
Preparation of standards for in situ sulfur isotope measurement in sulfides using femtosecond laser ablation MC-ICP-MS
Lu
Chen
,
Kaiyun
Chen
 ,
Zhian
Bao
,
Peng
Liang
,
Tiantian
Sun
and
Honglin
Yuan
*
,
Zhian
Bao
,
Peng
Liang
,
Tiantian
Sun
and
Honglin
Yuan
*
State Key Laboratory of Continental Dynamics, Department of Geology, Northwest University, Collaborative Innovation Center of Continental Tectonics, Xi'an 710069, China. E-mail: sklcd@nwu.edu.cn; Fax: +86 29 88303447; Tel: +86 29 88305924
First published on 1st November 2016
Abstract
We prepared a series of pressed powder tablets and chalcopyrite glass standards to correct mass bias when using standard–sample bracketing for in situ sulfur isotope measurements. A femtosecond laser ablation coupled multi-collector inductively coupled plasma mass spectrometry (fsLA-MC-ICP-MS) method was used to determine the sulfur isotope composition of these standards. Chalcopyrite glass (YN411-m) was prepared by quickly quenching the fused chalcopyrite, which was melted at 1000 °C under a protective N2 atmosphere. Multiple experiments were conducted for homogeneity testing of YN411-m by fsLA-MC-ICP-MS and the external precision was 0.28‰ (n = 35). Mineral particles, pressed powder tablets, and chalcopyrite glass were used as bracketing standards when determining the δ34S of a natural chalcopyrite (GC). The results showed that the concentration, elemental composition, and crystal structure caused the matrix effect. Considering practicability, the melted glass was a more appropriate standard than the pressed powder tablet. We also found that the carrier gas flow rate, laser fluence, and spot size impacted the regularity of results. Therefore, we can obtain accurate δ34S by adjusting the laser and MC-ICP-MS parameters when using non-matrix matching standards. Furthermore, fsLA-MC-ICP-MS is advantageous for in situ sulfur isotope measurement because the sensitivity can be improved and it dramatically improves the spatial resolution (10–20 μm), which can be used to explain the origin of multi-genesis deposits by analyzing smaller mineral micro-areas, especially mineral fracture-filling sulfides later in mineralization.
1. Introduction
Sulfur is abundant in nature, existing in versatile states such as gaseous H2S and SO2, and solid sulfide, sulfate, elemental sulfur, and organic sulfur. The fractionation of sulfur isotopes during geological processes is distinct because of the large difference in their atomic masses and valence states. Generally, the sulfur isotope compositions are presented as δ34S:1,2| δ34S = [(34S/32Ssample)/(34S/32Sstandard) − 1] × 1000. |
δ 34S is stabilized at around 0‰, or a small positive amount, in mantle-derived rocks, while the isotopic composition of sulfur in crust rocks, having undergone magmatism, sedimentation, metamorphism, and supergenesis, has a large range of −50 to +35‰.1,3,4 Therefore, sulfur isotopes serve as key tracers in ore-forming material sources in geochemical processes and sulfur cycling in nature.5–9
Conventional bulk analytical techniques for sulfur isotope ratios involve the use of gas-source mass spectrometry (GS-MS) or a solution multi-collector inductively coupled plasma mass spectrometer (MC-ICP-MS).2,10–12 GS-MS requires sulfur to be converted from a solid powder to gaseous SO2 or SF6 before being subjected to mass spectrometry analysis. Generally, GS-MS is considered the standard method for precise and accurate sulfur isotope measurements, but requires time-consuming sample preparation and large samples (more than 10 mg for sulfide). Solution MC-ICP-MS can provide good accuracy, precision better than 0.2‰ (2SD),2,12,13 and sample demand is lower than GS-MS, needing 500 μg S. But the chemical purification of samples is complex, and the procedure of processing through resins may generate unnegligible resin originated blank (0.25 μg S).2 Recent studies have shown that solution MC-ICP-MS can be used to measure very low sulfur samples (e.g., glacier, snow, and rainwater samples), having a sulfur demand as low as 2 μg.14,15 Though bulk analytical techniques have very high precision, it is difficult to distinguish sulfur isotopes of zonation within a single grain or multi-genesis minerals in micro-areas. Therefore, in situ analytical techniques have been developed to resolve the problems. Previous research has combined laser ablation with GS-MS, but the spatial resolution was limited by the high GS-MS sample demand, usually ∼150 μm.16 In comparison, secondary ion mass spectrometry (SIMS) and LA-MC-ICP-MS not only provided high precision, but also achieved better spatial resolution. Ushikubo17 measured a standard pyrite, achieving a precision of 0.23‰ (2SD) with a primary beam size of ∼20 μm by SIMS. Kozdon achieved ±0.2‰ precision in pyrite (2SD) with a 10 μm spot size using SIMS.18 Since there is no 36Ar interference to 36S, this technique can be used to test δ36S, which cannot be accomplished using MC-ICP-MS. However, because SIMS is prone to serious matrix effects,17 this technique cannot use IAEA pressed powder tablets as standards to calibrate most natural sulfides. Predecessors have measured sulfur isotopes in pressed powder tablets of international standards by LA-MC-ICP-MS with a precision of 0.2–0.4‰ for IAEA-S-1 (Ag2S)13,19 and ∼0.2‰ for NBS123 (ZnS).13,20 Commonly used lasers are a 213 nm Nd:YAG solid state laser or 193 nm excimer ArF laser, and the spatial resolutions are 40–60 μm or larger.2,13,19–22 Most ablation modes are line scans, with the 32S signal ranging from several to dozens of volts.2,13,19–22 Due to its low sample demand, high analytic precision, and spatial resolution, LA-MC-ICP-MS is widely used.
