
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceValve interstitial cell contractile strength and metabolic state are dependent on its shape†
Ngoc Thien
Lam
a,
Timothy J.
Muldoon
b,
Kyle P.
Quinn
b,
Narasimhan
Rajaram
b and
Kartik
Balachandran
*b
aCell and Molecular Biology Program, University of Arkansas, Fayetteville, AR 72701, USA
bDepartment of Biomedical Engineering, University of Arkansas, 122 John A. White Jr. Engineering Hall, Fayetteville, AR 72701, USA. E-mail: kbalacha@uark.edu; Fax: +1 479 575 5319; Tel: +1 479 575 3376
First published on 15th September 2016
Abstract
The role of valvular interstitial cell (VIC) architecture in regulating cardiac valve function and pathology is not well understood. VICs are known to be more elongated in a hypertensive environment compared to those in a normotensive environment. We have previously reported that valve tissues cultured under hypertensive conditions are prone to acute pathological alterations in cell phenotype and contractility. We therefore aimed to rigorously study the relationship between VIC shape, contractile output and other functional indicators of VIC pathology. We developed an in vitro model to engineer VICs to take on the same shapes as those seen in normal and hypertensive conditions. VICs with longer cellular and nuclear shapes, as seen in hypertensive conditions, had greater contractile response to endothelin-1 that correlated with increased anisotropy of the actin architecture. These elongated VICs also demonstrated altered cell metabolism through a decreased optical redox ratio, which coincided with increased cellular proliferation. In the presence of actin polymerization inhibitor, however, these functional responses were significantly reduced, suggesting the important role of cytoskeletal actin organization in regulating cellular responses to abnormal shape. Overall, these results demonstrate the relationship between cell shape, cytoskeletal and nuclear organization, with functional output including contractility, metabolism, and proliferation.
Insight, innovation, integrationAbnormal hemodynamics experienced by the heart valve cells are associated with valvulopathy. This altered mechanical environment also leads to modulation in valve cellular shape and architecture. We hypothesized that alteration of cell shape may cause changes in cellular function that eventually prevents the proper function of the heart valve. To rigorously study this, we engineered valve interstitial cells with varying width-to-length ratios and quantified their contractility as well as metabolic activity. We observed an increase in contractile strength and decrease in metabolic redox ratio as cells became more elongated and their actin architecture became more aligned. These results suggested that valve interstitial cells modulated their contractile and metabolic function depending on their shape and arrangement of cytoskeletal elements. |
Introduction
Valvular heart disease accounts for approximately 3% of all cardiovascular pathologies and is associated with abnormal valve structure and function.1In vivo, the valves maintain their integrity and functionality by a complex interaction between the valve cusps, cells, their relative structures and the surrounding hemodynamic and mechanical environment.2 Significant prior work has focused on the role of the mechanical environment in potentiating cellular alterations and subsequent progression toward disease.3 However, the role of cellular shape and architecture in regulating the overall balance between health and disease in the context of the valve is not well understood.The cells and nuclei of the valve interstitial cells (VICs) within aortic valve cusps respond very rapidly to mechanical stimuli by dynamic elongation and deformation. For instance, VIC nuclear width-to-length aspect ratio (AR) almost instantaneously increased from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 1:5 when transvalvular pressure was increased from 0 to 90 mm Hg.4 This elongation was correlated with significant alterations in collagen fiber architecture.4 Similarly, cell and nuclear ARs significantly increased from 1:5 to 1:7 and 1:6 to 1:9, respectively, when aortic leaflet tissues were subjected to increasing strains from 10 to 20%.5 These aforementioned strains represented a transition from normotension to hypertension.6 Furthermore, transition from the systolic tension-free state to fully-closed diastolic tension resulted in a 1.5-fold increase in nuclear elongation of the VICs within the mitral valve.7 These observations thus suggest the possibility for a role for altered VIC shape and architecture in directly modulating cellular biology and function.
:2 to 1:5 when transvalvular pressure was increased from 0 to 90 mm Hg.4 This elongation was correlated with significant alterations in collagen fiber architecture.4 Similarly, cell and nuclear ARs significantly increased from 1:5 to 1:7 and 1:6 to 1:9, respectively, when aortic leaflet tissues were subjected to increasing strains from 10 to 20%.5 These aforementioned strains represented a transition from normotension to hypertension.6 Furthermore, transition from the systolic tension-free state to fully-closed diastolic tension resulted in a 1.5-fold increase in nuclear elongation of the VICs within the mitral valve.7 These observations thus suggest the possibility for a role for altered VIC shape and architecture in directly modulating cellular biology and function.
Although studies on the role of cell shape in modulating VIC behavior are few in number, there are, several reports in the literature on the effects of constraining cell shape in influencing function and biology in other cell types. The AR of ventricular myocytes decreased from 1:7 to 1:5 during concentric hypertrophy, while it increased up to 1:11 in eccentric hypertrophy.8 These alterations impaired cytoskeletal contractility and were therefore associated with heart failure.8 Human mesenchymal stem cells differentiated into chondrocytes when they were cultured on micropatterned substrates and exposed to transforming growth factor (TGF)-β, while they became smooth muscle cells if they were unpatterned and exposed to TGF-β.9 Additionally, the orientation of smooth muscle cell F-actin as well as nuclear elongation and chromatin density was found to be significantly altered as cell AR increased.10 Elongated smooth muscle cells also showed higher contractile strength in relation to its basal tone after stimulation with Endothelin-1 (ET-1).11 These studies clearly demonstrated that changes in cell shape can potentiate functional changes.
We hypothesized that by engineering the shape of single VICs to acquire the AR of cells seen in normal and pathological conditions, we could induce changes in cell architecture, subsequently potentiating acute alterations in cell function, specifically cell contractility, metabolism, proliferation and activation. Our hypothesis was rigorously tested using an engineered single VIC model with cells forced to adopt shape of 1:3, 1:5 and 1:7 aspect ratios with the surface area of the patterns maintained constant. We demonstrated that these aspect ratios corresponded to the steady-state shapes adopted by VICs when cyclically strained to 0%, 10% and 20%, respectively. Cytochalasin D, an inhibitor of actin polymerization, was used to study the causal relationship between changes in cell shape and changes in intracellular structure/function. Cytoskeletal and nuclear architecture were quantified and correlated with contraction measured via traction force microscopy, metabolic cofactor autofluorescence imaging, immunostaining and western blotting for detection of cellular proliferation and activation. We report here that cell shape significantly influenced cytoskeletal and nuclear architecture. Elongated cells generated greater active traction stresses, suggesting a greater capacity for mobility. These elongated cells also had a lower redox ratio, higher expression of activation marker α-SMA and proliferation markers Ki-67. These responses were significantly decreased with the addition of cytochalasin D. These results suggested that altered actin cytoskeletal architecture as a result of altered cell shape caused significant changes in cell function. These cells appeared more primed for increased contractility, proliferation and pathological activation.
Results
Engineering valve interstitial cell shape
Previous studies revealed that cells adopt different spreading areas depending on the stiffness of the underlying substrate.12 We therefore first quantified the average spreading area of isolated VICs on polydimethylsiloxane (PDMS) coated coverslips (Fig. S1A, ESI†), and showed that the cell spreading area had a mean of 1700 μm2. Reports in the literature have indicated that VICs adopt a wide variety of morphological shapes,13 but were typically confined to aspect ratios (AR) of 1:3 to 1:7 depending on the magnitude of cyclic hemodynamic pressure.5 We validated this same range of ARs in an in vitro VIC monolayer cyclic strain model (Fig. S1C, ESI†) and selected width-to-length ARs of 1:3 (23.8 × 71.4 μm), 1:5 (18.4 × 92.2 μm) and 1:7 (15.58 × 109.09 μm), representing the mechanical strain experienced during static, healthy and hypertensive hemodynamic conditions, respectively.5 Fibronectin was microcontact printed on PDMS-coated coverslips at these same aspect ratios and VICs were seeded at 1000 cells per cm2 coverslip area. Cells self-assembled and assumed the rectangular shape of these three aspect ratios (Fig. 1 A–C). Further details regarding this single cell model are included in the ESI.†
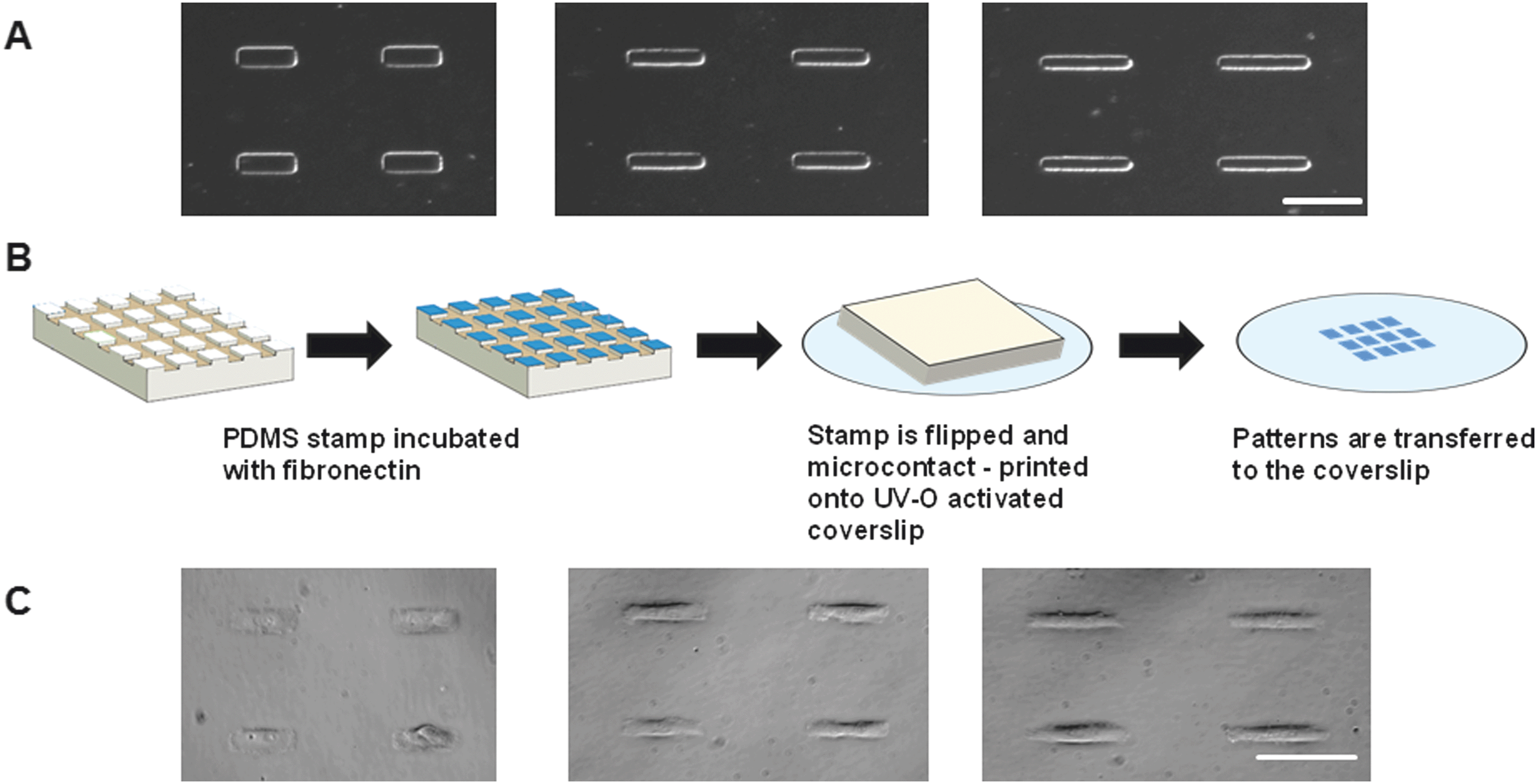 | ||
| Fig. 1 Single-cell culture model. (A) Photomasks of single-cell grid arrays with differing width-length aspect ratios. (Scale bar = 100 μm) (B) Schematic depicting microcontact printing protocol. (C) Single cell culture images (scale bar = 100 μm). | ||
Actin orientation and nuclear architecture and orientation varied as a function of cellular shape
Previous studies have reported that cellular structure was altered due to changes in the external mechanical boundary conditions.14 We therefore evaluated whether altered VIC shape resulted in changes in cytoskeletal and nuclear architecture. Phalloidin staining of F-actin, showed that the filaments became more prominent and aligned along the longitudinal direction of the cell as AR increased (Fig. 2A). Quantification of actin alignment using a previously developed technique14a revealed alignment to be statistically higher (p < 0.05) at an AR of 1:7 compared to 1:5 and 1:3 (Fig. 2C). In the presence of cytochalasin D, cells did not exhibit prominent actin stress fibers (Fig. 2A). Cells treated with cytochalasin D at 1:7 had significantly (p < 0.05) more aligned actin filaments compared to 1:3 and 1:5 cells. Overall, the actin orientation parameter was significantly reduced (p < 0.05) when the cells were treated with cytochalasin D.
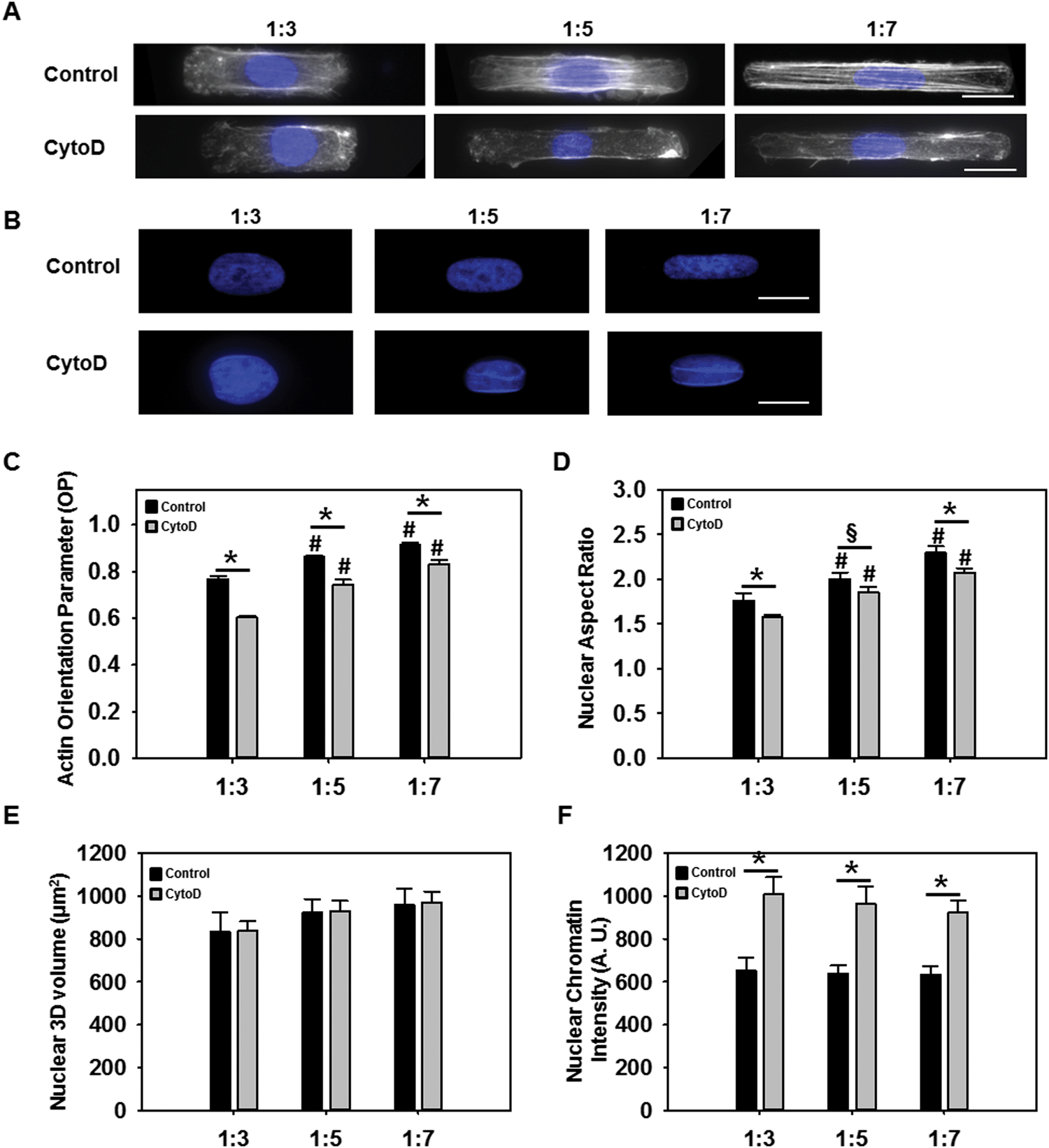 | ||
| Fig. 2 Actin orientation and nuclear morphology analysis of single cells. (A) Single VICs fluorescently stained with Phalloidin (white) and DAPI (blue) (scale bar = 10 μm). (B) Higher magnification DAPI images utilized for nuclear morphology analysis (scale bar = 5 μm) (C) Actin orientation parameter data. (D) Nuclear aspect ratio data. (E) Nuclear 3D volume data. (F) Nuclear chromatin density data. (*p < 0.05; #p < 0.05 with respect to 1:3). | ||
The actin cytoskeleton is thought to be stress-sensitive, allowing the cytoskeleton to detect extrinsic mechanical stimuli and dynamically remodel itself to accommodate the mechanical load.8,15 It has been suggested that extracellular forces are transmitted to the cell nucleus via the cytoskeleton causing substantial deformation in the nucleus which could contribute to changes in chromatin structure and later on transcriptional regulation.15,16 Nuclear staining using DAPI (Fig. 2B) indeed did indicate significantly increased nuclear elongation (p < 0.05) with increasing cellular AR (Fig. 2D). VIC nuclear ARs ranged from 1.7 to 2.2 for cell AR from 1:3 to 1:7, suggesting that the nucleus did not elongate to the same extent as the cell, probably due its higher mechanical rigidity.17 Analysis of nuclear 3D volume (Fig. 2E) showed that as cell AR increased, 3D volume was not significantly altered, suggesting that actin cytoskeletal modulation of nuclear AR occurred without any alteration of the nuclear volume. Average intensity of chromatin was also analyzed from DAPI stained images and revealed significant higher chromatin intensity in the presence of cytochalasin D (p < 0.05) (Fig. 2F).
Elongated VICs generated greater contractile traction
Cells generate tractions on their underlying substrate, that are thought to control cell shape and maintain cellular homeostasis, regulating diverse processes such as motility, differentiation and proliferation.18 As elongated VICs were typically found in higher and elevated mechanical stress environments,5 we hypothesized that VIC elongation would induce a greater capacity for contractile stress generation, that would be dependent on actin organization. This hypothesis was tested via traction force microscopy (TFM), wherein VICs with varying ARs, without and with cytochalasin D, were seeded on polyacrylamide substrates doped with fluorescent beads. Samples were first imaged prior to stimulation and sequentially stimulated with 50 nM of the vasoconstrictor endothelin-1 (ET-1) and a saturating dose of 100 μM of the vasodilator HA-1077 for 5 minutes each. Samples were imaged after each treatment, and traction stresses were calculated from the bead displacement images using the unstimulated image as the reference state.14b As expected, the highest VIC contractile (Fig. 3B) and relaxation (Fig. 3C) tractions were primarily localized to the longitudinal ends of the cells (Fig. 3A). VIC contractile strength, as computed by the traction stress applied by the cell on its substrate due to ET-1, significantly increased with increasing cellular elongation (Fig. 3B). Basal tone, as quantified from the response to HA-1077, was low at 1:3 AR and significantly higher at 1:5 AR (Fig. 3C). However there was no significant difference in basal tone between 1:5 and 1:7 ARs. When the actin filaments were disrupted with cytochalasin D, significantly reduced contractility and basal tone were observed (p < 0.05) (Fig. 3B and C). Taken as a whole, these results suggested that higher alignment and prominence of F-actin stress fibers potentiated by cellular elongation increased the ability of the VICs to generate an active contraction and maintain its basal cellular tone.
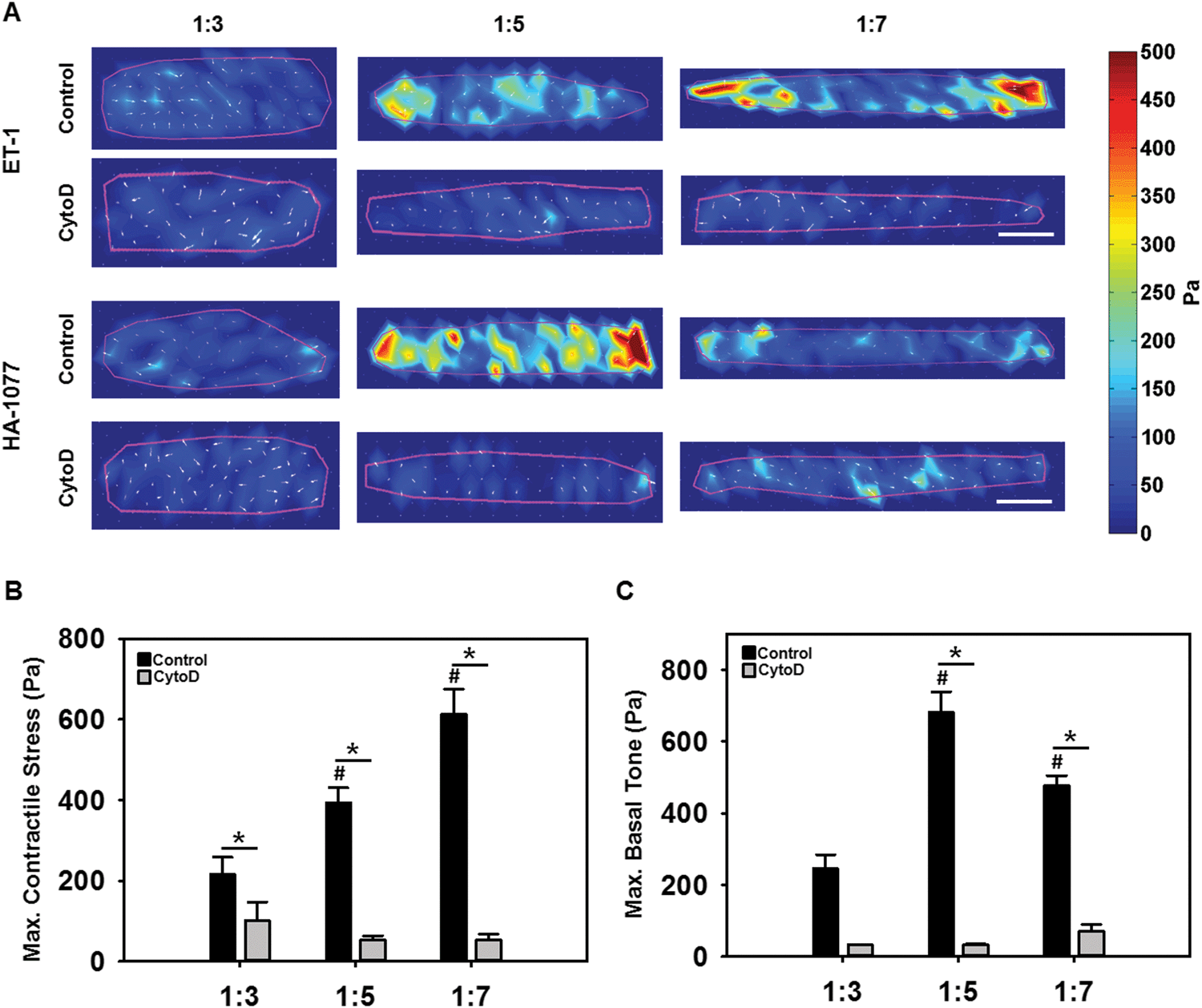 | ||
| Fig. 3 Traction force microscopy data. Representative constrained traction stress color maps representing (A) Peak contractile stress generation due to ET-1 and peak relaxation due to HA-1077 (scale bar = 10 μm). (B) Mean contractile traction due to ET-1. (C) Mean relaxation traction/basal tone due to HA-1077. (*p < 0.05; #p < 0.05 with respect to 1:3). | ||
Elongated VICs exhibited reduced metabolic redox ratio
Reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD) are coenzymes and electron carriers associated with metabolism in all eukaryotic cells. Many studies have demonstrated the potential of using the endogenous fluorescence of NADH and FAD as an indicator of cell metabolism.19 We evaluated whether an optical redox ratio of FAD/(NADH + FAD) fluorescence was altered as a function of cellular AR elongation and whether the presence of an intact actin cytoskeleton affected these responses. To address this question, we collected NADH and FAD fluorescence images and computed optical redox ratios from cells in each of the three (1:3, 1:5 and 1:7) ARs without and with cytochalasin D (Fig. 4A–C).
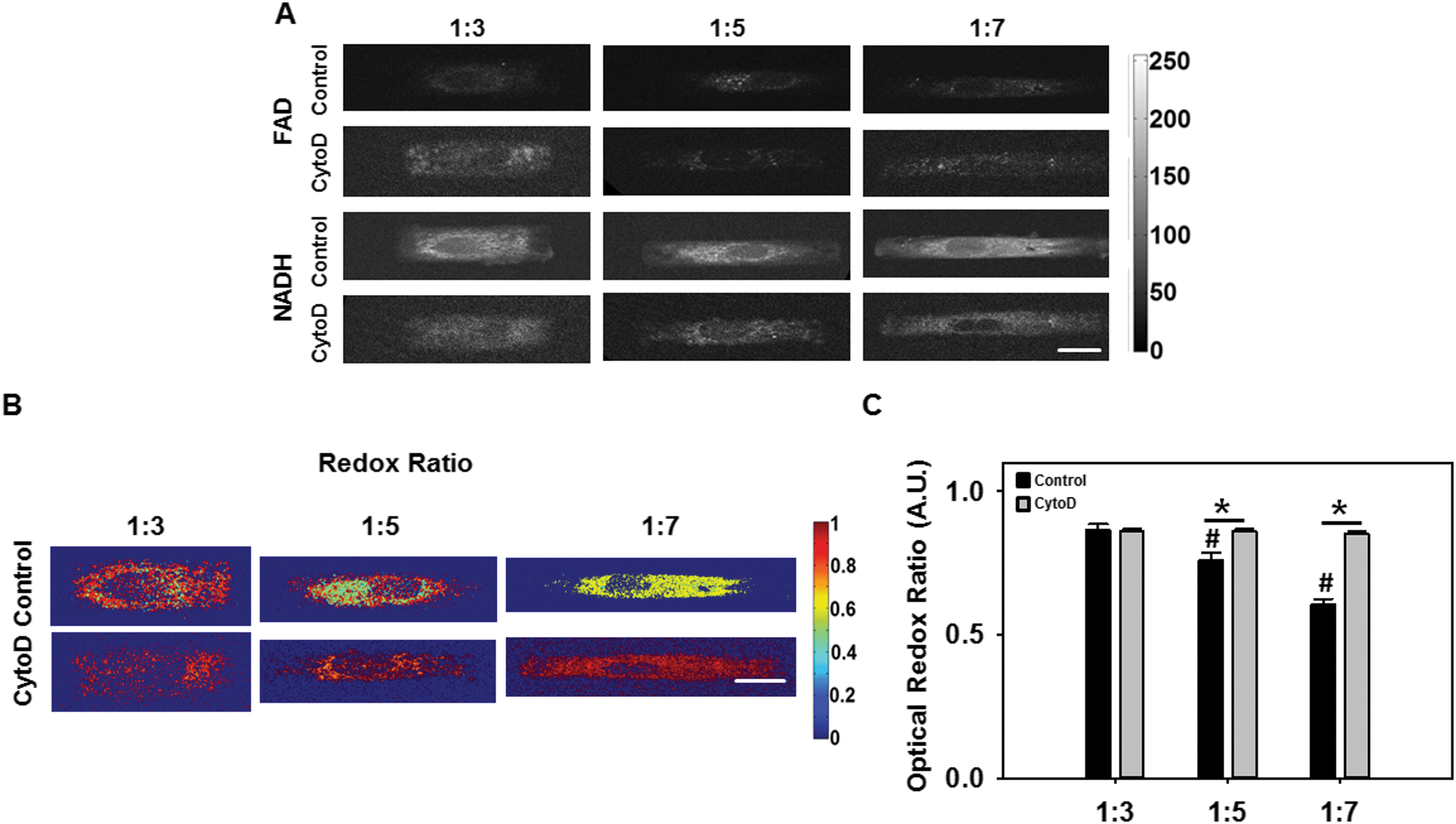 | ||
| Fig. 4 Redox imaging of single cell (A) Representative FAD and NADH fluorescence images (scale bar = 10 μm). (B) Representative color maps of VIC optical redox ratios (scale bar = 10 μm). (C) Mean redox ratio data for all VICs. (*p < 0.05; #p < 0.05 with respect to 1:3). | ||
Our results show that as VIC AR increased, there was a significant decrease (p < 0.05) in redox ratio (Fig. 4B). In the presence of cytochalasin D, redox ratio was significantly higher than VICs without cytochalasin D, and there was no significant difference as a function of cellular AR (p < 0.05). A decrease in the optical redox ratio was observed in mesenchymal stem cells undergoing osteogenic differentiation.20 Additionally, decreased redox ratios have been observed upon an increase in the proliferation of keratinocytes.21 In the context of the current study, our results thus suggest the possibilities that (I) elongated VICs with prominent actin fibers were more likely to be at the onset of the pathologic de-differentiation process, or that (II) elongated VICs were more proliferative, or both.
Elongated cells exhibited higher proliferation capability and were prone to pathological activation
We considered the hypothesis that VICs with longer ARs and lower redox ratios had a higher cell proliferation and activation potential. First, VICs at the three ARs (1:3, 1:5, 1:7) were analyzed using a known indicator of cellular proliferation. Ki-67 expression, as determined via immunostaining (Fig. 5A) and semi-quantitation (Fig. 5B), significantly increased as cell AR increased. Additionally, ERK-1/2 phosphorylation was also increased as cell AR increased (Fig. S3, ESI†). Addition of cytochalasin D significantly reduced the expression of Ki67 in all cases although higher expression was still found at increased ARs. Ki-67 is a nuclear protein that is associated with cell proliferation and these results suggested that VICs possess higher proliferation capability with increasing elongation only if the actin cytoskeleton was intact.
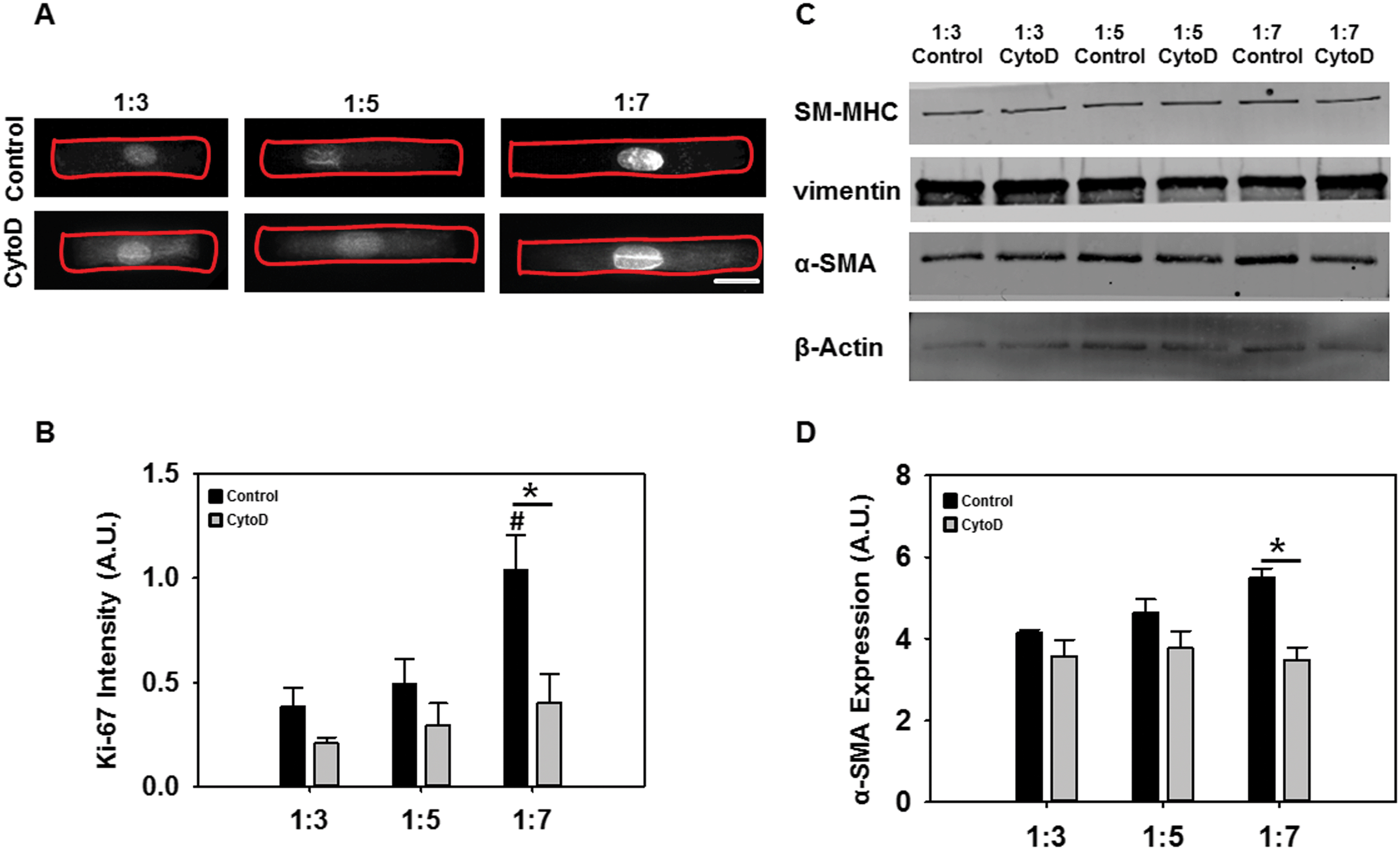 | ||
| Fig. 5 Immunofluorescent and western blotting data showing the proliferation and pathological activation of VICs. (A) Representative immunofluorescently stained VICs for Ki-67 (white) (scale bar = 10 μm). (B) Semi-quantitation of Ki-67 immunostains. (C) Representative western blot scan for VIC phenotype markers. (D) Densitometric analysis of α-SMA western blots. (*p < 0.05; #p < 0.05 with respect to 1:3). | ||
Next, we performed western blotting to detect the expression of VIC phenotype markers α-smooth muscle actin (α-SMA), smooth muscle myosin heavy chain (SM-MHC) and vimentin. α-SMA is a cytoskeletal isoform of actin which is usually found in activated VICs, SM-MHC is a smooth muscle-specific marker, and vimentin is typically an indicator of quiescent, non-activated VICs.22 Additionally, α-SMA-positive activated cells typically display features of myofibroblasts such as increased contraction and prominent stress fibers.14a As expected, cells at the longest ARs had significantly increased α-SMA expression while adding cytochalasin D reduced its expression (Fig. 5C and D). This particular reduction was statistically significant only at the 1:7 aspect ratio (Fig. 5D). There was no significant difference in expression of SM-MHC and vimentin (Fig. 5C). Overall, these results suggested that VIC elongation potentiated stronger proliferative responses than differentiation ones. VICs did demonstrate moderately increased activated myofibroblast phenotype at the highest level of cell elongation, and these responses were more pronounced in the presence of actin cytoskeletal stress fibers.
Discussion
We show in this work that altered actin cytoskeletal organization due to elongated VIC shape was an important factor in regulating VIC acute contractile function, metabolism, proliferation and phenotypic activation. While several other methods exist wherein cellular shape and structure could be modulated while maintaining cell–cell contact,11b,c,23 our single cell model represents the smallest functional unit of the VIC and thus an important first step in understanding the role of VIC shape on its biology and function. Shorter and wider VICs generated less contractile stress and were at a less biosynthetic and proliferative state, while elongated narrower VICs had greater contractile stress generation, more biosynthesis, increased proliferation and α-SMA activation, correlating with greater organization of the actin cytoskeleton. Our results suggest that VICs in a higher pressure environment in vivo, which were more elongated with typical ARs of 1:7,4,5 had a greater potential for increased cellular activity, proliferation and myofibroblast differentiation.
Since the actin cytoskeleton, was responsible for maintaining the cell shape and mechanical resistance to deformation, any change in cell shape was expected to change the actin organization and subsequent modulation of nuclear shape.24 Our results supported the notion that as the VIC elongated, cytoskeletal actin and cell nuclear significantly altered their organization and shape, respectively. Specifically, actin filaments became more prominent with increased anisotropy, while cell nuclei elongated and increased 2D area and AR, while conserving nuclear volume. Others have demonstrated that forces transduced to altered cell architecture could result in significant nuclear deformations.25 The primary mediator for these structural alterations is thought to be the actin filament.26 Previous work had demonstrated that vascular smooth muscle cells (vSMC) became more elongated and expressed higher α-SMA as well as more aligned stress fibers on engineered nanogrooves compared to unpatterned substrates.23 This altered F-actin cytoskeletal remodeling was known to significantly affect vSMC proliferation and migration ability.27 The increase in alignment and organization of actin that we observed in elongated VICs was thus thought to allow for generation of extra tension that might be needed for a functional response or migration.18b The causal relationship between cell shape, actin and nuclear shape was demonstrated when the addition of cytochalasin D to the single cell culture significantly mitigated changes in actin and nuclear structure. Our TFM results further supported our above-mentioned conclusion as elongated 1:7 AR cells which had more prominent actin filaments, generated greater contractile response to ET-1. It should be noted that the increased contractile strength did not result in a significant change in the basal tone of the 1:7 AR cells compared to the 1:5 cells, suggesting that the basal tension in these elongated cells was not increased as a result of shape elongation, but its active contractility was. When the actin filament network was disrupted, cells were no longer able to potently respond to ET-1 as well as HA1077.
The decrease in the optical redox ratio has been used previously to monitor precancerous transformations,28 cell proliferation during wound healing,21 and as an early indicator of cellular differentiation in mesenchymal stem cells.20 Our results show that the redox ratio could serve as an indicator of VIC proliferation and to a lesser extent, phenotypic activation. Redox ratio was significantly lowered in the 1:7 AR cells, which were also contractile and proliferative. Hence, an overall decrease in redox ratio could also be an indicator of increased macro molecule synthesis for proliferation.20 The clear correlation between reduced redox ratio and increased contractility and proliferative potential in elongated VICs suggests that the redox ratio might be an early indicator of dysfunction in the VIC, and merits future study.
We also observed altered Ki-67 and α-SMA protein expression as a result of the altered actin architecture potentiated by VIC shape change. A number of studies have reported that large scale changes in cell shape increased force transduction and affected nuclear chromatin condensation and subsequent gene expression.15,29 We observed similar responses in our current work. Chromatin density was statistically similar for all ARs. In the presence of actin inhibitor, chromatin density was significantly increased. A recent report on cancer cells did report highest levels of chromatin density in cells undergoing normal growth, while activated cancer cells showed lower levels of chromatin density.30 This same study reported that senescent cells, where there was no proliferation, had the lowest levels of chromatin density.30 Our results thus possibly suggest that elongated cells with more aligned actin filaments were likely to experience pathological activation (i.e. the cell differentiation process in which quiescent VIC exhibited features of myofibroblast). This notion was further supported by our western blot which showed the increased expression of α-SMA marker as ARs increased. When the cytoskeleton was disrupted, its expression was reduced. Overall, our results underlined the potential role of the cytoskeleton, and the actin network in particular, in acting as an ‘antenna’ for the VIC mechanobiological response – receiving external signals, processing and transducing downstream signals to the cell nucleus to exert appropriate responses.
The following limitations are to be noted. We have subjected a single VIC to different width-to-length ARs to study the correlation between cell elongation and its function. Our model does not recapitulate cell–cell contact, or the three-dimensional environment that exists within the valve interstitial milieu. However, as mentioned previously, being able to comprehend VIC behavior at its fundamental, functional, single-cell unit is an important first step toward understanding valve physiology as well as pathology. Our model also does not directly apply mechanical forces onto the VICs. However, we demonstrate (Fig. S1, ESI†) that the ARs employed in our study (1:3, 1:5, 1:7) are a reasonable approximation of the steady-state shape achieved by VICs that are cyclically stretched to 0%, 10% and 20%, respectively.
In summary, we demonstrate the strong correlation between VIC shape and its contractile and metabolic function. VIC shape change could be thus seen as an indication of potential changes in the cell activity and vice versa. We also propose the possibility of using redox ratio as one of the tools for early detection of the pathological state of VICs.
Materials and methods
Valve interstitial cell isolation
Fresh porcine hearts (3–6 months old) were obtained from Cockrum's Custom Meat Processing (Rudy, AR) and transported to our laboratory in cold Dulbecco's Phosphate Buffered Saline (dPBS; ThermoFisher, Waltham MA) supplemented with 1% antibiotic/antimycotic solution. Hearts were immediately dissected aseptically to reveal the aortic valve leaflets. Left, right and non-coronary leaflets were pooled and washed 2 times in Hank's Balanced Salt Solution (HBSS; ThermoFisher). Cells were isolated using collagenase digestion as described in previous protocols.31 Briefly, valve endothelial cells (VECs) were removed by incubating the leaflets in collagenase solution (60 U mL−1) for 30 min and discarding the digestate. Valve interstitial cells (VICs) were then isolated by incubation the leaflets in collagenase solution (60 U mL−1) at 37 °C for 2 hours with frequent agitation. The digestate was spun down in centrifuge at 250 g for 5 minutes at 4 °C and resuspended and cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 50 U mL−1 penicillin, 50 U mL−1 streptomycin, and 10 mM HEPES (all ThermoFisher). Fresh DMEM was exchanged every two to three days. Cells from passages 2–7 were used in all subsequent experiments.Photolithography
Photolithographic transparency masks were designed in AutoCAD (Autodesk Inc., San Rafael CA) and fabricated by CAD/Art Services Inc. (Bandon, OR). The design of the masks for the single cell studies consisted of rectangular features of constant surface area spaced 50 μm apart, with variable width-to-length aspect ratios (1:3 [23.80 × 71.41 μm]; 1:5 [18.43 × 92.19 μm]; 1:7 [15.58 × 109.09 μm]) in rectangular grids. The determination of the dimensions of the features is outlined in the ESI.† These aspect ratios represented the AR of cells that underwent 0% (undeformed), 10% (normal physiological stretch), and 20% (elevated) stretch, respectively.5 Each grid contained 12500 single cell features. Silicon wafers (Wafer World, West Palm Beach FL) were then spin coated with Su-8 2005 negative photoresist (MicroChem Corp, Westborough MA) and exposed to ultraviolet light through the photomask and developed using standard photolithography protocols.32 Silicon wafers were then subsequently silanized with tridecafluoro-1,1,2,2-tetrahydrooctyl-1-trichlorosilane (United Chemical Corp, Filmore CA) overnight in a vacuum dessicator.
Microcontact printing and cell culture
Polydimethylsiloxane (PDMS; Dow Corning, Midland MI) was poured over the above silicon wafer and allowed to cure for 3 hours at 70 °C. PDMS stamps with the desired features were then cut out. 50 μg mL−1 Fibronectin (Corning, Corning NY) was incubated on the PDMS stamps with aforementioned microscale raised rectangular features for 1 hour at room temperature and blown dry gently. The stamp was then placed in contact with ultraviolet-ozone treated PDMS-coated coverslips for 5 minutes, thereby transferring the pattern. Coverslips were then blocked for 10 minutes with 1% Pluronics F-127 (Sigma) to prevent non-specific cell binding onto the PDMS substrate. The coverslips were washed three times with dPBS and VICs were seeded at 1000 cells per cm2 coverslip area and cultured at 37 °C, 5% CO2. The justification for the seeding density is outlined in the ESI.† For samples that were treated with cytochalasin D, 0.1 μM of the drug was added to the cell 24 hours before terminating for sample processing.Fluorescent staining of actin and nucleus
After 48 hours of culture, cells were fixed with 4% paraformaldehyde (PFA), permeabilized using 0.5% Triton X-100 and stained for F-actin using Alexa Fluor 488 phalloidin (1:100; ThermoFisher) and for nuclei using 25 μg mL−1 4′,6-diamidino-2-phenylindole (DAPI; ThermoFisher) for 1 hour at room temperature. Samples were then mounted onto a glass slide using Prolong Gold mounting medium (ThermoFisher), allowed to dry overnight and imaged using a Nikon Ti epifluorescence microscope.
Analysis of actin and nuclear architecture
Actin alignment was quantified using a custom-written MATLAB script, and reported using the Orientation parameter (OP), where an OP of 1 indicated perfect anisotropy and an OP of zero indicated isotropy of the actin filaments.14a To characterize the 3D nuclear size and morphology, image volumes of DAPI-stained samples were acquired using a commercial multiphoton microscope (Ultima Investigator, Bruker Corp.) with a water immersion 20× objective (1.0 NA). The two-photon excitation wavelength was 755 nm, and emission was collected by a non-descanned detector with a 460(±20) nm bandpass emission filter. Digital voxel resolution was 0.5 μm in the axial and lateral resolution. Laser power and detector gain were initially optimized to prevent photobleaching and pixel saturation and remained consistent throughout the experiment. Using MATLAB, an intensity threshold of 250 was applied to the 13-bit intensity images to produce a nuclear mask from each image volume. Individual nuclei were identified based on pixel connectivity, and nuclear volume as well as average intensity within each nucleus were computed. Finally, a maximum intensity z-projection of the image volume was obtained and the aspect ratio was computed from the major and minor axes of an ellipse fit to each nucleus using the regionprops.m function.Traction force microscopy and analysis
VICs were seeded on 9 kPa polyacrylamide gels for traction force microscopy (TFM) using methods published by others.15,33 Polyacrylamide gels were prepared by mixing 40% acrylamide and 2% bis-acrylamide stock solutions with 2% v/v N-hydroxyethylacrylamide and 4% v/v Alexa Fluore 488-conjugated fluorescent beads (0.2 μm fluorospheres, ThermoFisher) in 50 mM HEPES. This mixture was sonicated for 20 minutes, followed by the addition of ammonium persulfate and tetramethylethylenediamine (TEMED) to allow polymerization. 15 μL of this mixture was pipetted onto silane-activated 35 mm coverslips.33 A clean 18 mm coverslip was put on top of the droplet to create a flat gel. This gel-coverslip was UV–O treated for 10 minutes before microcontact printing and cell seeding was performed as outlined earlier. Samples treated with cytochalasin D were cultured as mentioned earlier. At the end of the culture period, the sample was transferred to a heated imaging chamber (Warner Instruments, Hamden CT) and incubated in Tyrode's buffer (Sigma). Fluorescent images of the beads immediately beneath the patterned cells were recorded using the previously mentioned two-photon microscope with a 960 nm excitation wavelength and a 525 nm/45 nm emission filter. The change in the position of the fluorescent beads was quantified using a MATLAB script using an algorithm described by others.14b,15,34 Cell contractility was quantified by VIC response to 50 nM ET-1 (Sigma) after 5 minutes. VIC basal tone was quantified by VIC response to a 100 μM saturating dose of the rho-activated kinase inhibitor and vasodilator HA-1077 after 5 minutes. Tractions were calculated from the displacement field using a Fourier Transform method with mechanical properties of the gel assumed to be known (Young's Modulus = 9 kPa; Poisson's ratio = 0.5).34 As the traction stresses were mostly confined to the ends of the cell, each cell was then longitudinally divided into five equal sections and the mean traction stresses within the sections at the ends of the cell were calculated and reported.Two-photon redox imaging
For redox imaging, VIC samples were placed in a heated imaging chamber as before and imaged using a custom-built resonant-scanning multiphoton microscopy platform with a 40×, 0.8 NA water immersion objective (Nikon, Japan) and a MaiTai ultrafast Ti:Sapphire tunable laser source (Spectra-Physics, Santa Clara CA). The laser excitation source was tuned to 755 nm (NADH fluorescence) or 860 nm (FAD fluorescence) and images were acquired via non-descanned detectors with 460 nm/40 nm (NADH) or 525 nm/45 nm (FAD) bandpass filters, respectively. Laser power was kept constant at 30 mW at the sample plane and photomultiplier tube (PMT) gain was fixed at 90%. Images of a cuvette filled with 4 ng mL−1 Rhodamine B (Sigma) were acquired via a 607 nm/70 nm bandpass filter, under identical conditions for both NADH and FAD acquisitions, to account for possible day-to-day variation in laser intensity. Images were analyzed using a custom MATLAB script and redox ratio was calculated using the following equation on a per pixel basis:35[FAD] represents intensity of the FAD image normalized by the corresponding rhodamine intensity. [NADH] represents intensity of the NADH image normalized by the corresponding rhodamine intensity.
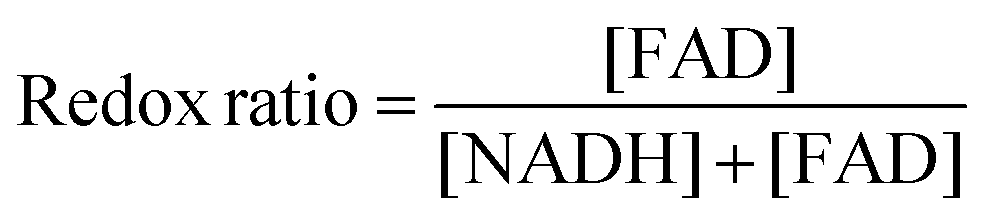
Immunofluorescence and western blotting for proliferation and activation markers
For Ki-67 immunostaining, cells were fixed and permeabilized with 4% PFA and 0.5% Triton-X100 for 10 minutes. An equal mixture of 5% bovine serum albumin (BSA; Sigma) and goat serum (Abcam, Cambridge United Kingdom) were used to block non-specific binding for 2 hours at room temperature. The cells were then incubated with Ki-67 antibody (Abcam) at a dilution of 1:500 and kept at 4 °C overnight. Alexa Fluor 488-conjugated goat-anti-rabbit antibody (1:200; ThermoFisher) and 50 μg mL−1 DAPI were applied to the cells and incubated for 1 hour before mounting the coverslips onto a glass slide using Prolong Gold antifade reagent for imaging.
For western blotting, cells were lysed with urea lysis buffer.14a Cell lysate was centrifuged at 10000 rpm at 4 °C and the supernatant was collected and quantified by BCA assay. 10 μg of total protein was loaded onto each well of a 4–15% polyacrylamide gel (Bio-Rad, Hercules CA) and separated by electrophoresis at 150 V. Proteins were then transferred to a polyvinyldifluoride (PVDF, EMD Millipore, Billerica MA) membrane, blocked in blocking buffer (Li-Cor, Lincoln NE) for 2 hours before probing with rabbit total ERK1/2 antibody (Cell Signaling Technology, Danvers MA; 1:50) and mouse phosphorylated ERK1/2 (Cell Signaling Technology; 1:500), rabbit anti-α-SMA (Abcam, 1:200), mouse anti-vimentin (Abcam, 1:1000), mouse anti-SM-MHC (Millipore, 1:20) or mouse β-actin (Abcam, 1:500). The membranes were kept at 4 °C overnight. Appropriate secondary antibodies (Li-Cor; 1:15000) were subsequently added to the membranes and incubated for 1 hour with gentle shaking, followed by washing with dPBS and distilled-deionized water. Membranes were then scanned using a Licor Odyssey scanner. Protein expression was quantified by obtaining the band intensity for the proteins of interest and normalizing it with the intensity obtained for β-actin protein or ERK1/2 protein in case of pERK1/2.
Statistical analysis
All data were first analyzed for normality using the Anderson–Darling method. All normally distributed data were subsequently analyzed by one- or two-way ANOVA followed by Holm–Sidak multiple pairwise comparisons. Data not normally distributed were analyzed using Mann–Whitney non-parametric methods. Unless otherwise specified, a p-value of less than 0.05 was used to indicate statistical significance. Data was plotted as mean with standard error bars. All results were from a sample size of at least 4 or more separate experiments.Acknowledgements
We acknowledge financial support from the Arkansas Biosciences Institute Grant 000943, the American Heart Association Predoctoral Fellowship 15PRE25710370 (NTL), the Vietnam Education Foundation Fellowship (NTL), the National Science Foundation under grant number CMMI-1452943 (KB), and the National Institutes of Health under grant number R00EB017723-01 (KPQ). We would like to thank the University of Arkansas High Density Electronics Center (HiDEC) and their staff for their technical assistance with the microfabrication work.References
- K. Maganti, V. H. Rigolin, M. E. Sarano and R. O. Bonow, Valvular heart disease: diagnosis and management, Mayo Clin. Proc., 2010, 85, 483–500, DOI:10.4065/mcp.2009.0706.
- (a) K. Balachandran, P. Sucosky and A. P. Yoganathan, Hemodynamics and mechanobiology of aortic valve inflammation and calcification, Int. J. Inflammation, 2011, 2011, 263870, DOI:10.4061/2011/263870; (b) S. T. Gould, S. Srigunapalan, C. A. Simmons and K. S. Anseth, Hemodynamic and cellular response feedback in calcific aortic valve disease, Circ. Res., 2013, 113, 186–197, DOI:10.1161/CIRCRESAHA.112.300154.
- (a) K. Balachandran, P. Sucosky, H. Jo and A. P. Yoganathan, Elevated cyclic stretch alters matrix remodeling in aortic valve cusps: implications for degenerative aortic valve disease, Am. J. Physiol.: Heart Circ. Physiol., 2009, 296, H756–H764, DOI:10.1152/ajpheart.00900.2008; (b) K. Balachandran, P. Sucosky, H. Jo and A. P. Yoganathan, Elevated cyclic stretch induces aortic valve calcification in a bone morphogenic protein-dependent manner, Am. J. Pathol., 2010, 177, 49–57, DOI:10.2353/ajpath.2010.090631 , ajpath.2010.090631 [pii]; (c) J. N. Warnock, S. C. Burgess, A. Shack and A. P. Yoganathan, Differential immediate-early gene responses to elevated pressure in porcine aortic valve interstitial cells, J. Heart valve Dis., 2006, 15, 34–41 Search PubMed , discussion 42; (d) W. D. Merryman, I. Youn, H. D. Lukoff, P. M. Krueger, F. Guilak, R. A. Hopkins and M. S. Sacks, Correlation between heart valve interstitial cell stiffness and transvalvular pressure: implications for collagen biosynthesis, Am. J. Physiol.: Heart Circ. Physiol., 2006, 290, H224–H231 CrossRef CAS PubMed; (e) C. Y. Yip, J. H. Chen, R. Zhao and C. A. Simmons, Calcification by Valve Interstitial Cells Is Regulated by the Stiffness of the Extracellular Matrix, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 936–942, DOI:10.1161/ATVBAHA.108.182394 [pii].
- H. Y. Huang, J. Liao and M. S. Sacks, In situ deformation of the aortic valve interstitial cell nucleus under diastolic loading, J. Biomech. Eng., 2007, 129, 880–889, DOI:10.1115/1.2801670.
- M. S. Sacks, W. David Merryman and D. E. Schmidt, On the biomechanics of heart valve function, J. Biomech., 2009, 42, 1804–1824, DOI:10.1016/j.jbiomech.2009.05.015 , S0021-9290(09)00265-6 [pii].
- C. H. Yap, H. S. Kim, K. Balachandran, M. Weiler, R. Haj-Ali and A. P. Yoganathan, Dynamic deformation characteristics of porcine aortic valve leaflet under normal and hypertensive conditions, Am. J. Physiol.: Heart Circ. Physiol., 2010, 298, H395–H405, DOI:10.1152/ajpheart.00040.2009 , 00040.2009 [pii].
- C. H. Lee, C. A. Carruthers, S. Ayoub, R. C. Gorman, J. H. Gorman, 3rd and M. S. Sacks, Quantification and simulation of layer-specific mitral valve interstitial cells deformation under physiological loading, J. Theor. Biol., 2015, 373, 26–39, DOI:10.1016/j.jtbi.2015.03.004.
- M. L. McCain and K. K. Parker, Mechanotransduction: the role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function, Pflugers Arch., 2011, 462, 89–104, DOI:10.1007/s00424-011-0951-4.
- (a) L. Gao, R. McBeath and C. S. Chen, Stem cell shape regulates a chondrogenic versus myogenic fate through Rac1 and N-cadherin, Stem Cells, 2010, 28, 564–572, DOI:10.1002/stem.308; (b) K. A. Kilian, B. Bugarija, B. T. Lahn and M. Mrksich, Geometric cues for directing the differentiation of mesenchymal stem cells, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4872–4877, DOI:10.1073/pnas.0903269107.
- T. Harkness, J. D. McNulty, R. Prestil, S. K. Seymour, T. Klann, M. Murrell, R. S. Ashton and K. Saha, High-content imaging with micropatterned multiwell plates reveals influence of cell geometry and cytoskeleton on chromatin dynamics, Biotechnol. J., 2015, 10, 1555–1567, DOI:10.1002/biot.201400756.
- (a) R. G. Thakar, Q. Cheng, S. Patel, J. Chu, M. Nasir, D. Liepmann, K. Komvopoulos and S. Li, Cell-shape regulation of smooth muscle cell proliferation, Biophys. J., 2009, 96, 3423–3432, DOI:10.1016/j.bpj.2008.11.074; (b) P. W. Alford, A. P. Nesmith, J. N. Seywerd, A. Grosberg and K. K. Parker, Vascular smooth muscle contractility depends on cell shape, Integr. Biol., 2011, 3, 1063–1070, 10.1039/c1ib00061f; (c) Z. Win, G. D. Vrla, K. E. Steucke, E. N. Sevcik, E. S. Hald and P. W. Alford, Smooth muscle architecture within cell-dense vascular tissues influences functional contractility, Integr. Biol., 2014, 6, 1201–1210, 10.1039/c4ib00193a.
