaging indeed showed that inhomogeneities arise across the microreactor, e.g., very fast moving cells near the inlet versus more immotile cells near the outlets (Video S1†), and formation of small cell clumps over time despite use of Triton X-100. Despite these inhomogeneities, however, the bactosensor cells removed from the microreactor and used for measurement reacted instantly to the analyte as we expected from physiologically active cells (Fig. 2a). Therefore, for practical purposes, the physiology of the cells in the microreactor was sufficiently homogenous and stable over time to allow multiple biosensor measurements. Advances shown on other microreactor systems, for example, monitoring of oxygen levels, pH or culture turbidity, or material surface modifications to further reduce cell adhesion,15,18 could be deployed to improve the bactosensor biochip and growth conditions.
In conclusion, the development reported here permits for the first time a relatively reproducible reporter signal from bactosensor cells for several consecutive days on different samples in an automated mode. By further control or measurement of the bactosensor cell quantity in the measurement cage,31 it should be possible to operate such bactosensor biochips on absolute reporter signal intensity without the need for constant reporter signal calibration with external standards, which is typically necessary for individual batch assays.33 One would expect that, when the physiology of individual cells is extremely similar, the amount of GFP formed as a result of exposure to arsenite during a precisely defined period of time should also become very similar between cells. Once this relation is known, it could be deployed to avoid constant new calibration of the GFP signal as a function of arsenite exposure. Even though PDMS-fabricated chips are not extremely robust in their operation, our results are an important step forward for the design of autonomously operating biosensor instruments,31 which might be placed in remote areas, marine environments or in industrial processes, and provide constant monitoring of specific target analytes. By multiplication of the design it should be possible to grow bactosensors with different analyte specificities in separate parallel independent microreactors and measurement cages, in order to achieve semi-continuous multi-analyte detection with a relatively small system. The example of an arsenic-responsive bactosensor we showed here may eventually be useful to improve the monitoring of arsenic levels in potable water sources,3,34 which continues to threaten the health of millions of people around the world.22
Author contributions
NB and SB fabricated the microfluidic reactors and performed the experiments. FT and MG provided the electronic valve control for the microreactor. NB, SB, HvL and PR designed the microfluidic reactor. JvdM designed the experimental work and wrote the manuscript.Acknowledgements
This work was supported by grants from the Swiss National Science Foundation Sinergia (CRSI20_122689/1 SINERGIA) and NanoTera programs (20NA21-143082, Envirobot), from the Gebert Rüf Foundation (GRS-070/13), and from the European Community (OCEAN-2013-614010, BRAAVOO).References
- J. R. van der Meer and S. Belkin, Nat. Rev. Microbiol., 2010, 8, 511–522 CrossRef CAS PubMed.
- A. Courbet, D. Endy, E. Renard, F. Molina and J. Bonnet, Sci. Transl. Med., 2015, 7, 289ra283, DOI:10.1126/scitranslmed.aaa3601.
- K. Siegfried, C. Endes, A. F. Bhuiyan, A. Kuppardt, J. Mattusch, J. R. van der Meer, A. Chatzinotas and H. Harms, Environ. Sci. Technol., 2012, 46, 3281–3287, DOI:10.1021/es203511k.
- B. Baumann and J. R. van der Meer, J. Agric. Food Chem., 2007, 55, 2115–2120 CrossRef CAS PubMed.
- J. W. Kotula, S. J. Kerns, L. A. Shaket, L. Siraj, J. J. Collins, J. C. Way and P. A. Silver, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4838–4843, DOI:10.1073/pnas.1321321111.
- T. Feher, V. Libis, P. Carbonell and J. L. Faulon, Front. Bioeng. Biotechnol., 2015, 3, 46, DOI:10.3389/fbioe. 2015.00046.
- H. U. Deepthike, R. Tecon, G. van Kooten, J. R. van der Meer, H. Harms, M. Wells and J. Short, Environ. Sci. Technol., 2009, 43, 5864–5870 CrossRef CAS PubMed.
- S. Daunert, G. Barrett, J. S. Feliciano, R. S. Shetty, S. Shrestha and W. Smith-Spencer, Chem. Rev., 2000, 100, 2705–2738 CrossRef CAS PubMed.
- R. Pedahzur, B. Polyak, R. S. Marks and S. Belkin, J. Appl. Toxicol., 2004, 24, 343–348 CrossRef CAS PubMed.
- K. Pardee, A. A. Green, T. Ferrante, D. E. Cameron, A. DaleyKeyser, P. Yin and J. J. Collins, Cell, 2014, 159, 940–954, DOI:10.1016/j.cell. 2014.10.004.