Most measurements used standard-sample bracketing to correct mass bias,2,21,22 with sulfur isotope compositions presented as values relative to a reference; thus, the standard is a key factor in sulfur measurement. Because the recognized standard, Vienna Cañon Diablo Troilite (VCDT), was used up, researchers are seeking for new sulfur isotope standards. Standards for LA-MC-ICP-MS can be natural minerals, pressed powder tablets, or any other type of artificial solid sample.13,23,24 Natural sulfide or sulfate minerals commonly have an inhomogeneous sulfur composition resulting from the various origins of sulfur and fractionation during mineral precipitation.25,26 Sulfides from magmatic deposits without later hydrothermal alteration may have a homogeneous sulfur isotope composition owing to the single sulfur resource and high ore-forming temperature. Bendall (2006) used a pyrite from a Kambalda magmatic deposit as the in-house standard, which had an external precision of 0.32‰ (2SD) measured by LA-MC-ICP-MS.22 Gilbert et al. (2014) also used a natural pyrite standard (PPP-1) as an isotopic reference material with a reproducibility of 0.2‰.27 In addition, metamorphosed massive sulfide deposits may contain homogeneous sulfides.28 The solid sulfide standards currently used in LA-MC-ICP-MS are IAEA series (Ag2S powder) and NBS123 (ZnS). IAEA powder was pressed into tablets for in situ sulfur isotope determination, but the matrix difference between the standard and sample caused diversity in the transmission, vaporization, and ionization efficiency of laser-generated aerosols.2,13,29 The majority of NBS123 crystals are too small (80–300 μm) for analysis by most LA-MC-ICP-MS equipment in line scan mode and, due to crystal defects in some sphalerite particles (NBS123), they showed a more heterogeneous ablation and vaporization behavior compared with tablets, resulting in poor precision.20 Most NBS123 were ground into powder and pressed into tablets. Furthermore, Pribil (2015) used a ZnS glass as the in-house standard, which showed good homogeneity.13
Higher spatial resolution is very important for geological sample analysis, especially of small-sized sulfides and fracture-filling sulfides during different mineralization stages. Compared with a nanosecond laser, a femtosecond laser has a much shorter pulse duration, shorter, even, than the thermal relaxation time of materials,30 so there is not enough time for the energy to transfer from electrons to the lattice and induce heating within and around the ablation volume to reduce thermal effects.31 Less thermal effects will result in less elemental fractionation,30,32,33 indicating that a femtosecond laser is more appropriate for in situ sulfur analysis. Beyond that, femtosecond lasers generate smaller and more uniform aerosol particles (usually on the nanoscale), which enhances the transmission efficiency and signal intensity.30–36 Therefore, the application of a femtosecond laser during in situ sulfur isotope analysis with a smaller ablation size is possible and will improve spatial resolution. For mass spectrometry analysis, isotopic interferences from molecular ions (e.g., 16O–16O+, 32S–1H+ and 17O–16O–1H+) and doubly charged ions (e.g., 64Zn2+) have a negative effect on sulfur isotope analysis.2,13,22 This problem is commonly resolved using sufficient mass resolution to enable the detection of S isotopes on the interference-free plateau. However, the sulfur plateau is too narrow compared with the mixed peak, and values might be significantly affected by instrument stability. In this study, we used a high-resolution MC-ICP-MS (Nu 1700) combined with a femtosecond laser to analyze sulfur isotopes. Nu 1700 is able to completely separate any interference peaks in 32S using medium resolution mode (resolution power = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and in 33S using a higher resolution mode (resolution power = 18000).
000) and in 33S using a higher resolution mode (resolution power = 18000).
We prepared several standards for in situ sulfur isotope analysis using different methods, and determined their δ34S values by GS-MS and solution MC-ICP-MS. We determined their feasibility as standards by comparing their matrix effects using fsLA-MC-ICP-MS. Moreover, the application of femtosecond laser and high-resolution MC-ICP-MS can significantly improve spatial resolution, which can be used to explain the origin of multi-genesis deposits by the analysis of smaller mineral micro-areas, especially the mineral fracture-filling sulfide during later mineralization.
2. Materials and methods
2.1. Instrumentation and analytical protocol
Most experiments were conducted at the State Key Laboratory of Continental Dynamics, Northwest University, Xi'an, China. Isotopic measurements were performed using a Nu Plasma 1700 MC-ICP-MS (Nu instruments, UK). A NWR UP Femto (Electro Scientific Industries, Inc., USA) femtosecond laser ablation system was used as the ablation source for in situ analysis. A JEOL JXA-8230 electron probe with 15 kV acceleration voltage, 20 nA beam current, 2 μm beam size and 30 ms cumulative time for each point was used for back-scattered electron images and major element analysis, including single and map analysis modes. Otherwise, a Delta V Plus gas-source mass spectrometer (GS-MS), housed in the analytical laboratory of Beijing Research Institute of Uranium Geology, China, was used to determine the δ34S of prepared sulfur-bearing materials. During this experiment, sulfide powder was mixed with Cu2O powder before being oxidized to SO2 under a vacuum of 2 × 10−2 Pa and temperature of 980 °C. SO2 was collected by the freezing method, and then was introduced into GS-MS to be tested.The NWR UP Femto femtosecond laser ablation system consisted of a Quantronix Ti:sapphire femtosecond laser amplifier Integra-HE and ESI NWR Femto laser ablation system. The laser output ultraviolet wavelength was 266 nm, which is the third harmonic of 800 nm, and its pulse duration was less than 130 fs. The instrument was equipped with a two-volume chamber that consisted of a large sample chamber (5000 cm3) and a high-sensitivity sample cell (1.6 cm3). Nu 1700 MC-ICP-MS was equipped with sixteen Faraday cups and three ion counters. Cup configurations for sulfur were H5 cup for 34S, Ax cup for 33S, and L4 cup for 32S. Serious interference from molecular ions, especially oxygen species, was present and mixed with the sulfur peaks. As shown in previous reports (Fig. 1a),13 sulfur isotopes were detected on the lighter interference-free plateau.2,13,22 The sulfur plateau was so narrow compared with the mixed peak that the obtained signal might be mixed with the interference signal, reducing the precision and accuracy. Nu 1700 was able to separate any interference peaks completely for 32S in medium resolution mode (resolution power = 10000) and for 33S in a higher resolution mode (resolution power = 18000). The determination of unmixed sulfur peaks ensures the stability and accuracy of results.
 | ||
| Fig. 1 Peak shapes for S isotopes with masses of 32 (green dotted line), 33 (black solid line), and 34 (red dotted line), using different resolutions. (a) Peak shapes obtained by Nu HR with pseudo-medium resolution and detected S isotopes on the interference-free plateau at lower mass.13 (b) Peak shapes of Nu 1700 with high resolution and interference from 16O–16O and 32S–1H were separated from 32S and 33S. (c) and (d) Peak shapes of Nu 1700 with medium resolution using solution and laser ablation to introduce samples. | ||
The properties of the laser systems and MC-ICP-MS are summarized in Table 1. Argon flow rates and voltage were adjusted to give the best signal stability and intensity in mass spectrometry. Ultra-high purity He with a flow rate of 0.7 L min−1 carried sample aerosol into the pipe, which was mixed with Ar in a gas-mixing chamber before entering the ICP. The argon gas flow rate depended on mass spectrometer conditions. Time-resolved software data acquisition mode with an integration time of 0.2 s was used to collect and integrate signals introduced by laser ablation. Each sample acquisition consisted of background collection for 30 s, followed by ablation signal collection for 70 s, and a wash time of 50 s to reduce memory effects. Due to the high sensitivity, a relatively low laser energy (fluence ranged from 0.5 to 3.5 J cm−2) and 10 Hz frequency were used. Since many experiments have shown that line scan ablation results in higher and more stable signal intensities, and generates better precision for S isotope ratios,2,21 a line scan ablation mode with an ablation speed of 2 μm s−1 and a frequency of 10 Hz was applied in this study. In general, the 32S signal would be 8–20 V with a spot size of 10–20 μm for most sulfides.
| Laser ablation setup | |
| Laser | NWR UP Femto (esi), 266 nm |
| Pulse length | <130 fs |
| Carrier gas flow rate | He, 0.7 L min−1 |
| Laser ablation mode | Line scan, 2 μm s−1 |
| Spot size | 10–20 μm |
| Frequency | 10 Hz |
| Energy density | 0.5–3.5 J cm−2 |
|
|
| Mass spectrometer setup | |
| MC-ICP-MS | Nu plasma 1700 |
| RF power | 1300 W |
| Acceleration voltage (HV1) | 6000 V |
| Cooling gas flow rate | 13 L min−1, Ar |
| Auxiliary gas flow rate | 0.8 L min−1, Ar |
| Carrier gas flow rate | ∼0.75–0.85 L min−1, He |
| Interface cones | Ni orifice |
|
|
| Data acquisition parameters | |
| Acquisition type | Static, time resolved analysis |
| Detector | Faraday cups |
| Cup configuration | 32S (L4), 33S (Ax), 34S (H5) |
| Resolution mode | 10000 RP (source slit, 200–260; α slit, 15 mm; β slit, 8 mm) |
| Signal analysis protocol | 0.2 s integration time, 1000 cycles |
As for solution MC-ICP-MS, a CETAC AridusIIautosampler and a quartz nebulizer with an uptake rate of 100 μL min−1 were used to produce wet plasma and introduce it into the ICP torch. Data acquisition type was static and signal-analysis protocol included 30 cycles with 8 s integration time per cycle. Two vials of pure water were used as the washer, with a total washing time of 140 s. The other parameters of MC-ICP-MS used in the solution method were same as those used in the in situ analysis, which are shown in the mass spectrometer setup of Table 1.
2.2. Reagents and samples
HNO3 and HCl were used as the acids in this work after sub-boiling preparation using a Savillex DST-1000. High-purity water was prepared using a Millipore Element (18.2 MΩ cm−1, Millipore Corporation, USA). All containers directly in contact with the solution and powder samples were cleaned for a 12 h period in 50% nitric acid and rinsed three times with 18 MΩ cm−1 Milli-Q water prior to use.Solid sulfur-bearing materials included some international sulfur reference materials (IAEA-S-1, IAEA-S-2, IAEA-S-3 and NBS123), a polymetal sulfide (MASS-1) and some natural chalcopyrite samples (Cpy-1, GC and YN411). IAEA series are Ag2S powders from the International Atomic Energy Agency (IAEA). NBS123 is a sphalerite standard with a particle size of 80–300 μm, obtained from the National Institute of Standards and Technology (NIST). MASS-1 from United States Geological Survey (USGS) is a pressed powder tablet of polymetallic sulfide produced by precipitating amorphous metal sulfides from solution with a Fe–Cu–Zn–S matrix,24 and its S concentration was 24% (data from Certificate of Analysis provided by USGS). GC is a copper ore from Guichi copper mine, Anhui, China. Cpy-1 and YN411 were chalcopyrites provided by the Institute of Geochemistry, Chinese Academy of Sciences, and Tangdan Copper Mine, Yunnan, China, respectively. GC was made into slices and the size of chalcopyrite ranged from tens of microns to several millimeters. Cpy-1 is a big chalcopyrite aggregation and was carved into small pieces, with a size of 5 millimeters, to make an epoxy resin target. YN411 consisted of pure chalcopyrite particles.
Sulfur-bearing solutions used in this work included Spex ((NH4)2SO4 solution from SSpex CertiPrep) and Alfa ((NH4)2SO4 solution from Alfa Aesar). They have a sulfur concentration of 10000 ppm and were diluted to 10 ppm as in-house standards. In addition, all the solid sulfur-bearing materials were digested and purified with reference to previously reported procedures2,14 for evaluation by solution MC-ICP-MS. GC, Cpy-1 and fused YN411 were sent to the analytical laboratory of Beijing Research Institute of Uranium Geology for secondary evaluation by GS-MS.
2.3. Preparation of pressed tablet and melted glass
The prepared solid sulfur-bearing materials included pressed powder tablets and glass chalcopyrite. With the exception of IAEA-S-1, IAEA-S-2, and IAEA-S-3 powders, NBS123 and GC were ground to a powder with a major size distribution of 2.5 μm, as determined by a Mastersizer 2000 Laser Particle Size Analysis System (Fig. 2a). Samples were ground using a ball mill (Retsch PM100) with agate vials using various agate milling ball numbers and sizes. The grinding process was taken from a study producing undiluted nanoparticulate pressed powder tablets for elemental analysis using LA-ICP-MS.23 The procedure for producing pressed powder tablets was as follows: (i) put the sample with a certain ratio of sample mass, ball mass, and water volume (1 g:17 g:2.5 mL) into an agate vial and grind for 60 min; (ii) after allowing to stand for 30 min, remove the upper water layer using a transfer pipette and dry the remaining sample at 70 °C; (iii) transfer the powder into a plastic pipe (4 mm diameter, 5 mm thickness) and press into a tablet by applying 15 MPa for 2 min using a Maekawa Press.
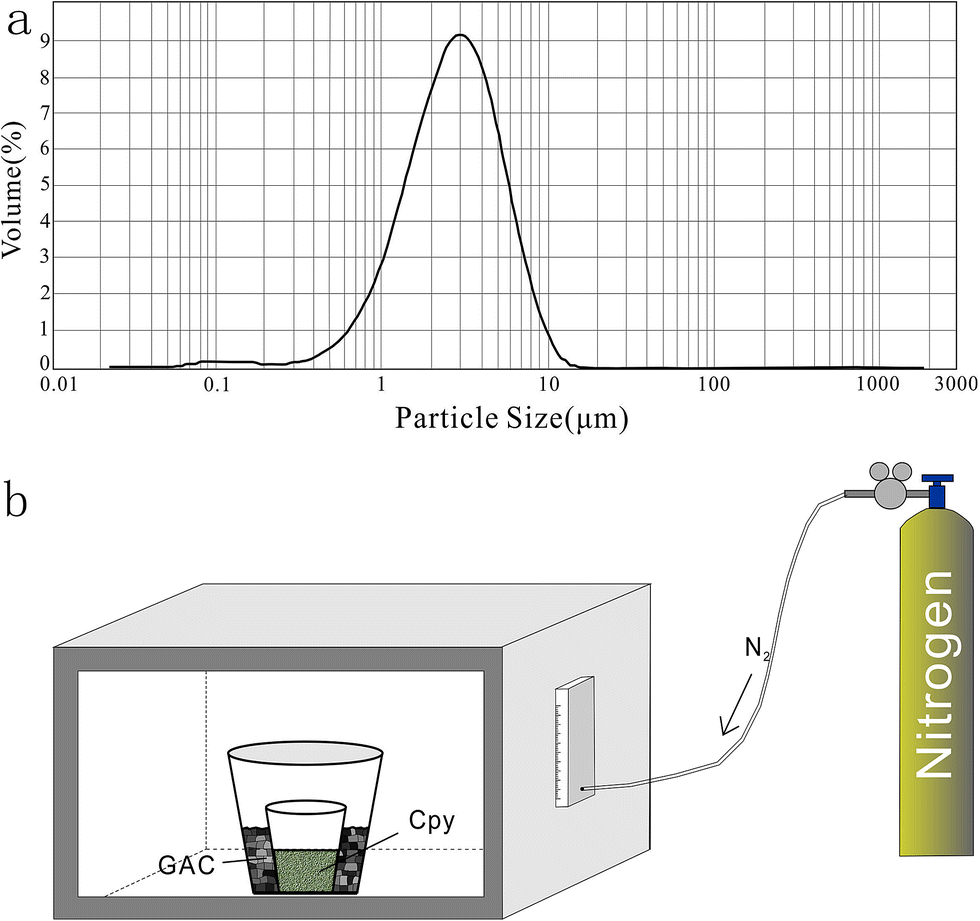 | ||
| Fig. 2 Producing processes of standards: (a) size distribution of powder ground by ball milling. (b) Procedure of melting chalcopyrite; GAC is granular active carbon and Cpy is chalcopyrite powder. | ||
YN411 (chalcopyrite) was converted into a sulfide glass (YN411-m) using the following procedure (Fig. 2b): (i) pure and unoxidized chalcopyrite was picked out under a stereoscope, cleaned with Milli-Q water, and dried in the air; (ii) pure chalcopyrite was ground down until the grain diameter was smaller than 15 μm; (iii) 1 g of the powder was compacted and sealed into a small ceramic crucible (Al2O3), and the operation was complete in a nitrogen-filled chamber; (iv) the small ceramic crucible was set inside a larger one loaded with granular active carbon and put into a KDF-S80 muffle furnace (applied temperature of 100–1150 °C, filled with nitrogen); (v) the chalcopyrite powder was melted for 3 min at 1000 °C under the protection of a nitrogen environment; (vi) the liquid melt was poured into iced high purity water with a small ceramic crucible as quickly as possible. The quenching process made the ceramic crucible easy to break, allowing the glass to be separated.
GC was made into slices with a thickness of hundreds of micrometers and stuck on a glass. Cpy-1 (chalcopyrite crystal) and YN411-m were made into epoxy resin targets. The surfaces of the slice and targets were polished.
2.4. Data processing and calculation
Instrument drift and mass bias were corrected using a sample–standard bracketing approach with repeated measurement of the standard before and after each sample.2,21 Since the most reported δ34S values of IAEA-S-1 were consistent (δ34SVCDT = −0.3‰),2,11,13,15,37 purified IAEA-S-1 solution and IAEA-S-1 pressed powder tablet were used as the bracketing standard for correcting the in-house standard (Spex solution) in solution analysis and IAEA-S-2 or IAEA-S-3 pressed powder tablets in in situ analysis respectively. The δ34S of samples relative to IAEA-S-1 was normalized to the Vienna Cañon Diablo Troilite (VCDT) scale using the following equation:| δ34SVCDT = [(δIAEA-S-134SVCDT/1000 + 1) × (δsample34SIAEA-S-1/1000 + 1) − 1] × 1000 |
δ IAEA-S-1 34SVCDT means the δ34S of IAEA-S-1 is related to VCDT (δ34SVCDT = −0.3‰), and δsample34SIAEA-S-1 means the δ34S of samples is related to IAEA-S-1.
Accurate δ34SVCDT values of the aforementioned solid sulfur-bearing materials were determined by solution MC-ICP-MS and GS-MS (Table 2). For replicate analyses, external reproducibility was reported at the 2SD (standard deviation). Error propagation should be considered when using an in-house standard to correct samples, using the error transfer formula, 2SD = √[(2SDsample)2 + (2SDstandard)2].2,20
| Name | Measured δ34SVCDT ± 2SD (‰) | Ref. δ34SVCDT ± 2SD (‰) | ||
|---|---|---|---|---|
| MC-ICP-MS (solution) | GS-MS | MC-ICP-MS (solution) | GS-MS | |
| a Published data: a(Craddock et al., 2008);2b(Das et al., 2012);15c(Han et al., 2013);38d(Ding, 2001);37e(Qi and Coplen, 2003);11f(Ono et al., 2012);39g(Fu et al., 2016).19 | ||||
| IAEA-S-2 | 22.25 ± 0.21 | 22.44 ± 0.43a | 22.64d | |
| 22.26 ± 0.42b | 22.67e | |||
| 22.30 ± 0.36c | 22.24 ± 0.27f | |||
| IAEA-S-3 | −32.28 ± 0.26 | −32.29 ± 0.45b | −32.05d | |
| −32.62 ± 0.62c | −32.58 ± 0.2f | |||
| NBS123 | 17.75 ± 0.17 | 17.77 ± 0.26a | ||
| GC | −0.79 ± 0.24 | −0.63 ± 0.12 | ||
| Cpy-1 | 4.12 ± 0.23 | 4.3 | ||
| YN411-m | 0.37 ± 0.24 | 0.53 ± 0.23 | ||
| MASS-1 | 5.60 ± 0.56g | |||
3. Results and discussion
3.1. Composition and homogeneity of melted glass
Fig. 3a presents the back-scattered electron image and major element analysis in map analysis mode of YN411-m. They clearly indicated that the sulfur concentration in an ablation area (20 × 150 μm) was homogeneous. Single and map analysis of Fe and Cu also showed good uniformity and an elemental composition similar to that of natural chalcopyrite. A small amount of Al existed in the cracks of YN411-m, resulting from contamination with polishing powder. The sulfur isotope homogeneity of the glass was evaluated by self-correcting (by setting YN411-m as the standard and samples simultaneously) using fsLA-MC-ICP-MS. As Fig. 3b shows, measurements were operated in four days' tests. Each session used its proper spot size range from 10 to 20 μm, and the same energy of 3 J cm−2. 35 values were calculated and the external precision of δ34S was 0.28‰ (2SD). | ||
| Fig. 3 (a) The left picture is the BSE image, and the right picture is the sulfur map analysis of YN411-m using an electron probe. (b) Results of long time self-correcting i.e. using YN411-m as the standard and samples simultaneously show the homogeneity of YN411-m. The different symbols represent four measurement sessions. Error bars for single analyses are internal precision (1 s). The grey area represents external precision (2SD) of 35 measurements. | ||
3.2. Sulfur isotopic compositions of pressed pellet and melted glass
Solution MC-ICP-MS and GS-MS were used to determine the sulfur isotope composition of the aforementioned samples (Table 2). The good uniformity of results among MC-ICP-MS, GS-MS, and reported values indicated that results obtained in this study were reliable. The former was carried out at the State Key Laboratory of Continental Dynamics using the same MC-ICP-MS (Nu 1700) equipment as in situ analysis. Solution MC-ICP-MS is a relatively mature technology for sulfur isotope measurement and showed good accuracy and reproducibility during multiple long experiments (Table 2). The δ34S result for in-house standard SAlfa (10 ppm (NH4)2SO4 solution), corrected by in-house standard Spex (10 ppm (NH4)2SO4 solution), from 141 data points of 27 experiments was δSpex34SAlfa = −11.77 ± 0.11‰ (2SD), demonstrating excellent reproducibility. The sulfur isotope composition of Spex (δ34SVCDT = 14.7 ± 0.1‰) was calibrated by IAEA-S-1 using MC-ICP-MS in multiple experiments. The δ34S of sulfur-bearing materials was tested relative to Spex and the error propagation from Spex was calculated by the formula mentioned in Section 2.4.Concentration and acidity effects are shown in Fig. 4. The 10 ppm SSpex solution and SAlfa solutions with diverse concentrations, ranging from 6.5 ppm to 20 ppm, were set as standard and samples respectively, and the δ34S values increased as the concentration of SAlfa increased (Fig. 4a). The values can be accurate within the error range (0.11‰) if the concentration difference was kept within 20%. The signal intensity of 10 ppm SAlfa solution in pure water and in 0.5% HNO3 to 2.5% HNO3 increased as the acidity increased (Fig. 4b). SSpex in pure water was set as the standard, and SAlfa in 0.5% HNO3 to 2.5% HNO3 was set as samples, respectively, causing the precision and accuracy to become worse than those of acidity match analysis (Fig. 4c). Both concentration and acidity effects could be explained as matrix effects, for which aerosols with different concentrations of sulfur and nitrate will affect the vaporization and ionization position, and the vaporization and ionization efficiency of sulfur in the plasma, resulting in instrument-induced isotopic fractionation.2,13,14
 | ||
| Fig. 4 Concentration and acidity effects of solution MC-ICP-MS. (a) The relationship between δ34S values and concentration ratios of samples and standards. (b) The acidity effects on signal intensity; (c) the effects of acidity on precision and accuracy. Error bars for single analyses are internal precision (1 s) and the grey area shows the external precision of multiple tests (2SD). | ||
3.3. Changes in δ34S caused by differences between the standard and samples for in situ analysis
Due to the complex elemental and physical composition of natural sulfide, it was difficult to make the matrix of the standard identical to that of the sample. Therefore, the study of effects ruled by sulfur concentration, chemical components, and physical structure was significant in developing an applicable standard. The following experiments were performed after testing the δ34S values of samples calibrated by the matrix matched standard to avoid instrument induced bias.The first experiment was designed to test the influence of spot size on δ34S values of natural sulfide. As Fig. 5 shows, GC and Cpy-1 were set as the standard and sample respectively. The δ34S values of Cpy-1 showed a tiny increase but still kept the accuracy within the error (δ34S = 4.21 ± 0.23‰) when the spot size was adjusted to 15 μm, 20 μm and 25 μm for the standard and sample simultaneously. This suggested that the spot size only had a slight effect on the δ34S values of sulfide minerals, and different spot sizes can be used to adjust signal intensities in the following experiment.
 | ||
| Fig. 5 The influence of spot size on δ34S values of Cpy-1. Error bars are external precision (2SD) for each experiment. The grey area represents accurate values within error determined by GS-MS and solution MC-ICP-MS in this work. | ||
The effects resulting from concentration differences were tested by NBS123 crystal analysis (Fig. 6). The spot size of the standard (NBS123) was 15 μm, and spot sizes of samples (NBS123) were changed from 10 μm to 25 μm, so varying signal intensity ratios can be obtained. Fig. 6 shows that with the difference in changes of signal intensities between NBS123 standards and NBS123 samples, δ34S values presented a trend similar to solution analysis (Fig. 4a). The measured δ34S values were positively associated with Isample/Istandard ratios and the slope became steeper when the Isample/Istandard ratio was less than 1. Only when Isample/Istandard ratios ranged from 0.7 to 1.5 could the deviation in δ34S values be less than 0.5%. Therefore, an appropriate spot size should be chosen for the standard and samples respectively to make the signal intensities consistent when the differences in sulfur concentration between standards and samples were significant. In general, signal intensity could reach 8–15 V using a spot size of 10 μm for pyrite and 15 μm for chalcopyrite, sphalerite, and Ag2S pressed powder pellets with a laser fluence of 0.5 J cm−2.
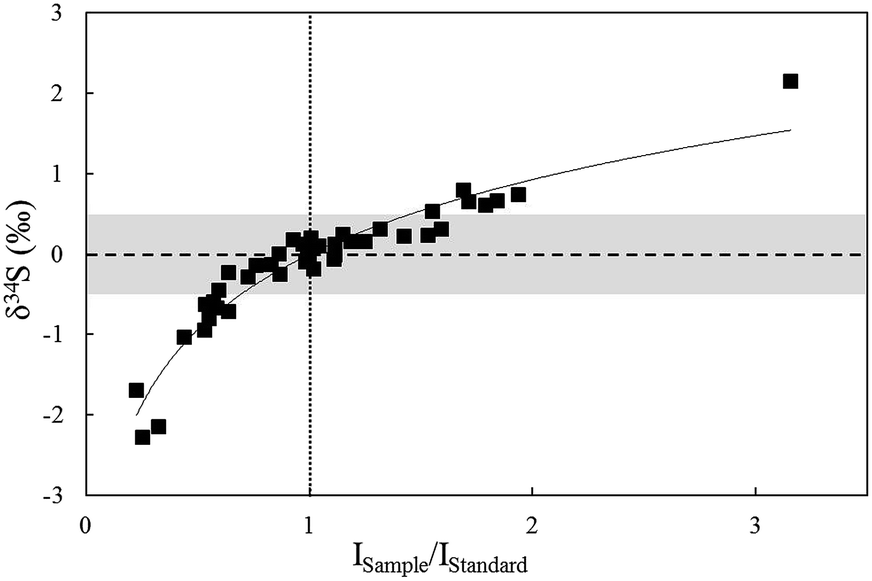 | ||
| Fig. 6 The relationship between δ34S values and concentration ratios of standards and samples for in situ S isotope ratio measurements by analysis of NBS123 with variant spot size. | ||
Matrix elements can affect vaporization and ionization efficiency in the plasma2,13 and may have their own unique influences on ionization conditions for sulfur isotopes, resulting in poor accuracy when using non-matrix-matched calibration. In this paper, research focused on Zn, which was studied in comparison with Cu and Fe as matrix elements, to explain the problems when NBS123 (ZnS) is used as a standard to calibrate other sulfides. It is well known that the interferences of 64Zn2+, 66Zn2+, and 68Zn2+, on 32S, 33S, and 34S (m/Δm = 4263, 3907, 6233, respectively) are difficult to resolve with the mass resolution currently available. Bendall (2006) found that the intensity of Zn2+ was less than 0.2% of that of the S peaks, meaning that its effects could be ignored using a New Wave LUV 213 laser coupled with a Neptune MC-ICP-MS.22 Under the same conditions with fsLA-MC-ICP-MS, including RF power (1300 W), cooling and auxiliary gas flow rate, Zn2+ productivity was tested by solution MC-ICP-MS. A method for sulfur measurement was carried out to test pseudo-S signals of a solution containing 10 ppm Zn, and the intensity of 64Zn2+ was 1.5‰ of the S peaks for a 10 ppm sulfur solution. This indicated that Zn2+ would have a large influence on δ34S values when the matrix contained an equal concentration of Zn and S. Fig. 7 shows the deviation of δ34S, which was less than 0.3‰ resulting from Cu and Fe, and reached up to 0.7‰ resulting from Zn when the solution contained an equal concentration of matrix elements and sulfur. Therefore, it was better to use matrix-matched standards when calibrating sphalerite samples.
