
A high-throughput mechanofluidic screening platform for investigating tumor cell adhesion during metastasis†
A.
Spencer
a,
C.
Spruell
a,
S.
Nandi
a,
M.
Wong
a,
M.
Creixell
a and
A. B.
Baker
*abc
aDepartment of Biomedical Engineering, University of Texas at Austin, Austin, TX, USA. E-mail: abbaker1@gmail.com
bInstitute for Cellular and Molecular Biology, University of Texas at Austin, Austin, TX, USA
cInstitute for Computational Engineering and Sciences (ICES), University of Texas at Austin, Austin, TX, USA
First published on 9th November 2015
Abstract
The metastatic spread of cancer is a major barrier to effective and curative therapies for cancer. During metastasis, tumor cells intravasate into the vascular system, survive in the shear forces and immunological environment of the circulation, and then extravasate into secondary tumor sites. Biophysical forces are potent regulators of cancer biology and are key in many of the steps of metastasis. In particular, the adhesion of circulating cells is highly dependent upon competing forces between cell adhesion receptors and the shear stresses due to fluid flow. Conventional in vitro assays for drug development and the mechanistic study of metastasis are often carried out in the absence of fluidic forces and, consequently, are poorly representative of the true biology of metastasis. Here, we present a novel high-throughput approach to studying cell adhesion under flow that uses a multi-well, mechanofluidic flow system to interrogate adhesion of cancer cell to endothelial cells, extracellular matrix and platelets under physiological shear stresses. We use this system to identify pathways and compounds that can potentially be used to inhibit cancer adhesion under flow by screening anti-inflammatory compounds, integrin inhibitors and a kinase inhibitor library. In particular, we identify several small molecule inhibitors of FLT-3 and AKT that are potent inhibitors of cancer cell adhesion to endothelial cells and platelets under flow. In addition, we found that many kinase inhibitors lead to increased adhesion of cancer cells in flow-based but not static assays. This finding suggests that even compounds that reduce cell proliferation might also enhance cancer cell adhesion during metastasis. Overall, our results validate a novel platform for investigating the mechanisms of cell adhesion under biophysical flow conditions and identify several potential inhibitors of cancer cell adhesion during metastasis.
Introduction
The metastasis of tumors is a key characteristic of malignant cancers and the ultimate cause of 90% of deaths in cancer patients.1,2 While metastasis is a critical determinant of patient survival, there are currently no clinically approved therapies that directly inhibit the metastatic process.3 Although there have been attempts to develop anti-metastatic compounds, these have yet to achieve significant success in large scale clinical trials.4 The metastatic cascade consists of sequential steps including intravasation, survival in the circulatory system, adhesion at the metastatic host organ site and extravasation.5,6 In recent years, the recognition of the importance of the pre-metastatic niche has added support for Paget's “seed and soil” hypothesis in which the interactions between circulating tumor cells (CTCs) and the local organ microenvironment facilitate organ specific metastasis.7,8 Within this process, the attachment of CTCs to endothelial cells in microvasculature is an essential, rate limiting step in the metastatic cascade, determining both the organ site of metastasis and providing initial attachment to facilitate extravasation.5 The interactions between circulating cancer cells and endothelial cells are dependent on adhesion receptors including members of the selectin and integrin families, CD44, CD164, galectin-3, VCAM-1 and many others.9–15A fundamental limitation in the development of new therapies to prevent metastatic cancer is a lack of in vitro systems that can accurately recapitulate the steps of cancer cell metastasis.16 During adhesion of CTCs under flow conditions, the biophysical forces of the circulation can dramatically alter the biochemical interactions of adhesion receptors with their ligands.10,17,18 Currently, assays for examining the steps of metastasis are most commonly carried out in the absence of the flow of the circulatory system or using low throughput flow chambers.16 Many studies have suggested these assays to be poorly predictive of the in vivo metastatic response making them unsuitable for drug discovery or large-scale mechanistic studies.19–21
Here, we present a device that enables the performance of high throughput in vitro screens for compounds that can inhibit cancer cell adhesion under physiological flow. Our system generates flow using a mechanofluidic mechanism similar to a cone-and-plate viscometer but parallelized to work in standard format 96-well culture plates. The high throughput cone-and-plate (HT-CAP) system uses multiple shafts with a low angle cone tip that can be rotated to apply shear stress to cells grown in a conventional 96-well plate. This well-plate format allows the system to interface effectively with a host of conventional assays, robotic pipetting and high throughput plate reading devices. We demonstrate that this system can be used as an effective assay for screening for compounds that alter cell adhesion under flow. In addition, we demonstrate that assays using this device are able to distinguish between moderately and highly metastatic cancer cell lines, and can identify known pathways involved in inflammatory and cancer cell adhesion. In contrast, identical assays performed in the absence of flow are not predictive, and often even lead to contrasting results when compared to studies in mice and human clinical trials. In addition, using high throughput experiments we identify relative importance of a broad range of kinases and integrins in the cancer cell adhesion under flow. Thus, this system is a promising tool for the pre-clinical discovery of new pathways and pharmacological inhibitors of cancer metastasis.