
3D cardiac μtissues within a microfluidic device with real-time contractile stress readout†
Aereas
Aung‡
a,
Ivneet Singh
Bhullar‡
a,
Jomkuan
Theprungsirikul
a,
Shruti Krishna
Davey
a,
Han Liang
Lim
a,
Yu-Jui
Chiu
b,
Xuanyi
Ma
a,
Sukriti
Dewan
a,
Yu-Hwa
Lo
c,
Andrew
McCulloch
a and
Shyni
Varghese
*a
aDepartment of Bioengineering, University of California-San Diego, La Jolla, CA, USA. E-mail: svarghese@ucsd.edu
bMaterials Science and Engineering Program, University of California-San Diego, La Jolla, California, USA
cDepartment of Electrical and Computer Engineering, University of California-San Diego, La Jolla, CA, USA
First published on 9th November 2015
Abstract
We present the development of three-dimensional (3D) cardiac microtissues within a microfluidic device with the ability to quantify real-time contractile stress measurements in situ. Using a 3D patterning technology that allows for the precise spatial distribution of cells within the device, we created an array of 3D cardiac microtissues from neonatal mouse cardiomyocytes. We integrated the 3D micropatterning technology with microfluidics to achieve perfused cell-laden structures. The cells were encapsulated within a degradable gelatin methacrylate hydrogel, which was sandwiched between two polyacrylamide hydrogels. The polyacrylamide hydrogels were used as “stress sensors” to acquire the contractile stresses generated by the beating cardiac cells. The cardiac-specific response of the engineered 3D system was examined by exposing it to epinephrine, an adrenergic neurotransmitter known to increase the magnitude and frequency of cardiac contractions. In response to exogenous epinephrine the engineered cardiac tissues exhibited an increased beating frequency and stress magnitude. Such cost-effective and easy-to-adapt 3D cardiac systems with real-time functional readout could be an attractive technological platform for drug discovery and development.
Introduction
Development of three-dimensional (3D) cell cultures has not only advanced regenerative medicine but also led to the development of physiologically relevant model systems to understand the interdependency between cells and their microenvironment as well as various cellular functions leading to tissue formation. Engineered 3D tissues also offer an invaluable technological platform for drug and small molecule discovery and development. Such systems could provide more physiologically relevant models than currently available monolayer cultures.1–9 Development of microtissues could also circumvent the limitations associated with pharmaceutical testing in animals or augment the efficiency and outcome of drug candidate screening.10,11 While animal models provide an in vivo environment with systemic readouts, drug testing in animals is time consuming, costly, and the acquired results are often unreliable due to the innate differences across species.12,13 Furthermore, from an ethical perspective, development of physiologically relevant in vitro platforms could reduce the dependency on animal usage for drug discovery and development. This need for cost-effective and efficient in vitro platforms to improve the drug development process led to the recent exploration of new technological platforms involving engineered tissue mimics.One of the organs that has been studied extensively is the heart. Over the past decade, many 3D in vitro models for cardiac drug screening have been developed using neonatal cells or human stem cell-derived cardiomyocytes.3,11,14–18 Microtissues in these static cultures have been shown to respond to the changes in environmental cues after electrical and pharmaceutical stimulation.17,18 While these static cultures are effective, it is attractive to integrate perfusion with engineered tissues to improve transport and diffusion of nutrients. Integration of such perfusion can be achieved by combining the microfabrication of engineered tissues with microfluidics. This also facilitates incorporation of tissue specific biochemical gradients and dynamic mechanical cues within the system. These systems could also be designed to provide real-time readouts. Such technological platforms, widely known as organ-on-chips, have been developed extensively in recent years.19,20
Specifically for cardiac applications, microfluidic heart-on-chip devices have emerged as one of the methods to achieve in vitro cardiac models that also allow for the monitoring of drug activity in real time within dynamic, perfusion-based cultures.21–26 Parker and colleagues have combined their muscular thin film (MTF) technology with microfluidics to create a heart-on-chip.22 One of the key features of functional cardiac tissues is their ability to beat. Hence, the contractile stresses generated by the microtissues could be used as readout to examine the response of the tissue to various environmental cues, including small molecules and drugs. The authors used cantilevers within the MTF platform to quantify the contractile stresses generated by cardiac cells. This system has further been extended to study pathophysiological changes in cardiac tissues.27,28 In another study, Mathur et al. incorporated iPSC-derived 3D cardiac tissues within a microfluidic system and demonstrated the application of such systems in drug screening.21 The researchers used calcium signaling and beating frequency as functional readouts.