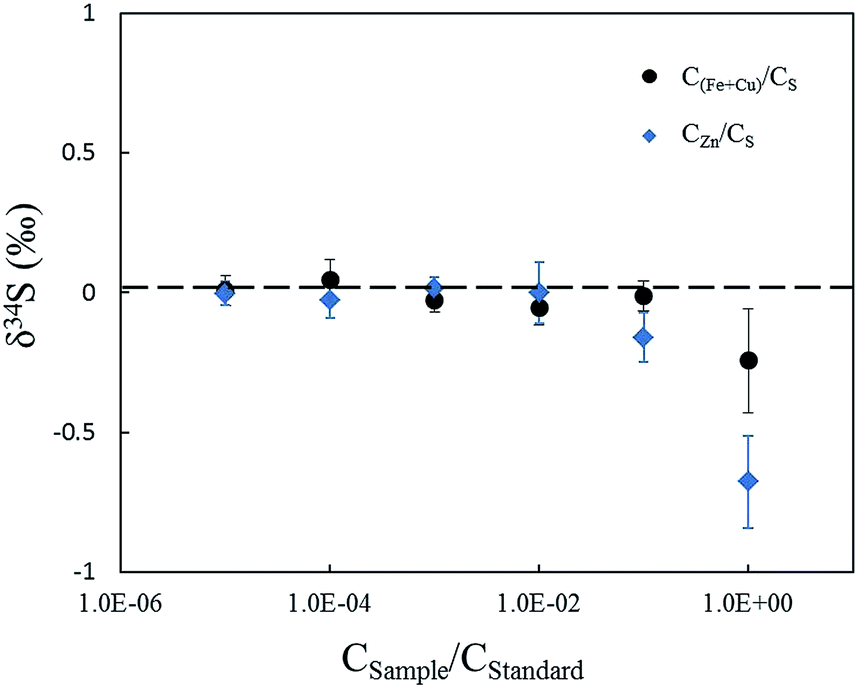 | ||
| Fig. 7 δ 34S results of solutions with different matrix types and concentrations. The abscissa represents matrix-to-sulfur concentration ratios. Zn element shows a greater effect than Cu and Fe elements. | ||
A difference in physical composition exists between synthetic standards and natural sulfides and it should be verified whether this is a reason for matrix effects. The δ34S of powder pressed pellets and natural sulfide crystals with the same chemical compositions were tested and compared using fsLA-MC-ICP-MS. Under the same laser ablation and mass spectrometry conditions, the spot size was 20 μm and the laser fluence was 0.4 J cm−2, the δ34S value of NBS123 powder pressed pellets measured by standard-sample bracketing, using the NBS123 mineral crystals as the bracketing standard, was −0.03 ± 0.22‰. The δ34SVCDT value of the GC (chalcopyrite) powder pressed pellet, using Cpy-1 (chalcopyrite) mineral crystals as the bracketing standard, was −0.50 ± 0.18‰, which was in agreement with the value measured by solution MC-ICP-MS and GS-MS and within the range of experimental error (−0.71 ± 0.20‰, Table 3). These results illustrated that the physical structure would not cause matrix effects. Previous studies confirmed that the initial aerosol particles ablated by femtosecond and nanosecond laser had different properties, including the difference in morphology, size and elemental composition,27,30,31 as well as different transfer and ionization efficiencies.40,41 However, aerosol particles are similar for different materials, such as metals, nonmetals, and sulfides, when generated by a femtosecond laser.30 Therefore, it can be inferred that femtosecond laser-induced aerosol sulfide particles from pressed powder and crystal with the same chemical compositions are uniform and would not cause matrix effects.
| Sample type | Sample | Standard | This work | Ref. | Laser parameters | |||
|---|---|---|---|---|---|---|---|---|
| δ 34SVCDT ± 2SD‰ | δ 34SVCDT ± 2SD‰ | Fluence (J cm−2) and spot size (μm) | ||||||
| fsLA-MC-ICP-MS | Bulk analysis | |||||||
| Sample | Standard | |||||||
| a Results measured by solution MC-ICP-MS. b Results reported in the previous paper using GS-MS.19 c Comprehensive results measured by solution MC-ICP-MS and GS-MS. | ||||||||
| Pressed powder pellets | IAEA-S-2 | IAEA-S-1 | 22.3 ± 0.3, n = 12 | 22.25 ± 0.21a | 0.3 | 15 | 0.3 | 15 |
| IAEA-S-3 | IAEA-S-1 | −32.3 ± 0.4, n = 16 | −32.28 ± 0.26a | 0.3 | 15 | 0.3 | 15 | |
| MASS-1 | IAEA-S-1 | 5.5 ± 0.5, n = 6 | 5.60 ± 0.56b | 0.3 | 15 | 0.3 | 15 | |
| Particles | NBS123 | IAEA-S-1 | 17.8 ± 0.4, n = 4 | 17.75 ± 0.17a | 3 | 15 | 3 | 15 |
| NBS123 | YN411 m | 17.7 ± 0.3‰, n = 5 | 3 | 20 | 3 | 20 | ||
| GC | NBS123 | −0.70 ± 0.2, n = 5 | −0.71 ± 0.20c | 3 | 25 | 3 | 20 | |
| Cpy-1 | GC | 4.4 ± 0.3, n = 8 | 4.21 ± 0.23c | 3 | 20 | 3 | 20 | |
| Sulfide melt | YN411-m | GC | 0.4 ± 0.2, n = 3 | 0.5 ± 0.26c | 3 | 15 | 3 | 20 |
| YN411-m | NBS123 | 0.6 ± 0.3, n = 19 | 3 | 20 | 3 | 20 | ||
The elemental composition and physical structure of chalcopyrite glass (YN411-m) is similar to that of natural chalcopyrite, as demonstrated by back-scattered electron imaging and major element analysis, while the biggest dissimilitude between melted glass and natural minerals was that the crystal structure of the former was predominantly broken by the procedure of fusion and quick solidification. An experiment was performed to determine the δ34S value of GC (natural chalcopyrite) using Cpy-1 (natural chalcopyrite), YN411-m (chalcopyrite glass) and IAEA-S-1 (Ag2S) as the bracketing standards with the spot size of 20 μm and laser fluence of 0.4 J cm−2. As shown in Fig. 8, the δ34S value of GC is −0.89 ± 0.56‰, within the measurement uncertainties, when the standard and sample matrix was natural chalcopyrite. Meanwhile, the δ34S value of GC was −1.6 ± 0.30‰ with YN411-m as the standard and 0.80 ± 0.26‰ with IAEA-S-1 as the standard, 0.7% lighter and 1.6% heavier, respectively, than the results of matrix matched analysis. These values suggested that differences in the crystal structure would cause matrix effects, but its impact was smaller than non-element matching powder pressed pellets (IAEA). According to the process used, in contrast to powder pressed pellets, melted glass was easy to preserve and polish, convenient to use, and not easily contaminated.