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) were cultured in MCDB-131 growth medium supplemented with 5% fetal bovine serum (FBS; Invitrogen), SingleQuots growth supplements (Lonza), L-glutamine and penicillin-streptomycin. THP-1 monocytic leukemia cells were cultured in RPMI 1640 growth medium supplemented with 10% FBS, 0.05 mM 2-mercaptoethanol, L-glutamine and penicillin-streptomycin. HCT116 colon cancer cells, MDA-MB-231 breast cancer cells, and MCF-7 breast cancer cells were cultured in DMEM growth medium supplemented with 10% FBS, L-glutamine and penicillin-streptomycin. All cells were cultured at 37 °C and 5% CO2.Calibration of cone alignment
Cone tips were aligned using pressure measurement film (Fujifilm) by raising the micro-stage lift until there was pressure from the cone tips on the film (Fig. S2†). Based on the pressure from each cone tips, vertical cone position would be adjusted until there was an even pressure produced from all 96 cone tips. Once cone tips were aligned they were glued in their vertical position with a heat-reversible epoxy.Leukocyte cell adhesion assay
HUVECs were seeded onto a 96-well optical-bottom plate (Nunc) at 2.75 × 105 cells per mL and cultured to confluence. HUVECs were treated with 10 ng mL−1 of TNF-α (Sigma, Peprotech) for 4 hours prior to the assay. THP-1 cells were fluorescently labeled with Qtracker® 655 (Life Technologies) according to protocol. THP-1 cells were washed twice, then suspended in phenol free media at a concentration of 3 × 105 cells per mL or 1 × 106 cells per mL for static and 0.5 dyns cm−2 assays, respectively and 100 μL of THP-1 cell solution was added to each well. The well plate was incubated for 15 minutes, non-adherent THP-1 cells were washed twice. The adherent cells were analyzed with a microplate reader, or fluorescently imaged. Static assays were performed through the same procedure except the THP-1 cell concentration was 3 × 105 cells per mL and there was no shear stress on the cells during adhesion.Cancer cell adhesion assay
HCT-116 cancer cells were seeded into 35 mm plates at a concentration of 7.5 × 105 cells per mL and cultured for 48 hours. Cells were then treated with the following anticancer drugs: niclosamide, marimastat, paclitaxel, doxorubicin, fluorouracil (Sigma-Aldrich), GM6001 (CalBioChem), and erlotinib (Santa Cruz Biotechnology) for 5 hours before the assay. Endothelial cells were stimulated with 10 ng mL−1 TNF-α for 4 hours before the assay. HCT-116 cells were trypsinized and allowed to recover for one hour prior to the assay. HCT-116 cancer cells were added to the 96-well plate at a concentration of 1 × 106 cells per mL. The cells were sheared for one hour with the shear stress device at 37 °C and 5% CO2. Non-adherent cells were washed from the well plate, and the remaining fluorescent cells were read in a microplate reader. Static assays were performed with the same procedure except HCT-116 cell concentration was 5 × 105 cells per mL and there was no shear stress on the cells during adhesion.Cell adhesion to extracellular matrix assay
Extracellular matrix components, Collagen I, Collagen II, Collagen IV, Laminin, Vitronectin (Sigma), and Tenascin C (R&D) were used to coat a 96-well plate according to their respective protocols. MCF-7/GFP and MDA-MB-231/GFP (Cell Biolabs, Inc.) were trypsinized and allowed to recover for 1 h. Breast cancer cells were added to the ECM coated well plate at a concentration of 5 × 105 cells per mL and sheared in the shear stress device for 30 min. Non-adherent cells were washed off and fluorescence was read in a plate reader.Cell adhesion-detachment assay
After completing an adhesion assay where cells were adhered at 0.5 dynes per cm2 of shear stress, cells were detached at increasing shear stresses. Non-adherent cells were removed and replaced with fresh media. The shear stress was increased and run for one minute. These steps were repeated for increasing shear stresses, depending on the cell type.Integrin inhibitor adhesion-detachment assay
MDA-MB-231 and MCF-7 cancer cells were trypsinized, and allowed to recover for 1 hour. Confluent endothelial cells or immobilized platelets in a 96-well plate were treated with integrin inhibitors directly before addition of cancer cells, and application of shear with the shear stress device. A shear stress of 0.5 dyns cm−2 was applied for 1 hour, and non-adherent cells were removed and the media was replaced. The fluorescent cells were read in a plate reader. The shear stress was increased and the procedure was repeated for 1, 2, 3, 4, 5, 10, and 20 dyns cm−2.Kinase inhibitor library
A kinase inhibitor library (EMD Calibiochem; Cat. no. 539744) was screened to test for the effect of kinases on cancer cell adhesion and immune cell adhesion. MDA-MB-231 cancer cells or THP-1 monocytes were treated with kinase inhibitors at 1 mM concentration for 1 hour prior to the assay. The treated cells were added to a well plate of endothelial cells or immobilized platelets, and a shear stress of 0.5 dyns cm−2 was applied for 1 hour. Detachment was measured in the same manner previously described.Statistical analysis
All results are shown as mean ± standard error of the mean. Comparisons between only two groups were performed using a 2-tailed Student's t-test. Differences were considered significant at p < 0.05. Multiple comparisons between groups were analyzed by 2-way ANOVA followed by a Tukey post-hoc test. A 2-tailed probability value <0.05 was considered statistically significant.Results
High-throughput assay to probe cell–cell interactions under flow
The system consists of 96 stainless steel shafts that have 2° angled cone machined on one end (Fig. 1A and Fig. S1†). The shafts were mounted into two parallel plates with bearings and a single gear per shaft. All of the gears interface to form a 96-shaft gearbox that is driven by the motor with adjacent shafts rotating in opposite directions. A 96-well plate is mounted on a micromanipulator lift table, allowing the well plate to be brought into accurate alignment with the cones. Photographs of the completed system are shown in Fig. 1A. Previously, we have performed an extensive set of simulations on the system that demonstrated a tight dependence on the apposition of the cone with the surface in order to get a uniform shear stress field (Fig. 1A).22 We aligned the shafts in the system using an iterative process in which we placed a pressure sensitive film between the cone tips and a flat surface (Fig. S2†). Finally, we adjusted shaft height to account for the thickness of the endothelial cells by changing the height to the lowest possible position that did not damage the endothelial layer on rotation of the cone (Fig. S3†). The archetypal adhesion assay procedure is that a confluent monolayer of cells is grown in a 96-well plate and then fluorescently labeled/fluorescent protein expressing cancer cells are added. The cells are then flowed over the monolayer through the rotation of the cone-tipped shaft, the plate is washed and then read for fluorescence of adhered cancer cells (Fig. 1B). We extended this assay in some cases by detaching the cells using increased levels of shear stress to measure the strength of adhesion. Most cancer cells arrest in the capillaries or between capillaries and post-capillary venules. In these regions shear stresses can be as low as 0.5 dynes per cm2 and the flow patterns can be tortuous. Using the cone-and-plate device we are able to control the shear stress experienced by the cells, as well as whether the shear stress is constant or oscillatory. The circular flow pattern in the wells created using the HT-CAP has a relatively large radius of curvature relative to the size of the endothelial cells in most of the well, so the cancer and endothelial cells may not experience much of a difference relative to linear flow patterns.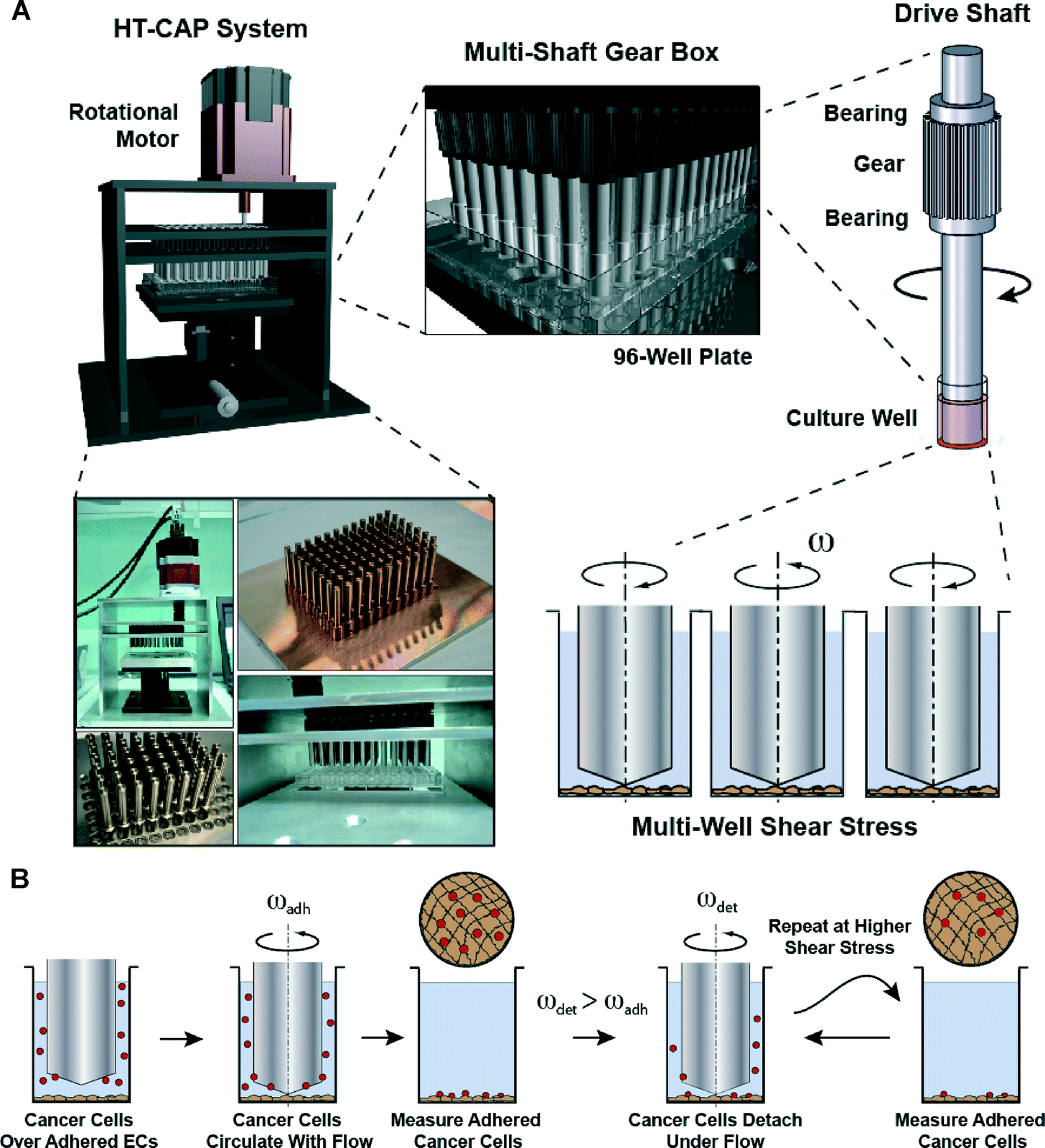 | ||
| Fig. 1 High throughput device for measuring cell adhesion under flow. (A) The mechanical design of the device includes a single shaft that is linked into a 96-shaft gearbox through precision gears. A lift table allows precise vertical position of the well plate relative to the cone-tipped shafts. Shown is CAD rendering of the device with inset showing the interface of the multi-shaft system with the 96-well plate. Also shown are pictures of the side view of the device, the drive shafts machined to have low angle cone tips linked in a 96-shaft gearbox and a side view of the shafts interfacing with a 96-well plate. Computational modelling was performed for the flow within the plate as the distance between the cone and the plate is varied. (B) Overall assay for a cell adhesion-detachment assay under flow within a 96-well plate. The cancer cells are adhered to a confluent monolayer of endothelial cells (ECs) under 0.5 dynes per cm2. The plate is washed and then the plate is read for the fluorescence of the adherent cancer cells. | ||
Optimization of flow adhesion assay and validation of within plate variability in the flow system
We first tested the system using THP-1 cell adhesion assay to a monolayer of endothelial cells (Fig. S4†). THP-1 cells are derived from a monocytic leukemia and have shared properties with monocytes and cancer cells. A confluent monolayer of endothelial cells was grown on a glass bottom 96-well plate and then stimulated with TNF-α to activate the endothelial cells. The THP-1 cells were fluorescently labeled with a dye and then applied to the endothelial cells under flow or static condition for 10 min. After the application of flow the plate was immediately removed from the system, washed and the fluorescence was read with a plate reader. We first examined the uniformity of the system performing the THP-1 cell adhesion assay under low (0.5 dynes per cm2) or high flow (12 dynes per cm2) with and without activation of the endothelial cells by TNF-α. Release of TNF-α by the primary tumor has been suggested to prepare the metastatic microenvironment for adhesion and extravasation of circulating tumor cells by upregulating adhesion molecules.23 As anticipated, high level of shear reduced THP-1 cell adhesion and activation with TNF-α increased the adhesion (Fig. S4†). The system was highly repeatable and uniform in the adhesion assay with a well-to-well variability comparable to the static adhesion assay. We next examined whether lysis of the cells after adhesion was necessary to obtain repeatable measurements of cell adhesion. We examined the difference between lysing the cells and just reading the plate fluorescence without lysing and found that for dye loaded cells this increased the signal from the fluorescence (Fig. S5A†). Finally, we examined the effect of THP-1 cell concentration on ability of the assay to detect increased cell adhesion to control or TNF-α stimulated cells (Fig. S5B†). Using these results we optimized the THP-1 cell concentration for the adhesion assay to the highest concentration of cells with a lysis step to increase the assay signal.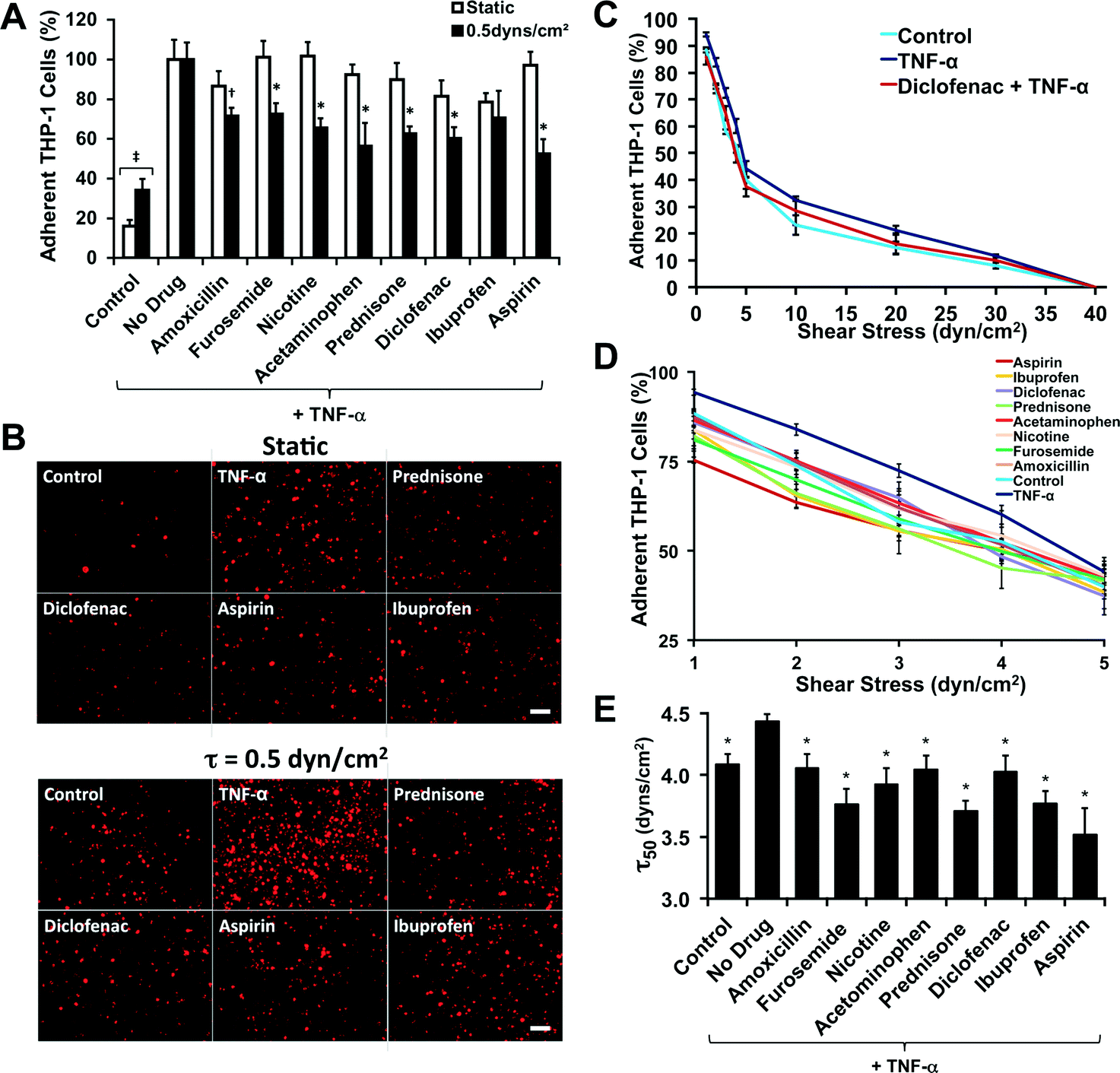 | ||
| Fig. 2 Validation of the assay with a drug screen using known anti-inflammatory compounds. (A) THP-1 cells were fluorescently labeled and flowed over a monolayer of confluent endothelial cells for 15 minutes. *p < 0.05 versus static assay and versus TNF-α treated cells; †p < 0.05 versus TNF-α treated cells. (B) Fluorescent images of cells adhered to the plate under the various anti-inflammatory cells. Bar = 100 μm. (C) Overall de-adhesion assay for the THP-1 cells under anti-inflammatory treatments. (D) Low shear stress range for the de-adhesion assay. (E) Shear stress under which 50% of the cells are lost from the surface (τ50), calculated from a logarithmic fit to the detachment curves. *p < 0.05 versus TNF-α only (“no drug”) treatment group. | ||
High throughput flow-based adhesion assays are predictive of in vivo anti-inflammatory activity
We fluorescently labeled THP-1 cells and treated them with various anti-inflammatory compounds (Table S1†) and then performed an adhesion assay to a confluent monolayer of TNF-α stimulated endothelial cells under static and flow conditions (0.5 dynes per cm2). This level of shear corresponds to that found in the pre-capillary arterioles vessels24 and in the range of shear stresses found in capillaries and post-capillaries venules.25 For most of the anti-inflammatory compounds there was not a statistically significant difference between the drug-treated groups and the non-treated group in the static assay (Fig. 2A, B and Fig. S6†). However, for the flow assay we found a significant reduction in the adhesion for almost all of the compounds tested (Fig. 2A and B). We probed strength of the adhesion of the leukocytes by performing an adhesion–deadhesion assay in which we first allowed cells to adhere under 0.5 dynes per cm2 of shear stress and then exposed the adherent cells to increasing bouts of shear stress ranging from 1 to 40 dynes per cm2. We found that the treatment with TNF-α (“no drug”) had stronger attachment with significantly fewer cells being released at shear stresses of 5, 10 and 20 dynes per cm2 than the non-TNF-α treated group (“control”). For example, Diclofenac led to an intermediate number of released cells. At the lower shear stresses there were also significant differences between the drug treatments and the TNF-α only treated cells (Fig. 2D). To create an index gauging the strength of adhesion, we performed a non-linear fit of the shear stress detachment and calculated the shear stress where 50% of the cell detached for each treatment (τ50; Fig. 2E).Flow-based cancer cell adhesion assays have contrasting results for the anti-metastatic properties of chemotherapeutic compounds in comparison to static adhesion assays
We next treated colon cancer cells (HCT-116) with chemotherapeutic agents (Table S2†) and then assayed their adhesion to TNF-α activated endothelial cells. We also included broad spectrum MMP inhibitors (Marimastat and GM6001) as a measure of the contribution of MMPs to the adhesion of cancer cells during attachment to the endothelium. We performed a static adhesion assay and a parallel assay in which the cancer cells adhered to endothelial cells under 0.5 dynes per cm2 of shear stress. In addition, to control for the effects of endothelial adaptation to flow, we pre-treated endothelial cells for 8 hours with 0.5 dynes per cm2 and then performed the adhesion assay. In the static assay, none of the compounds inhibited the adhesion of cancer cells (Fig. S7†). Both Marmistat and GM6001 significantly increased cancer cell adhesion in the static assay. In contrast, all of the compounds except GM6001 significantly inhibited the adhesion of cancer cells in the flow assay (Fig. S7†).High throughput, mechanofluidic adhesion assays differentiate between aggressively and non-aggressively metastatic breast cancer cells lines and reveal extracellular matrix specificity for different cancer types
The extracellular matrix (ECM) is a powerful regulator of cancer progression and can promote transformation and regulate metastasis.26 We examined two breast cancer cell lines that are known to be moderately metastatic (MCF-7) and strongly metastatic (MDA-MB-231). In a static adhesion assay, the less metastatic MCF-7 cell line adhered more than the strongly metastatic MDA-MB-231 cell line to all of types of ECM tested (Fig. 3A–C). In contrast, the adhesion assay under flow showed markedly lower adhesion overall and increased adhesion in the MDA-MB-231 cell line in comparison to the MCF-7 cell line (Fig. 3A–C and S8†). In a separate experiment, we explored whether our system could be used to measure the strength of adhesion by allowing the cells to adhere for 1 hour at 0.5 dynes per cm2 and then detaching the cells with increasing shear stresses up to 20 dynes per cm2 (Fig. 3D). This analysis allowed us to probe not only the adhesion of cells under flow but gauge the strength of adhesion of the cells. The MDA-MB-231 cell line had greater strength of adhesion at higher shear stresses for laminin over the MCF-7 cell line (Fig. 3E).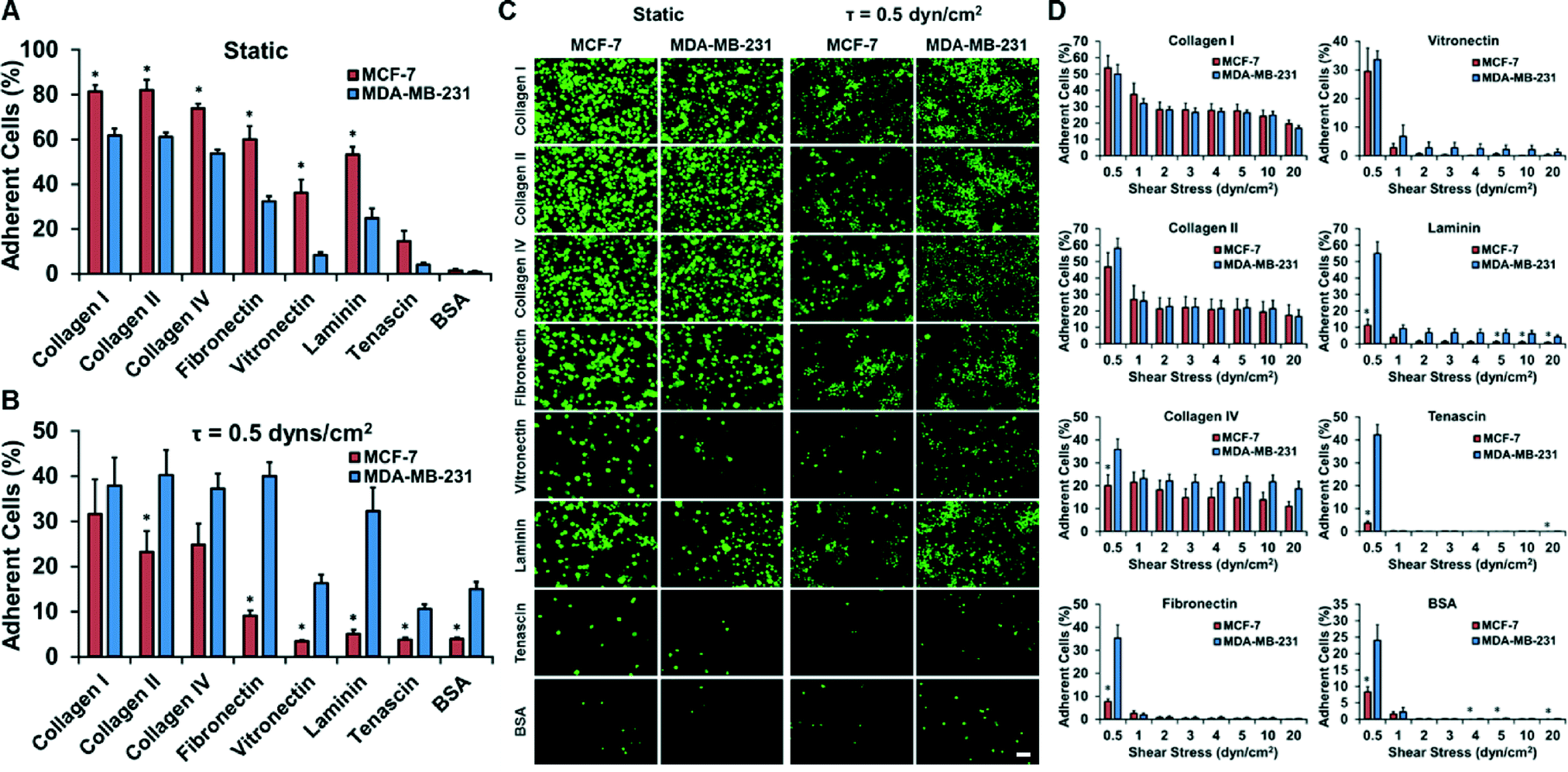 | ||
| Fig. 3 Adhesion assays of strongly metastatic (MDA-MB-231) or moderately metastatic (MCF-7) cell lines to extracellular matrix. (A) Cancer cell adhesion to extracellular matrix coated wells under static conditions. (B) Cancer cell adhesion to extracellular matrix coated wells under flow conditions in the system. (C) Images of adherent cancer cells under static and shear conditions. Bar = 100 μm. *p < 0.05 versus MCF-7 cell line under identical conditions. (D) Detachment assay on adherent cancer cells using increasing levels of shear stress. | ||
High throughput adhesion assays reveal integrin-dependent sensitivity of cancer–endothelial cell and cancer–platelet interactions
Targeting specific integrins is a promising emerging strategy for inhibiting the growth and metastasis of cancer.27 We probed the sensitivity of the breast cancer cell lines to a set of integrin inhibitors (Table S3†) to delineate the importance of specific integrins in the adhesion of different cell lines and under static and flow conditions. Additionally, we examined the implications of the integrin inhibitors on the strength of adhesion through a detachment assay. We found that under static conditions there was no difference in adhesion of MDA-MB-231 cells to endothelial cells with TNF-α treatment but there was a mild increase in adhesion of MCF-7 cells (Fig. 4A). Treatment with integrin inhibitors on the MDA-MB-231 cells led to significantly decreased adhesion with cilengitide treatment (an αvβ3 and αvβ5 integrin inhibitor) as well as several other integrin inhibitors (Table S4†). In addition, we examined the adhesion of MDA-MB-231 cells under oscillatory flow that varied sinusoidally from 1 to −1 dynes per cm2 of shear stress (Fig. S9†). The integrin dependence for adhesion under oscillatory flow was markedly different from steady flow and was, in general, less sensitive to the integrin inhibitors. The interaction of cancer cells with platelets has also been shown to regulate cancer metastasis.28,29 We took human platelets and immobilized them on the bottom of the wells of 96-well plate. We examined whether the shear stresses of the adhesion-deadhesion assay could remove the platelets and found that they were not visually altered by the shear stresses (Fig. S10†). We then performed a cancer cell adhesion assay on the immobilized platelets with treatment with the integrin inhibitors both under static and flow conditions (Fig. 4B).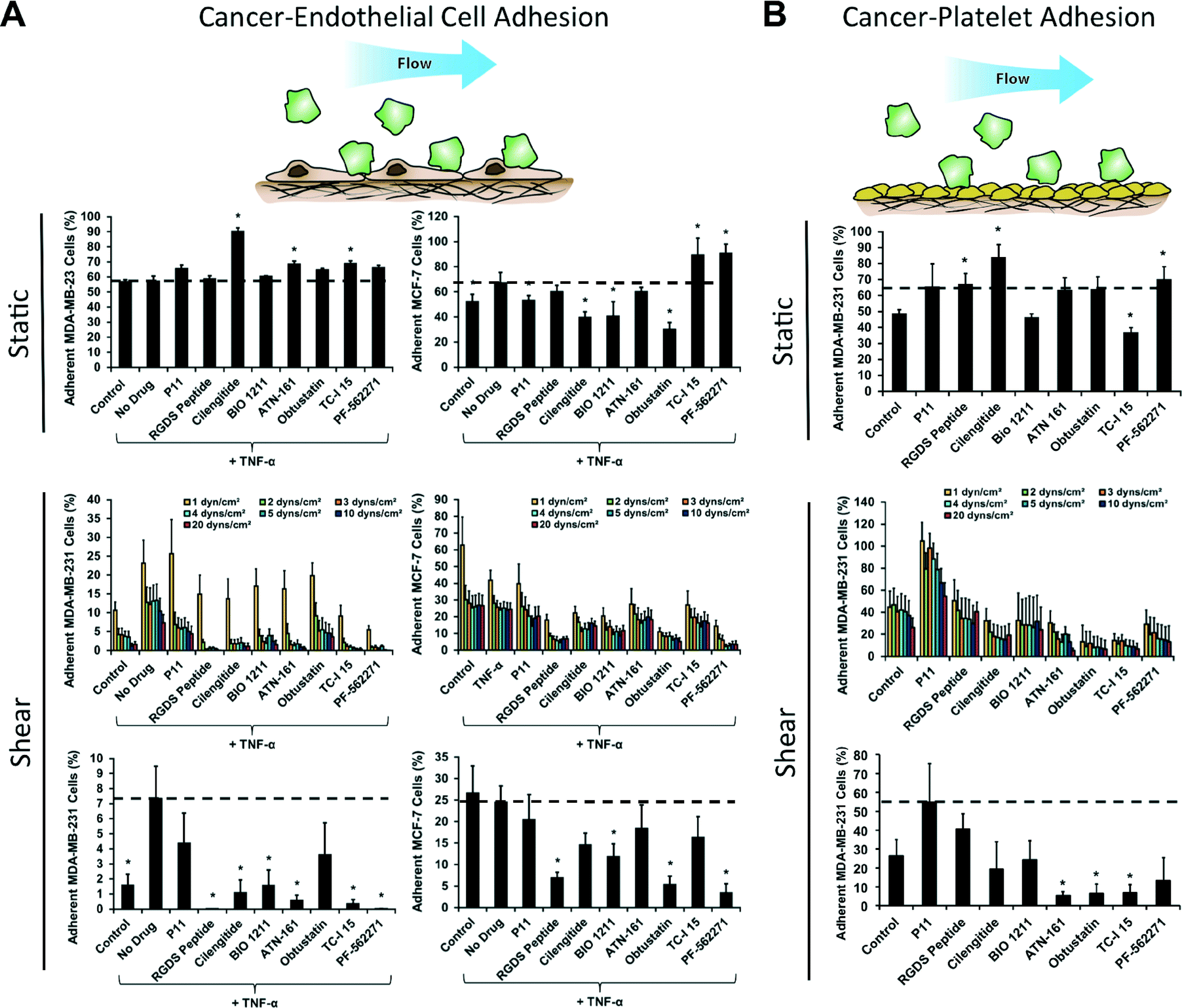 | ||
| Fig. 4 Screening of integrin inhibitors under static and flow conditions using a cancer cell de-adhesion assay. (A) Plots of relative adhesion of MDA-MB-231 or MCF-7 breast cancer cell lines under static conditions, the adhesion/detachment assay under 0.5 dynes per cm2 shear stress in the presence of integrin inhibitors, and the remaining adherent cells after 20 dynes per cm2 shear stress had been applied. (B) Plots of relative adhesion of MDA-MB-231 cells to immobilized platelets under static and flow. *p < 0.05 versus TNF-α treated cells. | ||
High throughput screening of a kinase inhibitor library using a mechanofluidic adhesion assay
Kinase inhibitors are the fastest growing class of approved anti-cancer agents and have a significant impact on the treatment of cancers primarily linked to a single kinase.30 We performed a high throughput screening assay on a kinase inhibitor library containing 80 compounds to elucidate which kinases are important for pathways involved in cancer cell adhesion. We examined three screens of drug activity including the ability to modify cancer cell adhesion to a monolayer of endothelial cells, cancer cell adhesion to platelets and leukocyte adhesion to endothelial cells (to assess potential effects on the immune response). Each assay was performed three times independently for each drug including an internal control. The drug treatments were rotated within the system to control for variability within the system. For each assay, we allowed the cells to adhere under 0.5 dynes per cm2 of shear stress and then exposed them to increased levels of shear stress to assess the strength of adhesion. The results of this screen were compared to a similar screen in the absence of fluid flow. The screen for cancer cell adhesion to endothelial cells revealed that many of the compounds did not significantly affect the adhesion of cancer cells under static conditions (Fig. 5A and B). However, during adhesion under flow conditions and for detachment of the cells by shear stress we found that many of the kinase inhibitors increased cancer cell adhesion significantly. This can be seen most strikingly in charts of a rank ordering for the compounds in which there are many molecules that increase adhesion strength of the cancer cells by 2–15 fold (Fig. S11†). In contrast, the static assay did not have changes greater than two-fold for either adhesion enhancement or inhibition. One notable finding was that many of the FLT-3 inhibitors had strong anti-adhesive effects on the cancer cells. In contrast to the results for adhesion to endothelial cells, cancer cell adhesion to platelets was reduced by most of the kinase inhibitors (including the FLT-3 inhibitors; Fig. 5A, and B). A second consistent finding was the role of the Akt/mTOR pathway in breast cancer cell adhesion, indicated by multiple Akt inhibitors and mTOR inhibitor rapamycin were found as hits. For most of these inhibitors, there was a minimal effect on adhesion in the static adhesion assay. In the immunotoxicity assay we did not see as great of change for adhesion for many of the compounds (Fig. 5A and B). In the three screens, an ideal compound would inhibit cancer cell adhesion to endothelial cells and platelets while having minimal effects on the adhesion of immune cells. The “hits” from the endothelial and platelet adhesion assays both under static and flow conditions are shown in Fig. 5C (Fig. S12 and Table S5†).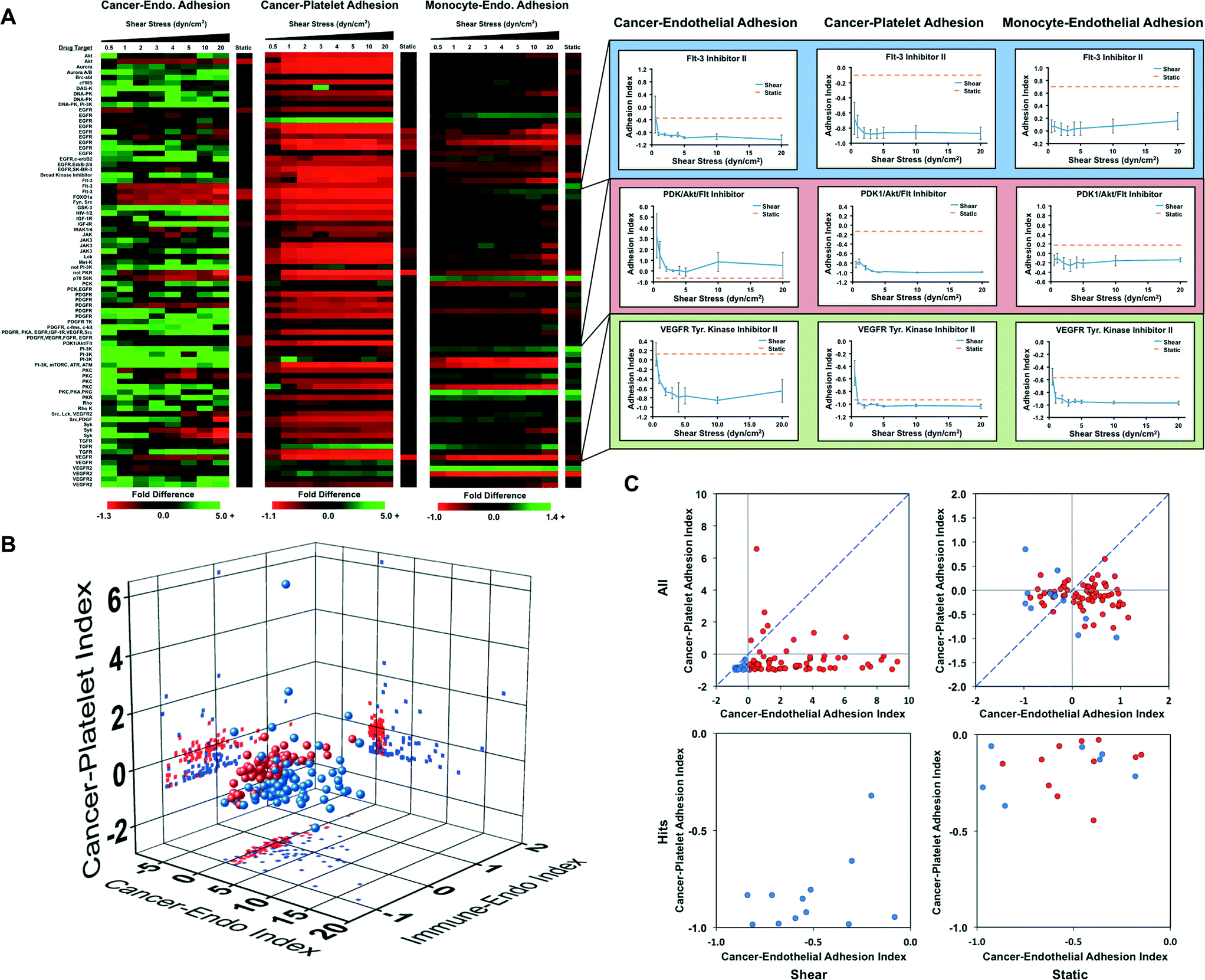 | ||
| Fig. 5 Screening for anti-metastatic properties of a kinase inhibitor library. (A) MDA-MB-231 cancer cells were treated with compounds from a kinase inhibitor library for 1 hour and then allowed to adhere to a confluent endothelial layer or immobilized platelets under static conditions or under 0.5 dynes per cm2 of shear stress. In addition, monocytic THP-1 cells were treated with the kinase inhibitors and subjected to the adhesion/de-adhesion assay to TNF-α activated endothelial cells. (C) 3D scatter plot showing the results of the three screens for the kinase inhibitors. The results of the static assay are shown in blue and the average adhesion index for the flow-based assay is shown in red. (D) Scatter plot of static adhesion indices for MDA-MB-231 cancer cell adhesion to endothelial cells and platelets. Compounds that had a negative adhesion index for both adhesion to endothelial cells and platelets in the flow-based adhesion assay are shown in blue. All other compounds in the flow-based assay are shown in red. | ||
Discussion
Accurate model in vitro systems for studying cancer metastasis would provide immense benefit to the scientific investigation on the mechanisms of the spread of cancer and to pharmaceutical researchers developing cancer therapeutics. We have developed a flexible platform for the study of cancer cell adhesion during metastasis. Using this system we have identified that inhibitors of the FLT-3 and AKT pathways block cancer cell adhesion to endothelial cells and platelets while leaving the adhesion of leukocytes to endothelial cells relatively unaffected. A number of other model systems have been created to simulate the adhesion of cancer cells during metastasis.31–36 Many of these have focused on using microfluidic chips to simulate flow or are adapted specifically for detailed imaging of a single metastasis event; however, these have not addressed the problems of creating a high throughput platform to perform practical drug screening assays. Microfluidic approaches for creating multichannel flow37 are promising but, in their current form, have challenges with respect to interfacing with robotic pipetting and culture systems, maintaining cell viability and phenotype in microchannels38 and interfacing with standard assay procedures such as plate reading, microscopy and immunostaining procedures. Here, we have shown that a mechanofluidic approach of creating flow within standard culture plates can overcome these limitations and has the potential to rapidly accelerate the study of shear stress mediated regulation of cell adhesion and the discovery of compounds that can prevent circulating cancer cell adhesion. This approach should have great utility in pre-clinical screening for compounds to treat metastasis by providing a more physiological model of cancer cell adhesion during metastasis.A consistent finding in our studies was that flow based adhesion assays have a greater degree of correlation with the known activities of therapeutic compounds. In our studies on inflammation, static assays failed to predict the activity of known anti-inflammatory compounds even at the relatively high doses used in the study. Similarly, a static adhesion assay could not accurately identify the comparative metastatic potential between cancer cell lines. The profiles of integrin inhibitor sensitively for cancer cell adhesion to endothelial cells or platelets were markedly different for assays with flow in comparison to a static assay. Thus, the biophysical forces present in the circulatory system are an important component in modifying the adhesion of cancer cells during the metastatic cascade. During an assay under flow there are additional forces on the forming bond during specific adhesion. If this force acts in the direction of a conformational change due to binding it may increase the rate of reaction, while if it acts in the opposite direction may serve to decrease the reaction rate.39 Both of these phenomena have been shown to occur in force-mediated detachment40 or shear stress enhanced adhesion.41 Moreover, the motion created within a flow-incorporating assay causes the circulating cell type to probe many cells in contrast to a single cell during settling in a static assay. An additional facet of an assay with shear stress is that the flow rate also changes the amount of time allowed for the reaction to occur before the motion of the cell brings the receptor-ligand pair out of opposition.42 Thus, markedly different kinetic requirements are needed for adhesion under flow in comparison to static conditions. In leukocytes, integrin mediated binding to endothelial cells is highly dependent on shear stress and the time scale of interactions. While rolling adhesion is mediated by selectins, firmer adhesion and arrest of motion can only be achieved after selectin-mediated rolling slows the cell to allow integrin-mediated adhesion.43 There is support for the theory that cancer cells can co-opt the mechanisms used in leukocyte adhesion either directly44 or by binding to circulating immune cells themselves.45 A leukocyte adhesion assay would be an appealing in vitro assay for discovering anti-inflammatory compounds as it assays a functional end point without having to measure specific inflammatory markers or perform further molecular assays. In contrast, our results demonstrate that using leukocyte adhesion under static conditions would not be as predictive of the actual in vivo inflammatory activity.
The ECM can serve as a barrier to metastasis and as a potential mechanism for the tissue specific metastasis of particular cancers.46 A recent study used a combinatorial assay to identify ECM combinations that were associated with metastatic versus primary cancer cell lines.47 We compared the adhesion of two cancer cells lines for adhesion to purified ECM proteins. Under static conditions the less metastatic MCF-7 cell line adhered more to nearly all the ECMs tested; however, under flow conditions the highly metastatic MDA-MB-231 cells adhered more than MCF-7. In addition, we observed that MCF-7 adhered to each other and formed clusters more than the MDA-MB-231 cells, particularly in the shear assay. One could hypothesize that these results are due to the increased cohesive strength of MCF-7 cells to each other in comparison to the MDA-MB-231 cells, while MDA-MB-231 cells may exhibit increased adhesive strength for ECM.48 In our detachment assays, the MDA-MB-231 cell detachment at 0.5 dynes per cm2 of shear stress (same as the adhesion flow) for many of the ECMs tested and at higher detachment shear stresses for collagen IV and laminin was significantly less than MCF-7 cells, indicating the MDA-MB-231 cells had adhered to the endothelial cells with greater strength. Interestingly, both collagen IV and laminin are found in high concentration in subendothelial basement membrane.49 Thus, our findings would support that, under flow conditions, the more aggressive cell line has increased binding to ECM molecules and the strength of these interactions are increased for the ECM found in the subendothelial basement membrane.50
The integrins are a family of adhesion receptors that are key molecules in tumorigenesis, progression and metastasis of tumors.27 The integrins αvβ3 and αvβ5 have been linked to cancer cell adhesion to endothelial cells.51,52 In addition, the αvβ3/αvβ5 integrin inhibitor cilengitide has been tested in phase II and III clinical trials for glioblastoma with mixed results.53 The integrin α4β1 is also known to serve as an adhesive ligand that binds to VCAM-1 on metastatic tumor cells and promotes lymph node metastasis.54 Integrin α1β1 has also been linked to the cancer cell invasion through tissues and metastasis.55,56 The mechanofluidic system identified that inhibitors to these integrins were very effective in blocking cancer cell adhesion to endothelial cells in the highly metastatic MDA-MB-231 cell where the static assay predicted higher adhesion or no change for these same inhibitors. This illustrates the importance of incorporating appropriate biomechanical forces in determining the relevance of the outcome of the drug screen. In the static case, these compounds would have been eliminated from the screen but using the flow assay we see profound differences that suggest these are compounds of interest. The role of platelets in cancer metastasis is supported by studies that have shown depletion, inhibitor or re-infusion of platelets can alter the dissemination of cancer.57 Platelets enhance many steps in extravasation including the adhesion of tumor cells to the vasculature, penetration of the endothelial monolayer and subsequent invasion into the tissue.28,29 Platelets and tumor cells form aggregates through crosslinking of platelet integrin αIIbβ3 with tumor cell integrins including αvβ3.58 In addition, other integrins including α3β1 and α5β1 have been implicated in tumor cell–thrombus interactions.59 Our screen for cancer cell adhesion to platelets supported a role for many of the β1 integrins as well as αvβ3/αvβ5 under flow conditions. In contrast only inhibition of α2β1 led to a significant reduction in cancer cell adhesion under static conditions. Thus, the mechanofluidic flow assay is better able to identify known platelet–cancer cell interactions than a static assay and may be useful in the study of the interaction of thrombosis with cancer metastasis.
We performed orthogonal drug screening of a kinase library for activity in regulating cancer–endothelial interaction, cancer–platelet interactions and leukocyte–endothelial adhesion. This screen yielded markedly different results from a static screen and identified “hits” that reduced cancer cell adhesion to endothelial cells and platelets while minimally affecting leukocyte adhesion. In the flow-based assay, we were also able to quantify the strength of adhesion by using progressively increasing bouts of shear stress. Thus, this assay yields significantly more information than the simple static assay. In looking at the data overall, it is clear that many of the kinase inhibitors cause increased strength of adhesion even at high shear levels. Thus, when looking at an indexed version of the data there is clearly a larger dynamic range of adhesion for the flow-based assays. Among the hits identified, we found many compounds with targets from drugs in clinical trials. One of the most consistent findings of our screen was that FMS-like tyrosine kinase 3 (FLT-3) inhibition led to reduced cancer cell adhesion to endothelial cells and platelets. Administration of FLT-3 ligand has been shown to reduce cancer metastasis in mouse models of cancer and FLT-3 inhibitors have reached phase III clinical trials for the treatment of acute myeloid leukemia (AML).60 The rationale for the development of the FLT-3 inhibitors was based on the role of FLT-3 in hematopoiesis, high expression in leukemia and its role in immunomodulation.61 Our studies have shown for the first time that pharmacologic inhibition of FLT-3 inhibits the adhesion of breast cancer cells to endothelial cells and platelets, and could therefore have potential as inhibitors of solid tumor metastasis. Our studies also identified several other kinases that had potential as inhibitors for metastasis including rapamycin and AKT inhibitors that are clinically approved or have targets in common with compounds in clinical trials. One unexpected result of the kinase library screen is that the vast majority of the small molecule inhibitors lead to increased adhesion of cancer cells and/or strengthening of their attachment (in many cases by over five-fold the baseline adhesion/adhesion strength). In contrast, relatively fewer inhibitors increased the adhesion of cancer cells to platelets. This is an important finding in our study as suggests that many compounds may increase cancer cell adhesion even if they have significant anti-proliferative properties. This property was only detected using a flow-based adhesion assay, suggesting there may be great utility for using the HT-CAP system to eliminate compounds with this undesirable property prior to starting pre-clinical animal studies.
Conclusions
In summary, we present a high throughput, flexible platform for measuring cell adhesion under flow conditions using a standard multi-well format and used this system to identify several candidate pathways for inhibiting cancer cell adhesion during metastasis. The utility of the HT-CAP mechanofluidic platform extends beyond the specific application in cancer metastasis shown here. The interface with standard wells allows the system to be used with virtually any well plate assay or high throughput system in which fluidic forces are relevant. Moreover, the cone and plate format allows the application of complex dynamic flow to simulate different physiological situations. Thus, the system has many potential applications including high throughput study of vascular mechanobiology, screening and studies of platelet activation/thrombosis and bacterial adhesion.Acknowledgements
The authors would like to acknowledge support through the Texas4000 Foundation, the Welch Foundation (F-1836), American Heart Association (10SDG2630139) and through the NIH Director's New Innovator Grant (1DP2 OD008716-01) to A. B. B. as well as a National Science Foundation Fellowship to A. S. The authors would like to thank Dr. Kevin Dalby (UT Austin) for critical review of the manuscript.References
- P. Mehlen and A. Puisieux, Nat. Rev. Cancer, 2006, 6, 449–458 CrossRef CAS PubMed.
- D. X. Nguyen, P. D. Bos and J. Massague, Nat. Rev. Cancer, 2009, 9, 274–284 CrossRef CAS PubMed.
- S. Valastyan and R. A. Weinberg, Cell, 2011, 147, 275–292 CrossRef CAS PubMed.
- A. M. Stock, G. Troost, B. Niggemann, K. S. Zanker and F. Entschladen, Curr. Pharm. Des., 2013, 19, 5127–5134 CrossRef CAS PubMed.
- A. F. Chambers, A. C. Groom and I. C. MacDonald, Nat. Rev. Cancer, 2002, 2, 563–572 CrossRef CAS PubMed.
- M. Labelle and R. O. Hynes, Cancer Discovery, 2012, 2, 1091–1099 CrossRef CAS PubMed.
- B. Psaila and D. Lyden, Nat. Rev. Cancer, 2009, 9, 285–293 CrossRef CAS PubMed.
- S. Paget, Lancet, 1889, 1, 571–573 CrossRef.
- G. Bendas and L. Borsig, Int. J. Cell Biol., 2012, 2012, 676731 Search PubMed.
- C. J. Dimitroff, M. Lechpammer, D. Long-Woodward and J. L. Kutok, Cancer Res., 2004, 64, 5261–5269 CrossRef CAS PubMed.
- A. M. Havens, Y. Jung, Y. X. Sun, J. Wang, R. B. Shah, H. J. Buhring, K. J. Pienta and R. S. Taichman, BMC Cancer, 2006, 6, 195 CrossRef CAS PubMed.
- R. M. Lafrenie, S. Gallo, T. J. Podor, M. R. Buchanan and F. W. Orr, Eur. J. Cancer, 1994, 30, 2151–2158 CrossRef.
- S. Mine, T. Fujisaki, C. Kawahara, T. Tabata, T. Iida, M. Yasuda, T. Yoneda and Y. Tanaka, Exp. Cell Res., 2003, 288, 189–197 CrossRef CAS PubMed.
- C. Strell, B. Niggemann, M. J. Voss, D. G. Powe, K. S. Zanker and F. Entschladen, Mol. Cancer Res., 2012, 10, 197–207 CrossRef CAS PubMed.
- L. G. Yu, N. Andrews, Q. Zhao, D. McKean, J. F. Williams, L. J. Connor, O. V. Gerasimenko, J. Hilkens, J. Hirabayashi, K. Kasai and J. M. Rhodes, J. Biol. Chem., 2007, 282, 773–781 CrossRef CAS PubMed.
- N. Pouliot, H. B. Pearson and A. Burrows, Investigating Metastasis Using In Vitro Platforms, Landes Bioscience, New York, 2013 Search PubMed.
- E. Bastida, L. Almirall, M. C. Bertomeu and A. Ordinas, Int. J. Cancer, 1989, 43, 1174–1178 CrossRef CAS.
- R. Giavazzi, M. Foppolo, R. Dossi and A. Remuzzi, J. Clin. Invest., 1993, 92, 3038–3044 CrossRef CAS PubMed.
- B. Felding-Habermann, R. Habermann, E. Saldivar and Z. M. Ruggeri, J. Biol. Chem., 1996, 271, 5892–5900 CrossRef CAS PubMed.
- K. Konstantopoulos, S. Kukreti and L. V. McIntire, Adv. Drug Delivery Rev., 1998, 33, 141–164 CrossRef CAS PubMed.
- U. Richter, Methods Mol. Biol., 2014, 1070, 47–56 CAS.
- C. Spruell and A. B. Baker, Biotechnol. Bioeng., 2013, 110, 1782–1793 CrossRef CAS PubMed.
- B. Psaila and D. Lyden, Nat. Rev. Cancer, 2009, 9, 285–293 CrossRef CAS PubMed.
- A. G. Koutsiaris, S. V. Tachmitzi and N. Batis, Microvasc. Res., 2013, 85, 34–39 CrossRef PubMed.
- A. G. Koutsiaris, S. V. Tachmitzi, N. Batis, M. G. Kotoula, C. H. Karabatsas, E. Tsironi and D. Z. Chatzoulis, Biorheology, 2007, 44, 375–386 Search PubMed.
- P. Lu, V. M. Weaver and Z. Werb, J. Cell Biol., 2012, 196, 395–406 CrossRef CAS PubMed.
- J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9–22 CrossRef CAS PubMed.
- K. Konstantopoulos and S. N. Thomas, Annu. Rev. Biomed. Eng., 2009, 11, 177–202 CrossRef CAS PubMed.
- N. M. Bambace and C. E. Holmes, J. Thromb. Haemostasis, 2011, 9, 237–249 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- S. Bersini, J. S. Jeon, G. Dubini, C. Arrigoni, S. Chung, J. L. Charest, M. Moretti and R. D. Kamm, Biomaterials, 2014, 35, 2454–2461 CrossRef CAS PubMed.
- M. B. Chen, J. A. Whisler, J. S. Jeon and R. D. Kamm, Integr. Biol., 2013, 5, 1262–1271 RSC.
- J. S. Jeon, S. Bersini, M. Gilardi, G. Dubini, J. L. Charest, M. Moretti and R. D. Kamm, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 214–219 CrossRef CAS PubMed.
- J. W. Song, S. P. Cavnar, A. C. Walker, K. E. Luker, M. Gupta, Y. C. Tung, G. D. Luker and S. Takayama, PLoS One, 2009, 4, e5756 Search PubMed.
- I. K. Zervantonakis, S. K. Hughes-Alford, J. L. Charest, J. S. Condeelis, F. B. Gertler and R. D. Kamm, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13515–13520 CrossRef PubMed.
- Q. Zhang, T. Liu and J. Qin, Lab Chip, 2012, 12, 2837–2842 RSC.
- C. G. Conant, M. A. Schwartz, J. E. Beecher, R. C. Rudoff, C. Ionescu-Zanetti and J. T. Nevill, Biotechnol. Bioeng., 2011, 108, 2978–2987 CrossRef CAS PubMed.
- S. Halldorsson, E. Lucumi, R. Gomez-Sjoeberg and R. M. T. Fleming, Biosens. Bioelectron., 2015, 63, 218–231 CrossRef CAS PubMed.
- L. S.-L. Cheung and K. Konstantopoulos, Biophys. J., 2011, 100, 2338–2346 CrossRef CAS PubMed.
- S. Chen and T. A. Springer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 950–955 CrossRef CAS PubMed.
- C. Zhu, T. Yago, J. Z. Lou, V. I. Zarnitsyna and R. P. McEver, Ann. Biomed. Eng., 2008, 36, 604–621 CrossRef PubMed.
- C. Dong and X. X. Lei, J. Biomech., 2000, 33, 35–43 CrossRef CAS PubMed.
- A. Zarbock and K. Ley, Microcirculation, 2009, 16, 31–42 CrossRef CAS PubMed.
- N. Reymond, B. B. d'Agua and A. J. Ridley, Nat. Rev. Cancer, 2013, 13, 858–870 CrossRef CAS PubMed.
- C. Chen and D. B. Khismatullin, Cancer Lett., 2014, 345, 75–84 CrossRef CAS PubMed.
- J. A. Joyce and J. W. Pollard, Nat. Rev. Cancer, 2009, 9, 239–252 CrossRef CAS PubMed.
- N. E. Reticker-Flynn, D. F. B. Malta, M. M. Winslow, J. M. Lamar, M. J. Xu, G. H. Underhill, R. O. Hynes, T. E. Jacks and S. N. Bhatia, Nat. Commun., 2012, 3 Search PubMed.
- R. Omidvar, M. Tafazzoli-Shadpour, M. A. Shokrgozar and M. Rostami, J. Biomech., 2014, 47, 3373–3379 CrossRef PubMed.
- P. S. Smith, J. C. Fanning and I. Aarons, Pathology, 1989, 21, 254–258 CrossRef CAS PubMed.
- C. H. Chou, J. D. Sinden, P. O. Couraud and M. Modo, PLoS One, 2014, 9, 16 Search PubMed.
- J.-X. Niu, W.-J. Zhang, L.-Y. Ye, L.-Q. Wu, Z.-H. Yang, G.-J. Zhu and J.-N. Lou, Zhonghua Zhongliu Zazhi, 2008, 30, 165–169 CAS.
- B. Felding-Habermann, T. E. O'Toole, J. W. Smith, E. Fransvea, Z. M. Ruggeri, M. H. Ginsberg, P. E. Hughes, N. Pampori, S. J. Shattil, A. Saven and B. M. Mueller, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1853–1858 CrossRef CAS PubMed.
- L. B. Nabors, T. Mikkelsen, M. E. Hegi, X. Ye, T. Batchelor, G. Lesser, D. Peereboom, M. R. Rosenfeld, J. Olsen, S. Brem, J. D. Fisher and S. A. Grossman, for the New Approaches to Brain Tumor Therapy (NABTT) Central Nervous System Consortium, A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306), Cancer, 2012, 118, 5601–5607 CrossRef CAS PubMed.
- B. Garmy-Susini, C. J. Avraamides, J. S. Desgrosellier, M. C. Schmid, P. Foubert, L. G. Ellies, A. M. Lowy, S. L. Blair, S. R. Vandenberg, B. Datnow, H. Y. Wang, D. A. Cheresh and J. Varner, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9042–9047 CrossRef CAS PubMed.
- T. C. Kuo, C. T. Tan, Y. W. Chang, C. C. Hong, W. J. Lee, M. W. Chen, Y. M. Jeng, J. Chiou, P. Yu, P. S. Chen, M. Y. Wang, M. Hsiao, J. L. Su and M. L. Kuo, J. Clin. Invest., 2013, 123, 1082–1095 CAS.
- C. Yang, M. Zeisberg, J. C. Lively, P. Nyberg, N. Afdhal and R. Kalluri, Cancer Res., 2003, 63, 8312–8317 CAS.
- S. Karpatkin, E. Pearlstein, C. Ambrogio and B. S. Coller, J. Clin. Invest., 1988, 81, 1012–1019 CrossRef CAS PubMed.
- A. S. Lonsdorf, B. F. Kramer, M. Fahrleitner, T. Schonberger, S. Gnerlich, S. Ring, S. Gehring, S. W. Schneider, M. J. Kruhlak, S. G. Meuth, B. Nieswandt, M. Gawaz, A. H. Enk and H. F. Langer, J. Biol. Chem., 2012, 287, 2168–2178 CrossRef CAS PubMed.
- A. Klepfish, M. A. Greco and S. Karpatkin, Int. J. Cancer, 1993, 53, 978–982 CrossRef CAS.
- T. Fischer, R. M. Stone, D. J. DeAngelo, I. Galinsky, E. Estey, C. Lanza, E. Fox, G. Ehninger, E. J. Feldman, G. J. Schiller, V. M. Klimek, S. D. Nimer, D. G. Gilliland, C. Dutreix, A. Huntsman-Labed, J. Virkus and F. J. Giles, J. Clin. Oncol., 2010, 28, 4339–4345 CrossRef CAS PubMed.
- M. Levis and D. Small, Leukemia, 2003, 17, 1738–1752 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5lc00994d |
| This journal is © The Royal Society of Chemistry 2016 |