- N. Buffi, D. Merulla, J. Beutier, F. Barbaud, S. Beggah, H. van Lintel, P. Renaud and J. R. van der Meer, Lab Chip, 2011, 11, 2369–2377, 10.1039/c1lc20274j.
- L. D. Knecht, P. Pasini and S. Daunert, Anal. Bioanal. Chem., 2011, 400, 977–989, DOI:10.1007/s00216-011-4835-4.
- F. K. Balagaddé, L. You, C. L. Hansen, F. H. Arnold and S. R. Quake, Science, 2005, 309, 137–140 CrossRef PubMed.
- A. Groisman, C. Lobo, H. Cho, J. K. Campbell, Y. S. Dufour, A. M. Stevens and A. Levchenko, Nat. Methods, 2005, 2, 685–689, DOI:10.1038/nmeth784.
- Z. Zhang, P. Boccazzi, H. G. Choi, G. Perozziello, A. J. Sinskey and K. F. Jensen, Lab Chip, 2006, 6, 906–913, 10.1039/b518396k.
- S. Cookson, N. Ostroff, W. L. Pang, D. Volfson and J. Hasty, Mol. Syst. Biol., 2005, 1, 2005–2024, DOI:10.1038/msb4100032.
- X. Luo, K. Shen, C. Luo, H. Ji, Q. Ouyang and Y. Chen, Biomed. Microdevices, 2010, 12, 499–503, DOI:10.1007/s10544-010-9406-5.
- A. Edlich, V. Magdanz, D. Rasch, S. Demming, S. Aliasghar Zadeh, R. Segura, C. Kahler, R. Radespiel, S. Buttgenbach, E. Franco-Lara and R. Krull, Biotechnol. Prog., 2010, 26, 1259–1270, DOI:10.1002/btpr.449.
- B. D. DeBusschere and G. T. Kovacs, Biosens. Bioelectron., 2001, 16, 543–556 CrossRef CAS PubMed.
- J. Stocker, D. Balluch, M. Gsell, H. Harms, J. S. Feliciano, S. Daunert, K. A. Malik and J. R. van der Meer, Environ. Sci. Technol., 2003, 37, 4743–4750 CrossRef CAS PubMed.
- D. K. Nordstrom, Science, 2002, 296, 2143–2144 CrossRef CAS PubMed.
- L. Rodriguez-Lado, G. Sun, M. Berg, Q. Zhang, H. Xue, Q. Zheng and C. A. Johnson, Science, 2013, 341, 866–868, DOI:10.1126/science.1237484.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- D. Merulla, V. Hatzimanikatis and J. R. van der Meer, Microb. Biotechnol., 2013, 6, 503–514, DOI:10.1111/1751-7915.12031.
- J. B. Andersen, C. Sternberg, L. K. Poulsen, S. P. Bjørn, M. Givskov and S. Mølin, Appl. Environ. Microbiol., 1998, 64, 2240–2246 CAS.
- C. Sternberg, B. B. Christensen, T. Johansen, A. Toftgaard Nielsen, J. B. Andersen, M. Givskov and S. Molin, Appl. Environ. Microbiol., 1999, 65, 4108–4117 CAS.
- G. Sezonov, D. Joseleau-Petit and R. D'Ari, J. Bacteriol., 2007, 189, 8746–8749, DOI:10.1128/JB.01368-07.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- B. Volkmer and M. Heinemann, PLoS One, 2011, 6, e23126, DOI:10.1371/journal.pone.0023126.
- S. E. Mainzer and W. P. Hempfling, J. Bacteriol., 1976, 126, 251–256 CAS.
- F. Truffer, N. Buffi, D. Merulla, S. Beggah, H. van Lintel, P. Renaud, J. R. van der Meer and M. Geiser, Rev. Sci. Instrum., 2014, 85, 015120, DOI:10.1063/1.4863333.
- H. M. Hegab, A. ElMekawy and T. Stakenborg, Biomicrofluidics, 2013, 7, 021502 CrossRef PubMed.
- A. Wackwitz, H. Harms, A. Chatzinotas, U. Breuer, C. Vogne and J. R. van der Meer, Microb. Biotechnol., 2008, 1, 149–157 CrossRef CAS PubMed.
- P. T. Trang, M. Berg, P. H. Viet, N. Van Mui and J. R. van der Meer, Environ. Sci. Technol., 2005, 39, 7625–7630 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6lc00119j |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |