
Assessment of the potential of a high frequency acoustomicrofluidic nebulisation platform for inhaled stem cell therapy†
Layla
Alhasan
abc,
Aisha
Qi
b,
Amgad R.
Rezk
b,
Leslie Y.
Yeo
b and
Peggy P. Y.
Chan
*bd
aDepartment of Biotechnology & Biological Science, RMIT University, Melbourne, Australia
bMicro/Nanophysics Research Laboratory, RMIT University, Melbourne, Australia
cDepartment of Biology, Education Faculty, Thi-Qar University, Thi-Qar, Iraq
dDepartment of Biomedical Engineering, Swinburne University of Technology, Hawthorn, Australia. E-mail: pchan@swin.edu.au; Tel: +61 3 9214 8276
First published on 16th November 2015
Abstract
Despite the promise of stem cell therapy for lung therapeutics and repair, there are few viable means for directly delivering stem cells to locally target the respiratory airways via inhalation. This is not surprising given the significant challenges in aerosolising stem cells, particularly given their susceptibility to damage under the large stresses involved in the nebulisation process. Here, we present promising results using a microfluidic acoustic nebulisation platform that is not only low cost and portable, but also its high MHz order frequencies are effective for preserving the structural and functional integrity of mesenchymal stem cells (MSCs) during the nebulisation process. This is verified through an assessment of the viability, structure, metabolic activity, proliferation ability and genetic makeup of the nebulised MSCs using a variety of assays, including cell viability staining, flow cytometry, reverse transcription and quantitative polymerase chain reaction, and immunophenotyping, thus demonstrating the platform as a promising method for efficient pulmonary stem cell delivery.
Insight, innovation, integrationDespite the promise of stem cell therapy for lung therapeutics and repair, there are few viable means for directly delivering stem cells to locally target the respiratory airways via inhalation. This is not surprising given the significant challenges in aerosolising stem cells, particularly given their susceptibility to damage under the large stresses involved in the nebulisation process. Here, we present promising results using a microfluidic acoustic nebulisation platform, that is not only low cost and portable, but also its high MHz order frequencies are effective for preserving the structural and functional integrity of mesenchymal stem cells (MSCs) during the nebulisation process. This is verified through biological assessments, thus demonstrating the platform as a promising method for efficient pulmonary stem cell delivery. |
Introduction
There is growing evidence that demonstrates the efficacy of stem cell based therapies for the treatment of a variety of lung diseases including chronic obstructive pulmonary disease (COPD), pulmonary and cystic fibrosis, and pulmonary hypertension.1,2 This is especially timely given the recognition of the urgency for new approaches to treat lung diseases in light of their increasing prevalence, in particular, COPD, which is anticipated to be among the top leading causes of global mortality in the next 5 years. Motivated by the shortage in donor lungs and the extremely high mortality rate (almost 50%) following lung transplantation, there are additionally an increasing number of studies examining the potential of stem cell administration to injured lungs, from the viewpoint of both an immodulatory role in attenuating inflammation as well as a regeneratory role in injury repair.3There are however far fewer studies on the development of methods to administer stem cells to the lungs. While there have been investigations to show the recruitment of the cells to the lungs following systemic (e.g., intravenous) administration, these depend critically on many factors.4 Direct lung delivery via aerosol inhalation, on the other hand, not only facilitates local targeting of the disease- or injury-specific region, but also allows easy and painless administration, involves fewer side effects and avoids the need for anaesthesia.
Nevertheless, there remain considerable challenges associated with stem cell aerosolisation, in particular the likelihood that the stem cells would survive and retain their function during the nebulisation process. Their fragility therefore rules out the use of a wide number of commonly used nebulisation techniques, particularly those that subject the aerosols to intense stresses (e.g., shear, cavitation and heat) such as jet (compressed air) and conventional ultrasonic nebulisers.5–7 In contrast, electrohydrodynamic atomisation has been shown to be a viable method for stem cell delivery,8–11 although the necessity for large (kV) DC potentials typically renders the electrospray impractical as a portable consumer inhalation device from a safety viewpoint.
In this work, we demonstrate the use of a novel high frequency acoustic nebulisation platform as an effective aerosolisation technique for inhaled mesenchymal stem cell (MSC) therapy. This is the first study that investigates the viability of stem cells under SAW nebulisation. These surface acoustic waves (SAWs)12,13 operate at sufficiently high frequencies (10–100 MHz order)14 in comparison to conventional bulk ultrasonic nebulisers (10 kHz to 1 MHz)15 such that the vibrational excitation that the therapeutic molecules or cells to be nebulised are subjected to occurs over a much shorter period compared to their hydrodynamic shear relaxation time scales, thus eliminating the risk of macromolecular denaturation or cell lysis.16,17 Additionally, the low powers (on the order of 1 W), which are one to two orders of magnitude less than those required by conventional ultrasonic nebulisers, not only eliminate the possibility of cavitation but also allow the possibility for miniaturised operation using a battery-powered operated portable handheld device.18 While the efficacy of the SAW nebulisation has been demonstrated for proteins (e.g., monoclonal antibodies for lung tumour inhibition19) and nucleic acids (e.g., plasmid DNA for influenza vaccination20), as well as for microfluidic mass spectrometry interfacing21–23 and protein substrate patterning,24 there is yet to be a demonstration of the efficacy of the SAW nebulisation platform for pulmonary stem cell delivery. We note that stem cells have been shown in a previous study to retain their viability and functionality under SAW excitation, but the intensity of the vibration that the cells were subjected to in that study (typically 100 mW order) was far below the critical levels required for nebulisation.25
Materials and methods
Materials
Dulbecco's Modified Eagle Medium (D-MEM) low glucose, GlutaMAX™ supplement, MSC qualified fetal bovine serum (FBS), gentamycin reagent solution, Dulbecco's phosphate-buffered saline (DPBS) without Ca2+ and Mg2+, TrypLE™ select enzyme, DNA-free™ kit, AlamarBlue® cell viability reagent, Quanti-iT™ PicoGreen® dsDNA assay kit, Calcein AM, propidium iodide (PI) and NP40 cell lysis buffer were purchased from Life Technologies Pty. Ltd (Mulgrave, VIC, Australia). Unless otherwise stated, all other chemicals were obtained from Sigma-Aldrich Pty. Ltd (Castle Hill, NSW, Australia).Cell culture
GIBCO® rat MSCs from the bone marrow of Sprague Dawley rats (Life Technologies, Mulgrave, VIC, Australia) were maintained in cell culture medium comprising D-MEM with GlutaMAX™ supplemented with 10% MSC qualified FBS and 5 μg ml−1 of gentamycin reagent solution at 37 °C in a humidified 5% CO2 incubator. Cells were rinsed with DPBS followed by trypsinisation with TrypLE™ after reaching 80% confluence. The harvested cells were rinsed with cell culture medium and reconstituted to a cell suspension solution containing 2 × 106 cells per ml.SAW nebuliser device fabrication and setup
A 0.5 mm thick 127.68° Y-axis rotated, X-axis propagating lithium niobate wafer (Roditi Ltd, London, UK), which constituted the piezoelectric substrate on which the SAWs are generated, was first cleaned according to standard procedures.26 The electrodes were then fabricated on the wafer using standard UV photolithography according to a protocol modified from Qi et al.27 In brief, the clean wafer was sputtered with a 5 nm layer of chromium followed by a 1.5 μm layer comprising 99% aluminium and 1% copper using an electron beam evaporator (Nanochrome II, Intlvac Corp., Niagara Falls, NY, USA). The wafer was then spun-coated with a thin layer (approximately 3 μm) of AZ1512 photoresist (Microchemicals GmBH, Ulm, Germany) followed by exposure to UV through a glass mask. The next step comprised the development of the photoresist layer and wet etching using an aluminium etchant ANPE 5-5-80-10 (Microchemicals GmBH, Ulm, Germany) to generate the interdigital transducer (IDT) electrode patterns, which, in this case, consisted of elliptical single-phase unidirectional transducers (SPUDTs)28 with an electrode finger width and spacing corresponding to a resonant frequency of approximately 30 MHz. The wafer containing the SPUDT was subsequently diced into microchips each with approximately 18 mm length and 12 mm width. A two-dimensional spatial distribution of the surface displacement amplitude near the focal point of the elliptical SPUDT was measured using a laser Doppler vibrometer (LDV) (UHF-120, Polytec GmBH, Waldbronn, Germany) and is shown in Fig. 1a; an in-depth investigation of the surface displacement patterns produced by various IDT designs can be found in the study of Shilton et al.28 | ||
| Fig. 1 (a) Representative two-dimensional spatial distribution of the surface displacement in the focal region of the SPUDT, obtained using laser Doppler vibrometry at an input power of 2.5 W. (b) Average surface displacement as a function of the input power to the device that corresponds to the nebulisation rates tabulated in Table 1. | ||
To affect the nebulisation process in order to generate the aerosol-laden MSCs, the edge of a SAW microchip was partially immersed into the MSC suspension contained in a Petri dish, as depicted in Fig. 2. Rayleigh SAWs were then generated by applying an input oscillating electrical signal to the SPUDT at the resonant frequency using a signal generator (SML01, Rhode & Schwarz, Munich, Germany) and amplifier (10W1000C, Amplifier Research, Souderton, PA, USA). Above, a critical power of 1.5 W, nebulisation ensued and the resultant aerosol mist containing the MSCs was collected for further characterisation.
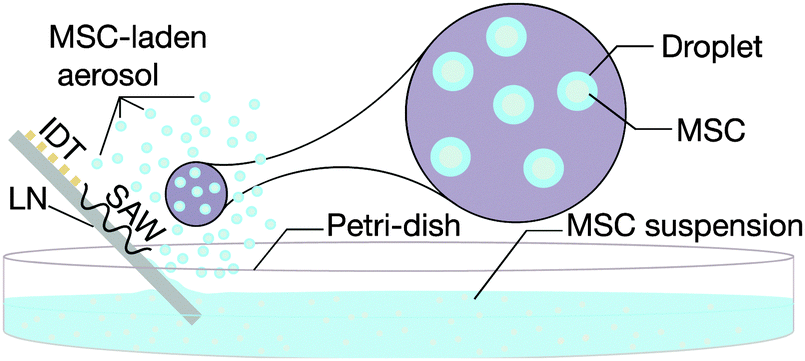 | ||
| Fig. 2 Schematic depiction of the SAW nebulisation setup for rapid aerosolisation of MSCs (not to scale). Power is supplied in the form of an oscillating electrical signal to the SPUDT electrodes photolithographically patterned onto a lithium niobate substrate. This causes the generation of a SAW, which when brought into contact with the MSC suspension in the Petri dish, nebulises the fluid to form an aerosol mist, each droplet of which is laden with the MSCs. | ||
Aerosol characterisation
We first characterised the SAW device to determine its nebulisation rate and the aerosol sizes under ambient conditions. The nebulisation flow rate of the SAW nebuliser device was first tuned by adjusting the input power (1.5–3 W) and the time for a fixed volume of solution to completely nebulise was subsequently measured. Aerosol droplet sizes were determined using laser diffraction (Spraytec, Malvern Instruments, Malvern, UK), specified as a median diameter from a volume-based size distribution; a mean value across 5 replicates was reported.Characterisation of the cell morphology, viability and proliferation
For preliminary testing, the nebulised MSCs were first collected and stained with Trypan blue. Cell counting was performed immediately to quantify the stained and unstained cells using an automated cell counter (Countess®, Life Technologies Pty. Ltd, Mulgrave, VIC Australia).The nebulised MSCs were then transferred to a new tissue culture well-plate at a seeding density of 5 × 103 cells per ml for re-culturing at 2 and 24 h post-nebulisation in order to determine their viability, metabolic activity and proliferation ability. The morphology of the cells was monitored using phase contrast microscopy and images were captured at various time points. The viability of the re-cultured MSCs was examined by staining the cells with Calcein AM and PI according to the manufacturer's instructions. The stained MSCs that were re-cultured for 2 h were visualised using laser scanning confocal microscopy (LSCM; Eclipse Ti, Nikon Instruments Inc., Melville, NY, USA) without further rinsing. The stained MSCs that were re-cultured for 24 h were rinsed using phosphate buffered saline (PBS) before LSCM visualisation.
For quantification, MSCs were nebulised as described above and were collected in a separate experiment. Some of these MSCs were immediately stained with Calcein AM and PI according to the manufacturer's instructions. The rest of the MSCs were transferred to a new tissue culture well-plate at a seeding density of 5 × 103 cells per ml for re-culturing for 24 h. The re-cultured cells were then trypsinised and stained with Calcein AM and PI. Both sets of stained cells were analysed using flow cytometry (FACSCanto™ II, BD Biosciences, North Ryde, NSW, Australia). As a positive control, MSCs were treated with 0.1% Triton X-100 for 2–3 min prior to staining.
The metabolic activity of the re-cultured MSCs was quantified using an AlamarBlue® assay according to Hoo et al.29 In brief, spent medium was removed from each well prior to rinsing with DPBS. AlamarBlue® dye (10%) diluted with cell culture medium was then added into each well and incubated at 37 °C in humidified 5% CO2 for 4 h. Fluorescence analysis was performed using a multi-mode microplate reader with an excitation wavelength of 570 nm and an emission wavelength of 600 nm (SpectraMax® Paradigm, Molecular Devices LLC, Sunnyvale, CA, USA).
The proliferation ability of the nebulised MSCs was determined by quantifying the deoxyribonucleic acid (DNA) content using a Picogreen® assay according to the study of Hoo et al.29 In brief, the MSCs were trypsinised after re-culturing for 1, 3, 5 and 7 days. The cells were then twice washed using cold DPBS followed by centrifugation for 5 min at 1200 rpm. The resultant cell pellet was subsequently collected for lysis using a NP40 cell lysis buffer for 30 min on ice and vortexed every 10 min; the cell lysates were subjected to a freeze-thaw cycle prior to centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C. Each 100 μl of clear lysate was aliquoted in a new 96-well plate and incubated for 5 min with 100 μl of the PicoGreen® reagent. Fluorescence analysis was subsequently performed using a multi-mode microplate reader at an excitation wavelength of 480 nm and an emission wavelength of 520 nm. The number of cells in each sample was determined by correlating the DNA content with a standard curve, obtained using cell lysates containing a known number of MSCs. Non-nebulised cells were used as control for all of the above experiments.
000 rpm for 10 min at 4 °C. Each 100 μl of clear lysate was aliquoted in a new 96-well plate and incubated for 5 min with 100 μl of the PicoGreen® reagent. Fluorescence analysis was subsequently performed using a multi-mode microplate reader at an excitation wavelength of 480 nm and an emission wavelength of 520 nm. The number of cells in each sample was determined by correlating the DNA content with a standard curve, obtained using cell lysates containing a known number of MSCs. Non-nebulised cells were used as control for all of the above experiments.
Reverse-transcription polymerase chain reaction (RT-PCR) and real-time polymerase chain reaction (qPCR)
The gene expression of nebulised MSCs and control MSCs was qualitatively determined using RT-PCR and quantitatively using qPCR with CD29, CD44, and CD106 as markers. Ribonucleic acid (RNA) was isolated from the MSCs using an RNAeasy mini kit (Qiagen, Chadstone, VIC, Australia) according to the manufacturer's instructions. The RNA quality and concentration were examined using a spectrophotometer (ND-1000, NanoDrop Technologies, Thermo Fisher Scientific Inc., Waltham, MA, USA). To eliminate any contaminating genomic DNA, the isolated RNA was treated with DNA-free™ kit according to the manufacturer's protocol as supplied. Complementary DNA (cDNA) generated using a QuantiTect Reverse Transcription kit (Qiagen, Chadstone, VIC, Australia) as per the manufacturer's instructions was subjected to PCR amplification using primers obtained from Geneworks Pty. Ltd (Thebarton, SA, Australia); the primer sequences are listed in Table 1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the housekeeping gene. For RT-PCR, the PCR products were size fractionated using 1.5% agarose gel electrophoresis.The qPCR analysis was performed using a Rotor-Gene Q (Qiagen, Chadstone, VIC, Australia) and a QuantiTect SYBR® Green PCR kit (Qiagen, Chadstone, VIC, Australia). After 10 min of denaturation at 95 °C, 40 PCR cycles were carried out at 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 15 s. The relative mRNA levels were calculated using the ΔΔCT method according to the study of Sarvi et al.30 The results were reported as fold change values compared to the non-nebulised cells.
Immunophenotyping
MSCs were nebulised as described previously and collected for further cultivation. After 24 h of cultivation, the cells were harvested and aliquoted at a concentration of 1 × 106 cells per sample. The cells were then washed with PBS and centrifuged at 1500 rpm three times and subsequently resuspended in buffer (0.1% bovine serum albumin (BSA) in PBS) containing antibodies and incubated on ice for 25–30 min. The MSCs were labelled with either the Alexa Fluor 488® conjugated integrin beta-1/CD29 antibody (Life Technologies, Mulgrave, VIC, Australia), FITC conjugated anti-CD44 antibody (Merck Millipore, Bayswater, VIC, Australia), or PE mouse conjugated anti-rat CD106 antibody (BioLegend, San Diego, CA, USA), followed by labelling with FITC conjugated goat anti-mouse IgG secondary antibodies (Merck Millipore, Mulgrave, VIC, Australia); the antibodies were diluted according to the manufacturer's protocols. The labelled MSCs were then washed twice in buffer (3% BSA in PBS), followed by resuspension in 500 μl of buffer (3% BSA in PBS) and flow cytometry analysis. 10000 events were acquired from each sample, and the results were plotted using flow cytometry data analysis software (Flowing Software, v2.5.1, http://www.flowingsoftware.com). The surface marker expression of the nebulised cells was compared to that of the non-nebulised cells (control).
Statistical analysis
All experiments were performed with at least 3 replicates. Unless otherwise stated, the results were reported as the average value ± standard deviation. Multiple groups of data were compared using one-way analysis of variance (ANOVA), while two groups of data were compared using Student's t-test; data are considered statistically significant when p < 0.05.Results and discussion
Aerosol characterisation
As shown in Fig. 3, the applied SAW power has a direct influence on the nebulisation rate, which increased as the applied power is ramped to approximately 350 μl min−1 at 3 W (p < 0.0001, n = 3); the corresponding surface displacement amplitude of the SAW is plotted in Fig. 1b in which we observe a linear response in the magnitude of the surface displacement with an increase in the power input, indicating that the device has not reached its power saturation limit. In any case, these observations are consistent with previous findings on the effect of SAW irradiation on the nebulisation flow rate, given that more acoustic energy is transmitted into the fluid to drive both streaming and the destabilisation of its interface, which, in turn, leads to its breakage into aerosol droplets that constitute the nebulised mist.18 | ||
| Fig. 3 SAW nebulisation rate (μl min−1) as a function of the applied power to the device. The data are represented in terms of a mean value (n = 3) ± standard error. | ||
Table 2 on the other hand shows the median droplet size of the aerosols containing the MSCs for various input powers to the device. We observe the median aerosol size across all input powers to be just slightly above 13 μm, which is similar to the size of a single MSC, thus indicating that most of the droplets contained a single cell. Given that aerosols with sizes >5 μm are expected to deposit due to inertial impaction in the upper respiratory tract,35,36 we envisage the potential use of the SAW device for inhaled stem cell delivery for tissue repair and regeneration within the extrathoracic (pharyngeal and laryngeal) regions.
| Applied power (W) | Median droplet size (μm) |
|---|---|
| 1.5 | 13.17 ± 0.29 |
| 2.0 | 13.33 ± 0.29 |
| 2.5 | 13.50 ± 0.50 |
| 3.0 | 13.50 ± 0.50 |
Cell morphology and viability
We subsequently conducted a preliminary study to optimise the SAW nebulisation in terms of maximising retention of the cell viability post-nebulisation. Here, the cells were examined using the Trypan blue assay immediately after being nebulised and the cell viability calculated by normalising the number of viable cells in the nebulised samples against the number of viable cells in the untreated samples. As shown by the data in Fig. 4, the number of viable cells decreases with increasing applied power, possibly due to the increase in the stresses that the cells are subjected to with increasing levels of acoustic radiation pressure. This observation is consistent with a previous study on using SAW radiation to drive osteoblast perfusion into tissue scaffolds (although not via nebulisation), in which the cell viability was found to decrease with increasing applied power.37 Nevertheless, it is encouraging to note that the cell viability normalised by that of the control cells remains relatively high—close to 90% even at 1.5 W, which, together with the 100 μl min−1 delivery at this power (Fig. 3), adequate for most stem cell therapeutic applications, demonstrates the potential efficacy of the device as a platform for pulmonary stem cell delivery. | ||
| Fig. 4 Post-nebulisation MSC viability as a function of the power applied to the SAW device, measured immediately after nebulisation using a trypan blue assay. The data are represented in terms of a mean value (n = 3) ± standard error. | ||
To minimise the loss in stem cell viability, the device was henceforth operated at the optimum applied power of 1.5 W. Fig. 5 shows representative LSCM images of the cells stained with Calcein AM and PI after being seeded on a new well-plate and allowed to cultivate for 2 and 24 h. At 2 h post-nebulisation, most of the MSCs were stained by Calcein AM (which appear in green), indicating that they remained viable after nebulisation. We observe these MSCs to have started to attach onto the well-plate. At 24 h post-nebulisation, it can be seen that the nebulised MSCs exhibited spindle-like morphologies similar to the untreated cells, indicating the retention of their structural integrity post-nebulisation. Furthermore, it can be seen that most of the cells were viable and were stained by Calcein AM. This was confirmed by the flow cytometry data wherein the representative dot plot of the Calcein AM intensity against the PI intensity in Fig. 6 shows that the majority of nebulised cells were stained with Calcein AM. Specifically, MSCs nebulised at 1.5 W exhibited a viability of 86.0 ± 4.2% immediately after nebulisation. Nonetheless, it appeared that the growth of the MSCs recovered after re-culturing for another 24 h, at which we observe no significant difference in the cell viability between the nebulised MSCs and untreated MSCs (p = 0.026, n = 4), thus verifying that the SAW nebulisation does not lead to adverse effects on the MSC viability.
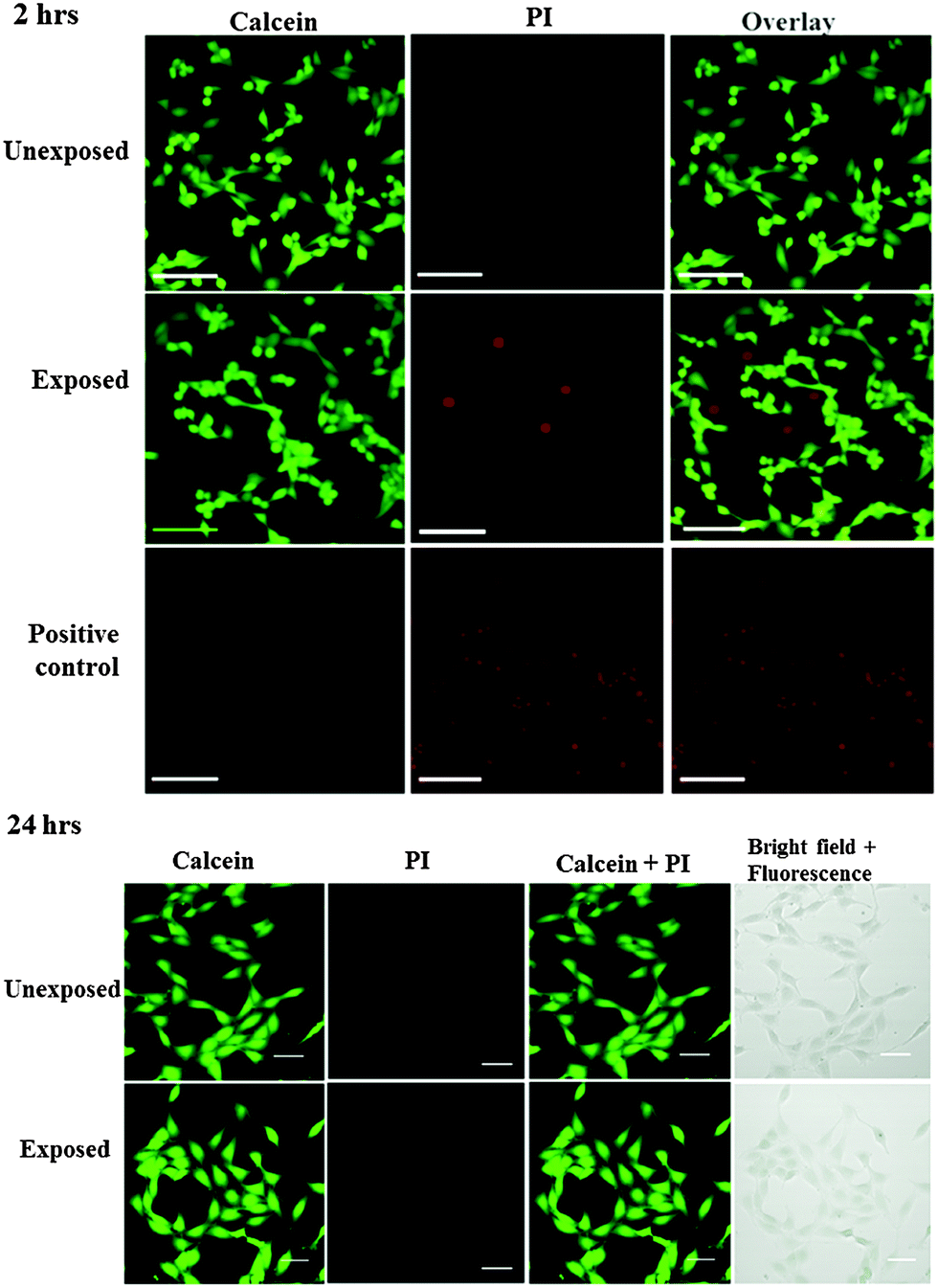 | ||
| Fig. 5 Viability of unexposed and nebulised MSCs, indicated by Calcein AM and PI staining. The cells were examined 2 h (top) and 24 h (bottom) post-nebulisation. Live cells stained by Calcein AM appear green and dead cells stained with PI appeared red. For the positive control, cells were treated with 0.1% Triton X-100 prior to staining. The scale bars represent a length of 50 μm. | ||
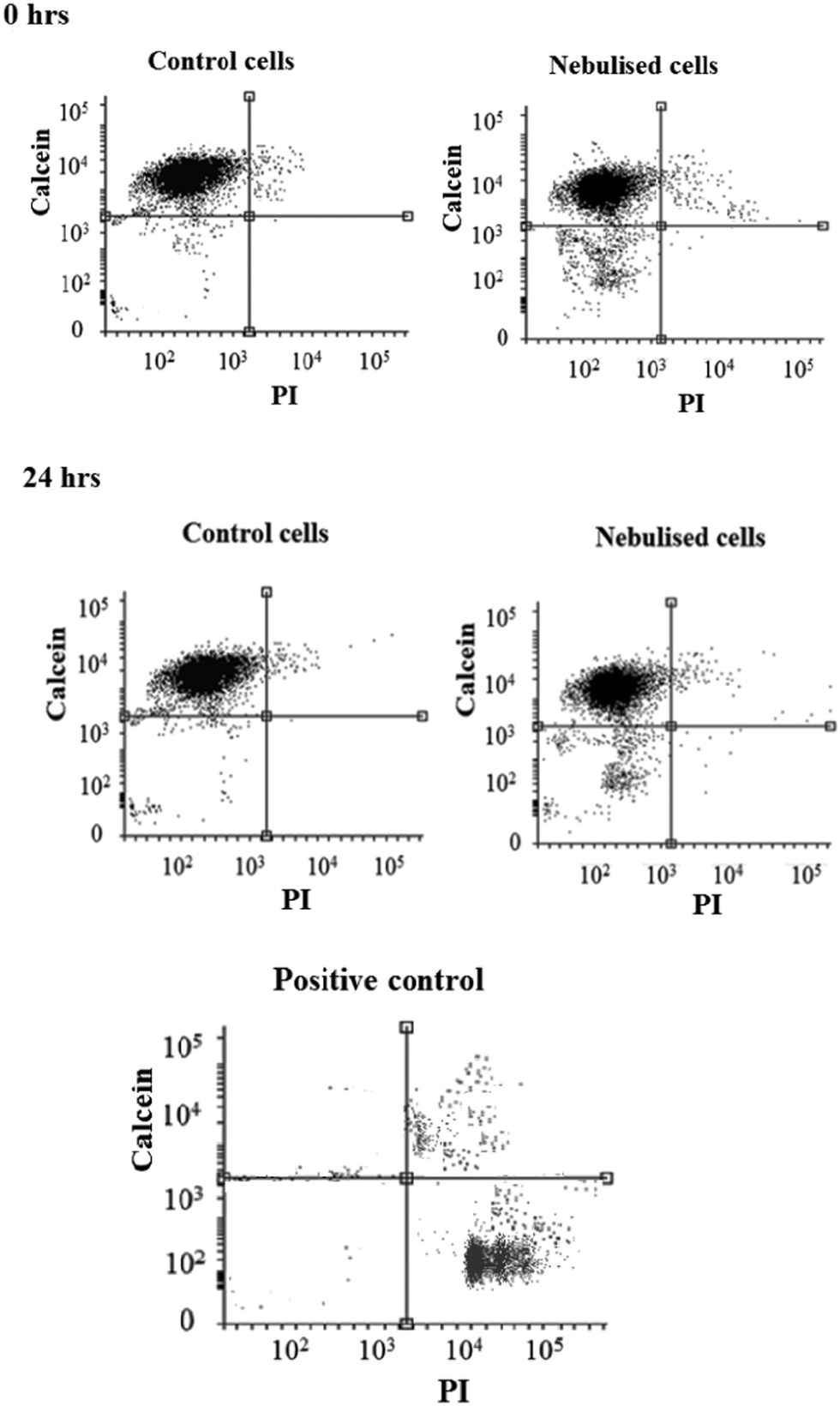 | ||
| Fig. 6 Flow cytometry analysis indicating the viability in the population of unexposed and nebulised MSCs stained with Calcein AM and PI immediately after nebulisation and at 24 h after nebulisation. For positive control, cells were treated with 0.1% Triton X-100 prior to staining. | ||
Cell metabolic activity and proliferation
Nebulised MSCs were seeded in a well-plate and cultured over a period of 7 days to assess their metabolic activity and proliferation ability at different time points compared to the untreated cells (control). As shown in the representative optical microscopy images in Fig. 7, both nebulised and untreated MSCs expanded and occupied more of the substrate area over time. In addition, we find that the morphology of nebulised MSCs remains similar to that of the untreated MSCs.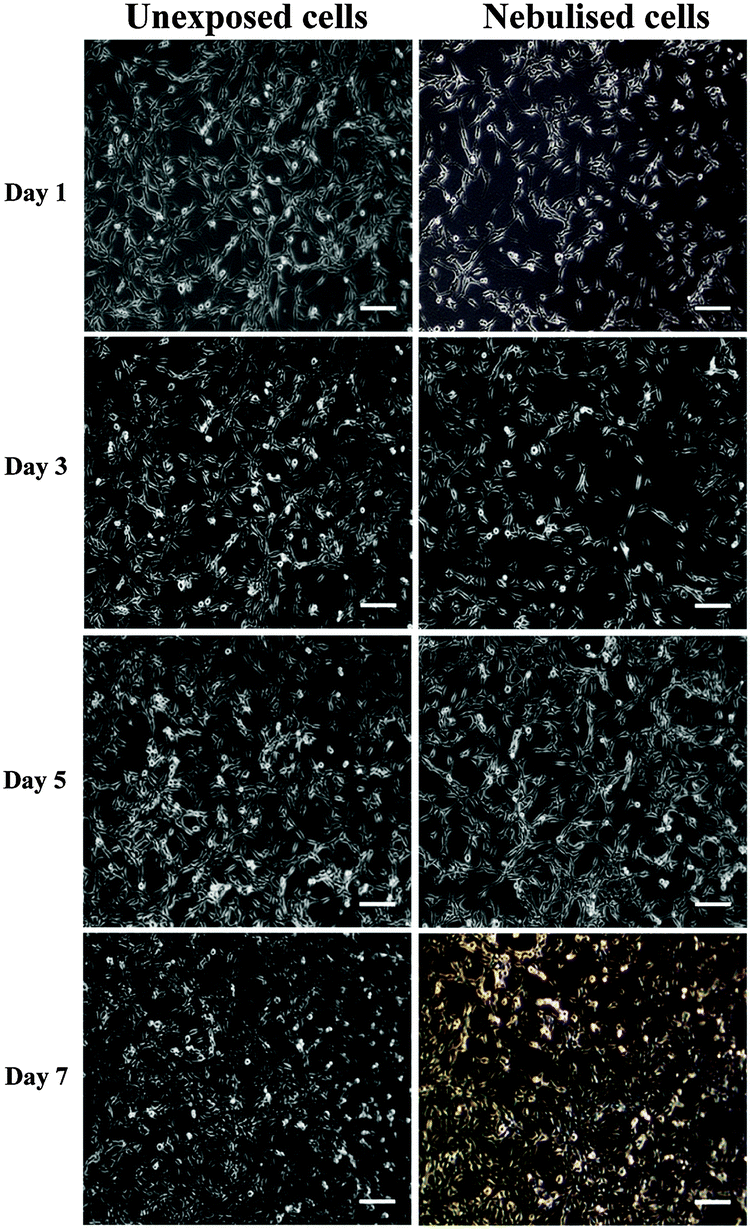 | ||
| Fig. 7 Phase contrast images of unexposed (left column) and nebulised (right column) MSCs acquired at different incubation times. The majority of cells exhibited a spindle-like morphology. Scale bars represent a length of 100 μm. | ||
To quantify the metabolic activity of nebulised cells, we carried out an AlamarBlue® assay wherein the cellular metabolic activity results in the chemical reduction of AlamarBlue®, manifesting as a measurable colour change in the redox indicator in the assay;38 an increase in the amount of reduced AlamarBlue® is therefore an indication of an increase in metabolic activity. Fig. 8 shows the metabolic activity of the nebulised MSCs over a period of 7 days, calculated by normalising the extent of AlamarBlue® reduced by the nebulised cells compared to that reduced by the untreated cells. It can be seen that the metabolic activity of the nebulised MSCs increased over time, regardless of the power at which the cells were nebulised, although we note that the metabolic activity of MSCs nebulised at 1.5 W was found to be the highest amongst the range of applied powers that were tested. This observation is however not surprising, given the decrease in cell viability with increasing power (Fig. 4) which is expected to influence the metabolic rate proportionally.
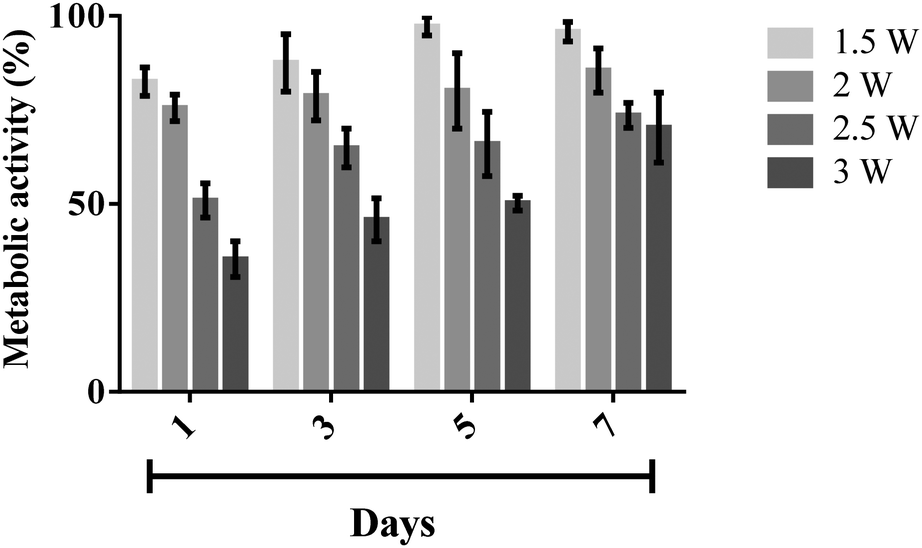 | ||
| Fig. 8 Metabolic activity of nebulised MSCs at day 1, 3, 5, and 7, determined using an AlamarBlue® assay, the results of which were normalised against the activity of the control, i.e., the unexposed cells (100% activity). The data are represented in terms of a mean value (n = 3) ± standard error. | ||
The strong correlation between the number of cells and DNA content allows us to determine the cellular proliferation rate with high accuracy. This was carried out by measuring the DNA content of the MSCs over time using a Quant-iT PicoGreen® dsDNA assay. Fig. 9 shows that the number of MSCs from both the nebulised (p < 0.0001, n = 3) and untreated (p < 0.0001, n = 3) samples gradually increased over a 7 day period. We note that though the nebulised cells proliferated slightly slower from day 5 (p = 0.16, n = 3), nonetheless, the nebulised cells were observed to maintain their proliferation ability throughout the entire test duration.
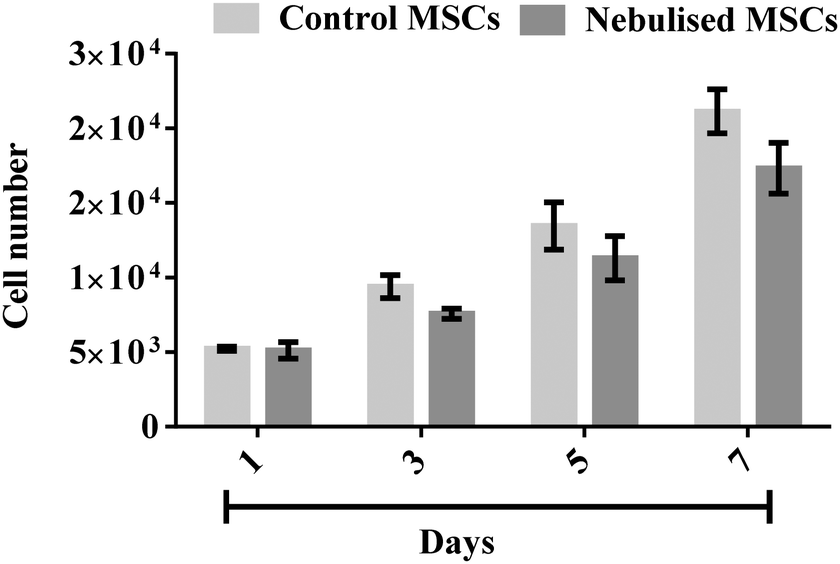 | ||
| Fig. 9 MSC proliferation after nebulisation at 1.5 W. The cell proliferation was determined by quantifying the DNA content using a Quant-iT PicoGreen® dsDNA assay. The data are represented in terms of a mean value (n = 3) ± standard error. | ||
Gene and protein expression
MSCs possess multi-lineage potential to differentiate into a number of cell types including osteocytes, adipocytes and chondrocytes. They are known to express surface markers CD29, CD44, and CD106 positively, with these markers often being considered indicators for MSC isolation as well as differentiation,39–42 and therefore used to assess the pluripotent phenotype and integrity of the MSCs prior to and after nebulisation at different time points. Fig. 10a shows the results from the RT-PCR analysis of the control and nebulised MSCs at 24 and 72 h wherein we observe both to express these markers, revealing that the nebulised cells are phenotypically similar to the control cells. The gene expression of the nebulised MSCs was further quantified using qPCR at different time points as shown in Fig. 10b. Compared to the control, the nebulised MSCs continued to express similar levels of CD29, CD44 and CD106 after 24 h (p = 0.92, n = 4; p = 0.6, n = 4 and p = 0.49, n = 4, respectively) and 72 h (p = 0.5, n = 4; p = 0.65, n = 4; p = 0.98, n = 4, respectively), suggesting that the SAW nebulisation did not lead to any alteration in the gene expression of the MSCs.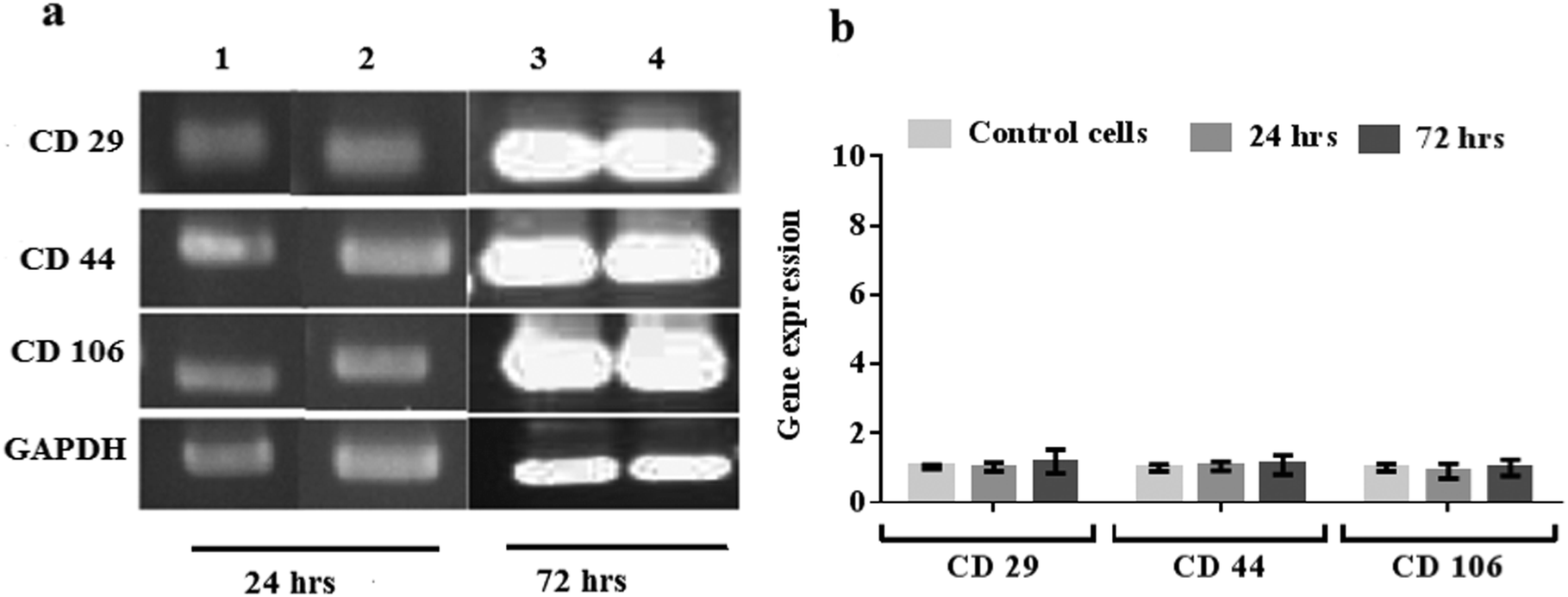 | ||
| Fig. 10 (a) RT-PCR and (b) qPCR analysis of nebulised MSCs, showing the insignificant fold change in the gene expression relative to unexposed MSCs after 24 and 72 h. The error bars indicate the standard deviation of values acquired across the triplicate (n = 3) data. In (a), lanes 1 and 3 represent the data for the unexposed cells (control) whereas lanes 2 and 4 represent those of the nebulised cells. | ||
This is further verified from the results of the flow cytometry analysis employed to determine the protein expression of cells in Fig. 11, which shows the nebulised MSCs having similar CD29, CD44 and CD106 positive cell counts compared to the control after 24 h (p = 0.30, n = 3; p = 0.69, n = 3 and p = 0.16, n = 3, respectively) and 72 h (p = 0.72, n = 3; p = 0.32, n = 3 and p = 0.83, n = 3, respectively). Taken together, the results above therefore confirm that the SAW nebulisation does not cause any detrimental effect to the phenotype and integrity of the MSCs even after taking into account the longer culture period post-nebulisation, thus further verifying the SAW device as a promising platform for stem cell nebulisation.
 | ||
| Fig. 11 Immunophenotypic profile of unexposed (control) and nebulised MSCs after incubation periods of 24 and 72 h. | ||
Conclusions
This study demonstrates the feasibility of generating MSC-laden aerosols using a SAW nebulisation platform without any deleterious effects on the cells. We found that an optimum power level of 1.5 W yielded a balance between maximising cell viability at an acceptable level of approximately 90% (measured after re-culturing for 24 h) whilst maintaining a practically useable delivery rate of 100 μL min−1 for inhalation. This is verified by post-nebulisation assessment of the structure, metabolic activity, proliferation and genetic makeup of the MSCs, which indicate that the SAW nebulisation process does not lead to any significant adverse effects on the cell metabolic rate, proliferation ability, and genotypic and phenotypic characteristics compared to their non-nebulised counterpart. Given the novelty of inhaled stem cell therapy, comparisons with other delivery methods are difficult at present, although we anticipate that more data for benchmarking will become available in the literature in the near future with the increasing interest in stem cell therapy given the absence of viable alternative treatment regimes for respiratory ailments to date. Nonetheless, the results in this work provide compelling evidence that the SAW nebulisation platform, with its inherent benefits such as low cost and portability, constitutes an attractive tool for the delivery of stem cells via inhalation for treatment and repair of lung functions.Acknowledgements
Funding for this work was provided through Australia Research Council Discovery Grants DP120102570 and DP140100805. LA is grateful for a PhD scholarship from the Ministry of Higher Education & Scientific Research/Iraq. ARR is grateful for a RMIT University Vice-Chancellor's Research Fellowship. LYY gratefully acknowledges support from a Future Fellowship from the Australian Research Council under grant FT13010672. The authors acknowledge the help of Dr Dodie Pouniotis (School of Medical Sciences, RMIT University) with the analysis of the flow cytometry data.References
- A. N. Lau, M. Goodwin, C. F. Kim and D. J. Weiss, Mol. Ther., 2012, 20, 1116–1130 CrossRef CAS PubMed.
- B. N. Gomperts and R. M. Strieter, Annu. Rev. Med., 2007, 58, 285–298 CrossRef PubMed.
- S. S. Iyer and M. Rojas, Expert Opin. Biol. Ther., 2008, 8, 569–581 CrossRef CAS PubMed.
- D. J. Weiss, J. K. Kolls, L. A. Ortiz, A. Panoskaltsis-Mortari and D. J. Prockop, Proc. Am. Thorac. Soc., 2008, 5, 637–667 CrossRef PubMed.
- L. Y. Yeo, J. R. Friend, M. P. McIntosh, E. N. T. Meeusen and D. A. V. Morton, Expert Opin. Drug Delivery, 2010, 7, 663–679 CrossRef CAS PubMed.
- K. M. G. Taylor and O. N. M. McCallion, Int. J. Pharm., 1997, 153, 93–104 CrossRef CAS.
- C. Harvey, M. O'Doherty, C. Page, S. Thomas, T. Nunan and D. Treacher, Eur. Respir. J., 1997, 10, 905–909 CAS.
- D. I. Braghirolli, F. Zamboni, P. C. Chagastelles, D. J. Moura, J. Saffi, J. A. P. Henriques, D. A. Pilger and P. Pranke, Biomicrofluidics, 2013, 7, 044130 CrossRef CAS PubMed.
- A. Abeyewickreme, A. Kwok, J. R. McEwan and S. N. Jayasinghe, Integr. Biol., 2009, 1, 260–266 RSC.
- S. Sahoo, W. C. Lee, J. C. H. Goh and S. L. Toh, Biotechnol. Bioeng., 2010, 106, 690–698 CrossRef CAS PubMed.
- C. Ye, Z. He, Y. Lin, Y. Zhang, J. Tang, B. Sun, M. Ma, J. Liu, L. Yang, H. Ren and B. Zhao, Biotechnol. Lett., 2015, 37, 449–456 CrossRef CAS PubMed.
- L. Y. Yeo and J. R. Friend, Annu. Rev. Fluid Mech., 2014, 46, 379–406 CrossRef.
- X. Ding, P. Li, S.-C. S. Lin, Z. S. Stratton, N. Nama, F. Guo, D. Slotcavage, X. Mao, J. Shi, F. Costanzo and T. J. Huang, Lab Chip, 2013, 13, 3626–3649 RSC.
- M. Kurosawa, T. Watanabe, A. Futami and T. Higuchi, Sens. Actuators, A, 1995, 50, 69–74 CrossRef CAS.
- M. P. Flament, P. Leterme and A. Gayot, Drug Dev. Ind. Pharm., 2001, 27, 643–649 CrossRef CAS PubMed.
- A. Qi, L. Yeo, J. Friend and J. Ho, Lab Chip, 2010, 10, 470–476 RSC.
- F. Guo, P. Li, J. B. French, Z. Mao, H. Zhao, S. Li, N. Nama, J. R. Fick, S. J. Benkovic and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 43–48 CrossRef CAS PubMed.
- A. Qi, J. R. Friend, L. Y. Yeo, D. A. V. Morton, M. P. McIntosh and L. Spiccia, Lab Chip, 2009, 9, 2184–2193 RSC.
- C. Cortez-Jugo, A. Qi, A. Rajapaksa, J. R. Friend and L. Y. Yeo, Biomicrofluidics, 2015, 9, 052603 CrossRef PubMed.
- A. Rajapaksa, J. Ho, A. Qi, T.-H. Nguyen, M. Tate, R. Bischof, D. Piedrafta, M. McIntosh, L. Yeo, E. Meeusen, R. Coppel and J. Friend, Respir. Res., 2014, 15, 60 CrossRef PubMed.
- J. Ho, M. K. Tan, D. B. Go, L. Y. Yeo, J. R. Friend and H.-C. Chang, Anal. Chem., 2011, 83, 3260–3266 CrossRef CAS PubMed.
- S. R. Heron, R. Wilson, S. A. Shaffer, D. R. Goodlett and J. M. Cooper, Anal. Chem., 2010, 82, 3985–3989 CrossRef CAS PubMed.
- Y. Huang, S. Yoon, S. Heron, C. Masselon, J. S. Edgar, F. Tureček and D. Goodlett, J. Am. Soc. Mass Spectrom., 2012, 23, 1062–1070 CrossRef CAS PubMed.
- M. Alvarez, J. R. Friend and L. Y. Yeo, Langmuir, 2008, 24, 10629–10632 CrossRef CAS PubMed.
- H. Li, J. Friend, L. Yeo, A. Dasvarma and K. Traianedes, Biomicrofluidics, 2009, 3, 034102 CrossRef PubMed.
- N. R. Glass, R. J. Shilton, P. P. Y. Chan, J. R. Friend and L. Y. Yeo, Small, 2012, 8, 1881–1888 CrossRef CAS PubMed.
- A. Qi, P. Chan, J. Ho, A. Rajapaksa, J. Friend and L. Yeo, ACS Nano, 2011, 5, 9583–9591 CrossRef CAS PubMed.
- R. Shilton, M. K. Tan, L. Y. Yeo and J. R. Friend, J. Appl. Phys., 2008, 104, 014910 CrossRef.
- S. P. Hoo, F. Sarvi, W. H. Li, P. P. Y. Chan and Z. Yue, ACS Appl. Mater. Interfaces, 2013, 5, 5592–5600 CAS.
- F. Sarvi, K. Jain, T. Arbatan, P. J. Verma, K. Hourigan, M. C. Thompson, W. Shen and P. Chan, Adv. Healthcare Mater., 2015, 4, 77–86 CrossRef CAS PubMed.
- A. Ode, A. Kurtz, K. Schmidt-Bleek, P. Schrade, P. Kolar, F. Buttgereit, K. Lehmann, D. Hutmacher, G. Duda and G. Kasper, Eur. Cells Mater., 2011, 22, 26–42 CAS.
- H. Qin, L.-D. Zhao, J.-H. Sun, L.-L. Ren, W.-W. Guo, H.-Z. Liu, S.-Q. Zhai and S.-M. Yang, Acta Oto-Laryngol., 2011, 131, 1136–1141 CrossRef CAS PubMed.
- H. Y. Lau and M. Bhatia, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 292, G1283–G1292 CrossRef CAS PubMed.
- S. Schäfer, A.-G. Calas, M. Vergouts and E. Hermans, J. Neuroimmunol., 2012, 249, 40–48 CrossRef PubMed.
- J. C. Sung, B. L. Pulliam and D. A. Edwards, Trends Biotechnol., 2007, 25, 563–570 CrossRef CAS PubMed.
- J. Heyder, J. Gebhart, G. Rudolf, C. F. Schiller and W. Stahlhofen, J. Aerosol Sci., 1986, 17, 811–825 CrossRef.
- H. Li, J. Friend, L. Yeo, A. Dasvarma and K. Traianedes, Biomicrofluidics, 2009, 3, 034102 CrossRef PubMed.
- G. K. Das, P. P. Y. Chan, A. Teo, J. S. C. Loo, J. M. Anderson and T. T. Y. Tan, J. Biomed. Mater. Res., Part A, 2010, 93, 337–346 Search PubMed.
- L. d. S. Meirelles and N. B. Nardi, Br. J. Haematol., 2003, 123, 702–711 CrossRef.
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143–147 CrossRef CAS PubMed.
- Y. A. Romanov, A. N. Darevskaya, N. V. Merzlikina and L. B. Buravkova, Bull. Exp. Biol. Med., 2005, 140, 138–143 CrossRef CAS PubMed.
- D. A. De Ugarte, K. Morizono, A. Elbarbary, Z. Alfonso, P. A. Zuk, M. Zhu, J. L. Dragoo, P. Ashjian, B. Thomas, P. Benhaim, I. Chen, J. Fraser and M. H. Hedrick, Cells Tissues Organs, 2003, 174, 101–109 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00206k |
| This journal is © The Royal Society of Chemistry 2016 |