Herein, we describe a novel microfluidic device capable of quantifying contractile stresses in situ. This device involves a tri-layer hydrogel system where a 3D cardiac cell-laden structure is sandwiched between two acellular hydrogels and is surrounded by microchannels. This was achieved by integrating 3D patterning technology, to encapsulate cells in a spatially controlled manner, along with a far-field approach to measure contractile stresses generated by the cardiac cells. In this proof-of-concept, we used neonatal mouse cardiomyocytes as a cell source. This system capitalizes on the physiological relevance of 3D structures and perfusion-based microfluidic systems while introducing a quantitative analysis to measure contractile stresses in real time.
Results
Fabrication and characterization of the device
To create a perfused cardiac tissue model with real-time contractile stress readouts, we created a microfluidic device with a vertical arrangement of tri-layer hydrogel structures. Fig. 1 summarizes the fabrication of the device including cell encapsulation. An integral part of the device is the 3D patterned cell-laden hydrogels sandwiched between two linearly elastic non-degradable hydrogels. In this proof-of-concept study, we used methacrylated gelatin (GelMA) hydrogel for cell encapsulation and polyacrylamide (PAm) hydrogels for the top and bottom layers as “stress sensors” for quantifying the contractile stresses generated by the encapsulated cardiac cells. Prior to cell encapsulation, an acellular device involving hydrogels embedded with fluorescent beads with different emission wavelengths was used to optimize and characterize the PAm-GelMA-PAm tri-layer hydrogels (Fig. 2 and S1†). The 3D patterned GelMA structures sandwiched between the PAm hydrogel layers can be designed to take any shape, which can be controlled by the photomask design. We have tested out elliptical and hexagonal structures. We incorporated 200 nm red fluorescent particles in GelMA solution prior to polymerization to visualize the structures within the device. For the PAm layer, hydrogels were polymerized in presence of 200 nm green fluorescent particles. The formed structures were imaged using a confocal microscope and the images were reconstructed to obtain a 3D representation of the device (Fig. 2 and S1†). The X–Z image sections illustrate the GelMA structures with red particles of approximately 120 μm thickness encased above and below by the PAm hydrogels of 70 μm thickness filled with green particles (Fig. 2A). The X–Y sections at the indicated Z positions demonstrate the planar structure of the PAm hydrogel along with the GelMA hydrogel (Fig. 2B and C). The 3D rendering of these structures in ESI†Fig. 1 shows the structures of GelMA and the tri-layer hydrogel composite.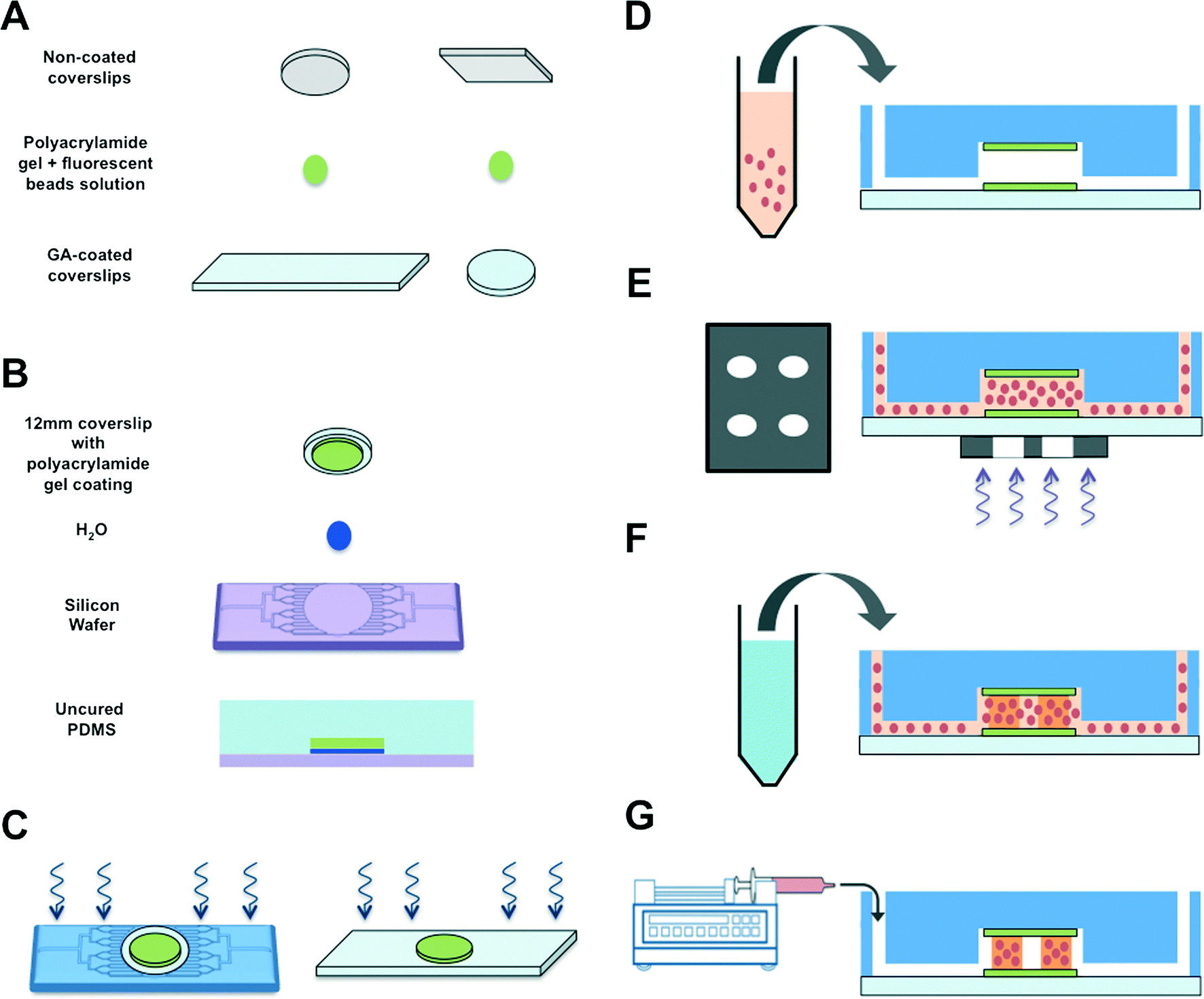 | ||
Fig. 1 Schematic of microfluidic device fabrication and 3D cell encapsulation. (A) Two PAm hydrogels were polymerized by sandwiching the precursor solution mixed with fluorescent nanoparticles between regular and GA-treated coverslips. (B) A small droplet of DI water was deposited onto a Teflon-coated silicon wafer prior to placing a PAm hydrogel tethered on a circular coverslip on top. PDMS solution containing the curing agent (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) was gently poured onto the construct and cured at 37 °C overnight. (C) The PDMS mold attached to the PAm hydrogel was removed from the wafer and inlet and outlet ports of the device were generated using a hole punch. This construct was bonded to a rectangular coverslip tethered to a PAm hydrogel using UV/Ozone treatment. Care was taken to align the two hydrogels during the process. The device was placed in 60 °C for an hour within a humidity chamber prior to moving to a 37 °C chamber overnight. (D) Cells mixed with GelMA, photoinitiator, and ascorbic acid were injected into the chamber. (E) A patterned transparency photomask was placed underneath the PAm hydrogels before exposing the region to UV light. (F) PBS solution was injected into the device to remove GelMA mixture in the non-polymerized region. (G) The device was attached to a syringe pump containing maintenance media to culture the cells encapsulated within patterned GelMA matrices. :1 ratio) was gently poured onto the construct and cured at 37 °C overnight. (C) The PDMS mold attached to the PAm hydrogel was removed from the wafer and inlet and outlet ports of the device were generated using a hole punch. This construct was bonded to a rectangular coverslip tethered to a PAm hydrogel using UV/Ozone treatment. Care was taken to align the two hydrogels during the process. The device was placed in 60 °C for an hour within a humidity chamber prior to moving to a 37 °C chamber overnight. (D) Cells mixed with GelMA, photoinitiator, and ascorbic acid were injected into the chamber. (E) A patterned transparency photomask was placed underneath the PAm hydrogels before exposing the region to UV light. (F) PBS solution was injected into the device to remove GelMA mixture in the non-polymerized region. (G) The device was attached to a syringe pump containing maintenance media to culture the cells encapsulated within patterned GelMA matrices. | ||
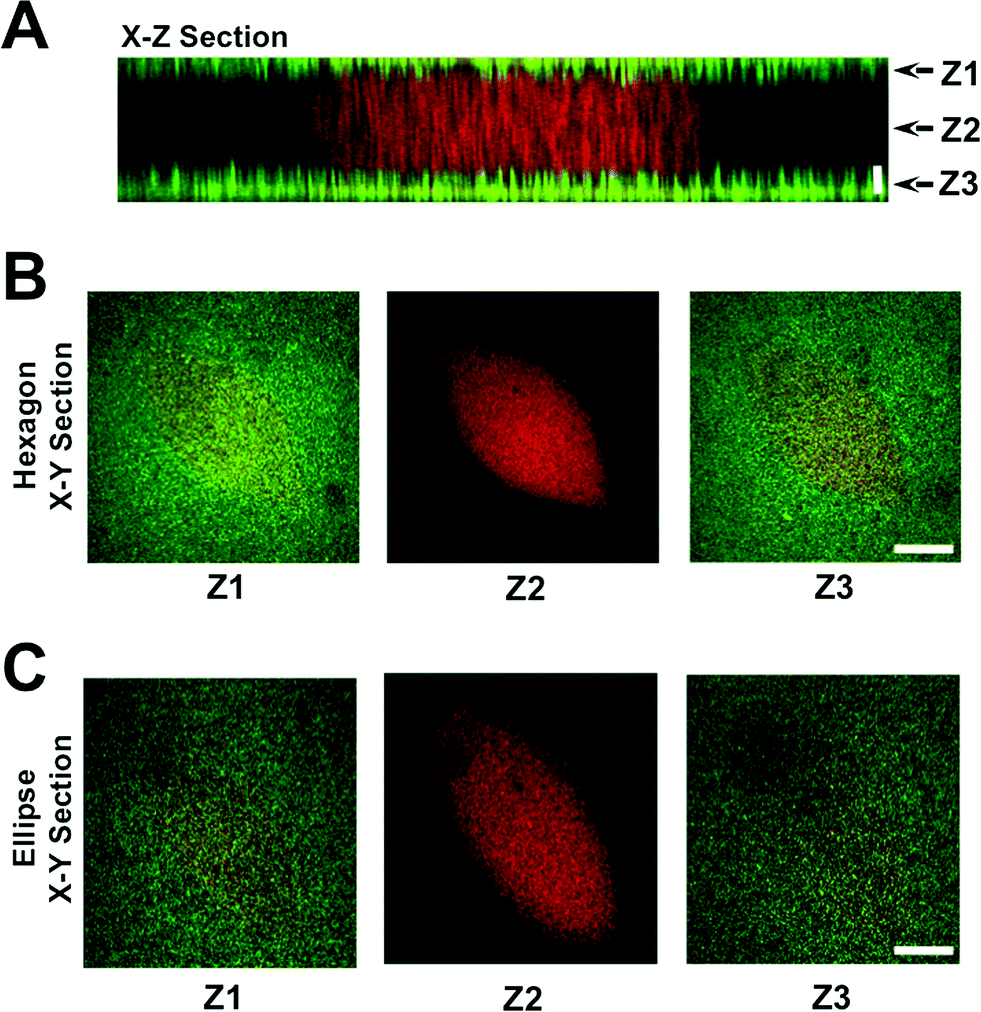 | ||
| Fig. 2 Characterization of tri-layer hydrogels. Z-stack images of the hydrogels (GelMA hydrogel sandwiched between acellular PAm hydrogels) were obtained using a laser scanning confocal microscope. The X–Z cross-section (A) and the corresponding X–Y planes at different Z locations are shown for hexagon (B) and ellipse (C) geometries. GelMA and the PAm hydrogels were embedded with red and green fluorescent nanoparticles, respectively, to visualize these structures. Horizontal scale bar: 100 μm. Vertical scale bar: 40 μm. | ||
Encapsulation of cardiomyocytes within the device
The neonatal mouse cardiomyocytes were encapsulated within the GelMA hydrogels by photopolymerization, where the cardiomyocyte-laden GelMA structures are housed within the internal circular chamber (Fig. 1D–G). The cell-laden GelMA structures of ~120 μm thickness were sandwiched between two thin layers of polyacrylamide (PAm) hydrogels (~70 μm thickness) containing 200 nm fluorescent nanoparticles. The cell-laden structures were spatially patterned to maintain a horizontal distance of ~525 μm and a vertical distance of ~300 μm between the structures. The device was infused with cardiomyocyte maintenance media and the cell function was monitored as a function of time. The encapsulated cells were found to contract spontaneously starting day 2 and maintain a coordinated contraction for up to 14 days (maximum allowed experimental length time). Differential interference contrast (DIC) images of the GelMA structures at various culture time points are shown in Fig. 3A. We labeled the cells using CellMask, which stains all cells. Since the isolated cardiomyocytes also contain a small population of fibroblasts, we used Connexin-43, a cardiac specific marker to identify the encapsulated cardiomyocytes. Confocal sections of the stained cells indicated that the cells positive for Connexin-43 were located within the interior of the structures with the fibroblasts confined mostly on the periphery (Fig. 3B). For a 12 mm diameter inner chamber, we were able to create an array of ~200 cardiac microtissues of dimensions 400 μm × 133 μm × 120 μm.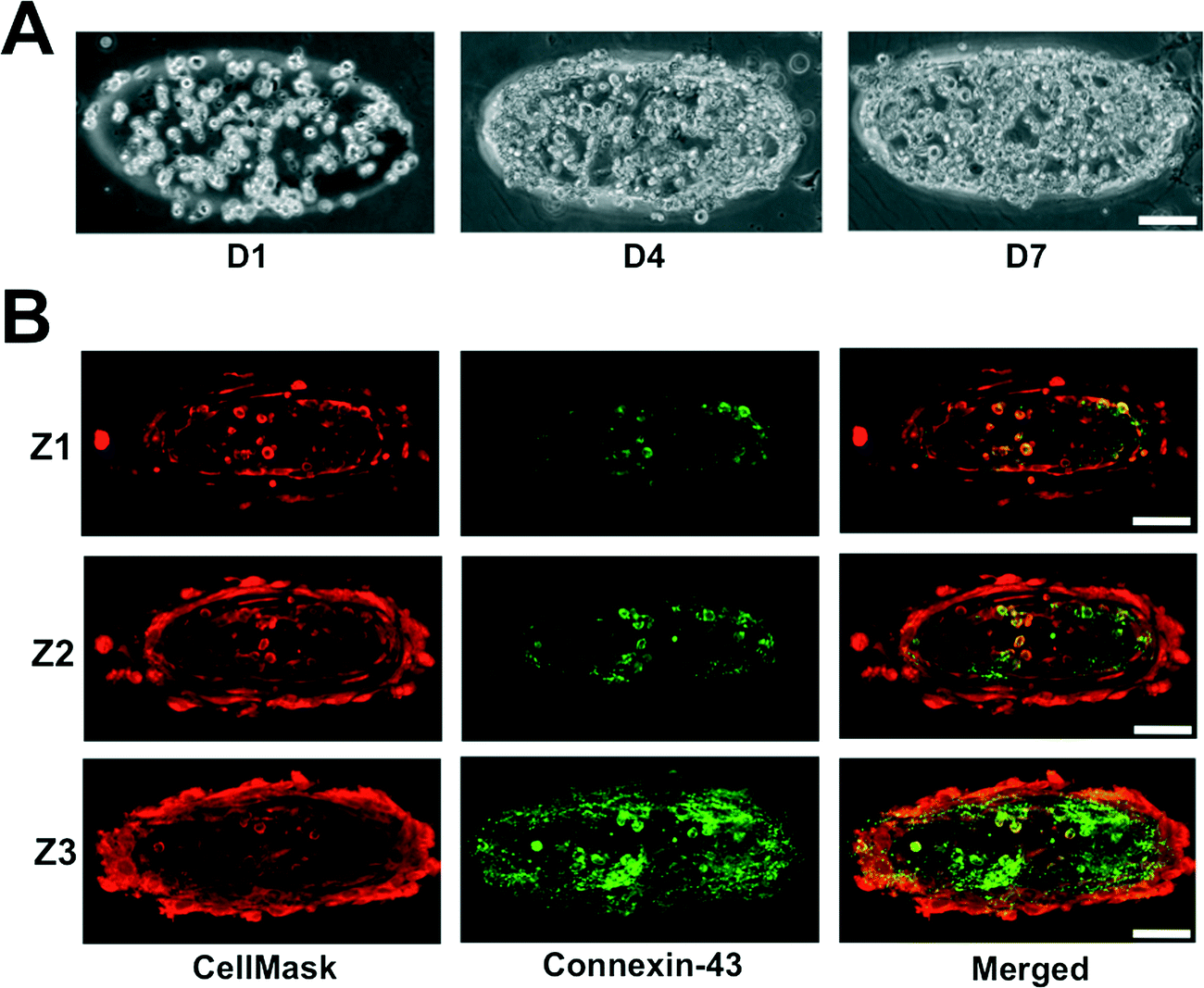 | ||
| Fig. 3 Characterization of encapsulated cardiomyocytes. (A) Cell density within the GelMA structures increased as a function of culture time. (B) X–Y confocal sections of cells ubiquitously stained with CellMask and immunostained for Connexin-43. Connexin-43 negative staining amongst the encapsulated cells suggests the presence of cardiac fibroblasts. The Confocal sections proceed from the top, Z1, to the bottom, Z3, of the GelMA structures. Scale bar: 100 μm. | ||
Real-time contractile stress measurements
Cardiomyocytes encased within the GelMA structures were cultured for 7 days within the microfluidics device prior to assessing the contractile stresses exerted by the cells onto the surrounding matrix. Since GelMA undergoes degradation in the presence of cells, we have used the stresses transduced into the adjacent PAm hydrogel layers surrounding the cell-laden GelMA structures to calculate the contractile stresses generated by the cells. For this, we first obtained the deformation of the PAm hydrogels, measured by the displacement of the embedded particles, in response to the contractile stresses generated by the encapsulated cells. The cells were then removed by using cell-dissolving solution, as described by us previously,29 and the structures were re-imaged. The position of the embedded fluorescent particles after the removal of the cells was used as a reference state. The inherent effect of the cell-dissolving solution on the PAm structures was determined by monitoring the changes in the position of the beads before and after exposing acellular structures to the cell-dissolving solution. Our analyses show there is no displacement of the beads after the cell-dissolving solution is introduced, suggesting minimal to no effect of the cell-dissolving solution on the hydrogel network (ESI†Fig. 2).The deformation of the fluorescent particles within the PAm hydrogel layers of the device in response to the beating cardiac cells encapsulated within the GelMA hydrogel is shown in Movie S1.† The region showing fluorescent particle movement corresponds to the ellipsoid pattern of the GelMA layer with encapsulated cardiomyocytes. The quantification of the deformation of the nanoparticles through displacement values is given in ESI† Movie S2. Using Particle Image Velocimetry (PIV), displacement fields were obtained from the movement of the particles and were used as boundary conditions for the finite element analysis to quantify the contractile stresses generated by the encapsulated cardiac cells. Fig. 4A illustrates the displacement field of the particles and the corresponding shear stresses generated on the PAm hydrogels by the encapsulated cells at various time points (T1–T4) during a contraction cycle of the cardiomyocytes. The shear stresses were found to be higher along the major axis of the ellipse compared to that along the minor axis. The peak stress value, calculated by taking a local average near the maximum stress value in the direction of the major axis of the ellipse, for oscillatory contraction cycles is depicted in Fig. 4B. Furthermore, our analyses showed non-zero stresses in the relaxed state of the contractile cycle indicating the presence of resting stresses within the structures. The resting stresses were determined by comparing the stress exerted by the cells during the relaxation phase of the contractile cycle to the absolute reference state obtained by removing the cells from the system. The shear stress heat maps and the corresponding peak stresses generated by the encapsulated cardiomyocytes as a function of time is shown in ESI†Fig. 3A and B. The shear stress heat maps suggest isolated cell contractions at day 2 and 4 while cells cohesively contract at days 7 and 12. The peak stress showed an increase in their value from day 2 to 7 (ESI†Fig. 3B). However, a drop in peak stress was observed at day 12 due to the degradation of the GelMA structure by the cells, which results in a change in the geometry of the GelMA structure. In addition, we have also quantified the variability in the contractile stresses across different microtissues within the same and different chips. While subtle differences in the peak stress values were observed among different chips, the values were on the order of 1 kPa and no statistical significance were observed (ESI†Fig. 3C).
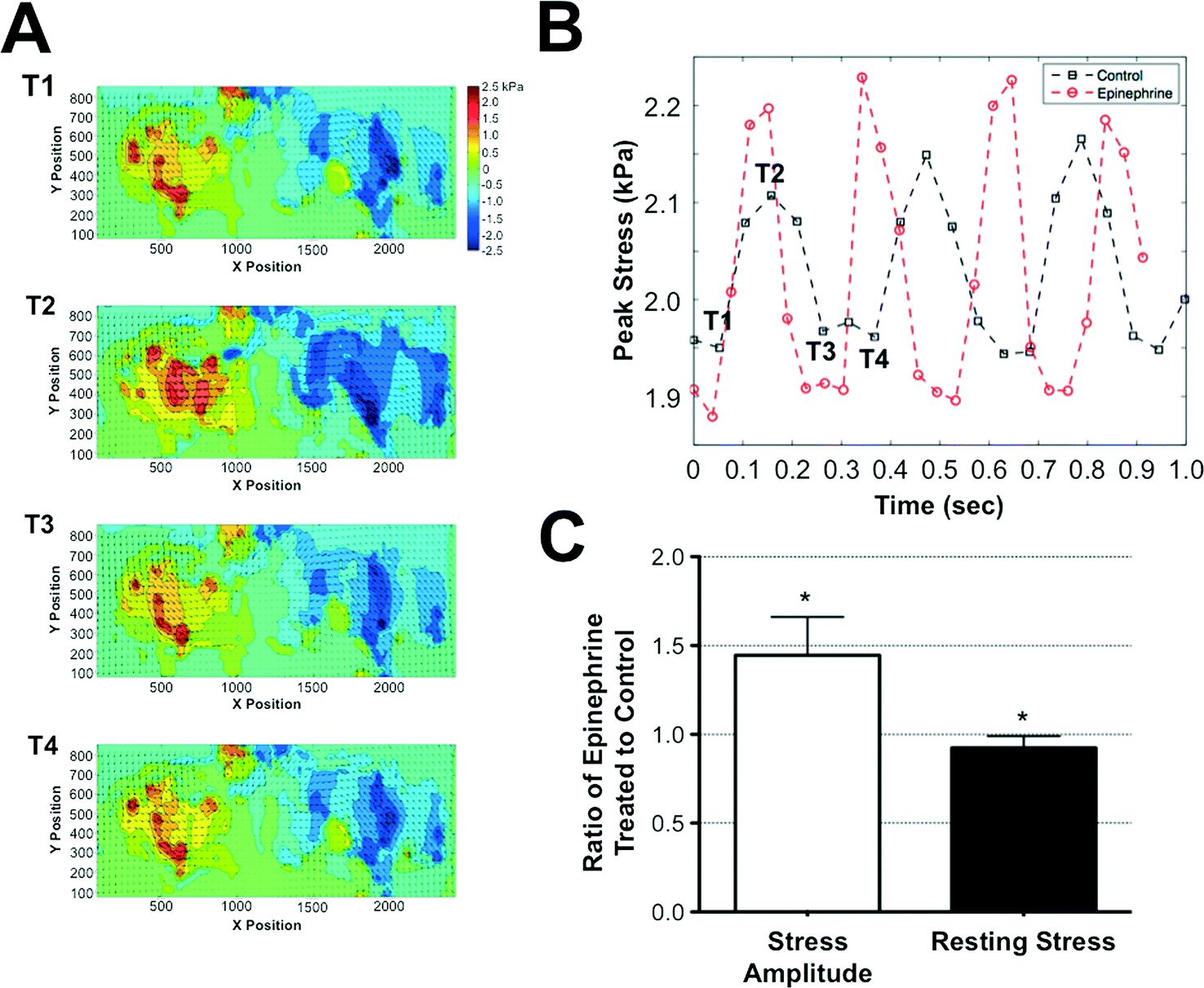 | ||
| Fig. 4 Quantification of contractile stresses generated by the encapsulated cardiomyocytes. (A) The displacements of the PAm hydrogel, shown as a vector field, overlaid onto the heat map of the shear stresses applied on the PAm hydrogel along the major axis of the GelMA ellipse structures. The positive and negative stresses indicate the direction of the shear stresses. Positive and negative means towards the right and left, respectively. The X- and Y- axis of the graphs indicate the physical location of the measured quantities while the values within the heat map are indicated by the color bar. The T1 to T4 represents the time points during the contraction–relaxation cycles of the beating tissue for which the stress heat maps shown in (A) are generated. The peak stresses associated with each heat map labeled with T1 through T4 are shown in (B). (B) A representative plot of the peak traction stress as a function of time in the absence (black squares) and presence (red circles) of epinephrine. The stresses along the major axis of the GelMA structures were used for determining the peak value. (C) The ratio of stress amplitude and resting stress in the presence and absence of epinephrine measured from multiple cell-laden structures. Values lower than or higher than 1 indicates a decrease or increase in stress amplitude and resting stress, respectively, while a value of 1 suggests the lack of change. * indicates statistically significant difference (p < 0.05) obtained from t-test in the measured stress amplitude and resting stresses from microtissues before and after addition of epinephrine. The contractile stresses were calculated from 4 different chips with 3–5 different microtissues in each chip. | ||
Cardiac-specific response to epinephrine
To validate the ability of this system to exhibit and detect cardiac-specific effects, we exposed the system to an environment supplemented with 0.1 μg ml−1 of epinephrine. Epinephrine is a molecule known to increase the frequency and amplitude of cardiac tissue contractions.30 Consistent with the known effects of epinephrine, the measured contractile stresses showed an increased frequency and amplitude of contraction cycles when the cells were exposed to epinephrine. While the presence of epinephrine increased the frequency and stress amplitude, the resting stress value observed at the relaxation phase of the contraction cycle was slightly lower when compared to the control cultures lacking epinephrine (Fig. 4C).Discussion
Organ-on-chip platforms have become increasingly popular to study cell- and tissue-microenvironment interactions in vitro and to investigate the effects of small molecule and environmental perturbations on cell/tissue toxicity and functions. Recently, we have developed a micropatterning technique to encapsulate and spatially organize cells.31 In this study, we have integrated this micropatterning technology with microfluidics to build a perfusion-based device containing arrays of viable 3D cardiac microtissues. A key component of the device is the tri-layer hydrogels—a cardiac cell-laden GelMA network sandwiched between two PAm hydrogels tethered onto the top and bottom surfaces of the flow chamber. We used this setup along with finite element analysis to determine the contractile stresses generated by the encapsulated cells.The cardiac cells were encapsulated within GelMA network, a collagen-derived matrix, as it provides the ligands necessary to foster cell-matrix interactions.32 The biodegradable nature of the GelMA also enables matrix remodeling and promotes cell–cell interactions. While GelMA network can provide adhesive sites for the cells to adhere and grow, the remodeling process causes time variant material properties and hence it is not amenable to estimate the contractile stresses generated by the cells by using traction stress measurements. Therefore, we adapted the tri-layer hydrogel system and determined the displacement of the particles embedded within the PAm hydrogel layers adjacent to the cell-laden GelMA layers. The displacement is a result of transduction of the stresses generated by the contracting cardiomyocytes. Since the PAm hydrogels exhibit linear elastic properties and maintain constant material properties during the course of the experimental studies, they are ideal candidates for traction stress measurements.33 Such far-field approaches have been employed by researchers for studying cancer cell motility.34 Also, in a study by Boudou et al. researchers have used a similar concept to measure contractile stresses by examining the stresses transduced by cardiac microtissues to attached microcantilevers.11
However, the utilization of a far-field approach requires one to address the artifacts associated with the potential decay of stress transduced with increasing distance from the contracting cells. To eliminate any such possibilities, we used a high cell density within each cell-laden GelMA structure. The use of high cell density ensures the presence of cells throughout the construct, and enables the measurement of the stresses generated by the entire 3D construct. This also allows the calculated stresses to be representative of the entire microtissue structure rather than one that is dominated by any single or a group of cells. Additionally, stresses were measured by examining the displacement of nanoparticles very close to the GelMA layer, minimizing the decay of transduced stress by minimizing the distance between stress generation and measurement. Further, each construct was compared only to itself before and after the introduction of epinephrine, thus eliminating variability among experiments and ensuring an accurate measurement of the small molecule effect on the cells. Furthermore, the significant distance between the structures ensured that the measured contractile stresses solely belong to individual structures and that the neighboring structures have no effect on the stress readout.
In this study, though the cell-laden GelMA structures were encased between two PAm hydrogels, we have only analyzed the deformations within the bottom PAm hydrogel layer due to imaging limitations. With an inverted microscope, the presence of the GelMA structures causes light diffraction, and thus the embedded fluorescent particles above the GelMA hydrogels could not be imaged with the resolution needed. Further, the thickness of the PDMS chamber exceeds the working distance of the microscope lenses that are used to image the particles from the top of the device. These limitations confine the analysis of traction stresses to the PAm hydrogel on either the top or bottom of the GelMA structures. Nevertheless, the PAm hydrogels used on the top and bottom surfaces had the same rigidity of ~8.5 kPa and similar thicknesses of ~70 μm to eliminate the asymmetrical effect of cells experiencing a rigid glass surface on the top while only experiencing a soft PAm hydrogel at the bottom. The large thicknesses of the PAm hydrogel layers also aid in eliminating the possibility of mechanical edge effects on the cardiomyocytes that could have been imparted by tethering the cell-laden GelMA layer directly to the rigid glass surfaces.29 The stress analyses showed the presence of non-zero stress even in the relaxed state and this resting stress could be attributed to the cell–matrix interactions.
The degradation of GelMA structures by the cells could have an effect on the contractile stress measurements. A change in the geometry of the cell laden GelMA structure is apparent at day 12 as evident from the shear stress heat map for the cardiomyocytes-laden GelMA structures. The circular pattern of the shear stress reflects the change in geometry from an ellipse structure to a cylindrical one. The degradation of GelMA structures can be slowed down through copolymerization of the GelMA with non-degradable materials such as poly(ethylene glycol diacrylate) and others.
The calculated contractile stress is lower than previously reported data using cardiac microtissues on a chip.22 This could primarily be attributed to the differences in tissue maturity as our measurements are carried out seven days post-encapsulation. Another possibility is that the modulus used for the contractile stress calculations could actually be lower than the actual modulus of the PAm hydrogels after device fabrication as repeated exposure of these hydrogels to UV light may induce increased network cross-linking. Hence, the calculated stresses could be an underestimation.
The potential of the device to respond to exogenous small molecule intervention and detect the corresponding changes in the contractile stresses was further verified by exposing the cells to epinephrine, an adrenergic neurotransmitter. Consistent with its known function, the encapsulated cardiac cells exhibited an increased amplitude and frequency of contraction with the addition of epinephrine. Further, investigating the changes in the resting stresses that the cardiomyocytes apply in the presence of drugs, we noticed a decrease in resting stress in the presence of epinephrine. This lower relaxed state in response to increased contractility could be the result of increased stretching of the GelMA material caused by the increased magnitude of contractile stress. Due to this system's ability to quantify stresses on the cellular level, monitoring such changes in resting stresses may lead to novel insights into the effects of drugs on cell–ECM interactions.