 | ||
| Fig. 8 δ 34SVCDT values of GC using Cpy-1, YN411-m, and IAEA-S-1 as bracketing standards. The grey area represents the external precision (2SD) of the measurement using natural chalcopyrite (Cpy-1 and GC) as the standard and sample. | ||
3.4. δ 34SVCDT values of prepared sulfur-bearing materials measured by fsLA-MC-ICP-MS
The reliability of measurements can be tested by comparison of measured values with a reference, as shown in Tables 2 and 3. IAEA-S-1 was set as the standard to calibrate IAEA-S-2, IAEA-S-3 and pressed powder of NBS123 with a spot size of 15 μm and laser fluence of 0.5 J cm−2 in one measurement to test δ34S and δ33S. The isotope plot of δ34S and δ33S (Fig. 9) had a slope of 0.518, a value similar to the mass-dependent fractionation line, which had a slope of 0.515,20,42 indicating an appropriate result. | ||
| Fig. 9 A plot of δ34S and δ33S for the sulfide reference material by laser ablation. IAEA-S-1 was set as the standard to calibrate IAEA-S-2, IAEA-S-3 and NBS123 pressed powder. | ||
A reported study suggested that particle evaporation and phase explosion are the major mechanisms of ablation in fs-LA, and that thermal effects are negligible at low laser fluence due to fast energy deposition and the formation of plasma after the laser pulse ends.30,31 Therefore, thermal effects in various samples are not the reason for matrix effects. Taking into account the above contrasting experiments, the concentration, elemental composition, and crystal structure were the reasons for the shifted values for non-matrix matching experiments. In addition, it was observed that carrier gas flow rate, laser fluence and spot size might result in sulfur isotope fractionation even though matrixes of the standard and sample were matched well. In particular, the results were accurate when the Ar and He flow rates were kept within a range from 0.72 L min−1 to 0.82 L min−1 and 0.65 L min−1 to 0.8 L min−1 respectively. The effects of spot size and laser fluence on pressed powder (IAEA) were larger than on natural crystals. Only laser fluence ranging from 0.3 to 1.5 J cm−2 can lead to accurate ratios when testing IAEA series, and the values (δ34S) increased with laser fluence. But for chalcopyrite crystals, the values can be accurate even with a fluence of 3 J cm−2. We believed that the impacts of laser fluence and spot size may result from different particles produced by femtosecond laser and transfer efficiency of the cell we used in this research. The way to avoid value deviation in the matrix matched test was to choose suitable Ar and He flow rates, low laser fluence, and suitable spot size according to the sulfur concentration of materials to keep high transfer efficiency. For the matrix unmatched test, how particles with different matrixes work in ICP is unclear, but we can test the degree of deviation from the true value for different matrixes and calibrate the results by the laws of laser fluence and spot size. Table 3 shows δ34SVCDT of aforementioned standards using fsLA-MC-ICP-MS, for which accurate values were obtained with matrix matching and unmatching using same or different operation parameters. When NBS123 and YN411-m are set as the sample and standard, it is easy to obtain correct values with the same laser parameters. For instance, considering the value in the fifth line of Table 3, the δ34SVCDT value of NBS123 was 17.7 ± 0.28‰ using YN411-m as the bracketing standard with parameters of 0.5 J cm−2 laser fluence, and 20 μm spot size. However, when IAEA is set as the standard to calibrate other materials, the matrix effects were dramatic, so the signal intensity and laser parameters should be coordinated. As shown in line 4 of Table 3, when the signal difference between NBS123 and IAEA-S-1 was 40%, the δ34SVCDT value of NBS123 against IAEA-S-1 was 17.8 ± 0.38‰ with parameters of 3 J cm−2 laser fluence, and 15 μm spot size. For the materials with similar signal intensities, different spot sizes were used to obtain correct values. The δ34SVCDT value of GC in the sixth line of Table 3, for example, was correct and obtained with 25 μm spot size for GC and 20 μm spot size for NBS123.
4. Conclusion
The fsLA-MC-ICP-MS technique showed obvious advantages for improving the sensitivity and spatial resolution in in situ sulfur isotope analysis. A signal of 8–20 V could be achieved with a spot size of 10–20 μm. A series of sulfur-bearing materials with different matrixes were prepared as in-house standards by powder-pressing and sulfide-melting under high temperatures with a protective N2 atmosphere. Back-scattered electron images and major element analysis in map analysis mode of YN411-m indicated that the concentration of sulfur in an ablation area was homogeneous. Multiple experiments were conducted for homogeneity tests of YN411-m by fsLA-MC-ICP-MS and the external precision was 0.28‰ (n = 35). We conducted a series of experiments to study the matrix effects of these sulfur-bearing materials and found that differences in concentration, elemental composition, and crystal structure between standards and samples caused the shifted values. Furthermore, differences in elemental composition made values worse than differences in the crystal structure, resulting in a value much closer to the reference when using YN411-m as the bracketing standard to calibrate chalcopyrite instead of IAEA-S-1. Considering the process used, melted glass was easy to preserve and polish, convenient to use, and not easily contaminated; more importantly, it had less matrix effects than the IAEA-S-1 powder pressed pellet and was, therefore, more suitable for in situ sulfur isotope analysis.Acknowledgements
Financial support for this work was jointly provided by the National Natural Science Foundation of China (Grant No. 41427804, 41421002, 41373004), the Program for Changjiang Scholars and Innovative Research Team in University (Grant No. IRT1281), and the MOST Research Foundation from the State Key Laboratory of Continental Dynamics (BJ08132-1).References
- H. G. Thode, Geochim. Cosmochim. Acta, 1961, 25, 150–174 CrossRef.
- P. R. Craddock, O. J. Rouxel, L. A. Ball and W. Bach, Chem. Geol., 2008, 253, 102–113, DOI:10.1016/j.chemgeo.2008.04.017.
- R. R. Seal, Rev. Mineral. Geochem., 2006, 61, 633–677, DOI:10.2138/rmg.2006.61.12.
- J. Hoefs, Stable Isotope Geochemistry, Springer Verlag, 3rd edn, 1987 Search PubMed.
- Y. Özen and F. Arık, Ore Geol. Rev., 2015, 70, 262–280, DOI:10.1016/j.oregeorev.2015.04.001.
- N. J. Hill, G. R. T. Jenkin, A. J. Boyce, C. J. S. Sangster, D. J. Catterall, D. A. Holwell, J. Naden and C. M. Rice, How the Neoproterozoic S-isotope record illuminates the genesis of vein gold systems: an example from the Dalradian Supergroup in Scotland, Geological Society, Special Publications, London, 2013, vol. 393, pp. 213–247, DOI:10.1144/sp393.9.
- M. Chen, Z. Zhang, M. Santosh, Y. Dang and W. Zhang, J. Asian Earth. Sci., 2015, 103, 115–128, DOI:10.1016/j.jseaes.2014.08.022.