- (a) T. Yeung, P. C. Georges, L. A. Flanagan, B. Marg, M. Ortiz, M. Funaki, N. Zahir, W. Ming, V. Weaver and P. A. Janmey, Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion, Cell Motil. Cytoskeleton, 2005, 60, 24–34, DOI:10.1002/cm.20041; (b) O. Chaudhuri, L. Gu, M. Darnell, D. Klumpers, S. A. Bencherif, J. C. Weaver, N. Huebsch and D. J. Mooney, Substrate stress relaxation regulates cell spreading, Nat. Commun., 2015, 6, 6364, DOI:10.1038/ncomms7365.
- A. C. Liu and A. I. Gotlieb, Characterization of cell motility in single heart valve interstitial cells in vitro, Histol. Histopathol., 2007, 22, 873–882 CAS.
- (a) K. Balachandran, P. W. Alford, J. Wylie-Sears, J. A. Goss, A. Grosberg, J. Bischoff, E. Aikawa, R. A. Levine and K. K. Parker, Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19943–19948, DOI:10.1073/pnas.1106954108; (b) G. J. Ye, Y. Aratyn-Schaus, A. P. Nesmith, F. S. Pasqualini, P. W. Alford and K. K. Parker, The contractile strength of vascular smooth muscle myocytes is shape dependent, Integr. Biol., 2014, 6, 152–163, 10.1039/c3ib40230d.
- M. Versaevel, T. Grevesse and S. Gabriele, Spatial coordination between cell and nuclear shape within micropatterned endothelial cells, Nat. Commun., 2012, 3, 671, DOI:10.1038/ncomms1668.
- K. N. Dahl, A. J. Ribeiro and J. Lammerding, Nuclear shape, mechanics, and mechanotransduction, Circ. Res., 2008, 102, 1307–1318, DOI:10.1161/CIRCRESAHA.108.173989.
- H. Lee, W. J. Adams, P. W. Alford, M. L. McCain, A. W. Feinberg, S. P. Sheehy, J. A. Goss and K. K. Parker, Cytoskeletal prestress regulates nuclear shape and stiffness in cardiac myocytes, Exp. Biol. Med., 2015, 240, 1543–1554, DOI:10.1177/1535370215583799.
- (a) C. C. DuFort, M. J. Paszek and V. M. Weaver, Balancing forces: architectural control of mechanotransduction, Nat. Rev. Mol. Cell Biol., 2011, 12, 308–319, DOI:10.1038/nrm3112; (b) P. W. Oakes, S. Banerjee, M. C. Marchetti and M. L. Gardel, Geometry regulates traction stresses in adherent cells, Biophys. J., 2014, 107, 825–833, DOI:10.1016/j.bpj.2014.06.045; (c) J. T. Parsons, A. R. Horwitz and M. A. Schwartz, Cell adhesion: integrating cytoskeletal dynamics and cellular tension, Nat. Rev. Mol. Cell Biol., 2010, 11, 633–643, DOI:10.1038/nrm2957.
- I. Georgakoudi and K. P. Quinn, Optical imaging using endogenous contrast to assess metabolic state, Annu. Rev. Biomed. Eng., 2012, 14, 351–367, DOI:10.1146/annurev-bioeng-071811-150108.
- K. P. Quinn, G. V. Sridharan, R. S. Hayden, D. L. Kaplan, K. Lee and I. Georgakoudi, Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation, Sci. Rep., 2013, 3, 3432, DOI:10.1038/srep03432.
- K. P. Quinn, E. C. Leal, A. Tellechea, A. Kafanas, M. E. Auster, A. Veves and I. Georgakoudi, Diabetic Wounds Exhibit Distinct Microstructural and Metabolic Heterogeneity through Label-Free Multiphoton Microscopy, J. Invest. Dermatol., 2016, 136, 342–344, DOI:10.1038/JID.2015.371.
- E. Rabkin, S. P. Hoerstrup, M. Aikawa, J. E. Mayer, Jr. and F. J. Schoen, Evolution of cell phenotype and extracellular matrix in tissue-engineered heart valves during in vitro maturation and in vivo remodeling, J. Heart Valve Dis., 2002, 11, 308–314 Search PubMed , discussion 314.
- S. Chaterji, P. Kim, S. H. Choe, J. H. Tsui, C. H. Lam, D. S. Ho, A. B. Baker and D. H. Kim, Synergistic effects of matrix nanotopography and stiffness on vascular smooth muscle cell function, Tissue Eng., Part A, 2014, 20, 2115–2126, DOI:10.1089/ten.tea.2013.0455.
- R. Vishavkarma, S. Raghavan, C. Kuyyamudi, A. Majumder, J. Dhawan and P. A. Pullarkat, Role of actin filaments in correlating nuclear shape and cell spreading, PLoS One, 2014, 9, e107895, DOI:10.1371/journal.pone.0107895.
- (a) F. Guilak, J. R. Tedrow and R. Burgkart, Viscoelastic properties of the cell nucleus, Biochem. Biophys. Res. Commun., 2000, 269, 781–786, DOI:10.1006/bbrc.2000.2360; (b) F. Guilak, Compression-induced changes in the shape and volume of the chondrocyte nucleus, J. Biomech., 1995, 28, 1529–1541 CrossRef CAS PubMed.
- A. J. Maniotis, C. S. Chen and D. E. Ingber, Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 849–854 CrossRef CAS.
- H. R. Kim, C. Gallant, P. C. Leavis, S. J. Gunst and K. G. Morgan, Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent, Am. J. Physiol.: Cell Physiol., 2008, 295, C768–C778, DOI:10.1152/ajpcell.00174.2008.
- (a) M. C. Skala, K. M. Riching, A. Gendron-Fitzpatrick, J. Eickhoff, K. W. Eliceiri, J. G. White and N. Ramanujam, In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19494–19499, DOI:10.1073/pnas.0708425104; (b) A. Varone, J. Xylas, K. P. Quinn, D. Pouli, G. Sridharan, M. E. McLaughlin-Drubin, C. Alonzo, K. Lee, K. Munger and I. Georgakoudi, Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues, Cancer Res., 2014, 74, 3067–3075, DOI:10.1158/0008-5472.CAN-13-2713.
- D. Zink, A. H. Fischer and J. A. Nickerson, Nuclear structure in cancer cells, Nat. Rev. Cancer, 2004, 4, 677–687, DOI:10.1038/nrc1430.
- J. H. Oh, A. Gertych and J. Tajbakhsh, Nuclear DNA methylation and chromatin condensation phenotypes are distinct between normally proliferating/aging, rapidly growing/immortal, and senescent cells, Oncotarget, 2013, 4, 474–493, DOI:10.18632/oncotarget.942.
- (a) J. T. Butcher and R. M. Nerem, Porcine aortic valve interstitial cells in three-dimensional culture: comparison of phenotype with aortic smooth muscle cells, J. Heart Valve Dis., 2004, 13, 478–485 Search PubMed , discussion 485–486; (b) R. A. Gould and J. T. Butcher, Isolation of valvular endothelial cells, J. Visualized Exp., 2010, 29, 2158, DOI:10.3791/2158 [pii].
- (a) R. S. Kane, S. Takayama, E. Ostuni, D. E. Ingber and G. M. Whitesides, Patterning proteins and cells using soft lithography, Biomaterials, 1999, 20, 2363–2376 CrossRef CAS PubMed; (b) D. Qin, Y. Xia and G. M. Whitesides, Soft lithography for micro- and nanoscale patterning, Nat. Protoc., 2010, 5, 491–502, DOI:10.1038/nprot.2009.234.
- M. Versaevel, T. Grevesse, M. Riaz, J. Lantoine and S. Gabriele, Micropatterning hydroxy-PAAm hydrogels and Sylgard 184 silicone elastomers with tunable elastic moduli, Methods Cell Biol., 2014, 121, 33–48, DOI:10.1016/B978-0-12-800281-0.00003-8.
- J. P. Butler, I. M. Tolic-Norrelykke, B. Fabry and J. J. Fredberg, Traction fields, moments, and strain energy that cells exert on their surroundings, Am. J. Physiol.: Cell Physiol., 2002, 282, C595–C605, DOI:10.1152/ajpcell.00270.2001.
- M. Skala and N. Ramanujam, Multiphoton redox ratio imaging for metabolic monitoring in vivo, Methods Mol. Biol., 2010, 594, 155–162, DOI:10.1007/978-1-60761-411-1_11.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ib00120c |
| This journal is © The Royal Society of Chemistry 2016 |