As seen in Fig. 3B, the cardiac culture contains a small amount of fibroblasts. During the primary cell isolation process, it is difficult to isolate pure cardiomyocytes. Staining for Connexin-43, a cardiac-specific gap junction protein,35 we see an even distribution of cardiomyocytes and fibroblasts, which are visualized through negative staining for Connexin-43. The presence of fibroblasts could have some anabolic influence as it has been shown that fibroblasts are critical to cardiac electrophysiological functions, even allowing for the propagation of electrical signal between cardiomyocytes separated by layers of fibroblasts.36,37
Conclusion
In summary, we developed a 3D “heart”-on-chip system in which we have successfully characterized contractile stresses generated by encapsulated cardiac cells in real time. We have shown the potential of the system to respond to exogenous small molecules such as epinephrine, demonstrating the promise of this device for small molecule drug testing. The ability of the tri-layer hydrogel device to support long-term cardiac cell culture and measure contractile stresses in real time allows for the study of short and long-term drug and small molecule effects on cardiac tissues, thereby improving on current models that lack real-time stress analysis. Also, replacing the neonatal mouse cardiac cells with cardiomyocytes derived from human-induced pluripotent stem cells (hiPSCs) could lead to a human-specific in vitro 3D cardiac system that has the potential to improve existing heart-on-chip disease models.23,27,28Material and methods
All experiments reported in the study were repeated at least twice independently with each experimental group containing a minimum of three biological replicates.Synthesis of gelatin methacrylate (GelMA)
Gelatin methacrylate (GelMA) was synthesized as described earlier.38 Briefly, 10 g of bovine skin gelatin (Sigma Aldrich, St. Louis, MO, USA) was mixed at 10% (wt/v) with 100 ml PBS and stirred at 60 °C until fully dissolved. Next, methacrylic anhydride (MA; Sigma Aldrich) was added to the solution at a rate of 0.5 ml min−1 for a total of 8 ml. The solution was then stirred for 60 minutes at 50 °C. After being diluted 2× with warm PBS, the solution was dialyzed against distilled water using 12–14 kDa cutoff dialysis tubing (Spectrum Laboratories, Rancho Dominguez, CA, USA) for one week (3 times per day water change) at 40 °C to remove the unreacted methacrylic anhydride and methacrylic acid from the solution. Next, the GelMA solution was frozen using liquid nitrogen and lyophilized in a freeze dryer for 4 days before being stored at −80 °C until usage. The dried GelMA was further purified using column chromatography with a Sephadex G-25 column (GE Healthcare Life Sciences, Pittsburgh, PA, USA) and lyophilized.Synthesis of lithium phenyl-2,4,6-trimethylbenoylphosphinate (LAP)
First, 2,4,6-trimethylbenzoyl chloride was mixed drop-by-drop with an equal molar solution of dimethyl phenylphosphonite under argon while stirring at room temperature. Next, the temperature of the reaction mixture was set to 50 °C after 18 hours. Then, lithium bromide mixed with 2-butanone was added to the reaction mixture in excess, causing precipitation within 10 minutes. After precipitation, the temperature was again cooled to room temperature and left for 4 hours. Next, to ensure complete removal of excess lithium bromide, the precipitate was collected by filtration and washed three times using 2-butanone. Finally, the product was dried using a vacuum to remove excess 2-butanone, yielding LAP.Fabrication of silicon mold
Firstly, microfluidic channels were photolithographically defined using NR9-1500PY negative photoresist (Futurrex, Frankling, NJ, USA) on 4 inches Si wafer. The Si mold master was etched by using deep reactive ion etching (DRIE) process. In the DRIE process, SF6 gas was flowed at 100 sccm during the 11 s of reaction time, followed by a passivation cycle when C4F8 gas was flowed at 80 sccm for 7 s. A 75 μm of etching depth was achieved under the etching rate of about 0.7 μm per cycle. After the DRIE process, the NR9-1500PY photoresist was removed by immersion in acetone for 4 hours before rinsing with methanol, isopropanol, and deionized water. The Si mold was then blown dry by nitrogen gas and silanized by vapor deposition of trichlorosilane (TCI Inc, Portland, OR, USA) to facilitate PDMS demolding.Synthesis of PAm hydrogels
12 mm round glass coverslips and 24 × 50 mm rectangular glass coverslips were first cleaned by treatment with 2.5 M NaOH for 20 minutes, and then rinsed with DI water and air dried. A silane solution was applied for 5 minutes to chemically functionalize the glass, and then the coverslips were once again rinsed with DI water and dried. Finally, a 0.75% glutaraldehyde solution was applied for 30 minutes followed by rinse and dry.Polyacrylamide (PAm) hydrogel precursor solution was synthesized by mixing together 6.25 μL of 40% (wt/v) acrylamide solution, 5.625 μL of 2% (wt/v) bisacrylamide solution, and 37.25 μL of phosphate buffered saline (PBS; Gibco, Billings, MT, USA). Fluorescent green beads were added to this 50 μL solution at a 1:100 dilution, and 0.6 μL of ammonium persulfate (APS) was added from a 10% wt/vol stock solution. Immediately prior to polymerization, 0.6 μL of 10% v/v tetramethylethylenediamine (TEMED) was added to the precursor solution. 4 μL of this solution was pipetted onto a new 22 × 22 mm square glass coverslip and the solution was sandwiched with the treated 12 mm round coverslip. Another 4 μL of the precursor solution was pipetted onto a treated 24 × 50 rectangular coverslip and sandwiched with a new, untreated 12 mm round coverslip. These were placed in a humidity chamber and allowed to polymerize for 20 minutes. A razor blade was used to clip the two coverslips apart. The PAm-coated glass coverslips were kept hydrated in water to preserve the PAm until further use. Prior to making the fluidics chamber, 4.0 μL of DI H2O was pipetted onto the circular chamber engraved on the etched silicon wafer, and the PAm hydrogel-tethered 12 mm round coverslip was placed on top of the water droplet, with the hydrogel in contact with the wafer.
PDMS solution preparation for fluidics chamber
Poly(dimethylsiloxane) (PDMS; Sylgard 184; Dow Corning, Midland, MI, USA) was obtained and prepared by mixing a 10:1 ratio by mass of base materials to the curing agent. This solution was thoroughly mixed for 3 minutes prior to it being poured gently on top of the silicone wafer containing PAm-coated 12 mm round glass coverslips in a 15 cm petri dish. Once the PDMS was level and an even distribution was achieved, it was degassed in a vacuum chamber for approximately 30 minutes to remove bubbles, if any. The PDMS was then incubated overnight at 37 °C for curing.
Primary neonatal cardiomyocyte isolation and culture
All animal procedures were done following the guidelines of an animal use protocol by the University of California at San Diego, Institutional Animal Care and Use Committee (IACUC). Neonatal mouse cardiomyocytes were acutely isolated from the one-day-old pups of CD-1 wild-type mice (Charles River Labs) as previously described.39 Briefly, hearts were surgically removed from one-day-old pups and digested for 12 hours in Hank's Balanced Salt Solution with 0.046% Trypsin at 4 °C. Hearts were treated with type II Collagenase for two to ten minutes at 37 °C. Isolated cells were pre-plated for 1.5 hours on tissue culture flasks in a humidified incubator at 37 °C with 5% CO2. Isolated cardiomyocytes were re-suspended in dark medium formulated by 75% DMEM and 25% M199 medium containing 10 mM HEPES, 10% horse serum and 5% fetal bovine serum, 1% 100× Penicillin/Streptomycin/L-Glutamine solution.3D μhearts-on-chip
To create the 3D cardiac microtissues on a chip, we integrated 3D photopatterning technology with microfluidics. The device can be roughly described as containing an inner circular chamber composed of tri-layer hydrogels with microchannels leading to and from the chamber. To this end, a photomask with the desired designs was generated and this photomask was used to fabricate corresponding silicone wafers. The overall device fabrication is given in Fig. 1.Poly-acrylamide (PAm) hydrogels embedded with 200 nm diameter green fluorescent particles were formed on glutaraldehyde-treated coverslips, as described in Synthesis of PAm hydrogels. Two such hydrogel-laden coverslips were generated, specifically a 12 mm diameter round coverslip and a 24 × 50 mm rectangular coverslip (Fig. 1A). The glutaraldehyde treatment was used to ensure strong bonding between the coverslip and the hydrogel. Next, the PAm hydrogel-tethered 12 mm round coverslip was placed onto the circular chamber engraved on the etched silicon wafer, with 4 μL water droplet in between the wafer and glass (Fig. 1B). A 10:1 ratio of base to curing agent of PDMS was degassed and then gently poured over the wafer and coverslip, and incubated overnight at 37 °C for curing. After complete curing, the PDMS was detached from the wafer. The detached PDMS is now embedded with the PAm hydrogel-tethered coverslip, with the hydrogel side exposed. We then punctured holes in the PDMS to provide inlet and outlet flow paths. Next, we subjected both the PDMS and the rectangular coverslip described in step 1A to UV-Ozone treatment (Fig. 1C). The PAm hydrogels attached on the PDMS mold and the rectangular coverslips were protected from deep UV by covering the hydrogels with 12 mm coverslips marked with dark ink. The circular depression within the PDMS was aligned with the hydrogel tethered onto the rectangular coverslip and pressed into contact. This was incubated at 37 °C overnight to allow the PDMS to chemically and irreversibly bond to the glass, thereby forming the device as depicted in Fig. 1D. The inlet and outlet openings of the fluidics device were sealed with tape to prevent the dehydration of the hydrogels within the device. Following bonding, PBS was flushed into the chamber on the subsequent day to equilibrate the hydrogel to physiological pH and osmolarity.