- M. C. Stam, P. R. D. Mason, C. Pallud and P. Van Cappellen, Chem. Geol., 2010, 278, 23–30, DOI:10.1016/j.chemgeo.2010.08.006.
- R. Tostevin, A. V. Turchyn, J. Farquhar, D. T. Johnston, D. L. Eldridge, J. K. B. Bishop and M. McIlvin, Earth Planet. Sci. Lett., 2014, 396, 14–21, DOI:10.1016/j.epsl.2014.03.057.
- F. Fourel, F. Martineau, M. Seris and C. Lecuyer, Geostand. Geoanal. Res., 2015, 39, 47–53, DOI:10.1111/j.1751-908x.2014.00297.x.
- H. P. Qi and T. B. Coplen, Chem. Geol., 2003, 199, 183–187, DOI:10.1016/s0009-2541(03)00075-5.
- R. Clough, P. Evans, T. Catterick and E. H. Evans, Anal. Chem., 2006, 78, 6126–6132, DOI:10.1021/ac060875h.
- M. J. Pribil, W. I. Ridley and P. Emsbo, Chem. Geol., 2015, 412, 99–106, DOI:10.1016/j.chemgeo.2015.07.014.
- G. Paris, A. L. Sessions, A. V. Subhas and J. F. Adkins, Chem. Geol., 2013, 345, 50–61, DOI:10.1016/j.chemgeo.2013.02.022.
- A. Das, C.-H. Chung, C.-F. You and M.-L. Shen, J. Anal. At. Spectrom., 2012, 27, 2088, 10.1039/c2ja30189j.
- S. Ono, B. Wing, D. Rumble and J. Farquhar, Chem. Geol., 2006, 225, 30–39, DOI:10.1016/j.chemgeo.2005.08.005.
- T. Ushikubo, K. H. Williford, J. Farquhar, D. T. Johnston, M. J. Van Kranendonk and J. W. Valley, Chem. Geol., 2014, 383, 86–99, DOI:10.1016/j.chemgeo.2014.06.006.
- R. Kozdon, N. T. Kita, J. M. Huberty, J. H. Fournelle, C. A. Johnson and J. W. Valley, Chem. Geol., 2010, 275, 243–253, DOI:10.1016/j.chemgeo.2010.05.015.
- J. Fu, Z. Hu, W. Zhang, L. Yang, Y. Liu, M. Li, K. Zong, S. Gao and S. Hu, Anal. Chim. Acta, 2016, 911, 14–26, DOI:10.1016/j.aca.2016.01.026.
- B. Bühn, R. V. Santos, M. A. Dardenne and C. G. de Oliveira, Chem. Geol., 2012, 312–313, 163–176, DOI:10.1016/j.chemgeo.2012.04.003.
- P. R. D. Mason, J. Košler, J. C. M. de Hoog, P. J. Sylvester and S. Meffan-Main, J. Anal. At. Spectrom., 2006, 21, 177–186, 10.1039/b510883g.
- C. Bendall, Y. Lahaye, J. Fiebig, S. Weyer and G. P. Brey, Appl. Geochem., 2006, 21, 782–787, DOI:10.1016/j.apgeochem.2006.02.012.
- D. Garbe-Schönberg and S. Müller, J. Anal. At. Spectrom., 2014, 29, 990, 10.1039/c4ja00007b.
- S. A. Wilson, W. I. Ridley and A. E. Koenig, J. Anal. At. Spectrom., 2002, 17, 406–409, 10.1039/b108787h.
- A. Fiege, F. Holtz, N. Shimizu, C. W. Mandeville, H. Behrens and J. L. Knipping, Geochim. Cosmochim. Acta, 2014, 142, 501–521, DOI:10.1016/j.gca.2014.07.015.
- Y. Kajiwara and H. R. Krouse, Can. J. Earth Sci., 1971, 8, 1397–1408 CrossRef CAS.
- S. E. Gilbert, L. V. Danyushevsky, T. Rodemann, N. Shimizu, A. Gurenko, S. Meffre, H. Thomas, R. R. Large and D. Death, J. Anal. At. Spectrom., 2014, 29, 1042–1051, 10.1039/c4ja00011k.
- D. E. Crowe and R. G. Vaughan, Am. Mineral., 1996, 81, 187–193, DOI:10.10/00on.
- X. P. Bian, T. Yang, A. J. Lin and S. Y. Jiang, Talanta, 2015, 132, 8–14, DOI:10.1016/j.talanta.2014.08.053.
- R. Glaus, R. Kaegi, F. Krumeich and D. Günther, Spectrochim. Acta, Part B, 2010, 65, 812–822, DOI:10.1016/j.sab.2010.07.005.
- M. E. Shaheen, J. E. Gagnon and B. J. Fryer, Spectrochim. Acta, Part B, 2015, 107, 97–109, DOI:10.1016/j.sab.2015.02.010.
- C. C. Garcia, H. Lindner, A. von Bohlen, C. Vadla and K. Niemax, J. Anal. At. Spectrom., 2008, 23, 470–478, 10.1039/b718845e.
- G. Steinhoefel, I. Horn and F. von Blanckenburg, Chem. Geol., 2009, 268, 67–73, DOI:10.1016/j.chemgeo.2009.07.010.
- M. Lazarov and I. Horn, Spectrochim. Acta, Part B, 2015, 111, 64–73, DOI:10.1016/j.sab.2015.06.013.
- H. Yuan, K. Chen, B. ZhiAn, C. Zong, M. Dai, C. Fan and C. Yin, Chin. Sci. Bull., 2013, 58, 3914–3921, DOI:10.1007/s11434-013-5804-4.
- K. Ikehata, K. Notsu and T. Hirata, J. Anal. At. Spectrom., 2008, 23, 1003, 10.1039/b801044g.
- T. Ding, S. Valkiers, H. Kipphardt, P. De Bievre, P. D. P. Taylor, R. Gonfiantini and R. Krouse, Geochim. Cosmochim. Acta, 2001, 65, 2433–2437 CrossRef CAS.
- S.-H. Han, Z. Varga, J. Krajkó, M. Wallenius, K. Song and K. Mayer, J. Anal. At. Spectrom., 2013, 28, 1919, 10.1039/c3ja50231g.
- S. Ono, N. S. Keller, O. Rouxel and J. C. Alt, Geochim. Cosmochim. Acta, 2012, 87, 323–340, DOI:10.1016/j.gca.2012.04.016.
- S. E. Gilbert, L. V. Danyushevsky, K. Goemann and D. Death, J. Anal. At. Spectrom., 2014, 29, 1024–1033, 10.1039/c4ja00012a.
- P. Sylvester, Laser-Ablation-ICPMS in the Earth Sciences, Canadian Mineral Association, 2001 Search PubMed.
- D. T. Johnston, Earth-Sci. Rev., 2011, 106, 161–183, DOI:10.1016/j.earscirev.2011.02.003.
| This journal is © The Royal Society of Chemistry 2017 |