Isolated primary neonatal cardiomyocytes were mixed with 8.5% (wt/v) GelMA solution containing 0.01% (wt/v) Ascorbic acid and 2 mM LAP. An encapsulation density of 40 million cells per mL was used for 3D patterning of the cells. The macromer solution containing the cells was injected into the fluidics chamber (Fig. 1D) and the photomask containing the ellipse or hexagon patterns was placed under the circular chamber of the device. The GelMA solution dispersed with the cells was photopolymerized using a UV light for 10–15 s with an excitation wavelength of 365 ± 40 nm (Fig. 1G). The regions of the hydrogel precursor solution exposed to UV light polymerized rapidly, thereby forming the structures with cells encapsulated within (Fig. 1E). The unreacted solution was removed from the fluidics chamber by injecting PBS in excess (Fig. 1F). Thus, the 3D heart-on-chip device with patterned cell-laden GelMA hydrogels sandwiched between two acellular PAm hydrogels was created (Fig. 1G). The device was then perfused with dark media and incubated at 37 °C and 10% CO2 culture conditions.
Time-lapse imaging of cardiomyocyte embedded structures during contraction
The contraction of the GelMA structures embedded with cardiomyocytes was imaged as a function of time (days 2, 4, 7, and 12) by using 40× UMPlanFl lenses mounted onto a spinning disk confocal microscope. The fluorescent particles at the top surface of the PAm hydrogel below the GelMA structures were acquired at ~20 frames per second for 90 s duration. Three fields of view were acquired to encompass the entire GelMA structure. The time-lapse images from these fields of view were temporally synchronized to account for latency in the deformations between each field. These images were then stitched using a custom Matlab algorithm to obtain the entire image containing the GelMA structures.To obtain the reference state, the embedded cells were removed from the GelMA structure afterwards using a cell dissolving solution comprised of ammonium hydroxide and Triton-X 100.29 After the removal of cells, the PAm gels embedded with beads were reimaged as previously mentioned to obtain the reference state. We obtained the displacement field using PIV by comparing the reference state to the images captured during the contraction of the cells.
Calculation of contractile stresses
To quantify the stresses generated by the cardiomyocytes, we utilized the propagation of stresses originating from the GelMA structures to the PAm hydrogels. Since GelMA is derived from collagen and is susceptible to degradation, the stresses cannot be accurately determined from the GelMA structures themselves due to their changing material properties. However, the inert nature of PAm hydrogels allows for an accurate calculation of stresses exerted by the cardiomyocytes. Since the PAm hydrogel is assumed to be in mechanical equilibrium, we used the Galerkin method from finite element analysis to solve an elastostatic equation in which the body force is neglected (eqn (1)).| σij,j = 0 | (1) |
 | (2) |
In eqn (1) and (2), σij represents the stress tensor, E, the elastic modulus, ν, the Poisson's ratio, ui, the displacement, and δij, the kronecker delta. We next designate the PAm hydrogel as a 3D block tethered at the bottom while the sides of the block are assumed to be stress-free. For the top surface of the 3D block, we imposed displacement boundary conditions in the x- and y-directions. We neglected the normal deformations since our results indicated negligible normal displacements thereby allowing us to impose a stress-free condition on the top surface in the z- direction. To obtain the input for the boundary condition at the top surface of the hydrogel, we quantified the displacement of the PAm hydrogel by tracking the fluorescent particles embedded within the network using particle image velocimetry (PIV).40 The FE mesh was constructed to have 100 elements in the x- and y-direction and 60 elements in the z-direction. With the described boundary conditions and governing equations, we calculated the stress tensor throughout the material.
We next obtained the traction stress vector on the top surface of the PAm hydrogel (i.e., at the GelMA-PAm interface). Due to the stress-free boundary condition imposed in the z-direction, only the stress tensor components, σ13 and σ23, contribute to the stress vector. Since the stresses in the direction of the major axis dominated the minor axis, we considered only the σ13 component of the stress tensor. To estimate the peak stress, we used the magnitude of the shear stresses and obtain a localized average of the values around the maximum shear stress. The average value was calculated from a square window of length 200 μm whose center was positioned at the maximum shear stress location.
Immunofluorescent staining within fluidics device
The microfluidic chip was removed from the 37 °C incubator and disconnected from the media input before being flushed with PBS twice to clean out remaining media within the circular chamber. Next, formaldehyde solution (1:10 dilution in PBS) was infused into the device and incubated for 10 minutes at room temperature. After flushing three times with PBS at 10 minute intervals, the cardiac cell-laden structures were incubated with collagenase type II solution (0.1% in PBS) for 15 minutes at room temperature. After flushing three times with PBS at 10 minute intervals, the structures was exposed to blocking buffer (3% bovine serum albumin + 0.1% Triton-X 100) and incubated for 45 minutes at room temperature. Anti-Connexin-43 rabbit antibody (Sigma Aldrich, St. Louis, MO, USA) diluted 1:400 in blocking buffer was then injected into the device and the device was incubated in a humidity chamber for 2 hours. After flushing three times with PBS at 10 minute intervals, the cell-laden structures were incubated with Alexa Fluor 488 goat anti-rabbit antibody (1:250 dilution in blocking buffer; Life Technologies, Carlsbad, CA, USA) for 2 hours. After flushing three times with PBS at 10 minute intervals, CellMask (Life Technologies, Carlsbad, CA, USA) was injected into the device and the inlet and outlet ports were sealed with tape prior to imaging.
Laser scanning confocal microscopy for imaging acellular hydrogels
The samples used for the 3D confocal imaging of the acellular GelMA patterned structures within the microfluidics device was made with green fluorescent particles embedded within the PAm hydrogels, and with red fluorescent particles in the GelMA hydrogel. An Olympus UMPlanFl 10x water immersion objective lens mounted onto a Leica SP5 microscope was used for imaging. The scan speed was set to 400Hz, with z-step size of 0.74 μm. The green particles were visualized using 488 nm laser, the 594 nm laser was to visualize the red beads. Sequential scanning was utilized to individually image the green and red fluorescent particles to minimize unwanted excitation of the channels.Acknowledgements
We thank the financial support from National Institutes of Health (NIH; R01 AR063184- 02). AA acknowledges the Ruth L. Kirschstein National Research Service Award NIH/NHLBI T32 HL 105373. The authors also acknowledge the University of California San Diego Neuroscience Microscopy Shared Facility funded through NS047101.References
- C. Heylman, A. Sobrino, V. S. Shirure, C. C. Hughes and S. C. George, Exp. Biol. Med., 2014, 239, 1240–1254 CrossRef PubMed.
- J. W. Haycock, Methods Mol. Biol., 2011, 695, 1–15 CAS.
- J. Kim, J. Park, K. Na, S. Yang, J. Baek, E. Yoon, S. Choi, S. Lee, K. Chun, J. Park and S. Park, J. Biomech., 2008, 41, 2396–2401 CrossRef PubMed.
- F. Wang, V. M. Weaver, O. W. Petersen, C. A. Larabell, S. Dedhar, P. Briand, R. Lupu and M. J. Bissell, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14821–14826 CrossRef CAS.
- M. Anders, R. Hansen, R. X. Ding, K. A. Rauen, M. J. Bissell and W. M. Korn, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1943–1948 CrossRef CAS PubMed.
- K. A. Beningo, M. Dembo and Y. L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 18024–18029 CrossRef CAS PubMed.
- E. Cukierman, R. Pankov, D. R. Stevens and K. M. Yamada, Science, 2001, 294, 1708–1712 CrossRef CAS PubMed.
- A. S. Meshel, Q. Wei, R. S. Adelstein and M. P. Sheetz, Nat. Cell Biol., 2005, 7, 157–164 CrossRef CAS PubMed.
- D. Walpita and E. Hay, Nat. Rev. Mol. Cell Biol., 2002, 3, 137–141 CrossRef CAS PubMed.
- A. Hansen, A. Eder, M. Bonstrup, M. Flato, M. Mewe, S. Schaaf, B. Aksehirlioglu, A. P. Schwoerer, J. Uebeler and T. Eschenhagen, Circ. Res., 2010, 107, 35–44 CrossRef CAS PubMed.
- T. Boudou, W. R. Legant, A. Mu, M. A. Borochin, N. Thavandiran, M. Radisic, P. W. Zandstra, J. A. Epstein, K. B. Margulies and C. S. Chen, Tissue Eng., Part A, 2012, 18, 910–919 CrossRef CAS PubMed.
- M. Philip, M. Benatar, M. Fisher and S. I. Savitz, Stroke, 2009, 40, 577–581 CrossRef CAS PubMed.
- A. A. Bottini and T. Hartung, Altex, 2009, 26, 3–16 Search PubMed.
- K. Baar, R. Birla, M. O. Boluyt, G. H. Borschel, E. M. Arruda and R. G. Dennis, FASEB J., 2005, 19, 275–277 CAS.
- P. Linder, J. Trzewik, M. Ruffer, G. M. Artmann, I. Digel, R. Kurz, A. Rothermel, A. Robitzki and A. Temiz Artmann, Med. Biol. Eng. Comput., 2010, 48, 59–65 CAS.
- A. Khademhosseini, G. Eng, J. Yeh, P. A. Kucharczyk, R. Langer, G. Vunjak-Novakovic and M. Radisic, Biomed. Microdevices, 2007, 9, 149–157 CrossRef PubMed.
- N. Thavandiran, N. Dubois, A. Mikryukov, S. Masse, B. Beca, C. A. Simmons, V. S. Deshpande, J. P. McGarry, C. S. Chen, K. Nanthakumar, G. M. Keller, M. Radisic and P. W. Zandstra, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E4698–4707 CrossRef CAS PubMed.
- M. Radisic, H. Park, H. Shing, T. Consi, F. J. Schoen, R. Langer, L. E. Freed and G. Vunjak-Novakovic, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 18129–18134 CrossRef CAS PubMed.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–754 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- A. Mathur, P. Loskill, K. Shao, N. Huebsch, S. Hong, S. G. Marcus, N. Marks, M. Mandegar, B. R. Conklin, L. P. Lee and K. E. Healy, Sci. Rep., 2015, 5, 8883 CrossRef CAS PubMed.
- A. Agarwal, J. A. Goss, A. Cho, M. L. McCain and K. K. Parker, Lab Chip, 2013, 13, 3599–3608 RSC.
- G. Khanal, K. Chung, X. Solis-Wever, B. Johnson and D. Pappas, Analyst, 2011, 136, 3519–3526 RSC.
- A. Grosberg, P. W. Alford, M. L. McCain and K. K. Parker, Lab Chip, 2011, 11, 4165–4173 RSC.
- W. Cheng, N. Klauke, H. Sedgwick, G. L. Smith and J. M. Cooper, Lab Chip, 2006, 6, 1424–1431 RSC.
- G. A. Giridharan, M. D. Nguyen, R. Estrada, V. Parichehreh, T. Hamid, M. A. Ismahil, S. D. Prabhu and P. Sethu, Anal. Chem., 2010, 82, 7581–7587 CrossRef CAS PubMed.
- M. L. McCain, S. P. Sheehy, A. Grosberg, J. A. Goss and K. K. Parker, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9770–9775 CrossRef CAS PubMed.
- G. Wang, M. L. McCain, L. Yang, A. He, F. S. Pasqualini, A. Agarwal, H. Yuan, D. Jiang, D. Zhang, L. Zangi, J. Geva, A. E. Roberts, Q. Ma, J. Ding, J. Chen, D. Z. Wang, K. Li, J. Wang, R. J. Wanders, W. Kulik, F. M. Vaz, M. A. Laflamme, C. E. Murry, K. R. Chien, R. I. Kelley, G. M. Church, K. K. Parker and W. T. Pu, Nat. Med., 2014, 20, 616–623 CrossRef CAS PubMed.
- A. Aung, Y. N. Seo, S. Lu, Y. Wang, C. Jamora, J. C. del Alamo and S. Varghese, Biophys. J., 2014, 107, 2528–2537 CrossRef CAS PubMed.
- R. F. Furchgott, Pharmacol. Rev., 1959, 11, 429–441 CAS , discussion 441–422.
- S. K. Davey, A. Aung, G. Agrawal, H. L. Lim, M. Kar and S. Varghese, Tissue Eng., Part C, 2015, 21, 1188–1196 CrossRef CAS PubMed.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Biomaterials, 2010, 31, 5536–5544 CrossRef CAS PubMed.
- J. P. Butler, I. M. Tolic-Norrelykke, B. Fabry and J. J. Fredberg, Am. J. Physiol., 2002, 282, C595–605 CrossRef CAS PubMed.
- R. J. Polackwich, D. Koch, R. Arevalo, A. M. Miermont, K. J. Jee, J. Lazar, J. Urbach, S. C. Mueller and R. G. McAllister, PLoS One, 2013, 8, e58138 CAS.
- B. J. Warn-Cramer, G. T. Cottrell, J. M. Burt and A. F. Lau, J. Biol. Chem., 1998, 273, 9188–9196 CrossRef CAS PubMed.
- G. Gaudesius, M. Miragoli, S. P. Thomas and S. Rohr, Circ. Res., 2003, 93, 421–428 CrossRef CAS PubMed.
- P. Kohl, P. Camelliti, F. L. Burton and G. L. Smith, J. Electrocardiol., 2005, 38, 45–50 CrossRef PubMed.
- H. Kang, Y. R. Shih, Y. Hwang, C. Wen, V. Rao, T. Seo and S. Varghese, Acta Biomater., 2014, 10, 4961–4970 CrossRef CAS PubMed.
- P. Sreejit, S. Kumar and R. S. Verma, In Vitro Cell. Dev. Biol.: Anim., 2008, 44, 45–50 CrossRef CAS PubMed.
- B. Alonso-Latorre, R. Meili, E. Bastounis, J. C. Del Alamo, R. Firtel and J. C. Lasheras, Conference proceedings : … Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Annual Conference, 2009, vol. 2009, pp. 3346–3349 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5lc00820d |
| ‡ Contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2016 |